
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMutation in the D1 domain of CD4 receptor modulates the binding affinity to HIV-1 gp120
Jiping Shao†
 *a,
Gezhi Liu†b and
Gang Lvacd
*a,
Gezhi Liu†b and
Gang Lvacd
aDepartment of Pathogen Biology, Hainan Medical University, Haikou 571199, P. R. China. E-mail: 2252290399@qq.com
bUniversity of Maryland, Maryland 20850, USA
cKey Laboratory of Translation Medicine Tropical Diseases, Hainan Medical University, Haikou 571199, P. R. China
dHainan Medical University-The University of Hong Kong Joint Laboratory of Tropical Infectious Diseases, Hainan Medical University, Haikou 571199, P. R. China
First published on 11th January 2023
Abstract
The gp120 surface subunit of HIV-1 envelope lycoprotein (Env) is the key component for the viral entry process through interaction with the CD4 binding site (CD4bs) of the primary receptor CD4. The point mutant was introduced into SD1, a CD4 D1 variant, to enhance the interaction with HIV-1 gp120.The three-dimensional structures of gp120 and SD1 were determined using homology modeling based on the results previously determined by X-ray crystallography. The binding models were carried out via protein–protein docking tools. The 5 best docking solutions were retained according to the docking scores and were used for structural assessment. Our results demonstrated the consistency between the 3D models of gp120 and SD1 predicted by molecular docking calculations and the co-crystallized data available. We first discovered that most residues in SD1 that interacted with gp120 were located within the region 6–94 of the first N-terminal D1 domain of CD4. SD1 bound to gp120 stably at which 15 residues formed 20 hydrogen bonds with 16 residues of gp120. Five pairs of electrostatic interactions between positively and negatively charged side chains of amino acids were identified in the SD1-gp120 interface, which showed an increased number of electrostatic interactions with gp120. The mutant in the D1 domain of human CD4 receptor could strengthen binding affinity with HIV-1 gp120 and might improve the interaction pattern of the neighboring residues. The sequence analysis of gp120 suggested that Asp186, Asn189, Arg191, Glu293, Phe318 and Tyr319 were located in the variable regions of gp120, which may be HIV-1 AE strain-specific amino acid residues. Together, the results presented in this study contributed to a better understanding of the changes in the interaction between the gp120 protein and the human host CD4 receptor associated with point mutation in the D1 domain. The stabilized derivative of human CD4 D1 should serve as a promising target for therapeutics development in HIV-1 vaccine and viral entry inhibitor and may warrant further investigation.
Introduction
Human immunodeficiency virus type 1(HIV-1) has caused AIDS outbreaks and remains a significant public health threat around the world. HIV-1 strains have been classified into four groups: M, N, O and P. HIV-1 group M accounts for almost half of all the AIDS epidemic worldwide, and is further divided into 9 subtypes (A, B, C, D, F, G, H, J and K), 3 sub-subtypes of A(A1, A2 and A3) and 2 sub-subtypes of F(F1 and F2). In addition to pure subtypes of HIV-1, more than 70 circulating recombinant forms (CRFs) have also been recognized.1,2 HIV-1 CRF01_AE, the first identified CRF subtype, represents a putative subtype of A/E recombinant originated from Central Africa and is now most prevalent in Southeast and East Asia.3 In Cambodia, Thailand and Vietnam, CRF01_AE is responsible for more than 95% of the infections. It was found in northern Thailand in 1989,4,5 then spread rapidly to neighboring regions in Asia.6–8 Of all national infections, it accounted for 42.5% in China, and 95% of those in Thailand. HIV-1 CRF01_AE was found in many regions of China, including Hainan, Guangdong and Guangxi since the early 2000s. It should draw more and more attention to make efforts in supervising and preventing the spread of HIV infection.9The envelope glycoprotein gp120 of HIV-1, present in the outer layer of the virus, is an essential component in a viral infection process. The process of gp120 binding to the human T-cell receptor CD4 is the first step of HIV-1 entry into the CD4+ host cells,10 which results in damaging the human immune system. Based on amino acid sequence analysis, gp120 is composed of five relatively conserved domains (C1–C5) and five variable domains (V1–V5) (Fig. 1), and also contains a highly conserved CD4bs for the primary cell surface receptor CD4, which is a broadly neutralizing antibody target. The conserved regions form the gp120 core consisting of the inner domain and the outer domain linked by the bridging sheet, while the variable regions form external solvent-exposed loops. The disulfide bonds of gp120 are mainly located in the core structure from which the variable loops and N-linked glycans protrude.11,12 The first and second variable regions (V1V2) are highly diverse in both sequence and length. The length of V1V2 is about 57 to 86 residues with amino acids (aa) sequence variations. V1V2 has two disulfide bonds; the V1 disulfide bond between cysteine residues 131 and 157 is located within the V2 disulfide bond between residues 126 and 196.13 The highly conserved disulfide bonds play key roles in maintaining the structure and function of proteins. The third hypervariable domain (V3) of gp120 has a disulfide-bonded structure, frequently glycosylated, and is critical for viral infection. The principal neutralizing determinant (PND) of the virus is mapped to a 10-amino acid-long sequence RGPGRAFVTI (residues 309–318), derived from the V3 loop region of HIV-1 gp120. The V3 region of HIV-1 subtype AE shows strong immunoreactivity. Both the variable V4 loop and the V5 loop are involved in neutralization escape, they also go through variations during early infection.14–17
 | ||
| Fig. 1 The structure and functional domains of HIV-1 CRF01_AE gp120 and CD4. (A) Schematic diagram of the gp120 functional domains were indicated. C1–C5 were colored in blue, and V1–V5 in light blue. Residues were numbered corresponding to their positions in HIV-1 CRF01_AE gp120. (B) The functional domains of CD4 were indicated. The arrangement of CD4 extracellular domains D1, D2, D3 and D4 were colored in pink, light purple, green and light blue, respectively. | ||
Human CD4 belongs to the immunoglobulin superfamily and is mainly expressed on the majority of normal peripheral blood monocytes, such as T-lymphocytes as well as various other cells of the immune system.18 It is a membrane glycoprotein of T lymphocytes with a molecular weight of 55 kDa, is also a primary receptor for HIV-1 entry through interactions with the gp120 subunit. It participates in postbinding events for viral infection and plays an important role in the process of HIV-1 infection. The CD4 antigen is comprised of four extracellular immunoglobulin domains (D1–D4, residues 1–371) in its ectodomain, and a single transmembrane domain (residues 372–395), and a short cytoplasmic tail (residues 396–433), but only D1 binds to HIV-1 gp120. The D1 domain corresponds to residues 1–97 of CD4, however, residues 98 and 99 interact with several residues within 1–97.D2 refers to amino acids 100–183, D3(184–297 aa) and D4(298–371 aa) of hCD4, from N- to C-terminal respectively.19
Homology modeling is an efficient and most preferred method of protein structure prediction that is used to determine 3D structure of a protein from its amino acid sequence based on its template. Homology modeling is considered to be the most accurate of the computational structure prediction methods and has a vast range of applications in structure based drug development, analysis of mutations and binding mechanisms, identification of active sites, designing of novel ligands, protein–protein docking simulations etc20. Protein–protein interactions (PPIs) are universal to life and play a crucial role in cellular functions and biological processes in all organisms. X-ray crystallography, isothermal titration calorimetry and other biophysical methods have been used to study PPIs. The identification of protein interactions can lead to a better understanding of fundamental cellular regulation, infection mechanisms, the development of several medication drugs and treatment optimization.21
Numerous mutagenesis and monoclonal antibody studies have shown that the D1 domain of CD4 directly interacts with gp120 for all HIV-1 isolates to enable virus–cell membrane fusion and viral entry, and is critical for gp120 binding. Since the single D1 domain alone is unstable, largely unfolded and has weak binding affinity for gp120.19,22 We hypothesized that the design of CD4 D1 with additional mutation might alter its affinity to cell receptor. In the present work, the point mutant was introduced into SD1, a new variant of CD4 D1-derived peptide, corresponded to the N-terminal D1 domain (amino acids 1–99) of human CD4.We performed computational experiments to predict the interactions between HIV-1 gp120 and SD1. We attempted to investigate the effect of the mutation impacted on protein-peptide binding affinity using computational methods. The online docking servers for protein–peptide interaction were utilized to find the binding sites and identify the key residues between the SD1 molecular and the gp120 protein.
Experimental
Sequence retrieval
Protein database is a collection of sequences from several sources, including GenBank, RefSeq and TPA, as well as SwissProt, PIR, PRF, and PDB etc. The amino acid sequences of protein are the fundamental determinants of biological structure and function. Some of these sequences come from laboratories that have done protein sequencing (primary data) and others are derived from genetic sequences (derived data), like the NCBI RefSeq records. The amino acid sequences of HIV-1 CRF01_AE gp120 and SD1 were retrieved from Los Alamos National Laboratory HIV database at http://www.hiv.lanl.gov and were used for further analysis in this study.Physicochemical characterization analysis
The physicochemical properties of a protein such as amino acid composition, solubility, stability, R and +R (total number of positive and negative residues), EI (extinction coefficient), II (instability index), AI (aliphatic index), theoretical pI (isoelectric point) and ionization constant (s) are essential. The physicochemical properties of HIV-1 CRF01_AE gp120 and SD1 were analyzed using the Expasy's ProtParam server at http://www.expasy.org/tools/protscale.html. Their theoretical pI, extinction coefficient, instability index, aliphatic index, and grand average hydropathy (GRAVY) were computed using the Expasy's ProtParam server.23,24Protein structure prediction
Based on the number of polypeptide chains, the secondary structure of a protein can be classified into 3 types: type-H(α-helix), type-E(β-sheet) and type-C(coil). SOPMA (Self-Optimized Prediction Method with Alignment) available at https://npsa-prabi.ibcp.fr/cgi-bin/npsa%20_automat.pl?%20page=%20npsa_%20sopma.html can be employed for calculating the secondary structural features of the selected protein sequences.25 Phyre2 uses advanced remote homology detection methods to build 3D models, and predict and analyze protein structure, function and mutations.26 This server is available at http://www.sbg.bio.ic.ac.uk/phyre2. Secondary structure and disorder prediction is on average 78–80% accurate, which means 78–80% of the residues are predicted to be in their correct state. This accuracy is only reached if there is a substantial number of diverse sequence homologues detectable in the sequence database. If the sequence has very few homologues, then the accuracy falls to approximately 65%.The secondary structure components of HIV-1 CRF01_AE gp120 and SD1 were determined using SOPMA and phyre2 with the NN algorithm.Protein modelling
Protein modeling plays more and more important roles in investigating biomedical mechanism of diseases and drug design. Successful model building requires at least one experimentally solved 3D structure template that has a significant amino acid sequence similarity to the target sequence. SWISS-MODEL (https://swissmodel.expasy.org) is the automated protein structure homology-modelling platform for generating 3D models of a protein using a comparative approach, and database of annotated models for key reference proteomes based on UniProtKB.27 This will enable comparative modeling to build 3D models of target proteins for the vast majority of amino acid sequences. Phyre2 uses the alignment of hidden Markov models via HHsearch1 and incorporates a new ab initio folding simulation called Poing2 to model regions of the targeted proteins with no detectable homology to known structures, which significantly improve accuracy of alignment and detection rate.26 The amino acid sequences of gp120 and SD1 were input in FASTA format, template selection, alignment and model building were done completely automated by the servers. Templates were ranked according to expected quality of the resulting models, as estimated by Global Model Quality Estimate (GMQE).28 Based on high score, lower e-value and maximum sequence identity, the top templates were selected to build 3D models.Protein–protein interactions
PPIs are necessary for the biological processes at the molecular level and play a crucial role for a better understanding of involved mechanisms of interacting pairs, functional domains and characterizing specific molecular interaction of host and pathogen. Thus, it is important to develop docking methods that can elucidate the details of specific interactions at the atomic level. The ZDOCK server is a Fourier transform based protein docking program and is freely available to all academic and non-profit users at http://zdock.umassmed.edu/.29 It provides a fast and effective method to produce models of protein–protein complexes and symmetric multimers via a user-friendly web interface. The ClusPro server (https://cluspro.org/) is a widely used tool for protein–protein docking. ClusPro employs a scoring function and combination with the energy function substantially increases the accuracy of docking, resulting in more near-native structures. The 3D structures of gp120 (PDB ID: 6IEQ) and CD4 D1 (PDB ID: 1CDY) were extracted from RCSB Protein Data Bank (RCSB PDB),a public resource of structure database that contains the three-dimensional crystal structure of proteins that were experimentally determined. These docking services were performed to predict the binding sites between the SD1 molecular and the gp120 protein. Docking with each energy parameter set resulted in ten models defined by centers of highly populated clusters of low-energy docked structures.30Results and discussion
Physicochemical properties
Physicochemical properties are key factors in controlling the protein–protein interactions, and the silico methods are available for the rapid calculation of physicochemical properties. The amino acid sequences of HIV-1 CRF01_AE gp120 and SD1 were obtained from the protein database, their physicochemical properties were calculated using the Expasy's Protparam tool in the ExPASy portal. The amino acid compositions of HIV-1 CRF01_AE gp120 and SD1 were shown in Table 1. The molecular formula of HIV-1 CRF01_AE gp120 was C2659H4205N745O792S33 and the theoretical pI value was 8.66 suggesting a moderately alkaline nature of the protein. The proportion of positively and negatively charged residues of gp120 was 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :47.The predicted aliphatic index was 82.82, and the grand average of hydropathicity (GRAVY) was −0.293, thus classifying the gp120 protein as hydrophilic. The estimated half-life was 30 h. Stability is critical for rational design of vaccines and antibodies, the instability index provides an estimate of a protein stability. The instability index of a protein is smaller than 40 is predicted as stable, a value above 40 indicates that it may be unstable. The instability index (II) of gp120 was computed to be 35.2 < 40, which meant it was stable.
:47.The predicted aliphatic index was 82.82, and the grand average of hydropathicity (GRAVY) was −0.293, thus classifying the gp120 protein as hydrophilic. The estimated half-life was 30 h. Stability is critical for rational design of vaccines and antibodies, the instability index provides an estimate of a protein stability. The instability index of a protein is smaller than 40 is predicted as stable, a value above 40 indicates that it may be unstable. The instability index (II) of gp120 was computed to be 35.2 < 40, which meant it was stable.
| HIV-1 CRF01_AE gp120 | SD1 | ||||
|---|---|---|---|---|---|
| Amino acid names and abbreviations | Number | Percentage % | Amino acid names and abbreviations | Number | Percentage % |
| Asn (N) | 55 | 10.2% | Lys (K) | 13 | 13.1% |
| Ile (I) | 46 | 8.5% | Leu (L) | 10 | 10.1% |
| Thr (T) | 44 | 8.2% | Ser (S) | 9 | 9.1% |
| Gly (G) | 37 | 6.9% | Asp (D) | 7 | 7.1% |
| Leu (L) | 35 | 6.5% | Gln (Q) | 7 | 7.1% |
| Val (V) | 35 | 6.5% | Ile (I) | 7 | 7.1% |
| Lys (K) | 32 | 5.9% | Asn (N) | 6 | 6.1% |
| Ala (A) | 29 | 5.4% | Glu (E) | 6 | 6.1% |
| Glu (E) | 28 | 5.2% | Gly (G) | 6 | 6.1% |
| Pro (P) | 26 | 4.8% | Val (V) | 6 | 6.1% |
| Ser (S) | 26 | 4.8% | Thr (T) | 5 | 5.1% |
| Arg (R) | 24 | 4.5% | Phe (F) | 4 | 4.0% |
| Asp (D) | 19 | 3.5% | Arg (R) | 3 | 3.0% |
| Cys (C) | 19 | 3.5% | Ala (A) | 2 | 2.0% |
| Gln (Q) | 19 | 3.5% | Cys (C) | 2 | 2.0% |
| Phe (F) | 17 | 3.2% | Pro (P) | 2 | 2.0% |
| Met (M) | 14 | 2.6% | Trp (W) | 2 | 2.0% |
| Tyr (Y) | 14 | 2.6% | His (H) | 1 | 1.0% |
| Trp (W) | 12 | 2.2% | Tyr (Y) | 1 | 1.0% |
| His (H) | 8 | 1.5% | Met (M) | 0 | 0.0% |
Considering the importance of the surface receptors of CD4 T lymphocytes, we downloaded the amino-acid sequence of SD1 from the protein sequence database for calculating the physico-chemical properties, such as molecular weight, net charge of protein, isoelectric point, molar extinction coefficient, grand average hydropathy, aliphatic index, and number of charged residues etc. Our results showed that the molecular formula of SD1 was C499H808N138O154S2, and the theoretical pI was 8.93. The number of lysine(Lys, K) was 13, which accounted for 13.1% of full protein, Leucine(Leu, L) made up approximately 10.1%, while other amino acids accounted for below 10%. The proportion of positively and negatively charged residues of SD1 was 16:13. The estimated half-life was 1.3 h. The instability index (II) was computed to be 37.30, which classified the SD1 peptide as stable. The aliphatic index for the selected protein sequence was found to be 86.57. GRAVY is used for the computational analysis of various physicochemical parameters for a given amino acid sequence. The value of the GRAVY index of SD1 was −0.639, which meant that it was hydrophilic and had a high affinity for water. These data could be used for optimizing methods of expression and characterization of the selected proteins.
Protein secondary structure prediction
The most common types of secondary structures are alpha helix, beta sheet, beta turn and random coil. The self-optimized prediction method with alignment (SOPMA) has been developed to predict protein secondary structure. Phyre2 uses advanced remote homology detection methods to analyze secondary structure and disorder prediction for a user's protein sequences. The secondary structure properties of HIV-1 CRF01_AE gp120 and SD1 were carried out using Phyre2 and SOPMA (Fig. 2). Subsequently, the secondary structure of gp120 was made up of 26.16% α-Helix, 5.19% β-turn, 26.35% extended strand and 42.3% random coil, respectively. The secondary structure of SD1 contained α-Helix, β-turn, extended strand and random coil, comprising 8.08%, 9.09%, 39.39% and 43.43%, respectively. An increased number of extended strands and random coils of proteins were correlated with protein antigenic epitopes formation. As seen in Fig. 2B1 and B2, 228 of 539 amino acid residues localized to the random coil indicating that it was the dominant secondary structure of gp120, while β-turn was the least. As showed in Fig. 2C1 and C2, 46 of 99 amino acid residues localized to the random coil indicating that it was the dominant type of secondary structure of SD1, which proved that it might be potential to form highly antigenic epitopes.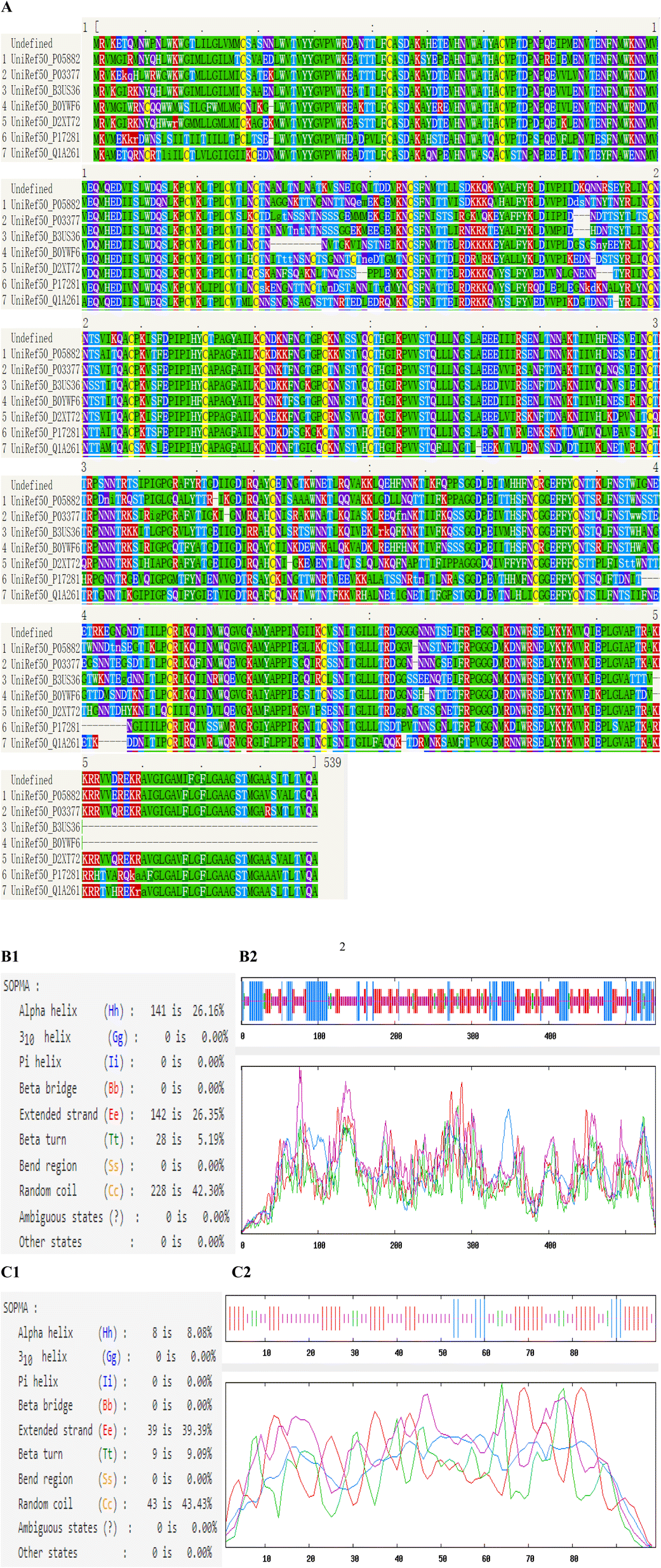 | ||
| Fig. 2 The predicted secondary structure results of HIV-1 CRF01_AE gp120 and SD1. (A) Conservation analysis of the gp120 protein sequence. The amino acids were colored based on their conservation grades and conservation levels. (B1 and C1) The predicted secondary structure elements of gp120 and SD1 after computing the query sequences, α-helix, extended strand, β-turn and random coil were colored in blue, red, green and yellow, respectively. (B2 and C2) The corresponding positions of α-helix, extended strand, β-turn and random coil in gp120 and SD1. | ||
Phyre2 uses the alignment of hidden Markov models via HHsearch to significantly improve accuracy of alignment and detection rate. Our results showed that the secondary structure and disorder prediction of gp120 was 78% accurate, which indicated that 78% of the aa residues were predicted to be in their correct state as shown in Fig. 2A. The structure identity of SD1 was 99%.The weakest region of helix prediction coincided with a relatively strong prediction of disorder. Disordered regions could often be functionally very important. Protein conservation analysis based on amino acid sequence alignment among species can identify regions of similarity that may be a consequence of evolutionary relationships between the sequences. Multiple Sequence Alignment (MSA) of the gp120 protein depicting the conserved domains obtained from Phyre2. The sequence analysis indicated that HIV-1 CRF01_AE gp120 consisted of five relatively conserved domains (C1–C5) and five variable domains (V1–V5), and most of the amino acids in the gp120 protein were highly conserved in different species (Fig. 2), these results were in agreement with previous results.15–17
3D models of proteins
In order to form stable and biologically active structures, the individual elements of secondary structure of a protein can pack against one another to form the proper tertiary structure, this depends on the amino acid sequence and the atomic details of the structure. The amino acid sequences of gp120 and SD1 were input in FASTA format, template alignment and model building were done completely automated by the servers. The three-dimensional structures of gp120 and SD1 were determined by homology modeling of the separate domain structures based on results previously determined by X-ray crystallography and were visualized using the program PyMOL. The gp120 core contained three domains: the outer domain, the inner domain, and the bridging sheet, the inner and outer domains were fixed by a four-strand beta-sheet, termed the bridging sheet presented in Fig. 3A1. The monomeric gp120 consisted of five conserved regions (C1–C5) and five highly variable regions (V1–V5). The conserved regions and variable regions of HIV-1 CRF01_AE gp120 and SD1 were listed in Table 2. Four helices 1, 4, 5 and 6 were found in the conserved C1, C4 and C5 regions of gp120, two helices 2 and 3 were predicted in the V2 region and the C3 region, which exhibited a conformation consistent with the findings from previous reports.23,31 The SD1 peptide folded into a stable eight-stranded beta-sheet shown in Fig. 3A2.| HIV-1 CRF01_AE gp120 | SD1 | |||||||
|---|---|---|---|---|---|---|---|---|
| Conserved regions | Variable regions | Conserved regions | ||||||
| Helix | Strand | Coil | Helix | Strand | Coil | Helix | Strand | Coil |
| Residues 12–29 | Residues 33–39 | Residues 6–11 | Residues 152–155 | Residues 124–129 | Residues 130–151 | Residues 59–61 | Residues 1–6 | Residues 15–23 |
| 61–67 | 50–53 | 41–49 | 174–183 | 156–160 | 90–92 | 12–14 | 39–42 | |
| 86–113 | 226–230 | 55–60 | 193–197 | 166–171 | 24–28 | 46–53 | ||
| 328–334 | 245–249 | 71–82 | 295–299 | 184–192 | 35–38 | 56–58 | ||
| 340–356 | 257–264 | 119–123 | 308–311 | 300–307 | 43–45 | 75–77 | ||
| 415–426 | 273–277 | 207–217 | 317–320 | 394–409 | 68–74 | |||
| 473–483 | 286–291 | 231–244 | 410–414 | 467–472 | 82–86 | |||
| 509–515 | 360–363 | 335–339 | 93–98 | |||||
| 527–534 | 370–377 | 364–370 | ||||||
| 447–453 | 433–439 | |||||||
| 484–489 | 455–464 | |||||||
| 516–520 | 490–501 | |||||||
GMQE (Global Model Quality Estimate) is coverage dependent and gives an overall quality estimate between 0 and 1, indicating the accuracy of the model built with the specific alignment and template. The higher the number, the higher the reliability of the model is. It combines properties from the target-template alignment and the template structure to increase reliability of the quality estimation. The QMEAN (quality model energy analysis) Z-score reflects the degree of nativeness of the structure in the model. Values around 0 mean good quality agreement between the modeled structure and experimental structures of similar size. Values less than −4 indicate models with low quality. In this study, the predicted gp120 model had been obtained with a 0.7 GMQE scores and a −1.03 QMEAN Z-score which was higher than −4.0, and the GMQE and QMEAN Z-score of SD1 were 0.89 and −0.30, respectively. Our predicted model structures contented all the parameters destined to accredit these models being reliable and precise as seen in Fig. 3B1 and B2.
 | ||
| Fig. 3 The structural models of HIV-1 CRF01_AE gp120 and SD1. (A1) HIV-1 gp120 envelope glycoprotein could be organized into five conserved regions (C1–C5) and five highly variable domains (V1–V5), helix, β-sheets and loop were colored in cyan, magenta and salmon, respectively. The four β-strands of the bridging sheet that linked the inner and outer domains were indicated. (A2) The structural models of SD1, helix, β-sheets and loop were colored in warm pink, blue and forest green, respectively. (B1 and B2) GMQE reports. The x-axis showed protein length (number of residues), and the y-axis was the “QMEAN” score in the “Comparison” plot. Every dot represented one experimental protein structure. Black dots were experimental structures with a “QMEAN” score within 1 standard devation of the mean (|Z-score| between 0 and 1), experimental structures with a |Z-score| between 1 and 2 were grey. | ||
Models selection by docking algorithm
ClusPro performs the following three computational steps in the order given:32–34 (1) rigid body docking, (2) root-mean-square deviation (RMSD) based clustering the lowest energy 1000 docked structures, (3) refinement of selected structures using energy minimization and outputs the structures at the centers of the 10 most populated clusters. ClusPro employs more complex and more accurate scoring functions that includes a structure-based pairwise interaction, and the combination with the energy function, and both electrostatic and desolvation contributions etc, substantially increases the accuracy of docking, resulting in more near-native structures. Thus, the Fast Fourier Transform (FFT) based algorithm enabled docking of proteins could be efficiently calculated, the docking score values of the top 5 gp120/SD1 complexes with negative binding energy values were considered good seen in Table 3.| Complex | Members | Representative | Weighted score |
|---|---|---|---|
| 1 | 119 | Center | −597.6 |
| Lowest energy | −671.4 | ||
| 2 | 78 | Center | −618.6 |
| Lowest energy | −618.6 | ||
| 3 | 52 | Center | −575.1 |
| Lowest energy | −616.7 | ||
| 4 | 40 | Center | −541.1 |
| Lowest energy | −601.4 | ||
| 5 | 39 | Center | −531.1 |
| Lowest energy | −639 |
The ClusPro server generates four sets of models using the scoring schemes called (1) balanced, (2) electrostatic-favored, (3) hydrophobic-favored, and (4) van der Waals + electrostatics (Table 4).PIPER represents the interaction energy between two proteins with an expression of the form E = w1Erep + w2Eattr + w3Eelec + w4EDARS, where Erep and Eattr denote the repulsive and attractive contributions to the van der Waals interaction energy, and Eelec is an electrostatic energy term, and EDARS primarily represents desolvation contributions and is scaled to the magnitudes of protein–protein binding free energies. Our results indicated that w1 < 1.0 and w2 < 1.0 yielded “softening” of both repulsive and attractive van der Waals terms, w4 = 1.0 was the “neutral” choice, w3 = 600 in the balanced option of the parameter set was shown to generally provide very good results for gp120-SD1 complexes (Table 4).
| Coefficient set | Energy term weight coefficients | ||||||
|---|---|---|---|---|---|---|---|
| Erep | Eattr | Eelec | EDARS | ||||
| Balanced | 0.40 | −0.40 | 600 | 1.00 | |||
| Electrostatic-favored | 0.40 | −0.40 | 1200 | 1.00 | |||
| Hydrophobic-favored | 0.40 | −0.40 | 600 | 2.00 | |||
| van der Waals + electrostatics | 0.40 | −0.10 | 600 | 0.00 | |||
Model of gp120-CD4 complex
Many proteins form homooligomers and heterooligomers, containing two or more copies of at least one subunit type. The protein complexes structures are referred to as biological assemblies. We developed models for analyzing the protein–protein interactions between HIV-1 CRF01_AE gp120 and SD1 using homology modeling. The gp120 core was folded into an inner and outer domain, as well as a bridging sheet. The inner domain, which included the N and C termini of the protein, consisted of a two-helix, two-strand bundle with a five-stranded β-sandwich at its terminiproximal end and a projection at the distal end where the V1/V2 emanated. The outer domain was composed of a stacked double barrel, whose axis lay parallel to the axis of the inner domain bundle.17,31 The resulting four-stranded antiparallel β-sheet (β2–β3 and β20–β21) linked the outer and inner domains to form a third domain, the bridging sheet. Together with the V3 loop of gp120, the bridging sheet made up the binding site for coreceptor.14,16 SD1 formed a stable eight-stranded beta-sheet and could bind to HIV gp120 at the interfaces of the outer domain, the inner domain and the bridging sheet displaying high-affinity. On the left of the gp120/SD1 complex, SD1-1 bound to the C1, V1 and V3 regions of gp120; SD1-3 bound to the V2 and the C3 region of gp120. On the right of the gp120/SD1 complex, SD1-2 bound to the C2 and C4 regions and retained high binding affinity for HIV-1 gp120 core. SD1-4 could bind to gp120 and targeted the V3 hypervariable region, while SD1-5 only bound to the C1 conserved region as shown in Fig. 4. Here, our results revealed that one gp120 protein and five SD1 molecules assembled together to form a large protein complex, this assembly involved in protein–protein interaction. In this gp120/SD1 large complex, the 15 amino acid residues of SD1 that contacted 16 amino acids of gp120 were located in the N-terminus of CD4 D1 (residues 6–94). These contacts between gp120 and SD1 gave the gp120/SD1 complex a greater affinity. | ||
| Fig. 4 Structure of the gp120/SD1 complex. One gp120 protein and five SD1 molecules assembled together to form a large protein complex. The ribbon diagram showed gp120 in magenta, SD1-1 in blue, SD1-2 in cyan, SD1-3 in orange, SD1-4 in yellow and SD1-5 in green, respectively. | ||
The SD1 molecule interaction with gp120
Since the single D1 domain of human CD4 was unstable and was prone to aggregation, and had weak binding affinity for gp120.19,22 We assumed mutation may alter its affinity to gp120. To increase the affinity and structure stability of the isolated D1 domain, in the present work, single point mutant G47S was introduced into the D1 domain of human CD4, designated SD1. For the mutated residue serine was chosen based on the possible hydrogen bond formation. The model of tertiary structure of gp120 was docked with SD1 using the docking tool. The top 5 highest individual scores models were selected from the 10 overlapped results, and the docking output files were analyzed for hot spots and hydrogen bonds in protein–protein interactions using the program PyMOL software.CD4 binding to gp120 took place through its N-terminal extracellular D1 domains, the modes of SD1 into the binding sequence of gp120 were presented in Fig. 5 and Table 5. Our results showed that the residues Gln94, Gly6 and Lys7 from the SD1 molecule were bonded to the amino acids Asn189, Thr127 and Thr122 in N-terminal trans-activation domain (TAD) of gp120 (Fig. 5A). The hydrogen bonds between Asn189 and Gln94 made significant contributions to the stability of folded proteins. The additional five interactions were made by the residues Lys255, Asn265, Thr447, Arg453 and Glu293 located in the random coils and extended strand of gp120 with Gln40, Gln25, Asp88 and Arg58 located in the random coils and extended strand of SD1 (Fig. 5B), two ion pairs of the Glu293-Arg58 and Arg453-Asp88 stabilized the interaction between gp120 and SD1. The thermophilic study demonstrated the importance of surface side-chain ion pairs in protein stability.
 | ||
| Fig. 5 Ribbon diagram of HIV-1 CRF01_AE gp120 structural homology model bound with SD1 generated by docking tools. Gp120 was shown as bright yellow ribbons, SD1 was shown as bright green ribbons. Dotted lines represented hydrogen bonds. (A) The key residues Lys7, Gly6 and Gln94 of SD1 were highlighted in red, rose and pink, respectively, and the contact residues Thr122, Thr127 and Asn189 of gp120 were colored in red, rose and pink, respectively. (B) The key residues Gln40, Gln25, Asp88 and Arg58 of SD1 were depicted in cyan, red, blue and orange, respectively, and the contact residues Lys255, Asn265, Thr447, Arg453 and Glu293 of gp120 in rose, rose, red, blue and orange, respectively. (C) The key residues Arg59, Asp53 and Lys50 of SD1 were depicted in blue, rose and cyan, respectively, and the contact residues Asp369, Arg191 and Asp186 of gp120 in blue, rose and cyan, respectively. (D) The key residues Asn23, Ser47 and Phe43 of SD1 were depicted in blue, rose and cyan, respectively, and the contact residues Ser309, Phe318 and Tyr319 of gp120 in blue, rose and cyan, respectively. (E) The key residues Lys46 and Ler44 of SD1 were depicted in rose and blue, respectively, and the contact residues Thr49 and Thr50 of gp120 in rose and blue, respectively. | ||
| Complex | Residue pairs | Bond distance Å |
|---|---|---|
| 1 | Asn189 – Gln94 | 3.2 |
| Thr127 – Gly6 | 3.1 | |
| Thr122 – Lys7 | 3.5 | |
| 2 | Lys255 – Gln40 | 2.7 |
| Asn265 – Gln40 | 2.8 | |
| Thr447 – Gln25 | 3.2 | |
| Arg453 – Asp88 | 3.1 | |
| Glu293 – Arg58 | 3.0 | |
| 3 | Asp369 – Arg59 | 2.3 |
| Arg191 – Asp53 | 3.2, 2.4 and 2.3 | |
| Asp186 - Lys50 | 2.8 and 3.3 | |
| 4 | Ser309 – Asn23 | 3.1 and 2.0 |
| Phe318 – Ser47 | 3.1 | |
| Tyr319 – Phe43 | 2.3 | |
| 5 | Thr49 – Lys46 | 3.4 |
| Thr50 – Leu44 | 3.4 |
Two hot spots Phe43 and Arg59 in human CD4 for its interaction with gp120 had been widely studied. The central involvement of Phe43 and Arg59 was confirmed by the three-dimensional crystal structure of a truncated gp120 protein complexed with a soluble CD4 protein. Mutations of Phe43 and Arg59 to alanine or glycine impaired binding to gp120, which corroborated the importance of Phe43 and Arg59 for the interaction of CD4 with gp120.17,35 Our results revealed that the charged residues Asp369, Arg191 and Asp186 located in the peptides of gp120 made three electrostatic interactions with the charged residues Arg59, Asp53 and Lys50 located in the peptide 50–59 (KLNDRADSRR) of SD1 (Fig. 5C). This highly conserved salt bridge between Arg59 SD1 and Asp369 gp120 stabilized the conformation. Moreover, the Nη1 and Nη2 atoms of SD1 Arg59 formed double hydrogen bonds with the Oδ1 and Oδ2 atoms of HIV-1 gp120 Asp369. Our results proved that these charge pair electrostatic interactions made significant contributions to the stability of the gp120/SD1 complex, these findings were in agreement with earlier reports.23 The intermolecular hydrogen bonds and the insertion of Phe43 of CD4 into the hydrophobic cavity of the gp120 surface played a critical role in the gp120/CD4 complex. Mutagenesis and antibody-blocking studies mapped the gp120-binding site identified side chains of Phe43 and other nearby residues as important contributors to this interaction. Our results discovered that the key amino acid residues Asn23, Ser42 and Phe43 of SD1 could interact with the contact residues Ser-309, Phe-318 and Tyr-319 of gp120 (Fig. 5D). The most prominent interaction between gp120 and SD1 occurred with the CD4 binding site, the residue Phe43 of SD1 made tightly contact with gp120 to form a hydrophobic Phe43 cavity located at the interface between the inner and outer domains of gp120 with a unique protruding CD4-Phe43 structure surrounded by an intermolecular hydrogen-bond, these observed interactions was consistent with previous data.36,37 The residues Lys46 and Leu44 in SD1 targeted the residues Thr49 and Thr50 of gp120, which theoretically could introduce hydrogen bonds with the main-chain O atom of CD4 Lys46 and Leu44 via the side chain of threonine (Fig. 5E).
Previous studies discovered some proteins exhibited low stability and solubility due to the presence of cavities within or on the surfaces of proteins, which could lead to loss in van der Waals contacts and decreased stability. Protein stability increased linearly with increasing volume of the amino acid side chains of cavity filling mutations,38 and had proven effective in improving properties of proteins. It has been shown previously that only the D1 domain of CD4 made direct contacts with gp120, and the amino acids 40–60 of CD4 D1 played a vital role for binding to gp120 for all isolates examined, with the residues Phe43 and Arg59 being particularly important. However, our studies discovered that SD1 bound to gp120 stably at the amino acid residues 6–94, and SD1 had more different binding sites than the wild-type CD4 D1 peptide, and 15 residues located at SD1 formed 20 hydrogen bonds with 16 residues of gp120. The introduced residue Ser47 was in close contact with the residue Phe317 of gp120, and this mutant showed an increased number of hydrogen bonding with gp120 than the wild-type CD4 D1 peptide. Five salt bridges, Glu293-Arg58 and Arg444-Asp88 from the complex gp120/SD1-2, Asp368-Arg59, Arg192-Asp53 and Asp186-Lys50 from the complex gp120/SD1-3, increased the stability of the gp120/SD1 complex. These findings therefore supported our hypothesis that SD1 bound to HIV-1 gp120 with high affinity. The above analysis confirmed that the mutant may strengthen the interactions of gp120/SD1 and should stabilize the tertiary structure of the gp120/SD1 complex.
Conclusion
The envelope glycoprotein of HIV-1 consists of non-covalently associated gp120/gp41 subunits involving in receptor binding and fusion. The binding of HIV-1 gp120 to the cell surface receptor CD4 is the first step in the entry of the virus into target cells. Here we performed computational experiments to investigate molecular docking and analyzed the molecular interactions between HIV-1 CRF01_AE gp120 and SD1. Since a protein's function is largely determined by its structure, predicting a protein's structure from its amino acid sequence can be useful to understand its functions and its roles in biological process. Our results proved that alpha helical structure was mainly composed of methionine, alanine, leucine, glutamate and lysine amino acids; whereas the beta strand consisted of tryptophan, tyrosine, phenylalanine, valine, isoleucine, and threonine; furthermore, glycine and proline helped to build the relevant turns.The quality assessment of the 3D models of gp120 and SD1 suggested that the computational methods and experimental data were capable of generating gp120-SD1 models similar to the near-native gp120-CD4 D1. Our results demonstrated the consistency between the 3D models of gp120 and SD1 predicted by molecular docking calculations and the co-crystallized data available. The overall folding of the 3D structures of gp120 and SD1 were as good as the available crystal structure of gp120 and SD1 and therefore, reliable and suitable for further structure-based studies. Previous studies shown that the amino acids located in position 40–60 from the CD4 D1 domain made direct contacts with gp120. In the present study, we first discovered that SD1 bound to gp120 stably at the amino acid residues 6–94, and SD1 had more different binding sites than the wild-type CD4 D1 peptide. Hydrogen bonds contribute to the stability of the ligand–protein complex. Our studies found that the mutant in the peptide of SD1 showed an increased number of hydrogen bonding with gp120, and effectively enhanced the stability of the gp120/SD1 complex.
Multiple sequence alignments can show conserved regions within a protein family which are of structural and functional importance. We performed multiple sequence alignment to distinguish regions which are more highly conserved than others. The sequence analysis of gp120 suggested a certain degree of conservation, with some highly-conserved residues throughout evolution. The key amino acid residues Thr49, Thr50, Thr122, Thr127, Lys255, Asn265, Ser309, Asp369, Thr447 and Arg453 were highly conserved located in the conserved regions of gp120, while Asp186, Asn189, Arg191, Glu293, Phe318 and Tyr319 were located in the variable regions of gp120, which may be HIV-1 AE strain-specific amino acid residues.
In sum, these findings revealed the polar and nonpolar intermolecular interactions contributing to the HIV-1 gp120-CD4 binding stability. Our studies identified the key interaction residues between the SD1 molecular and the gp120 protein, which provided an ideal target for viral entry inhibition and broad neutralization activity. The new SD1 peptide should be a promising drug candidate for HIV-1 prevention and therapy and may warrant further investigation.
Author contributions
JP conceived and designed the research; JP and GZ wrote the manuscript; JP and GZ performed experiments; JP and GZ analyzed data. GL reviewed the manuscript.Conflicts of interest
No conflict of interest exists in the submission of this manuscript.Acknowledgements
We thank Dimiter S Dimitrov (NCI-Frederick, MD, USA) for the valuable technical assistance. This study was supported by research grants, Program for High-Level Talents of Hainan Province (Grant No: 2019RC213) and Hainan Provincial Key Research and Development Program (Grant No: ZDYF2022SHFZ077 and ZDYF2016126).References
- J. Louwagie, W. Janssens, J. Mascola, L. Heyndrickx, P. Hegerich, G. van der Groen, F. E. McCutchan and D. S. Burke, J. Virol., 1995, 69, 263–271 CrossRef CAS PubMed.
- D. L. Robertson, J. P. Anderson, J. A. Bradac, J. K. Carr, B. Foley, R. K. Funkhouser, F. Gao, B. H. Hahn, M. L. Kalish, C. Kuiken, G. H. Learn, T. Leitner, F. McCutchan, S. Osmanov, M. Peeters, D. Pieniazek, M. Salminen, P. M. Sharp, S. Wolinsky and B. Korber, Science, 2000, 288, 55–56 CrossRef CAS PubMed.
- M. L. Kalish, K. E. Robbins, D. Pieniazek, A. Schaefer, N. Nzilambi, T. C. Quinn, M. E. St Louis, A. S. Youngpairoj, J. Phillips, H. W. Jaffe and T. M. Folks, Emerging Infect. Dis., 2004, 10, 1227–1234 CAS.
- J. K. Carr, M. O. Salminen, C. Koch, D. Gotte, A. W. Artenstein, P. A. Hegerich, D. St Louis, D. S. Burke and F. E. McCutchan, J. Virol., 1996, 70, 5935–5943 CrossRef CAS PubMed.
- K. E. Nelson, D. D. Celentano, S. Suprasert, N. Wright, S. Eiumtrakul, S. Tulvatana, A. Matanasarawoot, P. Akarasewi, S. Kuntolbutra, S. Romyen, N. Sirisopana and C. Theetranont, JAMA, J. Am. Med. Assoc., 1993, 270, 955–960 CrossRef CAS PubMed.
- P. H. Kilmarx, S. Supawitkul, M. Wankrairoj, W. Uthaivoravit, K. Limpakarnjanarat, S. Saisorn and T. D. Mastro, AIDS, 2000, 14, 2731–2740 CrossRef CAS PubMed.
- Y. Feng, X. He, J. H. Hsi, F. Li, X. Li, Q. Wang, Y. Ruan, H. Xing, T. T. Lam, O. G. Pybus, Y. Takebe and Y. Shao, AIDS, 2013, 27, 1793–1802 CrossRef PubMed.
- F. E. McCutchan, A. W. Artenstein, E. Sanders-Buell, M. O. Salminen, J. K. Carr, J. R. Mascola, X. F. Yu, K. E. Nelson, C. Khamboonruang, D. Schmitt, M. P. Kieny, J. G. McNeil and D. S. Burke, J. Virol., 1996, 70, 3331–3338 CrossRef CAS PubMed.
- W. Deng, P. Fu, L. Bao, N. Vidal, Q. He, C. Qin, M. Peeters, E. Delaporte, J. M. Andrieu and W. Lu, AIDS, 2009, 23, 977–985 CrossRef PubMed.
- D. G. Myszka, R. W. Sweet, P. Hensley, M. Brigham-Burke, P. D. Kwong, W. A. Hendrickson, R. Wyatt, J. Sodroski and M. L. Doyle, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9026–9031 CrossRef CAS PubMed.
- D. Lyumkis, J. P. Julien, N. de Val, A. Cupo, C. S. Potter, P. J. Klasse, D. R. Burton, R. W. Sanders, J. P. Moore, B. Carragher, I. A. Wilson and A. B. Ward, Science, 2013, 342, 1484–1490 CrossRef CAS PubMed.
- J. P. Julien, A. Cupo, D. Sok, R. L. Stanfield, D. Lyumkis, M. C. Deller, P. J. Klasse, D. R. Burton, R. W. Sanders, J. P. Moore, A. B. Ward and I. A. Wilson, Science, 2013, 342, 1477–1483 CrossRef CAS PubMed.
- C. K. Leonard, M. W. Spellman, L. Riddle, R. J. Harris, J. N. Thomas and T. J. Gregory, J. Biol. Chem., 1990, 265, 10373–10382 CrossRef CAS PubMed.
- B. Chen, E. M. Vogan, H. Gong, J. J. Skehel, D. C. Wiley and S. C. Harrison, Nature, 2005, 433, 834–841 CrossRef CAS PubMed.
- C. C. Huang, F. Stricher, L. Martin, J. M. Decker, S. Majeed, P. Barthe, W. A. Hendrickson, J. Robinson, C. Roumestand, J. Sodroski, R. Wyatt, G. M. Shaw, C. Vita and P. D. Kwong, Structure, 2005, 13, 755–768 CrossRef CAS PubMed.
- C. C. Huang, S. N. Lam, P. Acharya, M. Tang, S. H. Xiang, S. S. Hussan, R. L. Stanfield, J. Robinson, J. Sodroski, I. A. Wilson, R. Wyatt, C. A. Bewley and P. D. Kwong, Science, 2007, 317, 1930–1934 CrossRef CAS PubMed.
- P. D. Kwong, R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski and W. A. Hendrickson, Nature, 1998, 393, 648–659 CrossRef CAS PubMed.
- P. Saha, B. Barua, S. Bhattacharyya, M. M. Balamurali, W. R. Schief, D. Baker and R. Varadarajan, Biochemistry, 2011, 50, 7891–7900 CrossRef CAS PubMed.
- D. Sharma, M. M. Balamurali, K. Chakraborty, S. Kumaran, S. Jeganathan, U. Rashid, P. Ingallinella and R. Varadarajan, Biochemistry, 2005, 44, 16192–16202 CrossRef CAS PubMed.
- M. T. Muhammed and E. Aki-Yalcin, Chem. Biol. Drug Des., 2019, 93, 12–20 CrossRef CAS PubMed.
- A. Singh, T. Dauzhenka, P. J. Kundrotas, M. J. E. Sternberg and I. A. Vakser, Proteins, 2020, 88, 1180–1188 CrossRef CAS PubMed.
- W. Chen, Y. Feng, R. Gong, Z. Zhu, Y. Wang, Q. Zhao and D. S. Dimitrov, J. Virol., 2011, 85, 9395–9405 CrossRef CAS PubMed.
- M. R. Wilkins, E. Gasteiger, A. Bairoch, J. C. Sanchez, K. L. Williams, R. D. Appel and D. F. Hochstrasser, Methods Mol. Biol., 1999, 112, 531–552 CAS.
- E. Gasteiger, A. Gattiker, C. Hoogland, I. Ivanyi, R. D. Appel and A. Bairoch, Nucleic Acids Res., 2003, 31, 3784–3788 CrossRef CAS PubMed.
- A. Sahay, A. Piprodhe and M. Pise, J. Genet. Eng. Biotechnol., 2020, 18, 44 CrossRef PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS PubMed.
- M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T. Gallo Cassarino, M. Bertoni, L. Bordoli and T. Schwede, Nucleic Acids Res., 2014, 42, W252–W258 CrossRef CAS PubMed.
- B. G. Pierce, K. Wiehe, H. Hwang, B. H. Kim, T. Vreven and Z. Weng, Bioinformatics, 2014, 30, 1771–1773 CrossRef CAS PubMed.
- W. Chen, Y. Feng, P. Prabakaran, T. Ying, Y. Wang, J. Sun, C. D. Macedo, Z. Zhu, Y. He, V. R. Polonis and D. S. Dimitrov, J. Virol., 2014, 88, 1125–1139 CrossRef PubMed.
- C. C. Huang, M. Venturi, S. Majeed, M. J. Moore, S. Phogat, M. Y. Zhang, D. S. Dimitrov, W. A. Hendrickson, J. Robinson, J. Sodroski, R. Wyatt, H. Choe, M. Farzan and P. D. Kwong, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2706–2711 CrossRef CAS PubMed.
- I. T. Desta, K. A. Porter, B. Xia, D. Kozakov and S. Vajda, Structure, 2020, 28, 1071–1081 CrossRef CAS PubMed.
- D. Kozakov, D. R. Hall, B. Xia, K. A. Porter, D. Padhorny, C. Yueh, D. Beglov and S. Vajda, Nat. Protoc., 2017, 12, 255–278 CrossRef CAS PubMed.
- S. Vajda, C. Yueh, D. Beglov, T. Bohnuud, S. E. Mottarella, B. Xia, D. R. Hall and D. Kozakov, Proteins: Struct., Funct., Bioinf., 2017, 85, 435–444 CrossRef CAS PubMed.
- P. D. Kwong, R. Wyatt, S. Majeed, J. Robinson, R. W. Sweet, J. Sodroski and W. Hendrickson, Structure, 2000, 8, 1329–1339 CrossRef CAS PubMed.
- U. Esser, R. F. Speck, K. C. Deen, R. E. Atchison, R. Sweet and M. A. Goldsmith, AIDS Res. Hum. Retroviruses, 2000, 16, 1845–1854 CrossRef CAS PubMed.
- S. W. de Taeye, A. T. de la Peña, A. Vecchione, E. Scutigliani, K. Sliepen, J. A. Burger, P. van der Woude, A. Schorcht, E. E. Schermer, M. J. van Gils, C. C. LaBranche, D. C. Montefiori, I. A. Wilson, J. P. Moore, A. B. Ward and R. W. Sanders, J. Biol. Chem., 2018, 293, 1688–1701 CrossRef CAS PubMed.
- R. Guo, Z. Cang, J. Yao, M. Kim, E. Deans, G. Wei, S. G. Kang and H. Hong, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 22146–22156 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |