
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom CdPb8(SeO3)4Br10 to Pb3(TeO3)Br4: the first tellurite bromide exhibiting an SHG response and mid-IR transparency†
Peng-Fei
Li
 ab,
Chun-Li
Hu
a,
Bing-Xuan
Li
a,
Jiang-Gao
Mao
ab and
Fang
Kong
*ab
ab,
Chun-Li
Hu
a,
Bing-Xuan
Li
a,
Jiang-Gao
Mao
ab and
Fang
Kong
*ab
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, P. R. China. E-mail: kongfang@fjirsm.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 1st November 2023
Abstract
Herein, we report two new selenite/tellurite bromide compounds, namely, CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4. The former crystallized in a centrosymmetric space group, while the latter is a non-centrosymmetric compound. Specifically, CdPb8(SeO3)4Br10 demonstrates high thermal stability (425 °C) and a wide optical transparency window (0.33–6.5 μm), while Pb3(TeO3)Br4 exhibits an second harmonic generation response approximately equivalent to that of KDP, appropriate birefringence (0.095@532 nm and 0.066@1064 nm), wide optical transparency window (0.33–6.5 μm) and high thermal stability (522 °C). These results indicated that lead tellurite bromides could be promising nonlinear optical material systems in the mid-IR wavelength.
Introduction
Nonlinear optical (NLO) crystals are widely used in various fields such as frequency conversion, optical modulation, optical switching and limiting, optical sensing, remote sensing, environmental monitoring, and free-space communications.1–6 Second-order nonlinear optical materials require compounds to crystallize in non-centrosymmetric (NCS) space groups.7–10 According to the ICSD database, the probability of obtaining a non-centrosymmetric structure is less than 20%.11–13 So, the rational design of inorganic compounds with non-centrosymmetric symmetry remains a challenging but beneficial task for researchers in chemical and materials science.Selenite and tellurite compounds are important systems for exploring NLO materials.14 The coordination environment of Se4+ and Te4+ is inherently polar due to the presence of stereo-chemically active lone pairs (SALP) of electrons, with oxygen ligands being located on one side of the cations.15,16 Researchers have made significant efforts to enhance the probability of obtaining NCS selenite and tellurite crystals. In this endeavor, one effective strategy employed is to combine different nonlinear active groups within a single structure.17 Known nonlinear active units include, but are not limited to, d0 transition metal octahedra (TiO6, MoO6, WO6, VO6, NbO6, etc.)11,18 and their derivatives (GaO6, GaO3F3, etc.),19–21 d10 transition metals with large polar displacement (Zn2+, Cd2+, Hg2+),22,23 and SALP cations (Pb2+, Bi3+, Sn2+, Sb3+, etc.).24,25 Some high-performance NLO materials have been explored, such as Pb2(SeO3)(NO3)2 (2 × KDP),26 NaNbO(SeO3)2 (7.8 × KDP),18 LiNbTeO5 (17 × KDP),27 Cd2Nb2Te4O15 (31 × KDP),28 β-BaTeW2O9 (1.5 × KTP)29 and TlSb3Te2O12 (37.2 × KDP).30 Furthermore, replacing the oxygen atoms in the coordination with VIIA anions is another effective approach for obtaining NCS compounds.31,32 For example, the first UV NLO selenite, Y3F(SeO3)4 (5.5 × KDP), was created through a fluorination control strategy.33 Ba(MoO2F)2(TeO3)2, containing partially fluorinated MoO6 octahedra, can demonstrate a large second harmonic generation (SHG) intensity (7.8 × KDP).34 Additionally, some halogenated selenite/tellurite compounds, such as Pb2GaF2(SeO3)2Cl (4.5 × KDP),35 Pb2Bi(SeO3)2Cl3 (13.5 × KDP),36 Cs(TiOF)3(SeO3)2 (5 × KDP),37 RbGa3F6(SeO3)2 (5.6 × KDP),38 BaF2TeF2(OH)2 (3 × KDP)39 and RbTeMo2O8F (27 × KDP),40 also exhibit excellent SHG effects. Based on literature research, it can be observed that most of the research on halogenated selenite/tellurite NLO materials focuses on F− and Cl−, while there is relatively less research on Br− and I−. Only the following selenites, Pb2NbO2(SeO3)2Br (1.4 × KDP),41 Pb2GaF2(SeO3)2Br (4.5 × KDP),41 Pb2Cd(SeO3)2Br2 (1.4 × KDP),17 and Pb3(SeO3)Br4 (1 × KDP),42 have been reported to exhibit SHG effects.
Generally, Br− and I− anions have higher polarizability and lower electronegativity compared to F− and Cl− anions.43 This leads to more favorable effects of Br− and I− anions on the SHG efficency.42,44,45 Additionally, heavier elements such as Br− and I− are more helpful for transparency in the mid-infrared (MIR) range.46 Based on this, we conducted research on halide selenite/tellurite compounds. To achieve high transparency in the MIR spectrum and increase the probability of obtaining NCS compounds, we have chosen the heavy element Pb(II) and d10 TM Cd(II) as counter cations. After numerous attempts, we have synthesized a new compound of CdPb8(SeO3)4Br10 through mild hydrothermal reactions. Unfortunately, CdPb8(SeO3)4Br10 crystallized in a centrosymmetric (CS) space group. Through structural analysis, we found that the cadmium is surrounded by four SeO3 groups and the polarity of the SALP Se4+ was almost cancelled out in the [Cd(SeO3)4]6− unit. In order to achieve the structural transformation from CS to NCS, such an unfavourable arrangement should be changed.47 We attempted to remove the CdO6 groups from the structure, and introduce larger Te4+ cations to support the framework. Ultimately, we successfully obtained the compound Pb3(TeO3)Br4, which crystallized in an NCS space group. It is worth noting that Pb3(TeO3)Br4 is the first reported NLO material with SHG activity and exhibits good transparency in the MIR region. Here, we provide a detailed description of their synthesis, crystal structures, thermal stability, and optical properties.
Results and discussion
CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4 were synthesized using a mild hydrothermal synthesis method. Details of the synthesis can be found in the ESI (ESI Experimental section†). Additionally, their powder X-ray diffraction (PXRD) patterns were recorded and found to perfectly match those of simulated data, indicating that the obtained samples are produced in pure phases (Fig. S1†). The crystallographic data of CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4 are listed in ESI Table S1.†CdPb8(SeO3)4Br10 crystallizes in the C2/c (No. 15) space group with a CS structure. Its asymmetric unit contains one Cd, four Pb, two Se, five Br, and six O atoms, totalling eighteen atoms. Only Cd(1) occupies a special position with an occupancy of 0.5. In the structure, Cd(1) is coordinated with six O atoms forming a CdO6 octahedron with Cd–O bond lengths in the range of 2.263–2.617 Å. The Se atoms are connected with three O atoms forming SeO3 trigonal pyramids with Se–O bond lengths ranging from 1.692 to 1.736 Å. The Pb atoms are connected with O and Br atoms, with Pb–Br and Pb–O bond lengths in the range of 3.022–3.193 Å and 2.435–2.734 Å, respectively. Bond valence calculations revealed that Cd(1), Pb(1)–Pb(4), and Se(1)–Se(2) exhibit bond valences of 1.744, 1.492–2.039, and 3.922–4.036, respectively. The deviation of Pb(1), Pb(2) and Pb(4) from their ideal oxidation states can be attributed to their fewer primary coordination bonds and longer secondary coordination bonds that are often overlooked in calculations. This phenomenon is a common occurrence in lead-containing compounds, as evidenced by some previously reported compounds.48 If longer Pb⋯Br distances were considered, the BVS of Pb(1)–Pb(4) can be raised to 1.885–2.039.
CdPb8(SeO3)4Br10 features a novel three-dimensional (3D) network structure composed of a lead oxybromide framework decorated with CdO6 octahedra and SeO3 trigonal pyramids (Fig. 1). One CdO6 octahedron is connected with four SeO3 groups via edge- and corner-sharing to form a [Cd(SeO3)4]6− unit (Fig. S2a†). Two Pb(1)O5Br1 and two Pb(2)O5Br1 units are connected via oxygen atoms to form Pb4O12Br4 tetramers (Fig. S2b†). The Pb(3)O2Br6 and Pb(4)O2Br3 units share bromine atoms to form a 3D network with four-membered polyhedral ring (4-MR) tunnels (Fig. S2c†). The [Cd(SeO3)4]6− units and Pb4O12Br4 tetramers are interconnected into one-dimensional (1D) chains along the b-axis (Fig. 1b and S2d†), which are located in the center of the 4-MR tunnels to support the framework.
 | ||
| Fig. 1 The 3D network structure (a) and the 1D chain component (b) of CdPb8(SeO3)4Br10. | ||
Pb3(TeO3)Br4 crystallizes in the NCS space group Pna21 (No. 33), isostructural with Pb3(TeO3)Cl4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 49 and Pb3(SeO3)Br4.42 The asymmetric unit of the compound consists of a total of 11 atoms, which include three Pb, one Te, four Br, and three O atoms. All these atoms are located in general positions. The Te(1) atom adopts a TeO3 trigonal pyramid coordination mode with Te–O bond lengths of 1.860–1.891 Å. The Pb atoms are coordinated with Br and O atoms with Pb–O and Pb–Br bond lengths in the range of 2.373–2.581 Å and 2.938–3.230 Å, respectively. Based on valence bond analysis, the oxidation states of lead and tellurium atoms are +2 and +4, respectively. The calculated values for Pb(1), Pb(2), Pb(3), and Te(1) are 1.797, 1.844, 1.640, and 3.984, respectively. Similarly, due to the neglect of longer secondary bonds involving Pb2+ cations, the calculated oxidation states tend to be underestimated.50 If longer Pb⋯Br and Pb⋯O distances were to be considered, the BVS of Pb(1)–Pb(3) can be raised to 1.943–1.967.
49 and Pb3(SeO3)Br4.42 The asymmetric unit of the compound consists of a total of 11 atoms, which include three Pb, one Te, four Br, and three O atoms. All these atoms are located in general positions. The Te(1) atom adopts a TeO3 trigonal pyramid coordination mode with Te–O bond lengths of 1.860–1.891 Å. The Pb atoms are coordinated with Br and O atoms with Pb–O and Pb–Br bond lengths in the range of 2.373–2.581 Å and 2.938–3.230 Å, respectively. Based on valence bond analysis, the oxidation states of lead and tellurium atoms are +2 and +4, respectively. The calculated values for Pb(1), Pb(2), Pb(3), and Te(1) are 1.797, 1.844, 1.640, and 3.984, respectively. Similarly, due to the neglect of longer secondary bonds involving Pb2+ cations, the calculated oxidation states tend to be underestimated.50 If longer Pb⋯Br and Pb⋯O distances were to be considered, the BVS of Pb(1)–Pb(3) can be raised to 1.943–1.967.
Pb3(TeO3)Br4 exhibits a 3D framework composed of Pb–O–Br skeletons modified by TeO3 groups (Fig. 2). The Pb(1)O2Br3, Pb(2)O1Br5, and Pb(3)O3Br3 polyhedra are connected via shared Br(1), Br(3), O(2) and O(3) atoms to form Pb3O4Br9 trimers. These Pb3O4Br9 trimers are interconnected through Pb–Br bonds to form a 1D chain with 4-MR tunnels along the c-axis. The TeO3 groups are embedded in the center of the tunnels and connected to Pb atoms through bridging O atoms. The 1D chains are further interconnected via Pb–Br bonds to form the 3D framework structure (Fig. S3†).
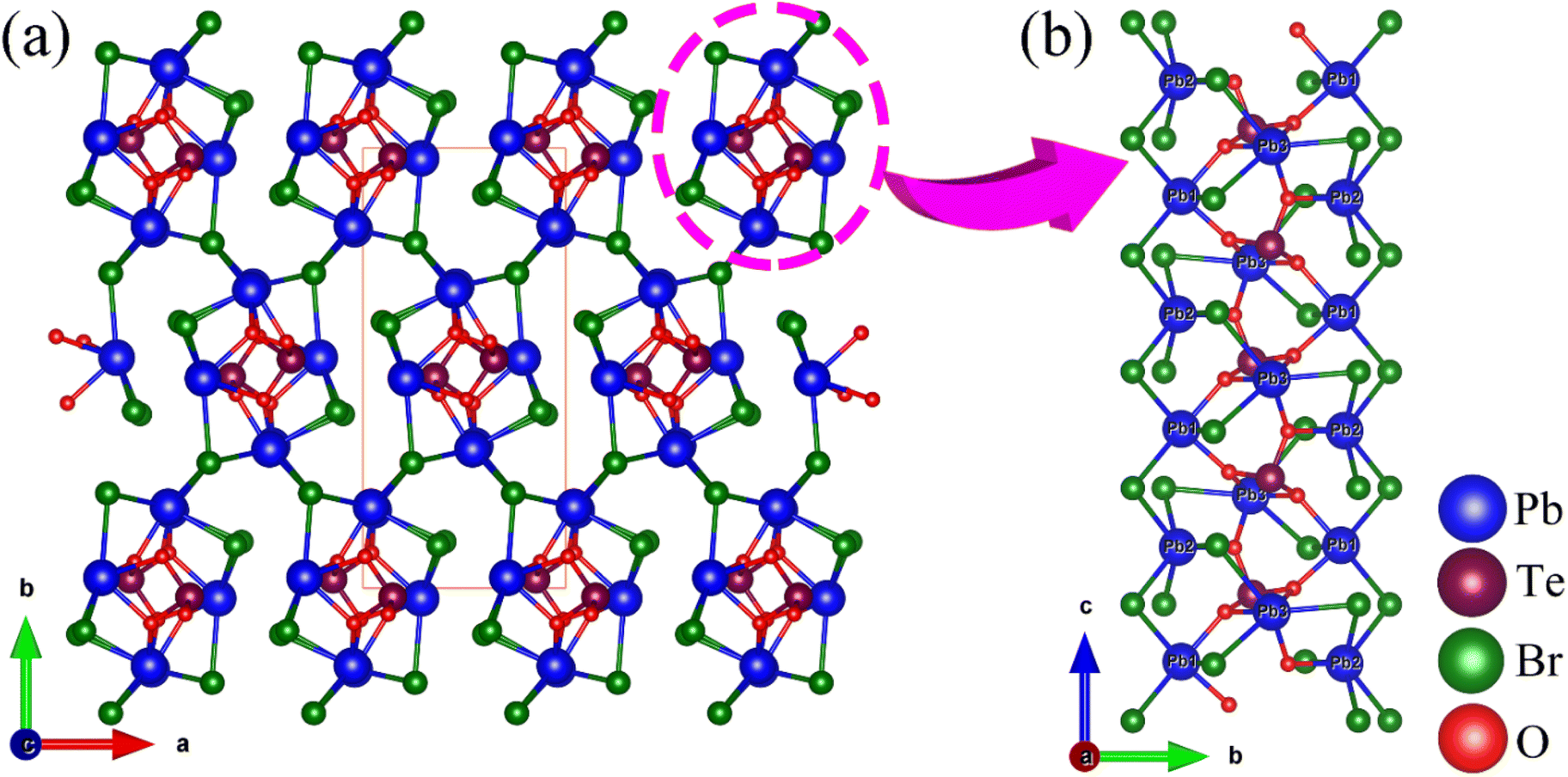 | ||
| Fig. 2 The 3D network structure (a) and the 1D chain component (b) of Pb3(TeO3)Br4. | ||
To figure out the relationships between the macroscopic symmetries and the arrangements of building blocks, the local dipole moments of CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4 have been calculated. As shown in Table S3,† the polarity of the four selenite groups around the CdO6 octahedron is almost cancelled out. Three quarters of the polarity of Pb(3)2Pb(4)2 4-MR has been counteracted by the CdO6 octahedron and Pb4O12Br4 tetramer. Although the 1D chain component of CdPb8(SeO3)4Br10 is polar, the polarities of two neighbouring chains have been cancelled out completely. As for Pb3(TeO3)Br4, the polarities of TeO3 groups can be superimposed at z-components. Although the polarities of Te and Pb polyhedra are opposite, the 1D chain component is also polar. Furthermore, the polarities of the neighboring chains have been superimposed too, which leads to the NCS and polar space group of Pb3(TeO3)Br4.
The thermal stability of CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4 was investigated through thermogravimetric analysis (TGA) in the temperature range of 25–1200 °C under an N2 atmosphere. As shown in Fig. S4,† CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4 can remain stable up to 425 °C and 522 °C, respectively. After reaching these temperatures, both compounds began to experience weight loss. At 1200 °C, CdPb8(SeO3)4Br10 exhibited a total weight loss of 88.95%, corresponding to the loss of all SeO2 and bromides of Cd and Pb, and partial loss of Pb oxides. Pb3(TeO3)Br4 showed a weight loss of 80.13% (calculated value: 80.02%), equivalent to the loss of one molecule of TeO2 and two molecules of PbBr2.
The UV-vis-NIR diffuse-reflectance spectra reveal that CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4 are nearly transparent between 500 and 2000 nm. The UV absorption cutoff edges for these two compounds are 326 nm and 328 nm, respectively. Their band gaps were determined to be 3.32 eV and 3.31 eV, respectively (Fig. S5†), which are comparable to previously reported compounds, such as Pb2NbO2(SeO3)2Br (3.17 eV),41 Ba(MoO2F)2(SeO3)2 (3.23 eV),34 Lu3F(SeO3)4 (3.57 eV),51 Ag2(TeO2F2) (3.22 eV)52 and Hg3(Te3O8)(SO4) (3.36 eV).22
The infrared spectra (IR) demonstrate that CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4 exhibit good transparency in the range of 4000–775 cm−1 (Fig. S6†). In particular, the absorption peaks at 400–455 cm−1 can be attributed to the vibration of the Pb–O and Cd–O bonds. The prominent absorption peaks at 605–775 cm−1 correspond to the bending and stretching vibrations of the Se–O and Te–O bonds, which are consistent with previous literature reports.17,50 In summary, CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4 exhibit a wide transparency range of 0.33–12.96 μm and 0.33–12.95 μm, respectively. Considering the deviation of the IR cutoff edge for powder and crystal measurements due to multiphonon absorption, 50% eliminations were made.53,54 So, the transparency range of CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4 should be 0.33–6.5 μm, covering an important atmospheric transparency window (3–5 μm) in the MIR region28,55 (Fig. 3). This indicates that lead tellurite bromides could be potential NLO material systems in the MIR wavelength.
 | ||
| Fig. 3 UV-vis-NIR diffuse reflectance and IR transmittance spectra of CdPb8(SeO3)4Br10 (a) and Pb3(TeO3)Br4 (b). | ||
Pb3(TeO3)Br4 possesses a large bandgap (3.31 eV), which tends to result in a high laser-induced damage threshold (LIDT). We measured the LIDT of Pb3(TeO3)Br4 using the reported powder method. The LIDT of Pb3(TeO3)Br4 is 21.5 MW cm−2, which is significantly higher than that of AgGaS2 (2.6 MW cm−2).
Since the structure of Pb3(TeO3)Br4 crystallizes in an NCS space group and contains two different nonlinear active units, it is necessary to detect its second-order nonlinear-optical response. A Q-switched Nd:YAG 1064 nm laser was chosen as the fundamental radiation, and the SHG signal was measured using the Kurtz–Perry method (ESI Experimental section†).56 The SHG measurement indicates that Pb3(TeO3)Br4 exhibits a frequency-doubling efficiency comparable to that of the commercial KDP (Fig. 4a). Compared with the SHG-inactive isomorphic compound Pb3(TeO3)Cl4,49 Pb3(TeO3)Br4 shows a stronger SHG response, primarily attributed to the more polarizable property of the Br− anion compared with the Cl− anion.42 In order to better understand the difference in the SHG effects between Pb3(TeO3)Cl4 and Pb3(TeO3)Br4, we compared the local dipole moments of the TeO3 groups and PbOnXm (X = Cl, Br) polyhedra, as well as the net dipole moments of these two compounds in their unit cells (Table S4†). The net dipole moment in the unit cell of Pb3(TeO3)Cl4 is 8.68 D, while the net dipole moment in the unit cell of Pb3(TeO3)Br4 is 10.062 D. The larger net dipole moment in Pb3(TeO3)Br4 results in a larger SHG response compared to Pb3(TeO3)Cl4. It is worth noting that Pb3(TeO3)Br4 is the first example of tellurite bromide that has been detected to exhibit the SHG effect.
 | ||
| Fig. 4 The oscilloscope traces of the SHG signals for the powder crystals (150–210 μm) of Pb3(TeO3)Br4 and KDP under laser irradiation at 1064 nm (a). The total and partial density of states (b), the calculated refractive indices and birefringence (c) and the electron density difference (EDD) maps (d) of Pb3(TeO3)Br4. | ||
To investigate the electronic structures and optical properties of CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4, theoretical calculations based on the density functional theory (DFT) method were performed. The state energies of the lowest conduction band and the highest valence band are listed in Table S4.† As shown in Fig. S7,† CdPb8(SeO3)4Br10 is an indirect bandgap compound with a calculated bandgap of 2.955 eV, while Pb3(TeO3)Br4 is a direct bandgap compound with a calculated bandgap of 3.134 eV. Due to the limitations of the DFT-GGA-PBE (GGA = generalized gradient approximation and PBE = Perdew−Burke−Ernzerhof) exchange–correlation functional, the calculated bandgaps are underestimated compared to the experimental values. Therefore, in the subsequent calculations, we employed a scissor operator with energy offsets of 0.365 eV and 0.176 eV, respectively.
The linear optical response characteristics of compounds CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4 were calculated based on the complex dielectric function ε(ω) = ε1(ω) + iε2(ω). Both compounds belong to biaxial crystals, where the three principal permittivity coefficients are unequal, ε1 ≠ ε2 ≠ ε3, and the refractive indices are also different, n1 ≠ n2 ≠ n3. For CdPb8(SeO3)4Br10, the refractive index order is n001 > n100 > n010. The calculated birefringence (Δn) values are 0.028@532 nm and 0.014@1064 nm (Fig. S8†), which are similar to those previously reported for compounds La2Hg3(SeO3)4(SO4)2(H2O)2 (0.013@532 nm and 0.008@1064 nm) and Ag2Cd(Se2O5)(Se0.3S0.7O4) (0.026@532 nm and 0.022@1064 nm) by our group.57 For Pb3(TeO3)Br4, the refractive index order is n100 > n010 > n001. The calculated birefringence (Δn) values are 0.095@532 nm and 0.066@1064 nm (Fig. 4b). It is worth noting that Pb3(TeO3)Br4 exhibits a similar birefringence to those of previously reported Te oxides, such as Y3(TeO3)2(SO4)2(OH)(H2O) (0.092@532 nm),15 AgAl(Te4O10) (0.104@532 nm),19 BaF2TeF2(OH)2 (∼0.078@300–700 nm),39 Rb[Te2O4(OH)5] (0.0545@1064 nm),58 Li2ZrTeO6 (0.064@1064 nm)59 and AgTeO2F (0.078@1064 nm).52
Partial density of states (PDOS) and total density of states (TDOS) calculations were performed for CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4. As shown in Fig. 4c, the lowest conduction bands of Pb3(TeO3)Br4 mainly originate from the unoccupied Pb-6p orbitals and Te-5p orbitals, while the highest valence bands are mainly contributed by the Br-4p and O-2p non-bonding states. As for CdPb8(SeO3)4Br10, the lowest conduction bands are mainly from the unoccupied Pb-6p orbitals while the highest valence bands are mainly contributed by the Br-4p non-bonding states (Fig. S9†). Therefore, the band gap of Pb3(TeO3)Br4 is mainly determined by the Pb and Te polyhedra, while the band gap of CdPb8(SeO3)4Br10 is mainly determined by the Pb–Br bonds.
It is worth noting that the states near the Fermi level have a significant impact on the linear and nonlinear properties of Pb3(TeO3)Br4. The main influencing factors are Pb-6p, Te-5p, O-2p, and Br-4p orbitals in this compound. This indicates that the optical properties of Pb3(TeO3)Br4 primarily originate from the synergistic interaction between the TeO3 groups and the lead oxybromide polyhedra. To further support this inference, we have studied the electron density difference (EDD) map of Pb3(TeO3)Br4. Fig. 4d shows that Te4+ possesses stereo-chemically active lone pair electrons and there is charge transfer from Pb atoms to O/Br atoms.
Conclusions
In summary, two new bromine-containing selenite/tellurite compounds, namely, CdPb8(SeO3)4Br10 and Pb3(TeO3)Br4, were obtained by a mild hydrothermal method in the Pb2+-Se4+O3/Te4+O3-Br− system. Interestingly, due to the difference in the arrangement of the polar building blocks, these two compounds exhibit two different lead oxybromide 3D structures. CdPb8(SeO3)4Br10 is crystallized in a CS space group of C2/c while Pb3(TeO3)Br4 is crystallized in an NCS space group of Pna21. Importantly, Pb3(TeO3)Br4 is the first reported tellurite bromide compound that exhibits SHG activity. It demonstrates an SHG intensity comparable to that of KDP, high thermal stability (522 °C), wide optical transparency window (0.33–6.5 μm) and appropriate birefringence (0.095@532 nm and 0.066@1064 nm). This work proved that lead tellurite bromides can be promising NLO material systems in the mid-IR wavelength.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22031009, 22375201, 21921001 and 91963105) and the NSF of Fujian Province (Grant No. 2023J01216).References
- L. Luo, L. Wang, J. Chen, J. Zhou, Z. Yang, S. Pan and J. Li, AIB3IIC3IIIQ8VI: A New Family for the Design of Infrared Nonlinear Optical Materials by Coupling Octahedra and Tetrahedra Units, J. Am. Chem. Soc., 2022, 144, 21916–21925 CrossRef CAS PubMed.
- P. F. Li, J. G. Mao and F. Kong, A survey of stereoactive oxysalts for linear and nonlinear optical applications, Mater. Today Phys., 2023, 37, 101197 CrossRef CAS.
- C. Yang, X. Liu, C. Teng, X. Cheng, F. Liang and Q. Wu, Hierarchical molecular design of high-performance infrared nonlinear Ag2HgI4 material by defect engineering strategy, Mater. Today Phys., 2021, 19, 100432 CrossRef CAS.
- M. Mutailipu, J. Han, Z. Li, F. Li, J. Li, F. Zhang, X. Long, Z. Yang and S. Pan, Achieving the full-wavelength phase-matching for efficient nonlinear optical frequency conversion in C(NH2)3BF4, Nat. Photonics, 2023, 17, 694–701 CrossRef CAS.
- P. F. Li, Y. P. Gong, C. L. Hu, B. Zhang, J. G. Mao and F. Kong, Four UV Transparent Linear and Nonlinear Optical Materials Explored from Pure Selenite Compounds, Adv. Opt. Mater., 2023, 2301426, DOI:10.1002/adom.202301426.
- H. Wang, Y. Chu, X. Pan, Z. Yang, S. Pan and J. Li, Double alkaline earth metals sulfide SrMgGeS4 with high laser-induced damage threshold and strong second-harmonic generation, Mater. Today Phys., 2023, 38, 101243 CrossRef CAS.
- A. Tudi, C. W. Xie, S. L. Pan and Z. H. Yang, Design of novel deep-UV nonlinear optical materials with one-dimensional functional module [BO2]∞ chain by fluorine-driven short phase-matching, Mater. Today Phys., 2022, 28, 100852 CrossRef CAS.
- H. Y. Wu, C. L. Hu, M. B. Xu, Q. Q. Chen, N. Ma, X. Y. Huang, K. Du and J. Chen, From H12C4N2CdI4 to H11C4N2CdI3: highly polarizable CdNI3 tetrahedron induced a shape enhancement of second harmonic generation response and birefringence, Chem. Sci., 2023, 14, 9533–9542 RSC.
- Y. Chu, H. Wang, T. Abutukadi, Z. Li, M. Mutailipu, X. Su, Z. Yang, J. Li and S. Pan, Zn2HgP2S8: A Wide Bandgap Hg-Based Infrared Nonlinear Optical Material with Large Second-Harmonic Generation Response, Small, 2023, 2305074, DOI:10.1002/smll.202305074.
- H. Chen, M. Y. Ran, S. H. Zhou, X. T. Wu, H. Lin and Q. L. Zhu, Simple yet extraordinary: super-polyhedra-built 3D chalcogenide framework of Cs5Ga9S16 with excellent infrared nonlinear optical performance, Chin. Chem. Lett., 2023, 34, 107838 CrossRef CAS.
- J. H. Wu, B. Zhang, T. K. Jiang, F. Kong and J. G. Mao, From Cs8Sb4Nb5O5F35 to Cs6Sb4Mo3O5F26: the first noncentrosymmetric fluoroantimonite with d0 transition metal, Chin. J. Struct. Chem., 2023, 42, 100016 CrossRef.
- M. Y. Ran, S. H. Zhou, W. B. Wei, B. X. Li, X. T. Wu, H. Lin and Q. L. Zhu, Rational Design of a Rare-Earth Oxychalcogenide Nd3[Ga3O3S3][Ge2O7] with Superior Infrared Nonlinear Optical Performance, Small, 2023, 19, 2300248 CrossRef CAS PubMed.
- L. Wang, D. Chu, D. Yin, C. Xie, Z. Yang, J. Li and S. Pan, Theoretical investigations on ternary defective diamond-like infrared nonlinear optical materials in Be-Ga-Se system, Mater. Today Phys., 2023, 38, 101245 CrossRef CAS.
- P. F. Li, C. L. Hu, B. X. Li, F. Kong and J. G. Mao, Y(HSeO3)(SeO3)(H2O)·(H2O) and Y2(SeO3)2(SeO4)(H2O)2·(H2O)0.75: Two yttrium selenites with a short UV cut-off edge explored from pure selenite compounds, J. Alloys Compd., 2023, 959, 170570 CrossRef CAS.
- P. F. Li, C. L. Hu, F. Kong, S. M. Ying and J. G. Mao, Y2(Te4O10)(SO4): a new sulfate tellurite with a unique Te4O10 polyanion and large birefringence, Inorg. Chem. Front., 2021, 8, 164–172 RSC.
- S. Cho, S. Park, Y. Kuk and K. M. Ok, Elucidating the structure-nonlinear optical property relationship of Te2O4(OH)2, Mater. Today Phys., 2023, 34, 101075 CrossRef CAS.
- Y. P. Gong, C. L. Hu, Y. X. Ma, J. G. Mao and F. Kong, Pb2Cd(SeO3)2X2 (X = Cl and Br): two halogenated selenites with phase matchable second harmonic generation, Inorg. Chem. Front., 2019, 6, 3133–3139 RSC.
- X. L. Cao, C. L. Hu, F. Kong and J. G. Mao, Explorations of New SHG Materials in the Alkali-Metal-Nb5+-Selenite System, Inorg. Chem., 2015, 54, 10978–10984 CrossRef CAS PubMed.
- P. F. Li, C. L. Hu, F. Kong and J. G. Mao, AAl(Te4O10) (A = Na, Ag) and K2Ga2(HTe6O16)(HTeO3): Three Aluminum/Gallium Tellurites with Large Birefringence and Wide Band Gap, Inorg. Chem., 2023, 62, 8494–8499 CrossRef CAS PubMed.
- J. Li, W. D. Yao, J. N. Li, X. H. Li, W. Liu and S. P. Guo, Partial substitution induced structural transformation and enhanced nonlinear optical properties of Na2GaxIn6-xSe10 (x = 3, 3.76), Mater. Today Phys., 2023, 32, 101007 CrossRef CAS.
- P. Wang, Y. Chu, A. Tudi, C. Xie, Z. Yang, S. Pan and J. Li, The Combination of Structure Prediction and Experiment for the Exploration of Alkali-Earth Metal-Contained Chalcopyrite-Like IR Nonlinear Optical Material, Adv. Sci., 2022, 9, 2106120 CrossRef CAS PubMed.
- P. F. Li, C. L. Hu, Y. P. Gong, F. Kong and J. G. Mao, Hg3(Te3O8)(SO4): a new sulfate tellurite with a novel structure and large birefringence explored from d10 metal compounds, Chem. Commun., 2021, 57, 7039–7042 RSC.
- J. Zhou, Z. Fan, K. Zhang, Z. Yang, S. Pan and J. Li, Rb2CdSi4S10: novel [Si4S10] T2- supertetrahedra-contained infrared nonlinear optical material with large band gap, Mater. Horiz., 2022, 10, 619–624 RSC.
- S. Han, A. Tudi, W. Zhang, X. Hou, Z. Yang and S. Pan, Recent Development of Sn(II), Sb(III)-based Birefringent Material: Crystal Chemistry and Investigation of Birefringence, Angew. Chem., Int. Ed., 2023, 62, e202302025 CrossRef CAS PubMed.
- Y. Long, X. Dong, L. Huang, H. Zeng, Z. Lin, L. Zhou and G. Zou, BaSb(H2PO2)3Cl2: An Excellent UV Nonlinear Optical Hypophosphite Exhibiting Strong Second-Harmonic Generation Response, Mater. Today Phys., 2022, 28, 100876 CrossRef CAS.
- C. Y. Meng, L. Geng, W. T. Chen, M. F. Wei, K. Dai, H. Y. Lu and W. D. Cheng, Syntheses, structures, and characterizations of a new second-order nonlinear optical material: Pb2(SeO3)(NO3)2, J. Alloys Compd., 2015, 640, 39–44 CrossRef CAS.
- K. C. Chen, C. S. Lin, G. Peng, Y. Chen, H. Z. Huang, E. Z. Chen, Y. X. Min, T. Yan, M. Luo and N. Ye, LiNbTeO5: A High-Performance Multifunctional Crystal Material with a Very Large Second-Harmonic Generation Response and Piezoelectric Coefficient, Chem. Mater., 2022, 34, 399–404 CrossRef CAS.
- Q. Wang, X. H. Dong, L. Huang, K. M. Ok, Z. E. Lin and G. H. Zou, Cd2Nb2Te4O15 : A Novel Pseudo-Aurivillius-Type Tellurite with Unprecedented Nonlinear Optical Properties and Excellent Stability, Small, 2023, 19, 2302797 CrossRef CAS PubMed.
- C. Li, Z. Gao, P. Zhao, X. Tian, H. Wang, Q. Wu, W. Lu, Y. Sun, D. Cui and X. Tao, Crystallographic Investigations into the Polar Polymorphism of BaTeW2O9: Phase Transformation, Controlled Crystallization, and Linear and Nonlinear Optical Properties, Cryst. Growth Des., 2019, 19, 1767–1777 CrossRef CAS.
- R. Robert, V. Balisetty, K. Mohanrao, M. Mannamala, S. Mangalassery, D. N. Rao and K. Vidyasagar, Syntheses, Crystal Structure, and Second Harmonic Generation Response of Noncentrosymmetric Layered Selenites and Tellurites of Antimony(V), ASb3Se2O12 (A = K, Rb, Cs, Tl; X = Se, Te), Inorg. Chem., 2023, 62, 7890–7897 CrossRef CAS PubMed.
- J. Y. Chung, H. Jo, S. Yeon, H. R. Byun, T. S. You, J. I. Jang and K. M. Ok, Bi3(SeO3)3(Se2O5)F: A Polar Bismuth Selenite Fluoride with Polyhedra of Highly Distortive Lone Pair Cations and Strong Second-Harmonic Generation Response, Chem. Mater., 2020, 32, 7318–7326 CrossRef CAS.
- X. Chen, Q. Jing and K. M. Ok, Pb18O8Cl15I5 : A Polar Lead Mixed Oxyhalide with Unprecedented Architecture and Excellent Infrared Nonlinear Optical Properties, Angew. Chem., Int. Ed., 2020, 59, 20323–20327 CrossRef CAS PubMed.
- P. F. Li, C. L. Hu, F. Kong and J. G. Mao, The First UV Nonlinear Optical Selenite Material: Fluorination Control in CaYF(SeO3)2 and Y3F(SeO3)4, Angew. Chem., Int. Ed., 2023, 62, e202301420 CrossRef CAS PubMed.
- M. L. Liang, Y. X. Ma, C. L. Hu, F. Kong and J. G. Mao, Ba(MoO2F)2(QO3)2 (Q = Se, Te): Partial Fluorination of MoO6 Octahedra Enabling Two Polar Solids with Strong and Phase Matchable SHG Response, Chem. Mater., 2020, 32, 9688–9695 CrossRef CAS.
- F. G. You, F. Liang, Q. Huang, Z. G. Hu, Y. C. Wu and Z. S. Lin, Pb2GaF2(SeO3)2Cl: Band Engineering Strategy by Aliovalent Substitution for Enlarging Bandgap while Keeping Strong Second Harmonic Generation Response, J. Am. Chem. Soc., 2019, 141, 748–752 CrossRef CAS PubMed.
- Y. J. Jia, X. Y. Zhang, Y. G. Chen, X. X. Jiang, J. N. Song, Z. S. Lin and X. M. Zhang, PbBi(SeO3)2F and Pb2Bi(SeO3)2Cl3: Coexistence of Three Kinds of Stereochemically Active Lone-Pair Cations Exhibiting Excellent Nonlinear Optical Properties, Inorg. Chem., 2022, 61, 15368–15376 CrossRef CAS PubMed.
- X. L. Cao, C. L. Hu, F. Kong and J. G. Mao, Cs(TaO2)3(SeO3)2 and Cs(TiOF)3(SeO3)2: structural and second harmonic generation changes induced by the different d0-TM coordination octahedra, Inorg. Chem., 2015, 54, 3875–3882 CrossRef CAS PubMed.
- C. Wu, X. X. Jiang, L. Lin, Z. S. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, AGa3F6(SeO3)2 (A = Rb, Cs): A New Type of Phase-Matchable Hexagonal Tungsten Oxide Material with Strong Second-Harmonic Generation Responses, Chem. Mater., 2020, 32, 6906–6915 CrossRef CAS.
- J. J. Zhou, H. P. Wu, H. W. Yu, S. T. Jiang, Z. G. Hu, J. Y. Wang, Y. C. Wu and P. S. Halasyamani, BaF2TeF2(OH)2: A UV Nonlinear Optical Fluorotellurite Material Designed by Band-Gap Engineering, J. Am. Chem. Soc., 2020, 142, 4616–4620 CrossRef CAS PubMed.
- Y. L. Hu, C. Wu, X. X. Jiang, Z. J. Wang, Z. P. Huang, Z. S. Lin, X. F. Long, M. G. Humphrey and C. Zhang, Giant Second-Harmonic Generation Response and Large Band Gap in the Partially Fluorinated Mid-Infrared Oxide RbTeMo2O8F, J. Am. Chem. Soc., 2021, 143, 12455–12459 CrossRef CAS PubMed.
- H. Zhao, P. Gong, X. Zhang, Z. Lin, Z. Hu and Y. Wu, Selenite bromide nonlinear optical materials Pb2GaF2(SeO3)2Br and Pb2NbO2(SeO3)2Br: synthesis and characterization, Dalton Trans., 2020, 49, 14046–14051 RSC.
- X. X. Wang, X. X. Jiang, H. M. Liu, L. Yang, Z. S. Lin, Z. G. Hu, X. G. Meng, X. G. Chen and J. G. Qin, Pb3(SeO3)Br4: a new nonlinear optical material with enhanced SHG response designed via an ion-substitution strategy, Dalton Trans., 2018, 47, 1911–1917 RSC.
- C. Shen, D. Sun, Y. Dang, K. Wu, T. Xu, R. Hou, H. Chen, J. Wang and D. Wang, (C4H10NO)PbX3 (X = Cl, Br): Design of Two Lead Halide Perovskite Crystals with Moderate Nonlinear Optical Properties, Inorg. Chem., 2022, 61, 16936–16943 CrossRef CAS PubMed.
- T. Wang, Y. G. Chen, Y. Guo, F. Wang, Q. Song, Y. J. Jia and X. M. Zhang, BaLiTe2O5X (X = Cl, Br): mixed alkali/alkaline-earth metal tellurite halides with [Te2O5]∞ chains, Dalton Trans., 2020, 49, 4914–4919 RSC.
- X. H. Li, Z. H. Shi, M. Yang, W. Liu and S. P. Guo, Sn7Br10S2 : The First Ternary Halogen-Rich Chalcohalide Exhibiting a Chiral Structure and Pronounced Nonlinear Optical Properties, Angew. Chem., Int. Ed., 2022, 61, e202115871 CrossRef CAS PubMed.
- D. Wang, Y. Zhang, Q. Shi, Q. Liu, D. Yang, B. Zhang and Y. Wang, Tellurate polymorphs with high-performance nonlinear optical switch property and wide mid-IR transparency, Inorg. Chem. Front., 2022, 9, 1708–1713 RSC.
- S. M. Pei, B. W. Liu, W. F. Chen, X. M. Jiang and G. C. Guo, Breaking the Bottleneck of Simultaneously Wide Band Gap and Large Nonlinear Optical Coefficient by “Pore Reconstruction” Strategy in Salt-inclusion Chalcogenide, Mater. Horiz., 2023, 10, 2921–2926 RSC.
- P. F. Li, F. Kong and J. G. Mao, M2IIM3IIIF3(Te6F2O16) (MII = Pb, Ba; MIII = Al, Ga): New mixed anionic tellurites with isolated Te6 coplanar rings, J. Solid State Chem., 2020, 286, 121288 CrossRef CAS.
- S. Y. Zhang, C. L. Hu, P. X. Li, H. L. Jiang and J. G. Mao, Syntheses, crystal structures and properties of new lead(II) or bismuth(III) selenites and tellurite, Dalton Trans., 2012, 41, 9532–9542 RSC.
- C. Bai, Y. Chu, J. Zhou, L. Wang, L. Luo, S. Pan and J. Li, Two new tellurite halides with cationic layers: syntheses, structures, and characterizations of CdPb2Te3O8Cl2 and Cd13Pb8Te14O42Cl14, Inorg. Chem. Front., 2022, 9, 1023–1030 RSC.
- C. Wu, L. H. Li, L. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, Enhancement of Second-Order Optical Nonlinearity in a Lutetium Selenite by Monodentate Anion Partial Substitution, Chem. Mater., 2020, 32, 3043–3053 CrossRef CAS.
- B. Zhang, J. H. Wu, C. L. Hu, Y. F. Li, F. Kong and J. G. Mao, From AgTeO2F and Ag2(TeO2F2) to Ag3F3(TeF6)(TeO2)12: the first silver tellurite oxyfluorides with linear and nonlinear optical properties, Inorg. Chem. Front., 2023, 10, 1328–1337 RSC.
- Z. Yang, C. Hu, M. Mutailipu, Y. Sun, K. Wu, M. Zhang and S. Pan, Oxyhalides: prospecting ore for optical functional materials with large laser damage thresholds, J. Mater. Chem. C, 2018, 6, 2435–2442 RSC.
- G. M. Li, Q. Liu, K. Wu, Z. H. Yang and S. L. Pan, Na2CdGe2Q6(Q = S, Se): two metal-mixed chalcogenides with phase-matching abilities and large second-harmonic generation responses, Dalton Trans., 2017, 46, 2778–2784 RSC.
- M. Y. Ran, A. Y. Wang, W. B. Wei, X. T. Wu, H. Lin and Q. L. Zhu, Recent progress in the design of IR nonlinear optical materials by partial chemical substitution: Structural evolution and performance optimization, Coord. Chem. Rev., 2023, 481, 215059 CrossRef CAS.
- S. K. Kurtz and T. T. Perry, A Powder Technique for the Evaluation of Nonlinear Optical Materials., J. Appl. Phys., 1968, 39, 3798–3813 CrossRef CAS.
- P. F. Li, C. L. Hu, F. Kong and J. G. Mao, Hg2(SeO3)(SO4): the first sulfate selenite with large birefringence explored from d10 transition metal compounds, Mater. Chem. Front., 2022, 6, 3567–3576 RSC.
- D. Wang, P. Gong, X. Zhang, Z. Lin, Z. Hu and Y. Wu, Centrosymmetric Rb[Te2O4(OH)5] and noncentrosymmetric K2[Te3O8(OH)4]: metal tellurates with corner and edge-sharing (Te4O18)12− anion groups, Inorg. Chem. Front., 2022, 9, 2628–2636 RSC.
- W. Lu, Z. Gao, X. Liu, X. Tian, Q. Wu, C. Li, Y. Sun, Y. Liu and X. Tao, Rational Design of a LiNbO3-like Nonlinear Optical Crystal, Li2ZrTeO6, with High Laser-Damage Threshold and Wide Mid-IR Transparency Window, J. Am. Chem. Soc., 2018, 140, 13089–13096 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis, PXRD patterns, crystal data, TG curves, UV-vis-NIR diffuse-reflectance spectra, IR spectra, and computational method. CCDC 2278543 for CdPb8(SeO3)4Br10 and 2278544 for Pb3(TeO3)Br4]. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi01937c |
| This journal is © the Partner Organisations 2023 |