
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of anisotropic crystals with π-conjugated rings toward giant birefringence
Yunqi
Zhao
ac,
Liangmeng
Zhu
ac,
Yanqiang
Li
ac,
Xiaojun
Kuang
 b,
Junhua
Luo
ac and
Sangen
Zhao
*ac
b,
Junhua
Luo
ac and
Sangen
Zhao
*ac
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China. E-mail: zhaosangen@fjirsm.ac.cn
bCollege of Chemistry and Bioengineering, Guilin University of Technology, Guilin, Guangxi 541004, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 25th May 2023
Abstract
Birefringent crystals play a significant role in linear optical devices due to their ability to modulate the polarization of light. Commercial birefringent crystals MgF2, α-BaB2O4, CaCO3, YVO4, TiO2, and LiNbO3 have been widely used as polarization devices in the past decades. A variety of crystals have been developed to meet the requirements of large birefringence and have great potential as optical functional crystals. However, a key question is how to balance the conflict between the energy gap and birefringence. In this review, aiming to better judge the integrated properties of optical crystals, we came up with the birefringent quality factor. We also summarised our recent findings on the birefringent crystals, which contain structural units similar to [B3O6]3− rings, from these perspectives, including crystal structure features, optical performances, and structure–property relationships. We summarised the strategy to achieve the balance between the energy gap and birefringence by adjusting the delocalized π-conjugation and the confined π-conjugation to improve the performance of birefringent crystals, which will open up a new window for the exploration of novel birefringent crystals.
 Yunqi Zhao | Yunqi Zhao received his Bachelor's degree from Dalian University of Technology in 2021. He is currently a Master's student at the University of Chinese Academy of Sciences. |
 Liangmeng Zhu | Liangmeng Zhu received his Bachelor's degree in Chemical Normal Major from Wenzhou University in 2022. He is currently a Master's student at the University of Chinese Academy of Sciences. |
 Yanqiang Li | Yanqiang Li is pursuing PhD degree at the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences. His PhD project focuses on nonlinear optical crystals and birefringence crystals. |
 Xiaojun Kuang | Xiaojun Kuang received his BSc in Chemistry from Nanchang University in 1999 and PhD in Inorganic Chemistry from Peking University in 2004. After his postdoctoral research in the University of Liverpool and University of Durham, he was appointed as an Associate Professor in the Chemistry College, Sun Yat-Sen University, in 2010 before he settled down in Guilin in 2013. His current research interests include the discovery of new oxide ion conductors, oxide and (oxy)nitride dielectrics, and their structure–property relationships. |
 Junhua Luo | Junhua Luo received his PhD from Fujian Institute of Research on the Structure of Matter (FJIRSM) in 2003, Chinese Academy of Science, followed by postdoctoral research at North Carolina State University and University of Florida from 2003 to 2005. From 2006 to 2009, he was funded by the Los Alamos National Laboratory at the Neutron Diffraction Center. He became Professor of chemistry at FJIRSM in 2009. His current research interests focus on development of photoelectric functional materials based on organic-inorganic hybrids. |
 Sangen Zhao | Sangen Zhao is a professor at Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences. He received his PhD degree from Technical Institute of Physics and Chemistry, Chinese Academy of Sciences in 2012, and then joined Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences. His current research interests are inorganic optoelectronic functional crystals, such as nonlinear optical crystals and birefringent crystals. He is a recipient of the “NSFC Excellent Young Scientists Fund in China. |
1 Introduction
Birefringence is a linear optical property that occurs in anisotropic crystals. In anisotropic crystals, birefringence will arise when the refractive index of light changes based on the direction of light.1 According to the difference in optical properties, crystals can be divided into two types, isotropic crystals and anisotropic crystals. In terms of symmetry, crystals with cubic symmetry belong to isotropic crystals. In consequence, they lack birefringence. In contrast, anisotropic crystals have more types of symmetry, including triclinic, monoclinic, orthorhombic, tetragonal, hexagonal, and trigonal symmetry. Therefore, they can generate birefringence. The crystals belonging to trigonal, hexagonal, and tetragonal systems are called uniaxial crystals, in which the unique optical axis coincides with the highest axis of symmetry. Birefringence can modulate polarized light in uniaxial crystals and spilt the incident light into ordinary ray (o) and extraordinary ray (e). They correspond to different refractive indices, no (ordinary refractive index) and ne (extraordinary refractive index) (Fig. 1). If no > ne, the crystal is called the negative uniaxial crystal. On the contrary, no < ne for the positive uniaxial crystal. The birefringence can be expressed in the following equation:| Δn = |no – ne| |
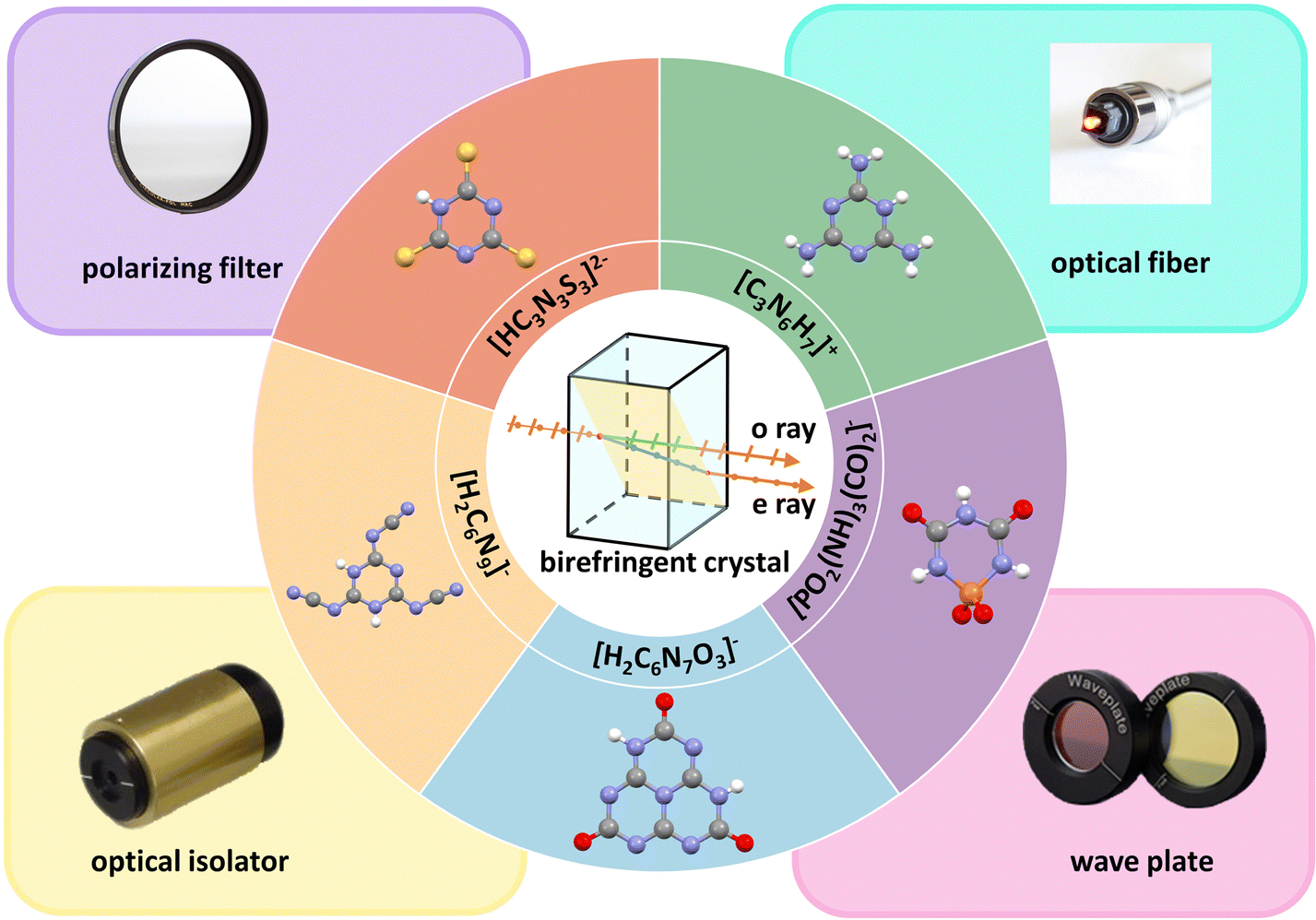 | ||
| Fig. 1 The principle of birefringence, structural units, and typical applications of commercial birefringent crystals. | ||
In recent decades, several commercial birefringent crystals have been developed, including MgF2,2α-BaB2O4 (α-BBO),3,4 calcite (CaCO3),5 YVO4,6 rutile (TiO2)7 and LiNbO3.8 Birefringent materials benefit to obtain phase matching in the specific wavelength range of the transparency and can be used to modulate the polarization-related light propagation. Because of these excellent optical properties, birefringent crystals are used in various optical devices, such as circulators, polarizers, phase compensators, wave plates, and optical isolators.9–15
The properties of the compound are closely associated with the structure of the microstructure group, so it is crucial to choose suitable units. In birefringent materials, planar groups containing π orbitals demonstrate better polarization anisotropy than the non-planar units.16–19 For example, α-BBO has a large birefringence (exp. Δn = 0.12@532 nm),20 and is transparent in the significant ultraviolet (UV) spectral region (λ < 400 nm). This is mainly attributed to the delocalized π-conjugated electron orbitals in the [B3O6]3− rings (Fig. 2).21–24 Recently, a number of birefringent crystals similar to α-BBO have been discovered, such as K2(HC3N3O3)·2H2O (exp. 0.19@514 nm),25 Rb2(HC3N3O3) (cal. 0.40@532 nm),26 K2Mg(H2C3N3O3)4·4H2O (cal. 0.38@800 nm),27 NaRb3(H2C3N3O3)4·3H2O (cal. 0.39@532 nm)28 and (C5H6ON)+(H2PO4)− (cal. 0.25@1064 nm).29
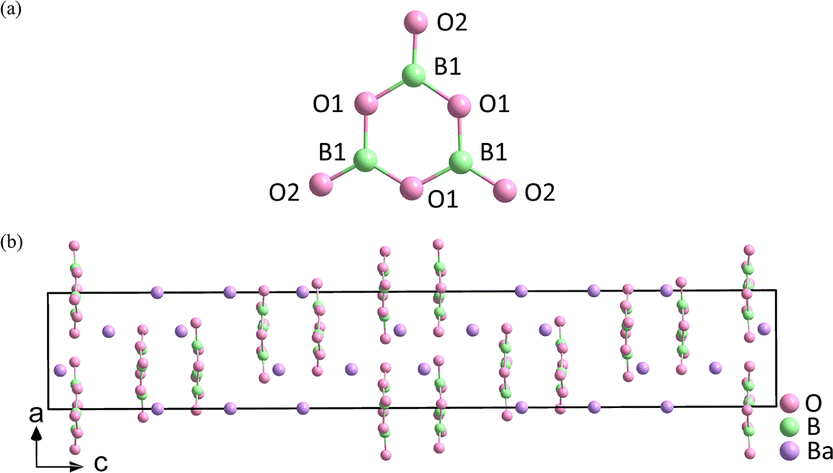 | ||
| Fig. 2 (a) [B3O6]3− ring. (b) Crystal structure of α-BBO. | ||
In order to make devices smaller, birefringent crystals should have large birefringence. From the structural perspective, π-conjugated rings are beneficial for increasing birefringence, and birefringence also depends significantly on the alignment and direction of the structural units.30 The more parallel the ring, the more anisotropic is enlarged, causing larger birefringence. On the other hand, the energy gap plays a decisive role in optical crystals’ applications, and thus keeping the balance of birefringence and energy gap is also a considerable problem.
In this review, we summarised our recent efforts in exploring birefringent crystals containing π-conjugated rings. In addition, the birefringent quality factor (BQF) is used to quantify the integrated optical performance, and it can be expressed in the following equation:
| BQF = Eg × Δn |
| Compounds | Space groups | Δn | Energy gap (eV) | BQF | Ref. |
|---|---|---|---|---|---|
| α-BaB2O4 |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c c |
0.12@532 nm | 6.56 | 0.79@546 nm | 20 |
| (C3N6H7)2SiF6·H2O | P21/n | 0.15@550 nm | 4.76 | 0.71@550 nm | 31 |
| (C3N6H8)PbBr4 | P21/c | 0.32@550 nm | 3.13 | 1.00@550 nm | 32 |
| Cs3Cl(HC3N3S3) | Pmc21 | 0.52@550 nm | 3.34 | 1.74@550 nm | 33 |
| CsH2C6N9·H2O |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
0.55@550 nm | 4.12 | 2.27@550 nm | 34 |
| Ba(H2C6N7O3)2·8H2O | Fdd2 | 0.24@550 nm | 4.10 | 0.98@550 nm | 35 |
| NaPO2(NH)3(CO)2 | P21/c | 0.28@550 nm | 6.50 | 1.82@550 nm | 36 |
2 Structure and optical properties of birefringent crystals with π orbitals
Herein, some structures like α-BBO are summarised, including (C3N6H7)2SiF6·H2O,31 MLAPbBr4 (MLA = melamine),32 Cs3Cl(HC3N3S3),33 CsH2C6N9·H2O,34 Ba(H2C6N7O3)2·8H2O,35 and NaPO2(NH)3(CO)2.36 In addition, from the perspective of the fundamental structural units, the polarizability anisotropy, and the gap from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) of structural units are summarised in Fig. 3. The polarizability anisotropy and HOMO–LUMO gap were calculated via the Gaussian 09 package37 with the hybrid B3LYP functional at 6-31G(d,p) level. After that, the calculation results were analysed by the Multiwfn 3.8 code.38 The polarizability anisotropy of all structural units exceeds that of the [B3O6]3−, providing the basis for large birefringence.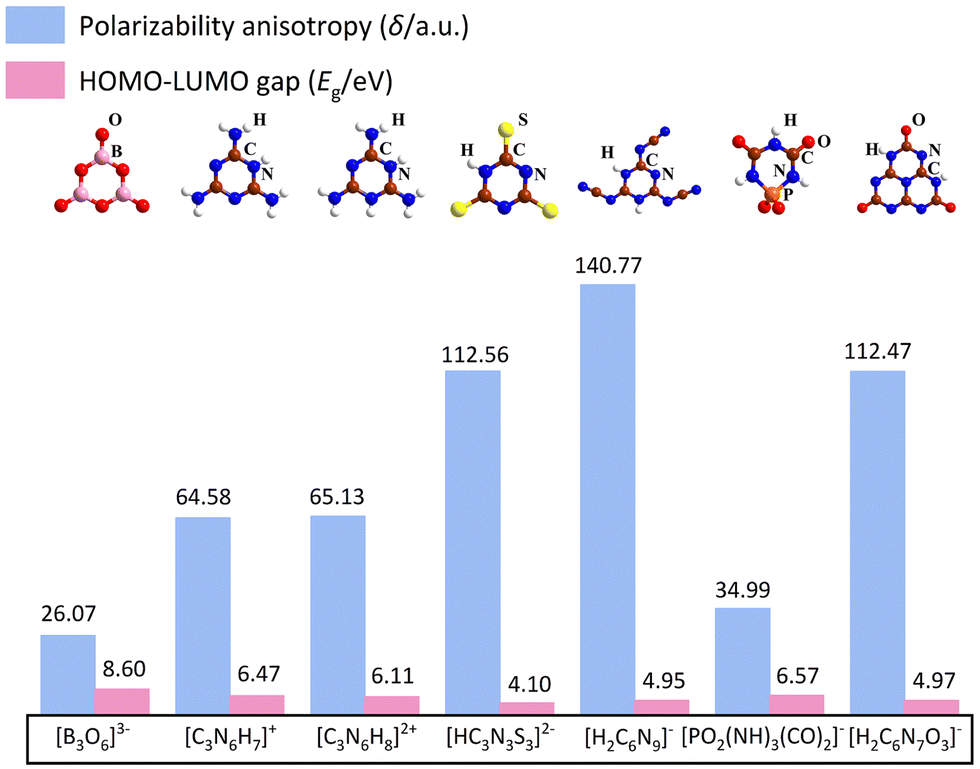 | ||
| Fig. 3 Polarizability anisotropy and HOMO–LUMO gap of [B3O6]3−, [C3N6H7]+, [C3N6H8]2+, [HC3N3S3]2−, [H2C6N9]−, [PO2(NH)3(CO)2]−, [H2C6N7O3]− anion units. | ||
(C3N6H7)2SiF6·H2O crystallizes in the monoclinic space group P21/n. The fundamental structural blocks contain a protonated melamine [C3N6H7]+, [SiF6]2− octahedron, and the H2O molecules. (C3N6H7)2SiF6·H2O combines the planar π-conjugated [C3N6H7]+ with the highly coordinated [SiF6]2− groups. The [SiF6]2− octahedra present in the gap along the c-axis connect the quasi-two-dimensional [C3N6H7]∞ chains (Fig. 4a). In addition, the H2O molecules with hydrogen bonds connect the adjacent [SiF6]2− octahedra (Fig. 4b). The [SiF6]2− octahedron is also connected to four [C3N6H7]+ cations by hydrogen bonds. The parallel structure group [C3N6H7]∞ chains lead to larger birefringence due to the π-conjugated [C3N6H7]+, which is similar to the [B3O6]3− functional units of α-BBO, showing high anisotropy. Inside the single [C3N6H7]∞ chain, adjacent [C3N6H7]+ cations are further linked by two N–H⋯N hydrogen bonds, resulting in the coplanar alignment (Fig. 4c). In addition, the highly symmetrical [SiF6]2− polyhedron further facilitates the aligned arrangement of each [C3N6H7]∞ chain. The Kubelka–Munk function F(R) = (1 − R)2/2R is used to calculate the absorption rate of light, where R is the reflectance.39 Additionally, the experimental energy gap can be inferred by extrapolating the linear part of the rising curve to zero. The large energy gap is 4.76 eV (Fig. 4d), suggesting the (C3N6H7)2SiF6·H2O crystal can be used in the UV region. The birefringence of (C3N6H7)2SiF6·H2O is 0.15@550 nm (Fig. 4e). Thus, the BQF is 0.71@550 nm. In accordance with the crystal structure and theoretical calculation, the birefringence and energy gap of (C3N6H7)2SiF6·H2O should be due to the parallel aligned protonated melamine [C3N6H7]+ groups and the highly symmetric [SiF6]2− octahedron.
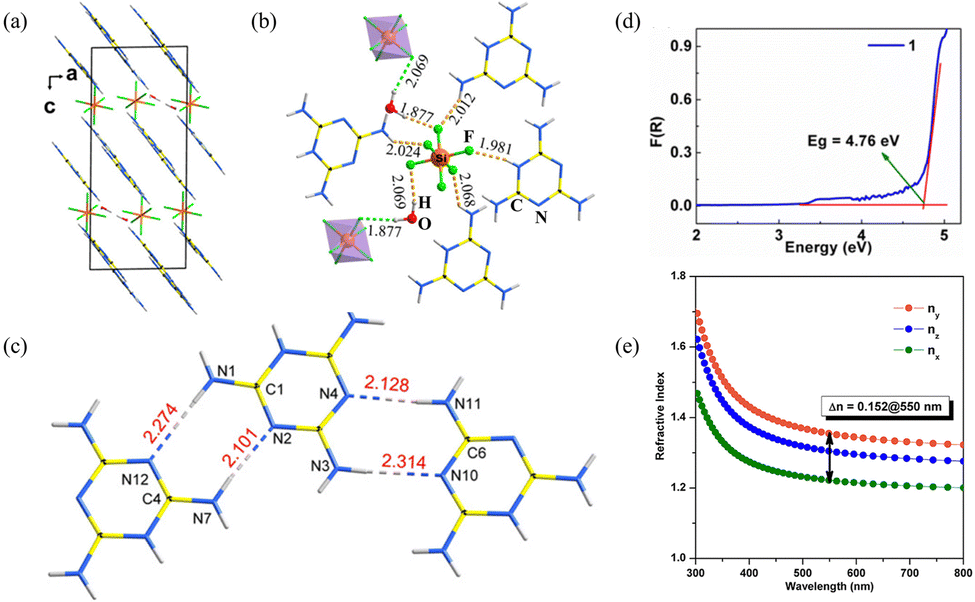 | ||
| Fig. 4 (a) Structure of (C3N6H7)2SiF6·H2O viewed along the b-axis. (b) The coordination environment of [SiF6]2− octahedra. (c) [C3N6H7]∞ chain formed by hydrogen bonds. (d) Absorption spectra. (e) Theoretically calculated refractive indices and birefringence of (C3N6H7)2SiF6·H2O.31 | ||
The MLAPbBr4 crystallizes in the centrosymmetric monoclinic space group of P21/c. The crystal structure of MLAPbBr4 is composed of melamine cations [C3N6H8]2+ (Fig. 5a) and PbBr6 octahedra (Fig. 5b). The structure combines the corrugated [PbBr4]∞ layers with the melamine cations to form a (110)-oriented perovskite framework (Fig. 5c). According to the absorption data, the energy gap of MLAPbBr4 is about 3.13 eV, meaning the optically transparent window can achieve to the UV spectral region of 374 nm. In addition, the experimental birefringence (0.32@550 nm) was calculated according to the formula R = Δn × T.40,41 Where R represents the optical path difference, T represents the thickness of the crystal, and Δn represents the birefringence. Hence, the BQF is 1.00@550 nm. The first-principles calculation is used to explore the potential mechanism of the large birefringence of MLAPbBr4. As shown in Fig. 5d, theoretical birefringence is 0.29@550 nm, which is in agreement with the experimental value. The HOMO and LUMO of MLAPbBr4 were calculated to show the origin of birefringence. Through Fig. 5e, for highly π-conjugated melamine cations, the HOMO shows an apparent anisotropy of electron densities, and the LUMO reflects highly distorted PbBr6 octahedra. Hence, the delocalized π-conjugated melamine cations and distorted PbBr6 octahedra coordinate to enlarge the birefringence of MLAPbBr4.
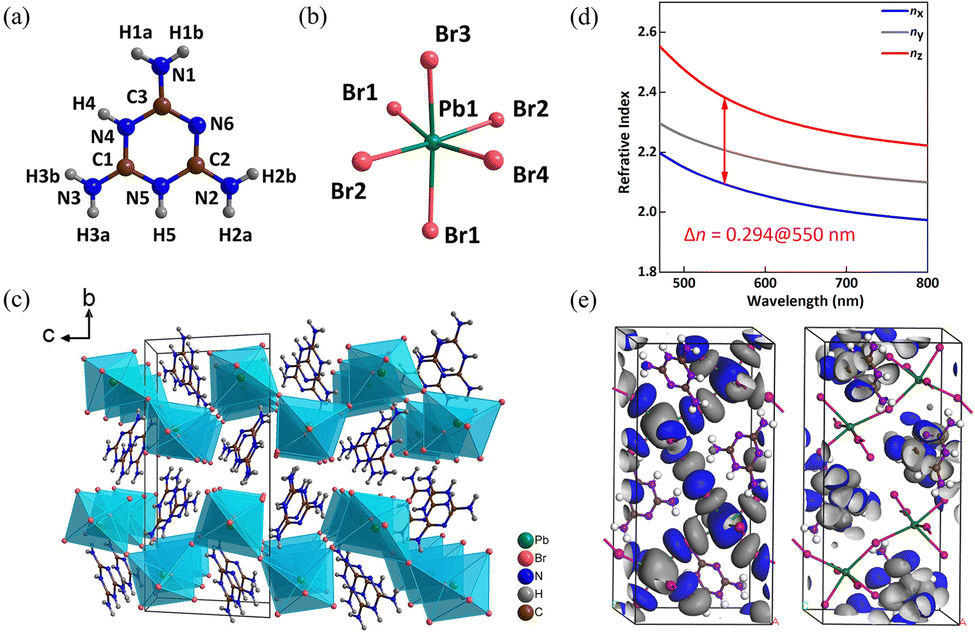 | ||
| Fig. 5 (a) Crystal structure of melamine cation. (b) Crystal structure of PbBr6 octahedron. (c) Viewed along the a-axis, the crystal structure of MLAPbBr4. (d) Theoretically calculated refractive indices and birefringence of MLAPbBr4. (e) HOMO and LUMO of MLAPbBr4.32 Copyright 2022, Wiley. | ||
Cs3Cl(HC3N3S3) crystallizes in the non-centrosymmetric space group of Pmc21. The crystal structure of Cs3Cl(HC3N3S3) is composed of [HC3N3S3]2− rings (Fig. 6a), which is similar to the α-BBO, and ClCs6 polyhedra (Fig. 6b). According to the Fig. 6c, the chains are parallel along the a axis, with the coplanar [HC3N3S3]2− rings occupy the interchain space. According to the UV-visible-near-infrared diffuse reflectance spectrum of Cs3Cl(HC3N3S3) powders, the absorption edge is near 371 nm with a corresponding energy gap of about 3.34 eV. The measured birefringence of Cs3Cl(HC3N3S3) is 0.52@550 nm. Thus, the BQF is 1.74@550 nm. To reveal the underlying mechanism of birefringence, first-principles calculations were calculated. As shown in Fig. 6d, the calculated Δn of Cs3Cl(HC3N3S3) is 0.60@550 nm, which is close to the measured birefringence. The HOMO and LUMO for crystal Cs3Cl(HC3N3S3) reflect the origin of birefringence (Fig. 6e and f). The HOMO is composed of the N 2p orbitals and S 3p orbitals, and the LUMO is mainly constituted by the π orbitals on the [C3N3] rings and the 3p orbitals of the S atoms. Therefore, from the perspective of structure–property relationships, the [HC3N3S3]2− structural unit is crucial to the birefringence of Cs3Cl(HC3N3S3) because of the delocalized π-conjugation. In addition, the [HC3N3S3]2− rings are parallel to the (100) plane, which enlarge the anisotropic. It makes a positive contribution to improving birefringence.
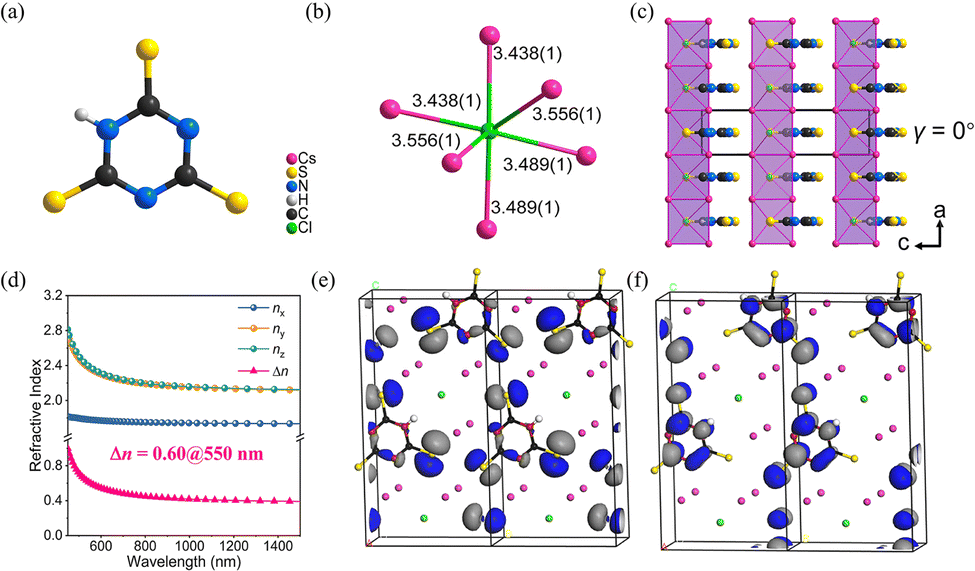 | ||
| Fig. 6 (a) [HC3N3S3]2− ring. (b) ClCs6 polyhedron. (c) The crystal structure of Cs3Cl(HC3N3S3), (γ) is the dihedral angle between the [HC3N3S3]2− rings and the (100) plane. (d) Theoretically calculated refractive indices and birefringence of Cs3Cl(HC3N3S3). (e) The HOMO of Cs3Cl(HC3N3S3). (f) The LUMO of Cs3Cl(HC3N3S3). (Black, red, green, yellow, purple and white balls represent C, N, Cl, S, Cs and H atoms, respectively).33 Copyright 2022, Wiley. | ||
CsH2C6N9·H2O crystallizes in the centrosymmetric space group of triclinic P. As the fundamental structural unit of CsH2C6N9·H2O, the [H2C6N9]− unit is made up of the three almost coplanar linear –(N–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N)– side arms and the planar π-conjugated [C3N3] ring (Fig. 7a). In addition, consistent with the reported protonated tricyanomelaminates, the H atoms in the [H2C6N9]− unit are bonded to the N atoms of the [C3N3] rings.42–44 As shown in Fig. 7b, CsH2C6N9·H2O has a pseudo-two-dimensional layered structure with Cs+ counter cations present in the interlayer space to maintain the overall charge balance. The [H2C6N9]− anion units are held in place by the hydrogen bonding network, which belongs to the nearly coplanar H2O molecules. The coplanarity of [H2C6N9]− anion units is an optimal arrangement for optical anisotropy and is conducive to the generation of large birefringence in CsH2C6N9·H2O, similar to the cases in α-BBO.45 According to the UV-visible-near-infrared diffuse reflectance spectrum, CsH2C6N9·H2O has an absorption edge at about 301 nm in the UV spectral region. This represents an experimental energy gap of about 4.12 eV. The experimental birefringence of CsH2C6N9·H2O (0.55@550 nm) was measured using a polarizing microscope.46 Hence, the BQF is 2.27@550 nm. The first-principles calculations were used to explore the microscopic origin of birefringence. According to the relationship between the wavelength and refractive index, the birefringence of CsH2C6N9·H2O is 0.52@550 nm, which agrees well with the measurement. In addition, there is a large anisotropy of refractive indices in CsH2C6N9·H2O (Fig. 7c). As shown in Fig. 7, the electron localization function (ELF)47,48 maps of CsH2C6N9·H2O have been presented, aiming to illustrate the contribution of Cs+, H2O molecules, and [H2C6N9]− units. In Fig. 7d, the HOMO consists mainly of N 2p orbitals. In addition, N 2p and C 2p orbitals of [H2C6N9]− predominate the LUMO (Fig. 7e). Especially, the π orbitals of the linear –(N–CN)– in the [H2C6N9]− and [C3N3] rings are parallel to one another and also parallel to the π orbitals in the adjacent [H2C6N9]− (Fig. 7f). Such a structural arrangement enhances microscopic anisotropy, thus increasing the birefringence. Therefore, the large birefringence of CsH2C6N9·H2O can be attributed to the delocalized π-conjugated orbitals in the [H2C6N9]− units.
N)– side arms and the planar π-conjugated [C3N3] ring (Fig. 7a). In addition, consistent with the reported protonated tricyanomelaminates, the H atoms in the [H2C6N9]− unit are bonded to the N atoms of the [C3N3] rings.42–44 As shown in Fig. 7b, CsH2C6N9·H2O has a pseudo-two-dimensional layered structure with Cs+ counter cations present in the interlayer space to maintain the overall charge balance. The [H2C6N9]− anion units are held in place by the hydrogen bonding network, which belongs to the nearly coplanar H2O molecules. The coplanarity of [H2C6N9]− anion units is an optimal arrangement for optical anisotropy and is conducive to the generation of large birefringence in CsH2C6N9·H2O, similar to the cases in α-BBO.45 According to the UV-visible-near-infrared diffuse reflectance spectrum, CsH2C6N9·H2O has an absorption edge at about 301 nm in the UV spectral region. This represents an experimental energy gap of about 4.12 eV. The experimental birefringence of CsH2C6N9·H2O (0.55@550 nm) was measured using a polarizing microscope.46 Hence, the BQF is 2.27@550 nm. The first-principles calculations were used to explore the microscopic origin of birefringence. According to the relationship between the wavelength and refractive index, the birefringence of CsH2C6N9·H2O is 0.52@550 nm, which agrees well with the measurement. In addition, there is a large anisotropy of refractive indices in CsH2C6N9·H2O (Fig. 7c). As shown in Fig. 7, the electron localization function (ELF)47,48 maps of CsH2C6N9·H2O have been presented, aiming to illustrate the contribution of Cs+, H2O molecules, and [H2C6N9]− units. In Fig. 7d, the HOMO consists mainly of N 2p orbitals. In addition, N 2p and C 2p orbitals of [H2C6N9]− predominate the LUMO (Fig. 7e). Especially, the π orbitals of the linear –(N–CN)– in the [H2C6N9]− and [C3N3] rings are parallel to one another and also parallel to the π orbitals in the adjacent [H2C6N9]− (Fig. 7f). Such a structural arrangement enhances microscopic anisotropy, thus increasing the birefringence. Therefore, the large birefringence of CsH2C6N9·H2O can be attributed to the delocalized π-conjugated orbitals in the [H2C6N9]− units.
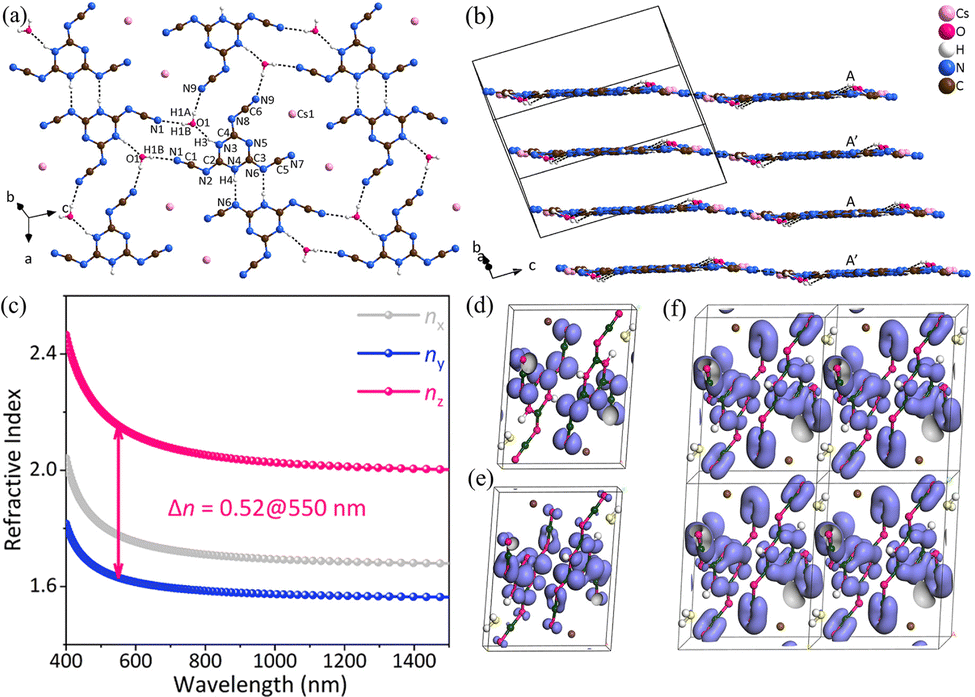 | ||
| Fig. 7 (a) [H4C6N9O]∞ single layer in CsH2C6N9·H2O. (b) The pseudo-two-dimensional layered structure of CsH2C6N9·H2O. (c) Theoretically calculated refractive indices and birefringence of CsH2C6N9·H2O. (d) The HOMO of CsH2C6N9·H2O. (e) The LUMO of CsH2C6N9·H2O. (f) π orbitals of (H2C6N9)− in CsH2C6N9·H2O, the fuchsia, yellow, brown, dark green and white spheres represent N, O, Cs, C and H atoms respectively.34 Copyright 2022, Wiley. | ||
Ba(H2C6N7O3)2·8H2O crystallizes in the non-centrosymmetric orthorhombic of the Fdd2 space group. The crystal structure of Ba(H2C6N7O3)2·8H2O consists of an independent [H2C6N7O3]− anion (Fig. 8a), one Ba2+ cation, and four lattice H2O molecules. In Fig. 8c, Ba(H2C6N7O3)2·8H2O exhibits a three-dimensional framework composed of π-conjugated [H2C6N7O3]− anions, Ba2+ cations, and H2O molecules. In Ba(H2C6N7O3)2·6H2O, each Ba2+ cation is shared by six H2O molecules and two [H2C6N7O3]− anions via Ba–O bonds. The UV-visible-near-infrared diffuse reflectance spectrum of Ba(H2C6N7O3)2·8H2O ranges from 200 nm to 1000 nm, the UV absorption edge of Ba(H2C6N7O3)2·8H2O is situated at λ = 302 nm, which corresponds to an energy gap of 4.10 eV. The measured birefringence of Ba(H2C6N7O3)2·8H2O is 0.24@550 nm. The theoretical value of birefringence is 0.22@550 nm, which agrees well with the experimental value. Hence, according to the energy gap and measured birefringence, the value of BQF is 0.98@550 nm. To investigate the cause of the large birefringence, the HOMO and LUMO patterns of Ba(H2C6N7O3)2·8H2O are shown in Fig. 8d and Fig. 8e. O 2p and N 2p orbitals are the main constituents of the HOMO. In comparison, the unoccupied π orbitals make up the LUMO components. In summary, due to the expanded π-conjugated delocalization in the [H2C6N7O3]− building blocks, it has an enhanced birefringence.
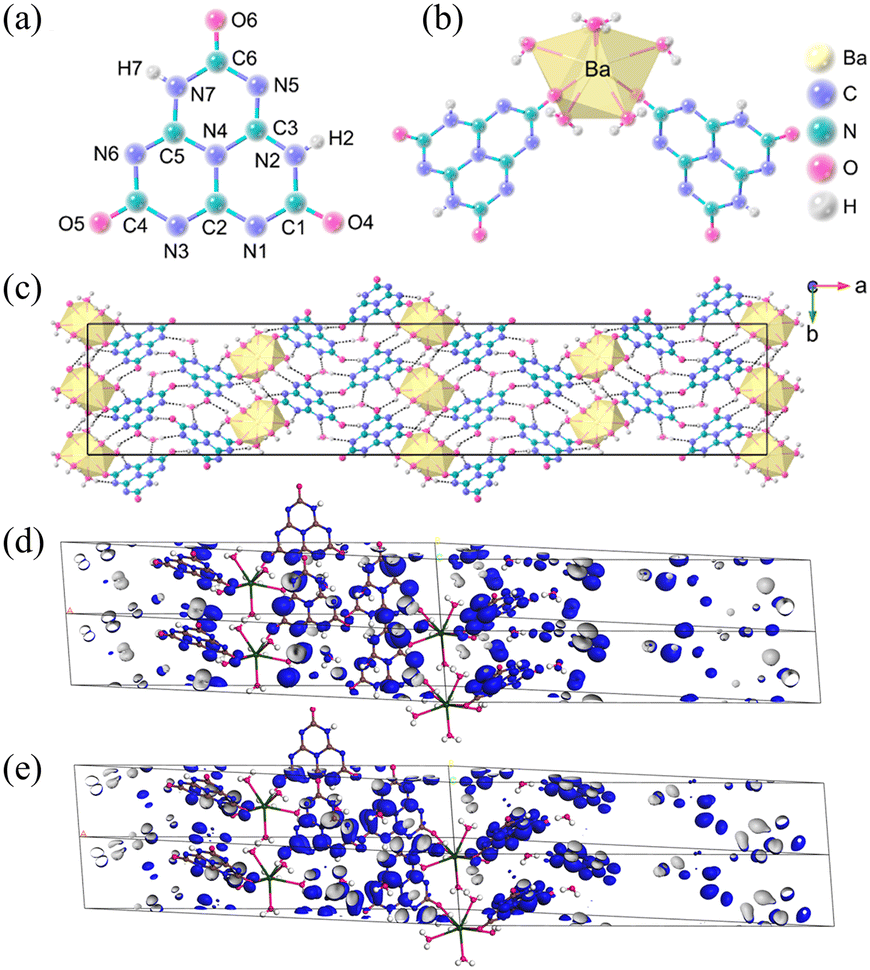 | ||
| Fig. 8 (a) The (H2C6N7O3)− anion. (b) The coordination environment of Ba2+. (c) Viewed along the crystallographic c-axis, the crystal structure of Ba(H2C6N7O3)2·8H2O, and the yellow polyhedra represent BaO8 polyhedra. (d) The HOMO of Ba(H2C6N7O3)2·8H2O. (e) The LUMO of Ba(H2C6N7O3)2·8H2O.35 Copyright 2022, Wiley. | ||
NaPO2(NH)3(CO)2 crystallizes in the monoclinic space group P21/c. As shown in Fig. 9a, the crystal structure of NaPO2(NH)3(CO)2 consists of six-membered rings [PO2(NH)3(CO)2] formed by direct covalent bonding of two π-conjugated planar triangles [CO(NH)2] and a non-π-conjugated tetrahedron [PO2N2]. The [PO2(NH)3(CO)2]− anions form a bond with the Na+ cations present in the vacancies (Fig. 9b). The UV−visible-near-infrared diffuse reflectance spectrum indicates that the energy gap is 6.50 eV. In addition, the experiment birefringence is 0.28@550 nm. Thus, the value of BQF is 1.82@550 nm. To investigate the fundamental relationship between optical performance and structure of NaPO2(NH)3(CO)2, first-principles calculations were used. In Fig. 9c, NaPO2(NH)3(CO)2 has a calculated birefringence of 0.28@550 nm and exhibits pronounced optical anisotropy, in general agreement with the measured value. The HOMO and LUMO were calculated to investigate further the relationship between optical properties and crystal structure at the molecular level (Fig. 9d and e). It demonstrates that the π-conjugated interactions have been partially decoupled by the PO2(NH)2 tetrahedron, and all of the π-conjugation is confined within the [PO2(NH)3(CO)2]− ring. Hence, in order to keep an effective balance between a large energy gap and large birefringence, the confined π-conjugation of [PO2(NH)3(CO)2]− units play a crucial role, and the integrated properties of the birefringent crystal are improved.
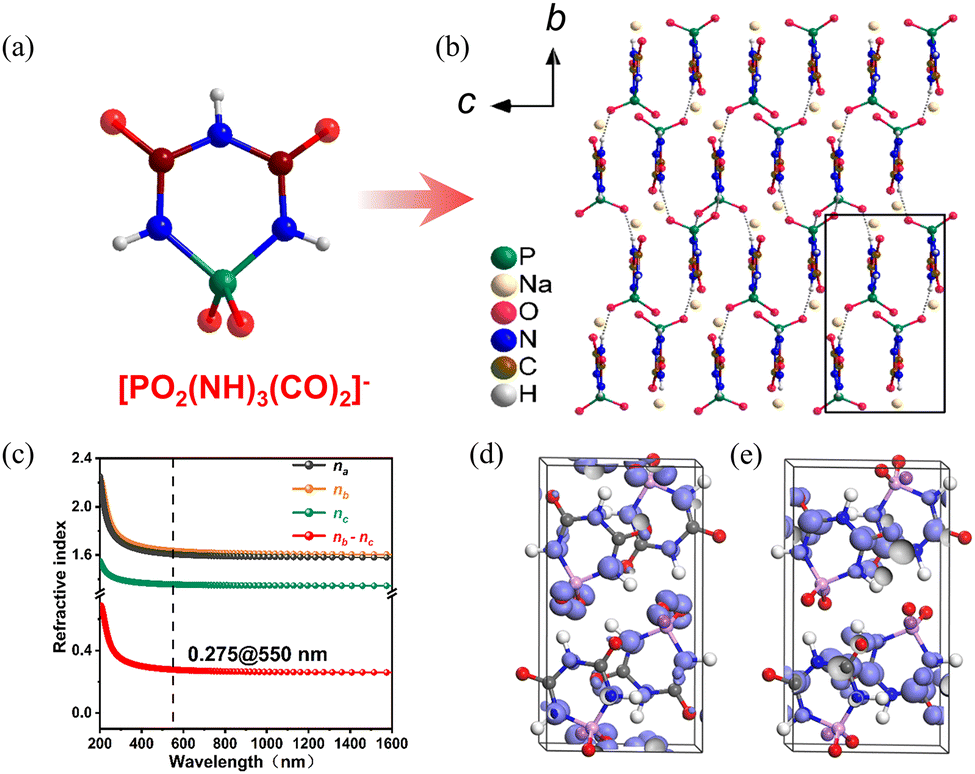 | ||
| Fig. 9 (a) [PO2(NH)3(CO)2]− anion. (b) Along the a-axis, the crystal structure of NaPO2(NH)3(CO)2. (c) Theoretically calculated refractive indices and birefringence of NaPO2(NH)3(CO)2. (d) The HOMO of NaPO2(NH)3(CO)2. (e) The LUMO of NaPO2(NH)3(CO)2.36 Copyright 2022, Elsevier. | ||
3 Conclusion
In this review, we have presented and discussed systematically the recent development of π-conjugated birefringent materials. We discussed the crystal structure, energy gap, birefringence, and the relationship between structure and property. For the birefringent materials containing π-conjugated groups, here are some perspectives and outlooks.(1) The delocalized π-conjugated C–N rings show strong hybridization between C and N atoms and contribute significantly to the birefringence of crystals. In addition, in order to achieve an effective balance between the large energy gap and the large birefringence, confined π-conjugation plays a critical role. Confined π-conjugation can partially decouple the π-conjugated interaction, and compared with the delocalized π-conjugation, confined π-conjugated birefringent crystals have a larger energy gap. At the same time, birefringent crystals with confined π-conjugation can maintain a suitable birefringence. Since birefringence and energy gap are important factors in the application of birefringent crystals, a reasonable balance between delocalized π-conjugation and confined π-conjugation is essential to improve the integrated performance of birefringent crystals.
(2) In addition, the birefringence depends significantly on the anisotropy of the structure, while the anisotropic structure depends on the arrangement of the unit. Therefore, the internal arrangement of the structural unit should be considered. An effective strategy to enhance birefringence is to induce an ordered arrangement of the π-conjugated unit.
(3) This paper reviews the synthesis of several large BQF crystals. According to the structure–property relationship, the optical properties of birefringent crystals depend on the microscopic groups and the arrangement of the groups within the crystal. Exploring suitable microscopic groups with large polarizability anisotropy and HOMO–LUMO gap and controlling their ordered arrangement increased the BQF of the birefringent crystals.
(4) Perovskite and anti-perovskite are novel structural systems in the development of birefringent crystals. They can increase the anisotropy and thus the birefringence through the optimal arrangement of cations. This is also an important direction in the study of birefringent crystals.
(5) The structure containing H2O will reduce the stability of the crystal. One way to improve the stability of crystals is to replace the H2O molecule with the halogen atom.
(6) With the continuous improvement in computing power and the development of theoretical calculation theory, bottom-up synthesis strategies can also be applied to the design and synthesis of birefringent crystals. The microscopic hyperpolarizability, polarizability anisotropy, and HOMO–LUMO gap can be calculated to select suitable groups, and then the different functional units can complement each other to form highly integrated birefringent crystals. This is also useful in the design and synthesis of novel birefringent crystals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22122507, 22193042, 21833010, 61975207, and 21921001), the Natural Science Foundation of Fujian Province (2022J02012), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (Y202069), the Fujian Institute of Innovation (FJCXY18010201) in the Chinese Academy of Sciences, the Key Research Program of Frontier Sciences of the Chinese Academy of Sciences (ZDBS-LY-SLH024), and the Ministry of Education/Guangxi Key Laboratory of Optical and Electronic Materials and Devices (20KF-11).References
- S. Niu, G. Joe, H. Zhao, Y. Zhou, T. Orvis, H. Huyan, J. Salman, K. Mahalingam, B. Urwin, J. Wu, Y. Liu, T. E. Tiwald, S. B. Cronin, B. M. Howe, M. Mecklenburg, R. Haiges, D. J. Singh, H. Wang, M. A. Kats and J. Ravichandran, Giant optical anisotropy in a quasi-one-dimensional crystal, Nat. Photonics, 2018, 12, 392–396 CrossRef CAS.
- M. J. Dodge, Refractive properties of magnesium fluoride, Appl. Opt., 1984, 23, 1980–1985 CrossRef CAS PubMed.
- G. Zhou, J. Xu, X. Chen, H. Zhong, S. Wang, K. Xu, P. Deng and F. Gan, Growth and spectrum of a novel birefringent α-BaB2O4 crystal, J. Cryst. Growth, 1998, 191, 517–519 CrossRef CAS.
- S. Wu, G. Wang, J. Xie, X. Wu, Y. Zhang and X. Lin, Growth of large birefringent α-BBO crystal, J. Cryst. Growth, 2002, 245, 84–86 CrossRef CAS.
- G. Ghosh, Dispersion-equation coefficients for the refractive index and birefringence of calcite and quartz crystals, Opt. Commun., 1999, 163, 95–102 CrossRef CAS.
- L. G. DeShazer, Improved midinfrared polarizers using yttrium vanadate, Proc. SPIE, 2002, 4481, 10–17 CrossRef.
- J. R. DeVore, Refractive Indices of Rutile and Sphalerite, J. Opt. Soc. Am., 1951, 41, 416–419 CrossRef CAS.
- D. E. Zelmon, D. L. Small and D. Jundt, Infrared corrected Sellmeier coefficients for congruently grown lithium niobate and 5 mol% magnesium oxide–doped lithium niobate, J. Opt. Soc. Am. B, 1997, 14, 3319–3322 CrossRef CAS.
- L. Wu, S. Patankar, T. Morimoto, N. L. Nair, E. Thewalt, A. Little, J. G. Analytis, J. E. Moore and J. Orenstein, Giant anisotropic nonlinear optical response in transition metal monopnictide Weyl semimetals, Nat. Phys., 2017, 13, 350–355 Search PubMed.
- L. H. Nicholls, F. J. Rodríguez-Fortuño, M. E. Nasir, R. M. Córdova-Castro, N. Olivier, G. A. Wurtz and A. V. Zayats, Ultrafast synthesis and switching of light polarization in nonlinear anisotropic metamaterials, Nat. Photonics, 2017, 11, 628–633 CrossRef CAS.
- G. A. Ermolaev, D. V. Grudinin, Y. V. Stebunov, K. V. Voronin, V. G. Kravets, J. Duan, A. B. Mazitov, G. I. Tselikov, A. Bylinkin, D. I. Yakubovsky, S. M. Novikov, D. G. Baranov, A. Y. Nikitin, I. A. Kruglov, T. Shegai, P. Alonso-Gonzalez, A. N. Grigorenko, A. V. Arsenin, K. S. Novoselov and V. S. Volkov, Giant optical anisotropy in transition metal dichalcogenides for next-generation photonics, Nat. Commun., 2021, 12, 854 CrossRef CAS PubMed.
- E. Collett, Field Guide to Polarization, SPIE, Bellingham, 2005 Search PubMed.
- X. Yang, L. Ma and D. Yan, Facile synthesis of 1D organic–inorganic perovskite micro-belts with high water stability for sensing and photonic applications, Chem. Sci., 2019, 10, 4567–4572 RSC.
- D. Yan, H. Yang, Q. Meng, H. Lin and M. Wei, Two-Component Molecular Materials of 2,5-Diphenyloxazole Exhibiting Tunable Ultraviolet/Blue Polarized Emission, Pump-enhanced Luminescence, and Mechanochromic Response, Adv. Funct. Mater., 2014, 24, 587–594 CrossRef CAS.
- S. Liu, Y. Lin and D. Yan, Hydrogen-bond organized 2D metal–organic microsheets: direct ultralong phosphorescence and color-tunable optical waveguides, Sci. Bull., 2022, 67, 2076–2084 CrossRef CAS PubMed.
- Y. Pan, S. Guo, B. Liu, H. Xue and G. Guo, Second-order nonlinear optical crystals with mixed anions, Coord. Chem. Rev., 2018, 374, 464–496 CrossRef CAS.
- Z. Zhang, Y. Wang, B. Zhang, Z. Yang and S. Pan, Polar Fluorooxoborate, NaB4O6F: A Promising Material for Ionic Conduction and Nonlinear Optics, Angew. Chem., Int. Ed., 2018, 57, 6577–6581 CrossRef CAS PubMed.
- F. Liang, L. Kang, P. Gong, Z. Lin and Y. Wu, Rational Design of Deep-Ultraviolet Nonlinear Optical Materials in Fluorooxoborates: Toward Optimal Planar Configuration, Chem. Mater., 2017, 29, 7098–7102 CrossRef CAS.
- Z. Du, Y. Zhou and S. Zhao, Synthesis, Crystal Structure and Birefringence Properties of Silver Cluster Compound Ag3B6O10I, Chin. J. Appl. Chem., 2023, 40, 229–235 Search PubMed.
- J. Liu, X. He, J. Xu, G. Zhou, S. Zhou, G. Zhao and S. Li, The study on properties of Sr2+-doped α-BBO crystal, J. Cryst. Growth, 2004, 260, 486–489 CrossRef CAS.
- M. T. Anderson and K. R. Poeppelmeier, Lanthanum copper tin oxide (La2CuSnO6): a new perovskite-related compound with an unusual arrangement of B cations, Chem. Mater., 1991, 3, 476–482 CrossRef CAS.
- M. Azuma, S. Kaimori and M. Takano, High-Pressure Synthesis and Magnetic Properties of Layered Double Perovskites Ln2CuMO6 (Ln = La, Pr, Nd, and Sm; M = Sn and Zr), Chem. Mater., 1998, 10, 3124–3130 CrossRef CAS.
- X. Dong, H. Wu, Y. Shi, H. Yu, Z. Yang, B. Zhang, Z. Chen, Y. Yang, Z. Huang, S. Pan and Z. Zhou, Na11B21O36X2 (X = Cl, Br): Halogen Sodium Borates with a New Graphene-Like Borate Double Layer, Chem. Eur. J., 2013, 19, 7338–7341 CrossRef CAS PubMed.
- Y. Wang, S. Pan, H. Yu, X. Su, M. Zhang, F. Zhang and J. Han, Cs4Mo5P2O22: a first Strandberg-type POM with 1D straight chains of polymerized [Mo5P2O23]6− units and moderate second harmonic generation response, Chem. Commun., 2013, 49, 306–308 RSC.
- D. Lin, M. Luo, C. Lin, F. Xu and N. Ye, KLi(HC3N3O3)·2H2O: Solvent-drop Grinding Method toward the Hydro-isocyanurate Nonlinear Optical Crystal, J. Am. Chem. Soc., 2019, 141, 3390–3394 CrossRef CAS PubMed.
- J. Lu, Y. Lian, L. Xiong, Q. Wu and L. Wu, How To Maximize Birefringence and Nonlinearity of π-Conjugated Cyanurates, J. Am. Chem. Soc., 2019, 141, 16151–16159 CrossRef CAS PubMed.
- X. Meng, F. Liang, K. Kang, J. Tang, Q. Huang, W. Yin, Z. Lin and M. Xia, A rich structural chemistry in π-conjugated hydroisocyanurates: layered structures of A2B(H2C3N3O3)4·nH2O (A = K, Rb, Cs; B = Mg, Ca; n = 4, 10) with high ultraviolet transparency and strong optical anisotropy, Dalton Trans., 2019, 48, 9048–9052 RSC.
- M. Aibibula, L. Wang and S. Huang, Rb3Na(H2C3N3O3)4·3H2O with Large Birefringence, ACS Omega, 2019, 4, 22197–22202 CrossRef CAS PubMed.
- J. Lu, X. Liu, M. Zhao, X. Deng, K. Shi, Q. Wu, L. Chen and L. Wu, Discovery of NLO Semiorganic (C5H6ON)+(H2PO4)−: Dipole Moment Modulation and Superior Synergy in Solar-Blind UV Region, J. Am. Chem. Soc., 2021, 143, 3647–3654 CrossRef CAS PubMed.
- Y. Li, C. Yin, X. Yang, X. Kuang, J. Chen, L. He, Q. Ding, S. Zhao, M. Hong and J. Luo, A Nonlinear Optical Switchable Sulfate of Ultrawide Bandgap, CCS Chem., 2021, 3, 2298–2306 CrossRef CAS.
- Y. Shen, Y. Zhou, X. Xue, H. Yu, S. Zhao and J. Luo, (C3N6H7)2SiF6·H2O: an ultraviolet birefringent crystal exceeding the intrinsic energy gap of an organic reagent, Inorg. Chem. Front., 2022, 9, 5226–5230 RSC.
- W. Huang, X. Zhang, Y. Li, Y. Zhou, X. Chen, X. Li, F. Wu, M. Hong, J. Luo and S. Zhao, A Hybrid Halide Perovskite Birefringent Crystal, Angew. Chem., Int. Ed., 2022, 61, e202202746 CAS.
- M. Li, X. Zhang, Z. Xiong, Y. Li, Y. Zhou, X. Chen, Y. Song, M. Hong, J. Luo and S. Zhao, A Hybrid Antiperovskite with Strong Linear and Second-Order Nonlinear Optical Responses, Angew. Chem., Int. Ed., 2022, 61, e202211151 CAS.
- Y. Li, X. Zhang, Y. Zhou, W. Huang, Y. Song, H. Wang, M. Li, M. Hong, J. Luo and S. Zhao, An Optically Anisotropic Crystal with Large Birefringence Arising from Cooperative π Orbitals, Angew. Chem., Int. Ed., 2022, 61, e202208811 CAS.
- Y. Li, W. Huang, Y. Zhou, X. Song, J. Zheng, H. Wang, Y. Song, M. Li, J. Luo and S. Zhao, A High-Performance Nonlinear Optical Crystal with a Building Block Containing Expanded π-Delocalization, Angew. Chem., Int. Ed., 2023, 135, e202215145 Search PubMed.
- Y. Zhou, X. Zhang, M. Hong, J. Luo and S. Zhao, Achieving effective balance between bandgap and birefringence by confining π-conjugation in an optically anisotropic crystal, Sci. Bull., 2022, 67, 2276–2279 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Gaussian Inc., Wallingford, CT, 2009, https://www.gaussian.com/ Search PubMed.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. Tauc, Absorption edge and internal electric fields in amorphous semiconductors, Mater. Res. Bull., 1970, 5, 721–729 CrossRef CAS.
- D. T. Baran, A. M. Sorensen, T. W. Honeyman, R. Ray and M. F. Holick, 1α,25-dihydroxyvitamin D3-induced increments in hepatocyte cytosolic calcium and lysophosphatidylinositol: Inhibition by pertussis toxin and 1β,25-dihydroxyvitamin D3, J. Bone Miner. Res., 1990, 5, 517–524 CrossRef CAS PubMed.
- L. Cao, G. Peng, W. Liao, T. Yan, X. Long and N. Ye, A microcrystal method for the measurement of birefringence, CrystEngComm, 2020, 22, 1956–1961 RSC.
- B. Jürgens, H. A. Höppe and W. Schnick, Synthesis, Crystal Structure and Properties of Rubidium Dihydrogentricyanomelaminate Semihydrate Rb[H2C6N9]·1/2 H2O, Z. Anorg. Allg. Chem., 2004, 630, 35–40 CrossRef.
- A. Nag, B. V. Lotsch, J. Schmedtaufder Günne, O. Oeckler, P. J. Schmidt and W. Schnick, Rare-Earth Tricyanomelaminates [NH4]Ln[HC6N9]2[H2O]7·H2O (Ln = La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy): Structural Investigation, Solid-State NMR Spectroscopy, and Photoluminescence, Chem. Eur. J., 2007, 13, 3512–3524 CrossRef CAS PubMed.
- A. Schwarzer, T. Saplinova and E. Kroke, Tri-s-triazines (s-heptazines)—From a “mystery molecule” to industrially relevant carbon nitride materials, Coord. Chem. Rev., 2013, 257, 2032–2062 CrossRef CAS.
- C. Chen, B. Wu, A. Jiang and G. You, A new-type ultraviolet SHG crystal β-BaB2O4, Sci. China, Ser. B: Chem., 1985, 28, 235–243 Search PubMed.
- B. E. Sørensen, A revised Michel-Lévy interference colour chart based on first-principles calculations, Eur. J. Mineral., 2012, 25, 5–10 CrossRef.
- A. D. Becke and K. E. Edgecombe, A Simple Measure of Electron Localization in Atomic and Molecular Systems, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- A. Savin, R. Nesper, S. Wengert and T. F. Fässler, ELF: The Electron Localization Function, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808–1832 CrossRef CAS.
| This journal is © the Partner Organisations 2023 |