
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, synthetic approach, in silico molecular docking and antibacterial activity of quinazolin-2,4-dione hybrids bearing bioactive scaffolds†
Aboubakr H. Abdelmonsef *a,
Mohamed Omara,
Huda R. M. Rashdanb,
Mohamed M. Tahaa and
Ahmed M. Abobakra
*a,
Mohamed Omara,
Huda R. M. Rashdanb,
Mohamed M. Tahaa and
Ahmed M. Abobakra
aChemistry Department, Faculty of Science, South Valley University, Qena 83523, Egypt. E-mail: aboubakr.ahmed@sci.svu.edu.eg
bChemistry of Natural and Microbial Products Department, Pharmaceutical and Drug Industries Research Institute, National Research Centre, Dokki, Cairo 12622, Egypt
First published on 21st December 2022
Abstract
Antimicrobial resistance (AMR) is one of ten global public health threats facing humanity. This created the need to identify and develop effective inhibitors as antimicrobial agents. In this respect, quinazolin-2,4-dione hybrids bearing N-heterocyclic cores such as pyrrolidine-2,5-dione, pyrazole and oxadiazole and/or bioactive scaffolds such as hydrazone, amide, sulfonamide, azomethine, and thiourea linkage are described for design, synthesis, antibacterial investigation, and in silico studies. The characterization of the target compounds was accomplished by elemental analysis and various spectroscopic data like FT-IR, 1H-NMR, 13C-NMR and MS. The antibacterial evaluation was achieved for the newly synthesized compounds using two G −ve bacteria (Escherichia coli ATCC 25955 and Pseudomonas aeruginosa ATCC 10145), and two G +ve bacteria (Bacillus subtilis ATCC 6633 and Staphylococcus aureus NRRL B-767). Synthesized compounds exhibited various activities against the tested pathogens, the results revealed that compound 3c exhibited a characteristic antimicrobial efficacy against all the tested pathogenic strains at a concentration lower than the tested standard drug ranging from 2.5 to 10 μg ml−1. Moreover, the molecular docking study against the target S. aureus tyrosyl-tRNA synthetase (PDB ID: 1JIJ) was carried out to investigate the mechanism of action of the prepared compounds, which is in line with an in vitro study. Most new compounds exhibited zero violation of Lipinski's rule (Ro5). These candidate molecules have shown promising antibacterial activity. Among these molecules, compound 3c with di-hydroxyl groups on two phenyl rings at position-4 exhibited a promising potent antibacterial inhibitory effect. Further SAR analysis reveals that a greater number of hydroxyl groups in an organic compound might be crucial for antibacterial efficacy. These findings demonstrate the potential activity of compound 3c as an antibacterial agent.
1. Introduction
Despite the profound success achieved to increase the number of antibiotics used to treat bacterial infections, the bacterial resistance to antibiotics is still an internationally-recognized problem.1 There is an urgent need to identify new bioactive molecules with high efficacy against bacterial diseases.Quinazolines represent an important class of nitrogen-heterocycles that participate in various pharmacological and biological activities, such as anti-cancer,2,3 anti-malarial,4 anti-inflammation,5 anti-microbial,6 anti-cholera7 and anti-covid19 8. In addition, the quinazoline derivatives are known to have drawn significant attention of in light of their biopharmaceutical significance.8 Currently, there are various approved drugs containing the quinazoline skeleton9 such as prazosin HCl, doxazosin, and terazosin HCl, as represented in Fig. 1.
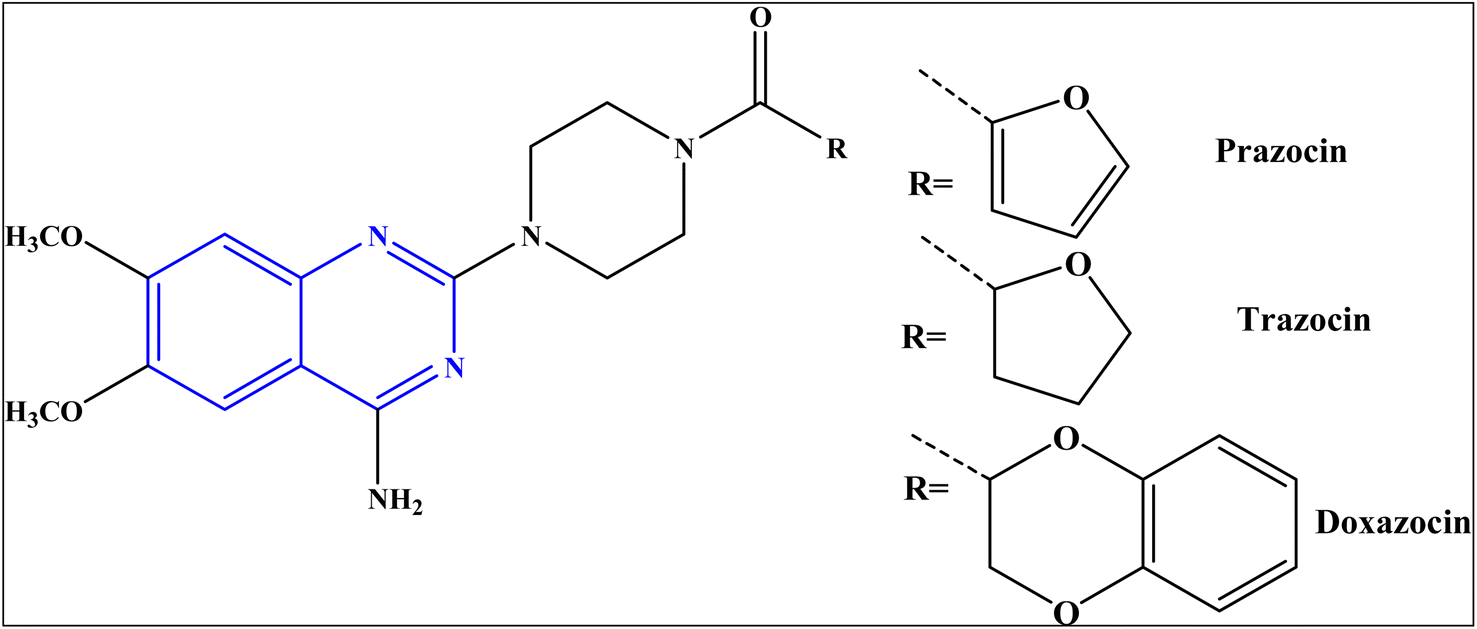 | ||
| Fig. 1 Approved marketed drugs containing quinazoline skeleton. | ||
On the other hand, compounds containing hydrazone, amide, sulfonamide, azomethine, and thiourea motifs are recognized for their high biological activity.10–12
As a result of the aforementioned facts and in continuation to our work on the development of a new class of heterocyclic molecules,2–5,7,13–21 we have strategically synthesized a new series of hybrid quinazolin-2,4-dione analogues having N-heterocyclic cores and/or bioactive scaffolds for the evaluation of antibacterial efficacy by in vitro studies. The newly synthesized products were structurally elucidated by means of elemental and spectroscopic analyses. Additionally, all of them were examined as antibacterial agents against G +ve and G −ve strains. Finally, the molecular docking studies22–24 were accomplished using PyRx software25 to examine the binding affinities of herein reported quinazolin-2,4-dione hybrid molecules toward S. aureus tyrosyl-tRNA synthetase. The pharmacokinetics and toxicity of these compounds were investigated using AdmetSAR, SwissADME, and mol inspiration.
In conclusion, the SAR study exhibited that the quinazoline skeleton was found to be essential for antibacterial activity as shown in Fig. 2. In addition, nitrogen heterocyclic scaffolds such as pyrrolidine-2,5-dione, pyrazole and oxadiazole enhanced the antimicrobial activity. Further, bioactive scaffolds such as hydrazone, amide, sulfonamide, azomethine, and thiourea linkage were found to possess promising antibacterial efficacy. Taken together, these quinazolindione skeletons attached to N-heterocyclic moieties and/or bioactive scaffolds were found to be unique templates for development of antibacterial agents.
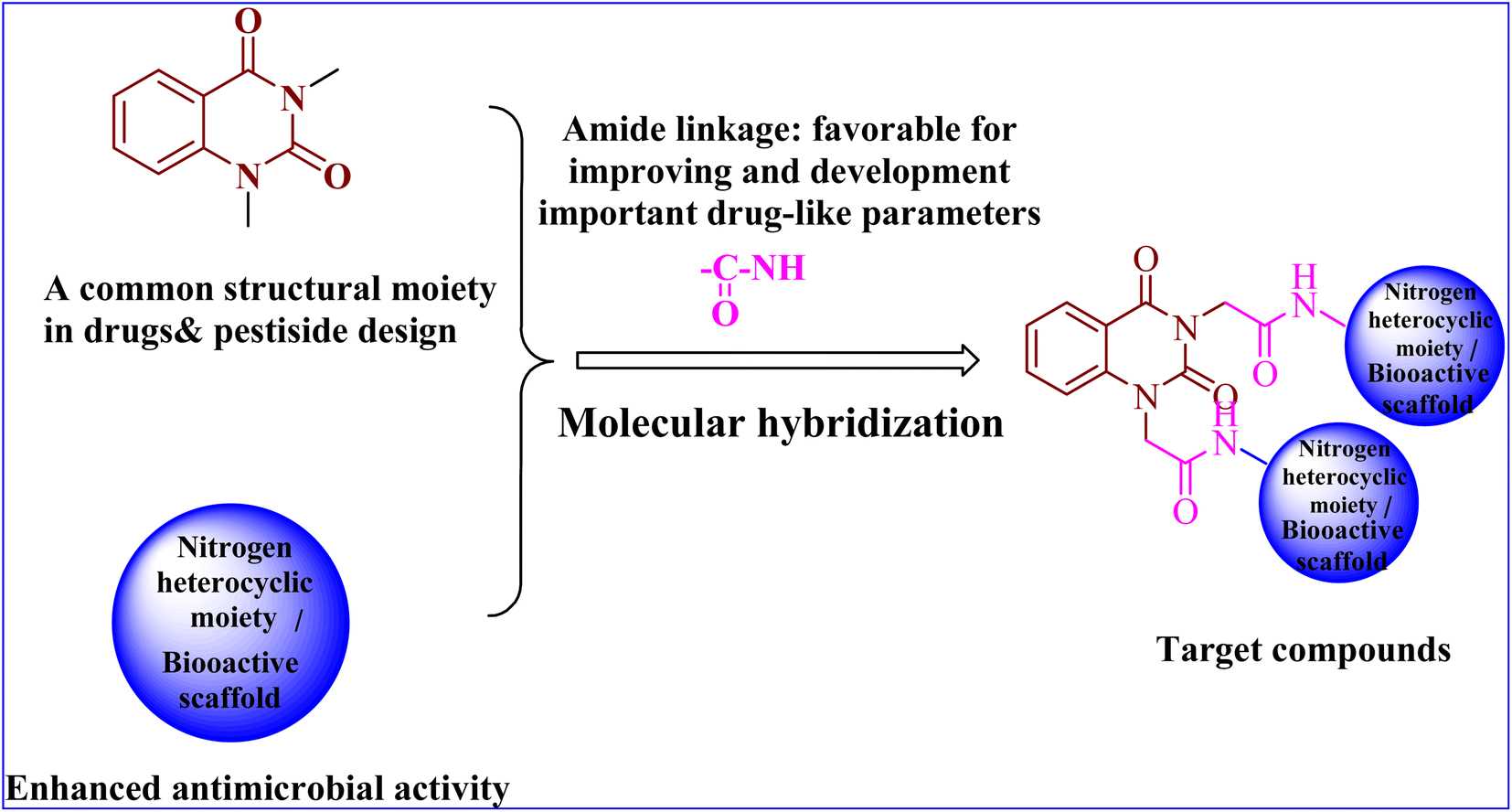 | ||
| Fig. 2 Design of the target compounds. | ||
2. Experimental
2.1 Chemistry
All chemicals and solvents were commercially available. Melting points (°C, uncorrected) of newly compounds were checked in capillary tubes by using MEL-TEMP II. Thin Layer Chromatography (TLC) technique was used for following reactions. The techniques FT-IR, NMR and MS spectra were recorded on Shimadzu 408, Bruker Vect. 22 and Mass 5988 Mass spectrometer, respectively. Elemental analyses were carried out at Cairo University, Egypt.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 1.27–1.32 (t, 6H, 2CH3), 4.22–4.29 (q, 4H, 2CH2), 6.99–8.26 (m, 4H, Ar-H); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 14.43, 42.91, 45.32, 61.56, 61.74, 115.17, 123.98, 128.60, 136.33, 168.10, 168.30; MS (electron ionization [EI]): m/z (%) = 334 [M+]; anal. calcd for C16H18N2O6: C, 57.48%; H, 5.43%; N, 8.38%. Found: C, 57.78%; H, 5.53%; N, 8.08%.O); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.25 and 4.31 (s, 4H, 2NH2), 4.56 and 4.73 (s, 4H, 2CH2), 7.24–8.10 (m, 4H, Ar-H), 9.25 and 9.32 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 42.74, 45.54, 114.94, 115.12, 115.58, 123.47, 128.28, 140.36, 150.29, 161.20, 166.56; MS (EI): m/z (%) = 306.28 [M+]; anal. calcd for C12H14N6O4: C, 47.06%; H, 4.85%; N, 4.61%. Found: C, 47.16%; H, 4.95%; N, 4.21%.O's), 1483 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 5.13 and 5.39 (s, 4H, 2CH2), 7.34–8.26 (m, 16H, Ar-H + 2NCH), 11.75 and 11.87 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 42.44, 45.26, 114.8, 115.12, 123.24, 127.36, 128.35, 128.97, 130.49, 134.62, 136, 140.67, 144.75, 150.87, 161.43, 168.34; MS (EI): m/z (%) = 482.5 [M+]; anal. calcd for C26H22N6O4: C, 64.72%; H, 4.60%; N, 17.42%. Found: C, 64.75%; H, 4.63%; N, 17.38%.O's), 1483 (CN), 844 (C–Cl stretching); 1H-NMR (400 MHz, TFA + CDCl3, δ, ppm) = 4.92 and 5.44 (s, 4H, 2CH2), 7.03–8.19 (m, 14H, Ar-H + 2NCH), 9.8 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 42.11, 45.21, 114.8, 115.12, 123.24, 127.36, 128.35, 128.97, 130.49, 134.62, 136, 140.67, 144.75, 150.87, 161.43, 168.34; MS (EI): m/z (%) = 550.09 [M+] and 552 ([M+] + 2) due to the presence of two chlorine atoms; anal. calcd for C26H20Cl2N6O4: C, 56.64%; H, 3.66%; Cl, 12.86; N, 15.24%. Found: C, 56.63%; H, 3.67%; Cl, 12.89; N, 15.20%.O's), 1421 (CN), 1963 (O–H bending); 1H-NMR (400 MHz DMSO-d6, δ, ppm) = 5.09 and 5.43 (s, 4H, 2CH2), 6.83 and 6.85 (s, 2H, NCH), 7.33–8.12 (m, 12H, Ar-H), 9.95 (s, 2H, 2OH), 11.54 and 11.64 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 42.12, 45.33, 114.8, 115.12, 123.24, 127.36, 128.35, 128.97, 130.49, 134.62, 136, 140.67, 144.75, 150.87, 161.43, 168.34; MS (EI): m/z (%) = 514.5 [M+]; anal. calcd for C26H22N6O6: C, 60.70%; H, 4.31%; N, 16.33%. Found: C, 60.74%; H, 4.34%; N, 16.30%.O), 1483 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 5.17 and 5.42 (s, 4H, 2CH2), 7.35 and 7.98 (s, 2H, 2NCH), 8.00–8.28 (m, 12H, Ar-H), 11.90 (s, 2H, 2NH); MS (EI): m/z (%) = 572.5 [M+]; anal. calcd for C26H20N8O8: C, 54.55%; H, 3.52%; N, 19.57%. Found: C, 54.58%; H, 3.56%; N, 19.53%.O), 1565, 1483 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 5.13 and 5.37 (s, 4H, 2CH2), 7.35–8.12 (m, 12H, Ar-H + 2NCH), 11.69 and 11.79 (s, 2H, 2NH); MS (EI): m/z (%) = 462 [M+]; anal. calcd for C22H18N6O6: C, 57.14%; H, 3.92%; N, 18.17%. Found: C, 57.15%; H, 3.96%; N, 18.13%.O); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 1.71–1.92 (m, 16H, 8CH2), 4.94 and 5.17 (s, 4H, 2CH2), 7.12–8.10 (m, 4H, Ar-H), 10.31 and 10.43 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 24.39, 28.67, 34.12, 43.20, 45.33, 114.24, 114.96, 115.29, 123.38, 127.77, 135.89, 140.82, 150.94, 161.40, 163.07, 164.38, 167.76; MS (EI): m/z(%) = 438 [M+]; anal. calcd for C22H26N6O4: C, 60.26%; H, 5.98%; N, 19.17%. Found: C, 60.31%; H, 5.99%; N, 19.14%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 1.60–1.66 (m, 10H, 5CH2), 2.25–2.42 (m, 10H, 5CH2), 4.93, 5.20 (s, 4H, 2CH2), 7.28–7.36 (m, 4H, Ar-H), 10.62, 10.77 (s, 2H, 2NH); MS (EI): m/z (%) = 466 [M+]; anal. calcd for C24H30N6O4: C, 61.79%; H, 6.48%; N, 18.01%. Found: C, 61.82%; H, 6.51%; N, 17.98%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 2.80 and 2.82 (s, 6H, 2CH3), 4.75 and 4.82 (s, 4H, 2CH2), 7.31–8.08 (m, 14H, Ar-H), 10.43 and 10.57 (s, 2H, 2NH); MS (EI): m/z(%) = 510 [M+]; anal. calcd for C28H26N6O4: C, 65.87%; H, 5.13%; N, 16.46%. Found: C, 65.90%; H, 5.17%; N, 16.42%.O's), 1542, 1483 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 2.29 (s, 6H, 2CH3), 2.34 (s, 6H, 2CH3), 4.96 and 5.38 (s, 4H, 2CH2), 7.24–8.11 (m, 12H, Ar-H), 10.94 and 10.31 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 14.01, 42.84, 45.06, 114.38, 114.98, 123.47, 126.51, 129.43, 135.41, 139.31, 140.83, 149.15, 150.9, 161.83, 169; anal. calcd for C30H30N6O4: C, 66.9%; H, 5.61%; N, 15.6%. Found: C, 67.02%; H, 5.64%; N, 15.3%.O's), 1558, 1485 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 2.27 and 2.37 (s, 6H, 2CH3), 4.82 and 5.30 (s, 4H, 2CH2), 7.09–8.12 (m, 10H, Ar-H), 11.02 and 11.11 (s, 2H, 2NH); MS (EI): m/z (%) = 522 [M+]; anal. calcd for C24H22N6O4S2: C, 55.16%; H, 4.24%; N, 16.08%, S, 12.27%. Found: C, 55.19%; H, 4.25%; N, 16.03%, S, 12.17%.O's), 1484, 1466 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 5.52 and 5.68 (s, 4H, 2CH2), 6.96–8.13 (m, 12H, Ar-H), 11.31 and 12.73 (s, 4H, 4NH); MS (EI): m/z (%) = 564 [M+]; anal. calcd for C28H20N8O6: C, 59.57%; H, 3.57%; N, 19.85%, Found: C, 59.61%; H, 3.59%; N, 19.81%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.89 and 5.15 (s, 4H, 2CH2), 7.33–8.13, (m, 12H, Ar-H), 11.11 and 11.23 (s, 4H, 4NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 42.68, 44.73, 115.1, 124.23, 128.51, 129.92, 135.76, 140.06, 150.8, 160.98, 164.94, 166; MS (EI): m/z (%) = 566.49 [M]+; anal. calcd for C28H18N6O8: C, 59.37%; H, 3.20%; N, 14.84%. Found: C, 59.34%; H, 3.16%; N, 14.80%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 2.21 and 2.25 (m, 8H, 4CH2), 3.27 (m, 4H, 4CH (sp3)), 4.76, 5.01 (s, 4H, 2CH2), 5.85 (m, 4H, 4CH (sp2)), 7.24–8.10 (m, 4H, Ar-H), 10.93, 11.03 (s, 2H, 2NH); MS (EI): m/z (%) = 574.55 [M+]; anal. calcd for C28H26N6O8: C, 58.53%; H, 4.56%; N, 14.63%; Found: C, 58.57%; H, 4.58%; N, 14.59%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 3.27 (d, 4H, 4CH), 4.83 (d, 4H, 4CH), 7.14–7.48 (m, 20H, Ar-H), 10.81 (s, 2H, 2NH); MS (EI): m/z (%) = 822.28 [M+]; anal. calcd for C48H38N6O8: C, 69.72%; H, 4.63%; N, 10.16%. Found: C, 69.76%; H, 4.67%; N, 10.13%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 1.85 (s, 6H, 2CH3), 4.64 and 4.88 (s, 4H, 2CH2) 7.15–8.09, (m, 4H, Ar-H), 10.21, 10.25, 10.31 and 10.35 (s, 4H, 4NH); anal. calcd for C16H18N6O6: C, 49.23%; H, 4.65%; N, 21.53%. Found: C, 49.25%; H, 4.66%; N, 21.51%.O's), 856 (C–Cl stretching);1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.14 and 4.16 (s, 4H, 2CH2), 4.67 and 4.91 (s, 4H, 2CH2), 7.15–8.11, (m, 4H, Ar-H), 10.49 (s, 4H, 4NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 41.25, 42.35, 44.49, 114.79, 123.32, 123.69, 128.57, 135.66, 140.1, 140.45, 150.83, 160.89, 164.72, 165.98; MS (EI): m/z (%) = 458.08 [M+] and 460.08 [M+ + 2], due to the presence of two chlorine atoms; anal. calcd for C16H16N6O6: C, 41.85%; H, 3.51, Cl, 15, 44%; N, 18.30%. Found: C, 41.88%; H, 3.56, Cl, 15, 44%; N, 18.25%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.14 and 4.16 (s, 4H, 2CH2), 4.67 and 4.91 (s, 4H, 2CH2), 7.15–8.11 (m, 4H, Ar-H), 10.49 (s, 4H, 4NH); MS (EI): m/z (%) = 514 [M+]; anal. calcd for C26H22N6O6: C, 60.70%; H, 4.31; N, 16.33%. Found: C, 60.70%; H, 4.34; N, 16.29%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.65 and 4.90 (s, 4H, 2CH2), 7.32–8.10 (m, 14H, Ar-H), 10.10, 10.13, 10.34 and 10.43 (s, 4H, 4NH); MS (EI): m/z (%) = 526 [M+]; anal. calcd for C22H18N8O8: C, 53.44%; H, 3.67; N, 17.00%. Found: C, 53.40%; H, 3.69; N, 16.96%.O's), 1070 (SO stretching); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.75 and 4.76 (s, 4H, 2CH2), 7.32–8.09 (m, 14H, Ar-H), 10.43, 10.44, 10.56 and 10.57 (s, 4H, 4NH); MS (EI): m/z(%) = 586 [M+]; anal. calcd for C24H22N6O8S2: C, 49.14%; H, 3.78; N, 14.33; S, 10.93%. Found: C, 49.17%; H, 3.83; N, 14.24; S, 10.91%.O's), 1484, 1439 (CN stretching); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 1.75 and 1.756 (s, 6H, 2CH3), 2.03 and 2.06 (s, 6H, 2CH3), 4.92 (s, 2H, CH2), 5.16 (s, 2H, CH2), 6.5 (s, 2H, sp2(CH)), 7.21–8.10 (m, 4H, Ar-H); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 16.38, 25.81, 44.27, 46.08, 52.01, 90.8, 114.2, 114.81, 123.36, 128.04, 128.77, 135.33, 140.59, 151.38, 156.42, 161.2, 163.46; MS(EI): m/z (%) = 434.17 [M+]; anal. calcd for C22H22N6O4: C, 60.82%; H, 5.10; N, 19.34%. Found: C, 60.86%; H, 5.14; N, 19.29%.O's), 1482, 1425 (CN stretching); 1H-NMR (400 MHz, CDCl3, δ, ppm) = 1.27 and 1.30 (s, 6H, 2CH3), 4.24 (s, 4H, 2CH2), 4.80 (s, 4H, 2CH2), 6.99–8.24 (m, 4H, Ar-H); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 14.14, 42.76, 45.03, 61.68, 62.1, 113.2, 115.35, 123.59, 129.41, 135.58, 139.73, 150.72, 161.21, 167.60, 167.80, 217.55; MS (EI): m/z (%) = 438.13 [M+]; anal. calcd for C20H18N6O6: C, 54.49%; H, 4.14; N, 19.17%. Found: C, 54.63%; H, 4.19; N, 19.13%.
O); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 1.27–1.32 (t, 6H, 2CH3), 4.22–4.29 (q, 4H, 2CH2), 6.99–8.26 (m, 4H, Ar-H); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 14.43, 42.91, 45.32, 61.56, 61.74, 115.17, 123.98, 128.60, 136.33, 168.10, 168.30; MS (electron ionization [EI]): m/z (%) = 334 [M+]; anal. calcd for C16H18N2O6: C, 57.48%; H, 5.43%; N, 8.38%. Found: C, 57.78%; H, 5.53%; N, 8.08%.O); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.25 and 4.31 (s, 4H, 2NH2), 4.56 and 4.73 (s, 4H, 2CH2), 7.24–8.10 (m, 4H, Ar-H), 9.25 and 9.32 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 42.74, 45.54, 114.94, 115.12, 115.58, 123.47, 128.28, 140.36, 150.29, 161.20, 166.56; MS (EI): m/z (%) = 306.28 [M+]; anal. calcd for C12H14N6O4: C, 47.06%; H, 4.85%; N, 4.61%. Found: C, 47.16%; H, 4.95%; N, 4.21%.O's), 1483 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 5.13 and 5.39 (s, 4H, 2CH2), 7.34–8.26 (m, 16H, Ar-H + 2NCH), 11.75 and 11.87 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 42.44, 45.26, 114.8, 115.12, 123.24, 127.36, 128.35, 128.97, 130.49, 134.62, 136, 140.67, 144.75, 150.87, 161.43, 168.34; MS (EI): m/z (%) = 482.5 [M+]; anal. calcd for C26H22N6O4: C, 64.72%; H, 4.60%; N, 17.42%. Found: C, 64.75%; H, 4.63%; N, 17.38%.O's), 1483 (CN), 844 (C–Cl stretching); 1H-NMR (400 MHz, TFA + CDCl3, δ, ppm) = 4.92 and 5.44 (s, 4H, 2CH2), 7.03–8.19 (m, 14H, Ar-H + 2NCH), 9.8 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 42.11, 45.21, 114.8, 115.12, 123.24, 127.36, 128.35, 128.97, 130.49, 134.62, 136, 140.67, 144.75, 150.87, 161.43, 168.34; MS (EI): m/z (%) = 550.09 [M+] and 552 ([M+] + 2) due to the presence of two chlorine atoms; anal. calcd for C26H20Cl2N6O4: C, 56.64%; H, 3.66%; Cl, 12.86; N, 15.24%. Found: C, 56.63%; H, 3.67%; Cl, 12.89; N, 15.20%.O's), 1421 (CN), 1963 (O–H bending); 1H-NMR (400 MHz DMSO-d6, δ, ppm) = 5.09 and 5.43 (s, 4H, 2CH2), 6.83 and 6.85 (s, 2H, NCH), 7.33–8.12 (m, 12H, Ar-H), 9.95 (s, 2H, 2OH), 11.54 and 11.64 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 42.12, 45.33, 114.8, 115.12, 123.24, 127.36, 128.35, 128.97, 130.49, 134.62, 136, 140.67, 144.75, 150.87, 161.43, 168.34; MS (EI): m/z (%) = 514.5 [M+]; anal. calcd for C26H22N6O6: C, 60.70%; H, 4.31%; N, 16.33%. Found: C, 60.74%; H, 4.34%; N, 16.30%.O), 1483 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 5.17 and 5.42 (s, 4H, 2CH2), 7.35 and 7.98 (s, 2H, 2NCH), 8.00–8.28 (m, 12H, Ar-H), 11.90 (s, 2H, 2NH); MS (EI): m/z (%) = 572.5 [M+]; anal. calcd for C26H20N8O8: C, 54.55%; H, 3.52%; N, 19.57%. Found: C, 54.58%; H, 3.56%; N, 19.53%.O), 1565, 1483 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 5.13 and 5.37 (s, 4H, 2CH2), 7.35–8.12 (m, 12H, Ar-H + 2NCH), 11.69 and 11.79 (s, 2H, 2NH); MS (EI): m/z (%) = 462 [M+]; anal. calcd for C22H18N6O6: C, 57.14%; H, 3.92%; N, 18.17%. Found: C, 57.15%; H, 3.96%; N, 18.13%.O); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 1.71–1.92 (m, 16H, 8CH2), 4.94 and 5.17 (s, 4H, 2CH2), 7.12–8.10 (m, 4H, Ar-H), 10.31 and 10.43 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 24.39, 28.67, 34.12, 43.20, 45.33, 114.24, 114.96, 115.29, 123.38, 127.77, 135.89, 140.82, 150.94, 161.40, 163.07, 164.38, 167.76; MS (EI): m/z(%) = 438 [M+]; anal. calcd for C22H26N6O4: C, 60.26%; H, 5.98%; N, 19.17%. Found: C, 60.31%; H, 5.99%; N, 19.14%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 1.60–1.66 (m, 10H, 5CH2), 2.25–2.42 (m, 10H, 5CH2), 4.93, 5.20 (s, 4H, 2CH2), 7.28–7.36 (m, 4H, Ar-H), 10.62, 10.77 (s, 2H, 2NH); MS (EI): m/z (%) = 466 [M+]; anal. calcd for C24H30N6O4: C, 61.79%; H, 6.48%; N, 18.01%. Found: C, 61.82%; H, 6.51%; N, 17.98%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 2.80 and 2.82 (s, 6H, 2CH3), 4.75 and 4.82 (s, 4H, 2CH2), 7.31–8.08 (m, 14H, Ar-H), 10.43 and 10.57 (s, 2H, 2NH); MS (EI): m/z(%) = 510 [M+]; anal. calcd for C28H26N6O4: C, 65.87%; H, 5.13%; N, 16.46%. Found: C, 65.90%; H, 5.17%; N, 16.42%.O's), 1542, 1483 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 2.29 (s, 6H, 2CH3), 2.34 (s, 6H, 2CH3), 4.96 and 5.38 (s, 4H, 2CH2), 7.24–8.11 (m, 12H, Ar-H), 10.94 and 10.31 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 14.01, 42.84, 45.06, 114.38, 114.98, 123.47, 126.51, 129.43, 135.41, 139.31, 140.83, 149.15, 150.9, 161.83, 169; anal. calcd for C30H30N6O4: C, 66.9%; H, 5.61%; N, 15.6%. Found: C, 67.02%; H, 5.64%; N, 15.3%.O's), 1558, 1485 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 2.27 and 2.37 (s, 6H, 2CH3), 4.82 and 5.30 (s, 4H, 2CH2), 7.09–8.12 (m, 10H, Ar-H), 11.02 and 11.11 (s, 2H, 2NH); MS (EI): m/z (%) = 522 [M+]; anal. calcd for C24H22N6O4S2: C, 55.16%; H, 4.24%; N, 16.08%, S, 12.27%. Found: C, 55.19%; H, 4.25%; N, 16.03%, S, 12.17%.O's), 1484, 1466 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 5.52 and 5.68 (s, 4H, 2CH2), 6.96–8.13 (m, 12H, Ar-H), 11.31 and 12.73 (s, 4H, 4NH); MS (EI): m/z (%) = 564 [M+]; anal. calcd for C28H20N8O6: C, 59.57%; H, 3.57%; N, 19.85%, Found: C, 59.61%; H, 3.59%; N, 19.81%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.89 and 5.15 (s, 4H, 2CH2), 7.33–8.13, (m, 12H, Ar-H), 11.11 and 11.23 (s, 4H, 4NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 42.68, 44.73, 115.1, 124.23, 128.51, 129.92, 135.76, 140.06, 150.8, 160.98, 164.94, 166; MS (EI): m/z (%) = 566.49 [M]+; anal. calcd for C28H18N6O8: C, 59.37%; H, 3.20%; N, 14.84%. Found: C, 59.34%; H, 3.16%; N, 14.80%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 2.21 and 2.25 (m, 8H, 4CH2), 3.27 (m, 4H, 4CH (sp3)), 4.76, 5.01 (s, 4H, 2CH2), 5.85 (m, 4H, 4CH (sp2)), 7.24–8.10 (m, 4H, Ar-H), 10.93, 11.03 (s, 2H, 2NH); MS (EI): m/z (%) = 574.55 [M+]; anal. calcd for C28H26N6O8: C, 58.53%; H, 4.56%; N, 14.63%; Found: C, 58.57%; H, 4.58%; N, 14.59%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 3.27 (d, 4H, 4CH), 4.83 (d, 4H, 4CH), 7.14–7.48 (m, 20H, Ar-H), 10.81 (s, 2H, 2NH); MS (EI): m/z (%) = 822.28 [M+]; anal. calcd for C48H38N6O8: C, 69.72%; H, 4.63%; N, 10.16%. Found: C, 69.76%; H, 4.67%; N, 10.13%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 1.85 (s, 6H, 2CH3), 4.64 and 4.88 (s, 4H, 2CH2) 7.15–8.09, (m, 4H, Ar-H), 10.21, 10.25, 10.31 and 10.35 (s, 4H, 4NH); anal. calcd for C16H18N6O6: C, 49.23%; H, 4.65%; N, 21.53%. Found: C, 49.25%; H, 4.66%; N, 21.51%.O's), 856 (C–Cl stretching);1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.14 and 4.16 (s, 4H, 2CH2), 4.67 and 4.91 (s, 4H, 2CH2), 7.15–8.11, (m, 4H, Ar-H), 10.49 (s, 4H, 4NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 41.25, 42.35, 44.49, 114.79, 123.32, 123.69, 128.57, 135.66, 140.1, 140.45, 150.83, 160.89, 164.72, 165.98; MS (EI): m/z (%) = 458.08 [M+] and 460.08 [M+ + 2], due to the presence of two chlorine atoms; anal. calcd for C16H16N6O6: C, 41.85%; H, 3.51, Cl, 15, 44%; N, 18.30%. Found: C, 41.88%; H, 3.56, Cl, 15, 44%; N, 18.25%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.14 and 4.16 (s, 4H, 2CH2), 4.67 and 4.91 (s, 4H, 2CH2), 7.15–8.11 (m, 4H, Ar-H), 10.49 (s, 4H, 4NH); MS (EI): m/z (%) = 514 [M+]; anal. calcd for C26H22N6O6: C, 60.70%; H, 4.31; N, 16.33%. Found: C, 60.70%; H, 4.34; N, 16.29%.O's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.65 and 4.90 (s, 4H, 2CH2), 7.32–8.10 (m, 14H, Ar-H), 10.10, 10.13, 10.34 and 10.43 (s, 4H, 4NH); MS (EI): m/z (%) = 526 [M+]; anal. calcd for C22H18N8O8: C, 53.44%; H, 3.67; N, 17.00%. Found: C, 53.40%; H, 3.69; N, 16.96%.O's), 1070 (SO stretching); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.75 and 4.76 (s, 4H, 2CH2), 7.32–8.09 (m, 14H, Ar-H), 10.43, 10.44, 10.56 and 10.57 (s, 4H, 4NH); MS (EI): m/z(%) = 586 [M+]; anal. calcd for C24H22N6O8S2: C, 49.14%; H, 3.78; N, 14.33; S, 10.93%. Found: C, 49.17%; H, 3.83; N, 14.24; S, 10.91%.O's), 1484, 1439 (CN stretching); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 1.75 and 1.756 (s, 6H, 2CH3), 2.03 and 2.06 (s, 6H, 2CH3), 4.92 (s, 2H, CH2), 5.16 (s, 2H, CH2), 6.5 (s, 2H, sp2(CH)), 7.21–8.10 (m, 4H, Ar-H); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 16.38, 25.81, 44.27, 46.08, 52.01, 90.8, 114.2, 114.81, 123.36, 128.04, 128.77, 135.33, 140.59, 151.38, 156.42, 161.2, 163.46; MS(EI): m/z (%) = 434.17 [M+]; anal. calcd for C22H22N6O4: C, 60.82%; H, 5.10; N, 19.34%. Found: C, 60.86%; H, 5.14; N, 19.29%.O's), 1482, 1425 (CN stretching); 1H-NMR (400 MHz, CDCl3, δ, ppm) = 1.27 and 1.30 (s, 6H, 2CH3), 4.24 (s, 4H, 2CH2), 4.80 (s, 4H, 2CH2), 6.99–8.24 (m, 4H, Ar-H); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 14.14, 42.76, 45.03, 61.68, 62.1, 113.2, 115.35, 123.59, 129.41, 135.58, 139.73, 150.72, 161.21, 167.60, 167.80, 217.55; MS (EI): m/z (%) = 438.13 [M+]; anal. calcd for C20H18N6O6: C, 54.49%; H, 4.14; N, 19.17%. Found: C, 54.63%; H, 4.19; N, 19.13%.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) 1688 (CO's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.12 and 4.16 (s, 4H, 2CH2), 5.37 and 5.64 (s, 4H, 2CH2), 8.00–8.85 (m, 4H, Ar-H); MS (EI): m/z (%) = 440.12 [M+]; anal. calcd for C18H16N8O6: C, 49.09%; H, 3.66; N, 25.45%. Found: C, 49.13%; H, 3.69; N, 25.41%.O's), 1483 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 5.14 and 5.39 (s, 4H, 2CH2), 7.34–8.49 (m, 24H, Ar-H), 11.75 to 11.84 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 44.59, 47.23, 114.80, 115.12, 123.24, 127.36, 128.35, 128.97, 130.49, 134.62, 136, 140.67, 144.75, 150.87, 161.43, 168.34; MS (EI): m/z(%) = 690 [M+]; anal. calcd for C40H30N6O6: C, 69.56%; H, 4.38; N, 12.17. Found: C, 69.56%; H, 4.39; N, 12.13.
N) 1688 (CO's); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 4.12 and 4.16 (s, 4H, 2CH2), 5.37 and 5.64 (s, 4H, 2CH2), 8.00–8.85 (m, 4H, Ar-H); MS (EI): m/z (%) = 440.12 [M+]; anal. calcd for C18H16N8O6: C, 49.09%; H, 3.66; N, 25.45%. Found: C, 49.13%; H, 3.69; N, 25.41%.O's), 1483 (CN); 1H-NMR (400 MHz, DMSO-d6, δ, ppm) = 5.14 and 5.39 (s, 4H, 2CH2), 7.34–8.49 (m, 24H, Ar-H), 11.75 to 11.84 (s, 2H, 2NH); 13C-NMR (100 MHz, DMSO-d6, δ, ppm) = 44.59, 47.23, 114.80, 115.12, 123.24, 127.36, 128.35, 128.97, 130.49, 134.62, 136, 140.67, 144.75, 150.87, 161.43, 168.34; MS (EI): m/z(%) = 690 [M+]; anal. calcd for C40H30N6O6: C, 69.56%; H, 4.38; N, 12.17. Found: C, 69.56%; H, 4.39; N, 12.13.2.2 In vitro antibacterial activity
Antimicrobial susceptibility and MIC of the newly molecules were determined against two G −ve and two G +ve bacteria. The studied pathogens were collected from Al-Azhar University, Egypt. They were cultivated in Mueller Hinton broth at 35 ± 2 °C for 24 h. The antimicrobial activity and MIC were performed as reported by Qader et al. (2021).2,27–362.3 Docking study
Docking studies37 were achieved on the new synthesized molecules 2–9a towards S. aureus tyrosyl-tRNA synthetase. The 3D structure of the target (PDB ID: 1JIJ) was retrieved from the Protein Data Bank (PDB).38 The 2D structures of the ligand molecules were sketched using ChemDraw Ultra 7.0, then converted to 3D structures using OpenBabel GUI tool.39 Before performing the docking approach, the 3D files of the ligand molecules and target were subjected to energy minimization by using AMBER Force Field40 in Open Babel and CHARMm Force Field41 in Discovery Studio 3.5, respectively, to obtain more stable confirmations. The molecular docking approach was performed by PyRx software tool.25 The 2D orientations of the docked molecules with the enzyme were generated by Discovery Studio visualizer software. Finally, the pharmacokinetics and ADMET properties of molecules were anticipated using AdmetSAR, SwissADME, and mol inspiration tools.3. Results and discussion
3.1 Chemistry
On the basis of our previous findings, and in demanding to develop new antibacterial drug candidates, this context deals with the design, synthesis and antibacterial assessment of the quinazolin-2,4-dione analogues 2–9a. In the present study, 2,2′-(2,4-dioxoquinazoline-1,3(2H,4H)-diyl)di(aceto-hydrazide) 2 was utilized as a versatile precursor starting for preparation of some new heterocyclic moieties attached to quinazolindione scaffold 3–9a. The structural elucidation of the compounds was ascertained from spectral and elemental analyses. The overall pathway adopted for the preparation of new class of quinazolin-2,4-dione analogues linked to N-heterocycles and/or bioactive scaffolds is represented in Schemes 1–3.As represented in Scheme 1, our starting 2 was synthesized through the reaction of the synthesized ester diethyl-2,2′-(2,4-dioxoquinazoline-1,3(2H,4H)-diyl)-diacetate 1 (ref. 26) with hydrazine hydrate in absolute ethanol, and its structure was confirmed by elemental and spectral analyses. The proton nuclear magnetic resonance (1H-NMR) spectrum of the ester 1 exhibited the characteristic triplet and quartet signals due to the ethoxy function of the ester groups at δ 1.28, 1.30, 4.22, and 4.29 ppm. Additionally, mass spectrum showed molecular ion peak at m/z = 334 corresponding to the chemical formula (C16H18N2O6). The fragment at m/z 162 referred to the quinazolin-2,4-dione ring.
 | ||
| Scheme 1 Synthesis of starting material 2. | ||
The infrared (IR) spectrum of compound 2 showed strong to medium intensity bands appearing at ν 3250 and 1700 cm−1 which attributed to presence of NH2 and CO groups. In addition, 1H-NMR spectrum revealed the disappearance of triplet and quartet signals related to the two ethyl groups of the starting di ester 1, in addition to existence of new characteristic singlet signals for two NH groups at δ 9.25 and 9.32 ppm, respectively. Furthermore, two singlet signals for two NH2 groups at δ 4.25 and 4.31 ppm, while the two methylene groups attached to quinazoline moiety appeared as singlet at δ 4.56 and 4.73 ppm and the four aromatic protons appeared multiplet in the range from δ 7.24–8.10 ppm. Mass spectrum recorded mass at m/z 306 which is corresponding to the chemical formula C12H14N6O4.
Various arylidene hydrazide derivatives (Schiff bases) 3a–e were synthesized in good to excellent yields by treatment of 2,2′-(2,4-dioxoquinazoline-1,3(2H,4H)-diyl)di(aceto hydrazide) 2 with various aromatic aldehydes such as, benzaldehyde, 4-hydroxybenzaldehyde, 4-chlorobenzaldehyde, 4-nitrobenzaldehyde and furfural, respectively (Scheme 2). The chemical structures of 3a–f were concluded from their IR, and 1H-NMR spectra, and elemental analysis. For instance, IR spectra of 3a–e represented characteristic absorption bands at 1570 cm−1 assigned for azomethine linkage. 1H-NMR spectra of all Schiff bases 3a–e showed a characteristic signal for azomethine proton (–NH–NCH–) appeared at δ 6.83–8.23 ppm, which is in interference with aromatic protons. Moreover, the value appeared at m/z 482 showed the mass of the synthesized molecules 3a.
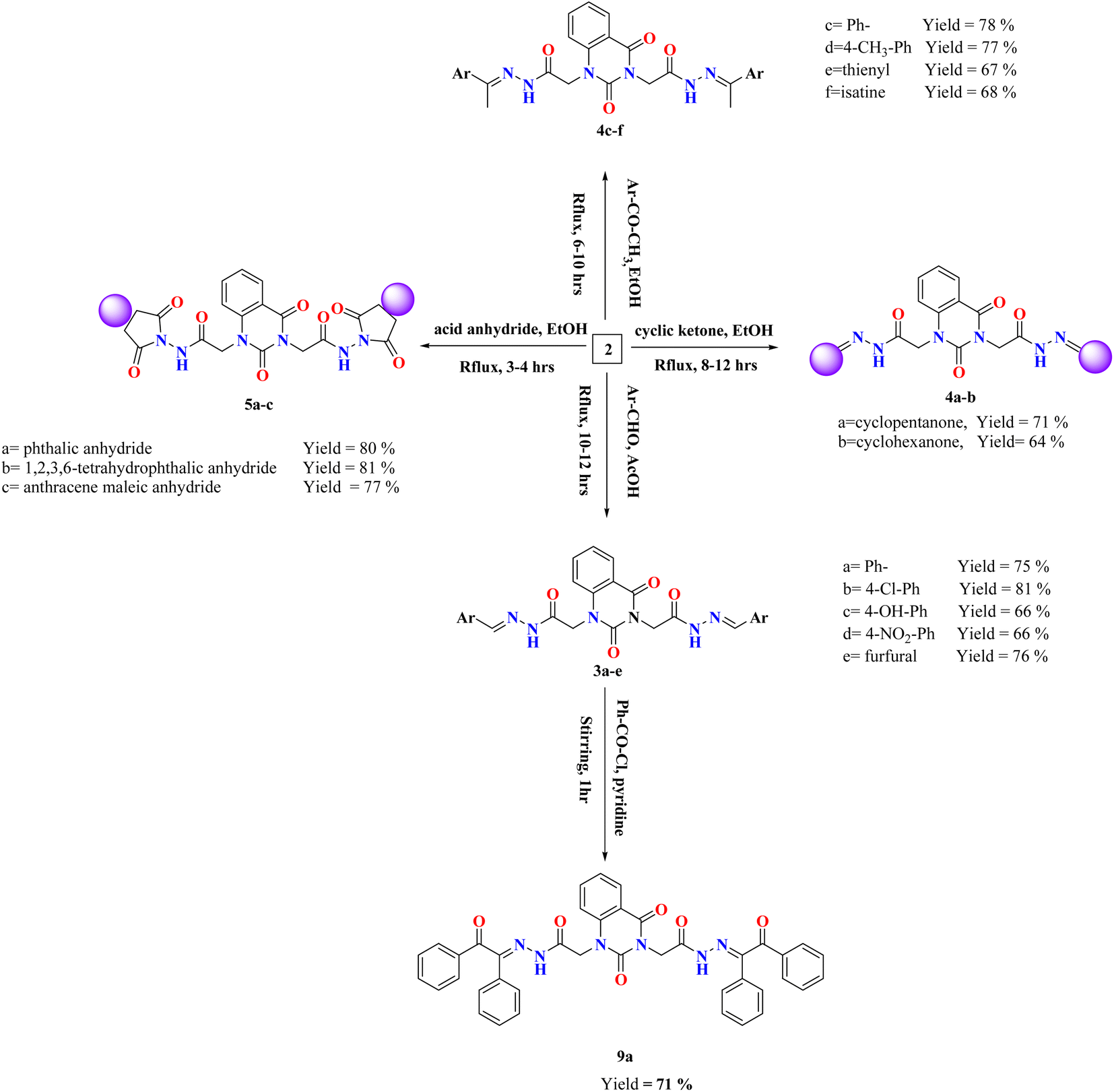 | ||
| Scheme 2 Synthesis of compounds 3–5 and 9a. | ||
The treatment of di-hydrazide 2 with cycloaliphatic ketones such as cyclo-pentanone and/or cyclohexanone produced the relevant hydrazones (–NH–NC-alicyclic ring) 4a–b, respectively.
Chemical shifts at the ranges of δ 1.71–2.39 and 1.60–2.52 ppm confirmed the presence of aliphatic CH2 groups in compounds 4a–b, respectively.
Reaction of (un)substituted acetophenones with of di-hydrazide 2 afforded hydrazone derivatives 4c–d, respectively. 1H-NMR of 4c, for example, showed new peaks at δ 2.80, 2.82 and 7.31–8.08 ppm related to two methyl groups and aromatic protons, respectively.
Condensation of the compound 2 with 2-acetylthiophene and Isatin yielded the comparable hydrazone derivatives 4e–f, respectively. IR spectrum of 4f, as an example, showed the presence of bands at ν 3051 and 1625 cm−1 characteristic for functional groups Ar-H and CN, respectively. 1H-NMR of 4f exhibited characteristic signals at δ 6.96–8.13 and 12.73 ppm assignable to the aromatic protons and NH group, respectively.
Upon condensation of 2 with acid anhydrides such as, phthalic anhydride, tetrachlorophthalic anhydride and/or anthracene maleic anhydride in glacial AcOH, the imide derivatives 5a–c, respectively, were afforded in good yields. 1H-NMR of 5a, for example, indicated the disappearance of two singlet peaks related to two amino groups of compound 2 and appearance of characteristic peak at the region δ 7.33–8.13 assigned for aromatic protons of 5a. Further, the mass spectral data of 5a exhibited a molecular ion peak at m/z 566 which agreed with the molecular formula C28H18N6O8.
The reactions of compound 2 with aliphatic/aromatic acid chlorides and/or benzene sulphonyl chloride in DMF afforded amide derivatives 6a–e, respectively. Compound 6a was structurally elucidated by spectral and elemental analyses. 1H-NMR of 6a displayed characteristic peaks at δ 2.78 ppm for two new methyl groups. For compound 6d, its 1H-NMR exhibited the characteristic (AMX) signals for thiophene ring at δ 7.32–8.09 ppm, which are in interference with aromatic protons.
Moreover, quinazolin-2,4-dione containing pyrazole and pyrazolone moieties 7a–b were synthesized based on condensation of 2 with active methylene compounds such as acetylacetone and ethyl acetoacetate via Knorr pyrazole reaction, as declared in Scheme 3. Cyclization of 7a–b was confirmed by spectral and microanalysis. Presence of various functional groups in the newly synthesized compound 7a–b was interpreted by IR and NMR spectra. For compound 7a, as example, IR spectrum showed band appearing at ν 1625 cm−1 which attributed to presence of functional group CN, along with the disappearance of absorption band assigned for NH2 group. On the other hand, 1H-NMR spectrum exhibited four singlet peaks at δ 1.75, 1.76, 2.03, 2.06 and ppm attributed to new four methyl groups in pyrazole rings, along with, two singlet peaks at δ 6.51 and 6.56 ppm assigned for two methine groups in pyrazole rings. In addition, the mass spectral data showed [M+] at m/z 434 which agreed with the molecular formula of the synthesized molecules 7a. A mechanism for the synthesis of 7a from compound 2 and 1,3-dicarbonyl compound (acetyl acetone) catalyzed by acetic acid is represented in Scheme 4. Initially, this mechanism involved a protonation of acetyl acetone, the produced carbanion then underwent a nucleophilic attack of the free doublet of nitrogen to yield the imine. On the other hand, the other nitrogen atom attacks the second carbonyl group of acetyl acetone which was protonated by the acid to furnish a second imine group. Finally, the diimine was deprotonated to yield the pyrazole product.
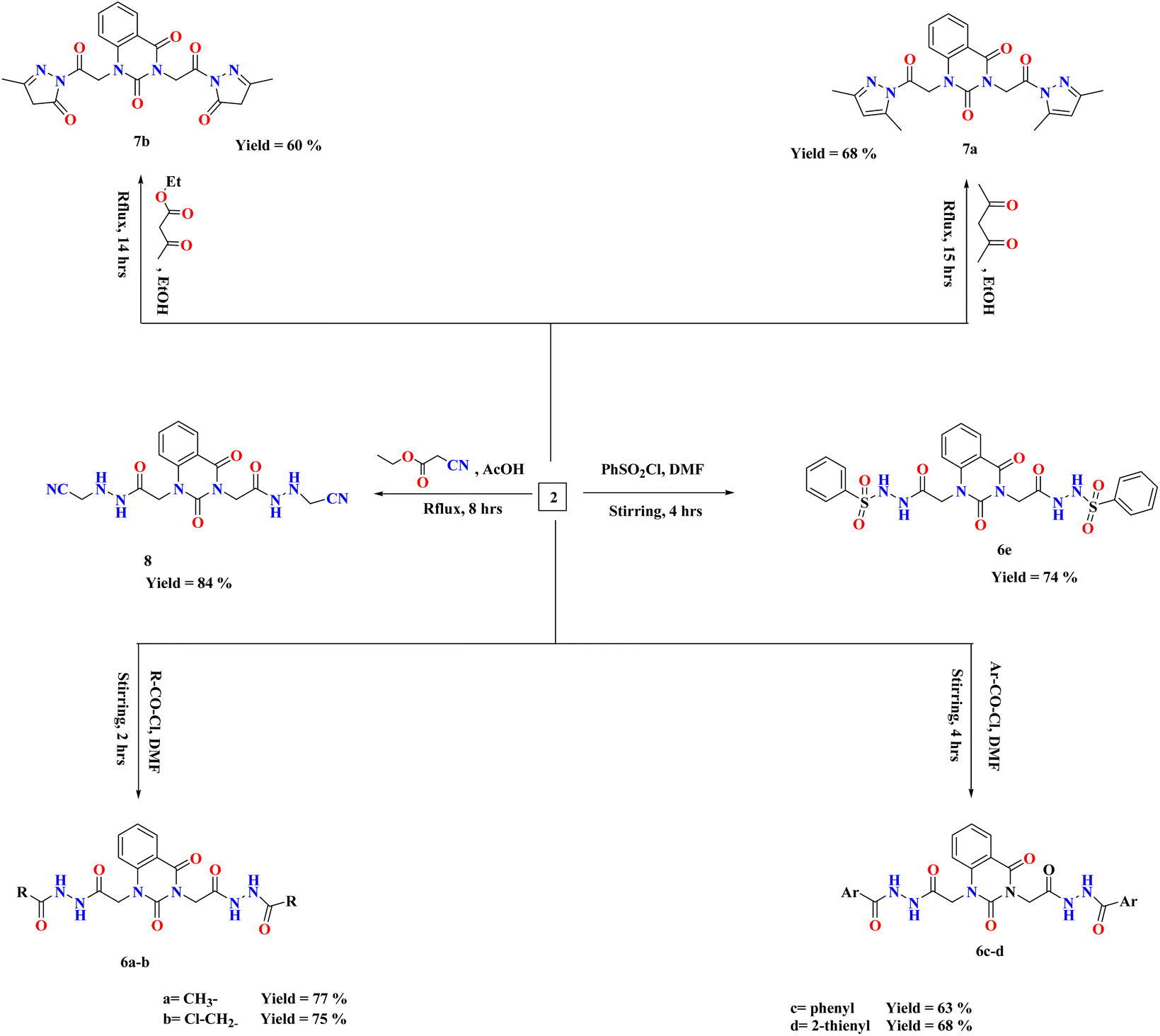 | ||
| Scheme 3 Synthesis of compounds 6–8. | ||
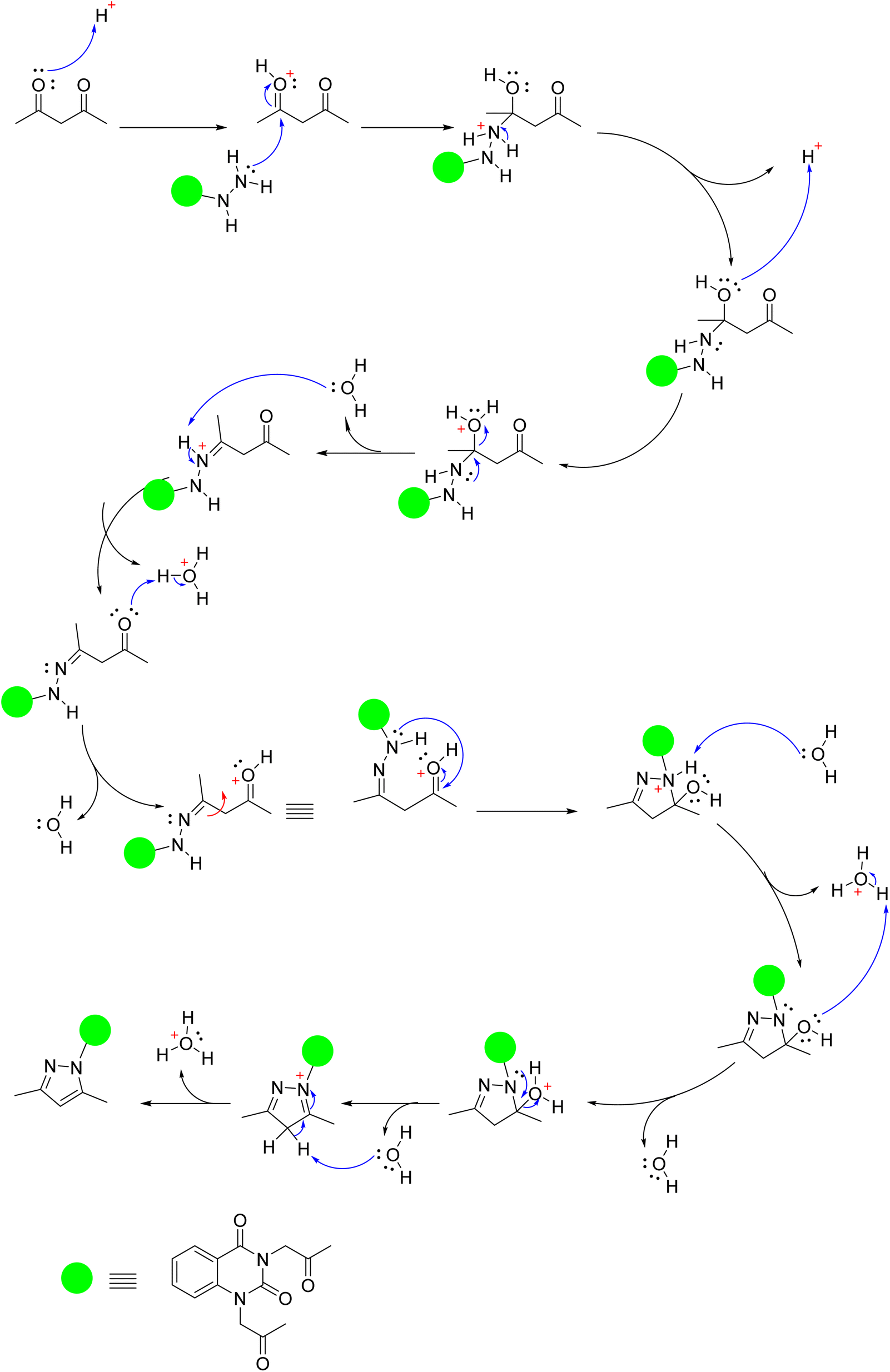 | ||
| Scheme 4 Plausible mechanism for the synthesis of compound 7a. | ||
Treatment of 2 with ethyl cyanoacetate in glacial AcOH afforded the desired compound 8, which was structurally confirmed by existence of sharp band at 2337 cm−1 assigned for cyano group in its IR spectrum. Further, the value appeared at m/z 440 showed the expected mass of product 8.
Else way, Schiff base 3a was then allowed to react with benzoyl chloride in pyridine to yield compound 9a. The latter product was structurally confirmed by 1H-NMR spectrum which exhibited the absence of a characteristic peak for azomethine proton (–NH–NCH–), along with increasing the signals of aromatic area according to presence of extra two phenyl rings.
The spectral data of all synthesized compounds are given as a ESI.† In addition, InChI code, SMILES and solubility of all compounds are also provided in ESI.†
3.2 Antibacterial activity
The spread of the microbial chemotherapeutic resistance is recognized as the most serious global health problem, which minimize the potency of the antibiotics. Therefore, herein, the antibacterial efficacy of a new series of heterocyclic compounds against various pathogenic microbes was determined. The minimum inhibitory concentration (MIC) of the tested molecules was mentioned in Table 1.| Sample no. | Minimum Inhibitory Concentration (MIC, μg mL−1) | |||
|---|---|---|---|---|
| Escherichia coli | Pseudomonas aeruginosa | Bacillus subtilis | Staphylococcus aureus | |
| 1 | 120 | 80 | ND | 40 |
| 2 | 40 | 20 | 120 | 40 |
| 3a | ND | ND | ND | ND |
| 3b | 40 | 60 | ND | ND |
| 3c | 2.5 | 5 | 10 | 5 |
| 3d | ND | ND | ND | ND |
| 3e | 20 | 40 | ND | ND |
| 4a | ND | ND | ND | ND |
| 4b | 120 | 40 | 80 | 80 |
| 4c | 20 | 20 | ND | ND |
| 4d | ND | ND | ND | ND |
| 4e | 10 | 40 | 80 | 120 |
| 4f | ND | ND | ND | ND |
| 5a | 20 | 15 | ND | ND |
| 5b | ND | ND | 20 | 15 |
| 5c | ND | ND | ND | ND |
| 6a | ND | ND | ND | ND |
| 6b | 10 | 15 | 7 | 12 |
| 6c | ND | ND | ND | ND |
| 6d | 40 | 80 | 40 | 40 |
| 6e | 120 | 40 | 80 | 80 |
| 7a | 140 | 160 | ND | 80 |
| 7b | 20 | 40 | 10 | 15 |
| 8 | 10 | 20 | 20 | 15 |
| 9a | 10 | 40 | 80 | 120 |
| Ciprofloxacin | 5 | 7 | 2.5 | 1.25 |
The lowest concentration with no observation of pathogenic microbial growth is known as MIC. It was noticed from the results that compound 3c revealed significant antimicrobial efficacy against all the tested pathogenic strains at low concentration compared to ciprofloxacin ranging from 2.5 to 10 μg ml−1. The results represented also, that compounds 6b and 8 exhibited high antibacterial potency towards all strains of tested pathogenic bacteria with low concentrations ranged from 5 to 20 μg ml−1. Meanwhile, compounds 4c, 5a declared significant antimicrobial effect at low concentrations against the two tested G −ve bacteria. Additionally, compound 5b showed strong antibacterial effect at relatively low concentrations against the two tested G +ve bacteria.
3.3 In silico docking study
To gain insight into molecular interactions and docking scores, the molecular docking study36,42,43 was carried out between the newly ligand molecules and the target enzyme. Herein, S. aureus tyrosyl-tRNA synthetase is a well-recognized attractive therapeutic target for antibacterial drug design.44 In the present study, the docking approach used S. aureus tyrosyl-tRNA synthetase to figure out the action mode of the ligand molecules as antibacterial agents. Target compounds 2–9a exhibited similar fitness to co-crystalized ligand and reference drug into active site of the target enzyme. The docking pattern was interestingly achieved similar to the reported position completed by the co-crystalized ligand with the active site of the target. The results showed that compound 3c exhibited the most promising bacterial inhibitory effect and the best binding affinity against the target (ΔG = −14.2 kcal mol−1). It engaged in five hydrogen bonds via its hydroxyl, azomethine, and carbonyl groups with the essential residues TYR36, ASP80, LYS84, ASP177, and GLN196. Considering the structure within this series, the SAR analysis demonstrated that the presence of two hydroxyl groups on para-positions of two phenyl rings (compound 3c) exhibited significantly greater inhibition effect. From the results tabulated in Table 2, compound 3c is considered as a highly antibacterial potency and might be act as an antibiotic. The 2D molecular interaction network between all ligand molecules 2–9a and S. aureus tyrosyl-tRNA synthetase are shown in Fig. 3.| No. | Binding energy (kcal mol−1) | Docked complex (amino acid–ligand) interactions | Distance (Å) |
|---|---|---|---|
| 2 | −8.6 | H-bonds | |
| GLY38:O–compound 2 | 2.24 | ||
| LYS84:NZ–compound 2 | 2.98 | ||
| LYS84:NZ–compound 2 | 2.95 | ||
| GLY193:N–compound 2 | 2.90 | ||
| GLN196:OE1–compound 2 | 2.32 | ||
| Arene-cation | |||
| PHE54–compound 2 | 5.12 | ||
| PHE54–compound 2 | 4.28 | ||
| HIS50–compound 2 | 5.65 | ||
| HIS50–compound 2 | 4.92 | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| 3a | −10.6 | H-bonds | |
| ASP40:N–compound 3a | 2.99 | ||
| ASP195:OD1–compound 3a | 2.32 | ||
| ASP195:OD2–compound 3a | 2.27 | ||
| Arene–arene | |||
| HIS47–compound 3a | 4.12 | ||
| Arene-cation | |||
| ARG88:NH1–compound 3a | 5.59 | ||
|
|||
| 3b | −9.1 | H-bonds | |
| ASP195:OD1–compound 3b | 2.52 | ||
| Arene-cation | |||
| LYS84:NZ–compound 3b | 5.28 | ||
| ARG88:NH1–compound 3b | 5.90 | ||
| Arene-sigma | |||
| LYS84:CG–compound 3b | 3.76 | ||
|
|||
| 3c | −14.2 | H-bonds | |
| TYR36:OH–compound 3c | 2.73 | ||
| ASP80:OD2–compound 3c | 2.25 | ||
| LYS84:NZ–compound 3c | 3.00 | ||
| ASP177:OD1–compound 3c | 2.18 | ||
| GLN196:OE1–compound 3c | 2.38 | ||
|
|||
| 3d | −8.8 | H-bonds | |
| GLY49:O–compound 3d | 2.32 | ||
| ASP40:OD1–compound 3d | 3.04 | ||
| VAL224:N–compound 3d | 2.88 | ||
| arene-cation | |||
| ARG88:NH1–compound 3d | 5.44 | ||
|
|||
| 3e | −9.9 | H-bonds | |
| ASP40:N–compound 3e | 3.10 | ||
| TYR170:OH–compound 3e | 2.92 | ||
| GLY193:N–compound 3e | 2.72 | ||
| GLN196:NE2–compound 3e | 2.85 | ||
| Arene–arene | |||
| HIS47–compound 3e | 4.59 | ||
| Arene-cation | |||
| HIS50–compound 3e | 5.12 | ||
| ARG88:NH1–compound 3e | 5.07 | ||
|
|||
| 4a | −10.0 | H-bonds | |
| TYR170:OH–compound 4a | 2.80 | ||
| GLN174:NE2–compound 4a | 3.06 | ||
| GLY193:N–compound 4a | 3.10 | ||
| ASP80:OD2–compound 4a | 2.25 | ||
| Arene-sigma | |||
| GLN196:CG–compound 4a | 3.26 | ||
|
|||
| 4b | −10.7 | H-bonds | |
| TYR170:OH–compound 4b | 2.99 | ||
| GLY193:N–compound 4b | 2.98 | ||
| Arene-sigma | |||
| GLN196:CG–compound 4b | 3.36 | ||
|
|||
| 4c | −10.8 | H-bonds | |
| TYR170:OH–compound 4c | 2.85 | ||
| GLY193:N–compound 4c | 2.76 | ||
| GLN196:NE2–compound 4c | 2.89 | ||
|
|||
| 4d | −10.3 | H-bonds | |
| ASP40:N–compound 4d | |||
|
|||
| 4e | −9.1 | H-bonds | |
| ASP40:N–compound 4e | 2.96 | ||
| LYS84:NZ–compound 4e | 3.10 | ||
| ASP195:OD1–compound 4e | 2.18 | ||
| Arene-cation | |||
| LYS84:NZ–compound 4e | 5.26 | ||
| LYS84:NZ–compound 4e | 4.77 | ||
|
|||
| 4f | −12.0 | H-bonds | |
| LYS84:NZ–compound 4f | 3.00 | ||
| TYR170:OH–compound 4f | 2.92 | ||
| GLY193:N–compound 4f | 2.77 | ||
| GLN196:NE2–compound 4f | 2.95 | ||
| Arene–arene | |||
| HIS47–compound 4f | 5.82 | ||
| HIS47–compound 4f | 4.52 | ||
| Arene-cation | |||
| ARG88:NH1–compound 4f | 5.29 | ||
| ARG88:NH1–compound 4f | 5.12 | ||
|
|||
| 5a | −11.9 | H-bonds | |
| LYS84:NZ–compound 5a | 3.14 | ||
| GLY193:N–compound 5a | 2.77 | ||
| GLY193:N–compound 5a | 3.02 | ||
| GLN196:NE2–compound 5a | 2.99 | ||
| Arene–arene | |||
| HIS47–compound 5a | 5.62 | ||
| Arene-cation | |||
| ARG88:NH1–compound 5a | 5.11 | ||
| ARG88:NH1–compound 5a | 5.16 | ||
|
|||
| 5b | −12.3 | H-bonds | |
| ARG88:NH1–compound 5b | 2.65 | ||
| GLY193:N–compound 5b | 1.99 | ||
| Arene-cation | |||
| LYS84:NZ–compound 5b | 5.19 | ||
|
|||
| 5c | −11.6 | H-bonds | |
| ARG88:NH1–compound 5c | 2.39 | ||
| ASP195:OD2–compound 5c | 2.58 | ||
|
|||
| 6a | −9.6 | H-bonds | |
| ASP40:N–compound 6a | 2.91 | ||
| LYS84:NZ–compound 6a | 2.80 | ||
| ARG88:NH1–compound 6a | 3.05 | ||
| TYR170:OH–compound 6a | 3.10 | ||
| ASP195:OD1–compound 6a | 2.39 | ||
| Arene-cation | |||
| HIS50–compound 6a | |||
|
|||
| 6b | −9.3 | H-bonds | |
| ASP40:N–compound 6b | 2.82 | ||
| LYS84:NZ–compound 6b | 3.19 | ||
|
|||
| 6c | −10.7 | H-bonds | |
| ASP40:N–compound 6c | 3.04 | ||
| LYS84:NZ–compound 6c | 2.85 | ||
| ARG88:NH1–compound 6c | 3.00 | ||
| TYR170:OH–compound 6c | 3.00 | ||
| GLY193:N–compound 6c | 2.80 | ||
| GLN196:NE2–compound 6c | 2.93 | ||
| Arene-cation | |||
| HIS50–compound 6c | 5.16 | ||
|
|||
| 6d | −10.2 | H-bonds | |
| ASP40:N–compound 6d | 3.00 | ||
| LYS84:NZ–compound 6d | 2.84 | ||
| TYR170:OH–compound 6d | 2.91 | ||
| GLY193:N–compound 6d | 2.79 | ||
| GLN196:NE2–compound 6d | 2.89 | ||
| Arene-cation | |||
| ARG88:NH2–compound 6d | 5.79 | ||
|
|||
| 7a | −10.3 | H-bonds | |
| LYS84:NZ–compound 7a | 2.80 | ||
| ARG88:NH1–compound 7a | 3.05 | ||
| Arene-cation | |||
| LYS84:NZ–compound 7a | 5.95 | ||
|
|||
| 7b | −8.5 | H-bonds | |
| ARG58:NH1–compound 7b | 2.55 | ||
| ARG58:NH1–compound 7b | 2.37 | ||
| Arene–arene | |||
| PHE306–compound 7b | 5.24 | ||
| PHE306–compound 7b | 5.27 | ||
|
|||
| 8 | −8.6 | H-bonds | |
| ASP195:OD2–compound 8 | 2.31 | ||
| GLN196:OE1–compound 8 | 2.30 | ||
|
|||
| 9a | −9.6 | H-bonds | |
| ASP195:OD1–compound 11a | 2.30 | ||
|
|||
| Reference drug | −9.0 | H-bonds | |
| GLY193:N–reference drug | 2.11 | ||
|
|||
| Co-crystalized ligand | −8.2 | H-bonds | |
| GLY38:O–co-crystalized ligand | 2.37 | ||
| ASP40:N co-crystalized ligand | 2.79 | ||
| GLY193:N–co-crystalized ligand | 3.07 | ||
| ASP195:OD1–co-crystalized ligand | 3.11 | ||
| TYR170:OH–co-crystalized ligand | 2.94 | ||
| ASP195:OD1–co-crystalized ligand | 2.23 | ||
| GLN196:NE2–co-crystalized ligand | 2.55 | ||
| VAL224:N–co-crystalized ligand | |||
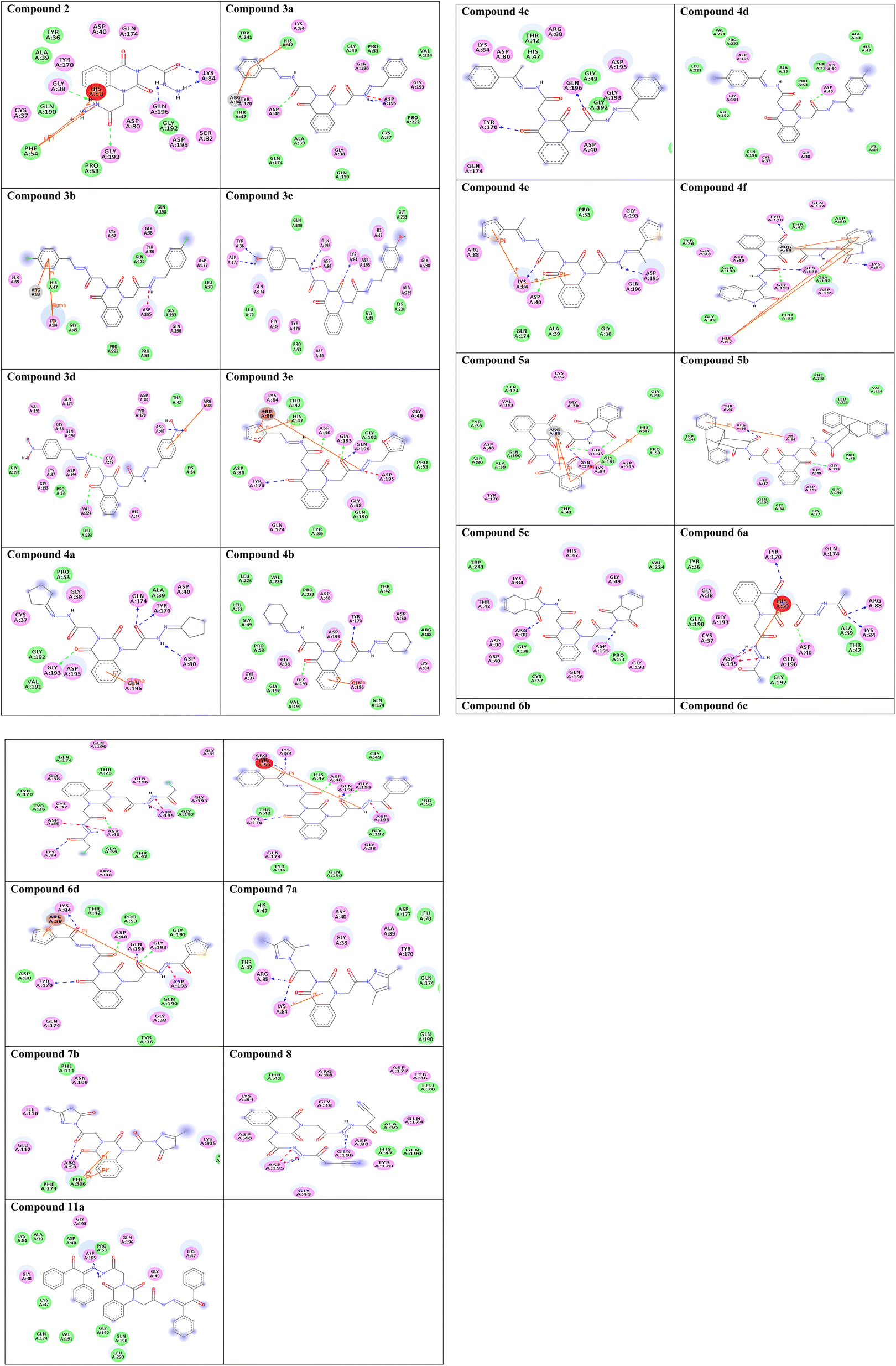 | ||
| Fig. 3 Two-dimensional (2D) orientation of complexes. H-bond interactions are represented in green, blue and pink dotted lines. Pi-stacking are represented in orange lines. | ||
Redocking of the co-crystalized ligand [2-amino-3-(4-hydroxy-phenyl)-propionylamino]-(1,3,4,5-tetrahydroxy-4-hydroxymethyl-piperidin-2-yl)-acetic acid was carried out to validate the docking process (RMSD < 2 Å) and the results showed similar fitness to the docked compounds. The co-crystalized ligand exhibited binding affinity of −8.2 kcal mol−1, and it showed H-bond interactions with the residues GLY38, ASP40, TYR170, GLY193, ASP195, GLN196, and VAL224, as shown in Fig. 4. In addition, the reference drug ciprofloxacin (binding affinity = −9.0 kcal mol−1) docked to the target though H-bond interaction with the residue GLY193 at the distance 2.11 Å, as presented in Fig. 4.
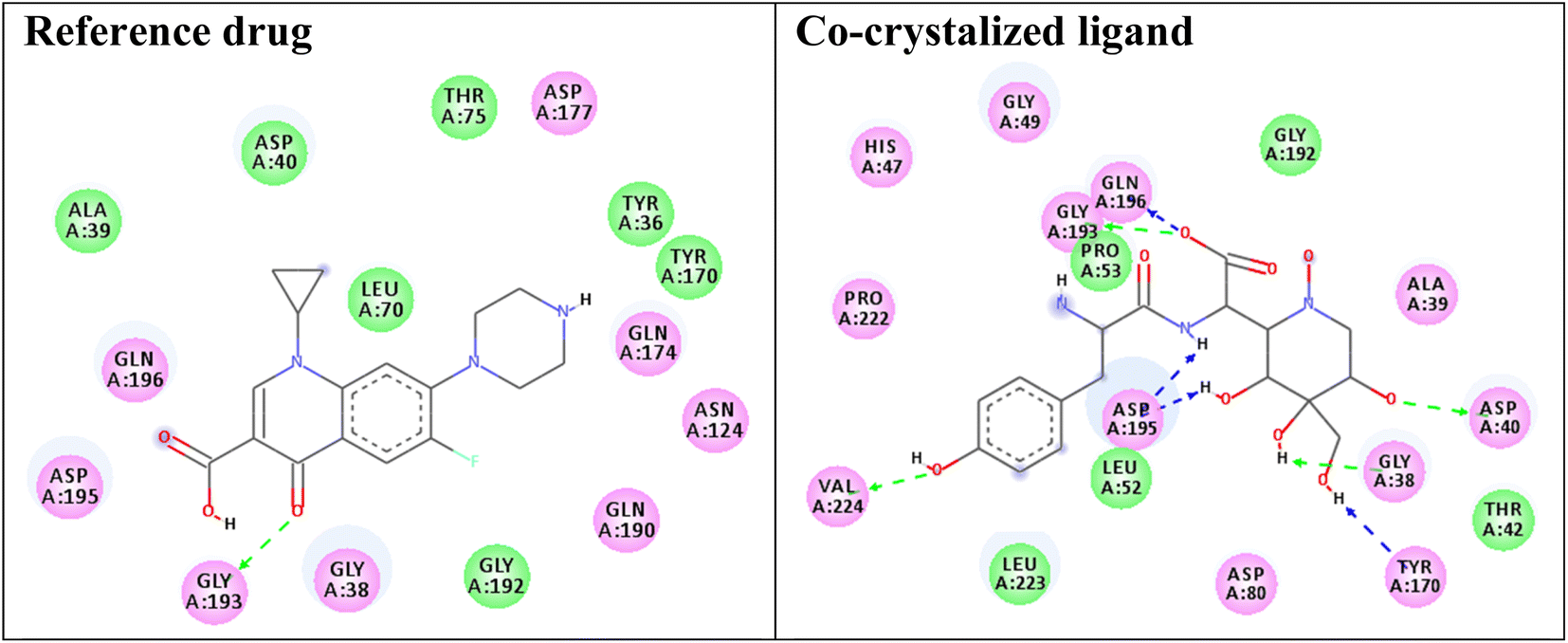 | ||
| Fig. 4 Two-dimensional (2D) orientation of the reference drug and co-crystalized ligand with the target enzyme S. aureus tyrosyl-tRNA synthetase. H-bond interactions are shown in green, and blue dotted lines. Pi-stacking interactions are represented in orange lines. | ||
The molecular and pharmacokinetics properties of the most active compound 3c and the known antibiotic, ciprofloxacin were calculated by SwissADME, admetSAR, and mol inspiration web servers. The bioavailability and physicochemical properties of 3c were evaluated using ADMETlab tool, by plotting radar showing 13 properties (Fig. 5). According to Lipinski's rule, most of the tested compounds satisfied with the Ro5 (no. of violations ≤ 1) and meet all criteria for good permeability and acceptable oral bioavailability, displayed rotatable bonds number in the range <10, which means they are flexible. Their HBA and HBD values were in the satisfied range, gave them higher solubility in cellular membranes. The logp values less than 5, reflected good lipophilicity character, as tabulated in Table 3. Furthermore, the ADMET parameters exhibited that the molecules had better Human Intestinal Absorption (% HIA) scores; indicating that they could be better absorbed by the human intestine. The target compounds 2–9a does not pass blood–brain barrier that indicating their good CNS safety profile. Finally, all showed negative AMES toxicity and carcinogenicity test; indicating their safety.
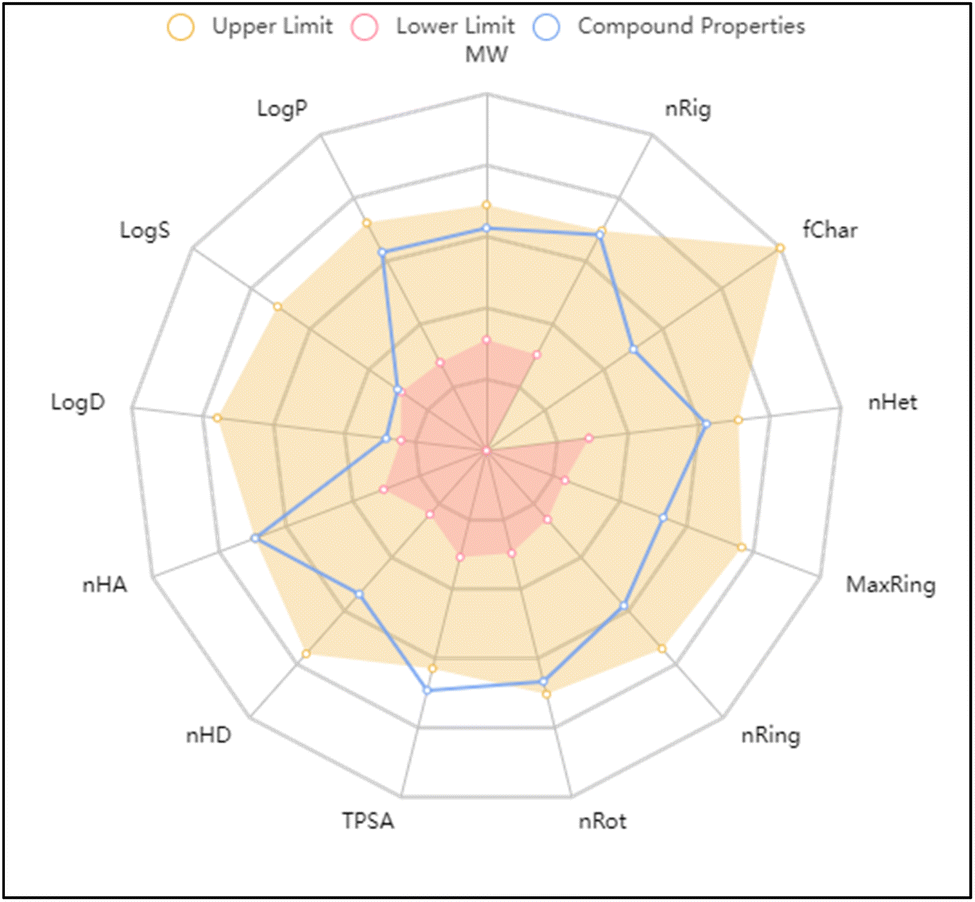 | ||
| Fig. 5 In silico ADMET properties of compound 3c predicted by ADMETlab tool. | ||
p, logarithm ratio of partition coefficient between n-octanol and water; TPSA, topological polar surface area; MW, molecular weight; HBA, number of hydrogen bond acceptors; HBD, number of hydrogen bond donors; N rotatable, number of rotatable bonds
| Molecular weight (g mol−1) | BBB permeant | GI absorption | % Human Intestinal Absorption (HIA+) | logp |
TPSA A2 | HBA | HBD | N rotatable | N violations | Bioavailability score | AMES toxicity | Carcino-genicity | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. range | 130–500 | <25 poor | ≤5 | ≤140 | 2.0–20.0 | 0.0–6.0 | ≤10 | ≤1 | Nontoxic | Non-carcinogenic | |||
| >80 high | |||||||||||||
| 2 | 306.28 | No | Low | 98.50 | −3.92 | 154.25 | 6 | 4 | 6 | 0 | 0.55 | Nontoxic | Non-carcinogenic |
| 3a | 482.49 | No | High | 92.12 | 2.91 | 126.93 | 6 | 2 | 8 | 0 | 0.55 | Nontoxic | Non-carcinogenic |
| 3b | 551.39 | No | High | 93.28 | 4.27 | 126.93 | 10 | 2 | 8 | 1 | 0.55 | Nontoxic | Non-carcinogenic |
| 3c | 514.50 | No | Low | 91.71 | 1.95 | 137.39 | 8 | 4 | 10 | 1 | 0.17 | Nontoxic | Non-carcinogenic |
| 3d | 572.49 | No | Low | 76.14 | 2.38 | 218.58 | 10 | 2 | 12 | 2 | 0.17 | Nontoxic | Non-carcinogenic |
| 3e | 462.41 | No | Low | 96.93 | 1.43 | 153.20 | 8 | 2 | 10 | 0 | 0.55 | Nontoxic | Non-carcinogenic |
| 4a | 438.48 | No | High | 86.38 | 1.62 | 126.93 | 6 | 2 | 8 | 0 | 0.55 | Nontoxic | Non-carcinogenic |
| 4b | 466.53 | No | High | 79.46 | 2.63 | 126.93 | 6 | 2 | 8 | 0 | 0.55 | Nontoxic | Non-carcinogenic |
| 4c | 510.54 | No | High | 93.88 | 2.74 | 126.93 | 6 | 2 | 10 | 1 | 0.55 | Nontoxic | Non-carcinogenic |
| 4d | 538.60 | No | High | 93.32 | 3.64 | 126.93 | 6 | 2 | 10 | 1 | 0.55 | Nontoxic | Non-carcinogenic |
| 4e | 522.60 | No | High | 96.80 | 2.54 | 126.93 | 6 | 2 | 10 | 1 | 0.55 | Nontoxic | Non-carcinogenic |
| 4f | 564.51 | No | High | 85.53 | 1.38 | 185.12 | 8 | 4 | 8 | 2 | 0.55 | Nontoxic | Non-carcinogenic |
| 5a | 566.48 | No | Low | 96.96 | 1.35 | 180.36 | 8 | 2 | 8 | 2 | 0.17 | Nontoxic | Non-carcinogenic |
| 5b | 822.83 | No | Low | 96.96 | 2.98 | 176.97 | 8 | 2 | 8 | 2 | 0.17 | Nontoxic | Non-carcinogenic |
| 5c | 574.54 | No | Low | 97.50 | −0.68 | 176.97 | 8 | 2 | 8 | 2 | 0.17 | Nontoxic | Non-carcinogenic |
| 6a | 390.35 | No | Low | 98.68 | −2.78 | 160.40 | 6 | 4 | 10 | 1 | 0.55 | Nontoxic | Non-carcinogenic |
| 6b | 459.24 | No | Low | 99.50 | −1.65 | 160.40 | 6 | 4 | 12 | 1 | 0.55 | Nontoxic | Non-carcinogenic |
| 6c | 514.49 | No | Low | 98.28 | 0.56 | 160.40 | 6 | 4 | 12 | 2 | 0.17 | Nontoxic | Non-carcinogenic |
| 6d | 526.54 | No | Low | 99.12 | 0.36 | 160.40 | 6 | 4 | 12 | 2 | 0.17 | Nontoxic | Non-carcinogenic |
| 7a | 434.45 | No | High | 99.29 | 1.55 | 113.78 | 6 | 0 | 6 | 0 | 0.55 | Nontoxic | Non-carcinogenic |
| 7b | 438.39 | No | Low | 99.40 | −1.05 | 143.50 | 8 | 0 | 6 | 1 | 0.55 | Nontoxic | Non-carcinogenic |
| 8 | 440.37 | No | Low | 98.05 | −3.95 | 207.99 | 8 | 4 | 12 | 1 | 0.55 | Nontoxic | Non-carcinogenic |
| 9a | 690.72 | No | Low | 96.54 | 4.28 | 161.08 | 8 | 2 | 14 | 2 | 0.17 | Nontoxic | Non-carcinogenic |
| Ref. drug | 331.35 | No | High | 97.95 | −70.0 | 74.57 | 5 | 2 | 3 | 0 | 0.55 | Nontoxic | Non-carcinogenic |
4. Conclusion
In summary, a series of hybrid structures 2–9a containing quinazolin-2,4-dione analogue attached to N-heterocyclic cores such as pyrrolidine-2,5-dione, pyrazole and oxadiazole and/or bioactive scaffolds such as hydrazone, amide, sulfonamide, azomethine, and thiourea linkage was designed as potential antibacterial agents, synthesized, and investigated for their antibacterial activity. Predominantly, the in vitro studies revealed that the compound 3c exhibited strong significant antibacterial efficacy against all the tested pathogenic strains at low concentrations comparing with the tested standard drug. The findings were also connected with the molecular docking studies which concluded that compound 3c showed good inhibitory activity against the target S. aureus tyrosyl-tRNA synthetase. The presence of two hydroxyl groups on phenyl rings at positions-4 in the latter compound 3c seems to be essential for antibacterial activity.Data availability
The data that support the findings of this study are included within the article and ESI.†Author contributions
A. H. A., M. O., H. R. M. R., M. M. T. and A. M. A. made a significant contribution to the work reported, whether that is in the study design, analysis, and interpretation. A. H. A. and M. O. are responsible for the synthesis of products. Finally, A. H. A., M. O. and H. R. M. R. took part in writing, revising or critically reviewing the article. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted.Conflicts of interest
The authors declare no conflict of interest.References
- M. B. Pisano, A. Kumar, R. Medda, G. Gatto, R. Pal, A. Fais, B. Era, S. Cosentino, E. Uriarte, L. Santana, F. Pintus and M. J. Matos, Molecules, 2019, 24, 1–12 CrossRef.
- H. R. M. Rashdan, A. H. Abdelmonsef, M. M. Abou-Krisha and T. A. Yousef, Molecules, 2021, 26, 7119 CrossRef CAS PubMed.
- A. H. Abdelmonsef and A. M. Mosallam, J. Heterocycl. Chem., 2020, 57, 1637–1654 CrossRef CAS.
- A. Haredi Abdelmonsef, M. Eldeeb Mohamed, M. El-Naggar, H. Temairk and A. Mohamed Mosallam, Front. Mol. Biosci., 2020, 7, 1–19 CrossRef.
- A. H. Abdelmonsef, M. A. Abdelhakeem, A. M. Mosallam, H. Temairk, M. ElNaggar, H. Okasha and H. R. M. Rashdan, J. Heterocycl. Chem., 2022, 59, 474–492 CrossRef CAS.
- H. R. M. Rashdan, H. Okasha, M. A. Abdelhakeem, A. M. Mosallam, H. Temairk, A. G. Alhamzani, M. M. Abou-Krisha, T. A. Yousef and A. H. Abdelmonsef, Egypt. J. Chem., 2022, 65, 189–199 Search PubMed.
- M. El-Naggar, M. E. Mohamed, A. M. Mosallam, W. Salem, H. R. Rashdan and A. H. Abdelmonsef, Evol. Bioinform., 2020, 16, 1–13 CrossRef.
- A. Ahmed, A. Ibrahim, A. Mosallam, M. Taha and H. Temairk, Egypt. J. Chem., 2022, 65, 189–199 Search PubMed.
- T. P. Selvam and P. V. Kumar, Arch. Pharmacal Res., 2011, 1, 1–21 Search PubMed.
- C. Zhao, K. P. Rakesh, L. Ravidar, W. Fang and H. Qin, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS.
- G. Verma, A. Marella, M. Shaquiquzzaman, M. Akhtar, M. R. Ali and M. M. Alam, J. Pharm. Bioallied Sci., 2014, 6, 69–80 CrossRef.
- S. R. Alizadeh and S. M. Hashemi, Development and therapeutic potential of 2-aminothiazole derivatives in anticancer drug discovery, Springer US, 2021, vol. 30 Search PubMed.
- S. M. Gomha, H. A. Abdelhady, D. Z. Hassain, A. H. Abdelmonsef, M. El-Naggar, M. M. Elaasser and H. K. Mahmoud, Drug Des., Dev. Ther., 2021, 2021, 659–677 CrossRef.
- H. R. M. Rashdan, M. El-Naggar and A. H. Abdelmonsef, Molecules, 2021, 26, 1–17 Search PubMed.
- H. R. M. Rashdan, I. A. Shehadi and A. H. Abdelmonsef, ACS Omega, 2021, 6, 1445–1455 CrossRef CAS.
- H. R. M. Rashdan and A. H. Abdelmonsef, Struct. Chem., 2022, 33, 1727–1739 CrossRef CAS.
- H. S. El-Sheshtawy, A. H. Abdelmonsef, S. M. Abboudy, A. M. M. Younes, M. M. Taha and M. A. Hassan, Polycyclic Aromat. Compd., 2017, 39, 279–286 CrossRef.
- A. A. Noser, A. H. Abdelmonsef, M. El-naggar and M. M. Salem, Molecules, 2021, 1–28 Search PubMed.
- A. M. El-Maghraby and A. H. Abdelmonsef, Egypt. J. Chem., 2020, 63, 1341–1358 Search PubMed.
- A. A. Noser, I. A. Shehadi, A. H. Abdelmonsef and M. M. Salem, ACS Omega, 2022, 7, 25265–25277 CrossRef CAS PubMed.
- A. H. M. Hussein, A.-B. A. El-Adasy, A. M. El-Saghier, M. Olish and A. H. Abdelmonsef, RSC Adv., 2022, 12, 12607–12621 RSC.
- R. Rondla, L. S. PadmaRao, V. Ramatenki, A. Haredi-Abdel-Monsef, S. R. Potlapally and U. Vuruputuri, Comput. Biol. Chem., 2017, 71, 224–229 CrossRef CAS PubMed.
- T. Dasari, B. Kondagari, R. Dulapalli, A. H. Abdelmonsef, T. Mukkera, L. S. Padmarao, V. Malkhed and U. Vuruputuri, J. Biomol. Struct. Dyn., 2017, 35, 3119–3139 CrossRef CAS PubMed.
- A. H. Abdelmonsef, R. Dulapalli, T. Dasari, L. S. Padmarao, T. Mukkera and U. Vuruputuri, Comb. Chem. High Throughput Screening, 2016, 19, 1–18 CrossRef PubMed.
- S. Dallakyan and A. J. Olson, in Chemical Biology, Springer, 2015, vol. 1263, pp. 243–250 Search PubMed.
- M. A. Hassan, M. A. Seleem, A. M. M. Younes, M. M. T. Taha and A.-B. Haredi Abdel-Monsef, Eur. J. Chem., 2013, 4, 121–123 CrossRef CAS.
- M. A. El-Hashash, S. M. Sherif, A. A. Badawy and H. R. Rashdan, Der Pharma Chem., 2014, 6, 23–29 Search PubMed.
- H. R. M. Rashdan, A. Abdel-Aziem, D. H. El-Naggar and S. Nabil, Acta Pol. Pharm. - Drug Res., 2019, 76, 469–482 CrossRef CAS.
- S. Ahmed, M. A. Abdel-Naby and A. F. Abdel-Fattah, Research Square, 2021, preprint, DOI:10.21203/rs.3.rs-704571/v1.
- M. Fadel, A. A. Hamed, A. M. Abd-Elaziz, M. M. E. Ghanem and A. M. Roshdy, Egypt. J. Chem., 2021, 64, 3511–3520 Search PubMed.
- E. K. Radwan, H. R. M. Rashdan, B. A. Hemdan, A. A. Koryam and M. E. El-Naggar, Environ. Sci. Pollut. Res., 2022, 1–15 Search PubMed.
- A. Sabt, M. T. Abdelrahman, M. Abdelraof and H. R. M. Rashdan, ChemistrySelect, 2022, 7, e202200691 CrossRef CAS.
- I. A. Shehadi, M. T. Abdelrahman, M. Abdelraof and H. R. M. Rashdan, Molecules, 2022, 27, 342 CrossRef CAS PubMed.
- H. R. M. Rashdan, M. T. Abdelrahman, I. A. Shehadi, S. S. El-Tanany and B. A. Hemdan, Molecules, 2022, 27, 3613 CrossRef CAS PubMed.
- H. R. M. Rashdan, I. A. Shehadi, M. T. Abdelrahman and B. A. Hemdan, Molecules, 2021, 26, 4817 CrossRef CAS PubMed.
- H. R. M. Rashdan and A. H. Abdelmonsef, J. Mol. Struct., 2022, 1268, 133659 CrossRef CAS.
- A. A. Noser, A. H. Abdelmonsef and M. M. Salem, Bioorg. Chem., 2022, 106299 Search PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks, A. D. Mackerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- I. A. Shehadi, H. R. M. Rashdan and A. H. Abdelmonsef, Comput. Math. Methods Med., 2020, 2020, 1–12 CrossRef.
- A. Haredi Abdelmonsef, Egypt. J. Med. Hum. Genet., 2019, 20, 1–14 CrossRef.
- Z. P. Xiao, T. W. Ma, M. L. Liao, Y. T. Feng, X. C. Peng, J. L. Li, Z. P. Li, Y. Wu, Q. Luo, Y. Deng, X. Liang and H. L. Zhu, Eur. J. Med. Chem., 2011, 46, 4904–4914 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06527d |
| This journal is © The Royal Society of Chemistry 2023 |