
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovery of new STAT3 inhibitors as anticancer agents using ligand-receptor contact fingerprints and docking-augmented machine learning†
Nour Jamal Jaradata,
Walhan Alshaer b,
Mamon Hatmalc and
Mutasem Omar Taha*a
b,
Mamon Hatmalc and
Mutasem Omar Taha*a
aDepartment of Pharmaceutical Sciences, Faculty of Pharmacy, University of Jordan, Amman 11492, Jordan. E-mail: mutasem@ju.edu.jo; Fax: +962 6 5339649; Tel: +962 6 5355000 ext. 23305
bCell Therapy Center, The University of Jordan, Amman 11942, Jordan
cDepartment of Medical Laboratory Sciences, Faculty of Applied Medical Sciences, The Hashemite University, P.O. Box 330127, Zarqa 13133, Jordan
First published on 3rd February 2023
Abstract
STAT3 belongs to a family of seven vital transcription factors. High levels of STAT3 are detected in several types of cancer. Hence, STAT3 inhibition is considered a promising therapeutic anti-cancer strategy. In this work, we used multiple docked poses of STAT3 inhibitors to augment training data for machine learning QSAR modeling. Ligand–Receptor Contact Fingerprints and scoring values were implemented as descriptor variables. Escalating docking-scoring consensus levels were scanned against orthogonal machine learners, and the best learners (Random Forests and XGBoost) were coupled with genetic algorithm and Shapley additive explanations (SHAP) to identify critical descriptors that determine anti-STAT3 bioactivity to be translated into pharmacophore model(s). Two successful pharmacophores were deduced and subsequently used for in silico screening against the National Cancer Institute (NCI) database. A total of 26 hits were evaluated in vitro for their anti-STAT3 bioactivities. Out of which, three hits of novel chemotypes, showed cytotoxic IC50 values in the nanomolar range (35 nM to 6.7 μM). However, two are potent dihydrofolate reductase (DHFR) inhibitors and therefore should have significant indirect STAT3 inhibitory effects. The third hit (cytotoxic IC50 = 0.44 μM) is purely direct STAT3 inhibitor (devoid of DHFR activity) and caused, at its cytotoxic IC50, more than two-fold reduction in the expression of STAT3 downstream genes (c-Myc and Bcl-xL). The presented work indicates that the concept of data augmentation using multiple docked poses is a promising strategy for generating valid machine learning models capable of discriminating active from inactive compounds.
1. Introduction
STAT3 is a member of the “signal transducers and activators of transcription STATs” family of oncogenic transcription factors. This family also includes STAT 1, 2, 3, 4, 5a, 5b and 6. These transcription factors remain latent in the cytoplasm until being activated by cytokines, e.g., interleukin-6 (IL-6) and growth factors (FGF, IGF and EGF) at which point they get phosphorylated, dimerize and move to the nucleus, where they begin activating the transcription of various genes involved in a variety of cellular processes.1,2 STAT3 affects genes associated with proliferation (e.g., Bcl-2, Bcl-xL, survivin, cyclin D1, c-Myc and Mcl-1), angiogenesis (e.g., Hif1 and VEGF) and epithelial–mesenchymal transition (e.g., vimentin, TWIST, MMP-9 and MMP-7).3,4 Fig. 1 summarizes the signaling pathway of STAT3.5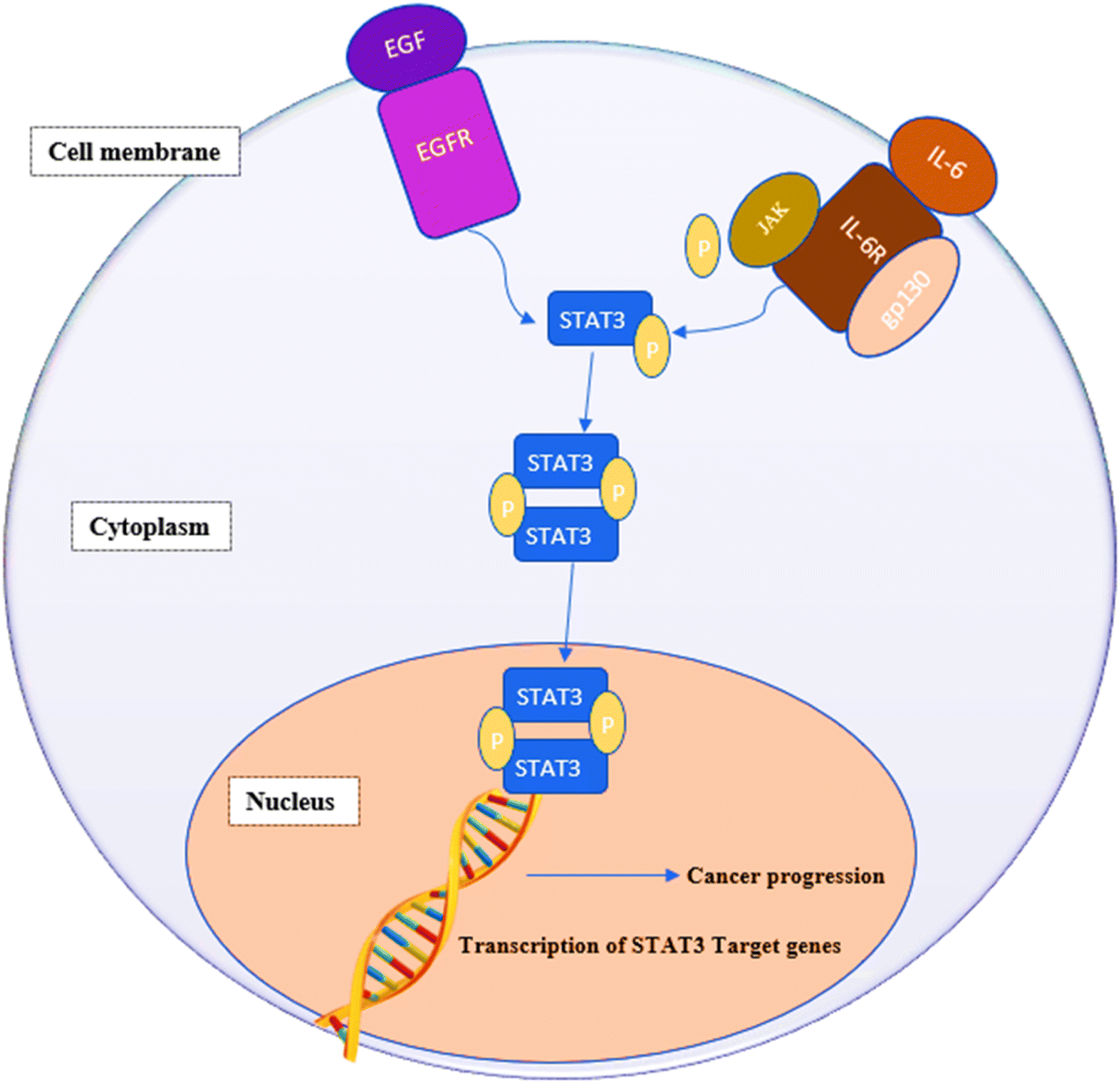 | ||
| Fig. 1 IL-6/STAT3 signaling pathway in cancer cells. IL-6 binds to membrane-bound IL-6 receptors α (IL-6R) and β (also known as gp130). The IL-6/IL-6R/gp130 complex activates phosphorylation of JAKs, followed by STAT3 phosphorylation and activation. Growth factors, such as FGF, IGF and EGF, can also phosphorylate STAT3 by binding to their membrane receptors. Phosphorylated STAT3 dimerizes and translocates into the nucleus where it binds to the promotor region of target genes and activates their transcription. | ||
STAT3 is extensively expressed in a variety of cancers such as human solid tumors.5,6 Blocking constitutively active STAT3 signaling causes tumor cells to die but has little effect on healthy cells. Additionally, STAT3 inhibition attenuates resistance to anticancer chemo- and radiotherapy.7 Furthermore, STAT3 inhibition prevents the transition of normal cells into tumor cells making this oncogenic protein an attractive target for cancer drug discovery.8–10
STAT3 inhibitors can be classified based on their mode of action into direct or indirect blockers. Direct inhibitors bind STAT3 domains, while indirect inhibitors affect STAT3 through cellular networks.9 Fig. 2 shows examples on potent STAT3 direct inhibitors.11–15
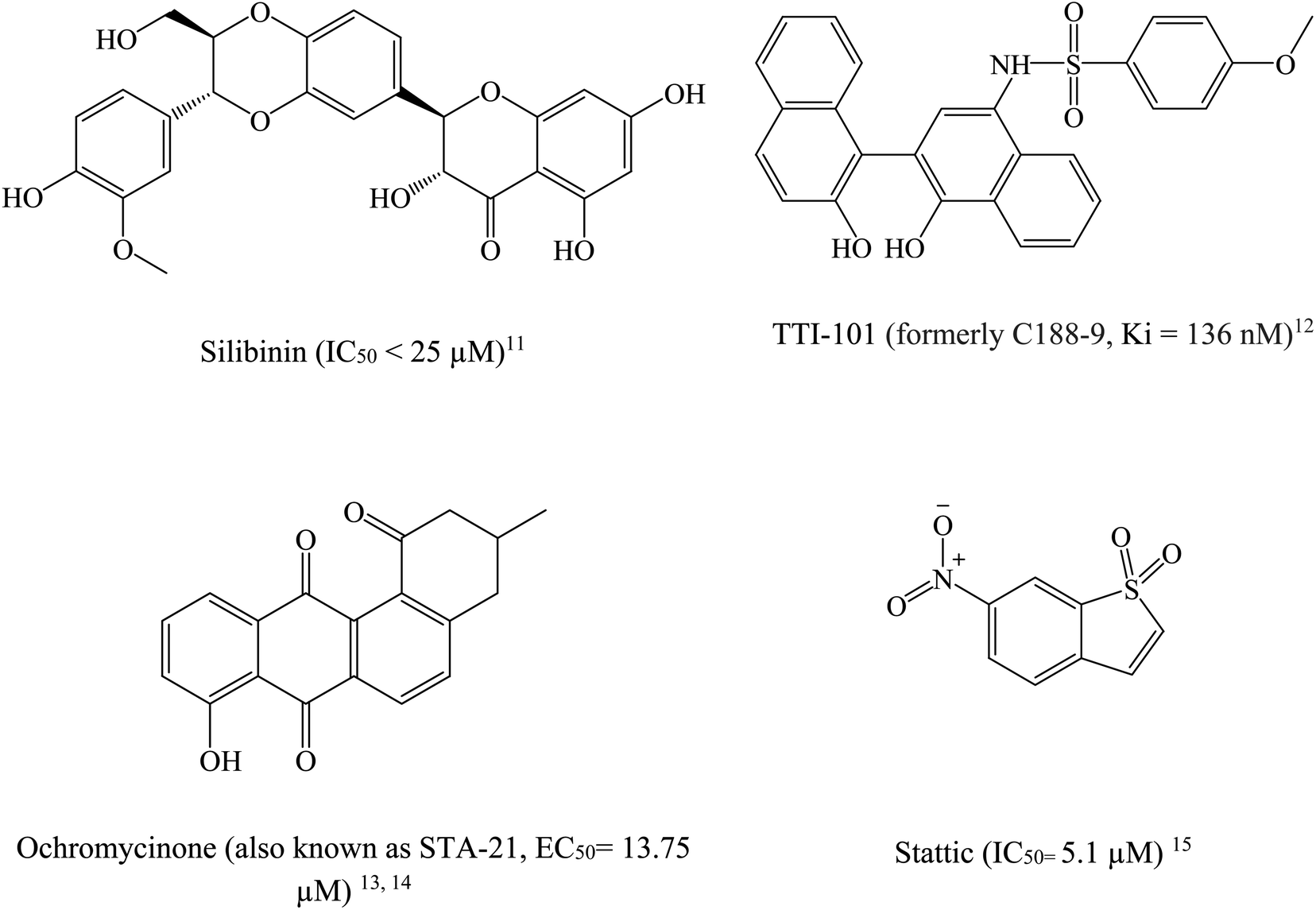 | ||
| Fig. 2 Chemical structures of some reported direct STAT3 inhibitors. | ||
Ligand–Receptor Contacts Fingerprint (LRCF) is a binary vector made up of bins filled with “ones” or “zeroes” corresponding to binding site atoms in the target protein that either engage or avoid a docked ligand pose.16–19
Machine learning (ML) in molecular modelling is the application of statistical approaches to learn and predict molecular properties.20,21 Some of the most often applied machine learning algorithms in drug design and discovery applications include: eXtreme Gradient Boosting (XGBoost);22 Random Forest (RF);21 Naive Bayesian (NB);23,24 k-nearest neighbors (kNN);17 Probabilistic Neural Networks (PNN)25,26 and multilayer perceptron MLP27(see ESI section SM2†).
The term data augmentation refers to methods to create additional training samples that will ultimately enhance machine learning model performance and reduce overfitting.28
In the current project we used numerous docked poses, generated by multiple docking engines and scoring functions for a list of active and inactive STAT3 ligands, to augment bioactivity ML classifiers. LRCFs and scoring function values were implemented as descriptors in ML models to classify STAT3 ligands into “active” or “inactive” categories. Since docking algorithms are usually successful in achieving enthalpically reasonable docked poses, especially for potent ligands, it can be reasonably assumed that ML-based agreement on a specific set of contact atoms inside the binding site (i.e., LRCFs) underlines their ability (i.e., the particular set of contact atoms) to classify docked virtual hits as being active or inactive.19
Upon testing many orthogonal MLs, the best performing MLs were paired with genetic function algorithm (GFA) to pinpoint particular descriptors (ligand–receptor contacts and/or scoring functions) that best explain bioactivity variation among training and testing compounds. The relative contribution of each descriptor in bioactivity class predictions was explained using Shapley values (SHAP).29,30 Subsequently, pharmacophore models were built based on GA-selected descriptors of consistent SHAP probabilities. Valid models were utilized as 3D search queries to look for novel STAT3 inhibitors from the NCI's database. Fig. 3 summarizes the workflow implemented in this study. High ranking hits were tested in vitro.
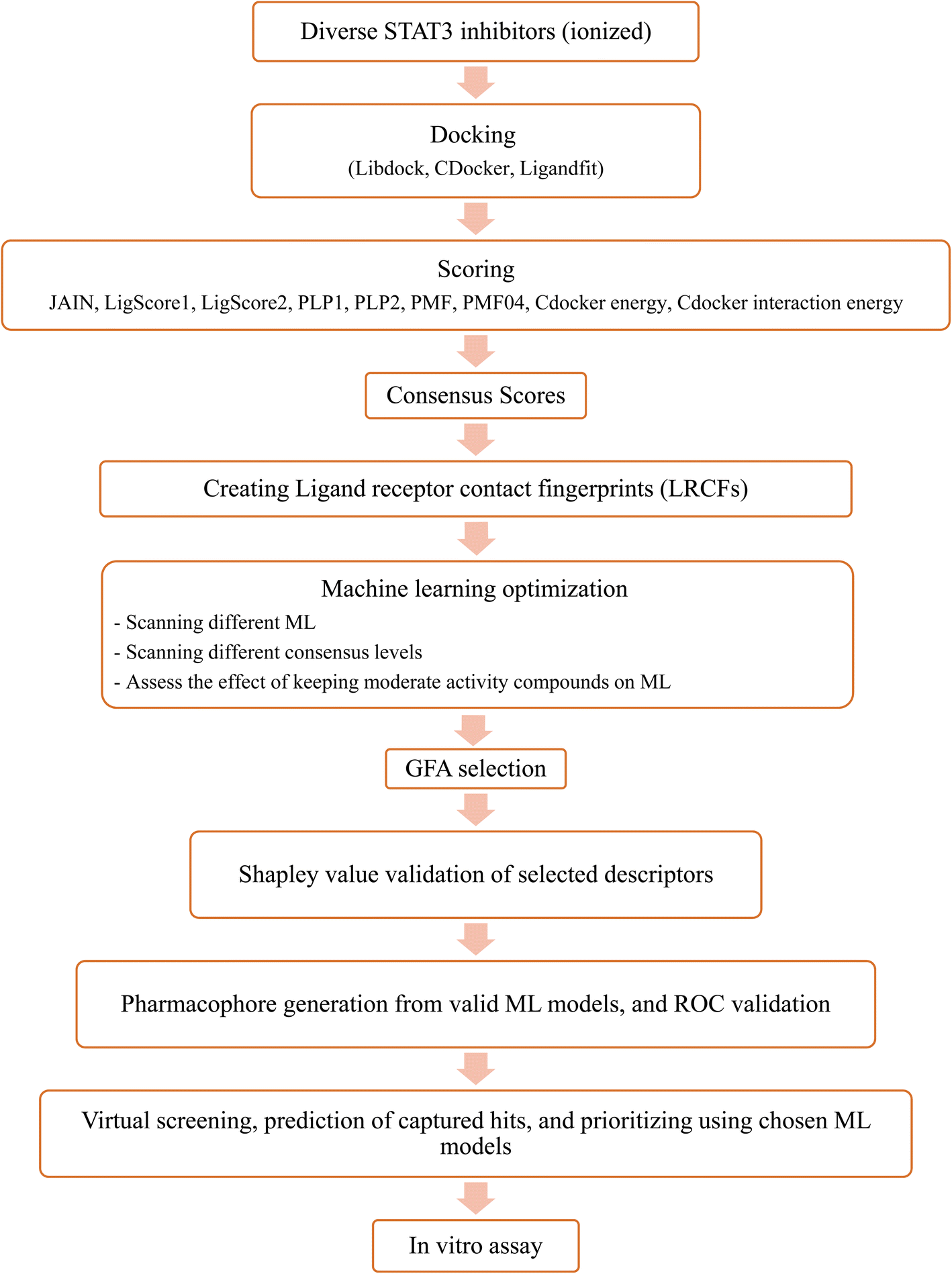 | ||
| Fig. 3 Summary of the workflow implemented in the current project. | ||
2. Materials and methods
The following software packages were used in this project:• BIOVIA DiscoveryStudio (Version 4.5), Biovia Inc. (https://www.3dsbiovia.com/), USA.
• In house built package to generate ligand–receptor contacts fingerprints written in Fortran.
• KNIME Analytics Platform (Version 4.3.3), https://www.knime.com/.
• CS ChemDraw Ultra (Version 7.0.1) Cambridge Soft Corp. (http://www.cambridgesoft.com), USA.
• Marvin View (ChemAxon Ltd., USA).
2.1 Data collection
STAT3 inhibitors were mined from the European Bioinformatics Institute database (ChEMBL) (https://www.ebi.ac.uk/chembl/).59–61 The collected compounds (935 antagonists) were carefully checked for errors, duplicate structures, and chirality. Erroneous structures, duplicates and racemic compounds were excluded, and only ligands that bind specifically to the SH2 domain were kept leaving 314 remaining inhibitors. These were divided into 116 actives (IC50 ≤ 5000 nM), 92 moderates (5000 nM < IC50 < 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nM), and 106 inactives (IC50 ≥ 20000 nM). Ligands were ionized as guided by Marvin View (ChemAxon Ltd., USA) at pH of 7.4. Accordingly, amine groups were protonated and assigned formal positive charge, while carboxylic acids, phosphoric acids, and sulfonamides were deprotonated and assigned formal negative charges. ESI Table S1† lists the chemical structures of modeled compounds in SMILE formats together with their reported bioactivities.
000 nM), and 106 inactives (IC50 ≥ 20000 nM). Ligands were ionized as guided by Marvin View (ChemAxon Ltd., USA) at pH of 7.4. Accordingly, amine groups were protonated and assigned formal positive charge, while carboxylic acids, phosphoric acids, and sulfonamides were deprotonated and assigned formal negative charges. ESI Table S1† lists the chemical structures of modeled compounds in SMILE formats together with their reported bioactivities.
2.2 Molecular modeling
2.3 Machine learning
All elements of machine learning (ML), such as scanning different learners, selecting descriptors using a genetic algorithm (GA), and evaluating models using accuracy, Cohen's kappa values, and Shapley values (SHAP), were done using graphical programming within the KNIME analytics platform (Version 4.3.3).To evaluate different MLs, LRCFs and scoring functions values (LigScore1, LigScore2, PLP1, PLP2, PMF, PMF04, JAIN, CDocker-energy, and CDocker-interaction energy) were considered as the independent variables (descriptors), while the corresponding activity classes (active, inactive and intermediate) were considered as the response. Six orthogonal MLs were scanned, namely, Random Forests (RF),21 eXtreme Gradient Boosting (XGBoost),22 Naive Bayes (NB),49 Probabilistic Neural Network (PNN),25,26 k-Nearest Neighbors (kNN),17 and Multilayer Perceptron (MLP).27
The classification power of each ML was judged based on its ability to correctly classify the docked poses of training and testing ligands into actives, inactives and intermediates. Two ML success criteria were considered, namely, accuracy (eqn (1))50 and Cohen's Kappa values (eqn (2)).51
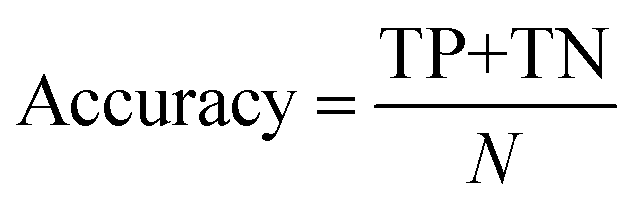 | (1) |
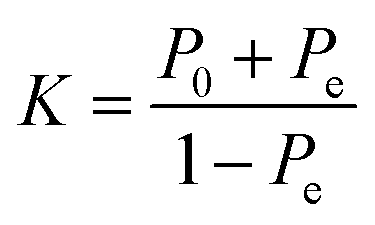 | (2) |
Evaluation against the training set involve removing 20% (i.e., leave-20%-out or 5-fold cross-validation) of the data points (i.e., docked poses), then building the particular ML-QSAR model from the remaining 80% data. The model is then used for classifying the removed 20% compounds. The process is repeated until all training data points are removed from the training list and predicted at least once. Accuracy and Cohen's Kappa values were calculated based on comparing classification results with actual bioactivity classes. Evaluation against the testing set involved calculating the accuracy or Cohen's Kappa values of the particular ML-QSAR model by comparing its predicted classification results with the actual bioactivity classes of the external testing set.52,53 Details about MLs are provided in ESI Section SM2.†
Two subsequent GA-phases were implemented in the current project: An initial preliminary simplistic phase was performed with population size and genetic iterations of 50 and 100, respectively, to narrow down the number of descriptors from 471 to 50. A subsequent more thorough GFA phase was performed on this list of descriptors with population size and genetic iterations of 500 and 5000, respectively, to refine the descriptors into a range of 10 to 20 variables.
2.4 Pharmacophore generation from docked poses
The Receptor–Ligand Pharmacophore Generation Protocol of Discovery Studio 4.5 was used to extract a maximum of 10 pharmacophore models from a docked pose of 115 (most potent within the testing list, IC50 = 136 nM, ESI Table S1†) selected because it has the highest probability contributions towards “Active” label by GFA-selected descriptors as determined by SHAP analysis (within the respective optimal ML model, see Section 3.4). This protocol (i.e., Receptor–Ligand Pharmacophore Generation Protocol) selects certain subsets from ligand–receptor binding interactions and translates them into pharmacophore models. The following settings were implemented in this protocol: binding site hydration water molecules were kept, range of allowed number of features = 4 to 6. The generated pharmacophores were ranked according to rules-based selectivity scoring function.56 Maximum charge–charge interaction distance = 8.0 Å (if the distance between a charged feature in the ligand and its nearest protein counterpart is longer than this value the electrostatic features will be removed). Maximum hydrogen bond distance = 4.0 Å (if the distance between two hydrogen-bonded heavy atoms is larger than this value then no hydrogen bonding feature will be added). Maximum hydrophobic distance = 5.5 (this is the maximum distance in Å between the center of a hydrophobic feature in a ligand and the nearest hydrophobic residue to permit adding hydrophobic feature). Maximum exclusion volume distance = 4.0 Å (this setting generates pharmacophore models without exclusion volumes). Minimum interfeature distance = 1.0 Å (this is the minimum distance between features in Å). The generated pharmacophores were validated using receiver operating characteristic (ROC)50,57,58 against a list of active (116) and inactive (106) STAT3 inhibitors extracted from ChEMBL59–61 with a maximum of 100 conformers per ligand.2.5 In silico screening for new STAT3 inhibitors and bioactivity prediction using ML models
Optimal pharmacophore models were employed as 3D search queries to screen the national cancer institute (NCI) list of compounds. Screening was performed employing the ‘‘Search 3D Database’’ protocol implemented within Discovery Studio (Version 4.5). Top 100 hits of highest fit values against each pharmacophore were docked into STAT3 protein (PDB code: 6njs) using docking-scoring settings that were used with the corresponding models (mentioned in section 2.2.1 to 2.2.2). Subsequently, the docked poses were filtered according to docking scoring consensus level (i.e., ≥1) and RMSD filter (2.0 Å) (see Section 2.2.3). Corresponding LRCFs and scoring values were substituted in the respective ML models (RF or XGBoost) to predict the activity label of each docked pose/conformer. This resulted in a situation where each screened compound yielded a set of poses that are assigned either “active” or “inactive” labels. Therefore, a threshold was defined to consider certain hit molecule as being promising or not. It was decided to define such a threshold based on predicted active/inactive poses ratios within the corresponding testing set. The least active-to-inactive ratio of unequivocally documented active inhibitor was used as threshold for prioritizing hits.19 Table 6 shows the percentages of active poses of testing compounds as predicted by the top two ML (i.e., XGboost and RF). Hits predicted to exceed the proposed active/inactive ratio threshold were requested from the NCI. However, only subset of the requested compounds were readily available from the NCI.2.6 Bioassay of NCI hits
Acquired hits from the NCI were bioassayed by two methods: (i) MTT cytotoxicity test62 against a panel of cell lines, and (ii) polymerase chain reaction (PCR) to detect the expression of STAT3-downstream genes: c-Myc, and Bcl-xl (see ESI Section SM3†).3. Results and discussion
Mining ChEMBL database for STAT3 inhibitors identified 930 inhibitors. Following data curation (deleting duplicates and indirect inhibitors) furnished 314 direct STAT3 inhibitors, out of which 116 ligands had IC50 values ≤ 5000 nM were labeled as “active”, 92 compounds of IC50 values ranging from 5000 to 20000 nM were labeled as “intermediate”, and 106 ligands of IC50 values ≥ 20000 nM were allocated “inactive” labels.
3.1 Scanning different docking-scoring consensus levels and machine learners
The collected compounds were docked into the binding pocket of STAT3 (PDB code: 6njs) using three docking engines (LibDock, LigandFit and CDocker). Docked poses were pooled and scored by 9 docking scoring functions (generally orthogonal, see ESI Table S3† for cross correlation matrix). Consensus scoring was performed in such a way that if a docked pose scored within the top 20% of a particular scoring function it receives the vote of this scoring function. Summing up votes of different scoring functions yields consensus scoring of the particular docked pose.19 However, we opted to remove closely similar docked poses/conformers to avoid noise leading to machine learning over-fitting errors.67 Accordingly, any cluster of docked poses within RMSD ≤ 2.0 Å was represented by a single pose (of highest consensus score) in subsequent steps. Table 1 details the counts of docked poses before and after RMSD filtrations for the ionized docked ligands.| Count of poses | ||||
|---|---|---|---|---|
| Actives | Intermediate | Inactive | Total | |
| Docked poses before RMSD filtering | 18143 |
14381 |
16022 |
48546 |
| Docked poses after RMSD filtering | 13408 |
10993 |
10794 |
35195 |
Clearly from Table 1, although the RMSD filtering reduced the number of docked poses, still, the remaining poses are significant augmentation of the collected modelled compounds (314 collected ligands were augmented to 35195 docked poses).
We propose that the convergence of high-quality docked poses (i.e., reasonable docking scoring consensus) of active ligands on specific, distinct binding site contacts, while the same contacts are avoided by docked poses of inactive or intermediately-active ligands, underlines the significance of these contact points as discriminators of bioactivity classes. However, it is necessary to identify the best possible docked poses that (i) augment training and testing data and (ii) define significant discriminatory binding site contacts. Moreover, it is necessary to identify optimal machine learner(s) for this purpose.
Therefore, we scanned the effects of escalating docking-scoring consensus levels and orthogonal machine learners on the classification capacities of corresponding ML models as reflected by their Cohen's Kappa values (Fig. 4).51 However, it must be mentioned that escalating the level docking-scoring consensus reduces the counts of docked training and testing compounds and corresponding counts of docked poses, which might undermine our intended data augmentation leverage via multiple docked poses.
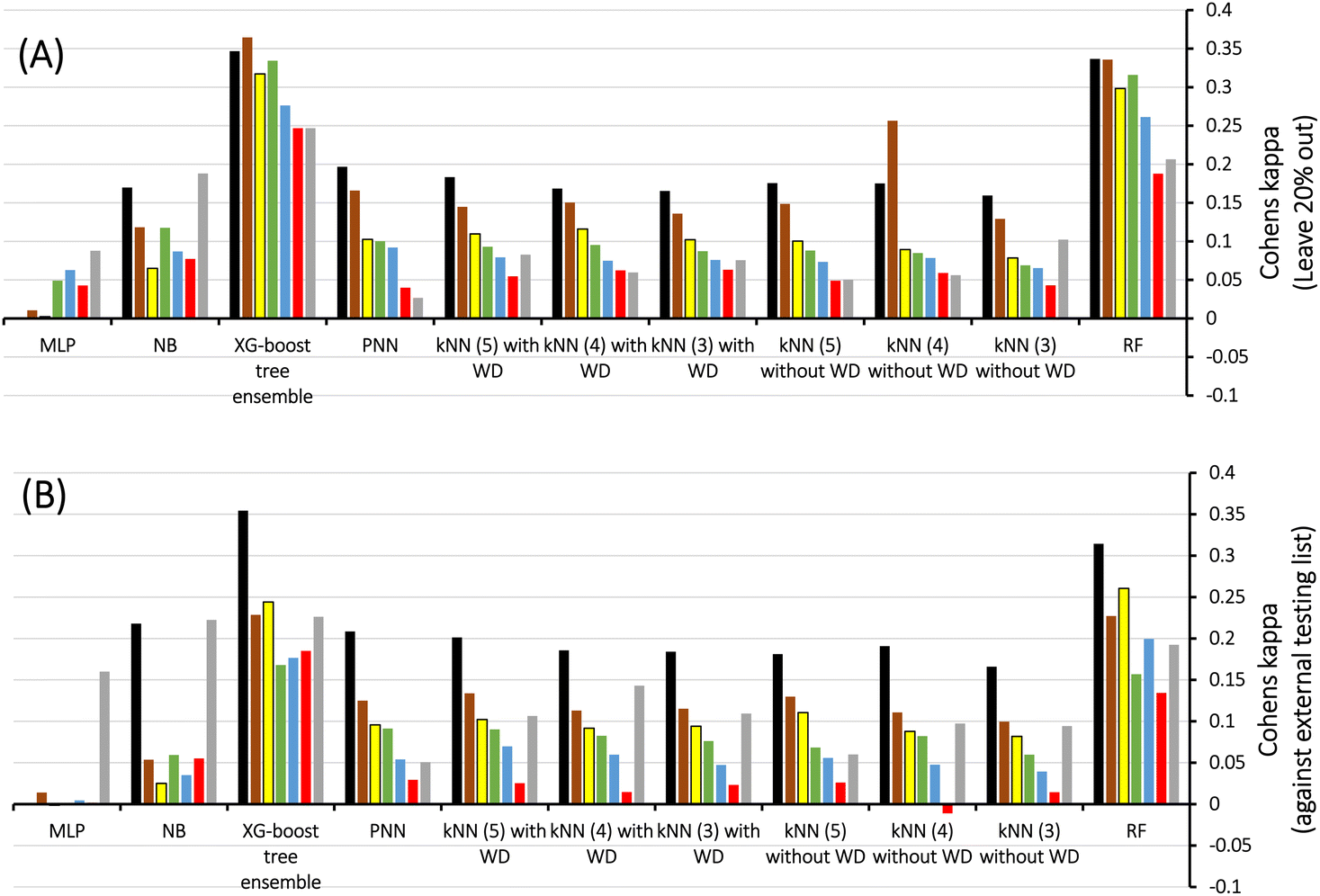 | ||
Fig. 4 Scanning Cohen's Kappa values against different MLs using LRCFs and scoring functions generated for docked poses of: (A) Training compounds, (B) Testing compounds, WD encode for kNN weighted distances, numbers in brackets correspond to count of nearest neighbors. Consensus scoring levels are color coded as follows:  at least 1, at least 1,  at least 2, at least 2,  at least 3, at least 3,  at least 4, at least 4,  at least 5, at least 5,  at least 6 and at least 6 and  at least 7 consensus level. at least 7 consensus level. | ||
Fig. 4 shows Cohen's Kappa values for several machine learners (RF, kNN XGBoost, PNN, NB, and MLP) at different docking-scoring consensus levels for training and testing sets. Clearly, RF and XGBoost were the best performing machine learners. Additionally, the results of the training and testing data highlight a consensus level of “at least one” (≥1) docking-scoring function to achieve best results among training and testing data. Interestingly, the corresponding counts of docked poses (Table 2) suggest significant data augmentation at this scoring consensus level (i.e., ≥1). That is, 249 training compounds were augmented to 16317 docked poses (Table 2), while 61 testing compounds were augmented to 4017 docked poses.
| Count of docked poses (corresponding count of compounds in brackets) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Level of consensus score | Training | Testing | ||||||
| Actives | Inter-mediate | Inactives | Total | Actives | Inter-mediate | Inactives | Total | |
| ≥1 | 7637(91) | 4303(73) | 4377(85) | 16317(249) |
1792(22) | 1134 (18) | 1091(21) | 4017(61) |
| ≥2 | 5854(88) | 2967(72) | 3243(81) | 12064 (241) |
1347(21) | 742(18) | 720(21) | 2809(60) |
| ≥3 | 4453(88) | 2335(72) | 2240(76) | 9028(236) | 1065(21) | 563 (17) | 740 (20) | 2368 (58) |
| ≥4 | 3815(88) | 1896(70) | 1970(75) | 7681(233) | 918(21) | 485(18) | 472(19) | 1875(58) |
| ≥5 | 3114(88) | 1460(71) | 1364(68) | 5938(227) | 740(21) | 336(17) | 418(18) | 1494(56) |
| ≥6 | 2236(87) | 910(64) | 782(56) | 3928(207) | 519(21) | 195(16) | 243(14) | 957(51) |
| ≥7 | 831(86) | 313(49) | 284(46) | 1428(181) | 206(22) | 78(13) | 57(11) | 341(46) |
| ≥8 | 59(37) | 32(10) | 25(9) | 116(56) | 15 (9) | 7(3) | 3(2) | 25 (14) |
| ≥9 | 12(12) | 4(3) | 3(3) | 19(18) | 3(2) | 1(1) | 1(1) | 5 (4) |
Table 3 shows the influence of incorporating/deleting intermediately-active compounds on resulting ML models. Unsurprisingly, deleting this class enhanced the corresponding ML models. This is not unexpected since moderate compounds create noise in ML models and hence should be hard to classify. Accordingly, it was decided to exclude moderate compounds from subsequent ML modeling.
| Learner | Intermediate activity class | Accuracy | Cohen's Kappa | ||
|---|---|---|---|---|---|
| L20% outa | Testingb | L20% outa | Testingb | ||
| a L20% out: leave 20% out cross-validation for accuracy and Cohen's Kappa.b Testing: accuracy and Cohen's Kappa determined against the testing set (marked with a in Table S1). | |||||
| XGboost | With | 0.60 | 0.60 | 0.35 | 0.35 |
| Without | 0.77 | 0.76 | 0.49 | 0.46 | |
| RF | With | 0.60 | 0.58 | 0.34 | 0.31 |
| Without | 0.77 | 0.76 | 0.47 | 0.45 | |
3.2 Veracity of the selected docking scoring settings
To evaluate the veracity of the docking-scoring consensus level ≥1, we compared the docked poses of a co-crystallized ligand (PDB code: KQV), at scoring consensus ≥1, with crystallographic pose of the same ligand. Interestingly, out of 48 docked poses of scoring consensus ≥1, 5 were of RMSD ≤ 2.00 Å from the experimental bound pose, while 10 were of RMSD ≤ 2.50 Å. Fig. 5 shows the best docked poses compared to the crystallographic bound pose of KQV. These results highlight the ability of docking-scoring consensus level ≥ 1 to reproduce the crystallographic pose among its solutions.19,68–70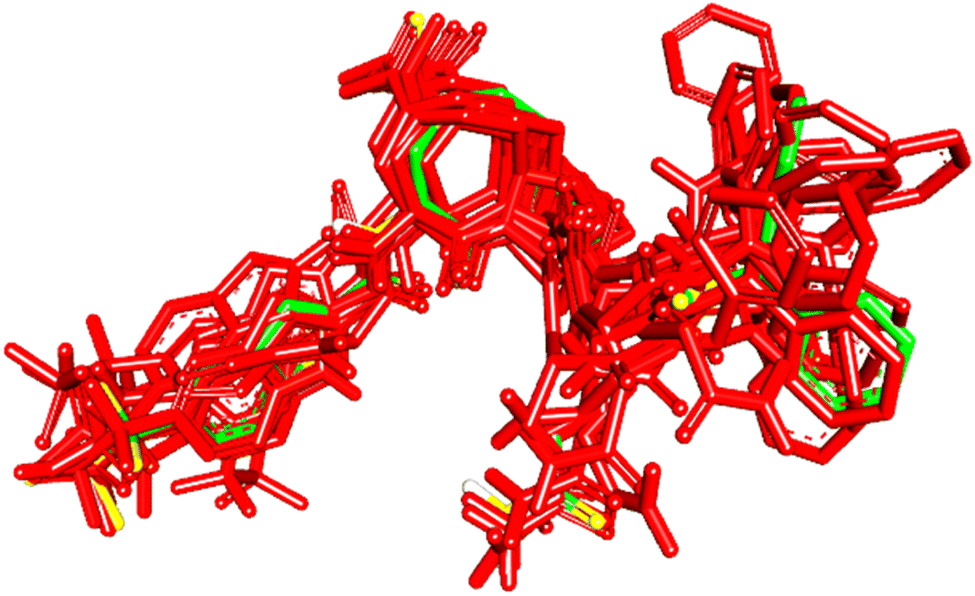 | ||
| Fig. 5 High-ranking docked poses (red) compared to crystallographic bound pose (green) of KQV (PDB code: 6NJS). | ||
3.3 Building genetic algorithm–machine learning models
In order to identify important descriptors that control bioactivity category within training and testing docked poses (at docking-scoring consensus ≥1), we felt it was necessary to couple optimal MLs with genetic function algorithm (GFA). GFA-ML modeling commenced by pooling the values of nine docking-scoring functions (see experimental part) with 471 ligand/binding site contact points as descriptors. The bioactivity class (i.e., active or inactive) was enlisted as dependent response variable. Descriptors were allowed to compete within the context of GFA tournaments using Cohen's Kappa of the resulting models as GFA fitness criteria.54 The GFA-ML models were validated by external testing as well as internal leave-20%-out cross validation. Table 4 shows the selected descriptors and statistical results of the two top classifiers.| Learner | Features selectora | Descriptorsb | Accuracy | Cohen's Kappa | ||
|---|---|---|---|---|---|---|
| L20% outc | Testingd | L20% outc | Testingd | |||
| a GFA: genetic function algorithm, SHAP: the SHapley Additive exPlanations.b Amino acid and water heavy atom contacts are coded according to the protein databank, while hydrogen atoms are coded according to Discovery Studio 4.5. LigScore2, PMF, PMF04, Cdocker energy, Cdocker interaction energy, represent scoring values.c L20% out: leave 20% out cross-validation for accuracy and Cohen's Kappa.d Testing: accuracy and Cohen's Kappa determined against the testing set (Table S1 under ESI). | ||||||
| Xgboost | GFA | Ligscore2, PLP2, PMF, PMF04, Cdocker energy, GLU 594 HG2, SER 613 HN, TRP 623 CZ2, VAL 637 HA, GLN 643 HG1, GLY 656C, TYR 657 HE1, LYS 658 HG1, MET 660 HB1, MET 660 HE2, MET 660 O, PRO 669 CB, HOH 32 OH2, HOH 60H2, HOH 107 OH2 | 0.744 | 0.734 | 0.414 | 0.404 |
| GFA + SHAP | PLP2, PMF, Cdocker energy, SER 613 HN | 0.714 | 0.719 | 0.348 | 0.368 | |
| RF | GFA | PMF04, Cdocker energy, Cdocker interaction energy, SER 613 HG, GLN 633 HE22, PRO 639 CD, TYR 640 HE1, TYR 657 OH, LYS 658 CG, ILE 659 HG11, ALA 662 HA, HOH 32 OH2, HOH 37H2, HOH 70 OH2, HOH 107 OH2, HOH 107H2, HOH 170 OH2, HOH 255H1, HOH 255H2, HOH 269H2 | 0.749 | 0.735 | 0.404 | 0.392 |
| GFA + SHAP | Cdocker energy, Cdocker interaction energy | 0.634 | 0.635 | 0.169 | 0.181 | |
Clearly from Table 4 and comparison with Table 3, it was possible to successfully reduce the number of features from ca. 471 to ca. 20 using genetic selection with negligible loss in models' accuracies and Cohen's kappa values highlighting the significance of the shortened lists of descriptors. Nevertheless, it is still hard to infer the role of each descriptor in predicting the bioactivity class of a particular compound based on GFA-ML models in Table 4. For example, it is hard to tell how certain LRCF (e.g., e.g., TYR 640 HE1) contributes to the bioactivity class within the context of GFA-RF ML model (in Table 4). Therefore, we decided to implement Shapley additive explanations (SHAP) to explain the relative contributions of individual descriptors in bioactivity class predictions for each GFA-ML model,29,55 as in Fig. 6.
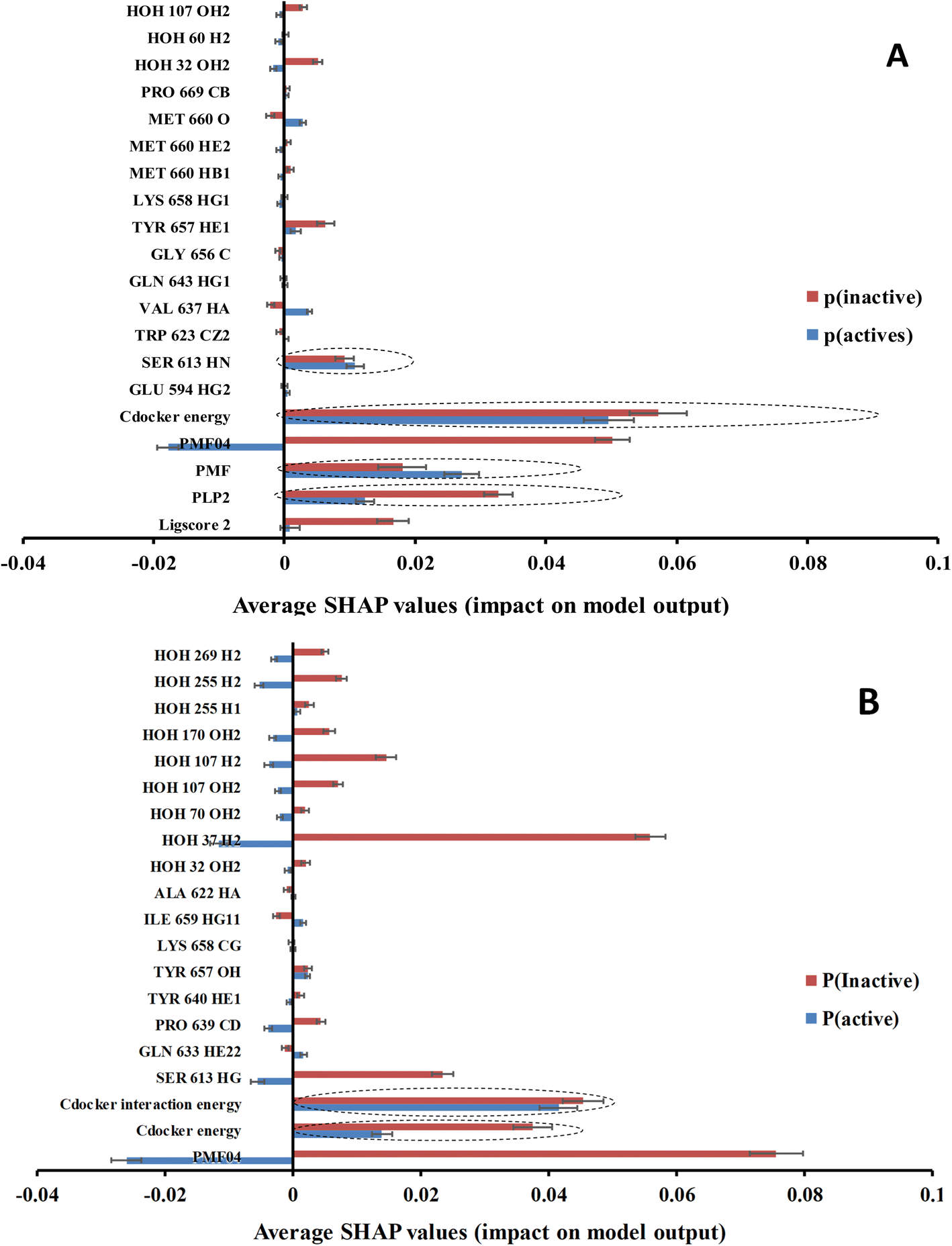 | ||
Fig. 6 SHAP probability contributions of descriptors emerging in optimal (A): GA-XGBoost and (B): GA-RF models within the testing set compounds. Average probability contribution for “inactive” prediction among inactive compounds are represented with red ( ) bar, average probability contribution for “active” prediction among active compounds are represented with ( ) bar, average probability contribution for “active” prediction among active compounds are represented with ( ) bar. Error bars represent the standard error of the average. SHAP-consistent features are encircled with blue dotted lines. ) bar. Error bars represent the standard error of the average. SHAP-consistent features are encircled with blue dotted lines. | ||
In the context of cheminformatics machine learning, SHAP values enable the identification and prioritization of features that control bioactivity prediction regardless to ML model.29 SHAP value of a particular feature for certain compound indicates how much this feature has contributed to the deviation of the prediction of that compound from mean prediction. Each average SHAP value in Fig. 6 was calculated as the mean of SHAP values of the particular descriptor across active or inactive testing compounds.
Interestingly, Fig. 6 shows that only few GFA-selected descriptors have average SHAP probability contributions consistent with corresponding bioactivity categories, i.e., they yielded positive probabilities towards the “active” label classification within active testing compounds and likewise showed positive probability contributions towards “inactive” label within the inactive testing category. These are encircled in Fig. 6. Remarkably, deleting inconsistent descriptors from GA-ML models in Table 4 caused only moderate detrimental effects on the corresponding predictive qualities (see GFA + SHAP selectors in Table 4).
3.4 Building pharmacophore models
GFA-selected descriptors of consistent SHAP probabilities were used to select a single docked pose for the most potent inhibitor in the testing list (115, IC50 = 136 nM, ESI Table S1†) to be used as template for pharmacophore building. The selected pose is characterized with the highest probability contributions towards the “active” label among other docked poses of 115 based on SHAP-consistent descriptors. It can be reasonably argued that this pose represents the most probable way by which 115 binds within STAT3 binding site according to the considered ML model. Corresponding pharmacophore models are then generated using the Ligand–Receptor Pharmacophore Generation protocol within Discovery Studio (see Section 2.4).For example, the docked pose in Fig. 8A is of the highest probability contributions by GFA-selected, SHAP-consistent, descriptors within the XGBoost model (Fig. 6A and Table 4, namely, PLP2, PMF, Cdocker energy and SER 613 HN). Hypo-1 pharmacophore (Fig. 8A) was extracted based from this pose via the Ligand-Receptor Pharmacophore Generation protocol. Fig. 7A shows its corresponding ROC curve.
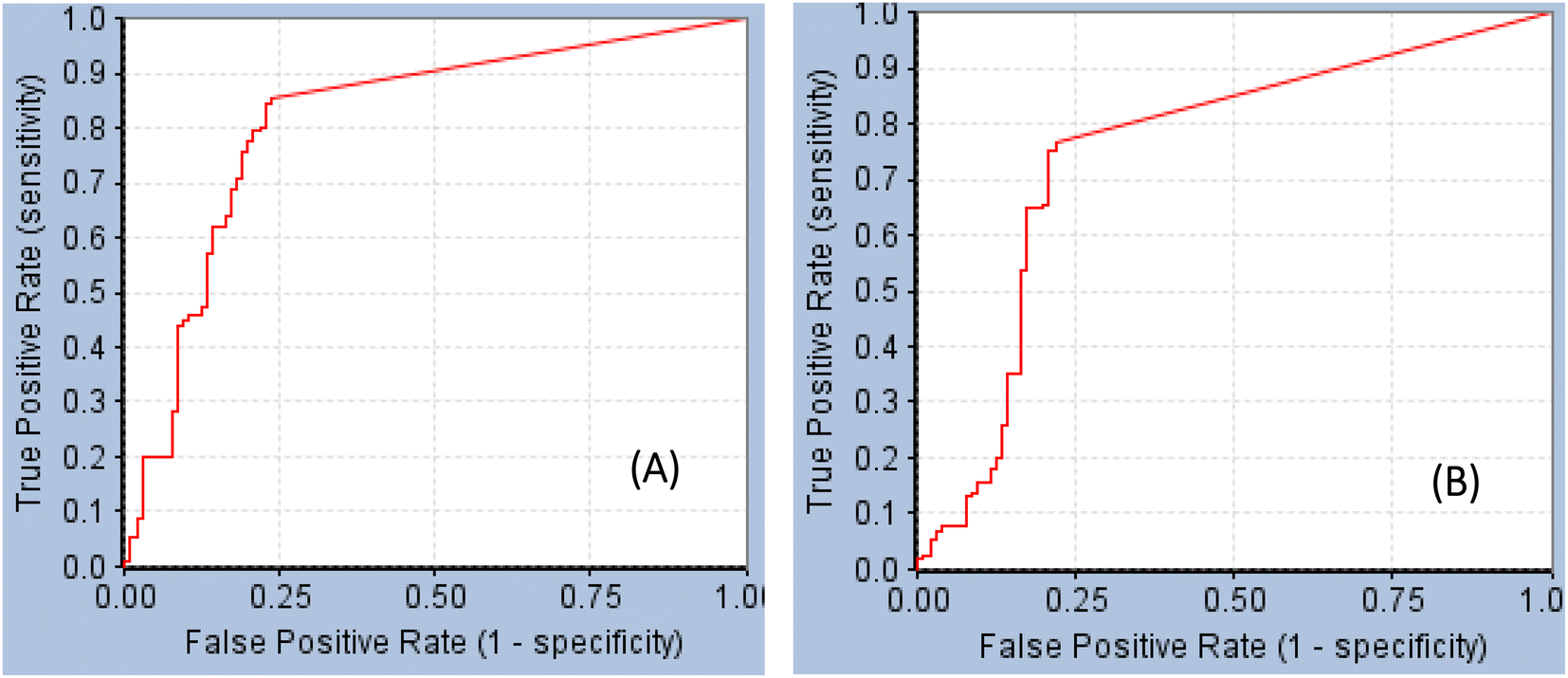 | ||
| Fig. 7 ROC curves of (A) Hypo-1 (AUC = 0.82, sensitivity: 0.85, specificity: 0.76), (B) Hypo-2 (AUC = 0.75, sensitivity: 0.77, specificity: 0.78).71 | ||
Clearly from Fig. 8A, the electrostatic interaction anchoring the docked pose's phosphate group and the guanidine of Arg609 is represented by two overlapping negative ionizable features (NegIon) in Hypo-1. Similarly, the hydrogen bond connecting the terminal hydroxyl of Ser613 to the phosphate ester oxygen atom of 115 via a bridging water molecule (H2O107) is represented in Hypo-1 by hydrogen bond acceptor (HBA) feature. Likewise, the close proximity between the central pyrrolidine ring in the docked ligand pose to the hydrophobic side chain of Val637 indicates mutual hydrophobic attraction that was represented by hydrophobic (Hbic) feature in Hypo-1. Interestingly, although the carboxylic acid side chain of Glu638 is rather flexible and assumes two distinct conformational states in the crystallographic structure of STAT3, it seems that both conformers play critical role in ligand binding: The central amide group of docked 115 (Fig. 8A) is hydrogen-bonded to the carboxylic acid of one of Glu638 conformers, while the other major conformer of this amino acid is hydrogen bonded to the terminal hydroxyl of docked 115 via bridging water molecule (H2O4). The two interactions involving Glu638 conformers are represented in Hypo-1 by two hydrogen-bond donor features. This is a rare example where a flexible amino acid residue plays critical role in ligand binding via more-than-one conformer.
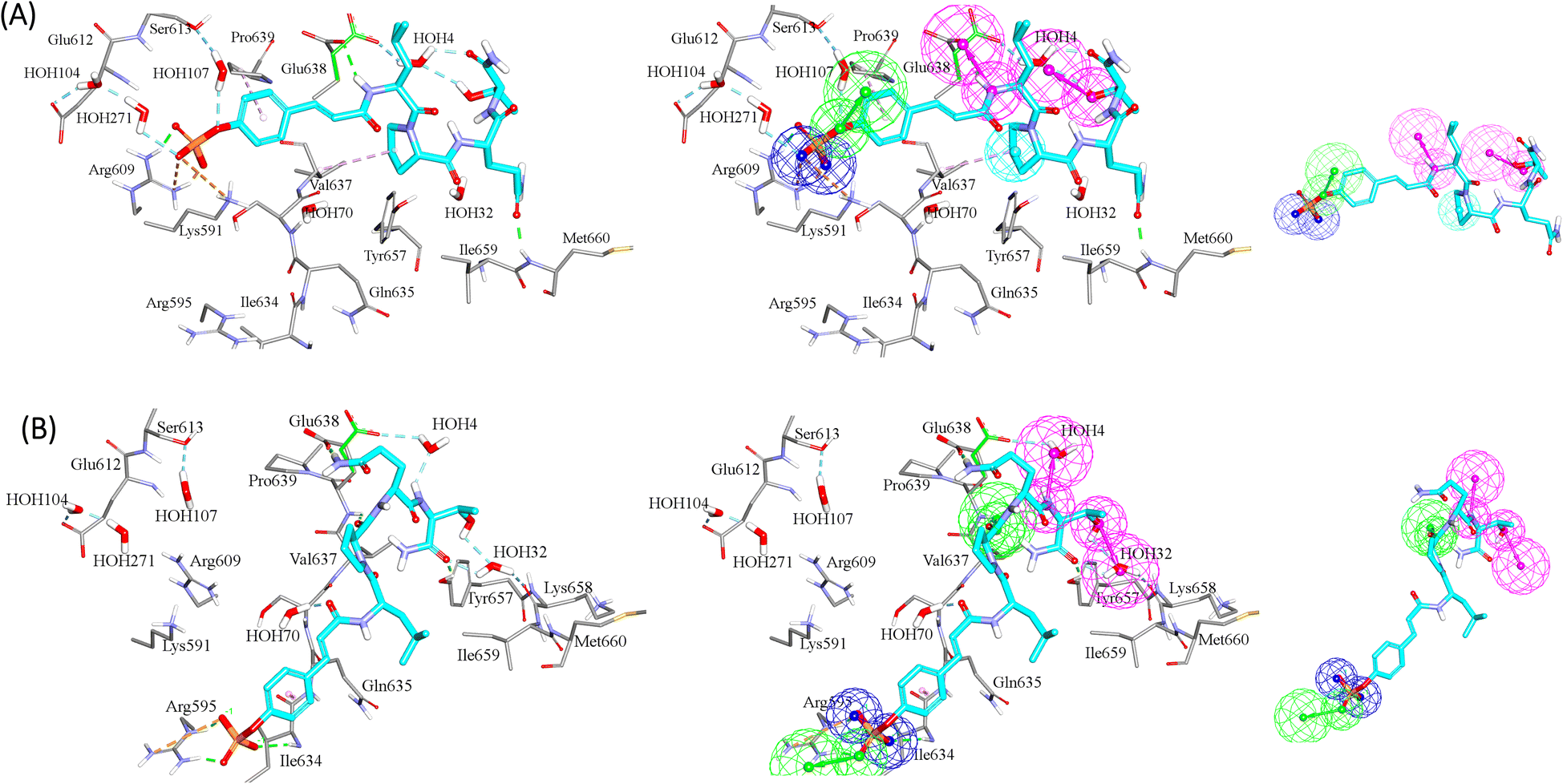 | ||
| Fig. 8 Steps to build pharmacophore models (A) Hypo-1 and (B) Hypo-2 based on SHAP-consistent features identified among GA/XGBoost (for Hypo-1) and GA/RF (for Hypo-2) selected-descriptors. The left images show the docked pose of 115 having the highest “Active” label probability (i.e., among other poses) as contributed by SHAP-consistent features. Hydrogen bonds are shown as green and light blue dotted lines, while hydrophobic interactions are shown as pink dotted lines. The middle images show the pharmacophore hypothesis fitted onto the docked pose. Images to the right show the resulting pharmacophore models. Hydrogen-bond donor (HBD) features are shown as vectored pink spheres, hydrogen bond acceptor (HBA) features are shown as vectored green spheres, hydrophobic (Hbic) features are shown as blue spheres, negative ionizable features are shown as dark blue spheres. | ||
Fig. 8B shows Hypo-2, which corresponds to the docked pose of 115 having the highest probability contributions by GA-selected and SHAP-consistent descriptors within the RF model (Fig. 6B and Table 4, namely, Cdocker energy and Cdocker interaction energy). These descriptors were used in the same way as in Hypo-1 case to generate Hypo-2 (Fig. 8B). Fig. 7 shows its corresponding ROC curve. Clearly from Fig. 8B, the electrostatic and hydrogen bonding interactions tying the terminal phosphate of 115 with the guanidine side chain of Arg595 are represented in Hypo-2 by two overlapping NegIon and a single HBA features. Meanwhile, the hydrogen-bonding connecting the terminal hydroxyl of docked 115 to the peptidic NH of Lys658 via bridging water molecule (H2O32) is represented by HBD feature in Hypo-2. Likewise, the hydrogen bonding interactions anchoring the NH and carbonyl oxygen atoms of the ligand's central amides to the carboxylic acid side chain (one of the conformers via bridging H2O4) and peptidic NH of Glu638 are represented by HBD and HBA, respectively, in Hypo-2.
Clearly from the figure the two pharmacophores, Hypo-1 and Hypo-2, represent significantly discrete binding modes.
3.5 Comparison with pharmacophores extracted from crystallographic complexes
To further validate our ML-generated pharmacophores we decided to compare their performances with naïve counterparts derived from crystallographic complexes. Towards this end, we implemented the Ligand–Receptor Pharmacophore Generation protocol of Discovery Studio (Version 4.5) to extract pharmacophore models from available STAT3 crystallographic structures complexed with SH2 ligands (PDB codes: 6njs and 6nuq). The resulting models were validated by ROC analysis against the same set of actives and inactives used for validating our ML-generated models Hypo-1 and Hypo-2. Only crystallographic pharmacophores of ROC-AUC exceeding 0.70 were kept for comparison with our ML-based pharmacophores.ESI Fig. S1 and S2† show the ROC curves and pharmacophoric features of successful crystallographic pharmacophores. Although the crystallographic pharmacophores were on par with their ML-based counterparts vis-à-vis ROC performances (ESI Fig. S1†), our ML pharmacophores exhibited much better abilities to classify active STAT3 ligands into potent (IC50 < 1.0 μM) and less potent (IC50 ≥ 1.0 μM) inhibitors, as in Fig. 9.
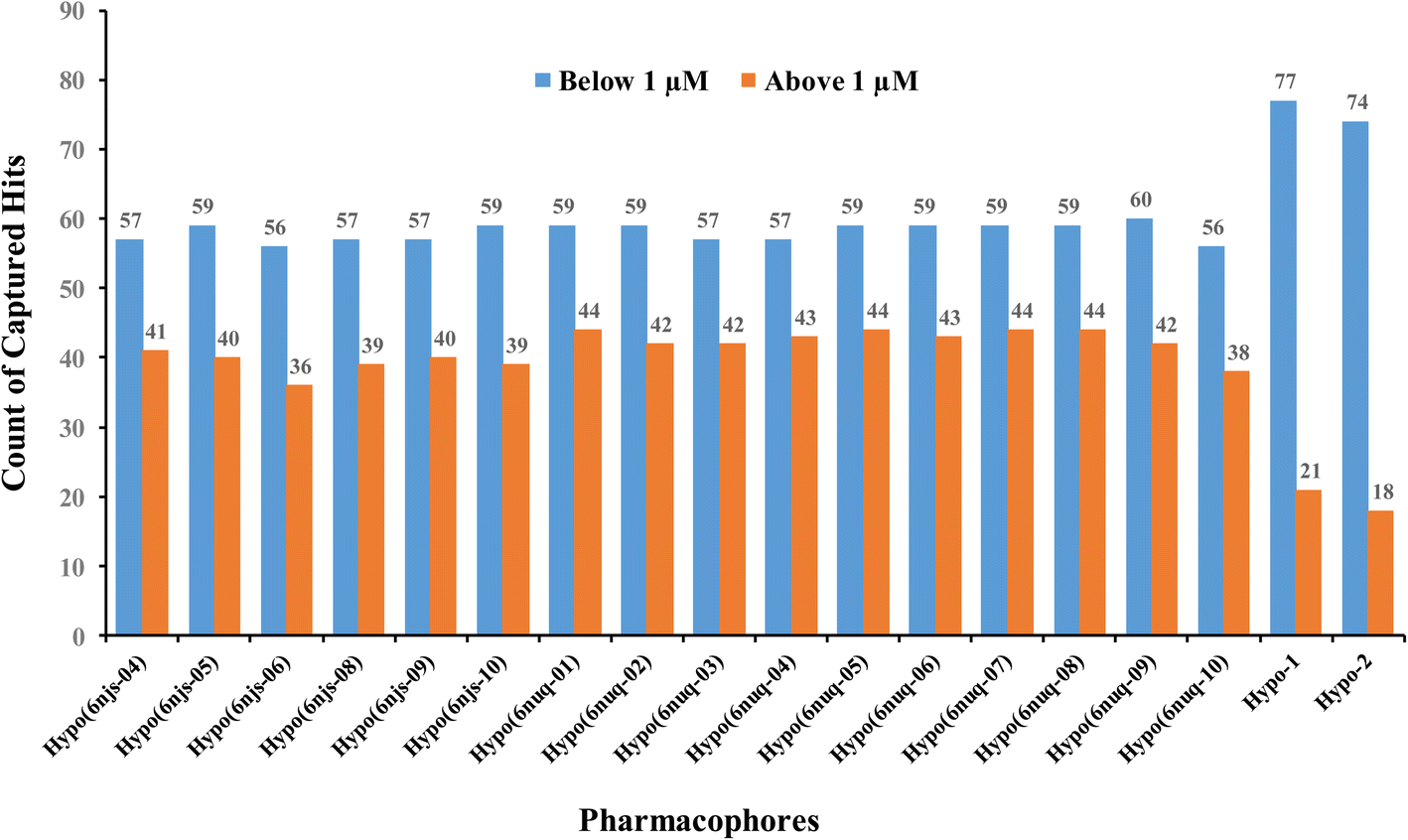 | ||
| Fig. 9 Counts of STAT3 inhibitors of IC50 ≥ or < 1.0 μM captured by successful crystallographic pharmacophores (of ROC-AUC > 0.70) compared to our ML-generated pharmacophores. | ||
This behavior is not unexpected, since the GA-selected and SHAP consistent features used for building pharmacophores Hypo-1 and Hypo-2 were selected by supervised modelling, whereby the bioactivity category dictates which descriptors are significant enough to be selected. These should be able to discriminate highly potent category (IC50 < 1.0 μM) from less potent category (IC50 ≥ 1.0 μM) within the “active” group. To test this theory, we evaluated the significance of difference between the two categories vis-à-vis descriptors used for building Hypo-1 and Hypo-2. ESI Table S4† summarizes the results. As expected, three out of 5 descriptors used for building Hypo-1 and Hypo-2, namely, PLP2, PMF and Cdocker interaction energy, were significantly different between the two groups (of IC50 < 1.0 μM and IC50 ≥ 1.0 μM). Moreover, these same descriptors provided the highest SHAP probability contributions for selecting template poses for building Hypo-1 and Hypo-2.
3.6 In silico screening of the NCI database for new STAT3 inhibitors
The primary use of pharmacophores and related ML models is scaffold hopping, i.e., the identification of new chemotypes with similar biological profiles. Thus, Hypo-1, and Hypo-2 were employed as 3D search queries to screen the NCI list for new STAT3 inhibitors. High ranking hits (according to their fit values58,72) were docked, scored (consensus score of at least 1) and RMSD-filtered utilizing the same settings implemented for the training and testing sets. The resulting docked poses were then used to generate corresponding LRCFs in exactly the same manner as in the training and testing sets. Subsequently, the resulting LRCFs and corresponding scoring values were substituted in the best ML models, namely, GFA-Xgboost, GFA-RF, however, using features selected by the combination of SHAP and GFA (GFA + SHAP selector in Table 4) to predict the activity label of each docked pose/conformer. As a result, each screened compound produced a collection of poses that were either labelled as “active” or “inactive”, as in Table 5. The ratio of docked poses/conformers anticipated to be “active” compared to those predicted to be “inactive” forced us to propose a threshold by which to regard a specific screened molecule as being promising or not. Examining the active/inactive ratios within the active compounds within the testing set is the most logical approach to create such a threshold.19 It is reasonable to presume that an acceptable threshold for discovering potentially new active hits is the least active-to-inactive ratio among well documented active inhibitors. Table 6 shows the percentages of docked poses of active testing set molecules that were correctly labelled as “active” by the two ML models. Clearly, compound 250 (IC50 = 5 μM, Table S1 under ESI†) fulfils the threshold requirements: It exhibits the least predicted active-to-inactive ratio of docked poses among other inhibitors in the testing set for both learners, and therefore, can be used to discriminate actives among screened compounds for the respective ML models. Incidentally, compound 312 failed to achieve sufficient data augmentation as it only produced one docked pose for ML modelling thus it was neglected in the decision related to activity threshold.| Hitsa | NCI Code | Captured Byb | Predicted number of active and inactive docked poses | |||||
|---|---|---|---|---|---|---|---|---|
| Xgboost | RF | |||||||
| Active poses | Inactive poses | Percent active posesc | Active poses | Inactive poses | Percent active posesc | |||
| a Chemical structures are shown in Fig. S3.b 1 represents XGboost-GFA model or Hypo-1, 2 represents RF-GFA model or Hypo-2.c Determined by dividing the number of active poses by the total number of poses (active + inactive). ND: not determined. | ||||||||
| 317 | 3590 | 1,2 | 183 | 31 | 85.5 | 78 | 30 | 72.2 |
| 318 | 20261 |
1 | 40 | 77 | 34.2 | ND | ND | ND |
| 319 | 59407 |
1 | 205 | 34 | 85.8 | ND | ND | ND |
| 320 | 65832 |
1 | 65 | 56 | 53.7 | ND | ND | ND |
| 321 | 745104 |
1,2 | 3 | 3 | 50.0 | 5 | 1 | 33.3 |
| 322 | 72868 |
1 | 56 | 45 | 55.4 | ND | ND | ND |
| 323 | 77028 |
1 | 52 | 64 | 44.8 | ND | ND | ND |
| 324 | 77029 |
1 | 47 | 64 | 42.3 | ND | ND | ND |
| 325 | 82523 |
1 | 152 | 20 | 88.4 | ND | ND | ND |
| 326 | 98711 |
1 | 48 | 71 | 40.3 | ND | ND | ND |
| 327 | 100791 |
1 | 70 | 38 | 64.8 | ND | ND | ND |
| 328 | 107137 |
1 | 102 | 41 | 71.3 | ND | ND | ND |
| 329 | 107139 |
2 | ND | ND | ND | 45 | 11 | 80.4 |
| 330 | 267431 |
1,2 | 40 | 48 | 45.5 | 48 | 37 | 56.5 |
| 331 | 289523 |
1 | 76 | 28 | 73.1 | ND | ND | ND |
| 332 | 338310 |
2 | ND | ND | ND | 129 | 34 | 79.1 |
| 333 | 341076 |
1,2 | 217 | 39 | 84.8 | 189 | 29 | 86.7 |
| 334 | 341077 |
1,2 | 209 | 41 | 83.6 | 180 | 37 | 82.9 |
| 335 | 363007 |
2 | ND | ND | ND | 119 | 42 | 73.9 |
| 336 | 372667 |
1,2 | 158 | 26 | 85.9 | 131 | 40 | 76.6 |
| 337 | 373233 |
1 | 53 | 55 | 49.1 | ND | ND | ND |
| 338 | 380962 |
1 | 45 | 33 | 57.7 | ND | ND | ND |
| 339 | 645793 |
2 | ND | ND | ND | 40 | 23 | 63.5 |
| 340 | 651016 |
2 | ND | ND | ND | 52 | 33 | 61.2 |
| 341 | 669269 |
1,2 | 135 | 54 | 71.4 | 124 | 33 | 79.0 |
| 342 | 722969 |
2 | ND | ND | ND | 51 | 20 | 71.8 |
| Compoundsa | Predicted number of active and inactive docked poses | |||||
|---|---|---|---|---|---|---|
| Xgboost | RF | |||||
| Active poses | Inactive poses | % Active posesb | Active poses | Inactive poses | % Active posesb | |
| a Compounds' numbers and bioactivities are as in Table S1.b Determined by dividing the number of poses labeled as “active” by the total number of poses (labeled as “active” and “inactive”).c The percent active poses of this compound (IC50 = 5000 nM) was used as threshold to classify screened compounds into potential active and inactive STAT3 inhibitors in both GA-RF and GA-XGboost models. | ||||||
| 10 | 105 | 14 | 88.2 | 98 | 21 | 82.4 |
| 22 | 113 | 21 | 84.3 | 99 | 35 | 73.9 |
| 34 | 86 | 22 | 79.6 | 88 | 20 | 81.5 |
| 49 | 76 | 20 | 79.2 | 73 | 23 | 76.0 |
| 54 | 60 | 29 | 67.4 | 57 | 32 | 64.0 |
| 66 | 76 | 20 | 79.2 | 75 | 21 | 78.1 |
| 72 | 98 | 23 | 81.0 | 87 | 34 | 71.9 |
| 78 | 77 | 2 | 97.5 | 65 | 14 | 82.3 |
| 107 | 54 | 17 | 76.1 | 58 | 13 | 81.7 |
| 113 | 57 | 6 | 90.5 | 56 | 7 | 88.9 |
| 115 | 90 | 2 | 97.9 | 73 | 19 | 79.3 |
| 116 | 67 | 22 | 75.3 | 65 | 24 | 73.0 |
| 126 | 97 | 2 | 98.0 | 80 | 19 | 80.8 |
| 127 | 66 | 1 | 98.5 | 55 | 12 | 82.1 |
| 133 | 51 | 2 | 96.2 | 41 | 12 | 77.4 |
| 135 | 65 | 0 | 100 | 56 | 9 | 86.2 |
| 146 | 82 | 5 | 94.3 | 73 | 14 | 83.9 |
| 148 | 54 | 3 | 94.7 | 45 | 12 | 78.9 |
| 156 | 90 | 7 | 92.8 | 85 | 12 | 87.6 |
| 162 | 62 | 5 | 92.5 | 59 | 8 | 88.1 |
| 250c | 10 | 32 | 23.8 | 24 | 18 | 57.1 |
| 312 | 0 | 1 | 0 | 1 | 0 | 100 |
Being above the proposed activity threshold of either ML models, it can be argued that hits 317–342 (Table 5) have promising potential as active STAT3 inhibitors. Accordingly, they were acquired from the national cancer institute (NCI) for in vitro evaluation. ESI Fig. S3† shows the chemical structures of the evaluated hits.
3.7 In vitro bioassay of captured hits
To study the anti-STAT3 inhibitory effects of captured hits we decided to use pyrimethamine as role model. Pyrimethamine, a well-known antimicrobial and antimalarial agent,73 is reported to be indirect selective STAT3 inhibitor65 that acts by blocking dihydrofolate reductase (DHFR) enzyme.63 Pyrimethamine is currently investigated as potential clinically useful STAT3 inhibitor.74 Therefore, we decided to assess the cytotoxic profiles of pyrimethamine (at 10 μM) against 10 cell lines (available within our stock) to identify cells that rely on STAT3 for their survival. The scanned cell lines were normal fibroblasts, HEK-293, 3T3, PANC1, DU145, U87, MDA-MB-231, A549, doxorubicin resistant and sensitive MCF7. Eventually, five cell lines were selected: HEK-239, MCF-7, U87, MDA-MB-231 and Fibroblasts. Of them, normal fibroblasts were found to be least susceptible to pyrimethamine with viability exceeding 90% (Table 7), while HEK293 cells were rather sensitive with viability ≲ 50%. On the other hand, MDA-MB231, U87 and MCF-7 cells were found to exhibit moderate sensitivities to pyrimethamine with viability range from 70–80% (Table 7).| Compound | HEK-293 | MCF-7 | U87 | MDA-MB-231 | Fibroblasts |
|---|---|---|---|---|---|
| a Standard STAT3 inhibitors. | |||||
| 317 | 0 | 7 | 3 | 0 | 0 |
| 318 | 8 | 14 | 4 | 0 | 0 |
| 319 | 60 | 46 | 21 | 36 | 7 |
| 320 | 26 | 8 | 23 | 10 | 0 |
| 321 | 27 | 9 | 3 | 0 | 4 |
| 322 | 11 | 22 | 7 | 23 | 9 |
| 323 | 2 | 13 | 13 | 2 | 0 |
| 324 | 0 | 13 | 7 | 6 | 0 |
| 325 | 38 | 13 | 38 | 8 | 30 |
| 326 | 0 | 3 | 1 | 15 | 10 |
| 327 | 3 | 3 | 0 | 0 | 11 |
| 328 | 0 | 16 | 0 | 0 | 11 |
| 329 | 10 | 15 | 0 | 0 | 12 |
| 330 | 6 | 17 | 0 | 0 | 14 |
| 331 | 0 | 24 | 0 | 2 | 25 |
| 332 | 14 | 20 | 10 | 3 | 0 |
| 333 | 53 | 24 | 3 | 24 | 1 |
| 334 | 39 | 37 | 0 | 1 | 14 |
| 335 | 3 | 21 | 9 | 9 | 18 |
| 336 | 11 | 25 | 0 | 0 | 2 |
| 337 | 37 | 0 | 0 | 21 | 11 |
| 338 | 0 | 12 | 0 | 0 | 13 |
| 339 | 0 | 0 | 16 | 0 | 7 |
| 340 | 31 | 3 | 5 | 0 | 2 |
| 341 | 42 | 44 | 32 | 21 | 7 |
| 342 | 53 | 39 | 18 | 20 | 23 |
| Pyrimethaminea | 53 | 31 | 19 | 20 | 7 |
| Stattica | 88 | 92 | 90 | 87 | 81 |
Table 7 shows the cytotoxic profiles of captured hits compared to the standard STAT3 inhibitors pyrimethamine and stattic.14 Clearly, hits 319, 333 and 342 (structures in ESI Fig. S3†) mimicked the cytotoxic profile of pyrimethamine prompting us to further pursue their IC50 values against the HEK293 (selected because it is the most STAT3-sensitive cell line). Table 8 and Fig. 10A show their IC50 values, Hill Slopes and dose–response correlation r2 values against HEK293 cells. Interestingly, stattic caused lower cellular viabilities at higher concentrations (≥15 μM) compared to all three hits and pyrimethamine, which had their curves plateaued at approximately 40% viability regardless to their escalating concentrations. We propose this behaviour to be due to the fact that stattic exerts nonselective inhibitory profiles against a plethora of targets beside STAT3.75,76
| Hit | IC50a (μM) | Hill lope | r2b |
|---|---|---|---|
| a Each value represents the average of 7 trials, values in brackets represent standard deviation of measurements.b The goodness of fit correlation coefficient of the dose–response curve. | |||
| 319 | 3.50 × 10−2 (±0.004) | 3.7 | 0.93 |
| 333 | 6.74 (±3.55) | 0.8 | 0.95 |
| 342 | 0.44 (±0.10) | 2.8 | 0.91 |
| Stattic | 1.57 (±0.17) | 2.5 | 0.96 |
| Pyrimethamine | 5.12 (±1.19) | 1.8 | 0.86 |
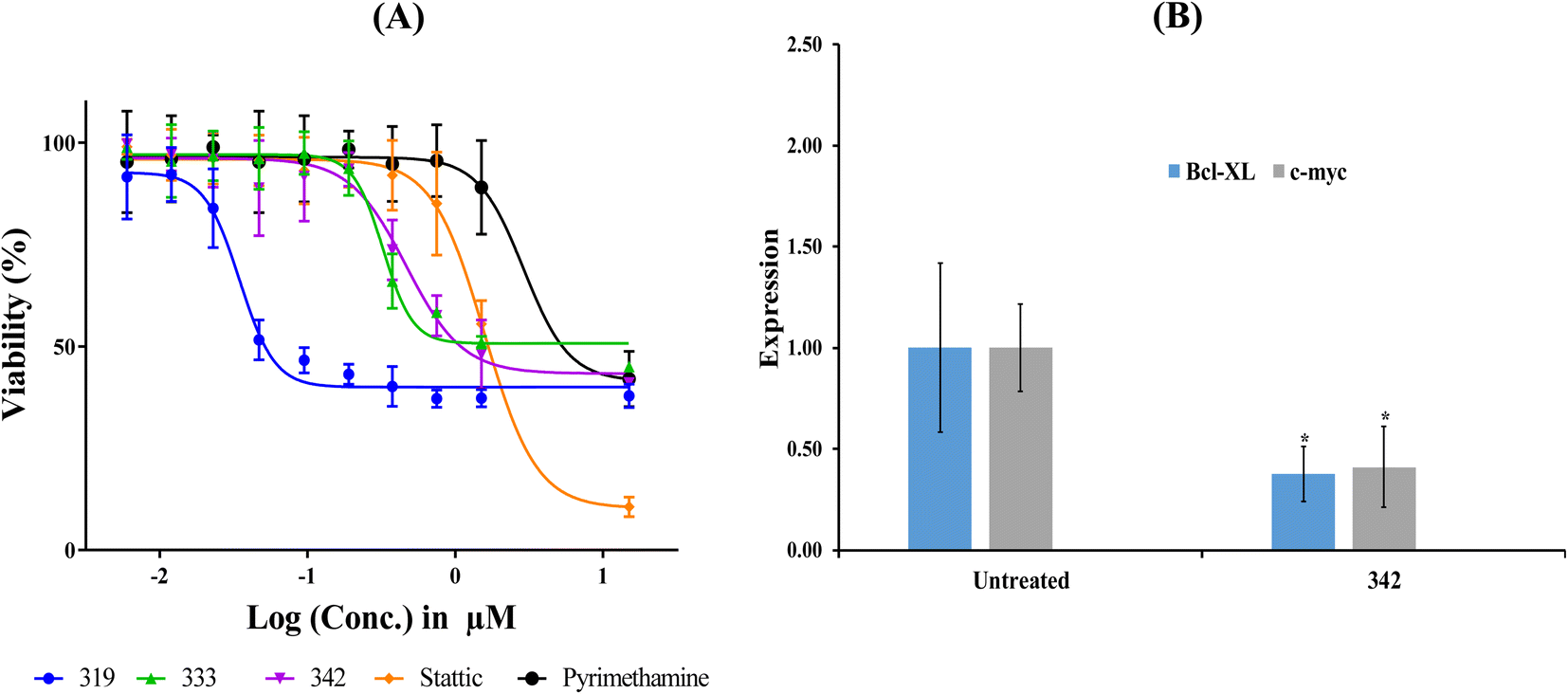 | ||
| Fig. 10 Bioactivity profiles of hit 342. (A) Dose-cellular viability curves of hit 342 compared to stattic and pyrimethamine against HEK 293 cells. (B) Expression of Bcl-xl and c-Myc genes following exposure to 342 at concentration corresponding to anticancer cytotoxic IC50 (see Table 8). Gene expression values were calculated using ΔΔCt method against housekeeping genes (actin-β and 18srRNA). *p < 0.05. | ||
Due to the fact that both 319 and 333 were reported to potently inhibit DHFR enzyme (IC50 values in nanomolar range)77,78 their STAT3 inhibition should be at least partially indirect,63,79 forcing us to exclude them from further evaluation. On the other hand, 342 has been reported to be totally devoid of DHFR inhibitory effects,80 and therefore, its bioactivity is attributable solely to direct binding to STAT3 SH2 domain.
The fact that cytotoxicity cannot be considered as unequivocal evidence of STAT3 inhibition; we opted for additional investigation using quantitative polymerase chain reaction (qPCR) to monitor the effect of 342 on the expression of c-Myc and Bcl-xl genes, both of which are downstream of STAT3. c-Myc and Bcl-xl are key regulators in cellular proliferation81 and evasion of apoptosis,82 respectively.
Fig. 10 shows the effect of 342, at its anti-HEK293 IC50 concentration (Table 8), on the expression of c-Myc and Bcl-xl. Clearly, 342 caused significant suppression of c-Myc and Bcl-xl. These results provide unequivocal evidence on the potent and statistically significant inhibitory effect of 342 against STAT3 at submicromolar levels (i.e., 440 nM).
Fig. 11 shows 342 and how it fits its corresponding capturing pharmacophore (Hypo-2) and how it docks into the binding pocket of STAT3 SH2 domain.
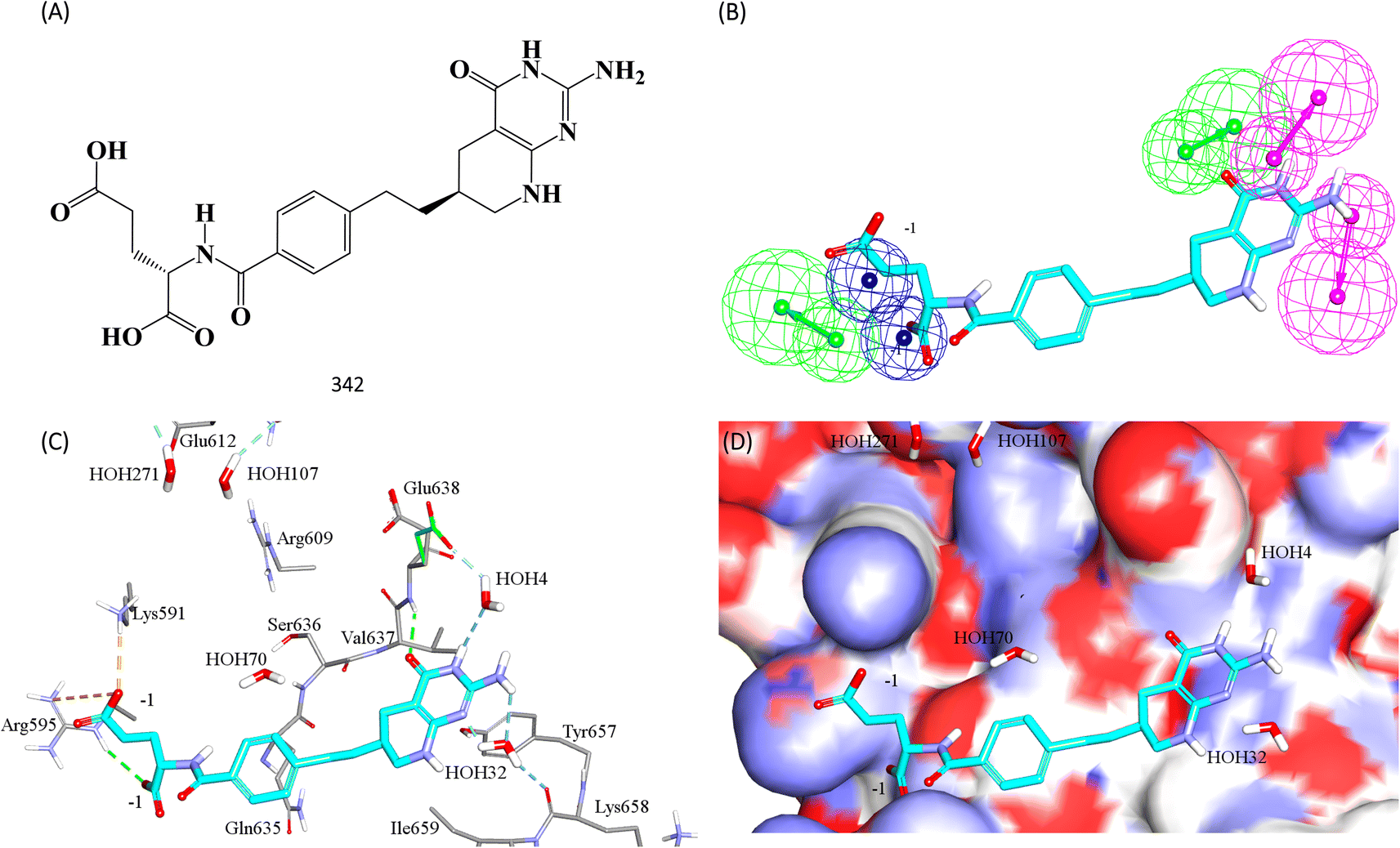 | ||
| Fig. 11 Hit 342 mapped against its capturing pharmacophore (A) structure of 342 (B) 342 fitted against Hypo-2, (C) 342 docked into STAT3 (PDB code: 6njs), (D) 342 docked into STAT3 with binding site covered with Connolly's surface. | ||
Principal component analysis (PCA) (Fig. 12) shows 342 to be significantly different chemotype compared to known potent STAT3 SH2 blockers (IC50 ≤ 5 μM) albeit drug-like and satisfies Lipinski's83 and Veber's84 rules.
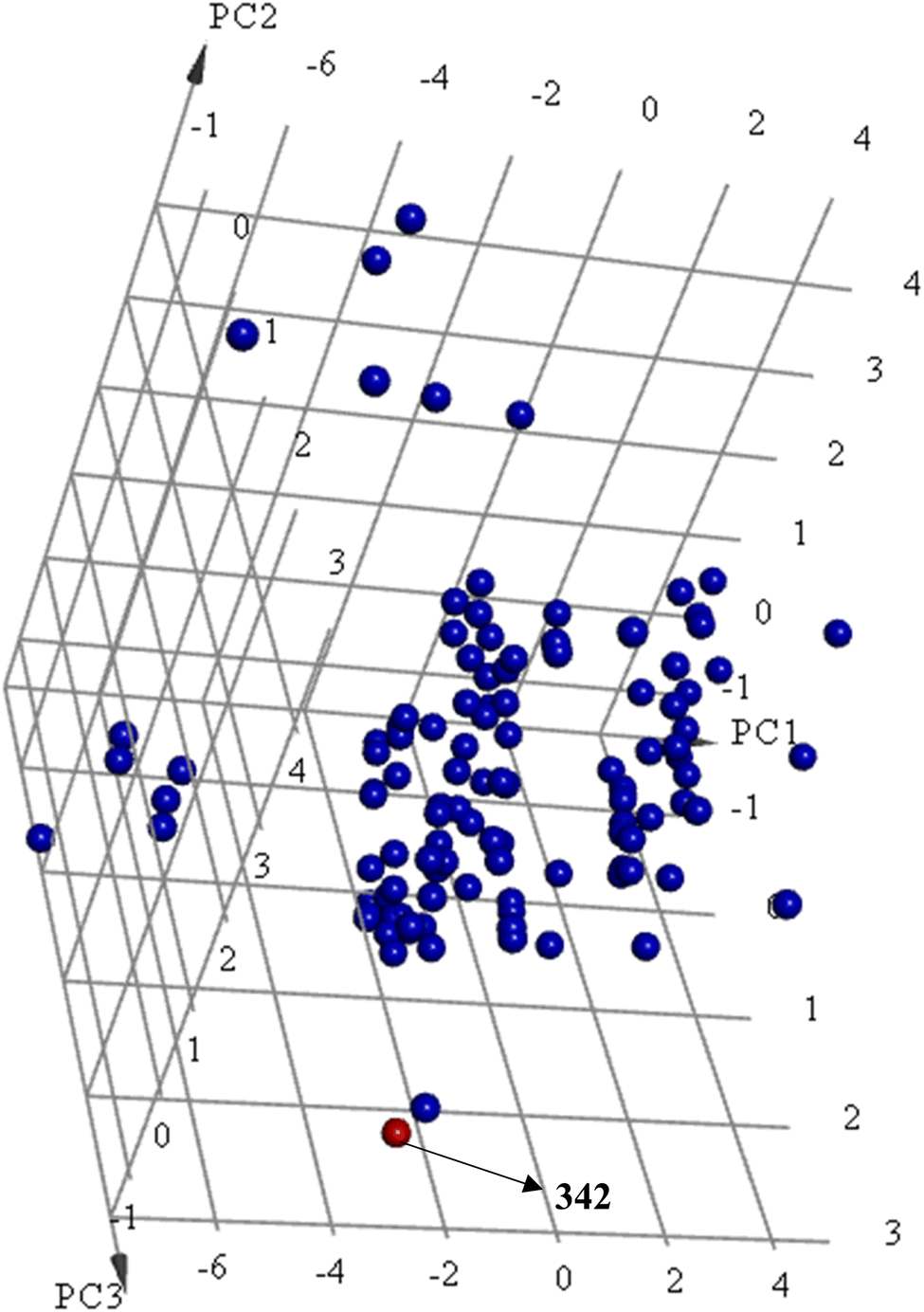 | ||
Fig. 12 Principal component analysis showing the relative distribution of captured active hit 342 (structure in Fig. 11 and bioactivity in Table 8, red spheres  ) compared to modeled active compounds (IC50 ≤ 5.0 μM, ESI Table S1,† blue spheres ) compared to modeled active compounds (IC50 ≤ 5.0 μM, ESI Table S1,† blue spheres  ). The top three principal components calculated for modeled compounds and captured hits are based on 11 descriptors (i.e., log(P), molecular weight, hydrogen bond donors and acceptors, rotatable bonds, number of atoms, number of rings, number of aromatic rings, molecular surface area, molecular polar surface area and molecular fractional polar surface area). Active hits are indicated in the figure with arrows. ). The top three principal components calculated for modeled compounds and captured hits are based on 11 descriptors (i.e., log(P), molecular weight, hydrogen bond donors and acceptors, rotatable bonds, number of atoms, number of rings, number of aromatic rings, molecular surface area, molecular polar surface area and molecular fractional polar surface area). Active hits are indicated in the figure with arrows. | ||
4. Conclusion
In conclusion, new STAT3 inhibitory lead of potent anti-STAT3 IC50 and novel chemotype was discovered using data augmentation algorithm based on computational sequence of docking, scoring, ligand-receptor contacts fingerprints. Optimal ML models and associated descriptors were translated into pharmacophore models. The resulting pharmacophores were validated by receiver operating characteristic (ROC) curve analysis and used as virtual search queries to screen the NCI database for promising STAT3 inhibitors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the Deanship of Academic Research at the University of Jordan for funding this project. The authors would also like to thank Dana Alqudah, Fadwa Daoud and Suha Wehaibi, from Cell Therapy Center for their technical assistance in biology testing experiments.References
- Y. S. Hu, X. Han and X. H. Liu, Curr. Top. Med. Chem., 2019, 19, 1305–1317 CrossRef CAS PubMed.
- L. Lin, S. Deangelis, E. Foust, J. Fuchs, C. Li, P.-K. Li, E. B. Schwartz, G. B. Lesinski, D. Benson, J. Lü, D. Hoyt and J. Lin, Mol. Cancer, 2010, 9, 217 CrossRef PubMed.
- S. R. Walker, M. Chaudhury and D. A. Frank, Mol. Cell. Pharmacol., 2011, 3, 13 CAS.
- K. Banerjee and H. Resat, Int. J. Cancer, 2016, 138, 2570–2578 CrossRef CAS PubMed.
- S. L. Furtek, D. S. Backos, C. J. Matheson and P. Reigan, ACS Chem. Biol., 2016, 11, 308–318 CrossRef CAS PubMed.
- J. Bosch-Barrera and J. A. Menendez, Cancer Treat. Rev., 2015, 41, 540–546 CrossRef CAS PubMed.
- P.-C. Shih, Life Sci., 2020, 242, 117241 CrossRef CAS PubMed.
- R. Catlett-Falcone, W. S. Dalton and R. Jove, Curr. Opin. Oncol., 1999, 11, 490 CrossRef CAS PubMed.
- D. Masciocchi, A. Gelain, S. Villa, F. Meneghetti and D. Barlocco, Future Med. Chem., 2011, 3, 567–597 CrossRef CAS PubMed.
- P. A. Johnston and J. R. Grandis, Mol. Interventions, 2011, 11, 18–26 CrossRef CAS PubMed.
- S. Verdura, E. Cuyàs, L. Llorach-Parés, A. Pérez-Sánchez, V. Micol, A. Nonell-Canals, J. Joven, M. Valiente, M. Sánchez-Martínez and J. Bosch-Barrera, Food Chem. Toxicol., 2018, 116, 161–172 CrossRef CAS PubMed.
- L. Zhang, Y. Wang, Y. Dong, Z. Chen, T. K. Eckols, M. M. Kasembeli, D. J. Tweardy and W. E. Mitch, Am. J. Physiol. Renal Physiol., 2020, 319, F84 CrossRef CAS PubMed.
- C. Brotherton-Pleiss, P. Yue, Y. Zhu, K. Nakamura, W. Chen, W. Fu, C. Kubota, J. Chen, F. Alonso-Valenteen and S. Mikhael, J. Med. Chem., 2020, 64, 695–710 CrossRef PubMed.
- J. Schust, B. Sperl, A. Hollis, T. U. Mayer and T. Berg, Chem. Biol., 2006, 13, 1235–1242 CrossRef CAS PubMed.
- K.-R. Feng, F. Wang, X.-W. Shi, Y.-X. Tan, J.-Y. Zhao, J.-W. Zhang, Q.-H. Li, G.-Q. Lin, D. Gao and P. Tian, Eur. J. Med. Chem., 2020, 201, 112428 CrossRef CAS PubMed.
- M. O. Taha, M. Habash, Z. Al-Hadidi, A. Al-Bakri, K. Younis and S. Sisan, J. Chem. Inf. Model., 2011, 51, 647–669 CrossRef CAS PubMed.
- N. J. Jaradat, M. A. Khanfar, M. Habash and M. O. Taha, J. Comput.-Aided Mol. Des., 2015, 29, 561–581 CrossRef CAS PubMed.
- M. M. Hatmal, S. Jaber and M. O. Taha, J. Comput.-Aided Mol. Des., 2016, 30, 1149–1163 CrossRef CAS PubMed.
- M. M. Hatmal, O. Abuyaman and M. Taha, Comput. Struct. Biotechnol. J., 2021, 19, 4790–4824 CrossRef CAS PubMed.
- A. Zhavoronkov, Q. Vanhaelen and T. I. Oprea, Clin. Pharmacol. Ther., 2020, 107, 780–785 CrossRef PubMed.
- J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, B. Li, A. Madabhushi, P. Shah, M. Spitzer and S. Zhao, Nat. Rev. Drug Discovery, 2019, 18, 463–477 CrossRef CAS PubMed.
- L. Zhang and C. Zhan, 2017.
- A. Lavecchia, Drug Discovery Today, 2015, 20, 318–331 CrossRef PubMed.
- I. Wickramasinghe and H. Kalutarage, Soft Comput., 2021, 25, 2277–2293 CrossRef.
- N. Varuna Shree and T. Kumar, Brain Inform., 2018, 5, 23–30 CrossRef CAS PubMed.
- M. Hajmeer and I. Basheer, J. Microbiol. Methods, 2002, 51, 217–226 CrossRef CAS PubMed.
- P. Gupta and N. K. Sinha, in Soft Computing and Intelligent Systems, ed. N. K. Sinha and M. M. Gupta, Academic Press, San Diego, 2000, pp. 337–356, DOI:10.1016/B978-012646490-0/50017-2.
- S. C. Wong, A. Gatt, V. Stamatescu and M. D. McDonnell, 2016.
- R. Rodríguez-Pérez and J. Bajorath, J. Comput.-Aided Mol. Des., 2020, 34, 1013–1026 CrossRef PubMed.
- A. Ghorbani and J. Zou, 2019.
- T. Hu, J. E. Yeh, L. Pinello, J. Jacob, S. Chakravarthy, G.-C. Yuan, R. Chopra and D. A. Frank, Mol. Cell. Biol., 2015, 35, 3284–3300 CrossRef CAS PubMed.
- Z. Ren, X. Mao, C. Mertens, R. Krishnaraj, J. Qin, P. K. Mandal, M. J. Romanowski, J. S. McMurray and X. Chen, Biochem. Biophys. Res. Commun., 2008, 374, 1–5 CrossRef CAS PubMed.
- Y. Belo, Z. Mielko, H. Nudelman, A. Afek, O. Ben-David, A. Shahar, R. Zarivach, R. Gordan and E. Arbely, Biochim. Biophys. Acta, Gen. Subj., 2019, 1863, 1343–1350 CrossRef CAS PubMed.
- E. Nkansah, R. Shah, G. W. Collie, G. N. Parkinson, J. Palmer, K. M. Rahman, T. T. Bui, A. F. Drake, J. Husby and S. Neidle, FEBS Lett., 2013, 587, 833–839 CrossRef CAS PubMed.
- K. L. Cheung, F. Zhang, A. Jaganathan, R. Sharma, Q. Zhang, T. Konuma, T. Shen, J.-Y. Lee, C. Ren and C.-H. Chen, Mol. Cell, 2017, 65, 1068–1080 CrossRef CAS PubMed.
- S.-L. Paiva, Nat. Rev. Drug Discovery, 2020, 19, 19–20 CrossRef CAS PubMed.
- S. Becker, B. Groner and C. W. Müller, Nature, 1998, 394, 145–151 CrossRef CAS PubMed.
- G. La Sala, C. Michiels, T. Kükenshöner, T. Brandstoetter, B. Maurer, A. Koide, K. Lau, F. Pojer, S. Koide and V. Sexl, Nat. Commun., 2020, 11, 1–16 CrossRef PubMed.
- L. Pinzi and G. Rastelli, Int. J. Mol. Sci., 2019, 20, 4331 CrossRef CAS PubMed.
- S. N. Rao, M. S. Head, A. Kulkarni and J. M. LaLonde, J. Chem. Inf. Model., 2007, 47, 2159–2171 CrossRef CAS PubMed.
- C. M. Venkatachalam, X. Jiang, T. Oldfield and M. Waldman, J. Mol. Graphics Modell., 2003, 21, 289–307 CrossRef CAS PubMed.
- G. Wu, D. H. Robertson, C. L. Brooks III and M. Vieth, J. Comput. Chem., 2003, 24, 1549–1562 CrossRef CAS PubMed.
- A. Krammer, P. D. Kirchhoff, X. Jiang, C. Venkatachalam and M. Waldman, J. Mol. Graphics Modell., 2005, 23, 395–407 CrossRef CAS PubMed.
- A. N. Jain, J. Comput.-Aided Mol. Des., 1996, 10, 427–440 CrossRef CAS PubMed.
- C. Y.-C. Chen, J. Biomol. Struct. Dyn., 2009, 27, 271–282 CrossRef CAS PubMed.
- G. Šinko, Chem.-Biol. Interact., 2019, 308, 216–223 CrossRef PubMed.
- Y. Wu and C. L. Brooks III, J. Chem. Inf. Model., 2021, 61, 5535–5549 CrossRef CAS PubMed.
- R. D. Clark, A. Strizhev, J. M. Leonard, J. F. Blake and J. B. Matthew, J. Mol. Graphics Modell., 2002, 20, 281–295 CrossRef CAS PubMed.
- D. Berrar, Encyclopedia of Bioinformatics and Computational Biology, ABC of Bioinformatics, 2018, p. 403 Search PubMed.
- J. Kirchmair, P. Markt, S. Distinto, G. Wolber and T. Langer, J. Comput.-Aided Mol. Des., 2008, 22, 213–228 CrossRef CAS PubMed.
- M. L. McHugh, Biochem. Med., 2012, 22, 276–282 CrossRef.
- A. Vehtari, A. Gelman and J. Gabry, Stat. Comput., 2017, 27, 1413–1432 CrossRef.
- P. K. Kondeti, K. Ravi, S. R. Mutheneni, M. R. Kadiri, S. Kumaraswamy, R. Vadlamani and S. M. Upadhyayula, Epidemiol. Infect., 2019, 147(e260), 1–11 Search PubMed.
- D. Rogers and A. J. Hopfinger, J. Chem. Inf. Model., 1994, 34, 854–866 CrossRef CAS.
- R. Rodríguez-Pérez and J. Bajorath, J. Med. Chem., 2019, 63, 8761–8777 CrossRef PubMed.
- M. A. Al-Sha’er and M. O. Taha, Curr. Comput.-Aided Drug Des., 2021, 17, 511–522 CrossRef PubMed.
- N. Triballeau, F. Acher, I. Brabet, J.-P. Pin and H.-O. Bertrand, J. Med. Chem., 2005, 48, 2534–2547 CrossRef CAS PubMed.
- R. Shahin and M. O. Taha, Bioorg. Med. Chem., 2012, 20, 377–400 CrossRef CAS PubMed.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis, E. Cibrián-Uhalte, M. Davies, N. Dedman, A. Karlsson, M. P. Magariños, J. P. Overington, G. Papadatos, I. Smit and A. R. Leach, Nucleic Acids Res., 2017, 45(D1), D945–D954 CrossRef CAS PubMed.
- M. Davies, M. Nowotka, G. Papadatos, N. Dedman, A. Gaulton, F. Atkinson, L. Bellis and J. P. Overington, Nucleic Acids Res., 2015, 43, W612–W620 CrossRef CAS PubMed.
- S. Jupp, J. Malone, J. Bolleman, M. Brandizi, M. Davies, L. Garcia, A. Gaulton, S. Gehant, C. Laibe and N. Redaschi, Bioinformatics, 2014, 30, 1338–1339 CrossRef CAS PubMed.
- D. C. Marks, L. Belov, M. W. Davey, R. A. Davey and A. D. Kidman, Leuk. Res., 1992, 16, 1165–1173 CrossRef CAS PubMed.
- L. N. Heppler, S. Attarha, R. Persaud, J. I. Brown, P. Wang, B. Petrova, I. Tošić, F. B. Burton, Y. Flamand and S. R. Walker, J. Biol. Chem., 2022, 298, 101531–101548 CrossRef CAS PubMed.
- A. Shastri, C. Schinke, A. V. Yanovsky, T. D. Bhagat, O. Giricz, L. Barreyro, J. Boultwood, A. Pellagati, Y. Yu and J. R. Brown, Blood, 2014, 124, 3602 CrossRef.
- M. W. Khan, A. Saadalla, A. H. Ewida, K. Al-Katranji, G. Al-Saoudi, Z. T. Giaccone, F. Gounari, M. Zhang, D. A. Frank and K. Khazaie, Cancer Immunol. Immunother., 2018, 67, 13–23 CrossRef CAS PubMed.
- X. Rao, X. Huang, Z. Zhou and X. Lin, Biostat. bioinforma. biomath., 2013, 3, 71 Search PubMed.
- V. Bulavas, V. Marcinkevičius and J. Rumiński, Informatica, 2021, 32, 441–475 CrossRef.
- K. E. Hevener, W. Zhao, D. M. Ball, K. Babaoglu, J. Qi, S. W. White and R. E. Lee, J. Chem. Inf. Model., 2009, 49, 444–460 CrossRef CAS PubMed.
- M. O. Taha and M. A. AlDamen, J. Med. Chem., 2005, 48, 8016–8034 CrossRef CAS PubMed.
- N. S. Pagadala, K. Syed and J. Tuszynski, Biophys. Rev., 2017, 9, 91–102 CrossRef CAS PubMed.
- H. Bertrand and N. Triballeau, Receptor, 2005, 2534–2547 Search PubMed.
- M. O. Taha, Virtual Screening, 2012, 1, 1–16 Search PubMed.
- M. Bamigboye and I. Ejidike, Nat. Appl. Sci. J., 2019, 2, 30–37 Search PubMed.
- J. R. Brown, S. R. Walker, L. N. Heppler, S. Tyekucheva, E. A. Nelson, J. Klitgaard, M. Nicolais, Y. Kroll, M. Xiang and J. E. Yeh, Am. J. Hematol., 2021, 96, E95–E98 CrossRef CAS PubMed.
- D. K. Poria, N. Sheshadri, K. Balamurugan, S. Sharan and E. Sterneck, J. Biol. Chem., 2021, 296, 100220–100228 CrossRef CAS PubMed.
- Y. Xia, G. Wang, M. Jiang, X. Liu, Y. Zhao, Y. Song, B. Jiang, D. Zhu, L. Hu and Z. Zhang, OncoTargets Ther., 2021, 14, 4047 CrossRef PubMed.
- A. Gangjee, N. Zaveri, S. F. Queener and R. L. Kisliuk, J. Heterocycl. Chem., 1995, 32, 243–247 CrossRef.
- P. Kumar, R. L. Kisliuk, Y. Gaumont, M. G. Nair, C. M. Baugh and B. T. Kaufman, Cancer Res., 1986, 46, 5020–5023 Search PubMed.
- T. Loughran, L. Zickl, T. L. Olson, V. Wang, D. Zhang, H. L. Rajala, Z. Hasanali, J. M. Bennett, H. M. Lazarus and M. R. Litzow, Leukemia, 2015, 29, 886–894 CrossRef CAS PubMed.
- E. C. Taylor, P. J. Harrington, S. R. Fletcher, G. P. Beardsley and R. G. Moran, J. Med. Chem., 1985, 28, 914–921 CrossRef CAS PubMed.
- J. D. Gordan, C. B. Thompson and M. C. Simon, Cancer Cells, 2007, 12, 108–113 CrossRef CAS PubMed.
- F. Zhou, Y. Yang and D. Xing, FEBS J., 2011, 278, 403–413 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07007c |
| This journal is © The Royal Society of Chemistry 2023 |