
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfide organic polymers as novel and efficient metal-free heterogeneous Lewis acid catalysts for esterification reactions†
M.
Melero
 ,
U.
Díaz
* and
F. X.
Llabrés i Xamena
*
,
U.
Díaz
* and
F. X.
Llabrés i Xamena
*
Instituto de Tecnología Química, Universitat Politècnica de València, Consejo Superior de Investigaciones Científicas, 46022, Valencia, Spain. E-mail: udiaz@itq.upv.es; fllabres@itq.upv.es
First published on 3rd April 2023
Abstract
Herein, we report on the synthesis of four organosulfide-based covalent organic polymers prepared via click processes, consisting of either SN2 (SOP-1 and SOP-3) or thiol–yne (SOP-2 and SOP-4) coupling reactions. Formation of the SOPs in high yields is confirmed by solid-state 13C NMR and FTIR spectroscopies, while the sulfur contents of SOP-type materials confirm the expected C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S molar ratio for the formation of stoichiometric products. CO2 adsorption isotherms reveal the porosity of SOPs, with specific surface areas of up to 180 m2 g−1 and strongly dependent on the ligands used. The catalytic activity of SOPs is evaluated for carboxylic acid esterification reactions, obtaining high conversions and efficient recyclability. The proposed reaction mechanism consists of the activation of carboxylic acid by hypervalent S⋯O (nO → σS*) interactions with sulfur centres of the SOPs, which increase the electrophilic character of the carboxylic carbon and facilitate the addition of the alcohol. Thus, SOPs constitute a novel class of metal-free heterogeneous Lewis acid (organo)catalysts.
:S molar ratio for the formation of stoichiometric products. CO2 adsorption isotherms reveal the porosity of SOPs, with specific surface areas of up to 180 m2 g−1 and strongly dependent on the ligands used. The catalytic activity of SOPs is evaluated for carboxylic acid esterification reactions, obtaining high conversions and efficient recyclability. The proposed reaction mechanism consists of the activation of carboxylic acid by hypervalent S⋯O (nO → σS*) interactions with sulfur centres of the SOPs, which increase the electrophilic character of the carboxylic carbon and facilitate the addition of the alcohol. Thus, SOPs constitute a novel class of metal-free heterogeneous Lewis acid (organo)catalysts.
Introduction
Covalent organic polymers (COPs) are a class of supramolecular compounds formed by self-assembly of organic building units through covalent bonds.1–3 They have recently emerged as new molecular platforms for designing promising organic materials. Within this group, different families exist, including (i) covalent organic frameworks (COFs),4–6 which are crystalline porous materials constructed with light elements through strong covalent bonds (e.g., C–C, C–N, and C–O), as well as structural heteroatoms such as B (boronate, borosilicate and borazine compounds); (ii) porous aromatic frameworks (PAFs),7 which are porous solids containing rigid frameworks exhibiting exceptionally high surface areas and constructed from carbon–carbon-linked aromatic building units; (iii) hypercrosslinked polymers (HCPs),8 another class of microporous organic materials prepared by post-cross-linking of polystyrenic networks with high surface areas.COPs can incorporate a large variety of constituents with intrinsic properties, such as stereochemical,9,10 electronic,11–14 magnetic,15,16 redox,17–19 optic or catalytic20,21 active sites. In this way, advanced materials have been prepared with many different applications in gas purification and separation,22 heterogeneous catalysis, biomedicine23 and other nanotechnological applications. In particular, heteroatom coordinated compounds24–26 containing main group elements, such as B, Si, Ge, P, S or N, have attracted increasing attention in recent years. For instance, COF-5 was the first mesoporous boronic material formed via the condensation reaction between hexahydroxy triphenylene (HHTP) and 1,4-benzene-diboronic acid (BDBA), exhibiting an eclipsed boron nitride structure.27
In this sense, the preparation of novel sulfur organic polymers with improved properties is intensely being sought for further applications. Organosulfide compounds28–31 contain at least one carbon–sulphur–carbon bond. These compounds have shown potential application in many fields, such as batteries, lubricants, pigments, polymers and catalysis.32,33 The most common synthetic method for sulfides (or thioethers) is based on the alkylation of thiols through the SN2 reaction in the presence of bases, using thiolates as a nucleophile.34,35 Alternatively, organosulfides are also prepared by thiol–ene or thiol–yne coupling reactions, through the addition of a thiol to an alkene or alkyne.36,37 This reaction is often catalyzed by thermal initiators, such as 2,2′-azobis(2-methylpropionitrile) (AIBN), or by photoinitiators, such as 2,2-dimethoxy-2-phenylacetophenone (DMPA). Following these procedures, few sulfide organic polymers (SOPs)38,39 have been prepared. The catalytic properties of organosulfide polymers have only recently been explored in reactions in which the polymer was used as a support for transition metal ions (e.g., Co, Ni or Mo) in hydrogenation and dehydrogenation processes, such as isobutane to isobutene conversion.40 In contrast, metal-free SOPs have been largely unexplored in the area of heterogeneous catalysis.
Herein, we will show that metal-free SOPs have high potential as heterogeneous catalysts for reactions of industrial interest, such as esterification of carboxylic acids, which provide interesting products including pesticides, flavorings, fragrances and biodiesel derivatives.41–43 Esterification is an important reaction in organic synthesis.44,45 In this regard, solid acid catalysts based on zeolites, metal–organic frameworks (MOFs), or sulfonated covalent organic frameworks (COFs), have attracted much interest in recent years46–56 as more sustainable alternatives to the use of conventional mineral acids.
In our quest for exploring the potential of SOPs as heterogeneous catalysts, four different organosulfide-based compounds, namely SOP-1, SOP-2, SOP-3, and SOP-4 (see Scheme 1) have been synthesized in very high yields by coupling aromatic dithiols (1,4-dimercaptobenzene 1, or bismuthiol 2) with suitable aromatic tris-substituted aryl compounds (1,3,5-tris(bromomethyl)benzene 3, or 1,3,5-triethynylbenzene 4). Coupling reactions consisted of click synthesis through SN2 (SOP-1 and SOP-3) or thiol–yne reactions (SOP-2 and SOP-4). The solids obtained have been characterized by X-ray diffraction (XRD), thermogravimetric analysis (TGA), Fourier transform infrared spectroscopy (FTIR), elemental analysis (EA), solid-state 13C NMR, scanning and transmission electron microscopy (FESEM and TEM) and CO2 isothermal adsorption. Herein, we will show that the as-prepared compounds constitute a novel family of active and reusable heterogeneous catalysts for the esterification of carboxylic acids.
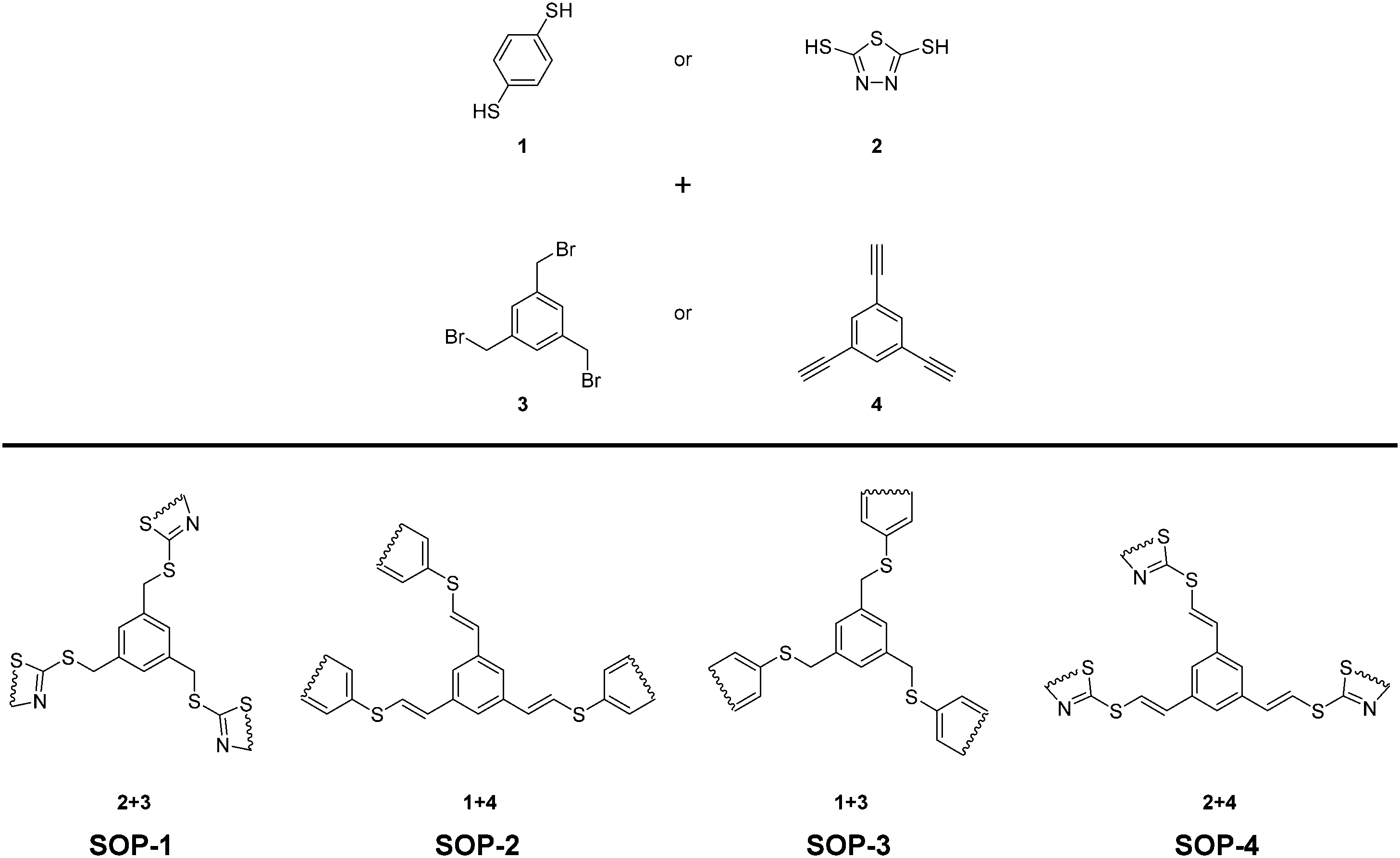 | ||
| Scheme 1 Schematic illustration of the synthetic procedures and the idealized structures of the obtained SOPs. | ||
Results and discussion
Synthesis and characterization
In this work, SOP-1 and SOP-3 materials were synthesized by reacting 1,3,5-tris(bromomethyl) benzene with either bismuthiol or 1,4-dimercaptobenzene monomers in methanol or acetonitrile at room temperature. NaOH was used to assist deprotonation of thiol groups to the corresponding thiolate to trigger SN2 coupling polymerization (Scheme 2). The coupling reaction was complete within 2 hours, and the resulting solids were isolated by filtration and washed with water and acetone to afford white powders with 92 and 93% yields, respectively for SOP-1 and SOP-3.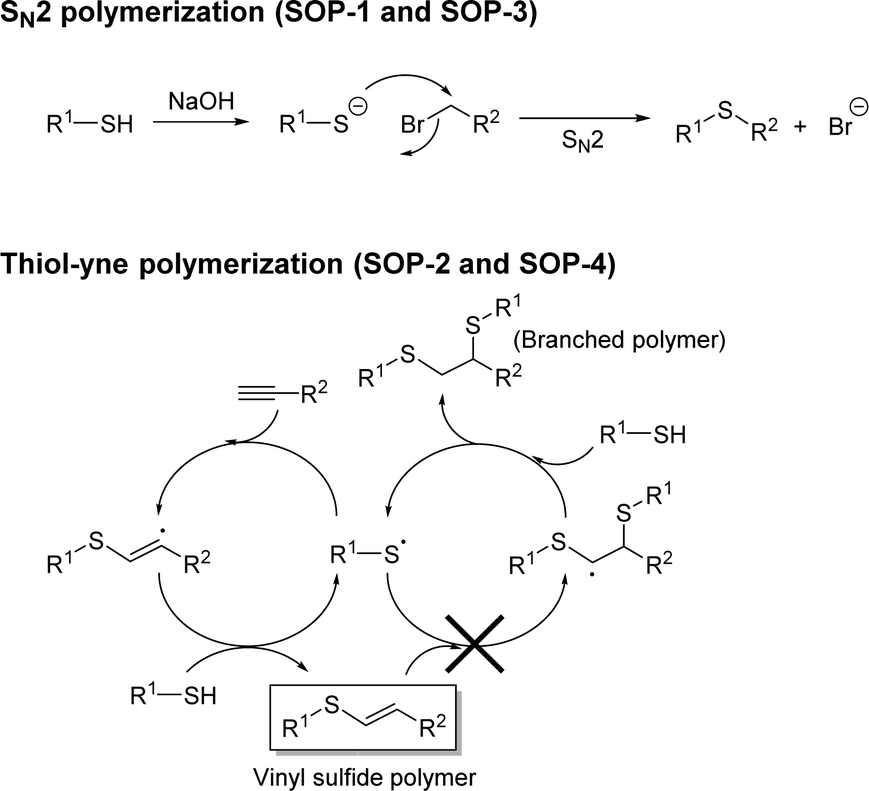 | ||
| Scheme 2 SN2 and thiol–yne click polymerization mechanisms used for the preparation of SOPs. | ||
For the synthesis of SOP-2 and SOP-4, 1,3,5-triethynyl benzene was used, which was coupled with the two sulfur-containing monomers via thiol–yne click polymerization in toluene at 80 °C. Yellow or brown powders were obtained after 24 h with 91 and 80% yield, respectively for SOP-2 and SOP-4.
Elemental analysis was used to determine the experimental C:S molar ratio of the four SOPs, as shown in Table 1. Comparison of the experimental and expected C:S ratios, together with the disappearance of characteristic –SH, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, C–H and C–Br FT-IR absorption bands (see below), confirmed the full assembly of the organic units in all four compounds during the polymerization process. Note that the data for SOP-2 and SOP-4 agree with the formation of vinyl sulfides and exclude the formation of branched polymers by the addition of two thiol groups to each ethynyl group in a two-step process (see Scheme 2), which would result in a much lower C:S molar ratio (viz. 5.0 and 2.0 for SOP-2 and SOP-4, see the last column in Table 1).
C, C–H and C–Br FT-IR absorption bands (see below), confirmed the full assembly of the organic units in all four compounds during the polymerization process. Note that the data for SOP-2 and SOP-4 agree with the formation of vinyl sulfides and exclude the formation of branched polymers by the addition of two thiol groups to each ethynyl group in a two-step process (see Scheme 2), which would result in a much lower C:S molar ratio (viz. 5.0 and 2.0 for SOP-2 and SOP-4, see the last column in Table 1).
:S molar ratios. The last column corresponds to the C:S molar ratio expected for a branched polymer (i.e., addition of two thiols to each ethynyl group)
| Catalyst | C (wt%) | S (wt%) | N (wt%) | H (wt%) | C:S exp. |
C:S calc. vinyl sulfide |
C:S calc. branched |
|---|---|---|---|---|---|---|---|
| SOP-1 | 41.0 | 37.6 | 10.9 | 2.6 | 2.9 | 2.7 | — |
| SOP-3 | 67.4 | 25.7 | — | 3.9 | 7.0 | 7.0 | 5.0 |
| SOP-3 | 63.2 | 27.6 | — | 4.4 | 6.0 | 6.0 | — |
| SOP-4 | 43.5 | 38.2 | 10.5 | 2.1 | 3.3 | 3.0 | 2.0 |
According to the X-ray diffractograms (not shown), all SOP compounds were amorphous. This is not surprising, since the presence of the sulfide bonds breaks the planarity of the structural units, thus hampering the formation of a regular stacking network similar to that found in many lamellar COFs.
Fig. 1 compares the FTIR spectra of SOP-2 and its monomers 1 and 4, while the spectra of other SOPs and monomers are shown in the ESI† (Fig. S1). The characteristic C–H, CC and S–H stretching bands of the monomers are observed at 2562 cm−1 and at 3282/2109 cm−1 for 1 and 4, respectively. These bands are not observed in the spectrum of SOP-2, whereas a new absorption band appears at 1686 cm−1 that is attributed to the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) mode of the vinyl sulfide groups of the resulting polymer. These spectral changes clearly demonstrate that the thiol groups of 1 react with the triple bonds of 4 during the thiol–yne polymerization.
C) mode of the vinyl sulfide groups of the resulting polymer. These spectral changes clearly demonstrate that the thiol groups of 1 react with the triple bonds of 4 during the thiol–yne polymerization.
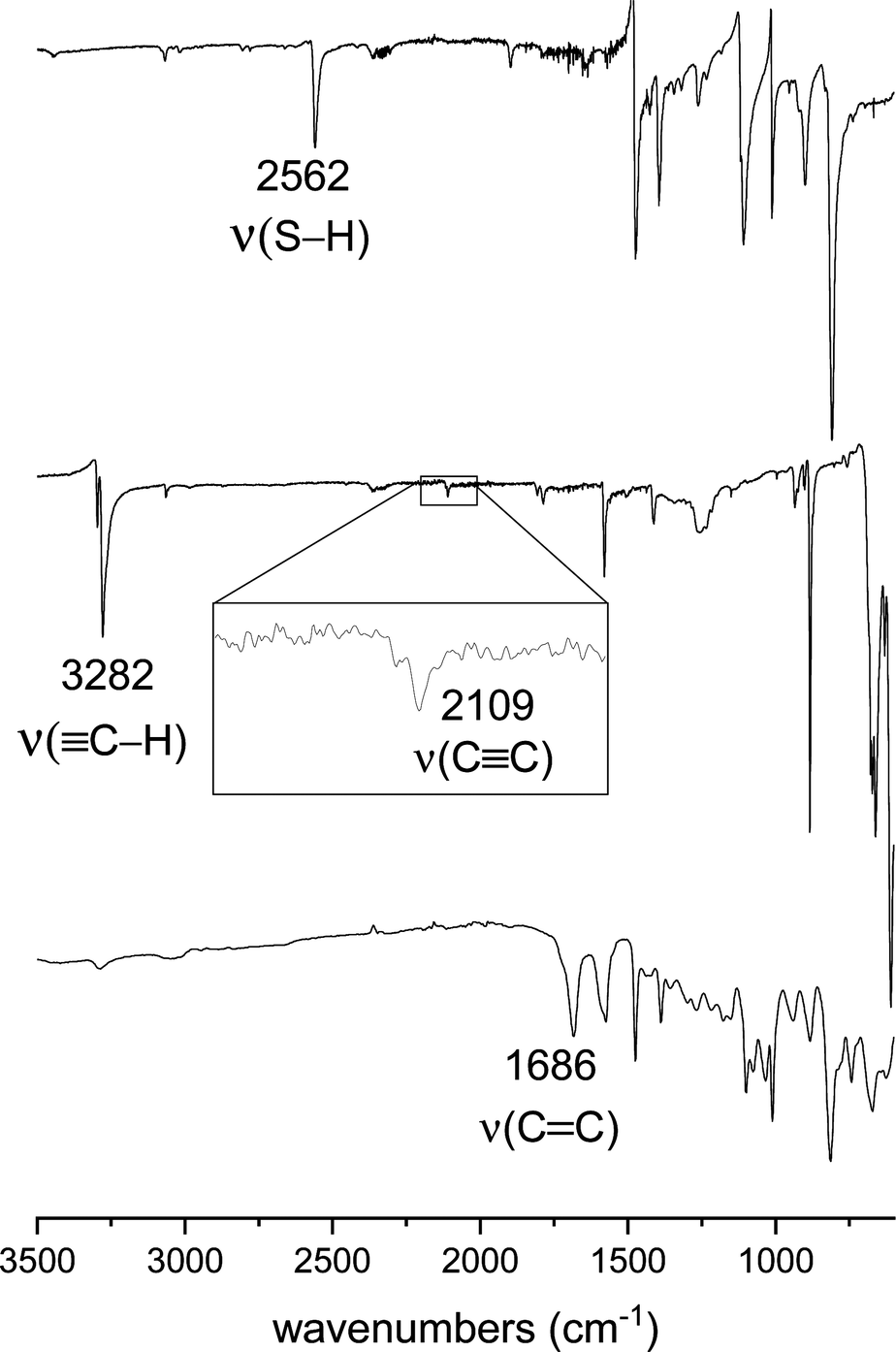 | ||
| Fig. 1 FTIR spectra of (from bottom to top) SOP-2 and its monomers, 1,3,5-triethynylbenzene (4) and 1,4-dimercaptobenzene (1). | ||
The lack of solubility of SOPs 1–4 in common organic solvents precluded their characterization by liquid-state NMR. Thus, solid-state CP-MAS 13C NMR spectroscopy was instead used. In all cases, the corresponding spectra of SOPs 1–4 shown in Fig. 2, Fig. S2 and S3 (ESI†) present signals in the range of 120–140 ppm. These chemical shifts correspond collectively to all sp2 carbon atoms associated with structural aromatic moieties. Note that in the spectra of SOP-2 and SOP-4, the peaks corresponding to the CC–S vinly sulfide groups strongly overlap with the signals coming from aromatic ring units. Accordingly, a broad band is observed in the 120–140 ppm range, resulting in a more complex envelope than in the spectra of SOP-1 and SOP-3, in which these vinyl sulfide moieties are absent. Also note that no additional peaks are observed at 45–50 ppm in the spectra of SOP-2 and SOP-4, where signals corresponding to the two-step addition of two thiols to each ethynyl group would be expected. This indicates that the formation of the branched polymer does not take place to a significant extent, in agreement with the elemental analysis discussed above. The spectra of SOP-1 and SOP-3 show additional signals at 38.8 ppm that correspond to the benzylic carbons. Finally, a minor signal appears at 185.2 ppm in the spectrum of SOP-1, indicative of a small fraction of free –SH groups associated with point defects. Note that the presence of these uncoordinated thiol groups was not evidenced due to the relatively small intensity of the ν(SH) absorption band in pure bismuthiol (2472 cm−1) in the corresponding FTIR spectrum (see Fig. S1, ESI†).
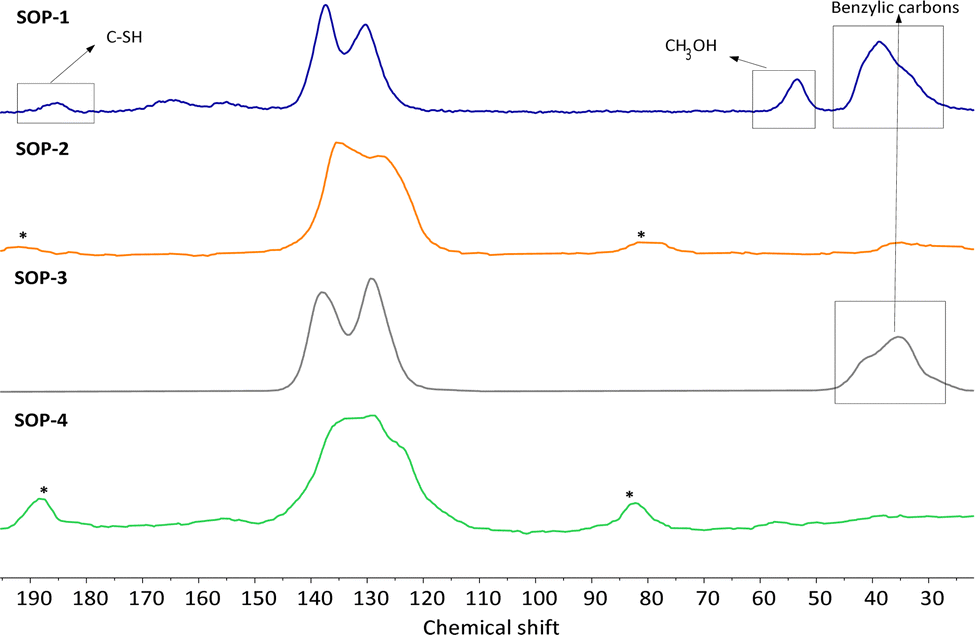 | ||
| Fig. 2 13C CP/MAS NMR spectra and deconvolution of SOP-type materials. Asterisks correspond to spinning bands. | ||
Thermal stability of the SOPs was evaluated by thermogravimetric analysis (TGA) under an air flux, as shown in Fig. 3. In all cases, degradation of the organic units started in the 260 to 380 °C range, with the polymers derived from bismuthiol (SOP-1 and SOP-4) exhibiting lower thermal stability than SOP-2 and SOP-3. This fact would be associated with the intrinsic higher stability of benzylic units included in the framework compared with the lower stability characteristic of nitrogen–sulfur aromatic heterocycles.
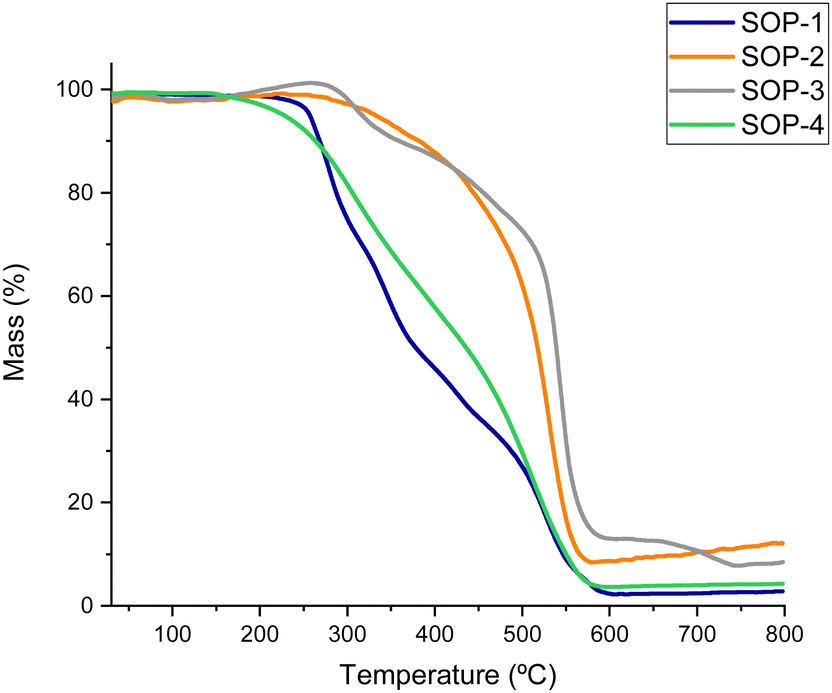 | ||
| Fig. 3 Thermogravimetric analysis (TGA) of SOPs. | ||
The porosity of the SOPs was analyzed by CO2 physical adsorption at 0° up to 1 bar, as shown in Fig. 4. The corresponding specific surface areas were estimated from the isotherms by using the Dubinin–Astakhov equation.57 The measured CO2 capacities (at 1 bar) corresponded to calculated surface areas of 44, 180, 15 and 80 m2 g−1 for SOPs 1–4, respectively. That is, the two compounds containing vinyl sulfide moieties are better adsorbents for CO2 than the other two SOPs prepared from 1,3,5-tris(bromomethyl)benzene. This fact is associated with the probable formation of more ordered rigid networks (SOP-2 and SOP-4), which would imply the generation of more regular microporous internal cavities with higher CO2 adsorption capacity. In the case of SOP-type materials based on non-conjugated flexible linkages (SOP-1 and SOP-3), sulphide organic polymers with more marked internal disorganizations were probably obtained. It is interesting to point out that none of the SOPs were able to adsorb any significant amount of N2 at low temperature (77 K) or H2O (at room temperature). Thus, compounds such as SOP-2 could have great potential for the selective separation of gas mixtures, including N2/CO2 or CO2 capture from humid air,58 which are currently being explored in our lab.
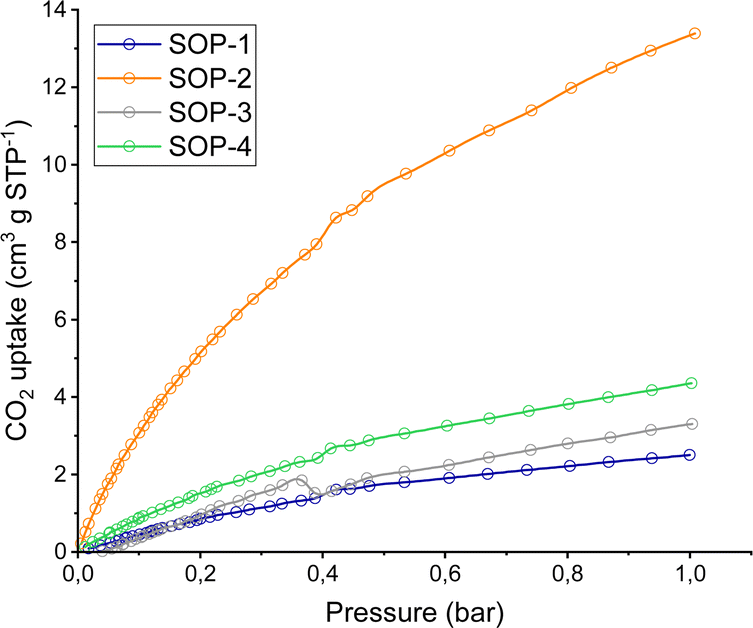 | ||
| Fig. 4 CO2 uptake isotherms of SOPs at 1 bar and 0 °C. | ||
Finally, Fig. 5 shows the representative scanning electron microscopy (SEM) images of SOPs 1–4. Uniform discrete spherical particles with a size of about 500 and 700 nm were formed in all cases, which tended to aggregate in the case of SOP-3. The surface of the resulting SOP materials was generally not smooth, but the spherical particles were decorated with smaller particles.
 | ||
| Fig. 5 SEM images of SOPs (a) and TEM images of SOPs immersed in 2-propanol (b). | ||
Catalytic performance of SOPs
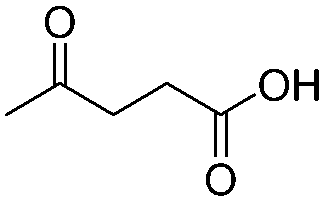
To start exploring the potential of SOPs as heterogeneous catalysts, we have first considered the esterification of levulinic acid with ethanol over SOPs 1–4 (Scheme 3). The results obtained are summarized in Table 2. To our delight, all the SOPs catalyze the esterification reaction, affording levulinate ester as the sole product in good yields. The best performing catalysts were SOP-1 and SOP-3; i.e., the two SOPs derived from 1,3,5-tris(bromomethyl) benzene ligands, for which conversion of levulinic acid above 93% was achieved after 24 h of reaction. | ||
| Scheme 3 Esterification reaction step. | ||
| Catalyst | Substrate | Conversion (mol%) | TOF (h−1) |
|---|---|---|---|
| a Reaction conditions: carboxylic acid (1 mmol), ethanol (0.9 mL, 15 eq.), 6 mol% catalyst based on S, stirring at 78 °C. | |||
| SOP-1 |
|
95 | 0.74 |
| SOP-2 | 78 | 0.73 | |
| SOP-3 | 93 | — | |
| SOP-4 | 40 | — | |
| SOP-1 |
|
||
The time-conversion plots obtained for the esterification of levulinic acid over SOP-1 and SOP-2 are shown in Fig. 6. Turnover frequencies (TOF) were calculated at a low conversion of the carboxylic acid and assuming that the active sites for this reaction are the sulfide centers connecting the structural units of the SOPs (see discussion below). The obtained TOFs were very similar for both materials (0.74 h−1 and 0.73 h−1 for SOP-1 and SOP-2, respectively) and revealed a very respectable catalytic performance of the SOPs in the esterification reaction.
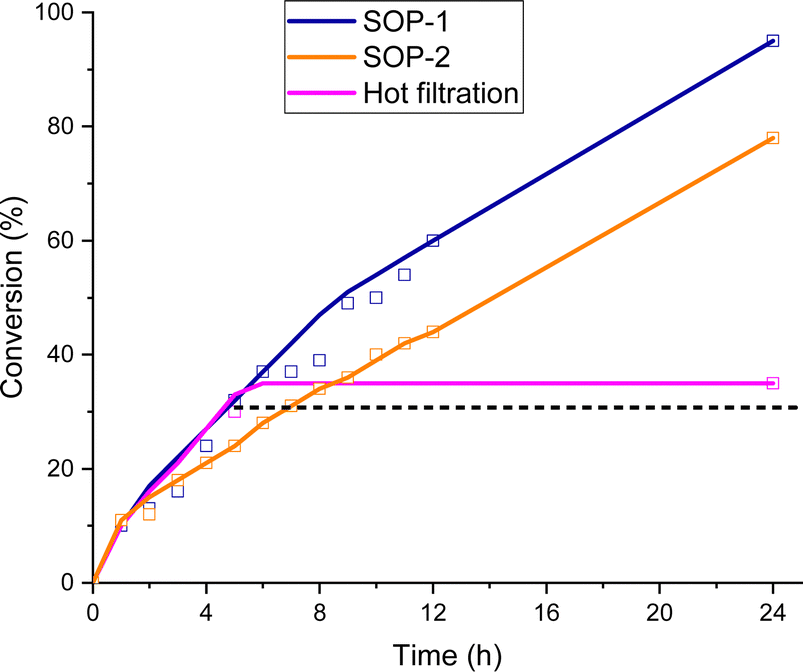 | ||
| Fig. 6 Time-conversion of levulinic acid to ethyl levulinate over SOP-1 and SOP-2. | ||
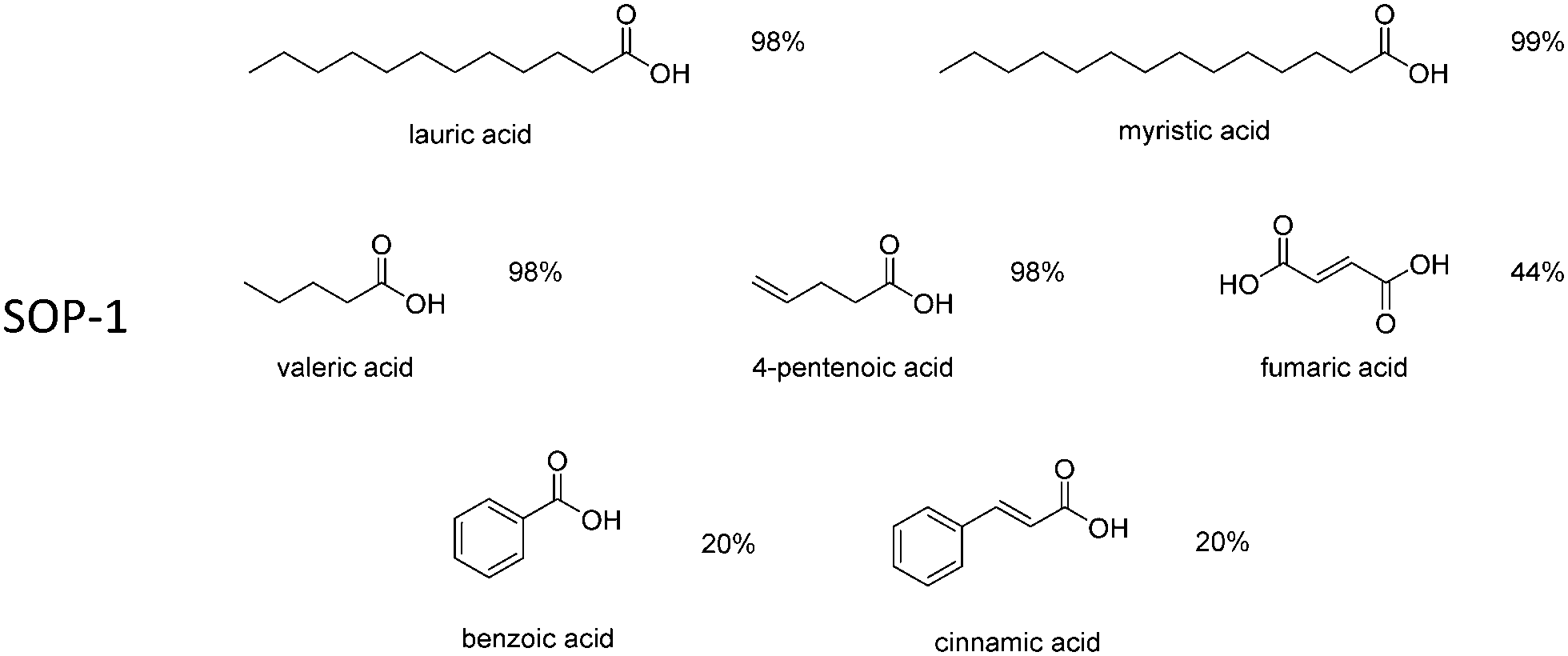
Encouraged by these results, the scope of the reaction was further evaluated for the esterification of various carboxylic acids with ethanol, using SOP-1 as the catalyst. Long chain fatty acids (namely, lauric and myristic acid) were quantitatively esterified in less than 24 h. Excellent ester yields were also attained within 24 h for other alkyl carboxylates (valeric and 4-pentenoic acids), but the reaction was considerably slower for substrates bearing phenyl rings, such as benzoic and cynnamic acids. This is probably due to the steric hindrance introduced by the aromatic ring, which could make it difficult to access the catalytic site of the SOP. Ethyl ester conversion for fumaric acid was lower (44%) because, in general, esterification of dicarboxylic acids occurred in both carboxylic acid groups of these substrates, therefore, necessitating more reaction time to reach complete conversion.
Note that, as commented above, all SOPs are highly hydrophobic, they do not adsorb any sensible amounts of moisture or other atmospheric contaminants in the open air (see TGA curves in Fig. 3), while isothermal H2O adsorption experiments revealed the lack of adsorbed water even on SOPs activated under vacuum. This hydrophobicity is likely beneficial for the esterification reaction, since the water generated as a by-product during the reactions is rapidly removed from the solid catalyst and the equilibrium is shifted towards product formation.
The reusability of SOPs is demonstrated in Fig. 7 for the esterification of lauric acid over SOP-1, although all the catalysts and substrates studied here showed analogous behavior. The catalytic activity was preserved almost completely for the first three catalytic runs, while a slightly lower activity was observed in the 4th cycle: the conversion at 5 h of reaction decreased from 30% in the first three runs down to 14% in the 4th cycle. Nevertheless, complete conversion of lauric acid was still achieved in the 4th run when the reaction time was extended to 32 h. These results showed that the slight decrease of the catalytic activity after the 3rd run is most likely due to the partial blocking of the active sites by adsorbed reactants and products not completely removed during the washing process between two consecutive runs, which accumulate on the surface of the catalyst. In fact, after the 1st catalytic cycle, the recovered catalyst was analyzed by elemental analysis, which shows a slight increase of % C content (see Table S1, ESI†). For this, after the 4th catalytic cycle, the catalyst was filtered and washed with a 10% HCl solution in ethanol (3 × 10 mL), then dried at 100 °C in a vacuum and used directly in a consecutive reaction cycle. Due to this final washing step, it was possible to recover the initial reactivity of SOP-type materials associated with the elimination of adsorbed organic compounds.
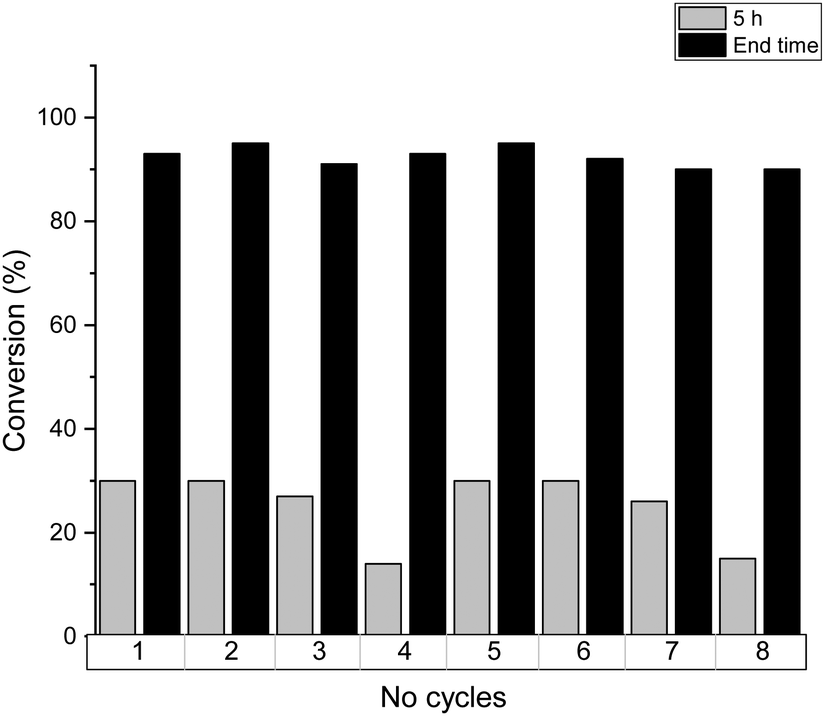 | ||
| Fig. 7 Reusability of the SOP-1 catalyst for the lauric acid esterification at 5 hours and end time. After 4th cycle, the catalyst was filtered and washed with a 10% HCl solution in ethanol (3 × 10 mL), then dried at 100 °C in a vacuum and used directly in a consecutive cycle. | ||
Moreover, the leaching test by hot filtration of the reaction slurry did not show any loss of active sites from the solid catalyst, being analysed by percolating waters, which did not contain active sulphur moieties from elemental analysis, confirming the heterogeneity of the catalytic system. Therefore, leaching tests corroborated the stability of SOP-type materials since the achieved conversion remained constant after the hot filtration step (Fig. 6). Finally, different characterization results of the used catalysts, from elemental analysis and NMR spectroscopy, were carried out and compared with the as-synthesized materials, evidencing that no substantial alteration was detected in the composition and physicochemical nature of the SOP-type materials after their use during several catalytic reaction cycles (Fig. S4 and Table S1, ESI†).
Origin of the catalytic activity of the SOP compounds: reaction mechanism
The catalytic properties of transition metal sulfides are well known since more than a century,59 as well as several organosulfur compounds that can be used as ligands or promoters, including sulfoxides, sulfonamides, N-sulfinyl ureas, thioureas, sulfoximines, etc.60 In particular, the use of sulfides as (metal-free) catalysts is also well documented,61 but it usually requires first the conversion of the sulfide into a sulfur ylide by either reacting with a carbene or carbenoid, or by alkylation and deprotonation with a base. After that, the in situ formed sulfur ylide is contacted with the desired reagents, and the starting sulfide is recovered at the end of the reaction. Applications for sulfur ylides as organocatalysts include epoxidations, aziridations and cyclopropanations. However, beyond the ylide route the existing reports on direct application of sulfides are very scarce, and they usually participate as additives together with a Lewis acid catalyst. For instance, in the Morita–Baylis–Hilman reaction together with TiCl4, or in electrophilic thiolation of alkynes in the presence of TMSOTf as the Lewis acid co-catalyst.62 Therefore, to the best of our knowledge, the reactivity of SOPs observed herein has never been described before.First, we wanted to know whether the catalytic activity observed here for the esterification reaction was a fortunate case of the SOP compounds, or if it can be generalized to other sulfide compounds. To this end, we carried out the esterification of lauric acid in the presence of either dimethyl- or diphenylsulfide as catalysts under the same reaction conditions used for the SOPs. Although the yields of ethyl laurate obtained were low (17% and 20% after 24 h of reaction for Me2S and Ph2S, respectively), they were still clearly above that obtained in a blank experiment (9% conversion), so both compounds showed a definite catalytic activity. Therefore, the catalytic activity observed for SOPs 1–4 can most likely be extended to other sulfide compounds as well.
Furthermore, SOP-type materials were compared with other homogeneous catalysts, such as p-toluene sulfonic acid, evidencing that higher conversions in a short time were achieved, although with the disadvantage of not being able to reuse it. Comparison with other heterogeneous solid catalysts, such as H-BEA zeolite (39% yield) or UiO-66-(COOH)2 MOF-type material (90% yield) is also shown in Table 3. It can be deduced that SOPs exhibited a high reactivity, selectivity and efficient recyclability for esterification reactions through a proposed electrophilic mechanism based on hypervalent S⋯O interactions.
In order to understand the origin of this catalytic activity, we have considered various possible reaction mechanisms. First, it is well known that the esterification reaction can be catalyzed by Brønsted acids (such as mineral acids or sulfonic-based compounds). However, potentiometric acid–base titration experiments (see Fig. S5, ESI†) ruled out the presence of any significant amount of acid sites in the SOPs, which could arise from uncoordinated –SH groups. We then considered a nucleophilic mechanism, in which the sulfide groups of SOPs could activate the carboxylic acid through the addition and elimination of water with the formation of a thiocarboxylate zwittterion intermediate. This mechanism would be somewhat related to that found for other Lewis bases, such as DMAP in the Steglich or Yamaguchi esterification mechanisms.63 However, this mechanism was soon discarded by ab initio computational calculations with model clusters, which evidenced that sulfide atoms have no tendency at all to attack the carboxylate carbon atom.
We thus propose an alternative electrophilic mechanism to explain the catalytic activity observed in SOPs (and, to a minor extent, in other sulfide compounds), based on hypervalent S⋯O (nO → σS*) interactions between the carboxylic acid and the sulfide centers. In this way, the sulfur atoms of the SOP would accept electron density from the CO oxygen atom, thus acting as Lewis acid centers in the broad sense of the term. This would increase the electrophilic character of the carboxylate carbon atom, facilitating the ensuing attack by the alcohol and formation of the ester product. In order to corroborate this matter, impregnation of butyric acid on two solid supports (SOP-type material and inert silica) was performed, observed in the obtained IR experimental tests a significant bathochromic shift of the characteristic vibration band (CO) of butyric acid for the SOP, confirming the effective interaction between our catalyst and the carboxylic acids used as reaction reagents. This fact would corroborate the existence of effective interactions between carboxylic groups and the sulfide active moieties of the SOPs, which would act as Lewis acid sites (Fig. S6, ESI†).
Hypervalent interactions are well known to occur in other main groups elements, such as Si, P, I, Al, C or B, and the properties of these hypervalent compounds have been successfully used in catalysis.64–70 In the case of sulfur, hypervalent S⋯O interactions are also reported, and they are known to control the molecular assembly of sulfur compounds in solution and in the solid state, as well as the structure and reactivity of isolated molecules. In proteins, nonbonded S⋯O hypervalent interactions between methionine and cystine (with S–S disulfide bridge) are also important factors controlling the protein folding architecture and reactivity,73 with calculated interaction energies on the order of 3.2–2.5 kcal mol−1.74
Therefore, based on the formation of hypervalent S⋯O interactions between the sulfide groups of the SOPs and the carboxylate reaction substrate, we propose the reaction mechanism shown in Scheme 4.
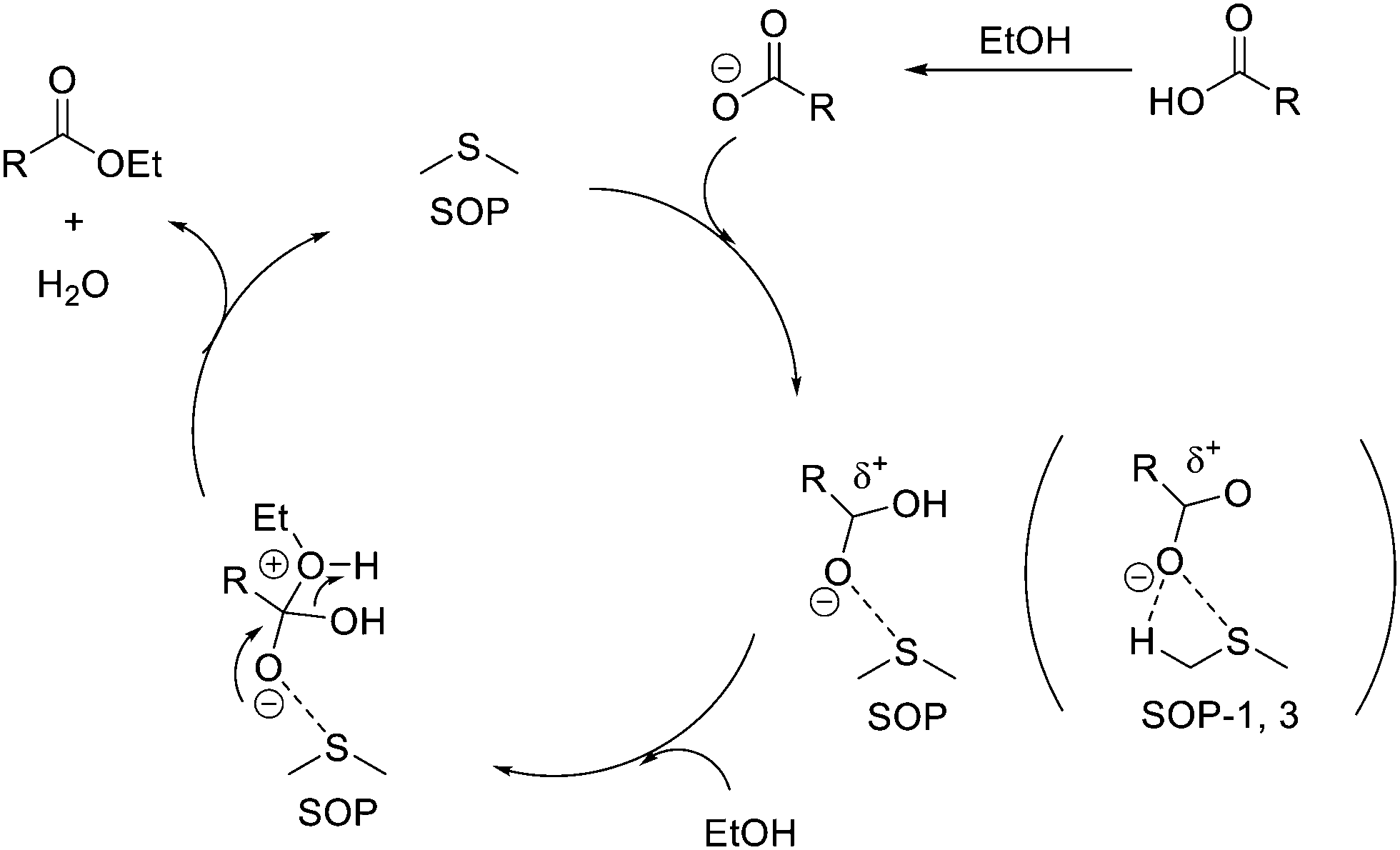 | ||
| Scheme 4 Proposed hypervalent S–O interaction mechanism. | ||
According to the above mechanism, SOPs would act as Lewis acid catalysts (electrophilic mechanism), activating the carboxylic group and increasing the electrophilic character (δ+) of the carbon atom through S⋯O hypervalent interactions. This would facilitate the addition of the alcohol and formation of the ester upon elimination of a water molecule and recovering the intact sulfide. Note also that the presence of available alpha H atoms in both SOP-1 and SOP-3 could further stabilize the hypervalent interaction through additional C–O⋯H contacts. These alpha H atoms are not available in SOP-2 and SOP-4, and this might explain their lower catalytic activity with respect to SOP-1 and SOP-3 (Table 2). We have currently undertaken computational studies to lend support to the proposed mechanism, which is also backed up by results on other Lewis acid catalyzed reactions with carbonyl compounds (viz., Paal–Knorr and carbonyl–ene isomerization) that will be presented elsewhere.
Experimental
Materials and methods
1,3,5-Triethynylbenzene and 1,4-dimercaptobenzene were purchased from ABCR and Alfa Aesar, respectively. Bismuthiol, 1,3,5-tris(bromomethyl)benzene, levulinic acid, valeric acid, benzoic acid, lauric acid, 4-pentenoic acid, trans-cinnamic acid, myristic acid and 2-methylvaleric acid were purchased from Sigma Aldrich. Sodium hydroxide, solvents and PTFE syringe filters were purchased from Scharlab.Synthesis of compounds
Characterization techniques
CHN content was determined with a Carlo Erba 1106 elemental analyzer. Thermogravimetric and differential thermal analysis (TGA-DTA) were performed in an air stream with a Mettler Toledo TGA/SDTA 851E analyzer. Solid state MAS-NMR spectra were obtained at room temperature under magic angle spinning (MAS) in a Brucker AV-400 spectrometer. IR spectra were determined with a Bruker Tensor 27 FT-IR spectrometer. Catalytic results were obtained in an Agilent 8860 GC with a carbowax column. The adsorption isotherms were measured in a Micromeritics ASAP 2010 instrument using approximately 200 mg of the adsorbent placed in a sample holder that was immersed in a liquid circulation thermostatic bath for precise temperature control. Before each measurement, the sample was treated overnight at 673 K under vacuum. CO2 adsorption isotherms were then acquired at 273 K.Catalytic experiments
Conclusion
In this article, we reported, for the first time, the synthesis via click processes of four hydrophobic organosulfide materials named SOP-1–4. SOP-1 and SOP-3 have been synthesized through SN2 reactions, while SOP-2 and SOP-4 by thiol–yne reactions. Characterization data, specifically elemental analysis, 13C NMR and IR confirmed the effective incorporation and assembly of the organic sulfide units used in the synthesis to form a fully coordinated structure of the SOP family. SEM and TEM images showed a morphology based on uniform discrete spherical particles without crystallinity, although CO2 sorption experiments evidenced an appreciable and accessible specific surface area. Sulfide-based materials have been shown to exhibit high reactivity, selectivity and reusability for esterification reactions through a proposed electrophilic mechanism based on hypervalent S⋯O interactions, mimicking similar catalytic routes observed in proteins. Definitively, this novel family of sulfide organic polymers opens the door to the generation of new types of heterogeneous catalysts based on sulfur units acting as effective electrophilic sites. Other applications, such as adsorption–separation processes, should also be considered, taking into account the high hydrophobicity exhibited by SOP-type materials.Author contributions
M. Melero: investigation, data curation, formal analysis, methodology, visualization, and writing – original draft; U. Díaz: conceptualization, data curation, validation, funding acquisition, project administration, supervision, and writing – review & editing; F. X. Llabrés i Xamena: conceptualization, data curation, validation, funding acquisition, project administration, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Science and Innovation (PID2020-112590GB-C21 and CEX2021-001230-S grants funded by MCIN/AEI/10.13039/501100011033) is gratefully acknowledged. This study forms part of the Advanced Materials Program and was supported by MICIN with funding from European Union NextGeneration (PRTR-C17.I1) and by the Generalitat Valenciana (MFA/2022/003). M. M. acknowledges financial support from FPI PhD fellowship PRE2018-084071.Notes and references
- A. Bhunia, V. Vasylyeva and C. Janiak, From a supramolecular tetranitrile to a porous covalent triazine-based framework with high gas uptake capacities, Chem. Commun., 2013, 49, 3961–3963 RSC.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Porous, Fluorescent, Covalent Triazine-Based Frameworks Via Room-Temperature and Microwave-Assisted Synthesis, Adv. Mater., 2012, 24, 2357–2361 CrossRef CAS PubMed.
- G. Mukherjee, J. Thote, H. B. Aiyappa, S. Kandambeth, S. Banerjee, K. Vanka and R. Banerjee, A porous porphyrin organic polymer (PPOP) for visible light triggered hydrogen production, Chem. Commun., 2017, 53, 4461–4464 RSC.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, An Azine-Linked Covalent Organic Framework, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed.
- S. Ding, M. Dong, Y. Wang, Y. Chen, H. Wang, C. Su and W. Wang, Thioether-Based Fluorescent Covalent Organic Framework for Selective Detection and Facile Removal of Mercury(II), J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed.
- J. Zhang, X. Han, X. Wu, Y. Liu and Y. Cui, Multivariate Chiral Covalent Organic Frameworks with Controlled Crystallinity and Stability for Asymmetric Catalysis, J. Am. Chem. Soc., 2017, 139, 8277–8285 CrossRef CAS PubMed.
- Y. Tian and G. Zhu, Porous Aromatic Frameworks (PAFs), Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- C. D. Wood, B. Tan, A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Kjimyak, N. L. Campbell, R. Kirk, E. Stöckel and A. I. Cooper, Hydrogen Storage in Microporous Hypercrosslinked Organic Polymer Networks, Chem. Mater., 2007, 19, 2034–2048 CrossRef CAS.
- C. Tan, X. Han, Z. Li, Y. Liu and Y. Cui, Controlled Exchange of Achiral Linkers with Chiral Linkers in Zr-Based UiO-68 Metal–Organic Framework, J. Am. Chem. Soc., 2018, 140, 16229–16236 CrossRef CAS PubMed.
- M. Li, F. Wang, Z. Gu and J. Zhang, Synthesis of homochiral zeolitic metal–organic frameworks with amino acid and tetrazolates for chiral recognition, RSC Adv., 2017, 7, 4872–4875 RSC.
- S. Royuela, J. Almarza, M. J. Mancheño, J. C. Pérez-Flores, E. G. Michel, M. M. Ramos, F. Zamora, P. Oncón and J. L. Segura, Synergistic Effect of Covalent Bonding and Physical Encapsulation of Sulfur in the Pores of a Microporous COF to Improve Cycling Performance in Li-S Batteries, Chem. – Eur. J., 2019, 25, 12394–12404 CrossRef CAS PubMed.
- Z. Li, H. Zhou, F. Zhao, T. Wang, X. Ding, B. Han and W. Feng, Three-dimensional Covalent Organic Frameworks as Host Materials for Lithium-Sulfur Batteries, Chin. J. Polym. Sci., 2020, 38, 550–557 CrossRef CAS.
- Y. Meng, G. Lin, H. Liao and C. Wang, Impregnation of sulfur into a 2D pyrene-based covalent organic framework for high-rate lithium–sulfur batteries, J. Mater. Chem. A, 2018, 6, 17186–17191 RSC.
- D. Wang, N. Li, Y. Hu, S. Wan, M. Song, G. Yu, Y. Jin, W. Wei, K. Han, G. Kuang and W. Zhang, Highly Fluoro-Substituted Covalent Organic Framework and Its Application in Lithium–Sulfur Batteries, ACS Appl. Mater. Interfaces, 2018, 10, 42233–42240 CrossRef CAS PubMed.
- G. Lorusso, J. W. Sharples, E. Palacios, O. Roubeau, E. K. Brechin, R. Sessoli, A. Rossin, F. Tuna, E. J. L. Mclnnes, D. Collison, M. Evangelisti and A. Dense, Metal-Organic Framework for Enhanced Magnetic Refrigeration, Adv. Mater., 2013, 25, 4653–4656 CrossRef CAS PubMed.
- S. He, T. Zeng, S. Wang, H. Niu and Y. Cai, Facile Synthesis of Magnetic Covalent Organic Framework with Three-Dimensional Bouquet-Like Structure for Enhanced Extraction of Organic Targets, ACS Appl. Mater. Interfaces, 2017, 9, 2959–2965 CrossRef CAS PubMed.
- Y. Zhang, S. N. Riduan and J. Wang, Redox Active Metal– and Covalent Organic Frameworks for Energy Storage: Balancing Porosity and Electrical Conductivity, Chem. – Eur. J., 2017, 23, 16419–16431 CrossRef CAS PubMed.
- W. Zhang, D. Banerjee, J. Liu, H. T. Schaef, J. V. Crum, C. A. Fernandez, R. K. Kukkadapu, Z. Nie, S. K. Nune, R. K. Motkuri, K. W. Chapman, M. H. Engelhard, J. C. Hayes, K. L. Silvers, R. Krishna, B. P. McGrail, J. Liu and P. K. Thallapally, Redox-Active Metal-Organic Composites for Highly Selective Oxygen Separation Applications, Adv. Mater., 2016, 28, 3572–3577 CrossRef CAS PubMed.
- J. M. Moreno, A. Velty and U. Díaz, MOFs based on 1D structural sub-domains with Brønsted acid and redox active sites as effective bi-functional catalysts, Catal. Sci. Technol., 2020, 10, 3572 RSC.
- A. Bhunia, S. Dey, J. M. Moreno, U. Díaz, P. Concepcion, K. Hecke, C. Janiak and P. Van Der Voort, A homochiral vanadium-salen based cadmium bpdc MOF with permanent porosity as an asymmetric catalyst in solvent-free cyanosilylation, Chem. Commun., 2016, 52, 1401 RSC.
- J. R. Cabrero-Antonino, T. García, P. Rubio-Marqués, J. A. Vidal-Moya, A. Leyva-Pérez, S. S. Al-Deyab, S. I. Al-Resayes, U. Díaz and A. Corma, Synthesis of Organic−Inorganic Hybrid Solids with Copper Complex Framework and Their Catalytic Activity for the S-Arylation and the Azide−Alkyne Cycloaddition Reactions, ACS Catal., 2011, 1, 147–158 CrossRef CAS.
- B. P. Biswal, H. D. Chaudhari, R. Banerjee and U. K. Kharul, Chemically Stable Covalent Organic Framework (COF)-Polybenzimidazole Hybrid Membranes: Enhanced Gas separation through Pore Modulation, Chem. – Eur. J., 2016, 22, 4695–4699 CrossRef CAS PubMed.
- L. Feng, C. Qian and Y. Zhao, Recent Advances in Covalent Organic Framework-Based Nanosystems for Bioimaging and Therapeutic Applications, ACS Mater. Lett., 2020, 2, 1074–1092 CrossRef CAS.
- R. Tao, X. Shen, Y. Hu, K. Kang, Y. Zheng, S. Luo, S. Yang, W. Li, S. Lu, Y. Jin, L. Qiu and W. Zhang, Phosphine-Based Covalent Organic Framework for the Controlled Synthesis of Broad-Scope Ultrafine Nanoparticles, Small, 2020, 1906005 CrossRef CAS PubMed.
- J. Roeser, D. Prill, M. J. Bojdys, P. Fayon, A. Trewin, A. N. Fitch, M. U. Schmidt and A. Thomas, Anionic silicate organic frameworks constructed from hexacoordinate silicon centres, Nature, 2017, 9, 977–982 CAS.
- S. Ashraf, Y. Zuo, S. Li, C. Liu, H. Wang, X. Feng, P. Li and B. Wang, Crystalline Anionic Germanate Covalent Organic Framework for High CO2 Selectivity and Fast Li Ion Conduction, Chem. – Eur. J., 2019, 25, 13479–13483 CrossRef CAS PubMed.
- L. K. Ritchie, A. Trewin, A. Reguera-Galan, T. Hasell and A. I. Cooper, Synthesis of COF-5 using microwave irradiation and conventional solvothermal routes, Microporous Mesoporous Mater., 2010, 132, 132–136 CrossRef CAS.
- Y. Gui, J. Li, C. Guo, X. Li, Z. Lu, Z. Huang and A. New, Chiral Organosulfur Catalyst for Highly Stereoselective Synthesis of Epoxides, Adv. Synth. Catal., 2008, 350, 2483–2487 CrossRef CAS.
- R. Nishiyori, A. Tsuchihashi, A. Mochizuki, K. Kaneko, M. Yamanaka and S. Shirakawa, Design of Chiral Bifunctional Dialkyl Sulfide Catalysts for Regio-, Diastero-, and Enantioselective Bromolactonization, Chem. – Eur. J., 2018, 24, 16747–16752 CrossRef CAS PubMed.
- Y. Liu, Q. Li, H. Zhu, X. Feng, C. Peng, W. Huang, J. Li and B. Han, Direct Sulfide-Catalyzed Diasteroselective [4 + 1] Annulations of ortho-Quinone Methides and Bromides, J. Org. Chem., 2018, 83, 12753–12762 CrossRef CAS PubMed.
- Q. Li, X. Zhang, R. Zeng, Q. Dai, Y. Liu, Z. Shen, H. Leng, K. Yang and J. Li, Direct Sulfide-Catalyzed Enantioselective Cyclopropanations of Electron-Deficient Dienes and Bromides, Org. Lett., 2018, 20, 3700–3704 CrossRef CAS PubMed.
- S. Otsuka, K. Nogi, T. Rovis and H. Yorimitsu, Photoredox-Catalyzed Alkenylation of Benzylsulfonium Salts, Chem. – Asian J., 2019, 14, 532–536 CrossRef CAS PubMed.
- Z. Li, H. Zhou, F. Zhao, T. Wang, X. Ding, B. Han and W. Feng, Three-dimensional Covalent Organic Frameworks as Host Materials for Lithium-Sulfur Batteries, Chin. J. Polym. Sci., 2020, 38, 550–557 CrossRef CAS.
- S. Tanaka, Y. Furusho and T. Endo, Radical ring-opening polymerization of five-membered cyclic vinyl sulfone using p-toluenesulfonyl halides, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 222–227 CrossRef CAS.
- S. Gürsoy, A. Cihan, M. B. Koçak and Ö. Bekaroğlu, Synthesis of New Metal-Free and Metal-Containing Phthalocyanines with Tertiaryoy Quaternary aminoethyl Substituents, Monatsh. Chem., 2001, 132, 813–819 CrossRef.
- L. Yang, L. Han, H. Ma, P. Liu, H. Shen, C. Li, S. Zhang and Y. Li, Synthesis of Alkyne-functionalized Polymers via Living Anionic Polymerization and Investigation of Features during the Post-“thiol-yne” Click Reaction, Chin. J. Polym. Sci., 2019, 37, 841–850 CrossRef CAS.
- A. B. Lowe, Thiol-yne ‘click’/coupling chemistry and recent applications in polymer and materials synthesis and modification, Polymer, 2014, 55, 5517–5549 CrossRef CAS.
- N. Naga, H. Tanaka and K. Moriyama, Synthesis of network polymers from multifunctional aromatic thiol compounds, J. Electrochem. Soc., 2019, 166, 3079–3083 CrossRef.
- J. Y. Zhang, L. B. Kong, L. Z. Zhan, J. Tang, H. Zhan, Y. H. Zhou and C. M. Zhan, Sulfides organic polymer: Novel cathode active material for rechargeable lithium batteries, J. Power Sources, 2007, 168, 278–281 CrossRef CAS.
- G. Wang, C. Li and H. Shan, Highly Efficient Metal Sulfide Catalysts for Selective Dehydrogenation of Isobutane to Isobutene, ACS Catal., 2014, 4, 1139–1143 CrossRef CAS.
- D. Li, C. Li, L. Zhang, H. Li, L. Zhu, D. Yang, Q. Fang, S. Qiu and X. Yao, Metal-Free Thiophene-Sulfur Covalent Organic Frameworks: Precise and Controllable Synthesis of Catalytic Active Sites for Oxygen Reduction, J. Am. Chem. Soc., 2020, 142, 8104–8108 CrossRef CAS PubMed.
- A. Enríquez, A. M. González-Vadillo, I. Martínez-Montero, S. Bruña, L. Leemans and I. Cuadrado, Efficient Thiol-Yne Click Chemistry of Redox-Active Ethynylferrocene, Organometallics, 2014, 33, 7307–7317 CrossRef.
- A. Yoshimura, Y. Takamachi, L. Han and A. Ogawa, Organosulfide-Catalyzed Diboration of Terminal Alkynes under Light, Chem. – Eur. J., 2015, 21, 13930–13933 CrossRef CAS PubMed.
- N. Iranpoor, H. Firouzabadi and S. Farahi, Sulfonated charcoal as a mild and efficient catalyst for esterification and trans-esterification reactions, J. Sulfur Chem., 2007, 6, 581–587 CrossRef.
- L. Guo, S. Jia, C. S. Diercks, X. Yang, S. A. Alshmimri and O. M. Yaghi, Amidation, Esterification, and Thioesterification of a Carboxyl-Functionalized Covalent Organic Framework, Angew. Chem., Int. Ed., 2020, 59, 2023–2027 CrossRef CAS PubMed.
- R. M. N. Kalla, M. Kim and I. Kim, Sulfonic Acid-Functionalized, Hyper-Cross-Linked Porous Polyphenols as Recyclable Solid Acid Catalysts for Esterification and Transesterification Reactions, Ind. Eng. Chem. Res., 2018, 57, 11583–11591 CrossRef CAS.
- T. M. M. Marso, C. S. Kalpage and M. Y. Udugala-Ganehenege, Application of Chromium and Cobalt Terephtalate Metal Organic Frameworks as Catalysts for the Production of Biodiesel from Calophyllum inophyllum Oil in High Tield Under Mild Conditions, J. Inorg. Organomet. Polym. Mater., 2020, 30, 1243–1265 CrossRef CAS.
- S. Maiti, A. R. Chowdhury and A. K. Das, Benzoselenadiazole-based nanoporous Covalent Organic Polymer (COP) as efficient room temperature heterogeneous catalyst for biodiesel production, Microporous Mesoporous Mater., 2019, 283, 39–47 CrossRef CAS.
- J. Chen, R. Liu, H. Gao, L. Chen and D. J. Yeb, Amine-functionalized metal-organic frameworks for the transesterification of triglicerides, J. Mater. Chem. A, 2014, 2, 7205–7213 RSC.
- F. G. Cirujano, A. Corma and F. X. Llabrés i Xamena, Conversion of levulinic acid into chemicals: Synthesis of biomass derived levulinate esters over Zr-containing MOFs, Chem. Eng. Sci., 2015, 124, 52–60 CrossRef CAS.
- J. Kou and L. B. Sun, Fabrication of Metal-Organic Frameworks inside Silica Nanopores with Significantly Enhanced Hydrostability and Catalytic Activity, ACS Appl. Mater. Interfaces, 2018, 10, 12051–12059 CrossRef CAS PubMed.
- F. G. Cirujano, A. Corma and F. X. Llabrés i Xamena, Zirconium-containing metal organic frameworks as solid acid catalysts for the esterification of free fatty acids: Synthesis of biodiesel and other compounds of interest, Catal. Today, 2015, 257, 213–220 CrossRef CAS.
- L. R. Macgillivray, Metal-Organic Frameworks: Design and Application, Wiley, 2010, p. 368 Search PubMed.
- F. G. Cirujano and F. X. Llabrés i Xamena, Tuning the Catalytic Properties of UiO-66 Metal–Organic Frameworks: From Lewis to Defect-Induced Brønsted Acidity, J. Phys. Chem. Lett., 2020, 11, 4879–4890 CrossRef CAS PubMed.
- F. Drache, F. G. Cirujano, K. D. Nguyen, V. Bon, I. Senkoyska, F. X. Llabrés i Xamena and S. Kaskei, Anion Exchanged and Catalytic Functionalization of the Zirconium-Based Metal-Organic Framework DUT-67, Cryst. Growth Des., 2018, 18, 5492–5500 CrossRef CAS.
- C. Caratelli, J. Hajek, F. G. Cirujano, M. Warroquier, F. X. Llabrés i Xamena and V. Van Speybroeck, Nature of active sites on UiO-66 and beneficial influence of water in the catalysis of Fischer esterification, J. Catal., 2017, 352, 401 CrossRef CAS.
- Y. H. Hu and E. Ruckenstein, Applicability of Dubinin–Astakhov equation to CO2 adsorption on single-walled carbon nanotubes, Chem. Phys. Lett., 2006, 425, 306–310 CrossRef CAS.
- R. van der Jagt, A. Vasileiadis, H. Veldhuizen, P. Shao, X. Feng, S. Ganapathy, N. C. Habisreutinger, M. A. van der Veen, C. Wang, M. Wagemaker, S. van der Zwaag and A. Nagai, Synthesis and Structure-Property Relationships of Polyimide Covalent Organic Frameworks for Carbon Dioxide Capture and (Aqueous) Sodium-Ion Batteries, Chem. Mater., 2021, 33, 818–833 CrossRef CAS PubMed.
- R. R. Chianelli, G. Berhault and B. Torres, Unsupported transition metal sulfide catalysts: 100 years of science and application, Catal. Today, 2009, 147, 275–286 CrossRef CAS.
- S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chiral Organosulfur Ligands/Catalysts with a Stereogenic Sulfur Atom: Applications in Asymmetric Synthesis, Chem. Rev., 2017, 117(5), 4147 CrossRef CAS PubMed.
- E. M. McGarrigle, E. L. Mylers, O. Illa, M. A. Shaw, Sl Riches and V. K. Aggarwal, Chalcogenides as Organocatalysts, Chem. Rev., 2007, 107(12), 5841–5883 CrossRef CAS PubMed.
- Y. Liang, J. Ji, X. Zhang, Q. Jiang, J. Luo and X. Zhao, Enantioselective Construction of Axially Chiral Amino Sulfide Vinyl Arenes by Chiral Sulfide-Catalyzed Electrophilic Carbothiolation of Alkynes, Angew. Chem., Int. Ed., 2020, 59, 4959 CrossRef CAS PubMed.
- A. Sakakura, K. Kawajiri, T. Ohkubo, Y. Kosugi and K. Ishihara, Widely Useful DMAP-Catalyzed Esterification under Auxiliary Base- and Solvent-Free Conditions, J. Am. Chem. Soc., 2007, 129, 14775–14779 CrossRef CAS PubMed.
- J. Tateiwa and A. Hosomi, Pentacoordinate Organosilicate-Catalyzed Michael Addition of β-Keto Esters to 3-Buten-2-one, Eur. J. Org. Chem., 2001, 1445–1448 CrossRef CAS.
- S. G. Nelson, T. J. Peelen and Z. Wan, Catalytic Asymmetric Acyl Halide-Aldehyde Cyclocondensations. A Strategy for Enantioselective Catalyzed Cross Aldol Reactions, J. Am. Chem. Soc., 1999, 121, 9742–9743 CrossRef CAS.
- S. G. Nelson, B. K. Kim and T. J. Peelen, Lewis Acidity Expressed in Neutral Electron-Rich Aluminium(III) Complexes: An Example of Ligand-Defined Catalysis, J. Am. Chem. Soc., 2000, 122, 9318–9319 CrossRef CAS.
- S. G. Nelson, C. Zhu and X. Shen, Catalytic Asymmetric Acyl Halide-Aldehyde Cyclocondensation Reactions of Substituted Ketenes, J. Am. Chem. Soc., 2004, 126, 14–15 CrossRef CAS PubMed.
- K. Maruoka and T. Ooi, The Synthetic Utility of the Hypercoordination of Boron and Aluminum, Chem. – Eur. J., 1999, 5, 829–833 CrossRef CAS.
- T. Ooi, D. Uraguchi, N. Kagoshima and K. Maruoka, Hypercoordination of Boron and Aluminium: Synthetic Utility as Chelating Lewis Acids, J. Am. Chem. Soc., 1998, 120, 5327–5328 CrossRef CAS.
- T. Ooi, N. Kagoshima and K. Maruoka, Fluorine-Assisted Selective Alkylation to Fluorinated Epoxides and Carbonyl Compounds: Implication of Pentacoordinate Trialkylaluminium Complexes, J. Am. Chem. Soc., 1997, 119, 5754–5755 CrossRef CAS.
- C. R. Patil, P. S. Niphadkar, V. V. Bokade and P. N. Joshi, Esterification of levulinic acid to ethyl levulinate over bimodal micro-mesoporous H/BEA zeolite derivatives, Catal. Commun., 2014, 43, 188–191 CrossRef CAS.
- F. Wang, Z. Chen, H. Chen, T. A. Goetjen, P. Li, X. Wang, S. Alayoglu, K. Ma, Y. Chen, T. Wang, T. Islamoglu, Y. Fang, R. Q. Snurr and O. K. Farha, Interplay of Lewis and Brønsted Acid Sites in Zr-Based Metal-Organic Frameworks for Efficient Esterification of Biomass-Derived Levulinic Acid, ACS Appl. Mater. Interfaces, 2019, 11, 32090–32096 CrossRef CAS PubMed.
- M. Iwaoka and N. Isozumi, Hypervalent Nonbonded Interactions of a Divalent Sulfur Atom. Implications in Protein Architecture and the Functions, Molecules, 2012, 17, 7266–7283 CrossRef CAS PubMed.
- M. Iwaoka, S. Takemoto, M. Okada and S. Tomoda, Weak Nonbonded S⋯X (X = O,N, and S) Interactions in Proteins. Statistical and Theoretical Studies, Bull. Chem. Soc. Jpn., 2002, 75, 1611–1625 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qm01127a |
| This journal is © the Partner Organisations 2023 |