
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolution behavior and encapsulation properties of fatty acid–elastin-like polypeptide conjugates†
Tingting Zhang a,
Frédéric Perucha,
Amélie Webera,
Katell Bathanyb,
Martin Fauquignona,
Angela Mutschlera,
Christophe Schatza and
Bertrand Garbay*a
a,
Frédéric Perucha,
Amélie Webera,
Katell Bathanyb,
Martin Fauquignona,
Angela Mutschlera,
Christophe Schatza and
Bertrand Garbay*a
aUniv. Bordeaux, CNRS, Bordeaux INP, LCPO UMR 5629, F-33600 Pessac, France. E-mail: bertrand.garbay@bordeaux-inp.fr
bUniv. Bordeaux, CNRS, Bordeaux INP, CBMN UMR 5248, F-33600 Pessac, France
First published on 12th January 2023
Abstract
Developing new biomaterials is an active research area owing to their applications in regenerative medicine, tissue engineering and drug delivery. Elastin-like polypeptides (ELPs) are good candidates for these applications because they are biosourced, biocompatible and biodegradable. With the aim of developing ELP-based micelles for drug delivery applications we have synthesized 15 acyl-ELP compounds by conjugating myristic, palmitic, stearic, oleic or linoleic acid to the N-terminus of three ELPs differing in molar mass. The ELP–fatty acid conjugates have interesting solution behavior. They form micelles at low temperatures and aggregate above the cloud point temperature (Tcp). The critical micelle concentration depends on the fatty acid nature while the micelle size is mainly determined by the ELP block length. We were able to show that ELPs were better hydrated in the micelles than in their individual state in solution. The micelles are stable in phosphate-buffered saline at temperatures below the Tcp, which can vary between 20 °C and 38 °C depending on the length or hydrophilicity of the ELP. Acyl-ELP micelles were loaded with the small hydrophobic molecule Nile red. The encapsulation efficiency and release kinetics showed that the best loading conditions were achieved with the largest ELP conjugated to stearic acid.
Introduction
The development of new biomaterials for biomedical applications is a rapidly growing area of research. For most of them, the target use will be drug delivery, tissue-engineering and regenerative medicine. Polypeptide-based materials are good candidates for such applications because they are biosourced, biocompatible, biodegradable, and if carefully designed, non-toxic and weakly immunogenic.1 Among them, recombinant polypeptides are of particular interest because they can be produced at an industrial level using GMP standards, contrarily to the polypeptides extracted from biological tissues which can be contaminated by virus or prions.2,3Elastin-like polypeptides (ELPs) are a class of polypeptides derived from the hydrophobic domain of mammalian elastin.4 Usually, they are made of repetitions of the pentapeptide sequence Val-Pro-Gly-X-Gly, where X can be any natural amino acid but proline. An interesting feature of ELPs is that they are thermosensitive, being soluble in aqueous solutions below their cloud point temperature (Tcp), and aggregating into polymer-rich particles when heated above their Tcp.5,6 The Tcp depends on the chain length, the nature of the guest residue Xaa (e.g. polar, charged, hydrophobic), the polypeptide concentration, and the presence of salts in the solution.7,8 Recombinantly produced ELP are preferred over the chemically synthesized ones, because they have controlled sequence and can be of higher molar masses.9,10 Regarding their interesting biological and physico-chemical properties, ELPs were used in recent years to develop hydrogels for tissue engineering and innovative drug delivery systems.10
Two different strategies have been used for drug delivery using ELPs. In the first one, the drug is covalently linked to the polypeptides, either by fusion at the gene level for therapeutic peptides or proteins, or by conjugation of the therapeutic molecules on purified ELPs. However, some hydrophobic drugs cannot be conjugated, or lose their activity after conjugation. In this case, a second strategy can be used where ELPs are engineered to self-assemble into nanoparticles, such as micelles or polymersomes, which can sequestrate hydrophobic molecules in their apolar compartments. Of particular interest, amphiphilic block polypeptides obtained by association of two ELP blocks having different Tcps can be genetically synthesized. These polypeptide structures showed self-assembly properties (micelles, vesicles) when incubated in aqueous buffers at intermediate Tcp between those of each corresponding block, namely the hydrophobic block forming the core of the micelles, whereas the hydrophilic block forms the corona.11 It was later shown that the length of the two polypeptide blocks and the hydrophilic/hydrophobic mass ratio influence the size of the micelles.12,13 However, it has been also demonstrated that ELP diblocks formed rather loose micelles, with a relatively high level of hydration of the hydrophobic core, thereby preventing the formation of well-segregated micelles.14–16 Consequently, the encapsulation of hydrophobic drugs is quite inefficient through this approach.17 Furthermore, micelles obtained from amphiphilic ELPs are generally not very stable at low polypeptide concentrations leading to micelle disassembly when diluted in biological fluids after injection.
Here, we propose to drastically increase the stability of ELP-based micelles by grafting lipid moieties to the ELP blocks. Two different strategies were developed in the past. In the first one, recombinantly expressed ELPs were purified and chemically modified on specific amino acids selected as guest residues of the ELP sequence. Following this approach, cholesterol was grafted on lysine residues18 and oleic acid on methionine residues.19 In both cases, the grafting of lipid chains to the ELP backbone yielded brush-like lipoproteins that can self-assemble into micelles with ELP–cholesterol,18 or polymersomes with ELP–oleic acid.19 However, these materials are highly hydrophobic owing to the number of lipid molecules grafted per ELP, and therefore difficult to handle in biological media. More recently, a short synthetic lipid (O-octadecylhydroxylamine) was also successfully graft to the N-terminal serine of an ELP via a pH-responsive oxime bond.20 We also reported the synthesis of polyisoprene–ELP copolymers that can self-assemble into nanoparticles capable of encapsulating hydrophobic molecules, but the assemblies aggregated in physiological buffer.21
A second elegant strategy developed in Chilkoti's and Mozhdehi's groups relies on the lipidation of the ELP in vivo during its recombinant expression. Indeed, by fusing a specific peptide sequence at the N-terminus of ELPs, and by coexpressing the polypeptide and a yeast N-myristoyltransferase (NMT) in Escherichia coli (E. coli) they could obtain ELP–C14 conjugates capable to form micelles for drug delivery to mammalian cells.17 The in vivo synthetic chemistry was then adapted to produce ELP–C14 conjugates containing a β-sheet-forming peptide,22 and to conjugate a myristic acid analog with an ELP.23 In a slightly different approach, an ELP containing a sterol binding site at its C-terminal domain was recombinantly expressed and purified allowing cholesterol to be grafted in vitro to obtain ELP–cholesterol conjugates.24 Although interesting, these approaches are rather limited regarding the yields of purified conjugates, i.e. only 40 mg L−1 culture for C14–ELP17 and 5–10 mg L−1 for ELP–cholesterol.24 These yields are an order of magnitude lower than those typically obtained for unmodified ELPs. Another limitation comes from the small repertoire of natural lipids that can be grafted to ELPs using this strategy. Indeed, for fatty acids (FAs), the approach is restricted to C14 or analogs of comparable length because of the specificity of the NMT enzyme. Longer FAs such as palmitate or stearate cannot be used, nor can unsaturated FAs.
In this work, we report the synthesis of 15 ELP–FA conjugates with variation in the length and saturation of the FA moiety as well as the length of the ELP block through a straightforward approach where the carboxyl group of FA was condensed with the primary amine of the N-terminal methionine of the ELPs. Acylated ELPs thus obtained were characterized, and their self-assembly in a biologically relevant buffer was studied to elucidate the respective roles of FA structure and ELP length on micelle properties. The drug loading capacity of this new class of micelles was also evaluated.
Experimental
Materials
Myristic acid (Fluka Chemistry, 98%), palmitic acid (Riedel-de HaënTM, 98%), stearic acid (Sigma-Aldrich, 98.5%), oleic acid (Sigma-Aldrich, 99%), linoleic acid (Sigma-Aldrich, 99%), O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU, Novabiochem), Nile red (Carl Roth), pyrene (Alfa Aesar, 98%), samarium(III) acetate hydrate (Sigma-Aldrich, 99.9%), sodium thiosulfate (Sigma-Aldrich, 99%), N,N-diisopropylethylamine (DIPEA, Sigma-Aldrich, 99.5%), N,N-dimethylformamide (DMF, VWR chemicals, 99.9%), tetrahydrofuran (Acros organics, 99.6%), diethyl ether (VWR international, 100%), acetone (VWR international, 99%), acetonitrile (VWR international, 99.95%), formic acid (Thermo Fisher Scientific, 99%), methanol (VWR international, 100%), hydrogen peroxide (Acros organics, 35 wt% solution in water), toluene (VWR international, 100%), trifluoroacetic acid (Sigma-Aldrich, 99%), acetic acid (Sigma-Aldrich, 99.8%), phosphate buffer saline 10× (PBS 10×, Euromedex) were used without further purification. Ultrapure water (18 MΩ cm) was obtained by passing in-house deionized water through a Millipore Milli-Q Biocel A10 purification unit. Bacto Tryptone (Sigma), yeast extract (Sigma), ampicillin (Sigma) and isopropyl β-D-thiogalactopyranoside (IPTG, VWR chemicals) were used for cell culture.Production and purification of ELPs
The amino acid sequences of the ELPs used are MW[(VPGVG)(VPGMG)(VPGVG)2]5,10,20. To simplify they will be termed M20, M40 and M80, respectively. Production and purification of these ELPs were performed as already described.25,26 Briefly, a single E. coli colony was cultured overnight at 37 °C in a rotary shaker at 200 rpm in 50 mL of lysogeny broth (LB) medium (1% Bacto Tryptone, 1% yeast extract, 0.5% NaCl) containing 100 μg per mL ampicillin. Thereafter, this seed culture was inoculated into 0.95 L of LB medium supplemented with glucose (1 g L−1) and ampicillin (100 μg mL−1), and cells were cultivated at 37 °C. When the optical density at 600 nm (OD600) reached a value close to 0.8, isopropyl β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM, and the temperature of the incubator was decreased to 25 °C. After 12 h the culture was harvested by centrifugation at 6000g and 4 °C for 15 min, and the cell pellet was suspended with 10 mL per g wet weight in phosphate buffer saline (PBS) buffer. Thereafter, cells were lyzed by sonication and insoluble debris were removed by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g and 4 °C for 30 min. The cleared lysate was subjected to three successive cycles of Inverse Transition Cycling (ITC).27 Briefly, ELP was precipitated at 30 °C for M40 and M80, at 40 °C for M20, and then centrifuged at 10000g and 35 °C for 30 min (“warm spin”). The ELP-containing pellets were dissolved in cold water and the insoluble proteins were eliminated by centrifugation for 15 min at 10000g and 4 °C (“cold spin”). Thanks to the incorporation of a tryptophan residue at the N-terminal end of the ELP sequences, the protein content of the purified solutions was measured by spectrophotometry at 280 nm with a NanoDrop 1000 (ThermoScientific). Finally, purified ELPs were dialyzed against ultrapure water at 4 °C (Spectra Por7, MWCO1000, Spectrum Laboratories), and then lyophilized.
000g and 4 °C for 30 min. The cleared lysate was subjected to three successive cycles of Inverse Transition Cycling (ITC).27 Briefly, ELP was precipitated at 30 °C for M40 and M80, at 40 °C for M20, and then centrifuged at 10000g and 35 °C for 30 min (“warm spin”). The ELP-containing pellets were dissolved in cold water and the insoluble proteins were eliminated by centrifugation for 15 min at 10000g and 4 °C (“cold spin”). Thanks to the incorporation of a tryptophan residue at the N-terminal end of the ELP sequences, the protein content of the purified solutions was measured by spectrophotometry at 280 nm with a NanoDrop 1000 (ThermoScientific). Finally, purified ELPs were dialyzed against ultrapure water at 4 °C (Spectra Por7, MWCO1000, Spectrum Laboratories), and then lyophilized.
In order to increase Tcp, oxidation of M80 was performed as previously described.25 The ELP was dissolved in 30% H2O2 and 1% AcOH in water, and stirred at 0 °C for 30 min. After quenching with a few drops of 1 M sodium thiosulfate aqueous solution, the reaction mixture was transferred to a 1000 g per mol MWCO dialysis tubing (Spectra/Por®) and dialyzed against ultrapure water for 48 h with water changes twice per day. The content of the dialysis bag was then lyophilized to yield oxidized M80 (MO80, 95% yield). 1H NMR (400 MHz, D2O): δ 4.4 ppm (160 H, αCH Val + αCH Pro), δ 4.17 ppm (t, 60 H, αCH Val guest residue, as reference peak), δ 3.72–3.97 ppm (br m, 478 H, 160 H δCH2 Pro + 320 H αCH2 Gly), δ 2.96 ppm (47 H, γCH2 Met), δ 2.73 (65 H, εCH3 Met), δ 1.98–2.32 (515 H = 140 H βCH Val + 42 H βCH2 Met + 160 H βCH2 Pro + 160 H γCH2 Pro), δ 0.9–1.05 ppm (br m, 837 H, γCH3 Val).
The yields of purified ELP given in mg L−1 of culture were 29 ± 8 (n = 3) for M20, 83 ± 15 (n = 17) for M40 and 116 ± 22 (n = 5) for M80.
Synthesis of ELP–fatty acids
The first step was the activation of the carboxylic acid of the fatty acids. Myristic, palmitic, stearic, oleic and linoleic acid solutions prepared at 3 mM in anhydrous DMF were stirred for 1 h at RT. The activation of carboxylic acids was achieved by adding 7.4 mg (18 μmol) of HCTU and 4.6 μL (26 μmol) of DIPEA to the FA solution (18 μmol), and performing the reaction for 15 min in the dark. The second step was the conjugation of the activated fatty acid with the primary amine of the ELP. There is only one primary amine per polypeptide, located at the N-terminal methionine. For conjugation, 1.5 μmol of activated fatty acid was reacted with 1 μmol of ELP for 12 h in the dark. Then, the reaction product was precipitated by 10 volumes of acetone, collected by centrifugation for 5 min at 2500g and 20 °C, and then re-suspended in DMF. After a second precipitation step using 10 volumes of diethyl ether, the pellets were recovered by centrifugation and washed three times with 10 volumes of diethyl ether. After drying for 2 h at room temperature, the product was dissolved in cold water, and centrifuged at 2500g for 10 min at 4 °C to remove insoluble impurities. The clear supernatant was dialyzed (1000 g mol−1, Spectra/Por® membrane) against ultrapure water for 2 days at 4 °C. The product was then lyophilized and stored at −20 °C.Methods
Reversed-phase high performance liquid chromatography (RP-HPLC) was performed using an Ultimate 3000 system (Thermo Scientific) instrument equipped with a hydrophobic C18 stationary phase (ZORBAX Eclips Plus C18, 4.6 × 250 mm, 5 μm, Agilent Technologies), and a gradient of solvent A (H2O + 0.1% TFA)/solvent B (methanol + 0.1% TFA). The flow rate was 1 mL min−1. 35 μL of ELP or ELP–FA samples (70 μM) were injected, and the absorbance at 230 nm was recorded with a diode array detector (Thermo Scientific). The gradient program used is described in Table S1.† Data obtained by RP-HPLC were used to calculate the percentage of purity of the ELP–FA conjugates. This was achieved by integrating the area below the peak of the ELP–FA, and by subtracting the area under the peak, if any, of the unmodified ELP.Mass spectrometry analysis (ESI-MS) were performed on an ESI-QTOF (Q-TOF Premier, Waters, Manchester, UK). Lyophilized compounds were dissolved in acetonitrile/0.1% aqueous formic acid (1:1 v/v) to a final concentration of around 30 μM, and infused into the electrospray ionization source at a flow rate of 5 μL min−1. The mass spectrometer was operated in the positive mode with external calibration performed with a solution of the standard protein at a concentration of 1 μM in a mixture of acetonitrile and 0.1% aqueous formic acid (1:1 v/v). The ESI mass spectra showed the characteristic charge state distribution corresponding to the desired ELPs and their derivatives. Data were then processed with the maximum entropy deconvolution system MaxEnt (MaxEnt Software, Waters) to determine the MWs.
Cloud point temperature (Tcp) was determined by measuring the turbidity at 350 nm between 10 and 50 °C at a 1 °C min−1 scan rate. Data were collected with a Cary 100 UV-vis spectrophotometer equipped with a multicell thermoelectric temperature controller from Agilent Technologies. The Tcp was defined as the maximum of the first derivative of absorbance versus temperature.
Circular dichroism spectra were recorded on a Jasco J-1500 equipped with a Peltier temperature control accessory (JASCO, Hachioji, Japan). Each spectrum was obtained by averaging two scans collected at 50 nm min−1. The CD spectrum of pure water solution was subtracted from the average scan for each sample.
The critical micelle concentration (CMC) of ELP–FAs in PBS was determined as previously described.28,29 Briefly, solutions of ELP–FAs in PBS with mass concentrations varying from 10−6 mg mL−1 to 2 × 10−1 mg mL−1 were prepared by simple dispersion of the material in the buffer. Then, 1 μL of pyrene solution at 5 mM in THF was added into 500 μL of particle dispersion prepared at various concentrations, and incubated under stirring at 300 rpm for 10 h at 4 °C. The dispersions were characterized by fluorescence spectroscopy using a Jasco FP 8500 spectrofluorometer. The excitation wavelength was set at 319 nm, and the fluorescence emission of pyrene was recorded between 360 and 400 nm. Bandwidths of 5 nm and 2.5 nm were used for excitation and emission, respectively. The intensities of the first (I1) and third (I3) vibrionic band of pyrene were determined at 373 nm and 385 nm respectively. The I1/I3 ratio was plotted as a function of the ELP–FA concentration.
Dynamic light scattering (DLS) analysis of the ELP–FAs micelles was performed using a Nano ZS instrument (Malvern U.K.) working at a 90° angle detection. All samples were filtered before measurement through 0.22 μm PVDF filtration devices (Millex, Merck Millipore). The hydrodynamic radius (RH) and the polydispersity index (PDI) were derived from the autocorrelation functions using the cumulant method. The zeta potential was measured with the same apparatus using the M3-PALS technique.
The morphology of ELP–FA micelles was examined by transmission electron microscopy (TEM) after negative staining of the samples, so the particles appeared white on a dark background. Samples were dissolved in Tris buffer instead of PBS, the high salt concentration of the latter being not compatible with TEM experiments. 20 μL of a solution of ELP–fatty acid at 5 mg mL−1 in Tris buffer was first dropped on the carbon grid. After incubation for 5 min, the excess liquid was removed and the grid was dried at room temperature. 20 μL of a 2% samarium acetate solution (filtered with 0.45 μm CA device) were deposited on the grid. After 5 min at 15 °C the excess liquid was removed and the grid was air-dried. TEM analyses were performed at 80 kV acceleration voltage with a Hitachi H7650 microscope equipped with an Orius camera (Gatan, Paris, France). Pictures were acquired with the digital micrograph software.
Drug encapsulation was assessed using Nile red as a hydrophobic molecule model. 6 mg of Nile red were dissolved in 6 mL of DMSO to obtain a concentration of Nile red of 3 mM. Then, 2 μL of this solution were injected into 600 μL of a 1 mg mL−1 solution of M80–FA in PBS. The final concentration of the Nile red in the M80–FA solution was then 10 μM. This mixture was incubated for 24 hours at 10 °C with stirring, and then centrifuged at 18500g for 10 min at 4 °C to remove unencapsulated Nile red which aggregates due to its low solubility in aqueous solvents. The supernatant containing the nanoparticles was freeze-dried, and the powder was then dispersed in DMSO for fluorescence analysis (Jasco FP 8500). The excitation wavelength was set at 550 nm and the fluorescence emission at 626 nm. Bandwidths were set at 20 nm and 5 nm for excitation and emission, respectively. Each point was measured in triplicate. A calibration curve was performed in DMSO (Fig. S1†). The encapsulation efficiency (EE%) was defined as the concentration of Nile red incorporated into the micelles to its initial concentration.
For drug release experiments, 6 μL of a Nile red solution in DMSO at 1 mg mL−1 were injected in 1.8 mL of a M80–FA solutions at 1 mg mL−1 in PBS. The solution was incubated for 24 h at 10 °C under stirring at 300 rpm, and then centrifuged at 18500g for 10 min at 4 °C to remove unencapsulated Nile red. After measuring the initial fluorescence value between 605 and 615 nm (λex = 570 nm), 600 μL of solution were transferred into a Spectra-Por® Float-A-Lyzer® device (molar mass cut-off 1000 g mol−1), and dialyzed at 10 °C against 1 L of PBS. The fluorescence of the dialyzed sample was measured at different time points to determine the relative amount of Nile red released. To model the release kinetics of Nile red from ELP–FA micelles, the simple empirical equation developed by Korsmeyer–Peppas30 for the drug release from spherical polymeric particles was used. The Nile red-release profile kinetics below 60% of release was fitted with the equation Qt/Q∞ = ktn where Qt is the cumulative drug release at time t, Q∞ is the maximum of drug release at infinite, k is a constant describing characteristics of the polymer matrix and drug, and n is the diffusional exponent characteristic of the release mechanism.
Results and discussion
Synthesis and purification of ELP–FA conjugates
Our goal was to obtain a library of 15 ELP–FAs, differing in the length of the ELP block and the nature of the FA. The amino acid sequences of the ELP are MW[(VPGVG)(VPGMG)(VPGVG)2]5,10,20, and therefore they differ by their number of pentapeptide repetitions, from 20 to 80. Methionine and valine were encoded at a 1:3 ratio, respectively, as guest residues in the VPGXG repeat units. The presence of methionine residues allows oxidation reactions to modulate the cloud point temperature (Tcp), while valine residues were used as nonreactive hydrophobic residues to dilute the methionine content. We selected different FAs among those abundant in membranes phospholipids.31 Myristic (C14:0), palmitic (C16:0) and stearic acids (C18:0) were considered to assess the influence of the hydrophobic block length on the self-assembly properties, while oleic (C18:1) and linoleic (C18:2) acids are expected to provide insight into the effect of the unsaturation degree.
Main physico-chemical characteristics of the FAs used in this study are reported in Table S2.† The targeted ELP–FAs conjugates are listed in Table S3.† ELPs were designed in such a way that a single primary amine is present in the whole polypeptide sequence, namely at the N-terminal methionine (Fig. S2†). Then, the condensation of the carboxylic group of FA with this amine group is a simple way to afford ELP–FA conjugates (Scheme 1A). The condensation reaction was performed by using O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU) as activating agent, in presence of N,N-diisopropylethylamine (DIPEA). HCTU is a nontoxic, nonirritating and noncorrosive activating reagent that is typically used for solid-phase peptide synthesis in DMF.32 DIPEA is a sterically hindered organic base commonly used in amide coupling reactions between an activated carboxylic acid and a nucleophilic amine.33 The condensing reactions were performed overnight at room temperature. ELP–FA conjugates were recovered by precipitation in a non-solvent, and purified by repetitive centrifugation/washing cycles. Typical yields were >92% whatever the ELPs and FAs used. As an example of ELP–FA conjugates, the chemical structure of M80–C16 is shown in Scheme 1B.
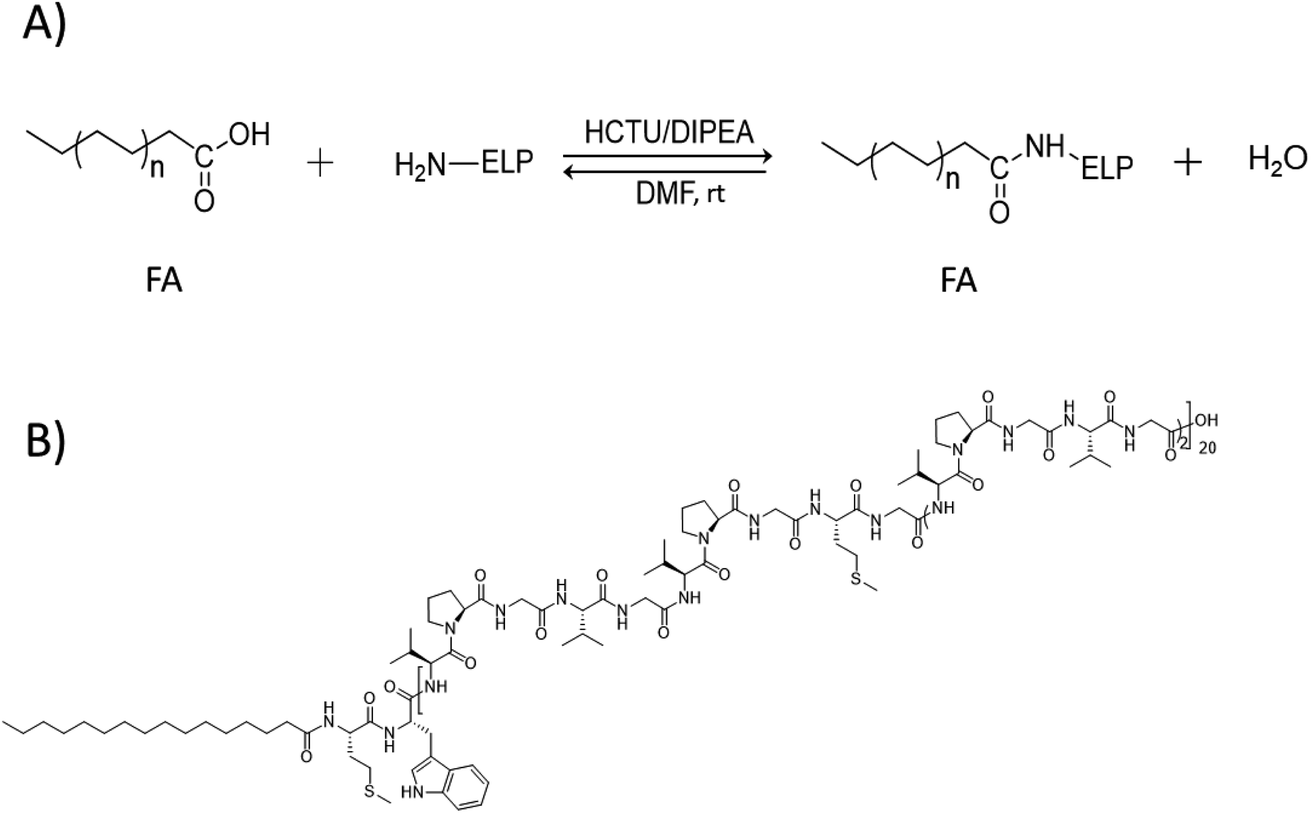 | ||
| Scheme 1 (A) Condensation reaction between FA and ELP. (B) Example of ELP–FA conjugate structure, here the M80–C16 conjugate. | ||
Characterization of ELP–FAs
The purity of ELP–FA conjugates was assessed by RP-HPLC. The chromatograms of five M40–FAs are shown in Fig. 1A (Fig. S3† for M20–FAs and M80–FAs). The retention times of all 15 conjugates are given in Table S4.†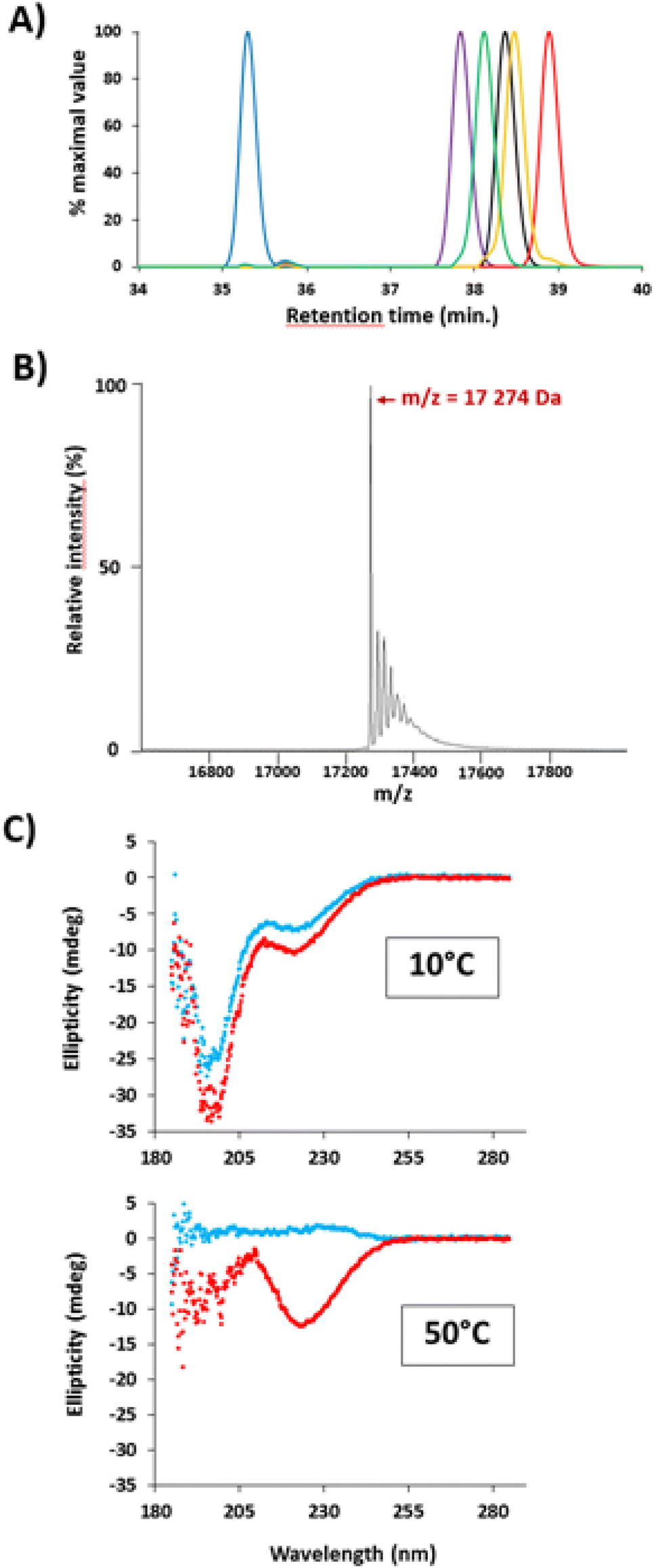 | ||
| Fig. 1 Characterization of ELP–FAs. (A) RP-HPLC traces for M40 (blue), M40–C14 (purple), M40–C16 (black), M40–C18 (red), M40–C18:1 (orange), and M40–C18:2 (green). (B) ESI-MS spectrum of M40–C16. (C) CD spectra in water of 10 μM solution of M40 (blue) and M40–C16 (red) at 10 °C and 50 °C. | ||
RP-HPLC analysis showed that the purification step was efficient, as judged by the absence of significant amounts of unmodified ELPs (purity > 96%, Table S4†). Retention times vary inversely with the ELP chain length: +4.2 min between M20 and M20–C18; +3.8 min between M40 and M40–C18; +2.6 min between M80 and M80–C18 (Table S4†). Thus, the impact of the acylation in terms of hydrophobic/hydrophilic ratios on the retention times was more pronounced for the shorter ELP M20 than for the larger M80. Retention times also increased systematically as the chain length increased from 14 to 18 carbons, with the longest retention times always measured with ELP–C18 conjugates. However, the presence of unsaturation in the C18 chain reduced the elution times due to a difference in hydrophobicity.34 A linear relationship between the retention times of ELP–FAs and the logP value of the FAs could be evidenced (Fig. S4†).
Successful acylation of ELPs was further verified by electrospray ionization-mass spectrometry (ESI-MS). A typical mass spectrum of M40–C16 is shown in Fig. 1B and the results obtained for the 15 ELP–FAs are summarized in Table S5.† The experimental mass values were in good agreement with the theoretical ones, with shifts of 210, 238, 266, 264, and 262 g mol−1 after acylation of ELP by C14, C16, C18, C18:1 and C18:2, respectively.
We then investigated if the secondary structure of ELPs was affected by the acylation with FAs. Aqueous solutions of M40 and M40–C16 (Fig. 1C) and M80 and M80–C16 (Fig. S5†) were analyzed by CD at 10 °C, that is, below the cloud point temperature (Tcp) of M40 and M80, and also at 50 °C, i.e. above Tcp. The spectra were then analyzed and the secondary structure determined using the BeStSel software.35,36 The spectra obtained for M40 and M80 at 10 °C were similar to those previously reported for ELPs below their Tcp, with a large negative peak around 190–200 nm, which is characteristic of random-coiled structures and a smaller negative peak around 220 nm.13,37,38 For these two ELPs, the prediction of secondary structures indicates a majority of antiparallel β-sheets (50–70%), and a smaller proportion of α-helix (12–17%). When heated above Tcp, there is a dramatic change in the secondary structure with the disappearance of the two negative peaks at 190–200 nm and 220 nm. Such a change can be ascribed to the disappearance of α-helix, and a moderate diminution of antiparallel β-sheets (−20% of total structures). In addition, the BeStSEl software predicts the apparition of β-turns in the structure, which is more important in M80 than in M40 because of the larger size of the polypeptide. The CD spectra recorded at low temperature for M40–C16 and M80–C16 conjugates are very similar to those recorded for unmodified ELPs, suggesting that ELP acylation did not lead to a profound reshuffle of the secondary structure of the polypeptides (Fig. 1C and S5†). A similar result was previously obtained for myristoylated ELPs, with no change in the secondary structure at low temperature compared to non-myristoylated ELPs.22 For M40–C16 and M80–C16, around 71–77% of the secondary structure was based on antiparallel β-sheets, and 21–23% on α-helix. After heating at 50 °C, the spectra dramatically changed with a large decrease in antiparallel β-sheets (around – 45% of total structures) concomitant with the apparition of β-turns. The observed changes in secondary structure after heating of ELP–FAs are therefore similar to those observed for unmodified ELPs, confirming that the ELP conformation is globally not altered by acylation. The only difference is that α-helix disappeared from the ELP structure upon heating, whereas they are conserved (21–26%) in ELP–FAs. This is in striking contrast to what we previously observed with polyisoprene-block-ELP copolymers, the secondary structure of the diblock at T < Tcp being similar to that of the unmodified ELP heated above the Tcp.21
We next performed turbidimetry experiments at different temperatures (T-scans) to measure the Tcps of the 15 ELP–FAs in PBS at four different concentrations. The absorbance of ELP–FA solutions at 350 nm was monitored when the temperature was ramped up and then down at a rate of 1 °C min−1 (Fig. 2A and S6†). The Tcp values determined during the temperature rise (Table S6†) were then plotted versus the ELP concentration in a semi-log scale (Fig. 2B). These experiments were performed in PBS because it is similar to human body fluids in terms of ionic strength, osmolarity, and pH. When the ELP–FA solutions are cooled, the optical density (OD) at 350 nm returns to its initial value with a temperature hysteresis of about 3–6 °C independent of FA (Fig. 2A and S6†). The origin of the hysteresis observed in T-scans of ELPs has been discussed elsewhere.39
 | ||
| Fig. 2 Thermoresponsivity of ELPs and ELP–FAs in PBS. (A) T-scans of M20–C18 prepared at different concentrations in PBS (plain lines for heating ramps, dashed lines for cooling ramps). Sample concentrations were 100 μM (green), 50 μM (blue), 25 μM (red) and 10 μM (black). (B) Cloud point temperatures of the three ELPs and the 15 ELP–FA conjugates as function of the ELP size given in DP (degree of polymerization) and the concentration. | ||
Data plotted in Fig. 2B first illustrates the inverse dependence of Tcp on the size of native ELPs, i.e. longer ELPs precipitate at lower temperatures. This is basically due to the decrease of the critical value of the Flory–Huggins interaction parameter (χc) with the ELP size.40 Fig. 2B also points out that ELP–FAs have Tcps 5–30 °C lower than the Tcp of the parent ELP, in agreement with the role of ELP hydrophobicity on the solution temperature behaviour.17 However, the length and unsaturation degree of FA have little effect on Tcp, probably because the overall hydrophilic–hydrophobic balance of ELP–FAs is not too dependent on the FA. Therefore, if a specific Tcp is targeted for a biological application the size and/or hydrophobicity of the ELP should be considered prior to the type of FA. In an attempt to increase the Tcp of the conjugates in PBS, we oxidized M80 to replace thioethers groups of methionine residues by sulfoxide groups as we have previously shown that this can significantly increase the Tcp value.25,41 Characterizations of the oxidized M80 (MO80) are shown in Fig. S7.† After palmitoylation of MO80 the Tcp was significantly higher (+18 °C) than that measured for M80–C16 (Fig. S8†), confirming that methionine oxidation is also a means to adjust Tcp of ELP–FAs.
An important finding was that the acylation of ELPs strongly decreased the magnitude of Tcp variation with ELP–FA concentration in comparison to native ELPs (Fig. 2B). This can be understood by considering that ELP–FAs have self-assembly properties i.e. they form micellar structures in solution at T < Tcp. Chilkoti's group previously showed that the conjugation of small hydrophobic molecules with logP > 1.5 to a hydrophilic ELP confers self-assembly behavior to ELPs, and that Tcps values were then almost independent of the conjugate concentration. They hypothesized that the aggregation was controlled by the local ELP concentration within the self-assembled structures and not by the total concentration of the conjugate.42
Solution self-assembly of ELP–FA
The results obtained from turbidimetry analyses suggest that ELP–FAs can self-assemble below the critical temperature, which is consistent with previous studies showing the self-assembly of ELP–lipids into micelles.17,22 We therefore investigated the self-assembly behavior of ELP–FAs. First, the critical micelle concentration (CMC), a characteristic of each ELP conjugate was determined using pyrene as a fluorescent hydrophobic probe.28,29,43 CMC measurements were performed in PBS to mimic biological fluids, and at 15 °C to limit dehydration of the ELP moiety and its subsequent phase separation. Importantly, ELP–FAs could be directly self-assembled in PBS without the need for co-solvents (DMSO, DMF) which are typically used with amphiphilic block copolymers. Thus, the activation energy of the ELP–FA self-assembly must be low and the structures formed are expected to be at equilibrium.Fig. 3A shows the variation of the I1/I3 ratio obtained for M80–C16, where I1 and I3 correspond to the intensity of the first and third vibronic peaks of pyrene.44,45 The graphs for the other 14 compounds are shown in Fig. S9.†
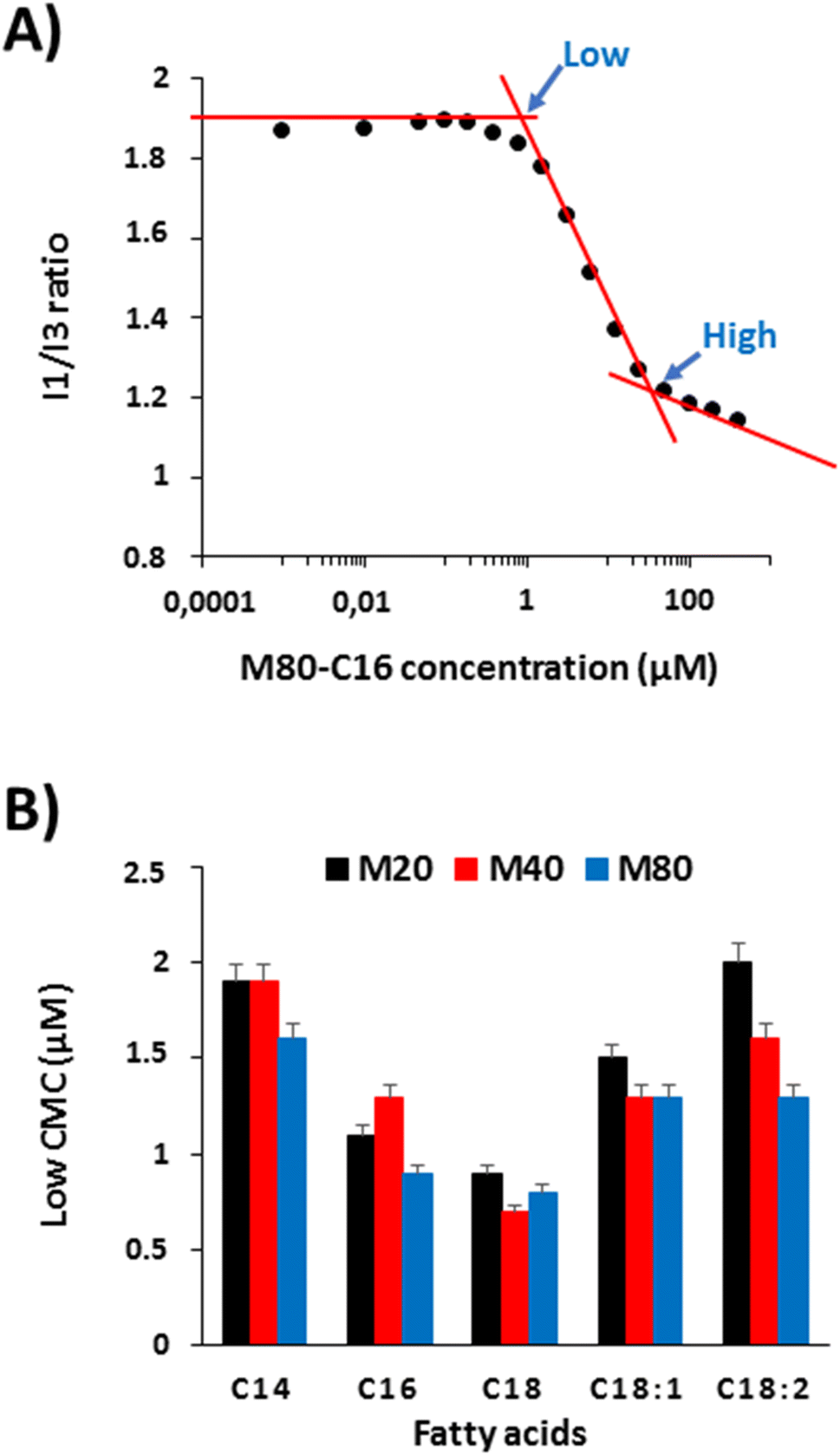 | ||
| Fig. 3 CMC of ELP–FAs. (A) Variation of the I1/I3 ratio for M80–C16 as a function of the ELP–FA concentrations in PBS at 15 °C. Two self-assembly regimes are depicted as low and high CMC. (B) Low CMC values for M20–FAs, M40–FAs, and M80–FAs. Data are mean value + SD (n = 3). | ||
The sigmoidal aspect of the I1/I3 plot is typical of surfactants or amphiphilic block copolymers.43 However, it is remarkable that the sigmoid almost spans two decades of concentration to complete the self-assembly process, from premicellar aggregates to micellar structures. This can be ascribed not only to the large molecular weights of the ELP–FAs but also to the moderate hydrophilicity of the ELP blocks. For the determination of the CMC, some authors have used the point marked as “high” in Fig. 3A, which corresponds to the interception of the second and third linear part of the plot.46,47 The high CMC value represent the concentration at which any additional monomers will be incorporated into micelles. On the contrary, the so-called “low” CMC obtained from the interception of the first and second parts of the plot, corresponds to the formation of premicellar aggregates. The latter is considered more suitable for surfactants with a low CMC, below 1 mM.48 We measured the two CMC values for the 15 ELP–FAs studied (Table S7†). Low CMC values varied between 0.7 and 2 μM, and high CMC values between 11 and 49 μM. These values are quite low compared to molecular surfactants for which CMC are typically in the mM range,49 thereby confirming that ELP–FA are rather hydrophobic compounds.
To better understand the effect of ELP size and fatty acid nature, we plotted the low CMC values of the 15 compounds (Fig. 3B). As expected, the CMC decreases with increasing the length of the saturated fatty acid chain due to increased hydrophobicity. The lowest values were always measured with C18-containing conjugates regardless of the size of the ELP moiety. The presence of unsaturation in the C18 chain increases the CMC as the hydrophobicity decreases when the number of unsaturated sites increases. Regarding the influence of the ELP size for a given FA, the lowest CMC was generally obtained for ELP M80, and the highest for ELP M20. However, the differences in CMC values between two ELPs acylated by the same FA were much smaller than those between ELP acylated by different FAs. Thus, the driving force for ELP–FA self-assembly at 15 °C, i.e. below the Tcp, is mainly controlled by the hydrophobicity of the FA. Finally, the “low” CMC values measured for the 15 ELP–FAs are less than 3 μM, i.e., well below the 10–100 μM concentrations used to determine Tcp by turbidimetry. This means that the cloud point determined at Tcp does not fully correspond to the transition of ELP–FA unimers to aggregates, but rather to the secondary aggregation of premicellar structures already formed at low temperature.
In order to investigate this hypothesis, the suspensions of ELP–FAs were analyzed by DLS in PBS at 15 °C. The concentration was set to 5 mg mL−1 (i.e., 150–580 μM) to have sufficient scattering intensity and to be well above the “high” CMC (Table S7†). A typical result obtained for M80–C16 in PBS at 15 °C is shown in Fig. 4A.
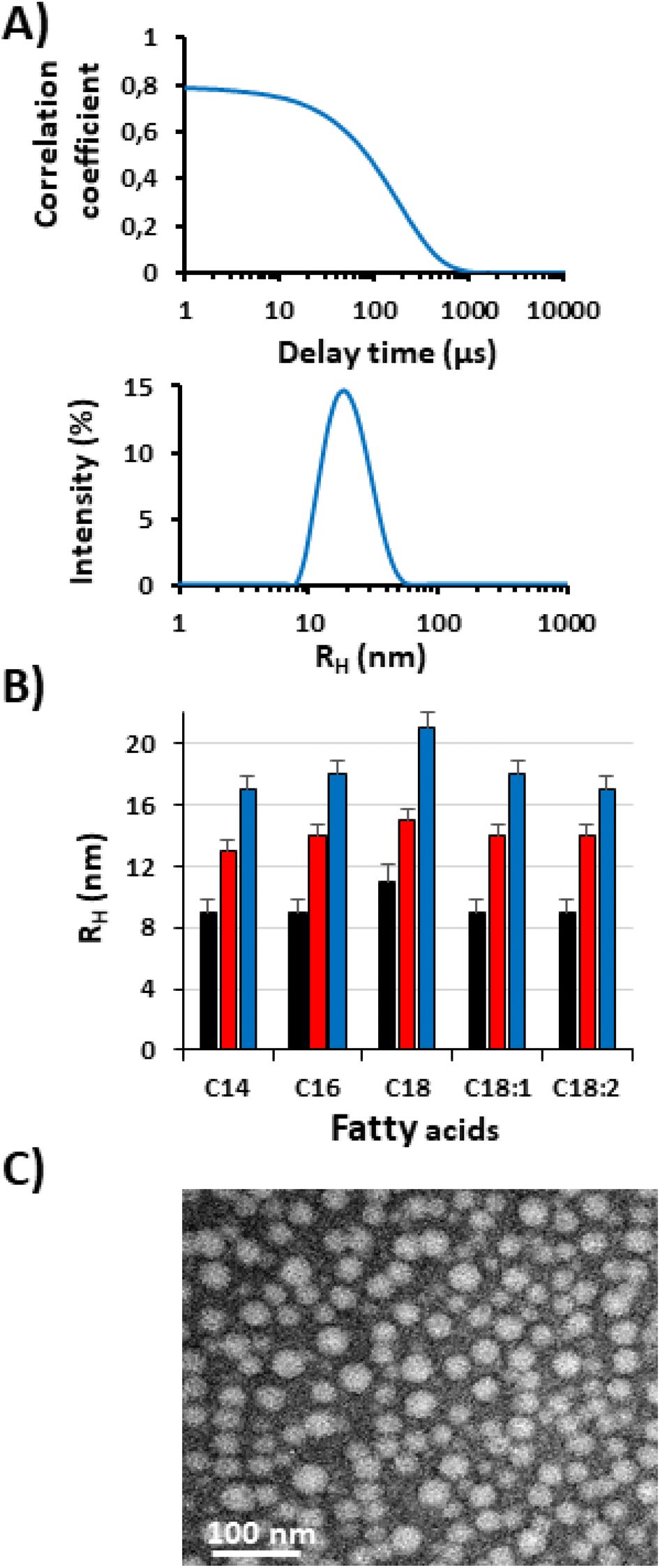 | ||
| Fig. 4 Self-assembly of ELP–FAs. (A) DLS autocorrelation function at 15 °C and intensity-weighted distribution of the hydrodynamic radius (RH) of the M80–C16 suspension in PBS at 5 mg mL−1 (150 μM). (B) RH values (% intensity) of the 15 ELP–FAs measured at 15 °C (5 mg mL−1 in PBS). Data are mean value + SD (n = 3). (C) TEM image of M40–C16 micelles with negative staining. | ||
At 15 °C, M80–C16 formed nanoparticles with a hydrodynamic radius (RH) of about 18 nm, which is characteristic of micellar structures. The RH and PDI values of the 15 conjugates are shown in Table S8.† The PDI was consistently less than 0.2 and in most cases less than 0.1, emphasizing the absence of large aggregates in solution.
TEM imaging shows that micelles have a spherical morphology with sizes consistent with DLS analysis (Fig. 4C). Most likely, the FA segments form the core of the micelles and the ELP chains the corona.
Fig. 4B shows that micelle size is largely determined by the ELP block, which is not surprising given the respective size of ELP and FA blocks (Table S3†). For a given FA the RH of the M80–FA is always the largest, and the RH of the M20–FA the smallest. The FA has a less pronounced effect, with a slight increase in micelle radius from C14 to C16 and C18. The presence of an unsaturation has an opposite effect, the kink due to the cis-double bond limits the FA stretching in the micelle core, thus contributing to decrease the overall micelle size, as previously shown for oleic and stearic acid-based imidazolium surfactants.50 Our results are in sharp contrast with those previously published by Luginbuhl et al. for myristoylated ELPs of 40, 80 and 120 repeats of VPGXG where X is 90% Ala and 10% Val (90A).17 The authors showed that the RH of the self-assembled nanoparticles was inversely proportional to the length of the ELP. This was explained by the observation that the short ELP–C14 formed rod-shaped micelles, whereas the longer ELP–C14 formed spherical micelles. We cannot offer a definitive explanation for these opposing results, but some differences in the design of the ELPs between the two studies should be pointed out. In the work of Luginbuhl et al., the N-terminal sequence of each ELP contains the NMT recognition sequence, which corresponds to the peptide Gly-Leu-Tyr-Ala-Ser-Lys-Leu-Phe Ser-Asn-Leu. This peptide is rather hydrophilic and contains one positive charge on the lysine residue at physiological pH. In addition, the overall hydrophobicity of the sequence VPGAG used to build the ELP90A series (GRAVY = 0.72) is lower than that of our ELPs made of repeats of VPGVG and VPGMG (GRAVY = 1.085). Altogether, we can hypothesize that the two series of ELP–FAs have different hydrophilic/hydrophobic ratios resulting in specific self-assembly properties.
Modeling of ELP–FA micelles was attempted using a star model developed by Halperin for polymer micelles consisting of a small insoluble core and an extended soluble corona,51 which is justified here by the large difference in block size (Table S3†). The micelle formation is driven by the tendency of the system to lower its interfacial energy but more specifically the micellar growth is controlled by the increasing confinement free energy of chains in the swollen corona.51 It was shown that the overall radius of star-like micelles scales as NB4/25NA3/5 with NB and NA the polymerization degree of the insoluble (FA) and soluble (ELP) blocks (NA ≫ NB). As ELPs cannot be fully approximated to flexible polymer chains in good solvents like PEG in water, the experimental micelle sizes obtained by DLS were fitted to NB4/25NAν with ν an adjusting parameter relative to the chain conformation of the ELP block in the micelle corona. The difference of unit length (a) in the ELP and FA was considered (aELP = 0.365 nm,52 aFA = 0.25 nm) to normalize NB in peptide units. A good linear adjustment was found for the C14–C16–C18 and C18–C18:1–C18:2 series of ELP–FA with respective ν values of 0.47 and 0.45 (Fig. S10†). This is in good agreement with the results of Fluegel et al.52 who studied by DLS at 20 °C the conformation of ELP based on (Val-Pro-Gly-Val-Gly)n repeats with n varying from n = 20 up to n = 120. They found that the hydrodynamic radius scales as N0.43 in 20 mM NaCl with N the number of peptides repeat units (data plotted from Table 1 in ref. 52). The ELP concentration was similar to that used in our study (5 mg mL−1). The value of ν they found is slightly lower than that obtained here from the micelle radius fitting. This could mean that ELP chains are better hydrated in the micelles than in their individual state in solution, possibly due to their stretching in the corona. Nonetheless, both ν values are below 0.5 (theta conditions) meaning that ELPs in water are not in good solvent conditions according to Flory's theory.
The stability of the ELP–FAs micelles upon storage in PBS at 4 °C was also evaluated (Fig. S11†). Micelles made of M80–C16 were stable under these conditions, as shown by the constant RH and PDI values obtained during 40 days. The acylation of the ELP takes place at its N-terminus, the C-terminus is therefore exposed to the solvent. Since the pKa of the acid groups is lower than 4.0,53 negative charges must be present at the surface of the micelles in PBS buffer (pH 7.2), thus preventing aggregation by electrostatic repulsion. This was confirmed by zeta potential measurements, a potential of −15.4 mV was determined for M80–C16 micelles in water (Fig. S12†).
The effect of temperature on the micelle size of the M80–FAs was also evaluated by DLS at a concentration of 1 mg mL−1 (∼30 μM) to limit the aggregation near the cloud point (Fig. S13†). Below the Tcp, there is a single population of M80–FA micelles with RH around 20 nm, which when heated above Tcp quickly increased in size to form micron-sized objects, thus indicating that micelles aggregated upon dehydration of their ELP coronas. The Tcps determined by DLS were close to those measured by turbidimetry at 50 μM (±1 °C difference).
Encapsulation and release of a hydrophobic molecule
We next evaluated the potential of ELP–FA micelles to encapsulate and release a hydrophobic molecule. Our interest being focused on the role of the fatty acyl chain length on the encapsulation properties of the micelles, we used M80–FAs because they form larger micelles than the M40–FAs and M20–FAs (Fig. 4B, Table S8†). As a model of hydrophobic molecule, we chose Nile red (logP = 2.8), a well-known dye model for performing encapsulation and release experiments.
The encapsulation efficiency was first evaluated. It was estimated at 20.4%, 31.2% and 43.8% for M80–C14, M80–C16 and M80–C18, respectively. This showed that fatty acid chain length is an important factor for the encapsulation of Nile red in the M80–FA micelles. This is primarily due to differences in micelle size which increases with the fatty acid length (Table S8†). The encapsulation efficiencies of Nile red by M80–FA micelles (20–40%) were higher than that previously measured for the encapsulation of doxorubicin by a myristoylated ELP (3–5%).17 However, these figures cannot be directly compared because the hydrophobicity of the encapsulated molecules is quite different, the logP values being 2.8 and 1.3 for Nile red and doxorubicin, respectively. Thus, because of its higher hydrophobicity, Nile red has a better avidity for the hydrophobic core of ELP–FA micelles.
We next measured the release of Nile red from the ELP–FA micelles in sink conditions (Fig. 5). Drug release experiments were systematically performed below Tcp to avoid phase separation of ELF–FAs conjugates. After encapsulation of Nile red and removal of unencapsulated dye by centrifugation, the micelles were placed into dialysis devices and incubated for several hours in PBS buffer at 10 °C. The fluorescence intensity of the dialyzed samples was measured at different times to calculate the amount of Nile red released (Fig. 5A). We previously verified that Nile red does not fluoresce if it is not encapsulated in the micelles (Fig. S14†).
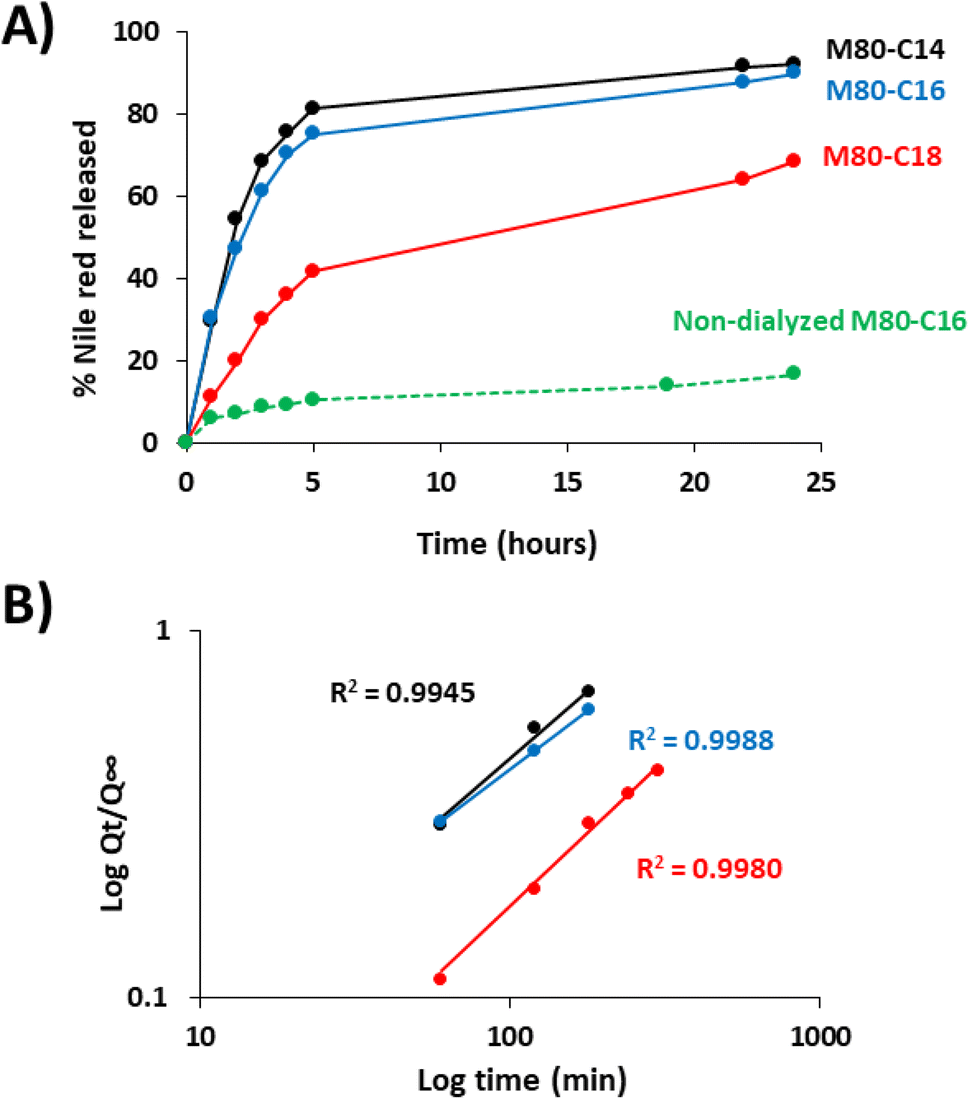 | ||
| Fig. 5 Release kinetics of Nile red from M80–FA micelles. (A) ELP–FA micelles loaded with Nile red were dialyzed against PBS at 10 °C. As a control, the fluorescence of a non-dialyzed suspension of Nile red-loaded M80–C16 was also monitored. Data are mean values of three measurements. (B) Fit of the experimental data with Korsmeyer–Peppas equation (Qt/Q∞ = ktn). M80–C14 (black), M80–C16 (blue) M80–C18 (red). | ||
The fastest kinetics were observed for the M80–C14 micelles, with 80% of the Nile red content released within the first five hours. Then, the release occurred at a slower rate with 90% of Nile red released after 24 h of dialysis. Similar results were obtained for M80–C16 micelles. In the case of M80–C18, only 40% of the encapsulated Nile red was released after 5 hours of dialysis. After 24 h, no plateau was reached and around 30% of the initial Nile red content was still detected in these micelles, in agreement with the stronger affinity of the dye with the more hydrophobic core.
Drug release from pharmaceutical forms has been generally described by kinetic models involving the released amount of drug (Q) as a function of the time (t). Some equations of the Q(t) profiles are commonly used as fitting for modelization, such as zero order, first order, Hixson–Crowell, Higuchi, or Korsmeyer–Peppas models.54 To modelize the release kinetic of Nile red from ELP–FA micelles, we tested these models and found that the simple empirical equation developed by Korsmeyer–Peppas for determining the release profile of a drug from spherical polymeric particles,30 Qt/Q∞ = ktn gave the best results with R2 above 0.99 (Fig. 5B). In this equation Qt is the cumulative drug release at time t, Q∞ is the maximum of drug release at infinite time, k is a constant depending on the characteristics of the polymer matrix and drug, and n is the diffusional exponent characteristic of the release mechanism. Using this model, we determine the k and n values for the three M80–FAs. Interestingly, we found that the exponent values n were higher than 0.5 (Table S9†). In the case of pure Fickian release from spheres, this n value is equal to 0.43 as previously demonstrated.55 For exponent values 0.5 < n < 1, the release mechanism is explained by an anomalous transport (non-Fickian diffusion), strongly suggesting a specific interaction between the Nile red and the fatty acid core of the micelles. Finally, these experiments demonstrate that the length of the fatty acid chain is of great importance both for the loading capacity of the ELP–FA micelles, but also for the release kinetics of the hydrophobic molecule. More specifically, the data obtained show that the presence of two additional carbons in the fatty acid radically modifies the encapsulation properties. We therefore consider the possibility of refining the design of ELP–FA to obtain micellar systems with good control of their loading capacity and release kinetics.
Conclusions
In this work, we successfully conjugated five natural FAs varying in length and unsaturation degree onto three ELPs using a simple coupling protocol that can be easily scaled up to large quantities. These biobased ELP–FA conjugates have been chemically characterized and their behavior in aqueous solution was studied. We have shown that ELP–FAs have amphiphilic properties, as shown by the formation of small micelles with RH between 9 nm and 22 nm in PBS at low temperature (<20 °C). The micelle size is primarily determined by the length of the ELP block. The star-like micelle model developed by Halperin adequately describes the scaling behavior of ELP–FA micelles but the exponent related to the ELP blocks was found to be smaller than 3/5 due to the relatively low water-solubility of ELPs in general. The micellization of ELP–FAs at low temperatures did not preclude their further phase separation upon heating which is the hallmark of ELPs. Acylation of ELPs greatly decreased the Tcp of ELPs in relation to the higher hydrophobicity of ELP–FAs, but no significant difference was found between the different FAs tested. The micelles can encapsulate small hydrophobic molecules like Nile red. Both the encapsulation efficiency and release kinetics can be tuned by changing the composition of the ELP–FAs. Depending on their Tcp, two applications can be proposed. First, when Tcp is above body temperature, the obtained micelles could be used for the delivery of therapeutic drugs after injection into the plasma. Second, when the Tcp is below 37 °C, the micelles containing the encapsulated drug can aggregate shortly after their injection into a tissue and thus form a drug depot.Author contributions
The manuscript was written through contributions of all authors. TZ achieved all the synthesis and most of the self-assembly experiments. FP, CS and BG conceptualized the study. Interpretation of DLS and modelization by CS and MF. AW and KB performed and interpret the RP-HPLC and MS experiments, respectively. AM supervised the drug release experiment and interpret the results. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the China Scholarship Council and Université de Bordeaux (UB-CSC 2018) for the financial support of TZ. CNRS, Univ. Bordeaux and Bordeaux INP are acknowledged for their financial support. The microscopy was carried out in the Bordeaux Imaging Center, a service unit of the CNRS-INSERM and Bordeaux University, member of the national infrastructure France BioImaging. The authors thank Frédéric Rosu for his help with the CD experiments.Notes and references
- A. Varanko, S. Saha and A. Chilkoti, Adv. Drug Delivery Rev., 2020, 156, 133–187 CrossRef CAS PubMed
.
- T. Nemoto, M. Horiuchi, N. Ishiguro and M. Shinagawa, Arch. Virol., 1999, 144(1), 177–184 CrossRef CAS PubMed
- K. Cai, A. Gröner, H. O. Dichtelmüller, F. Fabbrizzi, E. Flechsig, R. Gajardo, I. von Hoegen, J. I. Jorquera, C. Kempf, T. R. Kreil, D. C. Lee, M. Moscardini, G. Pölsler and N. J. Roth, Transfusion, 2013, 53, 1894–1905 CrossRef CAS PubMed
- D. W. Urry, T. L. Trapane and K. U. Prasad, Biopolymers, 1985, 24, 2345–2356 CrossRef CAS PubMed
- D. W. Urry, Prog. Biophys. Mol. Biol., 1992, 57, 23–57 CrossRef CAS PubMed
- B. Li, D. O. Alonso and V. Daggett, J. Mol. Biol., 2001, 305, 581–592 CrossRef CAS PubMed
- D. E. Meyer and A. Chilkoti, Biomacromolecules, 2004, 5, 846–851 CrossRef CAS PubMed
- D. W. Urry, B. Haynes and R. D. Harris, Biochem. Biophys. Res. Commun., 1986, 141, 749–755 CrossRef CAS PubMed
- E. Garanger and S. Lecommandoux, Angew. Chem., Int. Ed., 2012, 51, 3060–3062 CrossRef CAS PubMed
- A. K. Varanko, J. C. Su and A. Chilkoti, Annu. Rev. Biomed. Eng., 2020, 22, 343–369 CrossRef CAS PubMed
- E. R. Wright and V. P. Conticello, Adv. Drug Delivery Rev., 2002, 54, 1057–1073 CrossRef CAS PubMed
- M. R. Dreher, A. J. Simnick, K. Fischer, R. J. Smith, A. Patel, M. Schmidt and A. Chilkoti, J. Am. Chem. Soc., 2007, 130, 687–694 CrossRef PubMed
- S. M. Janib, M. F. Pastuszka, S. Aluri, Z. Folchman-Wagner, P. Y. Hsueh, P. Shi, Y. A. Lin, H. Cui and J. A. Mackay, Polym. Chem., 2014, 5, 1614–1625 RSC
- W. Hassouneh, E. B. Zhulina, A. Chilkoti and M. Rubinstein, Macromolecules, 2015, 48, 4183 CrossRef CAS PubMed
- E. Garanger, S. R. MacEwan, O. Sandre, A. Brûlet, L. Bataille, A. Chilkoti and S. Lecommandoux, Macromolecules, 2015, 48, 6617–6627 CrossRef CAS
- K. Widder, S. R. MacEwan, E. Garanger, V. Núñez, S. Lecommandoux, A. Chilkoti and D. Hinderberger, Soft Matter, 2017, 13, 1816–1822 RSC
- K. M. Luginbuhl, D. Mozhdehi, M. Dzuricky, P. Yousefpour, F. C. Huang, N. R. Mayne, K. L. Buehne and A. Chilkoti, Angew. Chem., Int. Ed., 2017, 56, 13979–13984 CrossRef CAS PubMed
- L. Poocza, F. Cipriani, M. Alonso and J. C. Rodríguez-Cabello, ACS Omega, 2019, 4, 10818–10827 CrossRef CAS PubMed
- V. Ibrahimova, H. Zhao, E. Ibarboure, E. Garanger and S. Lecommandoux, Angew. Chem., Int. Ed. Engl., 2021, 60, 15036–15040 CrossRef CAS PubMed
- D. M. Scheibel, M. S. Hossain, A. L. Smith, C. J. Lynch and D. Mozhdehi, ACS Macro Lett., 2020, 9, 371–376 CrossRef CAS PubMed
- T. Zhang, F. Peruch, A. L. Wirotius, E. Ibarboure, F. Rosu, C. Schatz and B. Garbay, Polym. Chem., 2021, 12, 6030–6039 RSC
- D. Mozhdehi, K. M. Luginbuhl, J. R. Simon, M. Dzuricky, R. Berger, H. S. Varol, F. C. Huang, K. L. Buehne, N. R. Mayne, I. Weitzhandler, M. Bonn, S. H. Parekh and A. Chilkoti, Nat. Chem., 2018, 10, 496 CrossRef CAS PubMed
- M. S. Hossain, C. Maller, Y. Dai, S. Nangia and D. Mozhdehi, Chem. Commun., 2020, 56, 10281–10284 RSC
- D. Mozhdehi, K. M. Luginbuhl, M. Dzuricky, S. A. Costa, S. Xiong, F. C. Huang, M. M. Lewis, S. R. Zelenetz, C. D. Colby and A. Chilkoti, J. Am. Chem. Soc., 2019, 141, 945–951 CrossRef CAS PubMed
- R. Petitdemange, E. Garanger, L. Bataille, W. Dieryck, K. Bathany, B. Garbay, T. J. Deming and S. Lecommandoux, Biomacromolecules, 2017, 18, 544–550 CrossRef CAS PubMed
- M. Dai, E. Georgilis, G. Goudounet, B. Garbay, J. Pille, J. C. M. van Hest, X. Schultze, E. Garanger and S. Lecommandoux, Polymers, 2021, 13, 1470 CrossRef CAS PubMed
- D. E. Meyer and a Chilkoti, Nat. Biotechnol., 1999, 17, 1112–1115 CrossRef CAS PubMed
- A. Mohr, P. Talbiersky, H. G. Korth, R. Sustmann, R. Boese, D. Bläser and H. Rehage, J. Phys. Chem. B, 2007, 111, 12985–12992 CrossRef CAS PubMed
- L. Piñeiro, M. Novo and W. Al-Soufi, Adv. Colloid Interface Sci., 2015, 215, 1–12 CrossRef PubMed
- R. W. Korsmeyer and N. A. Peppas, J. Membr. Sci., 1981, 9, 211–227 CrossRef CAS
- S. K. Abbott, P. L. Else, T. A. Atkins and A. J. Hulbert, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 1309–1317 CrossRef CAS PubMed
- C. A. Hood, G. Fuentes, H. Patel, K. Page, M. Menakuru and J. H. Park, J. Pept. Sci., 2008, 14, 97–101 CrossRef CAS PubMed
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS
- J. S. Patton, B. Stone, C. Papa, R. Abramowitz and S. H. Yalkowsky, J. Lipid Res., 1984, 25, 189–197 CrossRef CAS
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Proc. Natl. Acad. Sci., 2015, 112, E3095–E3103 CrossRef CAS PubMed
- A. Micsonai, F. Wien, É. Bulyáki, J. Kun, É. Moussong, Y. H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Nucleic Acids Res., 2018, 46, W315–W322 CrossRef CAS PubMed
- T. Yamaoka, T. Tamura, Y. Seto, T. Tada, S. Kunugi and D. A. Tirrell, Biomacromolecules, 2003, 4, 1680–1685 CrossRef CAS PubMed
- F. G. Quiroz and A. Chilkoti, Nat. Mater., 2015, 14, 1164–1171 CrossRef CAS PubMed
- F. G. Quiroz, N. K. Li, S. Roberts, P. Weber, M. Dzuricky, I. Weitzhandler, Y. G. Yingling and A. Chilkoti, Sci. Adv., 2019, 5, eaax5177 CrossRef CAS PubMed
- D. Patterson, Macromolecules, 1969, 2, 672–677 CrossRef CAS
- J. R. Kramer, R. Petitdemange, L. Bataille, K. Bathany, A.-L. Wirotius, B. Garbay, T. J. Deming, E. Garanger and S. Lecommandoux, ACS Macro Lett., 2015, 4, 1283–1286 CrossRef CAS PubMed
- J. R. McDaniel, J. Bhattacharyya, K. B. Vargo, W. Hassouneh, D. A. Hammer and A. Chilkoti, Angew. Chem., Int. Ed., 2013, 52, 1683–1687 CrossRef CAS PubMed
- J. Aguiar, P. Carpena, J. A. Molina-Bolívar and C. Carnero Ruiz, J. Colloid Interface Sci., 2003, 258, 116–122 CrossRef CAS
- A. Yekta, J. Duhamel, P. Brochard, H. Adiwidjaja and M. A. Winnik, Macromolecules, 1993, 26, 1829–1836 CrossRef CAS
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS
- M. Frindi, B. Michels and R. Zana, J. Phys. Chem., 1992, 96, 6095–6102 CrossRef CAS
- O. Regev and R. Zana, J. Colloid Interface Sci., 1999, 210, 8–17 CrossRef CAS PubMed
- R. Zana, H. Lévy and K. Kwetkat, J. Colloid Interface Sci., 1998, 197, 370–376 CrossRef CAS PubMed
- E. Fuguet, C. Ràfols, M. Rosés and E. Bosch, Anal. Chim. Acta, 2005, 548, 95–100 CrossRef CAS
- A. Bhadani, K. Iwabata, K. Sakai, S. Koura, H. Sakai and M. Abe, RSC Adv., 2017, 7, 10433–10442 RSC
- A. Halperin, Macromolecules, 1987, 20, 2943–2946 CrossRef CAS
- S. Fluegel, K. Fischer, J. R. McDaniel, A. Chilkoti and M. Schmidt, Biomacromolecules, 2010, 11, 3216–3218 CrossRef CAS PubMed
- G. R. Grimsley, J. M. Scholtz and C. N. Pace, Protein Sci., 2009, 18, 247–251 CAS
- P. Costa and J. M. Sousa Lobo, Eur. J. Pharm. Sci., 2001, 13, 123–133 CrossRef CAS PubMed
- P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 23–36 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06603c |
| This journal is © The Royal Society of Chemistry 2023 |