
PtI4-catalyzed oxidative and hydrogenative dearomative [3 + 2] cycloaddition of 1H-indole N-tethered o-alkynylbenzaldehydes†
Dandan
Shang
a,
Rui
Hu
a,
Qing
Bao
a,
Jichao
Chen
a,
Lei
Yu
b,
Philip Wai Hong
Chan
 *b and
Weidong
Rao
*a
*b and
Weidong
Rao
*a
aJiangsu Co-Innovation Center for Efficient Processing and Utilization of Forest Resources, College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, China. E-mail: weidong@njfu.edu.cn
bSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: phil.chan@monash.edu
First published on 14th November 2022
Abstract
A synthetic method for the chemodivergent assembly of a diverse range of highly functionalized and architecturally challenging cyclohepta[b]indolines which relies on PtI4-catalyzed oxidative and hydrogenative dearomative [3 + 2] cycloaddition of 1H-indole N-tethered o-alkynylbenzaldehydes in a single operation is described. For the synthesis of a key structural feature that is found in a myriad of bioactive natural products and pharmaceutical compounds, the proposed cascade chemodivergent process delineates the first example of an in situ formed Pt-bound benzopyrylium intermediate that participates in [3 + 2] cycloaddition with the C(2)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(3) bond of an aromatic N-heterocycle as the 2π cycloaddition partner with exclusive exo-selectivity in a highly efficient manner. It also offers a unique instance of the resulting metallocarbene species generated in this manner undergoing either oxidation by phenylmethylsulfoxide or reduction by THF, with the solvent as both the hydride and alkyl reagent source. The chemodivergent protocol exhibits exceptional functional group tolerance and is amenable to the late-stage modification of a series of structurally complex bioactive natural products and drug molecules.
C(3) bond of an aromatic N-heterocycle as the 2π cycloaddition partner with exclusive exo-selectivity in a highly efficient manner. It also offers a unique instance of the resulting metallocarbene species generated in this manner undergoing either oxidation by phenylmethylsulfoxide or reduction by THF, with the solvent as both the hydride and alkyl reagent source. The chemodivergent protocol exhibits exceptional functional group tolerance and is amenable to the late-stage modification of a series of structurally complex bioactive natural products and drug molecules.
Introduction
The cyclohepta[b]indoline and -indole cores are both privileged structural scaffolds that are frequently present in numerous biologically active natural products and pharmaceutical compounds.1–9 The polycyclic ring system, for example, is found in the bioactive alkaloids ajmaline, kopsifoline A, actinophyllic acid, ervitsine, and aristolasol (Fig. 1).2–6 It is also the key structural component, for instance, in the drug molecules shown in Fig. 1, which are SIRT1-inhibitor IV, A-FABP inhibitor, and L1210 MLC cytotoxin.7–9 This wealth of bioactivities possessed by the compound class has led to a plethora of elegant [4 + 3] and [5 + 2] cycloaddition/annulation- and Cope-rearrangement-based synthetic methods for their assembly.1 The development of new synthetic protocols beyond these modes of reactivity and access to a potentially wider scope of structurally diverse cyclohepta[b]indoline scaffolds from readily accessible substrates have thus remained immensely important targets in chemical synthesis.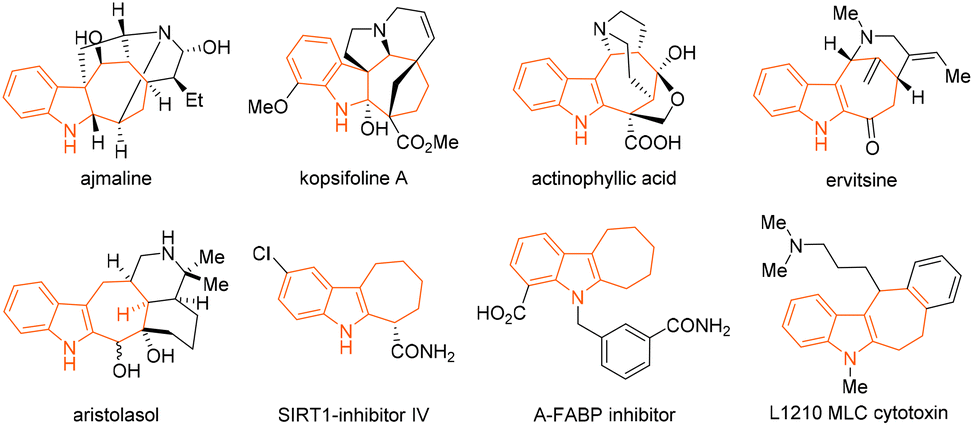 | ||
| Fig. 1 Examples of bioactive alkaloid natural products and drug molecules featuring the cyclohepta[b]indoline and -indole cores. | ||
The transition metal-catalyzed Huisgen-type [3 + 2] cycloaddition of o-alkynylbenzaldehydes with dipolarophiles has emerged in the last two decades as one of the most powerful strategies for the efficient and selective synthesis of a wide variety of polycyclic ring systems (Scheme 1a). A main reason for this is the ability of the postulated highly reactive in situ generated metal-bound benzopyrylium species to undergo pericyclization with a wide variety of dipolarophiles such as those of the alkene, allene, alkyne, and 1,n-diene compound classes.10–14 In addition to this is the demonstrated synthetic utility of the [3 + 2] cycloaddition protocol in retrosynthetic strategies to obtain a diverse array of natural products such as faveine methyl ester, taxamairin B, rosmaridiphenol and komaroviquinone.15–17 However, missing from this repertoire have been the [3 + 2] cycloaddition reactions of the in situ generated organometallic intermediate with an electron-rich aromatic CC bond of heteroarenes and access to a potentially wider scope of complex polycyclic products. To the best of our knowledge, the [4 + 2] cycloadditions of a metal-bound benzopyrylium species with isoxazoles and benzofurans as the 2π dipolarophile have remained the only two reported examples.18,19
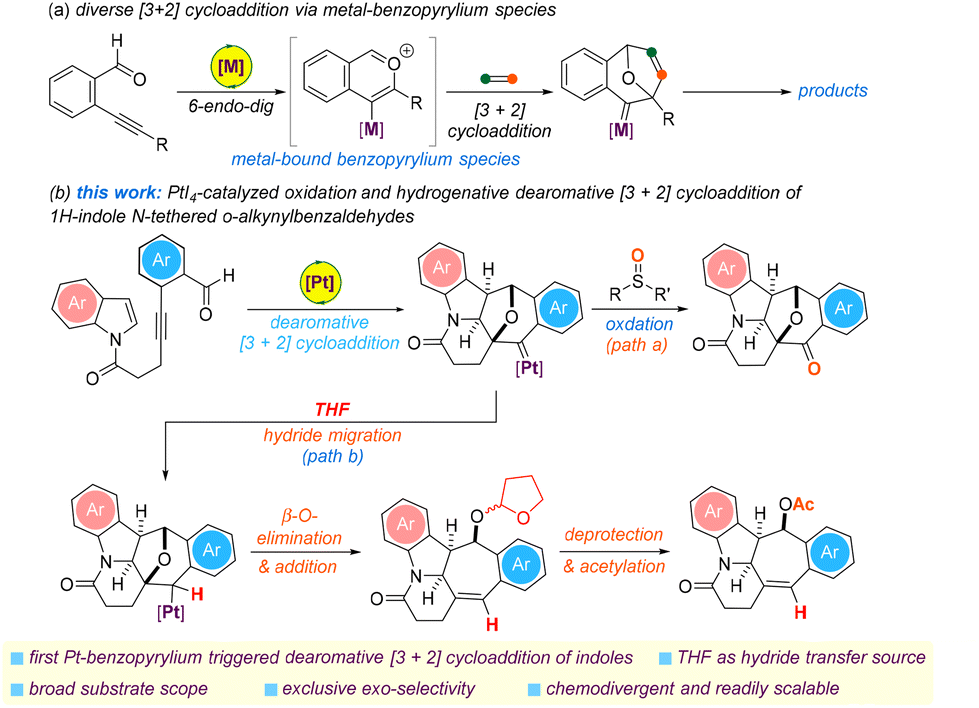 | ||
| Scheme 1 Transition metal-catalyzed [3 + 2] cycloaddition of o-alkynylbenzaldehydes. | ||
In this context and as part of an ongoing program exploring the new reactivities of the metal-bound benzopyrylium species and those of the 1H-indole compound class, we were drawn to the potential reactivity of o-alkynylbenzaldehydes containing a 1H-indolyl N-tether (Scheme 1b).20,21 We reasoned that a Pt(IV)-catalyzed cascade 6-endo-dig/dearomative Huisgen-type [3 + 2] cycloaddition of an N-heterocyclic substrate might be realized under appropriate conditions to give the corresponding platinum carbene species. In the presence of an appropriate sulfoxide as the oxidant or THF solvent as both a hydride and an alkyl source, it was anticipated that this would result in the corresponding hexacyclic and pentacyclic N-heterocycles shown in Scheme 1b, paths a and b.22,23 Herein, we disclose the details of this chemistry, which offers the first Pt(IV)-catalyzed expedient and chemodivergent approach to synthesize two structurally distinct and potentially useful benzo-fused cyclohepta[b]indoline derivatives with exclusive exo-selectivity.
Results and discussion
To evaluate the feasibility of our hypothesis, 1H-indole N-tethered o-alkynylbenzaldehyde 1a was chosen as the model substrate to establish the optimum reaction conditions, and the results are summarized in Table 1 and Table S1 in the ESI.† The substrates examined in this study were readily prepared from the corresponding 1H-indoles and 4-pentynoic acid by exploiting a condensation/Sonogashira coupling strategy following the known literature procedure.24 In a preliminary experiment, employing reaction conditions with [Rh(COD)Cl]2 as the catalyst (5 mol%) in water at 80 °C for 48 h was found to produce only the hydration product 2a in 28% yield (Table 1, entry 1).11a Suppression of the hydration adduct and delivery of the desired benzofused N-hexacyclic product 3a in 21% yield were achieved by switching the reaction solvent to toluene and using water (2 equiv.) as an additive (entry 2).11a,g,12,15 The structure and relative stereochemistry of 3a were unambiguously confirmed by X-ray crystallography analysis.25 Replacing H2O with the sulfoxides DMSO, DPSO and PMSO as the additives was found to lead to slightly improved product yields of 31, 39 and 44%, respectively (entries 3–5). Encouraged by these results, an evaluation of the catalytic performance of various other transition metal complexes with PMSO as the additive was performed. This revealed that while the AuBr3-mediated reaction of 1a resulted in an increase in product yield from 44 to 69%, the analogous transformation catalyzed by IPrAuNTf2 gave both 2a and 3a in 20 and 19% yield, respectively (entries 6 and 7). Our studies subsequently showed that repeating the reaction with Pt(II) salts PtBr2 and PtI2 and the Pt(IV) complex PtI4 resulted in superior catalytic efficiency and furnished product yields of 73–99% (entries 8–10). With PtI4 revealed to be the catalyst of choice, lowering its loading from 5 to 1 mol% in toluene at 80 °C for 12 h or room temperature for 3 days was found to give a similar outcome, affording near quantitative product yields in both instances (entries 11 and 12). In marked contrast, replacing toluene with THF as the solvent along with the omission of the sulfoxide additive was found to completely change the chemoselectivity outcome of the reaction (entry 13). Under these slightly modified reaction conditions, the THF-substituted adducts 4a, 5a and the alcohol 6a instead of 3a were obtained in respective yields of 36, 25 and 29% and a combined yield of 90%. The structures and relative stereochemistry of 4a and 5a were determined by X-ray crystallography analysis.25 However, the analogous reactions mediated by PtCl2, PtBr2 or PtBr4 in place of PtI4 as the catalyst were shown to be slightly less efficient, furnishing the corresponding adducts 4a, 5a and 6a in lower yields of 20–33% (entries 14–16).|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | t [h] | Yieldb (%) | ||||
| 2a | 3a | 4a | 5a | 6a | |||||
| a All reactions were performed with 1a (0.2 mmol), catalyst (5 mol%), additive (0.4 mmol), and 4 Å MS (200 mg) in solvent (2 mL) at 80 °C unless otherwise noted. b Isolated product yield. c Reaction was performed in the absence of 4 Å MS. d Reaction was performed with 1 mol% PtI4. e Reaction was performed with 1 mol% PtI4. | |||||||||
| 1c | [Rh(COD)Cl]2 | H2O | 48 | 28 | |||||
| 2c | [Rh(COD)Cl]2 | H2O | Toluene | 18 | 21 | ||||
| 3 | [Rh(COD)Cl]2 | DMSO | Toluene | 16 | 31 | ||||
| 4 | [Rh(COD)Cl]2 | DPSO | Toluene | 12 | 39 | ||||
| 5 | [Rh(COD)Cl]2 | PMSO | Toluene | 12 | 44 | ||||
| 6 | AuBr3 | PMSO | Toluene | 12 | 69 | ||||
| 7 | IPrAuNTf2 | PMSO | Toluene | 48 | 20 | 19 | |||
| 8 | PtBr2 | PMSO | Toluene | 12 | 85 | ||||
| 9 | PtI2 | PMSO | Toluene | 12 | 73 | ||||
| 10 | PtI4 | PMSO | Toluene | 12 | 99 | ||||
| 11 | PtI4d | PMSO | Toluene | 12 | 99 | ||||
| 12 | PtI4e | PMSO | Toluene | 72 | 97 | ||||
| 13 | PtI4 | THF | 12 | 36 | 25 | 29 | |||
| 14 | PtBr4 | THF | 12 | 33 | 23 | 20 | |||
| 15 | PtCl2 | THF | 12 | 28 | 25 | 21 | |||
| 16 | PtBr2 | THF | 12 | 31 | 27 | 24 | |||
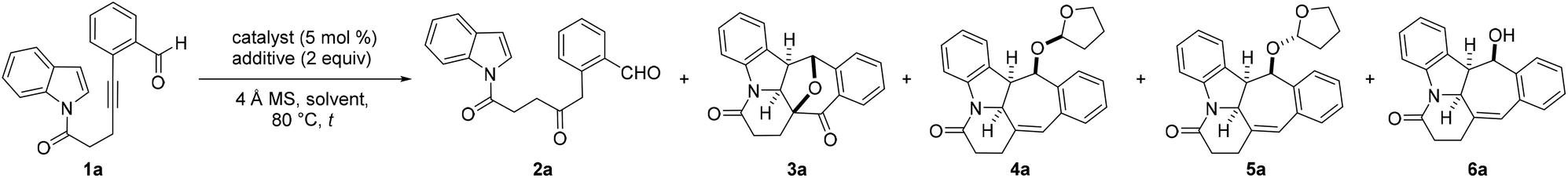
In view of the formation of two diastereomers 4a and 5a as well as the instability of 6a, the possibility of a cascade hydrogenative dearomative [3 + 2] cycloaddition/deacetalization/acetylation protocol in a two-pot, three-step manner was next examined. This led us to find that the polycyclic acetate product 7a could be afforded in 78% overall yield over three steps by following the synthetic procedure outlined in Scheme 2. This involved subjecting the crude reaction mixture obtained from the THF-enabled PtI4-catalyzed reaction of 1a to treatment with 10 mol% tetrabutylammonium tribromide (TBATB) in MeOH/THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) at room temperature for 1 h. Upon work-up, the ensuing crude reaction mixture was further treated with 5 mol% DMAP and Ac2O at room temperature for 1 h.
:1 v/v) at room temperature for 1 h. Upon work-up, the ensuing crude reaction mixture was further treated with 5 mol% DMAP and Ac2O at room temperature for 1 h.
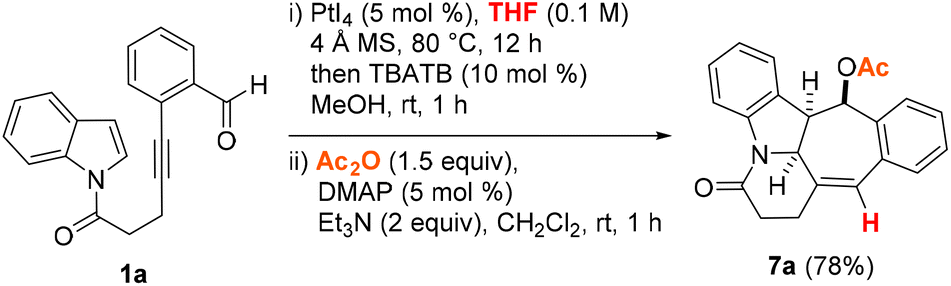 | ||
| Scheme 2 Cascade hydrogenative and dearomative [3 + 2] cycloaddition/deacetalization/acetylation of 1a. | ||
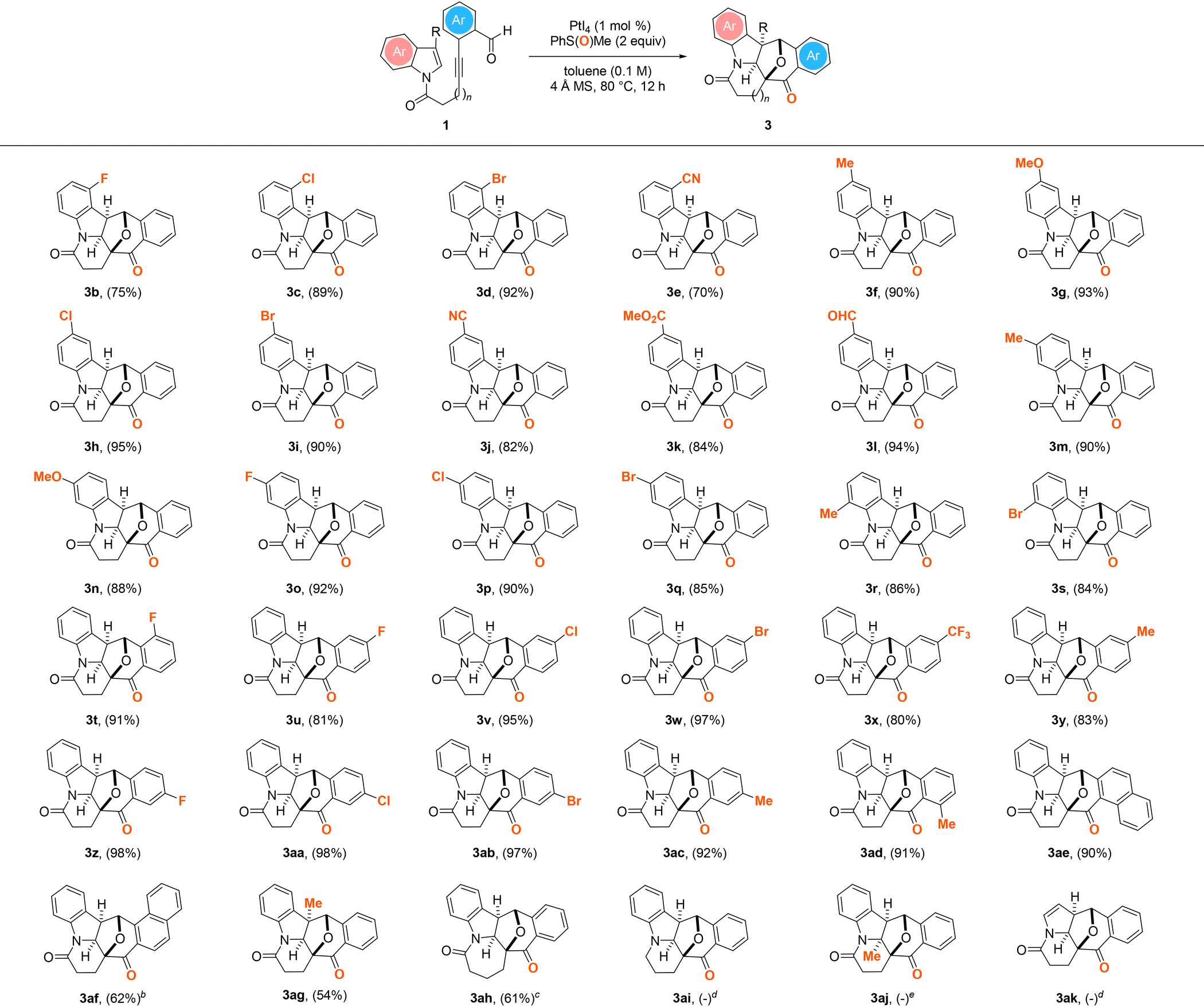
With the two sets of optimized reaction conditions outlined in Table 1, entry 11, and Scheme 2 established, the generality of the PtI4-catalyzed oxidative and dearomative [3 + 2] cycloaddition protocol with a set of 1H-indole N-tethered o-alkynylbenzaldehydes was first evaluated (Table 2). Overall, the reaction conditions were shown to be broad and a wide variety of structurally diverse cyclohepta[b]indolyl-embedded polycycles 3b–ah could be furnished with exclusive exo-selectivity in 54–98% yield from the corresponding substrates. Substrates possessing a diverse array of electron-withdrawing (1b–e, 1h–l, 1o–q, 1s) or -donating groups (1f,g, 1m,n, 1r) at various positions of the benzene ring of the 1H-indolyl motif were found to all react smoothly to give 3b–s in 75–95% yield. The reaction of 1l containing a pendant aldehyde functional group at the C5 position of the indolyl ring system is noteworthy as it provided 3l not only in 94% yield but also without the competitive formation of the condensation byproduct.26 Likewise, starting materials containing a halide (1t, 1u–w, 1z–ab), CF3 (1x) or methyl (1y, 1ac,ad) at the o-, m- or p-position of the benzaldehyde motif were demonstrated to afford the corresponding N-heterocyclic polycycles 3t–ad in 80–95% yield. The presence of a sterically demanding 1- or 2-naphthaldehyde moiety in the substrate, as in 1ae and 1af, was similarly found to be well tolerated, furnishing N-heptacycles 3ae and 3af in respective yields of 90 and 62%, although requiring a prolonged reaction time of 36 h in the case of the latter. Moreover, the PtI4-catalyzed reactions of substrates with a pendant 3-methylindolyl motif (1ag) or a three-methylene unit tether (1ah) were shown to readily undergo oxidative and dearomative [3 + 2] cycloaddition processes. In the case of the former, the corresponding product 3ag with an all-carbon quaternary center was delivered in 54% yield. Although it was found to proceed more efficiently with 5 mol% PtBr4 in place of PtI4 as the catalyst, the reaction of the latter also led to the delivery of the corresponding azepane-embedded polycycle in 61% yield. However, the analogous reactions of 1ai and 1ak were found to give a mixture of unidentifiable decomposition products under the optimized Pt(IV)-mediated oxidative dearomative [3 + 2] cycloaddition conditions. Likewise, the attempted PtI4-catalyzed oxidative dearomative [3 + 2] cycloaddition of 1aj was found to result in no reaction being observed and the recovery of the substrate in near quantitative yield.
a
| a All reactions were performed with 1 (0.2 mmol), 4 Å MS (200 mg), PMSO (0.4 mmol), and PtI4 (1 mol%) in toluene (2 mL) under an argon atmosphere at 80 °C for 12 h. Values in parentheses denote isolated product yield. b Reaction time = 36 h. c Reaction was performed with PtBr4 (5 mol%) in place of PtI4 as the catalyst. d Unidentifiable decomposition products were observed. e No reaction was observed based on TLC analysis and 1H NMR measurements of the crude reaction mixture. |
|---|
|
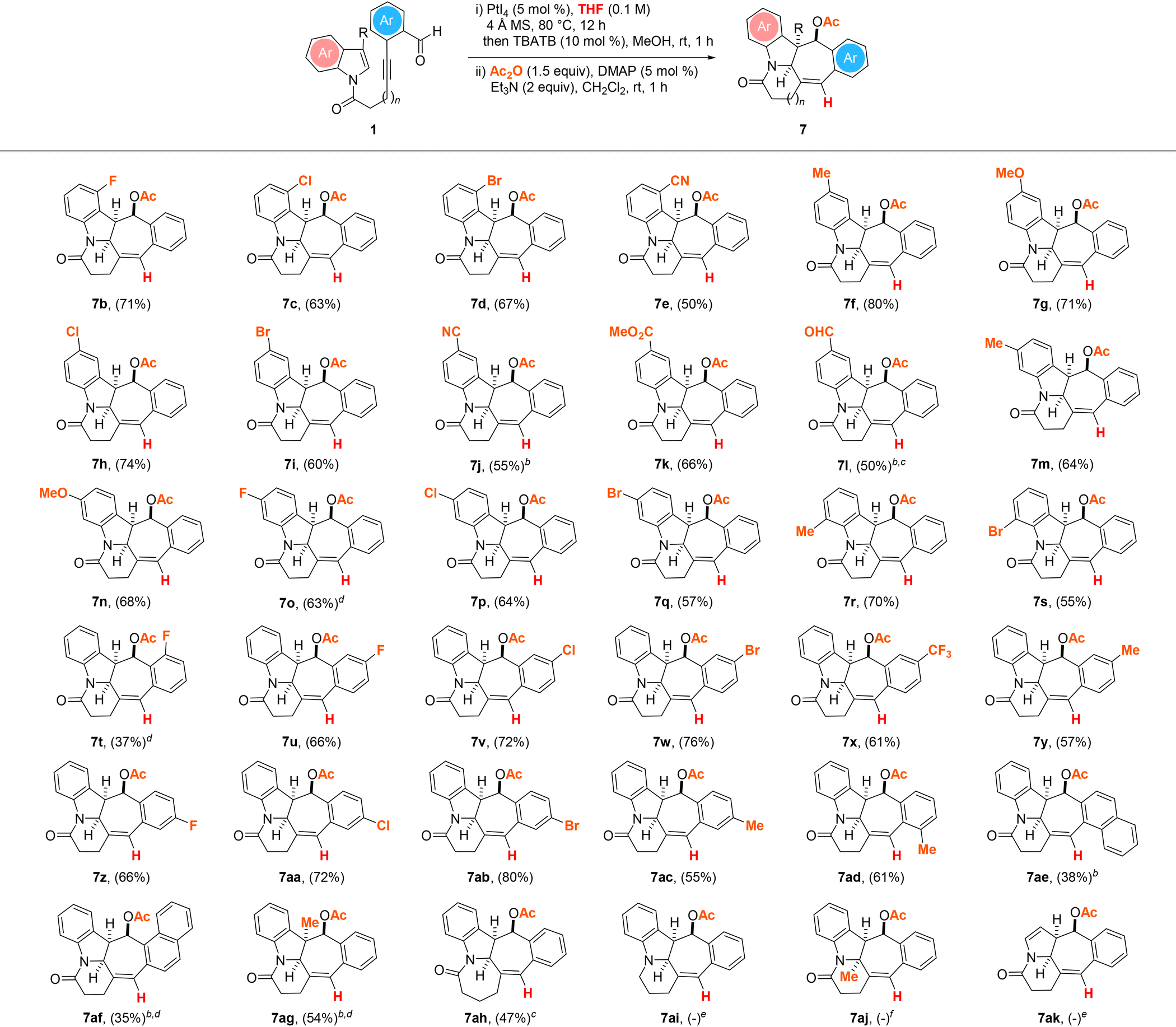
With these promising results in hand, we next sought to define the scope of the THF-enabled PtI4-catalyzed hydrogenative and dearomative [3 + 2] cycloaddition/deacetalization/acetylation cascade protocol with the same set of substrates (Table 3). In general, this revealed that experiments involving 1b–i, 1k, 1m,n, 1p–s and 1u–ad as the substrate tolerated the optimized reaction conditions to give the anticipated cyclohepta[b]indolyl-containing pentacyclic acetates 7b–i, 7k, 7m,n, 7p–s and 7u–ad in 50–80% overall yield over the three steps. The reactions of 1j, 1l, 1t and 1ae–ah were the only instances in which a slight modification of the optimized three-step synthesis procedure was required to achieve good product yields. Extending the reaction time from 12 to 48 h was deemed necessary to obtain the anticipated N-heterocyclic esters 7j and 7ae in respective overall yields of 55 and 38% over the three steps. In the case of the dialdehyde substituted substrate 1l, replacing PtI4 with 5 mol% PtBr4 as the catalyst along with a reaction time of 48 h was needed to afford 7l in 50% overall yield over the three steps. In the case of 1o, the assembly of the pentacyclic product 7o in 63% overall yield over the three steps from the substrate was found to be more efficient with 5 mol% PtBr2 as the catalyst. Likewise, experiments with the substrates 1af and 1ag were found to proceed more efficiently mediated by PtBr2 over an extended reaction time of 48 h, with the corresponding N-heterocycles 7af and 7ag obtained in 35 and 54% overall yields over the three steps. For the reaction of 1ah with a tether length increased by one more methylene group, its efficient conversion to the desired azepane-embedded pentacyclic product 7ah in 47% overall yield over the three steps was accomplished with PtBr4 as the catalyst. The structure and relative stereochemistry of the product were confirmed by X-ray crystallography analysis.25 In contrast, the analogous reactions of 1ai and 1ak products under the optimized Pt(IV)-mediated hydrogenative dearomative [3 + 2] cycloaddition conditions were found to give a mixture of unidentifiable decomposition products. Furthermore, the attempted PtI4-catalyzed hydrogenative dearomative [3 + 2] cycloaddition of 1aj was observed to lead to only the recovery of the substrate in near quantitative yield.
a
| a All reactions were performed with 1 (0.2 mmol), 4 Å MS (200 mg) and PtI4 (5 mol%) in THF (2 mL) under an argon atmosphere at 80 °C for 12 h, followed by treatment of the crude mixture with TBATB (10 mol%) and MeOH (2 mL) at room temperature for 1 h, work-up and treatment with Ac2O (0.4 mmol), DMAP (5 mol%), and Et3N (0.4 mmol) in CH2Cl2 (2 mL) at room temperature for 1 h. Values in parentheses denote isolated product yields. b Reaction time for the first step = 48 h. c Reaction was performed with PtBr4 (5 mol%) instead of PtI4 as the catalyst. d Reaction was performed with PtBr2 (5 mol%) instead of PtI4 as the catalyst. e Unidentifiable decomposition products were observed. f No reaction was observed based on TLC and 1H NMR analysis of the crude reaction mixture. |
|---|
|
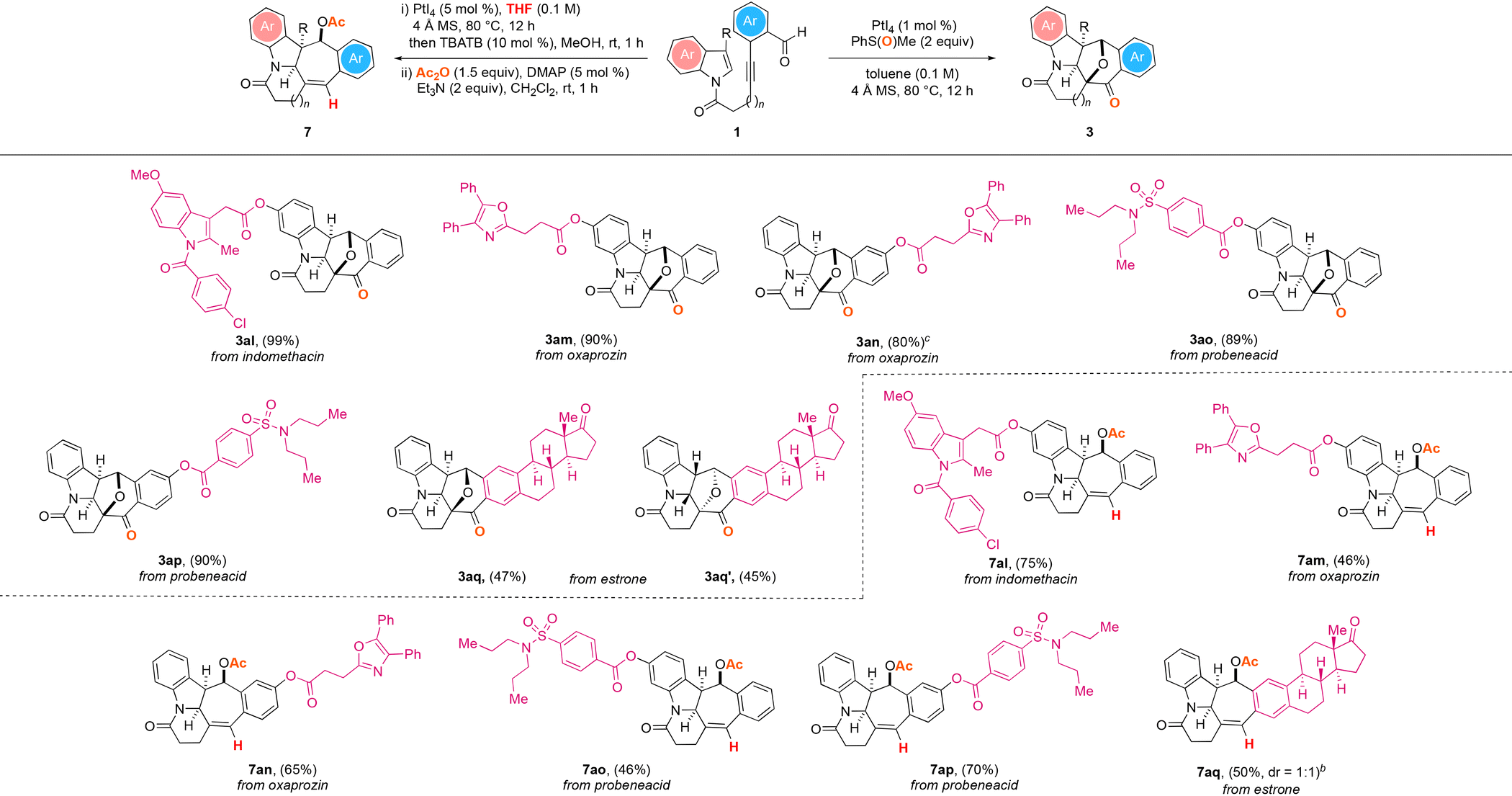
To demonstrate the potential synthetic utility of the two presented PtI4-catalyzed polycyclic ring formation protocols, the late-stage modification of the bioactive natural products and drug molecules illustrated in Table 4 was next examined. We were able to deconstruct the bioactive molecules indomethacin (1al), oxaprozin (1am and 1an), probenecid (1ao and 1ap) and estrone (1aq), and by subjecting the ensuing structurally complex substrates to the two sets of the optimized PtI4-catalyzed reaction conditions, we could obtain the corresponding N-heterocyclic products in good to excellent yields. The oxidative and dearomative [3 + 2] cycloaddition reactions of the compounds were found to produce the corresponding natural product and drug molecule derivatives 3al–3ap in 80–99% yield. For the estrone derived substrate 1aq, of the two separable enantiomers 3aq and 3aq′ with eight stereocenters obtained from the reaction, the structure and absolute configuration of the latter were also confirmed by X-ray crystallography analysis.25 The hydrogenative and dearomative [3 + 2] cycloaddition/deacetalization/acetylation cascade protocol of the starting materials was likewise revealed to deliver the corresponding natural product and drug molecule analogues 7al–7aq in 46–75% overall yield over the three steps.
| a All reactions were performed with 1 (0.2 mmol), 4 Å MS (200 mg), PMSO (0.4 mmol), and PtI4 (1 mol%) in toluene (2 mL) under an argon atmosphere at 80 °C for 12 h; or with 1 (0.2 mmol), 4 Å MS (200 mg) and PtI4 (5 mol%) in THF (2 mL) under an argon atmosphere at 80 °C for 12 h, followed by the treatment of the crude mixture with TBATB (10 mol%) and MeOH (2 mL) at room temperature for 1 h, then work-up and treatment with Ac2O (0.4 mmol), DMAP (5 mol%), and Et3N (0.4 mmol) in dichloromethane (2 mL) at room temperature for 1 h. b Isolated yields. c Reaction was performed in 1,2-dichloroethane. |
|---|
|
To illustrate the scalability as well as further highlight the potential synthetic utility of the two presented PtI4-catalyzed polycyclic ring formation protocols, the reaction of 1a on the 3.5 mmol scale (1.05 g) was initially explored (Scheme 3a). Under the oxidative and dearomative [3 + 2] cycloaddition reaction conditions at a catalyst loading of 0.5 mol%, we were pleased to find that this delivered 3a in 97% yield (1.08 g). Similarly, in an analogous experiment, the hydrogenative and dearomative [3 + 2] cycloaddition/deacetalization/acetylation cascade protocol at a catalyst loading of 3 mol% provided 7a in 66% overall yield (798 mg) over the three steps. The synthetic utility of the two PtI4-catalyzed polycyclic ring formation methods was first showcased by the selective reduction of 3a with NaBH4 and DIBAL-H (Scheme 3b). This gave the corresponding polycyclic secondary alcohols 8 and 9 in respective yields of 99% and 45%. Subjecting 3a to typical Grignard reaction conditions with methyl magnesium bromide afforded the polycyclic tertiary alcohol 10 exclusively as the only product in 75% yield and its structure was confirmed by X-ray crystallography analysis.25 Likewise, the selective oxidation of 7a by N-iodosuccinimide (NIS) and 2-iodoxybenzoic acid (IBX) was found to provide the indolyl-embedded polycyclic adducts 11 and 12 in 48 and 81% yields, respectively, with the structure of the latter also confirmed by X-ray crystallography.25 In a final transformation, treatment of 6a with PPh3/TBAI in 1,2-dichloroethane at 80 °C for 2 h was found to furnish the partially hydrogenated indolizin-7(1H)-one 13 in 72% yield resulting from a deoxychlorination/elimination/isomerization cascade.27
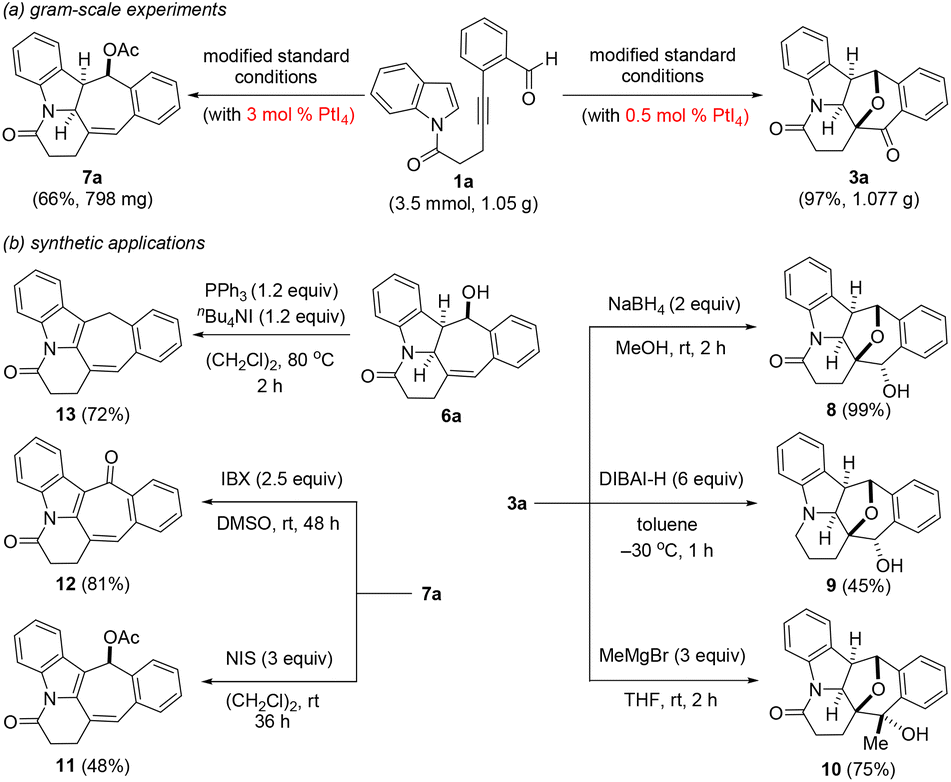 | ||
| Scheme 3 Gram-scale experiments and selected transformations. | ||
To gain a better understanding of the PtI4-catalyzed hydrogenative and dearomative [3 + 2] cycloaddition reaction mechanism, the following series of control experiments were performed (Scheme 4). To determine whether the reduction step proceeded via a hydride shift or H˙ atom transfer pathway, the PtI4-mediated reactions of 1a under the optimized conditions in the presence of the radical scavenger BHT or TEMPO were first examined. This revealed that the presence of two equivalents of BHT did not have a detrimental effect on the course of the reaction as the N-heterocyclic product 7a was furnished in 84% yield (Scheme 4a). On the other hand, decomposition was observed in an analogous control experiment of 1a under the optimized PtI4-catalyzed reaction conditions and two equivalents of TEMPO. As a comparison, a similar outcome was found, with a mixture of unidentifiable decomposition products obtained upon subjecting the THF-addition adduct 4a to the same experimental conditions with TEMPO but in the absence of the platinum(IV) salt (Scheme 4b). These results hinted at (1) the instability of the intermediacy of the THF-incorporated addition adduct and (2) a pathway involving H˙ atom transfer from THF being less likely to be operative. In a final control experiment, the role of THF as the hydride transfer source was supported by repeating the optimized PtI4-mediated hydrogenative and dearomative [3 + 2] cycloaddition/deacetalization/acetylation protocol with 1a in d8-THF as the solvent (Scheme 4c). This revealed that the deuterated adduct d1-7a was obtained as the only product in 74% yield and with complete deuterium incorporation at the alkenyl carbon center of the cycloheptene ring motif based on 1H NMR measurements of the crude reaction mixture.
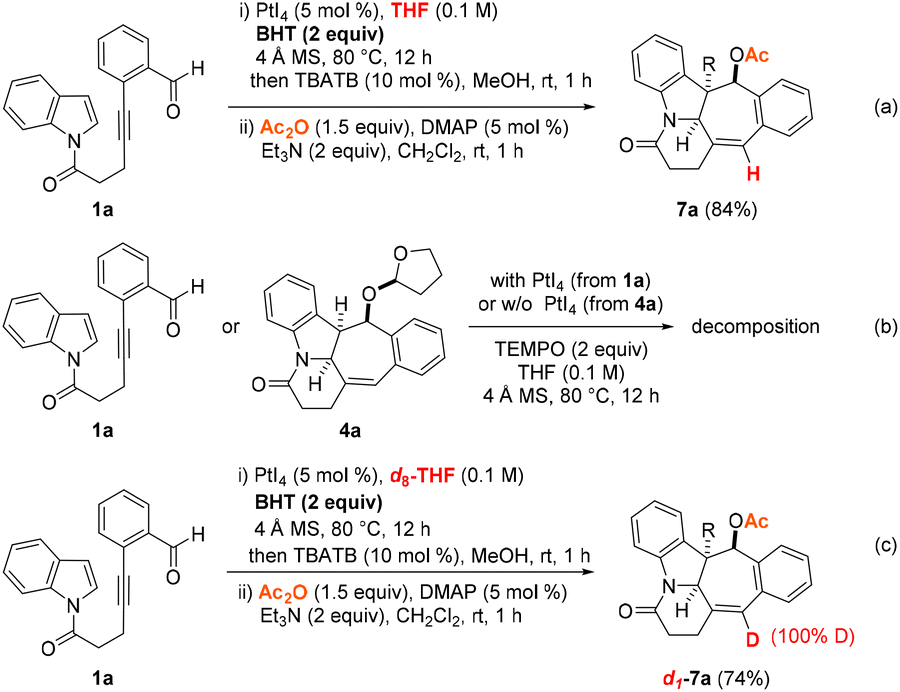 | ||
| Scheme 4 Control experiments with 1a. | ||
On the basis of our experimental findings vide supra, a tentative mechanism for the presented PtI4-catalyzed oxidative and hydrogenative dearomative [3 + 2] cycloaddition is proposed as shown in Scheme 5.10 This could initially involve the coordination of PtI4 to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond of 1a, which generates the Pt(IV)-complex A that undergoes 6-endo-dig cyclization to deliver the Pt-bound benzopyrylium intermediate B or its resonance structure C. Subsequent intramolecular dearomative [3 + 2] cycloaddition between the highly reactive oxonium moiety and the C(2)C(3) bond of the pendant electron-rich indolyl motif in B would afford the platinum carbene species D. A divergence in chemoselectivity is thought to occur at this juncture depending on the judicious choice of the solvent and oxidant. In the presence of phenylmethylsulfoxide, oxidation of this newly formed platinum carbene species via the cationic alkyl platinum intermediate E would furnish the desired product 3a with the expulsion of phenylmethyl sulfide and regeneration of the Pt(IV) salt (path a).22 Alternatively, in the absence of the oxidant and with THF as the solvent, the transient and highly reactive platinum carbene species D could undergo hydrogenation involving facile hydride migration from the reaction medium to the organometallic intermediate. This would provide alkyl platinum intermediate F along with the tetrahydrofuran cation.20a An ensuing β-oxygen elimination followed by migration of the Lewis acidic metal from the carbene carbon to the oxygen center in F would produce the oxyplatinum intermediate G. Nucleophilic addition of the pentacyclic metal species to the tetrahydrofuran cation would furnish the THF-substituted diastereomers 4a and 5a as well as regenerate the platinum(IV) catalyst (path b). Concomitant with this could be the competitive formation of 6a due to the hydration of the oxyplatinum species G by trace amounts of water that might be present in THF. A final sequence involving the deacetalization of 4a and 5a which is promoted by the introduction of a catalytic amount of TBATB followed by acetylation of the resulting alcohol intermediate 6a with acetic anhydride would afford the product 7a.
C bond of 1a, which generates the Pt(IV)-complex A that undergoes 6-endo-dig cyclization to deliver the Pt-bound benzopyrylium intermediate B or its resonance structure C. Subsequent intramolecular dearomative [3 + 2] cycloaddition between the highly reactive oxonium moiety and the C(2)C(3) bond of the pendant electron-rich indolyl motif in B would afford the platinum carbene species D. A divergence in chemoselectivity is thought to occur at this juncture depending on the judicious choice of the solvent and oxidant. In the presence of phenylmethylsulfoxide, oxidation of this newly formed platinum carbene species via the cationic alkyl platinum intermediate E would furnish the desired product 3a with the expulsion of phenylmethyl sulfide and regeneration of the Pt(IV) salt (path a).22 Alternatively, in the absence of the oxidant and with THF as the solvent, the transient and highly reactive platinum carbene species D could undergo hydrogenation involving facile hydride migration from the reaction medium to the organometallic intermediate. This would provide alkyl platinum intermediate F along with the tetrahydrofuran cation.20a An ensuing β-oxygen elimination followed by migration of the Lewis acidic metal from the carbene carbon to the oxygen center in F would produce the oxyplatinum intermediate G. Nucleophilic addition of the pentacyclic metal species to the tetrahydrofuran cation would furnish the THF-substituted diastereomers 4a and 5a as well as regenerate the platinum(IV) catalyst (path b). Concomitant with this could be the competitive formation of 6a due to the hydration of the oxyplatinum species G by trace amounts of water that might be present in THF. A final sequence involving the deacetalization of 4a and 5a which is promoted by the introduction of a catalytic amount of TBATB followed by acetylation of the resulting alcohol intermediate 6a with acetic anhydride would afford the product 7a.
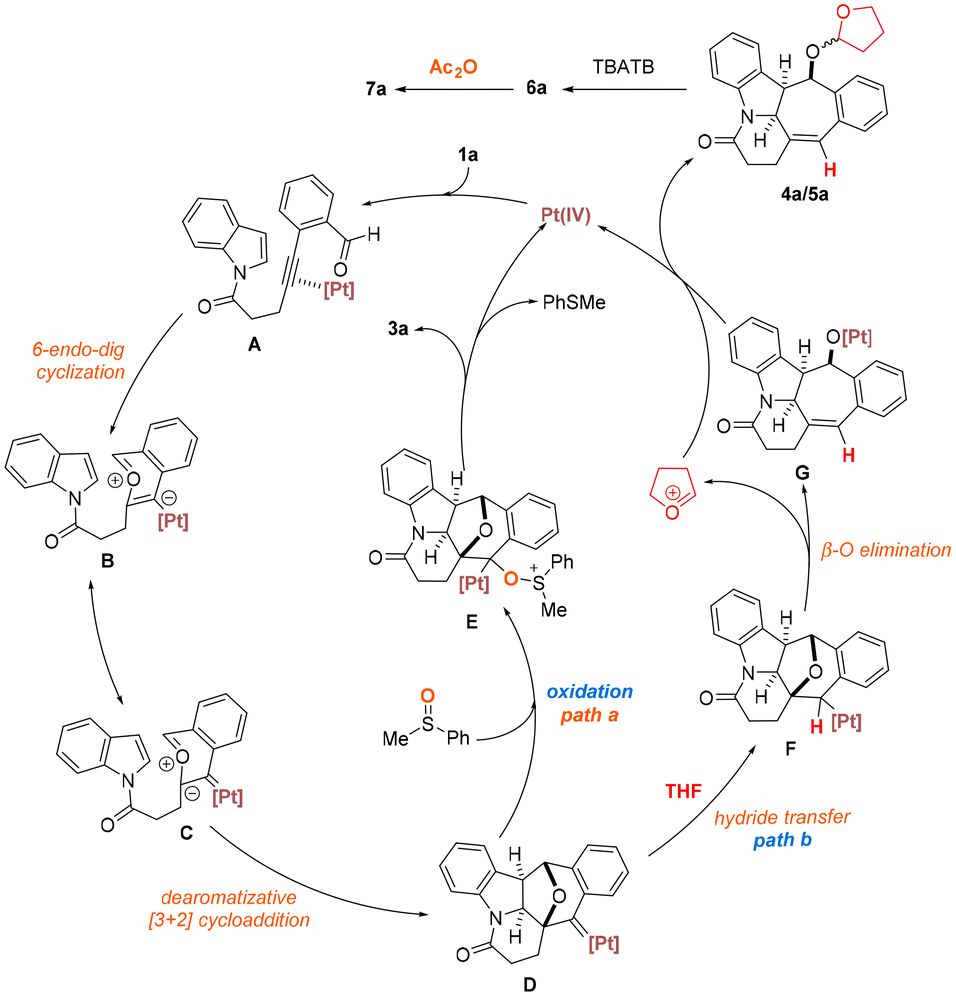 | ||
| Scheme 5 Proposed reaction mechanism for the PtI4-catalyzed oxidative and hydrogenative dearomative [3 + 2] cycloaddition represented by 1a. | ||
Conclusions
In summary, we have developed the first PtI4-catalyzed oxidative and hydrogenative dearomative [3 + 2] cycloaddition of readily accessible 1H-indole N-tethered o-alkynylbenzaldehydes. The protocol provides a practical and convenient method for the chemoselective synthesis of two structurally distinct benzofused cyclohepta[b]indolines with exclusive exo-selectivity in a highly efficient manner. It offers a unique instance in platinum catalysis of an in situ formed Pt-bound benzopyrylium species that subsequently engages in a dearomative [3 + 2] cycloaddition with the C(2)C(3) bond of the 1H-indole tether as the 2π cycloaddition partner. Our studies suggest that the key to achieving the observed chemodivergence was the ability to control the reactivity of the premised in situ generated platinum carbene species generated from the dearomative [3 + 2] cycloaddition step. Thus, the observed product selectivity was achieved by exploiting the respective oxidative and hydrogenative reaction conditions offered by PMSO and THF. The practicality of the oxidative and hydrogenative dearomative [3 + 2] cycloaddition methods is shown by the gram-scale preparation of two examples of the privileged scaffold at low catalyst loadings of 0.5 and 3 mol%, respectively. In addition to this is the successful late-stage modification of a diverse array of structurally complex natural products and drug molecules with this catalytic chemodivergent protocol. We envision that the present synthetic method will assist future efforts focused on the new dearomative cycloaddition chemistry of an in situ formed metal-bound benzopyrylium species with other members of the aromatic compound family, which is ubiquitous and includes commodity chemicals.
Author contributions
W. R. and P. W. H. C. conceived and directed the project. W. R. and D. S. designed the experiments and analysed the data. D. S., R. H., Q. B., J. C. and Y. L. performed all the experiments. All the authors contributed to the preparation and writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the Jiangsu Specially Appointed Professor Plan (to W. R.) and a Discovery Project Grant (DP210103425) from the Australian Research Council (to P. W. H. C.).References
- For recent reviews, see:
(a) E. Stempel and T. Gaich, Cyclohepta[b]indoles: A Privileged Structure Motif in Natural Products and Drug Design, Acc. Chem. Res., 2016, 49, 2390–2402 CrossRef CAS PubMed
; (b) J. Gierok, L. Benedix and M. Hiersemann, An Update on Cyclohepta[b]indoles, Eur. J. Org. Chem., 2021, 3748–3758 CrossRef CAS
-
(a) R. B. Woodward, Neuere Entwicklungen in der Chemie der Naturstoffe, Angew. Chem., 1956, 68, 13–20 CrossRef CAS
- T.-S. Kam and Y. Choo, Kopsifolines A–F: a New Structural Class of Monoterpenoid Indole Alkaloids from Kopsia, Helv. Chim. Acta, 2004, 87, 991–998 CrossRef CAS
-
(a) A. R. Carroll, E. Hyde, J. Smith, R. J. Quinn and G. Guymer, Actinophyllic Acid, a Potent Indole Alkaloid Inhibitor of the Coupled Enzyme Assay Carboxypeptidase U/Hippuricase from the Leaves of Alstonia actinophylla (Apocynaceae), J. Org. Chem., 2005, 70, 1096–1099 CrossRef CAS
- M. Andriantsiferana, R. Besselièvre, C. Riche and H. P. Husson, Structure De L′Érvitsine Alcaloïde α-Acylindolique D'Un Type Nouveau, Tetrahedron Lett., 1977, 18, 2587–2590 CrossRef
- J. C. Quirion, C. Kan-Fan, I. R. C. Bick and H. P. Husson, Aristolasol and Aristolasene: Indole Alkaloids from Aristotelia Australasica, Phytochemistry, 1988, 27, 3337–3339 CrossRef CAS
-
(a) A. D. Napper, J. Hixon, T. McDonagh, K. Keavey, J.-F. Pons, J. Barker, W. T. Yau, P. Amouzegh, A. Flegg, E. Hamelin, R. J. Thomas, M. Kates, S. Jones, M. A. Navia, J. O. Saunders, P. S. DiStefano and R. Curtis, Discovery of Indoles as Potent and Selective Inhibitors of the Deacetylase SIRT1, J. Med. Chem., 2005, 48, 8045–8054 CrossRef CAS
- T. Barf, F. Lehmann, K. Hammer, S. Haile, E. Axen, C. Medina, J. Uppenberg, S. Svensson, L. Rondahl and T. Lundbaäk,
N-Benzyl-Indolo Carboxylic Acids: Design and Synthesis of Potent and Selective Adipocyte Fatty-Acid Binding Protein (A-FABP) Inhibitors, Bioorg. Med. Chem. Lett., 2009, 19, 1745–1748 CrossRef CAS PubMed
- B. Joseph, D. Alagille, J. Mérour and S. Léonce, Synthesis and in Vitro Cytotoxic Evaluation of N-Substituted Benzo[5,6]cyclohepta[b]indoles, Chem. Pharm. Bull., 2000, 48, 1872–1876 CrossRef CAS PubMed
- For selected reviews, see:
(a) L. Chen, K. Chen and S. Zhu, Transition-Metal-Catalyzed Intramolecular Nucleophilic Addition of Carbonyl Groups to Alkynes, Chem, 2018, 4, 1208–1262 CrossRef CAS
-
(a) K. K. Gollapelli, V. B. Patil, A. Vinaykumar and R. Chegondi, Rh(I)-Catalyzed Stereoselective Desymmetrization of Prochiral Cyclohexadienones via Highly exo-Selective Huisgen-type [3 + 2] Cycloaddition, Chem. Sci., 2021, 12, 1544–1550 RSC
- A. K. Gupta, C. Y. Rhim, C. H. Oh, R. S. Mane and S.-H. Han, Gold Nanoparticle-Catalysed [3 + 2] Dipolar Cycloaddition of 1,6-Allenynebenzaldehydes: Construction of Polycyclic Ring Systems, Green Chem., 2006, 8, 25–28 RSC
- N. Kim, Y. Kim, W. Park, D. Sung, A. K. Gupta and C. H. Oh, Gold-Catalyzed Cycloisomerization of o-Alkynylbenzaldehydes with a Pendant Unsaturated Bond: [3 + 2] Cycloaddition of Gold-Bound 1,3-Dipolar Species with Dipolarophiles, Org. Lett., 2005, 7, 5289–5291 CrossRef CAS PubMed
- Z. Cao, H. Zhu, X. Meng, L. Tian, X. Sun, G. Chen and J. You, Gold-Catalyzed Reaction of ortho-Alkynylarylaldehydes with Conjugated Dienes: An Efficient Access to Highly Strained Tetracyclic Bridgehead Olefins, Chem. – Eur. J., 2016, 22, 9125–9129 CrossRef CAS
- C. H. Oh, S. M. Lee and C. S. Hong, Pt-Catalyzed Hydrative Cyclization of 2-Enynylbenzaldehydes and Its Application to Faveline Synthesis, Org. Lett., 2010, 12, 1308–1311 CrossRef CAS
- Q. T. Le, L. Guo, S. L. Lee, J. Lee and C. H. Oh, Gold-Catalyzed Synthesis of Icetexane Cores: Short Synthesis of Taxamairin B and Rosmaridiphenol, Org. Lett., 2020, 22, 9225–9228 CrossRef CAS
- C. H. Oh, L. Piao, J. J. Jung and J. Kim, A Formal Synthesis of Komaroviquinone: Use of a Pt-Catalyzed Hydrative Cyclization Reaction, Asian J. Org. Chem., 2016, 5, 1237–1241 CrossRef CAS
- S. S. Giri and R.-S. Liu, Copper-Catalyzed [4 + 2]-Cycloadditions of Isoxazoles with 2-Alkynylbenzaldehydes To Access Distinct α-Carbonylnaphthalene Derivatives: C(3,4)- versus C(4,5)-Regioselectivity at Isoxazoles, ACS Catal., 2019, 9, 7328–7334 CrossRef CAS
- G. Dyker, D. Hildebrandt, J. Liu and K. Merz, Gold(III) Chloride Catalyzed Domino Processes with Isobenzopyrylium Cation Intermediates, Angew. Chem., Int. Ed., 2003, 42, 4399–4402 CrossRef CAS
-
(a) R. Hu, J. Chen, Z. Wang, D. Shang, L. Yu, P. W. H. Chan and W. Rao, THF-Enabled PtBr2-Catalyzed Desymmetric Hydrogenative [3 + 2] Cycloaddition of 2-Alkynylbenzaldehyde-Tethered Cyclohexadienones, Org. Chem. Front., 2022, 9, 3577–3584 RSC
-
(a) J. Sang, L. Feng, R. Hu, J. Chen, D. Shang, Q. Bao and W. Rao, Sc(OTf)3-Catalyzed C2-Selective Cyanation/Defluorination Cascade of Perfluoroalkylated 3-Indolylmethanols and Application to the Synthesis of 3-Fluoro(perfluoroalkyl)-β-carbolines, Org. Lett., 2021, 23, 7666–7671 CrossRef CAS PubMed
- C. A. Witham, P. Mauleón, N. D. Shapiro, D. Benjamin, B. D. Sherry and F. D. Toste, Gold(I)-Catalyzed Oxidative Rearrangements, J. Am. Chem. Soc., 2007, 129, 5838–5839 CrossRef CAS PubMed
-
(a) C. Tortoreto, T. Achard, W. Zeghida, M. Austeri, L. Guéné and J. Lacour, Enol Acetal Synthesis through Carbenoid C–H Insertion into Tetrahydrofuran Catalyzed by CpRu Complexes, Angew. Chem., Int. Ed., 2012, 51, 5847–5851 CrossRef CAS PubMed
- A. Umehara, H. Ueda and H. Tokuyama, Condensation of Carboxylic Acids with Non-Nucleophilic N-Heterocycles and Anilides Using Boc2O, J. Org. Chem., 2016, 81, 11444–11453 CrossRef CAS
- CCDC 2189547 (3a), 2156622 (4a), 2156624 (5a), 2189548 (3aq′), 2156625 (7ah), 2189549 (10), and 2157370 (12) contain the supplementary crystallographic data for this paper.†.
- M. Shiri, M. A. Zolfigol and H. G. Kruger, Bis- and Trisindolylmethanes (BIMs and TIMs), Chem. Rev., 2010, 110, 2250–2293 CrossRef CAS PubMed
- J. Chen, J.-H. Lin and J.-C. Xiao, Halogenation through Deoxygenation of Alcohols and Aldehydes, Org. Lett., 2018, 20, 3061–3306 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral data, and crystallographic data. CCDC 2189547 (3a), 2156622 (4a), 2156624 (5a), 2189548 (3aq′), 2156625 (7ah), 2189549 (10), and 2157370 (12). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qo01520j |
| This journal is © the Partner Organisations 2023 |