
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral self-recognition in a bispericyclic cyclodimerisation reaction of 1-azadienes†
Adrián
López-Francés
 a,
Xabier
del Corte
a,
Zuriñe
Serna-Burgos
a,
Jesús M.
de los Santos
a,
Abel
de Cózar
*bc and
Javier
Vicario
*a
a,
Xabier
del Corte
a,
Zuriñe
Serna-Burgos
a,
Jesús M.
de los Santos
a,
Abel
de Cózar
*bc and
Javier
Vicario
*a
aDepartment of Organic Chemistry I, Faculty of Pharmacy, University of the Basque Country, UPV/EHU, Paseo de la Universidad 7, 01006 Vitoria-Gasteiz, Spain. E-mail: javier.vicario@ehu.eus
bDepartment of Organic Chemistry I, Donostia International Physics Centre (DIPC), University of the Basque Country, UPV/EHU, Paseo Manuel de Lardizabal, 3, 20018 Donostia-San Sebastián, Spain. E-mail: abel.decozar@ehu.eus
cIkerbasque, Basque Foundation for Science, Plaza Euskadi 5, 48009, Bilbao, Spain
First published on 16th October 2023
Abstract
Hermaphroditism of molecules: as in nature some species behave as male or female depending on the environment, herein we report a bispericyclic dimerisation of cyclic 1-azadienes where a molecule can behave as either diene or dienophile, depending on its location at the transition state. In a symmetrical reactive complex, here represented by an arbitrary reference system, a molecule that is positioned on top acts as the diene unit, while the dienophile partner is the one situated at the bottom. In addition, a strong chiral self-recognition phenomenon is observed, where each enantiomer within a racemic mixture of chiral 1-azadienes exclusively recognises itself. In order to shed some light into the understanding of the chiral self-recognition effect, an extensive DFT study of the reaction pathway is provided, concluding that a combination of attractive π-stacking forces and repulsive steric interactions is at the origin of the high stereospecificity of the reaction.
Introduction
The discovery of molecular chirality by Louis Pasteur in the 19th century is probably one of the most amazing experiments in the history of science,1 with enormous consequences in the understanding of the mechanisms of life. Nowadays, more than one-and-a-half centuries after Pasteur's experiment, it is widely agreed that receptors in living cells are able to discriminate between the two specular images of both enantiomers of one molecule, which has been defined as “chiral recognition”. The driving forces leading to chiral recognition are still not fully understood, but it is clear that non-covalent interactions are involved in the process. Electrostatic forces, hydrogen bonding, steric hindrance, π–π-stacking or dipolar interactions are the most common attractive or repulsive effects engaged in the chiral recognition phenomenon.2Particularly interesting in this domain of chemical science, “chiral self-recognition” is an intriguing phenomenon of “narcissism of molecules” that occurs when one of the two enantiomers within a racemic mixture exclusive and selectively recognises itself. The complementary event where an enantiomer recognises its opposite is defined as “chiral self-discrimination”. A limited number of examples of self-recognition or self-discrimination have been described thus far and, to the best of our knowledge, most of them are related to non-covalent interactions, mainly in solution,3,4 or gas phase5 and by the formation of metallic complexes6 or supramolecular structures.7 However, reports implying a chiral self-recognition effect within a racemic mixture leading to the formation of covalent bonds, where each enantiomer reacts selectively itself (or with its opposite), are scarce and, in those examples, the mechanisms accounting for the observed selectivity remain to be established. For example, in the design of first-generation light-driven molecular motors, Feringa and co-workers described the McMurry self-coupling of racemic cyclic ketones where a diastereomerically pure mixture of (R,R)- and (S,S)-olefins is formed instead of the expected statistical mixture.8 In addition, the thermal dimerisation of chiral allenes furnishes trans-cyclobutanes with a moderate degree of chiral self-recognition.9 Nevertheless, in both cases, the explanation for this exceptional behaviour is not discussed in detail, and it seems clear that a combination of attractive π-stacking interactions and repulsive steric effects is the main factor involved in the chiral self-recognition outcome, in view of the numerous π-systems present in the structure of the substrates involved in these examples.
Stereoselective cycloaddition reactions are probably some of the most interesting processes to be chosen for the study of the chiral recognition or chiral discrimination phenomenon, considering the diverse non-covalent interactions involved in their theorised transition states.10 In particular, bispericyclic Diels–Alder dimerisation is a striking process, where a 4π-system of two identical molecules can act as both the diene and dienophile species, of which the most simple example is the cyclopentadiene dimerisation reaction, widely studied by Caramella.11 In the same context, an analogous study of bispericyclic cycloheptatriene dimerisation has been recently reported by Jørgensen and Houk.12 An elegant example of bispericyclic Diels–Alder dimerisation is the [4 + 2] cyclodimerisation of ortho-quinols, applied for the synthesis of diverse natural products.13 As in nature in several hundred species of fish, angiosperms or invertebrate animals, experiencing life both as a male and as a female is the reproductive norm,14 we can consider bispericyclic reactions as a kind of “hermaphroditism of molecules”.
Among the very many established approaches for the construction of N-heterocycles, the aza-Diels–Alder reaction, analogous to all-carbon Diels–Alder cycloaddition, is one of the most efficient methods, leading to the formation of substituted pyridine derivatives.10,15 In this context, as a part of our ongoing research on the chemistry of 1-azadienes, in 2011 we reported an example of a cyclodimerisation reaction of 1-azadienes derived from α-aminoacids, where our 4π-system acted as both azadiene and dienophile species.16 Within a different research field, in 2006, we reported an efficient multicomponent protocol for the synthesis of unsaturated 3-amino-γ-lactam derivatives II making use of amines, aldehydes and pyruvate derivatives as starting materials.17 More recently, we have developed an enantioselective organocatalytic system for this transformation, using chiral phosphoric acids as catalysts, and we have extended this strategy to the preparation of phosphorus- and fluorine-substituted γ-lactam substrates.18 Key features of the structure of those substrates II are the presence of a reactive enamine moiety and a chiral stereocentre at the 5-membered ring. Taking advantage of those two attributes, we have used unsaturated 3-amino-γ-lactam as substrate in diverse stereoselective reactions.19 At the intersection of both fields, and considering that unsaturated 3-amino-γ-lactam substrates II can be easily obtained through a simple multicomponent reaction at a multigram scale and from readily available starting materials, we thought that they could be excellent precursors for the preparation of rigid 1-azadienes I embedded in a γ-lactam structure, through a simple olefination reaction of the enamine moiety (Fig. 1). The locked pseudo-cis configuration of the 4π-system would enhance the reactivity towards dienophiles and, in addition, the presence of a chiral centre at the 5-membered ring may allow the use of the 1-azadiene moiety in diastereoselective cycloaddition reactions, leading to highly functionalised bicyclic substrates.
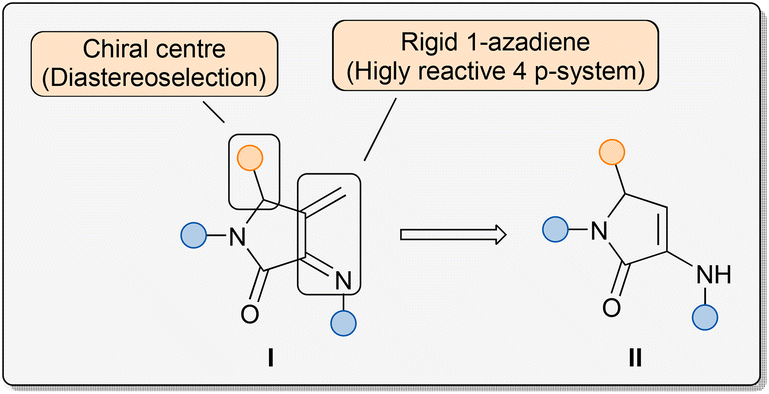 | ||
| Fig. 1 Features of γ-lactam-derived rigid 1-azadienes I from γ-lactam-based enamines II. | ||
Continuing with our ongoing research on the synthesis of 1-azadienes and their applications in aza-Diels–Alder reactions,16,20 herein we report the generation of chiral rigid 1-azadienes derived from γ-lactams and their spontaneous cyclodimerisation reaction through a highly stereoselective bispericyclic process, where a strong chiral self-recognition phenomenon is observed. Moreover, strong evidences of the self-recognition effect and the forces driving this phenomenon are provided through an extensive DFT study of the reaction pathway.
Results and discussion
Taking into account the considerations mentioned above, the starting 3-amino-γ-lactam derivatives 1 were prepared following a known procedure,18,19 consisting of a multicomponent reaction of benzaldehyde, amines and ethyl pyruvate in the presence of a Brønsted acid catalyst (see ESI†).In our previous report on the synthetic potential of γ-lactam derivatives, β-phosphorated cyclic enamine substrates proved to be ineffective in Horner–Wadsworth–Emmons reactions, providing vinylogous nucleophilic addition products instead of the expected 1-azadienes.18b Keeping in mind these results, we projected the formation of the azadiene moiety through a Mannich reaction with Eschenmoser's salt followed by the elimination of dimethylamine. For this reason, the functionalisation of substituted γ-lactam substrates 1 with Eschenmoser's salt was accomplished in very good yields using a slight excess of triethylamine as a base in refluxing chloroform (Scheme 1). Following this procedure, several functionalised γ-lactams derived from para-halogen-substituted anilines (2a–c), p-toluidine (2d), simple aniline (2e) and p-anisidine (2f) were prepared in good to excellent yields (61–96%). Unfortunately, the use of γ-lactams 1 derived from anilines bearing electron withdrawing substituents such as NO2 or CF3 did not provide the functionalised substrates, which may be due to a deactivation of the enamine moiety. Considering the assorted reactivity usually observed in similar 3-amino-γ-lactam derivatives 1,19 functionalised γ-lactams 2 were extensively characterised on the basis of their 1H, 13C, 19F NMR, 2D NMR spectra, IR and HRMS (see ESI†). As usual, the multiplicity of the carbons was verified by DEPT experiments and could be also deduced from the interactions observed in the HSQC spectrum. Dimethylaminomethyl-substituted γ-lactams 2 may be suitable starting materials for the generation of chiral 1-azadienes. Unfortunately, the direct elimination of trimethylamine by the classical treatment with methyl iodide or Me2SO4 failed to provide the target conjugated alkene. For this reason, the replacement of the dimethylamino moiety by a better leaving group was performed by the treatment of functionalised γ-lactams 2 in neat acetic anhydride at room temperature, leading in a few minutes to the formation of substituted γ-lactams 3 (Scheme 1). Purification of γ-lactams 3 by chromatography was unfeasible, due to the fast decomposition of the substrates under exposure to silica or alumina. However, we were able to isolate and fully characterise p-tolyl-derived lactam 3d (R = p-MeC6H4) straight from the crude reaction mixture as red crystals. In view of the low stability of these substrates, acetoxy-substituted γ-lactams 3 were used without purification in the further steps, after the elimination of acetic acid and the excess of acetic anhydride under low pressure.
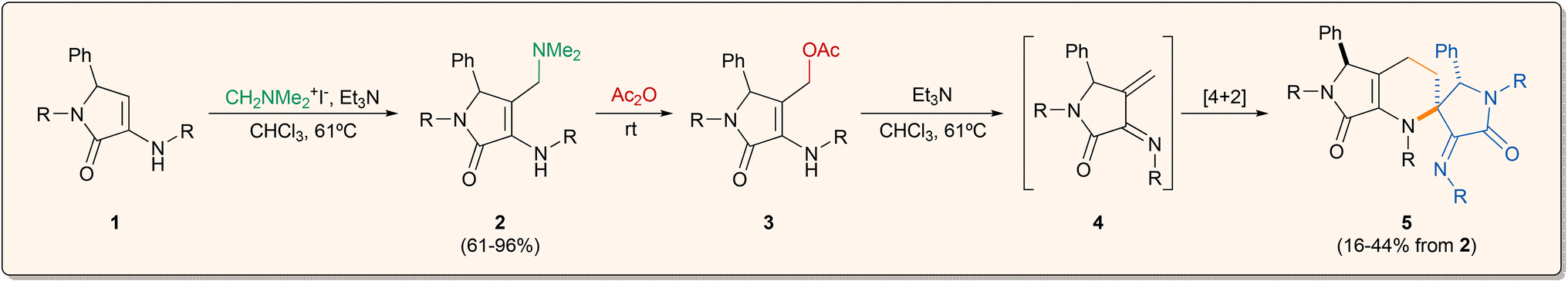 | ||
| Scheme 1 Cyclodimerisation of in situ generated 1-azadienes 4. aIsolated yield for a 50% maximum. | ||
In order to generate the target azadiene substrates, the elimination of acetic acid was promoted upon treatment of acetoxy-substituted γ-lactams 3 with freshly distilled triethylamine as the basic source. Surprisingly, under those conditions, only spirocyclic tetrahydropyridines 5 were obtained as single diastereomers. Although the isolation of azadiene species 4 was not feasible, we theorised that the reaction may proceed through the dimerisation of intermediate 4 by a regio- and diastereo-specific aza-Diels–Alder cycloaddition mechanism, where the α,β-unsaturated imine system acts both as diene and dienophile source (Scheme 1).
The reaction was applied to six γ-lactam derivatives 2 derived from different anilines (Table 1). The reaction using γ-lactams 2a–c (R = p-BrC6H4, p-ClC6H4, p-FC6H4), having halogen atoms at the aniline moieties, provided spirocyclic tetrahydropyridines 5a–c in moderate to good yields (16–38%) as a single diastereomer (Table 1, entries 1–3). Moreover, the use of γ-lactam 2d derived from p-toluidine (R = p-MeC6H4) furnished a single diastereomer of the corresponding spirocyclic substrate 5d in good yield (Table 1, entry 4). To our surprise, the in situ generation of 1-azadiene 4e derived from simple unsubstituted aniline (R = Ph) provided a complex mixture resulting from the decomposition of the azadiene species (Table 1, entry 5). Interestingly, the presence of a strong electron-donating group at the aromatic ring in γ-lactam 2f (R = p-MeOC6H4) led to the formation of spirocyclic tetrahydropyridine 5f in good yield (42%) as a mixture of diastereomers (dr = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Table 1, entry 6). Substrates 5, obtained from the cyclodimerisation of in situ generated 1-azadienes 4, were extensively characterised on the basis of their 1H, 13C, 19F NMR, 2D NMR spectra and HRMS (see ESI†). The most characteristic signals for the spirocyclic substrates 5 in the 1H NMR spectrum are the two singlets at δH ∼5.5 and 4.9 ppm, corresponding to the two CH groups at both stereogenic carbons of the two γ-lactam rings, and four multiplets, two of them at δH ∼2.3 ppm and the other two at δH ∼1.5 ppm, attributed to the two CH2 groups at the tetrahydropyridine ring, the multiplicity of which is consistent with their diastereotopic character. In the 13C NMR spectrum, the formation of substrates 5 is evident from the presence of two signals at δC ∼70 and 65 ppm corresponding to the two CH groups at the two lactam rings, while the two methylene groups appear at δC ∼25 and 18 ppm and the quaternary carbon at the spirocycle shows a chemical shift at δC ∼65 ppm.
:1) (Table 1, entry 6). Substrates 5, obtained from the cyclodimerisation of in situ generated 1-azadienes 4, were extensively characterised on the basis of their 1H, 13C, 19F NMR, 2D NMR spectra and HRMS (see ESI†). The most characteristic signals for the spirocyclic substrates 5 in the 1H NMR spectrum are the two singlets at δH ∼5.5 and 4.9 ppm, corresponding to the two CH groups at both stereogenic carbons of the two γ-lactam rings, and four multiplets, two of them at δH ∼2.3 ppm and the other two at δH ∼1.5 ppm, attributed to the two CH2 groups at the tetrahydropyridine ring, the multiplicity of which is consistent with their diastereotopic character. In the 13C NMR spectrum, the formation of substrates 5 is evident from the presence of two signals at δC ∼70 and 65 ppm corresponding to the two CH groups at the two lactam rings, while the two methylene groups appear at δC ∼25 and 18 ppm and the quaternary carbon at the spirocycle shows a chemical shift at δC ∼65 ppm.
Remarkably, although spirocyclic tetrahydropyridines 5 contain three stereogenic carbons and racemic functionalised γ-lactams 1 are used as starting materials, a single diastereomer is formed in the case of substrates 5a–d. With the aim of fully understanding the mechanism of the cycloaddition process and the nature of the generation of the stereocentres, a single crystal of spirocyclic tetrahydropyridine 5a was isolated from a mixture dichloromethane/hexane and X-ray diffraction experiments were performed in order to determine the relative configuration of the chiral carbons (Fig. 2). The crystal structure of 5a revealed a 2S*,3S*,5′S* relative configuration. Moreover, in order to get more solid evidence for the relative configuration of the stereocentres, a second X-ray structure was obtained also for spirocycle 5b, providing the same configuration as 5a (see ESI†). In congruity with this configuration, there must be an exclusive reaction within the racemic mixture between 1-azadienes 4 owning the same configuration (either R with R or S with S) through a stereospecific endo transition state relative to the imine substituent.
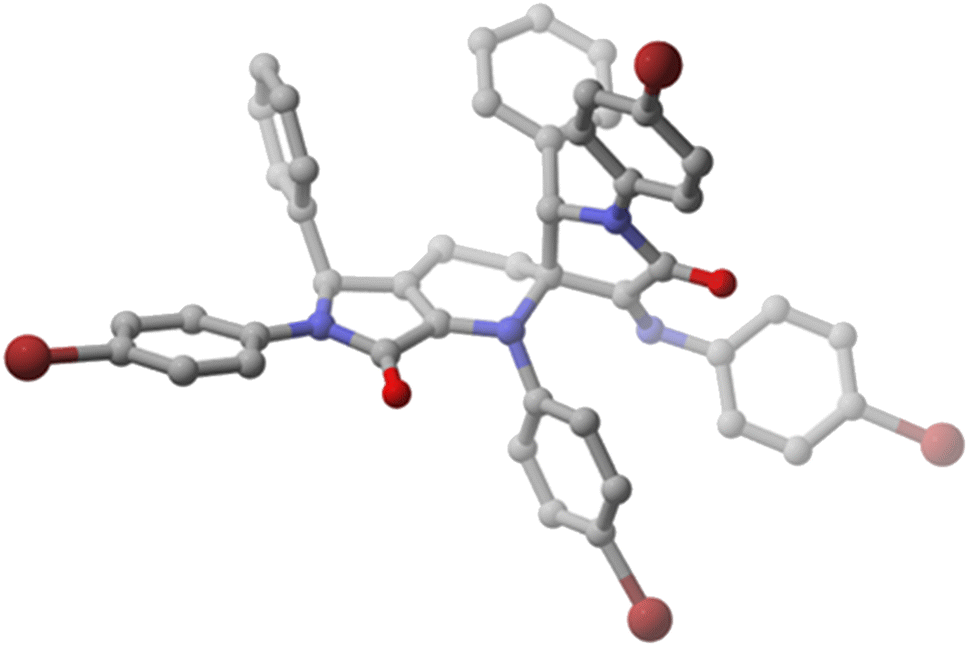 | ||
| Fig. 2 X-ray structure of functionalised spirocyclic tetrahydropyridine 5a (H, white; C, grey; O, red; N, blue; Br, brown) (2S,3S,5′S enantiomer shown). | ||
Wishing to firmly comprehend the cycloaddition process, we performed an NMR study of the reaction using p-bromoaniline-derived γ-lactam 2a as a model (see ESI†). After generating the acetylated substrate 3a, trimethylamine was added at rt and a temperature ramp-up regimen with NMR spectra acquired at 5-degree intervals was performed. Throughout the initial temperature increments (from rt to 50 °C), no significant changes were observed. Consequently, a temperature of 55 °C was determined as the optimum for the generation of 1-azadiene 4a. Then a 1H NMR spectrum was recorded every 30 minutes and the course of the reaction and the side products formed were analysed. Our conclusion is that, once the 1-azadiene species is formed, it reacts extremely fast avoiding the determination of the presence of any intermediate clearly. However, at a certain point, we were able to detect the signals corresponding to the vinylic protons of the 1-azadiene species at δH ∼5.8 ppm that disappear very fast, confirming that 1-azadiene 4a is the real intermediate in this reaction. We could also conclude that, as soon as the 1-azadiene is formed, it reacts instantaneously with itself and a minor part of 1-azadiene suffers from degradation leading to a complex mixture derived from reactions at the highly reactive conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and/or the hydrolysis of imine moiety. For these reasons, we theorise that the different yields obtained in the scope depend on the stability of the 1-azadiene (faster decomposition than dimerisation, lower yield; slower decomposition than dimerisation, higher yield).
C bond and/or the hydrolysis of imine moiety. For these reasons, we theorise that the different yields obtained in the scope depend on the stability of the 1-azadiene (faster decomposition than dimerisation, lower yield; slower decomposition than dimerisation, higher yield).
In order to get a more in-depth understanding of the empirical results and despite the complexity of the system involved in our reaction (88 to 104 atoms), we decided to perform computational calculations within the DFT framework. We focused on understanding the excellent chiral self-recognition towards homo-spirocycles 5a–d stemming from the reaction of two γ-lactams bearing the same absolute configuration, as well as the selectivity loss observed for 1-azadiene 4f (R = p-MeOC6H4) where both homo- and heterodimeric cycloadducts were isolated (experimental dr = 2:1, Scheme 1).
In view of the NMR experiments that point out the fast reactivity of the diene species once it is formed (vide supra), we hypothesized that the chiral self-recognition phenomenon would start already within the acetylated precursors 3a and 3f. In order to prove this theory, an initial optimisation of the structures of 3a and 3f was performed. Our calculations show that the phenyl substituent in position 5 and the acetyl group are placed perpendicular to the five-membered ring (see Fig. 3A), thus affecting the possible intermolecular interactions related to the self-recognition phenomenon. Therefore, we considered three possible approaches for the dimer formation: (a) homo-acetylated aggregate AAhomo, where the two phenyl groups are in outside positions (i.e. R-3a/f interacts with R-3a/f, and S-3a/f interacts with S-3a/f); (b) hetero-acetylated aggregate AAhetero, where one of the phenyl groups is placed in an inside position (i.e. R-3a/f interacts with S-3a/f); furthermore we considered (c) an additional homo-acetylated aggregate AAhomo-inin where the two phenyl groups are in an inside position. Note that in the calculations only R enantiomers will be considered as reference homo-aggregates for clarity. We evaluated the relative stability of the different acetylated aggregates using Gibbs binding free energies as follows:
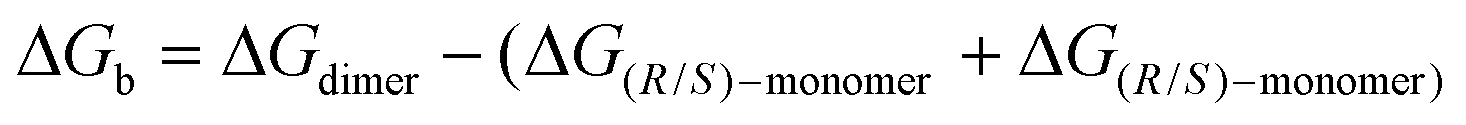 | (1) |
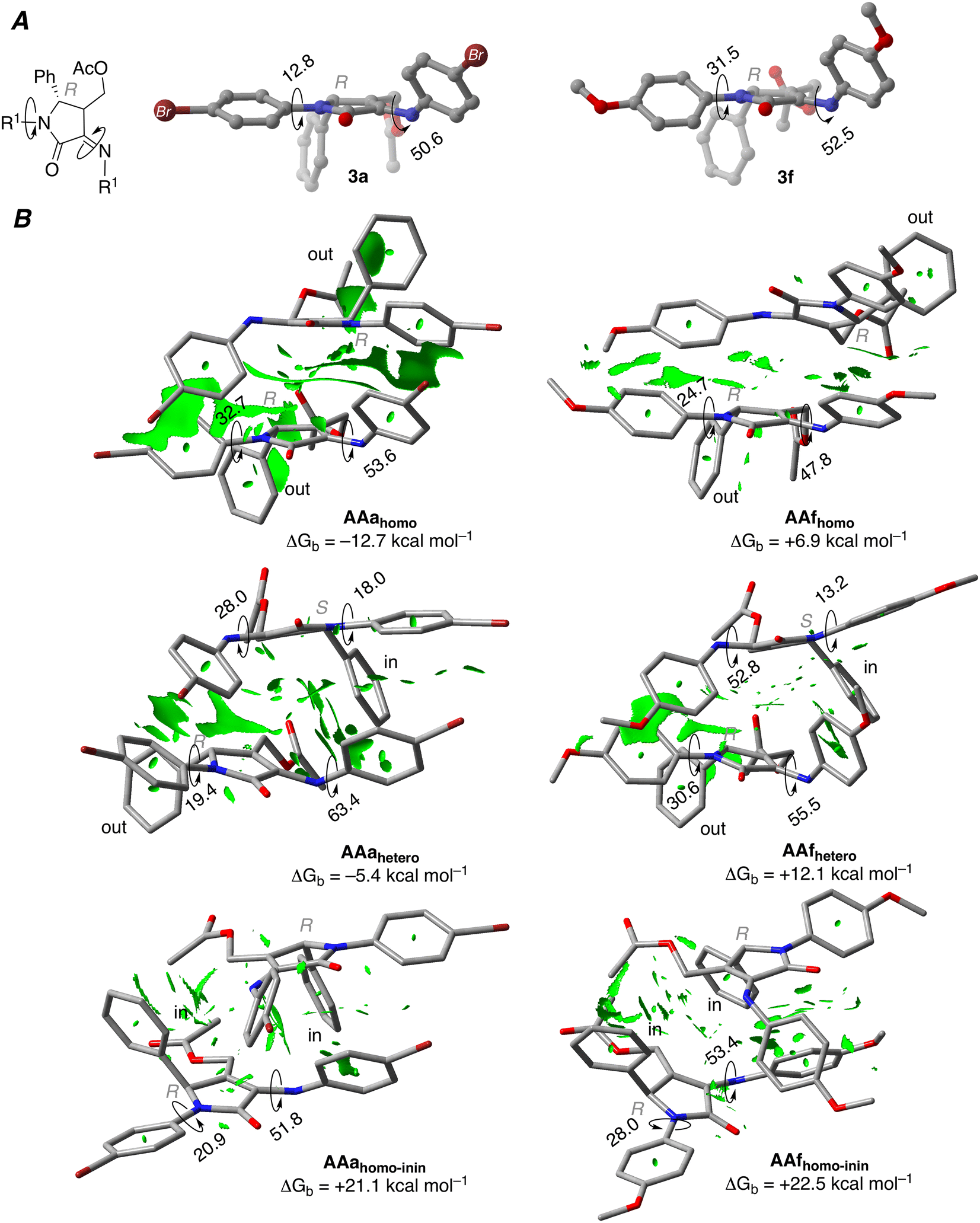 | ||
| Fig. 3 (A) Main geometrical features of compounds 3a and 3f computed at M06-2X-GD3 (PCM)/6-31G* level of theory. (B) Contour plots of the reduced density gradient isosurfaces (RGD, density cutoff = 0.20 au) for the acetylated aggregates AAa and AAf including Gibbs binding free energies (ΔGb) computed at M06-2X-GD3 (PCM)/6-31+G**//M06-2X-GD3 (PCM)/6-31G* level of theory. Dihedral angles are in degrees. The green surfaces indicate attractive non-covalent interactions. | ||
The computed ΔGb values for brominated compounds 3a (Fig. 3B) show that formation of AAahomo is strongly favoured compared to AAahetero (Gibbs binding energies of −12.7 and −5.4 kcal mol−1, respectively). These results indicate the energetic penalty associated with the phenyl and AcO substituents placed preferentially interacting with another R-enantiomer, placing the substituents in an outer position; and the same is expected for S-enantiomers. In fact, the formation of AAahomo-inin is strongly disfavoured, showing a Gibbs binding energy of +21.1 kcal mol−1.
In order to have more insights about these aggregates, we further analysed the bonding mechanism of these later dimers using a canonical energy decomposition analysis (EDA).21 Within this method, the interaction energy is decomposed into four terms: (i) classical electrostatic interactions, (ii) two-centre four-electron Pauli repulsions, (iii) stabilising orbital interactions and (iv) dispersion energy. The obtained results show that formation of AAahomo aggregates is favoured compared to AAahetero due to more stabilising dispersion interaction and less destabilising Pauli repulsion, pointing towards the relevance of the π–π interactions for this process.
When MeO-derived acetylated precursors 3f are considered, the calculations show that the aggregation process is not energetically favoured, as reflected by the positive ΔGb values. In this case, no self-recognition phenomenon can be addressed at this initial reaction stage. Next, we performed the optimisation of isolated γ-lactam dienes 4a and 4f. Geometrical inspection of these compounds indicates that the phenyl substituent in position 5 effectively blocks one of the prochiral faces of the azadiene moiety (Fig. 4). Analogously to the acetylated aggregate case, we anticipated that the formation of reactive complexes RChomo, where the two 5-phenyl substituents are placed in an outward position, would be favoured compared to RChetero counterparts, where one of the 5-phenyl moieties necessarily needs to be placed in an inward position. Our analysis shows that the binding strength depends on the nature of the R substituents, where Br-derived γ-lactam dimers RCa are more strongly bound compared to MeO-derived analogous RCf by ca. 8.0 kcal mol−1, in line with the results obtained for the acetylated aggregates (Table 2).
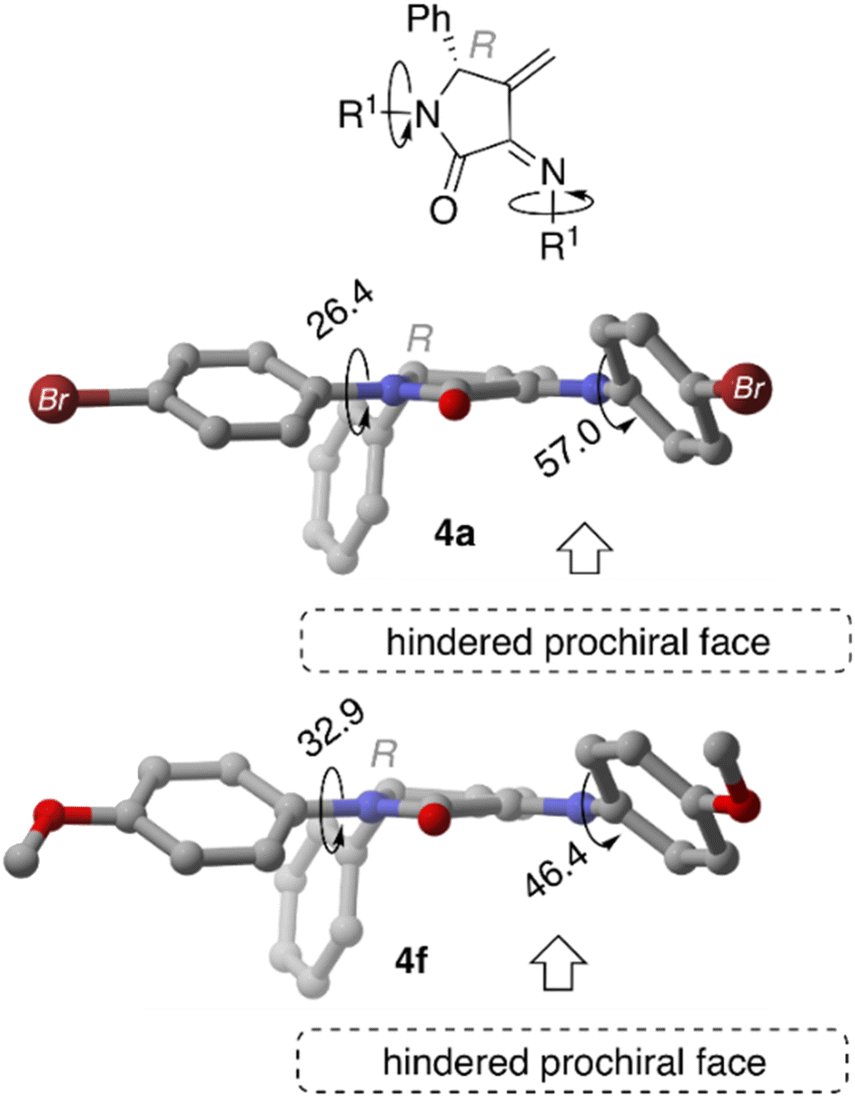 | ||
| Fig. 4 Main geometrical features of compounds 4a and 4f computed at M06-2X-GD3 (PCM)/6-31G* level of theory. | ||
| RCa | RCf | |
|---|---|---|
| Homo | −14.6 | −5.9 |
| Hetero | −9.4 | −1.4 |
Our canonical EDA calculations on the reactive complexes confirmed that the formation of RCa dimers is more favoured compared to RCf analogues, due to more stabilising electrostatic and dispersion interaction energies, which compensates a slightly more destabilising Pauli repulsion in the former (see ESI†). This is in agreement with the work reported by Houk et al.22 for model mono-substituted benzene dimers, where halogen substitution enhances π–π interactions compared to methoxy substitution due to more favourable electrostatic and dispersion interactions.
Our results also show the preferential formation of RChomo complexes compared to RChetero, a consequence of the presence of one 5-phenyl substituent in an inward position that prevents extended π–π interaction over the complete aromatic system (vide supra) that was observed in the acetylated precursors AA. Moreover, we observed that the less stabilising interactions observed for 4f are also reflected in a lower RChomo–RChetero discrimination (homo/hetero Gibbs binding free energies difference of 5.2 and 4.5 kcal mol−1 for RCa and RCf, respectively). We relate this fact to the lower chiral self-recognition observed for the synthesis of 5f.
Once the reactive complexes were analysed, we next calculated the key transition structures associated with the cyclodimerisation reaction of 4a and 4f that leads to spirocyclic tetrahydropyridines 5 (Fig. 5). Among all the transition structures analysed, the lowest Gibbs activation barriers are those associated with the imino moiety in endo approach, where at least one of the 5-substituents is placed in an outward position. In fact, the activation barrier associated with exo approaches lies about 10.0 kcal mol−1 above them (see ESI†).
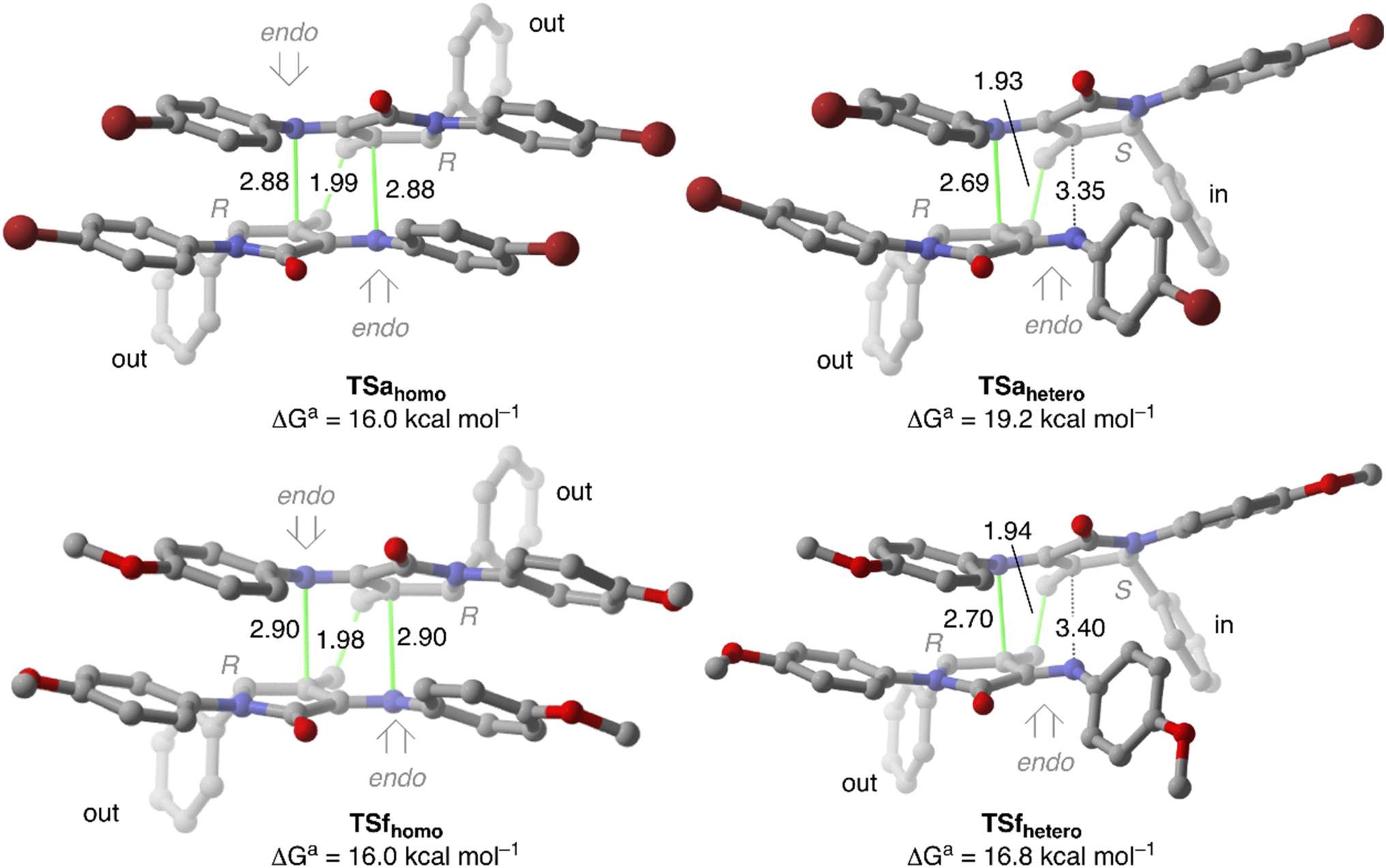 | ||
| Fig. 5 Main geometrical features and Gibbs activation energies computed for the less energetic transition structures associated with the Diels–Alder dimerisation of the 1-azadienes 4a and 4f computed at M06-2X-GD3 (PCM)/6-31+G**//M06-2X-GD3 (PCM)/6-31G* level of theory. Distances are in Å. | ||
From a geometrical point of view, both TSahomo and TSahetero are concerted but asynchronous where the carbon–carbon bond develops earlier than the carbon–nitrogen one. Remarkably, TSahomo has C2 symmetry, with two identical carbon–nitrogen distances. Moreover, the displacement vectors associated with the imaginary frequency correspond to C–C bond formation as with non-negligible in-phase vibration associated with formation of both N–C bonds. Therefore, it is not possible to identify which of the initial γ-lactams acts neither as diene nor as dienophile. In fact, TSahomo corresponds to a bispericyclic ambimodal [4 + 2]/[2 + 4] transition state analogous to that reported by Caramella et al.11 for the endo-dimerisation of cyclopentadiene. In that report, the authors state that, in such symmetrical TSs, the orbital interactions are maximised with a minimal structural deformation, thus leading to a decrease in the activation barrier.
In fact, our DFT calculations show a strong preference towards formation of homo-spirocycles 5a, as reflected by the Gibbs activation difference of 2.9 kcal mol−1 between TSahomo and TSahetero that is related to the higher stabilising interaction observed in RCahomo. It is important to notice that all our attempts to locate TSahomo-inin, where the dienophile fragment has the 5-substituent in an inside position, evolve towards TSahomo in few optimisation steps.
By including the previously computed Gibbs free activation energies in the Eyring–Polanyi equation23 and imposing the normalization conditions of eqn (3):
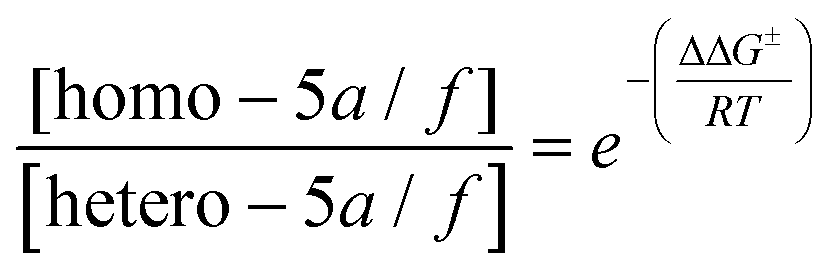 | (2) |
| [homo − 5a/f] + [hetero − 5a/f] = 100 | (3) |
:hetero-5a ratio of 99:<1, in perfect agreement with the experimental evidences.
Furthermore, we quantified the origin of that chiral self-recognition by using the activation strain (ASM) distortion/interaction model.24 ASM decomposes the electronic activation barrier into two terms: (i) strain energy that results from the deformation of the reactants and (ii) interaction energy between these deformed reactants. These analyses revealed that the interaction energy controls the strong preference towards homo-5a, mainly dominated by a lower Pauli repulsion and higher dispersion energy in TSahomo (see ESI†).
In order to further analyse that assessment, we extended our computational analysis to less sterically demanding brominated γ-lactams, where the phenyl group in position 5 is replaced by methyl 4g or isopropyl 4h groups (see ESI†). In these model systems, we observe similar homo-preference, thus pointing to the low relevance of the size of the 5-substituent on the selectivity.
On the contrary, the dimerisation of 4f is less stereo-selective (Gibbs activation difference of 0.8 kcal mol−1, theoretical homo-5f:hetero-5f ratio of 79:21). We relate this selectivity loss to the lower RChomo–RChetero discrimination for methoxy-substituted γ-lactam. ASM calculations reflect that TSfhomo preference is a consequence of lower Pauli repulsion, whereas almost equal dispersion energy stabilisation is observed in TSfhomo and TSfhetero, contrary to 5a case. Thus, mixtures of homo-5f and hetero-5f would be obtained (theoretical ratio of 80:20), in perfect agreement with the experimental results.
Conclusions
In conclusion, a stereospecific synthesis of γ-lactam-derived dimeric spirocycles is reported through a bispericyclic cyclodimerisation reaction of cyclic rigid 1-azadienes. Spirocycles possess unique three-dimensional structures that can provide a diverse range of chemical and biological properties. The presence of a spirocyclic motif can significantly impact the reactivity, stability, and physical properties of organic molecules, making them valuable in drug discovery and materials science.25 Moreover, a strong chiral self-recognition effect is observed in the cycloaddition process, where only one diastereomer is formed within a racemic mixture, as a consequence of the exclusive reaction of two chiral 1-azadienes with the same absolute configuration. Surprisingly, the presence of a p-MeOC6H4 substituent at the nitrogen atom of the 1-azadiene results in a diastereoselectivity loss. DFT calculations on the reaction mechanisms show that this chiral self-recognition can be related to the presence of more stabilising electrostatic and dispersion interaction energies of the homo-cycloadducts, compared to their hetero- counterparts, supporting the empirical results. Although several studies have been reported on bispericyclic dimerisation of all-carbon dienes, as far as we know, this is the first study of the analogous reaction of azadienes. In addition, this is also the first computational study of a chiral self-recognition phenomenon leading to the formation of covalent bonds.Author contributions
A. L.-F., A. d. C. and J. V. conceived the work. A. L.-F., X. d. C. and Z. S.-B. carried out the experimental work. A. L.-F., X. d. C. and A. d. C. carried out the computational work. A. d. C. and J. V. drafted the manuscript. All the authors contributed with comments, revising the manuscript and all have approved the final version. J. M. d. l. S., A. d. C. and J. V. directed the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by Ministerio de Ciencia, Innovación y Universidades (MCIU-Madrid) (PID2021-122558OB-I00, UE), and Eusko Jaurlaritza (GV, IT1701-22 and IT-1553-22; UPV-EHU) is gratefully acknowledged. The authors are grateful for technical and human support provided by SGIker (UPV/EHU/ERDF, EU). The authors thank SGI/IZO-SGIker of the UPV/EHU and the DIPC for the generous allocation of analytical and computational resources. A. L.-F. thanks the Basque Country Government for a predoctoral grant.References
- G. Vantomme and J. Crassous, Pasteur and chirality: A story of how serendipity favors the prepared minds, Chirality, 2021, 33, 597–601, DOI:10.1002/chir.23349.
- A. Berthod, Chiral Recognition Mechanisms, Anal. Chem., 2006, 78, 2093–2099, DOI:10.1021/ac0693823.
- Seminal work: T. Williams, R. G. Pitcher, P. Bommer, J. Gutzwiller and M. Uskokovic, Diastereomeric solute-solute interactions of enantiomers in achiral solvents. Nonequivalence of the nuclear magnetic resonance spectra of racemic and optically active dihydroquinine, J. Am. Chem. Soc., 1969, 91, 1871–1872, DOI:10.1021/ja01035a060.
- (a) V. Dašková, D. Padín and B. L. Feringa, Turning Enantiomeric Relationships into Diastereomeric Ones: Self-Resolving α-Ureidophosphonates and Their Organocatalytic Enantioselective Synthesis, J. Am. Chem. Soc., 2022, 144, 23603–23613, DOI:10.1021/jacs.2c10911; (b) Z. Szakács, Z. Sánta, A. Lomoschitz and C. Szántay Jr., Self-induced recognition of enantiomers (SIRE) and its application in chiral NMR analysis, Trends Anal. Chem., 2018, 109, 180–197, DOI:10.1016/j.trac.2018.07.020; (c) M. M. Safont-Sempere, P. Osswald, K. Radacki and F. Würthner, Chiral Self-Recognition and Self-Discrimination of Strapped Perylene Bisimides by π-Stacking Dimerization, Chem. – Eur. J., 2010, 16, 7380–7384, DOI:10.1002/chem.201001137; (d) S. Ghosh, A. Wu, J. C. Fettinger, P. Y. Zavalij and L. Isaacs, Self-Sorting Molecular Clips, J. Org. Chem., 2008, 73, 5915–5925, DOI:10.1021/jo8009424; (e) T. Kamada, N. Aratani, T. Ikeda, N. Shibata, Y. Higuchi, A. Wakamiya, S. Yamaguchi, K. S. Kim, Z. S. Yoon, D. Kim and A. Osuka, High Fidelity Self-Sorting Assembling of meso-Cinchomeronimide Appended meso-meso Linked Zn(II) Diporphyrins, J. Am. Chem. Soc., 2006, 128, 7670–7678, DOI:10.1021/ja0611137.
- A. Zehnacker and M. A. Suhm, Chirality Recognition between Neutral Molecules in the Gas Phase, Angew. Chem., Int. Ed., 2008, 47, 6970–6992, DOI:10.1002/anie.200800957.
- H. Jędrzejewska and A. Szumna, Making a Right or Left Choice: Chiral Self-Sorting as a Tool for the Formation of Discrete Complex Structures, Chem. Rev., 2017, 117, 4863–4899, DOI:10.1021/acs.chemrev.6b00745.
- (a) J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH Verlagsgesellschaft mbH, 1995. DOI:10.1002/3527607439; (b) G. M. Whitesides and B. Grzybowski, Self-Assembly at All Scales, Science, 2002, 295, 2418–2421, DOI:10.1126/science.1070821; (c) M. Petryk, K. Biniek, A. Janiak and M. Kwit, Unexpected narcissistic self-sorting at molecular and supramolecular levels in racemic chiral calixsalens, CrystEngComm, 2016, 18, 4996–5003, 10.1039/C6CE00256K.
- (a) M. K. J. ter Wiel, R. A. van Delden, A. Meetsma and B. L. Feringa, Light-Driven Molecular Motors: Stepwise Thermal Helix Inversion during Unidirectional Rotation of Sterically Overcrowded Biphenanthrylidenes, J. Am. Chem. Soc., 2005, 127, 14208–14222, DOI:10.1021/ja052201e; (b) M. K. J. ter Wiel, R. A. van Delden, A. Meetsma and B. L. Feringa, Increased Speed of Rotation for the Smallest Light-Driven Molecular Motor, J. Am. Chem. Soc., 2003, 125, 15076–15086, DOI:10.1021/ja036782o; (c) N. Harada, N. Koumura and B. L. Feringa, Chemistry of Unique Chiral Olefins. 3. Synthesis and Absolute Stereochemistry of trans- and cis-1,1′,2,2′,3,3′,4,4′- Octahydro-3,3′-dimethyl-4,4′-biphenanthrylidenes, J. Am. Chem. Soc., 1997, 119, 7256–7264, DOI:10.1021/ja970669e; (d) N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Light-driven monodirectional molecular rotor, Nature, 1999, 401, 152–155, DOI:10.1038/43646.
- (a) M. Christl, S. Groetsch and K. Günther, The Dimerization of Chiral Allenes: Pairs of Enantiomers and Pairs of Homomers Furnish Different Diastereomers, Angew. Chem., Int. Ed., 2000, 39, 3261–3263, DOI:10.1002/1521-3773(20000915)39:18<3261::AID-ANIE3261>3.0.CO;2-T; (b) M. Christl, D. Moigno, E.-M. Peters, K. Peters and H. G. von Schnering, Cycloallenes, 11. 1-Phenyl-1,2-cyclononadiene: Preparation and Dimerisation, Liebigs Ann., 1997, 8, 1791–1796, DOI:10.1002/jlac.199719970825; (c) W. R. Moore, R. D. Bach and T. M. Ozretich, Dimerization of racemic and optically active 1,2-cyclononadiene, J. Am. Chem. Soc., 1969, 91, 5918–5919, DOI:10.1021/ja01049a059.
- (a) Cycloaddition Reactions in Organic Synthesis, ed. S. Kobayashi and K. A. Jørgensen, Wiley, VCH Verlag, 2001, DOI:10.1002/3527600256; (b) K. N. Houk, F. Liu, Z. Yang and J. I. Seeman, Evolution of the Diels-Alder Reaction Mechanism since the 1930s: Woodward, Houk with Woodward, and the Influence of Computational Chemistry on Understanding Cycloadditions, Angew. Chem., Int. Ed., 2021, 60, 12660–12681, DOI:10.1002/anie.202001654.
- P. Caramella, P. Quadrelli and L. Toma, An Unexpected Bispericyclic Transition Structure Leading to 4 + 2 and 2 + 4 Cycloadducts in the Endo Dimerization of Cyclopentadiene, J. Am. Chem. Soc., 2002, 124, 1130–1131, DOI:10.1021/ja016622h , See also references therein.
- Q. Zhou, M. K. Thøgersen, N. M. Rezayee, K. A. Jørgensen and K. N. Houk, Ambimodal Bispericyclic [6 + 4]/[4 + 6] Transition State Competes with Diradical Pathways in the Cycloheptatriene Dimerization: Dynamics and Experimental Characterization of Thermal Dimers, J. Am. Chem. Soc., 2022, 144, 22251–22261, DOI:10.1021/jacs.2c10407.
- (a) E. Breitmaier, H. Kneifel, C. Poszich-Buscher and S. Rittich, The Diels-Alder Dimer of 6-Hydroxy-2,6-dimethyl-cyclohexa-2,4-dienone, an Unusual Metabolite in the Bacterial Degradation of 2,6-Xylenol, Angew. Chem., Int. Ed. Engl., 1991, 30, 202–203, DOI:10.1002/anie.199102021.v; (b) J. Gagnepain, F. Castet and S. Quideau, Total Synthesis of (+)-Aquaticol by Biomimetic Phenol Dearomatization: Double Diastereofacial Differentiation in the Diels-Alder Dimerization of Orthoquinols with a C2-Symmetric Transition State, Angew. Chem., Int. Ed., 2007, 46, 1533–1535, DOI:10.1002/anie.200604610; (c) S. Dong, J. Zhu and J. A. Porco, Enantioselective Synthesis of Bicyclo[2.2.2]octenones Using a Copper-Mediated Oxidative Dearomatization/[4 + 2] Dimerization Cascade, J. Am. Chem. Soc., 2008, 130, 2738–2739, DOI:10.1021/ja711018z; (d) P. A. Peixoto, M. El Assal, I. Chataigner, F. Castet, A. Cornu, R. Coffinier, C. Bosset, D. Deffieux, L. Pouysegu and S. Quideau, Bispericyclic Diels-Alder Dimerization of ortho-Quinols in Natural Product (Bio)Synthesis: Bioinspired Chemical 6-Step Synthesis of (+)-Maytenone, Angew. Chem., Int. Ed., 2021, 60, 14967–14974, DOI:10.1002/anie.202103410; (e) J. Lee, B. Kang, D. Kim and S. Chang, Alcohol-Incorporating Diels-Alder Dimerization of In Situ Formed ortho-Quinamine via Co-Nitrenoid Insertion, Org. Lett., 2022, 24, 5845–5850, DOI:10.1021/acs.orglett.2c02392.
- J. C. Avise and N. Trudy, Hermaphroditism: A Primer on the Biology, Ecology, and Evolution of Dual Sexuality, Columbia University Press, 2011, DOI:10.7312/columbia/9780231153867.001.0001.
- D. Boger and S. Weinreb, Hetero Diels-Alder Methodology, in Organic Synthesis, Academic Press, 1987. 0-12-110860-0 Search PubMed.
- J. Vicario, D. Aparicio and F. Palacios, A diastereoselective aza-Diels-Alder reaction of N-aryl-1-azadienes derived from α-amino acids with enamines, Tetrahedron Lett., 2011, 52, 4109–4111, DOI:10.1016/j.tetlet.2011.05.119.
- F. Palacios, J. Vicario and D. Aparicio, An Efficient Synthesis of Achiral and Chiral Cyclic Dehydro-α-Amino Acid Derivatives Through Nucleophilic Addition of Amines to β,γ-Unsaturated α-Keto Esters, Eur. J. Org. Chem., 2006, 2843–2850, DOI:10.1002/ejoc.200600092.
- (a) X. del Corte, A. Maestro, J. Vicario, E. Martínez de Marigorta and F. Palacios, Brönsted-Acid-Catalyzed Asymmetric Three-Component Reaction of Amines, Aldehydes, and Pyruvate Derivatives. Enantioselective Synthesis of Highly Functionalized γ-Lactam Derivatives, Org. Lett., 2018, 20, 317–320, DOI:10.1021/acs.orglett.7b03397; (b) X. del Corte, A. López-Francés, A. Maestro, E. Martínez de Marigorta, F. Palacios and J. Vicario, Brönsted Acid Catalyzed Multicomponent Synthesis of Phosphorus and Fluorine-Derived γ-Lactam Derivatives, J. Org. Chem., 2020, 85, 14369–14383, DOI:10.1021/acs.joc.0c00280.
- (a) A. López-Francés, X. del Corte, Z. Serna-Burgos, E. Martínez de Marigorta, F. Palacios and J. Vicario, Exploring the Synthetic Potential of γ-Lactam Derivatives Obtained from a Multicomponent Reaction-Applications as Antiproliferative Agents, Molecules, 2022, 27, 3624–3649, DOI:10.3390/molecules27113624; (b) X. del Corte, A. López-Francés, E. Martínez de Marigorta, F. Palacios and J. Vicario, Stereo- and Regioselective [3 + 3] Annulation Reaction Catalyzed by Ytterbium: Synthesis of Bicyclic 1,4-Dihydropyridines, Adv. Synth. Catal., 2021, 363, 4761–4769, DOI:10.1002/adsc.202100785.
- F. Palacios, J. Vicario and D. Aparicio, Aza-Diels-Alder reaction of α,β-unsaturated sulfinylimines derived from α-amino acids with enolethers and enamines, Tetrahedron Lett., 2007, 48, 6747–6750, DOI:10.1016/j.tetlet.2007.07.101.
- (a) T. A. Hamlin, P. Vermeeren, C. Fonseca Guerra and F. M. Bickelhaupt, Energy decomposition analysis in the context of quantitative molecular orbital theory, in Complementary Bonding Analysis, ed. S. Grabowsky, De Gruyter, Berlin, 2021, pp. 199–212. DOI:10.1515/9783110660074-008; (b) F. M. Bickelhaupt and E. J. Baerends, Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry, in Reviews in computational chemistry, ed. K. B. Lipkowitz and D. B. Boyd, Wiley, Hoboken, 2000, pp. 1–86. DOI:10.1002/9780470125922.ch1.
- S. E. Wheeler and K. N. Houk, Substituent Effects in the Benzene Dimer are Due to Direct Interactions of the Substituents with the Unsubstituted Benzene, J. Am. Chem. Soc., 2008, 130, 10854–10855, DOI:10.1021/ja802849j.
- H. Eyring and M. Polanyi, On Simple Gas Reactions, J. Phys. Chem. B, 1931, 12, 279–311, DOI:10.1524/zpch.2013.9023.
- (a) P. Vermeeren, T. A. Hamlin and F. M. Bickelhaupt, Chemical reactivity from an activation strain perspective, Chem. Commun., 2021, 57, 5880–5896, 10.1039/D1CC02042K; (b) F. M. Bickelhaupt and K. N. Houk, Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model, Angew. Chem., Int. Ed., 2017, 56, 10070–10086, DOI:10.1002/anie.201701486; (c) D. H. Ess and K. N. Houk, Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity, J. Am. Chem. Soc., 2007, 129, 10646–10647, DOI:10.1021/ja0734086; (d) F. M. Bickelhaupt, Understanding reactivity with Kohn-Sham molecular orbital theory: E2-SN2 mechanistic spectrum and other concepts, J. Comput. Chem., 1999, 20, 114–128, DOI:10.1002/(SICI)1096-987X(19990115)20:1<114::AID-JCC12>3.0.CO;2-L.
- (a) N. Moshnenko, A. Kazantsev, E. Chupakhin, O. Bakulina and D. Dar'in, Synthetic Routes to Approved Drugs Containing a Spirocycle, Molecules, 2023, 28, 4209–4256, DOI:10.3390/molecules28104209; (b) K. Hiesinger, D. Dar'in, E. Proschak and M. Krasavin, Spirocyclic Scaffolds in Medicinal Chemistry, J. Med. Chem., 2021, 64, 150–183, DOI:10.1021/acs.jmedchem.0c01473.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, complete characterisation and copies of 1H, 13C, 19F NMR and 2D NMR spectra for new compounds 1, 2, 3a and 5, crystallographic data for 2a, 5a and 5b, Cartesian coordinates, ASM and EDA energies, total energies, thermal corrections and number of imaginary frequencies of all the stationary points discussed in the article (pdf). CCDC 2125934, 2257284 and 2257535. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01562a |
| This journal is © the Partner Organisations 2023 |