
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel ABA block copolymers: preparation, temperature sensitivity, and drug release†
Jie Dou,
Shupei Yu,
Ojasvita Reddy and
Yuanwei Zhang *
*
Department of Chemistry and Environmental Science, New Jersey Institute of Technology, University Heights, Newark 07102, NJ, USA. E-mail: yuanwei.zhang@njit.edu
First published on 20th December 2022
Abstract
A new PEGylated macroiniferter was prepared based on the polycondensation reaction of polyethylene oxide (PEO), methylene diphenyl diisocyanate (MDI), and 1,1,2,2-tetraphenyl-1,2-ethanediol (TPED). The macroiniferter consists of PEO end groups and readily reacts with acrylamides (such as N-isopropylacrylamide, NIPAM) and forms ABA block copolymers (PEO-PNIPAM-PEO). This approach of making amphiphilic ABA block copolymers is robust, versatile, and useful, particularly for the development of polymers for biomedical applications. The resulting amphiphilic PEO-PNIPAM-PEO block copolymers are also temperature sensitive, and their phase transition temperatures are close to human body temperature and therefore they have been applied as drug carriers for cancer treatment. Two PEO-PNIPAM-PEO polymers with different molecular weights were prepared and selected to make temperature-sensitive micelles. As a result of the biocompatibility of these micelles, cell viability tests proved that these micelles have low toxicity toward cancer cells. The resultant polymer micelles were then used as drug carriers to deliver the hydrophobic anticancer drug doxorubicin (DOX), and the results showed that they exhibit significantly higher cumulative drug release efficiency at higher temperatures. Moreover, after loading DOX into the micelles, cellular uptake experiments showed easy uptake and cell viability tests showed that DOX-loaded micelles possess a better therapeutic effect than free DOX at the same dose.
1 Introduction
Synthetic block copolymers have a wide range of applications in everyday life, such as adhesives, fibers, and plastics.1,2 They have also received growing attention in the field of biomedical applications in recent years.3–5 Different advanced synthetic polymer chemistry techniques (mainly controlled/living radical polymerization), including atom transfer radical polymerization (ATRP), reversible addition fragmentation chain transfer polymerization (RAFT), nitroxide-mediated polymerization (NMP), etc., have evolved greatly in the last few decades. However, some challenges remain which limit their further application, especially in the biological field. For example, transition metal catalysts, such as Cu(I), Ru(II), Fe(II), etc., are required in ATRP reactions,6 most RAFT agents are unstable and require multiple synthetic steps to obtain,7 and NMP reactions always exhibit slow polymerization kinetics.8As a metal-free polymerization initiator, Otsu first proposed the iniferter concept in 1982 and established a new model for controlled/living radical polymerization.9 Iniferters are a type of organic compound that acts not only as an initiator but also as a chain transfer agent and terminator simultaneously during the radical polymerization.9
Iniferters can be divided into different categories: thermoiniferters and photoiniferters (which either thermally or photochemically dissociate into radicals), A–B type iniferters (asymmetric structures, which dissociate into radicals with different reactivities), C–C type iniferters (symmetric structures, which dissociate into two identical radicals), monomer or macromonomer iniferters, etc.
The mechanism of iniferter polymerization can be briefly explained as follows: the iniferter undergoes initiation, propagation and primary radical termination and transfer. Bimolecular termination and other chain transfer reactions are negligible in this polymerization, since chain transfer to the iniferter itself and/or primary radical termination take place.9,10 When an iniferter is used, polymerization involves direct insertion of monomers into the iniferter bonds, resulting in two iniferter fragments at the two chain ends. Additionally, various vinyl monomers, ranging from acrylates and styrene to vinyl pyrrolidone, could be polymerized11–19 under rather mild experimental conditions in the absence of metal catalysts and without multiple synthetic steps, making this method more feasible and eco-friendly,20 compared to ATRP20 and RAFT polymerizations.21 While the molecular weight and polydispersity of the obtained polymers are not easily controlled compared to ATRP, RAFT, and TEMPO polymerization systems, the process is more easily adapted to practical applications.22
1,2-Disubstituted tetraphenylethane is a type of thermal iniferter, where X can be different functional groups (for example OH,9 CN,23 C2H5,24 etc.22), resulting in the formation of polymers, as shown in Fig. S1.†
1,1,2,2-Tetraphenyl-1,2-ethanediol (TPED) (where X = OH) is a well studied 1,2-disubstituted tetraphenylethane based iniferter for controlled/living radical polymerization.20 When the OH groups are functionalized, for example, by reacting with isocyanate12,25 or α-bromoisobutyryl bromide, it can function as an iniferter for controlled/living radical polymerization,26 as reported in the literature.
By taking advantage of the hydroxyl groups, TPED can react with a variety of compounds, which broadens its potential in synthetic chemistry. Therefore, it has been selected as the focus of this research.
Since biomedical application was the ultimate goal of this research, polyethylene glycol (PEG) or polyethylene oxide (PEO) was selected in this research and introduced in the following context. PEG or PEO is a type of hydrophilic polymer and widely used as a biocompatible segment for drug delivery,27,28 diagnostics,29 tissue engineering applications, etc.30 It has been reported in the literature that PEO facilitates increased biocompatibility, prevents drug opsonization (also called “stealth properties”),31–33 and further increases the circulation time of therapeutic agents as a drug carrier. This type of polymer could function as a nanosized drug carrier, and it exhibits an enhanced permeability and retention (EPR) effect, resulting in improved effectiveness of nanosized drugs targeting tumor tissues, which has been proven not only in animal models but also in human cancer patients compared to free drugs.34–36 Moreover, these nanosized structures could also protect the drug from degradation in the physiological environment compared to free drugs.37
A variety of polymer based nanosized drug carriers have been developed in the past few years, including different types of stimuli-responsive polymers. Stimuli-responsive polymers refer to macromolecules that are sensitive to certain environmental triggers,38,39 such as temperature,40–43 pH,44–47 light,48–50 magnetic fields,51,52 enzymes,53–55 etc. They have been widely investigated and utilized in drug delivery applications in recent years. PNIPAM based thermo-responsive polymers56–61 are one of the most extensively studied structures for cancer chemotherapy, due to the fact that the intratumoral environment possess a higher temperature as compared to normal tissue. A subtle temperature change within cancer cells provides an ideal opportunity for thermoresponsive drug delivery.62
Here, we reported the design and synthesis of a functional macroiniferter (polymer based iniferter) to perform iniferter polymerization. In this work, 1,1,2,2-tetraphenyl-1,2-ethanediol (TPED) was used to prepare a polyurethane based macroiniferter, where PEO was incorporated into the macroiniferter by reacting with isocyanate to improve biocompatibility. N-Isopropylacrylamide (NIPAM), a vinyl monomer, was then inserted into the C–C iniferter bond of the macroiniferter by iniferter polymerization. Poly(N-isopropylacrylamide) (PNIPAM) chains are the hydrophobic middle blocks, and polyethylene oxide (PEO) is the hydrophilic block at the two chain ends. The polymerization kinetics and monomer conversion were characterized by time-dependent 1H NMR, and two PEO-PNIPAM-PEO polymers were selected for further application (see Fig. 1).
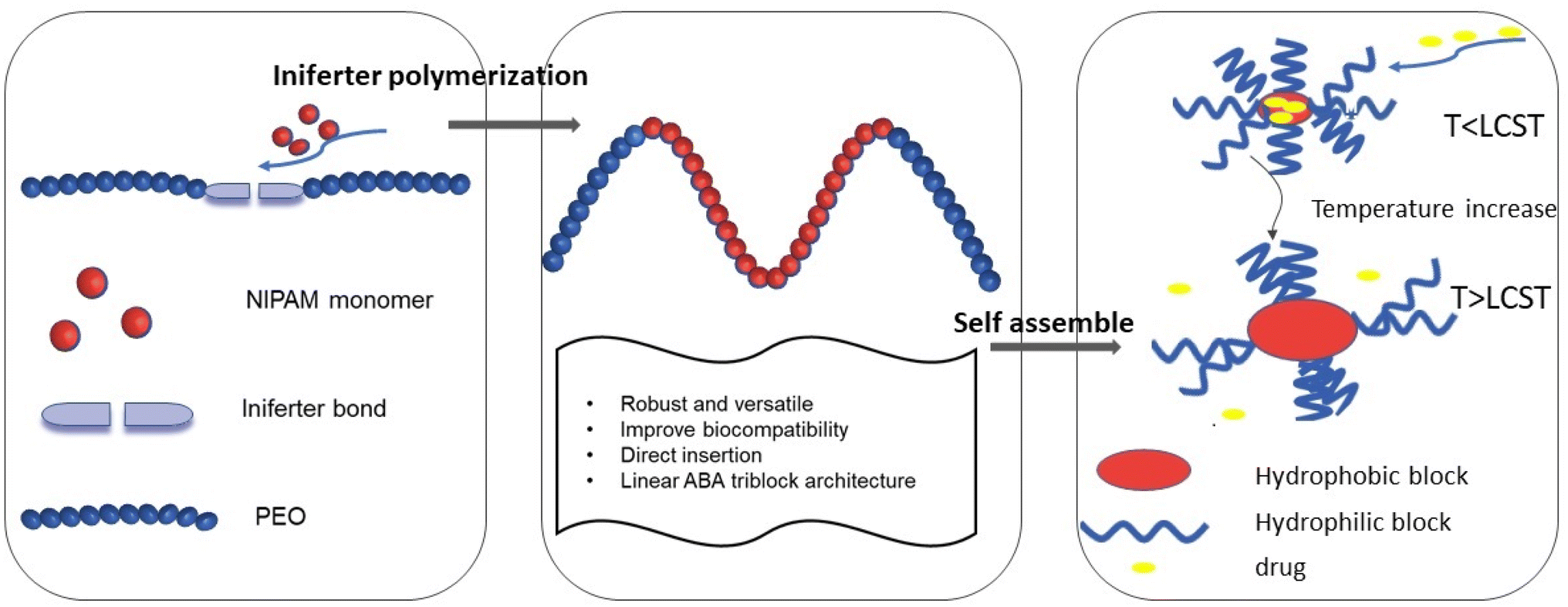 | ||
| Fig. 1 Illustration of preparing PEO-PNIPAM-PEO block copolymers and drug delivery application. | ||
Due to the amphiphilic nature of PEO-PNIPAM-PEO, the resultant block copolymers can self-assemble into micelles,33,63 where PNIPAM is the inner core, and PEO is the outer shell. In addition, the PNIPAM block is known to exhibit thermosensitivity and is well studied for drug release triggered by temperature.64–67 With these features, the hydrophobic anticancer drug doxorubicin (DOX) was loaded into the micelle core by hydrophobic interaction. The properties of PEO-PNIPAM-PEO micelles, including critical micelle concentration (CMC), size, and lower critical solution temperature (LCST), were characterized. The drug encapsulation efficiency (DEE) and drug loading content (DLC) were calculated, and the drug release profile was investigated under conditions below and above LCST, separately. The efficacy of this thermoresponsive micelle in DOX delivery was investigated in human cervical cancer HeLa cells, where both cell viability and cell imaging studies were conducted and showed a better therapeutic effect compared to free DOX.
2 Experimental section
2.1 Materials and reagents
Methylene diphenyl diisocyanate (MDI, 98%) was purchased from Acros Organics. Polyethylene oxide 2000 (PEO2000) was purchased from TCI and its number average molecular weight was confirmed by 1H NMR prior to reaction. Anhydrous tetrahydrofuran (THF, 99.8%, stabilized with 0.025% BHT) and anhydrous dimethyl sulfoxide (DMSO, 99.8%) were purchased from Alfa Aesar. 1,4-Diazabicyclo[2.2.2]octane (DABCO, >99%) was purchased from Sigma Aldrich. 1,1,2,2-Tetraphenyl-1,2-ethanediol (TPED, 98%) was purchased from Alfa Aesar. N-Isopropylacrylamide (NIPAM >98%, stabilized with MEHQ) monomer was purchased from TCI and it was recrystallized with hexane to remove the inhibitor prior to use. Doxorubicin hydrochloride (99%) was purchased from Achemblock. All reagents were used as received without further purification, unless otherwise specified.2.2 Synthesis
FTIR: N–H stretching (3300 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (1728 cm−1) and N–H bending (1529 cm−1) are the characteristic peaks of urethane. C–H stretching (2881 cm−1) and NCO (2270 cm−1) are the peaks of PEO repeating units and MDI, respectively. Macroiniferter: 1H NMR (500 MHz, DMSO) δ 7.47–7.22 (m, 18H), 7.21–6.92 (t, J = 7.73 Hz, 18H), 3.59–3.38 (s, 406H), 3.25–3.21 (s, 6H).
O stretching (1728 cm−1) and N–H bending (1529 cm−1) are the characteristic peaks of urethane. C–H stretching (2881 cm−1) and NCO (2270 cm−1) are the peaks of PEO repeating units and MDI, respectively. Macroiniferter: 1H NMR (500 MHz, DMSO) δ 7.47–7.22 (m, 18H), 7.21–6.92 (t, J = 7.73 Hz, 18H), 3.59–3.38 (s, 406H), 3.25–3.21 (s, 6H).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NIPAM = 1:500) is as follows. The macroiniferter (0.70 g, 1.0 equiv.) and NIPAM (7.40 g, 500.0 equiv.) were dissolved in anhydrous DMSO (w/w = 20%, 22 mL) and added into a three neck round bottom flask, then degassed 30 min before polymerization. Polymerization was carried out at 120 °C under a positive pressure of nitrogen. Aliquots were withdrawn at specific time intervals to monitor the reaction kinetics and completion (detailed discussion in Section 3.1), and the obtained polymers were purified with 15-fold cold diethyl ether three times and vacuum dried overnight before further characterization. Polymer structure, reaction kinetics and molecular weight were characterized by 1H NMR and gel permeation chromatography (GPC). The synthetic routes of the precursor, macroiniferter, and the resultant block copolymers PEO-PNIPAM-PEO are given in Fig. 2.
:NIPAM = 1:500) is as follows. The macroiniferter (0.70 g, 1.0 equiv.) and NIPAM (7.40 g, 500.0 equiv.) were dissolved in anhydrous DMSO (w/w = 20%, 22 mL) and added into a three neck round bottom flask, then degassed 30 min before polymerization. Polymerization was carried out at 120 °C under a positive pressure of nitrogen. Aliquots were withdrawn at specific time intervals to monitor the reaction kinetics and completion (detailed discussion in Section 3.1), and the obtained polymers were purified with 15-fold cold diethyl ether three times and vacuum dried overnight before further characterization. Polymer structure, reaction kinetics and molecular weight were characterized by 1H NMR and gel permeation chromatography (GPC). The synthetic routes of the precursor, macroiniferter, and the resultant block copolymers PEO-PNIPAM-PEO are given in Fig. 2.
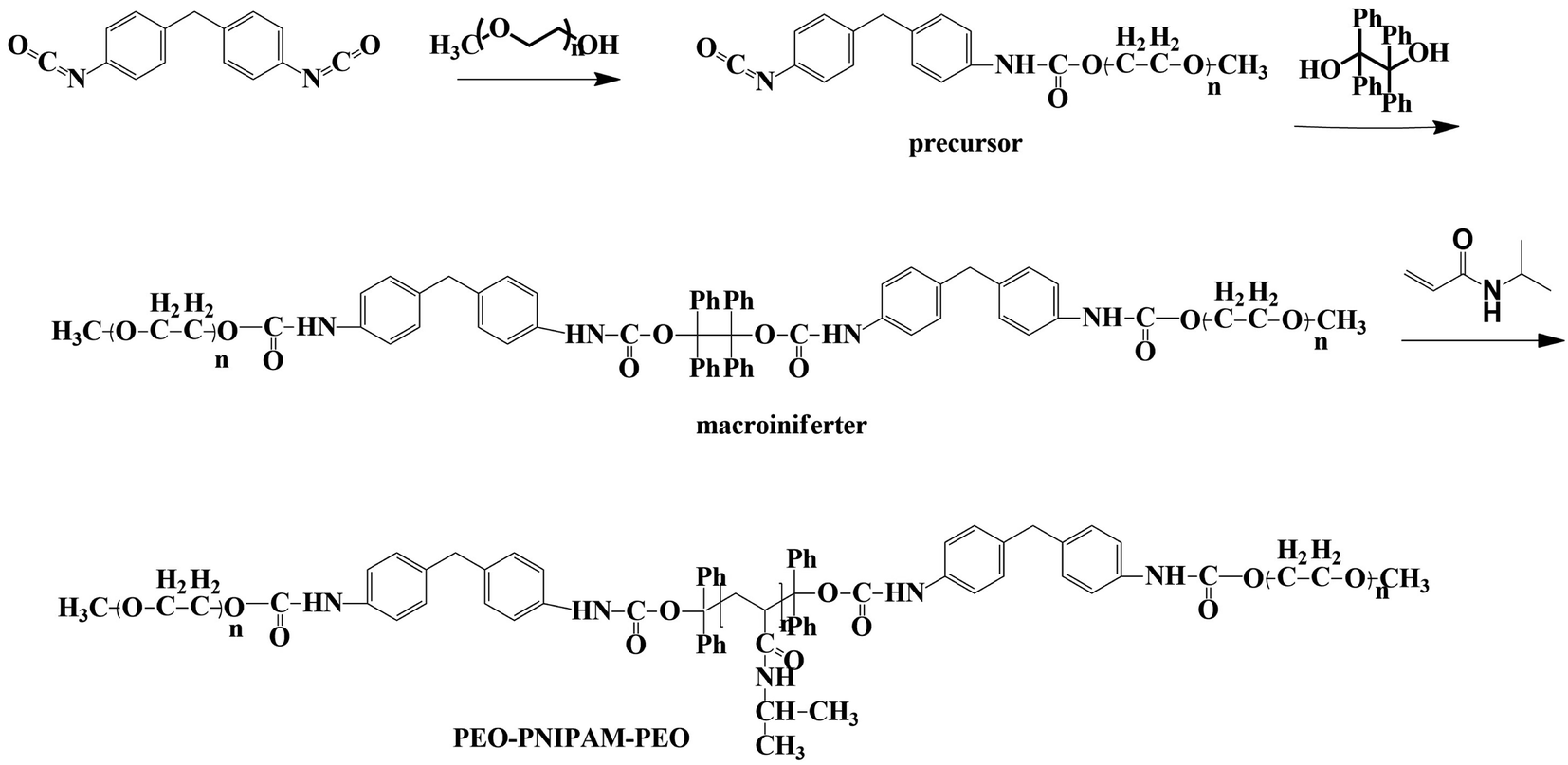 | ||
| Fig. 2 The chemical structures and synthetic routes of the precursor, macroiniferter, and final block copolymer PEO-PNIPAM-PEO. | ||
2.3 Polymer characterization
1H NMR measurements were carried out on a Bruker AVANCE spectrometer (500 MHz) in DMSO-d6. Infrared spectra were recorded using a Shimadzu Fourier Transform Infrared IRPrestige-21. Gel permeation chromatography (GPC) was performed on a Waters Empower system equipped with an RI detector, with a set of PLgel 5 μm guard, PLgel 5 μm MIXED-C and PLgel 10 μm MIXED-B columns. Measurements were carried out at 50 °C with DMF containing 1% tetrabutylammonium bromide (TBAB) as eluent, with a flow rate of 1.0 mL min−1. Polymethyl methacrylate (PMMA) was the standard for calibration, in the molecular weight range from 2000 to 340000 Da. P1 (Mn, GPC = 56500 g mol−1) and P2 (Mn, GPC = 91000 g mol−1) denote the two final PEO-PNIPAM-PEO polymers used for further analysis, as shown in Table 1.
| Entry | Feed ratio of macroiniferter:NIPAM |
Mn, GPC (g mol−1) | Mw, GPC (g mol−1) | PDI |
|---|---|---|---|---|
| P1 | 1:500 |
56500 |
76275 |
1.35 |
| P2 | 1:3000 |
91000 |
123760 |
1.36 |
2.4 Micelle preparation and characterization
2.4.2.1 Critical micelle concentration (CMC) measurement. The CMC is the minimum concentration for micelle formation and is also an important parameter representing micelle stability: below the CMC, the amphiphile is a unimer; above the CMC, the amphiphile will self-assemble into a micelle. Micelle formation is a thermodynamic process, where minimizing the interfacial free energy within the polymer–water system is the main driving force for micelle formation.69 The CMC was determined using Nile red as a fluorescent probe; Nile red is a hydrophobic structure that is not soluble in water, making it nonfluorescent in the aqueous phase; however, when Nile red is encapsulated in a hydrophobic core, its fluorescence intensity increases dramatically.70 The experimental procedure was carried out according to the literature with minor modifications.71,72 Micelle concentrations of MP1 and MP2 of 0.1 μg mL−1 to 1.0 mg mL−1 were prepared. 10 μL of Nile red solution (1.25 μM in acetone) was added into different centrifuge tubes and the acetone was evaporated in the dark overnight, then 1 mL of each micelle solution was added into the tubes and sonicated for 15 minutes and further incubated in the dark overnight. Subsequently, the solutions were filtered with 0.45 μm syringe filters, and then 100 μL of each solution of dye loaded micelles were added to a black 96-well plate. Fluorescence measurements were taken on a Tecan Infinite M200 Pro microplate reader. The excitation wavelength was 535 nm and the emission wavelength started at 585 nm. The maximum fluorescence intensity of the dye-loaded micelles of both MP1 and MP2 was recorded for further analysis.
2.4.2.2 Lower critical transition temperature (LCST) measurement. The LCST of different micelles was measured at different temperatures (from 25 °C to 45 °C) with a microplate reader (SpectraMax M2 Microplate reader, Molecular Devices). The LCST was defined as the temperature at which the transmittance decreased to 50% of its original value.73
2.4.2.3 Micelle size measurement. Dynamic light scattering (DLS) was used to measure the hydrodynamic diameter of the micelles (instrument: Malvern Zetasizer Nano-ZS equipped with a 4 mW 632.8 nm laser at a backscattering angle of 173°). Both MP1 and MP2 were prepared at a concentration of 1.0 mg mL−1, and measured in triplicate at three different temperatures: below LCST (25 °C), near LCST (36 °C), and above LCST (42 °C). The Z average sizes (also known as “cumulants mean”) were recorded.
2.5 Preparation and characterization of DOX-loaded polymeric micelles
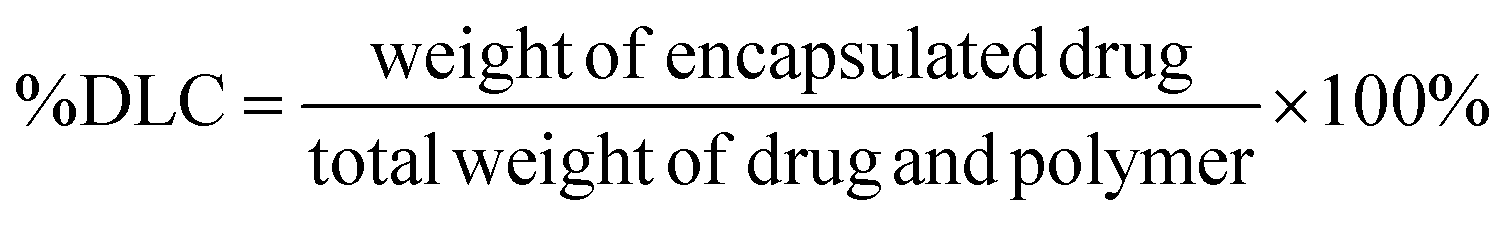 | (1) |
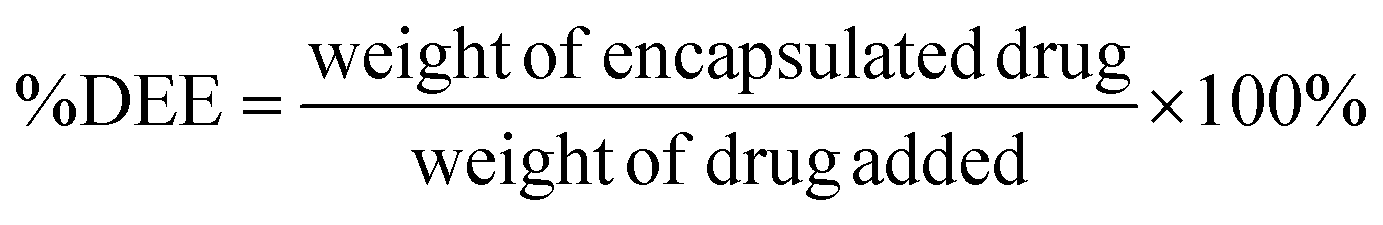 | (2) |
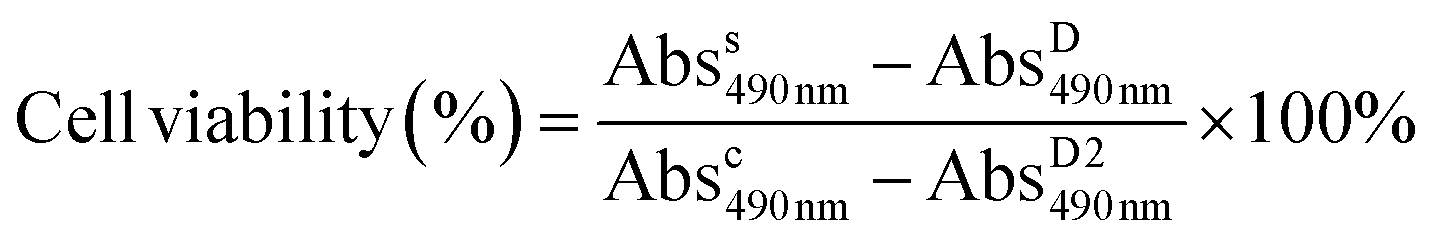 | (3) |
3 Results and discussion
3.1 Synthesis and characterization of polymers
The macroiniferter was synthesized by two-step polycondensation reactions: PEO first reacted with MDI to form an isocyanate-terminated precursor, and then the precursor further reacted with TPED to form the macroiniferter. The overall yield of the macroiniferter was 76%. The isocyanate-terminated precursor was confirmed by dibutylamine titration, where the NCO content decreased to 50%. Also, in the FTIR spectra (Fig. 3a), N–H stretching (3300 cm−1), CO stretching (1728 cm−1) and N–H bending (1529 cm−1) were the characteristic peaks of urethane, indicating that the reaction was conducted successfully. Furthermore, the C–H stretching (2881 cm−1) and NCO (2270 cm−1) are the peaks of the PEO repeating units and MDI, respectively. The presence of all of these peaks in the FTIR spectrum confirmed the precursor structure. The macroiniferter, the product of step 2, was characterized by FTIR and 1H NMR. In Fig. 3a, the isocyanate group (2270 cm−1) completely disappeared, indicating the completion of the reaction. The NMR spectrum of the macroiniferter is shown in Fig. 3b, where the characteristic peaks were found between 7.47 and 6.92 ppm for the aromatic rings and 3.59 and 3.21 ppm for the PEO end groups.
 | ||
| Fig. 3 The characterization of the precursor, macroiniferter and macroiniferter controlled/living radical polymerization. (a) The IR spectra of the precursor and macroiniferter, (b) the NMR spectrum of the macroiniferter, (c) time-dependent 1H NMR of the reaction mixture with a macroiniferter:NIPAM feed ratio of 1:500, (d) time-dependent 1H NMR of the reaction mixture with a macroiniferter:NIPAM feed ratio of 1:3000. | ||
To investigate the kinetics of iniferter polymerization, macroiniferter:NIPAM feed ratios equal to 1:500 and 1:3000 were chosen for polymerization and the processes were monitored by 1H NMR. The stacked reaction time-dependent 1H NMR spectra for 1:500 and 1:3000 (macroiniferter:NIPAM) feed ratios are shown in Fig. 3c and d.
By integrating the PEO repeating unit peaks (3.51 ppm) from the macroiniferter and vinyl proton peaks (around 5.40 ppm and 6.10 ppm) from the NIPAM monomer, the monomer conversion percentage was calculated, as shown in Fig. S7 and S8.†
4, 8, 24, 32, and 48 h were chosen as the specific time intervals for these two reactions. It is clearly seen that in both reactions the peak intensity of the vinyl proton peaks (around 5.40 ppm and 6.10 ppm) decreases with increasing time; however, the peaks at 1.04 ppm and 4.17 ppm change from sharp and well defined to a broad shape, which is a characteristic feature of polymer structures. More than 80% and 60% monomer conversion yields were achieved in 32 h and 48 h for 1:500 and 1:3000, respectively. Upon prolonging the reaction time, more NIPAM monomer was consumed, as shown in Fig. S7 and S8.†
Two final polymers (PEO-PNIPAM-PEO) with different molecular weights, denoted as P1 and P2, were characterized with NMR (including both 1H NMR and heteronuclear multiple quantum coherence (HMQC)) and GPC, as shown in Fig. S2–S6.† As presented in Table 1, the polydispersity indices (PDI) of P1 (Mn, GPC = 56500 g mol−1) and P2 (Mn, GPC = 91000 g mol−1) were 1.35 and 1.36, indicating that they were narrowly distributed polymers. The elution curve is shown in Fig. S4:† the elution time of P2 is shorter than that of P1, since the larger molecular weight polymer elutes first.79
3.2 Characterization of P1 and P2 micelles
The CMC is an important parameter that represents the stability of the micelles. Generally, the fundamental strategy to improve micelle stability is to improve intramicellar interaction, further reflected by a lower CMC.82–84
CMC is calculated using the Sigmoid–Boltzmann equation, as reported in the literature,85–88 where eqn (4) is the Boltzmann fitting function, where A1 is the maximum fluorescence intensity value, A2 is the minimum fluorescence intensity value, xθ is the center of the sigmoid curve, and dx represents the abrupt change in the dependent variable. The CMC value is defined as 10xθ.
| y = A2 + (A1 − A2)/(1 + exp((x − xθ)/dx)) | (4) |
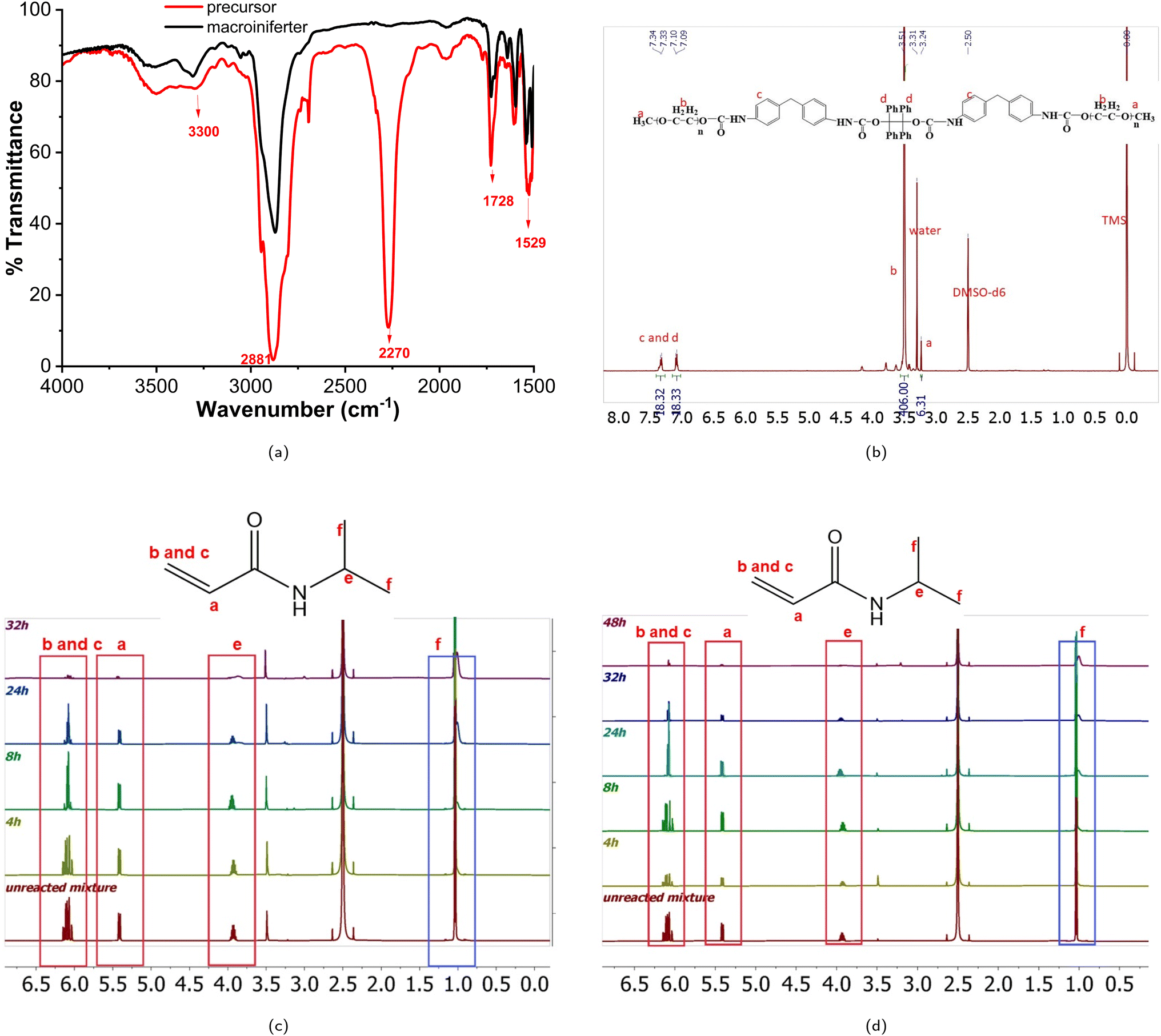
The CMC values of MP1 and MP2 are 0.67 mg mL−1 and 0.21 mg mL−1, respectively. The regression coefficient is 0.97 and 0.99 for MP1 and MP2, respectively. The Boltzmann fittings of MP1 and MP2 are shown in Fig. 4a and b.
 | ||
| Fig. 4 The micelle characterization of MP1 and MP2. (a) Nile red fluorescence intensity versus the logarithm of the MP1 concentration. (b) Nile red fluorescence intensity versus the logarithm of the MP2 concentration. (c) The LCST transition of MP1. (d) The LCST transition of MP2. (e) The diameter of MP1 at a concentration of 1.0 mg mL−1. (f) The diameter of MP2 at a concentration of 1.0 mg mL−1. | ||
MP2 has a lower CMC, owing to its higher molecular weight, i.e., longer hydrophobic blocks, compared to MP1. This indicates a thermodynamically stable self-association process, and this result was consistent with other research.89 Therefore, MP2 is a more stable micelle than MP1 at the same concentration level, so MP2 was selected for further drug and cell experiments.
Previous investigations showed that the LCST can be directly dependent on Mw,98 inversely dependent on Mw,99 and independent of Mw.100,101 In this study, the LCST of PEO-PNIPAM-PEO inversely depends on molecular weight slightly. Fig. 4c and d show the temperature-dependent transmittance of MP1 and MP2, respectively. The LCSTs of MP1 and MP2 were 35.9 °C and 35.6 °C, respectively. This phase transition can be explained as follows: when the temperature is below the LCST, the amide and urethane groups strongly interact with water via hydrogen bonding. Although the polymer–polymer interaction, including both hydrophobic and intermolecular hydrogen bonding interactions, exists, water is such a good solvent, and the polymer–water interaction significantly dominates the polymer–polymer interaction at lower temperature. With increasing temperature, the polymer–water interaction decreases, and the polymer–polymer interaction, including both hydrophobic interaction and inter/intramolecular hydrogen bonding, enhances dramatically, which is caused by the increase in entropy from water, and the phase transition occurs. The synergistic effect of hydrogen bonding and hydrophobic interaction contributes to the phase transition of amphiphilic polymers.102 The minor difference in LCST of MP1 and MP2 indicated that the phase transition upon heating is relatively stable, making it attractive for biomedical applications. Previously, Stöver and coworkers published a thorough study on the phase transition phenomenon of PNIPAM copolymers. PNIPAMs with different end groups ranging from hydrophilic amide to hydrophobic phenylamide, including 2-chloropropionamide (CP), N-isopropyl-2-chloropropionamide (i-PrCP), ethyl 2-chloropropionate (ECP), and N-phenyl-2-chloropropionamide (PhCP), were synthesized by ATRP polymerization. The LCSTs were measured using a UV-vis spectrophotometer equipped with a temperature-controlled and high-sensitivity differential scanning calorimeter (HS-DSC), and the LCST decreased with increasing Mw for all four series of samples, which is consistent with our study here.103
Moreover, similar structures have been studied in the literature: PEG-PNIPAM diblock copolymers are able to self-assemble into micelles (including “spherical”, “crew-cut”,108 “worm-like”,109 “lamellae”, etc.110), gels and vesicles, depending on the concentration, molecular weight, composition of the mixed solvent, salt, etc. PNIPAM-b-PEG-b-PNIPAM triblock copolymers have been found to form micelles and gels. Papagiannopoulos and coworkers presented a detailed morphology transition of PEO-b-PNIPAM copolymer vesicles with small angle neutron scattering, where a three level hierarchical structure below the LCST of PNIPAM was found: (i) individual unperturbed single coils, (ii) aggregates of coils and (iii) large aggregates.111
3.4 Drug encapsulation and efficiency
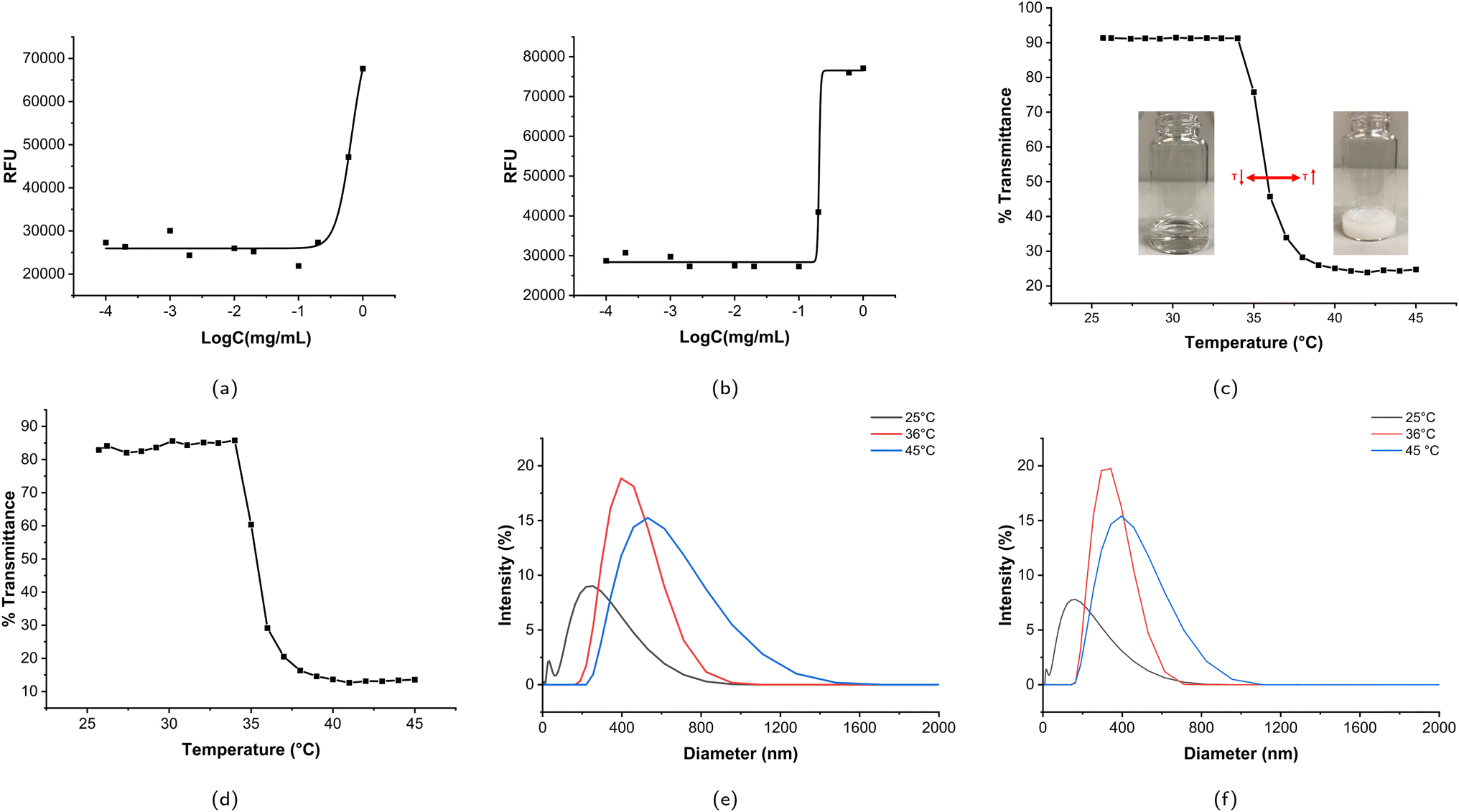
Since MP2 is more stable than MP1 (detailed discussion in Section 3.2.1), MP2 was used for drug and cell experiments, denoted as DOX@MP2. Doxorubicin (DOX) is a hydrophobic model drug for evaluating the drug encapsulation and drug release profile. The calibration curve was measured according to six different concentrations (see Fig. S9†). The drug loading content (DLC) and drug encapsulation efficiency (DEE) of DOX@MP2 are 3.1% and 12.5%, respectively. The relatively low drug encapsulation efficiency may be due to the relatively hydrophilic overall structure. The time-dependent drug release profiles at 25 °C and 38 °C are shown in Fig. 5a. Approximately 90% of encapsulated DOX was released at 38 °C within 80 h, which was significantly higher than at 25 °C. This result indicates that MP2 is temperature sensitive and that significant DOX could be released from the micelles above LCST. There was a small percentage of DOX entrapped within the micelles, due to the hydrophobic interaction between the isopropyl group and DOX. This smart drug delivery phenomenon can be explained by the following reasons: when the temperature is lower than LCST, the hydrophobic interaction between the micelle core and DOX is almost equal to the intermolecular/intramolecular interaction between polymer micelle chains and polymer–water hydrogen bonding; however, when the temperature increases, the polymer–polymer interaction is significantly stronger than the micelle–DOX interaction, so the DOX was released. Therefore, PEO-PNIPAM-PEO micelles are promising carriers for smart drug delivery. Similarly, by taking advantage of the unique thermoresponsive property, the PNIPAM-based hydrogel could be used to deliver the antibacterial peptide G (IIKK) 3I-NH2, as reported in the literature.112 | ||
| Fig. 5 Drug release and cell experiments. (a) Drug release below LCST and above LCST of DOX@MP2. (b) The cell viability of blank MP2 micelles. (c) The cell viability of free DOX and DOX@MP2 micelles. (d) The cellular uptake of free DOX. (e) The cellular uptake of DOX@MP2 micelles. | ||
3.5 Cell experiments
To evaluate the therapeutic efficacy of free DOX and DOX@MP2, HeLa cells were incubated with different concentrations of DOX, ranging from 0 to 40 μg mL−1. The MTS assay was used for quantitative measurement. As illustrated in Fig. 5c, both free DOX and DOX@MP2 presented a dramatic suppression effect on HeLa cells, with an increasing inhibition effect at increased concentration. More than 85% of cancer cells were killed by DOX@MP2 at an equivalent DOX concentration of 40 μg mL−1, and free DOX only suppressed approximately 55% of cells. The significant cytotoxicity difference between free DOX and DOX@MP2 at higher concentrations (from 5 to 40 μg mL−1) can be attributed to higher uptake of DOX@MP2 through endocytosis by HeLa cells, followed by temperature-induced release of DOX within the endosomal compartment.115,116
4 Conclusions
In this study, a novel macroiniferter with PEO groups was developed for the synthesis of ABA block copolymers, particularly for biomedical applications. The final PEO-PNIPAM-PEO triblock copolymer products were transformed into micelles, and their thermoresponsive transition temperatures (LCSTs) were around 36 °C, showing great potential in the delivery of human cancer drugs. The micelles were further used to encapsulate the anticancer drug doxorubicin. Cell experiments showed that polymeric micelles are biocompatible, and DOX-loaded micelles are highly active against HeLa cells, and they are effectively internalized in HeLa cells compared to free DOX at the same concentration. Overall, these results suggest that PEO-PNIPAM-PEO is an ideal drug carrier for anticancer drug delivery in biological applications.Abbreviations
| DOX | Doxorubicin |
| P1 | PEO-PNIPAM-PEO 1 (Mn, GPC = 56500 g mol−1) |
| P2 | PEO-PNIPAM-PEO 2 (Mn, GPC = 91000 g mol−1) |
| MP1 | Polymeric micelle 1 |
| MP2 | Polymeric micelle 2 |
| DOX@MP2 | DOX loaded polymeric micelle 2 |
Author contributions
J. D., conceptualization, investigation and methodology, data curation, writing original draft; S. Y., methodology, writing original draft; O. R., reviewing original draft; Y. Z., conceptualization, supervision, funding acquisition, project administration, and review & editing. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the New Jersey Health Foundation (Grant Number: PC 57-20 and PC 25-22), and the National Institutes of Health (1R21GM140438-01). O. R. would like to thank the NSF BioSMART REU program. The authors also thank Dr Xiaoyang Xu at NJIT for the assistance with micelle characterization.References
- Y. Zhang, X. Yu and Z. Cheng, Polymers, 2022, 14, 1208 CrossRef PubMed.
- H. Namazi, BioImpacts, 2017, 7, 73 CrossRef CAS PubMed.
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343–1355 CrossRef CAS PubMed.
- T. P. Lodge, Macromol. Chem. Phys., 2003, 204, 265–273 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- X.-P. Chen and K.-Y. Qiu, Macromolecules, 1999, 32, 8711–8715 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402 CrossRef CAS.
- R. B. Grubbs, Polym. Rev., 2011, 51, 104–137 CrossRef CAS.
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 CrossRef CAS.
- T. Otsu, M. Yoshida and T. Tazaki, Makromol. Chem., Rapid Commun., 1982, 3, 133–140 CrossRef CAS.
- Y. Zhang, L. Wang, Z. Zhang, Y. Zhang and X. Tuo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2161–2170 CrossRef CAS.
- G. Zhou, C. Ma and G. Zhang, Polym. Chem., 2011, 2, 1409–1414 RSC.
- G. Jiang, X. Tuo, D. Wang and Q. Li, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3248–3256 CrossRef CAS.
- A. Patel and K. Mequanint, J. Bioact. Compat. Polym., 2011, 26, 114–129 CrossRef CAS.
- H. Verma and T. Kannan, Polym. J., 2008, 40, 867–874 CrossRef CAS.
- K. Mequanint, A. Patel and D. Bezuidenhout, Biomacromolecules, 2006, 7, 883–891 CrossRef CAS.
- A. Patel and K. Mequanint, Macromol. Biosci., 2007, 7, 727–737 CrossRef CAS.
- S. Baek, B. Kim and B. Kim, Prog. Org. Coat., 2004, 49, 353–357 CrossRef CAS.
- K. Tharanikkarasu and B. K. Kim, Polym. Bull., 1998, 40, 675–681 CrossRef CAS.
- M. Kumar and T. Kannan, Polym. J., 2010, 42, 916–922 CrossRef CAS.
- H. Zhang, Eur. Polym. J., 2013, 49, 579–600 CrossRef.
- X. Li, J. Lu, W. Yang, Z. Fu and B. Han, Colloid Polym. Sci., 2014, 292, 257–265 CrossRef.
- G. Schulz and G. Wittig, Naturwissenschaften, 1939, 27, 456 CrossRef.
- E. Borsig, M. Lazar and M. Čapla, Macromol. Chem. Phys., 1967, 105, 212–222 CrossRef.
- K. Tharanikkarasu and G. Radhakrishnan, Eur. Polym. J., 1994, 30, 1351–1355 CrossRef.
- M. Kumar and T. Kannan, Polym. J., 2010, 42, 916–922 CrossRef.
- L. Mei, L. Fu, K. Shi, Q. Zhang, Y. Liu, J. Tang, H. Gao, Z. Zhang and Q. He, Int. J. Pharm., 2014, 468, 26–38 CrossRef CAS.
- X. Kong, K. Yu, M. Yu, Y. Feng, J. Wang, M. Li, Z. Chen, M. He, R. Guo and R. Tian, et al., Int. J. Pharm., 2014, 465, 378–387 CrossRef CAS.
- K. S. Kim, W. Park, J. Hu, Y. H. Bae and K. Na, Biomaterials, 2014, 35, 337–343 CrossRef CAS.
- J. Zhu, Biomaterials, 2010, 31, 4639–4656 CrossRef CAS.
- S. M. Tawfik, S. Azizov, M. R. Elmasry, M. Sharipov and Y.-I. Lee, Nanomaterials, 2020, 11, 70 CrossRef.
- A. A. D’souza and R. Shegokar, Expert Opin. Drug Delivery, 2016, 13, 1257–1275 CrossRef PubMed.
- S. Yadav, A. K. Sharma and P. Kumar, Front. Bioeng. Biotechnol., 2020, 8, 127 CrossRef PubMed.
- J. Fang, W. Islam and H. Maeda, Adv. Drug Delivery Rev., 2020, 157, 142–160 CrossRef.
- N. Alasvand, A. M. Urbanska, M. Rahmati, M. Saeidifar, P. S. Gungor-Ozkerim, F. Sefat, J. Rajadas and M. Mozafari, Multifunctional systems for combined delivery, Biosens. Diagn., 2017, 245–259 Search PubMed.
- Y. Zhang, Y. Huang and S. Li, AAPS PharmSciTech, 2014, 15, 862–871 CrossRef PubMed.
- S. S. Das, P. Bharadwaj, M. Bilal, M. Barani, A. Rahdar, P. Taboada, S. Bungau and G. Z. Kyzas, Polymers, 2020, 12, 1397 CrossRef CAS PubMed.
- P. Theato, B. S. Sumerlin, R. K. O’Reilly and T. H. Epps III, Chem. Soc. Rev., 2013, 42, 7055–7056 RSC.
- K. Bauri, M. Nandi and P. De, Polym. Chem., 2018, 9, 1257–1287 RSC.
- S. A. Mohammad, S. Dolui, D. Kumar, S. R. Mane and S. Banerjee, Biomacromolecules, 2021, 22, 3941–3949 CrossRef CAS.
- X. Huang, H. Mutlu, S. Lin and P. Theato, Eur. Polym. J., 2021, 142, 110156 CrossRef CAS.
- M. Li, H. Li, P. De and B. S. Sumerlin, Macromol. Rapid Commun., 2011, 32, 354–359 CrossRef CAS.
- P. De, S. R. Gondi and B. S. Sumerlin, Biomacromolecules, 2008, 9, 1064–1070 CrossRef CAS.
- Z. Miao, T. Kubo, D. Pal, B. S. Sumerlin and A. S. Veige, Macromolecules, 2019, 52, 6260–6265 CrossRef CAS.
- K. Bauri, S. G. Roy, S. Pant and P. De, Langmuir, 2013, 29, 2764–2774 CrossRef CAS.
- S. Pal, S. G. Roy and P. De, Polym. Chem., 2014, 5, 1275–1284 RSC.
- S. G. Roy, K. Bauri, S. Pal, A. Goswami, G. Madras and P. De, Polym. Int., 2013, 62, 463–473 CrossRef CAS.
- F. D. Jochum, L. Zur Borg, P. J. Roth and P. Theato, Macromolecules, 2009, 42, 7854–7862 CrossRef CAS.
- F. P. Nicoletta, D. Cupelli, P. Formoso, G. De Filpo, V. Colella and A. Gugliuzza, Membranes, 2012, 2, 134–197 CrossRef CAS PubMed.
- G. Stoychev, A. Kirillova and L. Ionov, Adv. Opt. Mater., 2019, 7, 1900067 CrossRef.
- G. Filipcsei, I. Csetneki, A. Szilágyi and M. Zrínyi, Oligomers-polymer composites-molecular imprinting, 2007, 137–189 Search PubMed.
- D. Szabo, G. Szeghy and M. Zrinyi, Macromolecules, 1998, 31, 6541–6548 CrossRef CAS.
- J. Hu, G. Zhang and S. Liu, Chem. Soc. Rev., 2012, 41, 5933–5949 RSC.
- P. D. Thornton, R. J. Mart and R. V. Ulijn, Adv. Mater., 2007, 19, 1252–1256 CrossRef.
- C. Wang, Q. Chen, Z. Wang and X. Zhang, Angew. Chem., 2010, 122, 8794–8797 CrossRef.
- U. Haldar, L. Ramakrishnan, K. Sivaprakasam and P. De, Polymer, 2014, 55, 5656–5664 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS.
- D. Roy, W. L. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093–5100 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172–1176 CrossRef CAS.
- K. Bauri, S. G. Roy, S. Arora, R. K. Dey, A. Goswami, G. Madras and P. De, J. Therm. Anal. Calorim., 2013, 111, 753–761 CrossRef CAS.
- A. Alsuraifi, A. Curtis, D. A. Lamprou and C. Hoskins, Pharmaceutics, 2018, 10, 136 CrossRef CAS.
- D. Lombardo, M. A. Kiselev, S. Magazù and P. Calandra, Advances in Condensed Matter Physics, 2015, vol. 2015 Search PubMed.
- Z. Ferjaoui, E. Jamal Al Dine, A. Kulmukhamedova, L. Bezdetnaya, C. Soon Chang, R. Schneider, F. Mutelet, D. Mertz, S. Bégin-Colin and F. Quilès, et al., ACS Appl. Mater. Interfaces, 2019, 11, 30610–30620 CrossRef CAS PubMed.
- Y. Yar, R. Khodadust, Y. Akkoc, M. Utkur, E. U. Saritas, D. Gozuacik and H. Y. Acar, J. Mater. Chem. B, 2018, 6, 289–300 RSC.
- J. Liang, Z. Zhang, H. Zhao, S. Wan, X. Zhai, J. Zhou, R. Liang, Q. Deng, Y. Wu and G. Lin, RSC Adv., 2018, 8, 15621–15631 RSC.
- H. Wei, X. Zhang, C. Cheng, S.-X. Cheng and R.-X. Zhuo, Biomaterials, 2007, 28, 99–107 CrossRef CAS.
- S. S. Patil and R. D. K. Misra, Mater. Technol., 2018, 33, 364–386 CrossRef CAS.
- S. C. Owen, D. P. Chan and M. S. Shoichet, Nano Today, 2012, 7, 53–65 CrossRef.
- M. C. Stuart, J. C. van de Pas and J. B. Engberts, J. Phys. Org. Chem., 2005, 18, 929–934 CrossRef.
- S. M. Sarett, T. A. Werfel, L. Lee, M. A. Jackson, K. V. Kilchrist, D. Brantley-Sieders and C. L. Duvall, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6490–E6497 CrossRef.
- G. Slor, A. R. Olea, S. Pujals, A. Tigrine, V. R. De La Rosa, R. Hoogenboom, L. Albertazzi and R. J. Amir, Biomacromolecules, 2021, 22, 1197–1210 CrossRef.
- J. Zhang, K. Feng, M. Cuddihy, N. A. Kotov and P. X. Ma, Soft Matter, 2010, 6, 610–617 RSC.
- R.-S. Lee, S.-W. Wang, Y.-C. Li and J.-Y. Fang, RSC Adv., 2015, 5, 497–512 RSC.
- G. Kwon, M. Naito, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, J. Controlled Release, 1997, 48, 195–201 CrossRef CAS.
- X. Liu, R. Ma, J. Shen, Y. Xu, Y. An and L. Shi, Biomacromolecules, 2012, 13, 1307–1314 CrossRef CAS PubMed.
- B. Razavi, A. Abdollahi, H. Roghani-Mamaqani and M. Salami-Kalajahi, Polymer, 2020, 187, 122046 CrossRef CAS.
- T. Zhao, X. Liu, S. Singh, X. Liu, Y. Zhang, J. Sawada, M. Komatsu and K. D. Belfield, Bioconjugate Chem., 2019, 30, 2312–2316 CrossRef.
- L. M. Nollet and L. S. De Gelder, Handbook of water analysis, CRC press, 2000 Search PubMed.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3–27 CrossRef.
- L. Yang, X. Wu, F. Liu, Y. Duan and S. Li, Pharm. Res., 2009, 26, 2332–2342 CrossRef.
- Y. Lu, E. Zhang, J. Yang and Z. Cao, Nano Res., 2018, 11, 4985–4998 CrossRef PubMed.
- A. Kapse, N. Anup, V. Patel, G. K. Saraogi, D. K. Mishra and R. K. Tekade, in Drug Delivery Systems, Elsevier, 2020, pp. 235–289 Search PubMed.
- Martin’s Physical Pharmacy and Pharmaceutical Sciences: Physical Chemical and Biopharmaceutical Principles in the Pharmaceutical Sciences, ed. A. N. Martin, P. J. Sinko and Y. Singh, Lippincott Williams & Wilkins, Baltimore, MD, 6th edn, 2011 Search PubMed.
- D. R. Perinelli, M. Cespi, N. Lorusso, G. F. Palmieri, G. Bonacucina and P. Blasi, Langmuir, 2020, 36, 5745–5753 CrossRef.
- L. Stopková, J. Gališinová, Z. Šuchtová, J. Čižmárik and F. Andriamainty, Molecules, 2018, 23, 1064 CrossRef.
- S. Mondal and S. Ghosh, J. Photochem. Photobiol., B, 2012, 115, 9–15 CrossRef PubMed.
- J. Aguiar, P. Carpena, J. Molina-Bolívar and C. Carnero Ruiz, J. Colloid Interface Sci., 2003, 258, 116–122 CrossRef.
- X. Dong, X. Guo, G. Liu, A. Fan, Z. Wang and Y. Zhao, Chem. Commun., 2017, 53, 3822–3825 RSC.
- M. K. Gupta, J. R. Martin, T. A. Werfel, T. Shen, J. M. Page and C. L. Duvall, J. Am. Chem. Soc., 2014, 136, 14896–14902 CrossRef PubMed.
- Z. Zhang, J. Wang, X. Nie, T. Wen, Y. Ji, X. Wu, Y. Zhao and C. Chen, J. Am. Chem. Soc., 2014, 136, 7317–7326 CrossRef.
- X. Xu, J. D. Flores and C. L. McCormick, Macromolecules, 2011, 44, 1327–1334 CrossRef.
- B. Wang, H.-J. Liu, T.-T. Jiang, Q.-H. Li and Y. Chen, Polymer, 2014, 55, 6036–6043 CrossRef.
- D. E. Bergbreiter, B. L. Case, Y.-S. Liu and J. W. Caraway, Macromolecules, 1998, 31, 6053–6062 CrossRef.
- Y. Lu, J. Yuan, F. Polzer, M. Drechsler and J. Preussner, ACS Nano, 2010, 4, 7078–7086 CrossRef.
- P. Muthiah, S. M. Hoppe, T. J. Boyle and W. Sigmund, Macromol. Rapid Commun., 2011, 32, 1716–1721 CrossRef.
- N. Nath and A. Chilkoti, Adv. Mater., 2002, 14, 1243–1247 CrossRef.
- Q. Liu, Z. Yu and P. Ni, Colloid Polym. Sci., 2004, 282, 387–393 CrossRef CAS.
- Z. Tong, F. Zeng, X. Zheng and T. Sato, Macromolecules, 1999, 32, 4488–4490 CrossRef CAS.
- E. I. Tiktopulo, V. N. Uversky, V. B. Lushchik, S. I. Klenin, V. E. Bychkova and O. B. Ptitsyn, Macromolecules, 1995, 28, 7519–7524 CrossRef.
- S. Fujishige, K. Kubota and I. Ando, J. Phys. Chem., 1989, 93, 3311–3313 CrossRef.
- X. Yin and H. D. H. Stöver, Macromolecules, 2002, 35, 10178–10181 CrossRef.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef.
- K. S. Schmitz, Introduction to Dynamic Light Scattering by Macromolecules, Academic Press, Cambridge, MA, USA, 1990, 978-0-32314035-5, https://www.elsevier.com/books/introduction-to-dynamic-light-scattering-by-macromolecules/ schmitz/978-0-12-627260-4 Search PubMed.
- P. Beaumont, A. Courtois, T. Richard, S. Krisa and C. Faure, Pharmaceutics, 2021, 13, 566 CrossRef PubMed.
- Y. Hiruta, Y. Kanda, N. Katsuyama and H. Kanazawa, RSC Adv., 2017, 7, 29540–29549 RSC.
- K. Liu, P. Pan and Y. Bao, RSC Adv., 2015, 5, 94582–94590 RSC.
- L. Zhang and A. Eisenberg, Polym. Adv. Technol., 1998, 9, 677–699 CrossRef CAS.
- P. Bhargava, Y. Tu, J. X. Zheng, H. Xiong, R. P. Quirk and S. Z. D. Cheng, J. Am. Chem. Soc., 2007, 129, 1113–1121 CrossRef CAS PubMed.
- O. V. Borisov and E. B. Zhulina, Macromolecules, 2003, 36, 10029–10036 CrossRef CAS.
- A. Papagiannopoulos, J. Zhao, G. Zhang, S. Pispas and A. Radulescu, Polymer, 2013, 54, 6373–6380 CrossRef CAS.
- M. Cao, Y. Wang, X. Hu, H. Gong, R. Li, H. Cox, J. Zhang, T. A. Waigh, H. Xu and J. R. Lu, Biomacromolecules, 2019, 20, 3601–3610 CrossRef CAS PubMed.
- H. Taymaz-Nikerel, M. E. Karabekmez, S. Eraslan and B. Kırdar, Sci. Rep., 2018, 8, 1–14 CAS.
- H. Lei, X. Wang and C. Wu, J. Mol. Graphics Modell., 2012, 38, 279–289 CrossRef CAS.
- X. Zhang, P. Yang, Y. Dai, P. Ma, X. Li, Z. Cheng, Z. Hou, X. Kang, C. Li and J. Lin, Adv. Funct. Mater., 2013, 23, 4067–4078 CrossRef CAS.
- J. Wang, N. Huang, Q. Peng, X. Cheng and W. Li, Mater. Chem. Phys., 2020, 239, 121994 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05831f |
| This journal is © The Royal Society of Chemistry 2023 |