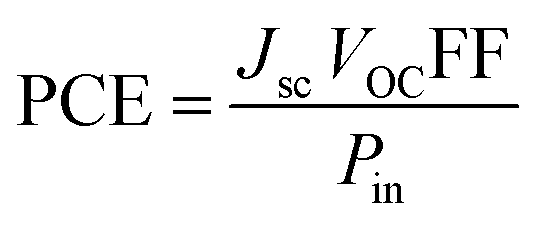DOI:
10.1039/D2RA07291B
(Paper)
RSC Adv., 2023,
13, 1640-1658
Novel A-π-D-π-A type non-fullerene acceptors of dithienyl diketopyrropopyrrole derivatives to enhance organic photovoltaic applications: a DFT study
Received
16th November 2022
, Accepted 16th December 2022
First published on 11th January 2023
Abstract
To boost the photovoltaic attributes of organic photovoltaic cells, seven dithienyl diketopyrropopyrrole (TDPP) donor-based A-π-D-π-A (acceptor-bridge-donor-bridge-acceptor) type molecules (TM1–TM7) were formulated by modifying the electron accepting ends of the reference molecule (TMR). Optical and quantum chemical parameters of seven synthesized molecules were investigated using density functional theory with the MPW1PW91/6-31G(d,p) functional. Several parameters that can be used to measure and improve the efficiency of solar cells have been analyzed and summed up. These parameters include binding energy of exciton, excitation energy of electron, reorganization energies, dipole moment, molecular electrostatic potential, charge mobility, wavelength of maximum absorption, open circuit voltage, short circuit current, fill factor, density of states, transition density matrices, as well as iso-surface and non-covalent interactions. Thus, all of our proposed structures are perceived to be superior to the reference in terms of the maximum possible solar energy yield in solar cells with bulk heterojunctions, as determined by analyses of our designed molecules for the aforementioned parameters.
1. Introduction
Considering the need to prevent the energy resources of the globe from being depleted, and to replace non-renewable energy sources with useful inexhaustible energy sources that could provide relief to society from undesirable and disease-creating pollution, there should be an alternative way of bringing into use a clean, continuous and renewable energy source. There is enough energy in the sun to power the entire planet for a year with just one hour of exposure. In order to take advantage of the beneficial aspects of solar energy, we endeavored to use this competent energy source in a way that would be sufficient to meet the energy needs of the entire world. Moreover the tunability, flexibility, mechanical adaptability, ease of preparation, wide range of absorption, and low weight of organic photovoltaic/solar cells (OPVs/OSCs) with bulk heterojunctions (BHJs) have attracted both the commercial and academic sectors.1–6 OSCs are susceptible to the incorporation of a variety of functional groups, which enables them to exhibit intriguing and novel optoelectronic properties.7,8 Due to their adaptable optoelectronic properties and increased efficiency of power conversion, organic photovoltaic and other such devices have garnered considerable interest.9–11
Since their invention in the mid-19th century, OPVs have been employed to transform solar energy into electricity.12 Initially, solar cells with thin layers of acceptor and donor were utilized, followed by solar cells with thick layers of acceptor and donor, but both of these were surpassed by BHJ solar cells in which both acceptor and donor layers are in maximum contact with each other, allowing for maximum interaction of both at a large interfacial area. In BHJ-type OSCs, the absorption of light generates a large number of electron–hole pairs that are tightly coupled to one another. These electron–hole pairs dissociate at the acceptor–donor interface and proceed towards their respective electrodes where they generate electricity.13–15
Due to their low reorganization energy and by association high mobility, fullerene acceptors (FAs) provide a remarkable 11% efficiency in OSCs. However, after discovering the drawbacks of FAs, their use has been avoided, and thus NFAs (non-fullerene acceptors) have been researched to overcome the drawbacks of FAs. Their assets include the outstanding tunability of Eg (band gaps), superior light-absorbing properties, thermal and photochemical stability, and increased lifetime. Furthermore, they have enhanced open-circuit voltages, reproducibility, as well as fill factor.16 Therefore, numerous attempts have been made to swap FA-based OSCs with NFA-based OSCs, such as BTP-Cl, ITC-2Cl, etc., which are currently being developed and utilized.17,18 In this regard, un-fused NFAs are acting as the premier contributors due to their two- to four-step and cost-effective synthetic routes, as well as high yield as opposed to their counterparts, the fused NFAs.19
The TDPP-RDN compound has already been engineered and scrutinized to evaluate the effectiveness and performance of OPVs.20 We chose this compound as our reference standard for the following reasons: it has a terminal functional group that accepts electrons and a dithienyl diketopyrropyrole (TDPP) core. The TDPP-RDN electron mobility reached 0.07 cm2 V−1 s−1. Diketopyrropopyrrole (DPP), which possesses good planarity, strong intermolecular contact, and good charge carrier mobility, served as the fundamental component (building block).21,22 The levels of energy for the molecule were evaluated. Using cyclic voltammetry, the calculated energies of the highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbital for TDPP-RDN were −5.39 eV and −3.85 eV, respectively. In combination with cyclic voltammetric data, the calculated HOMO and LUMO energy levels for TDPP-RDN were −5.61 eV and −3.83 eV, respectively. Maximum absorption for the TDPP-RDN molecule was measured at 710 nm in chlorobenzene, with a band gap Eg of 1.54 eV. Due to the lack of a single crystal structure, the exact molecular packing and arrangement of TDPP-RDN in thin films is unknown.
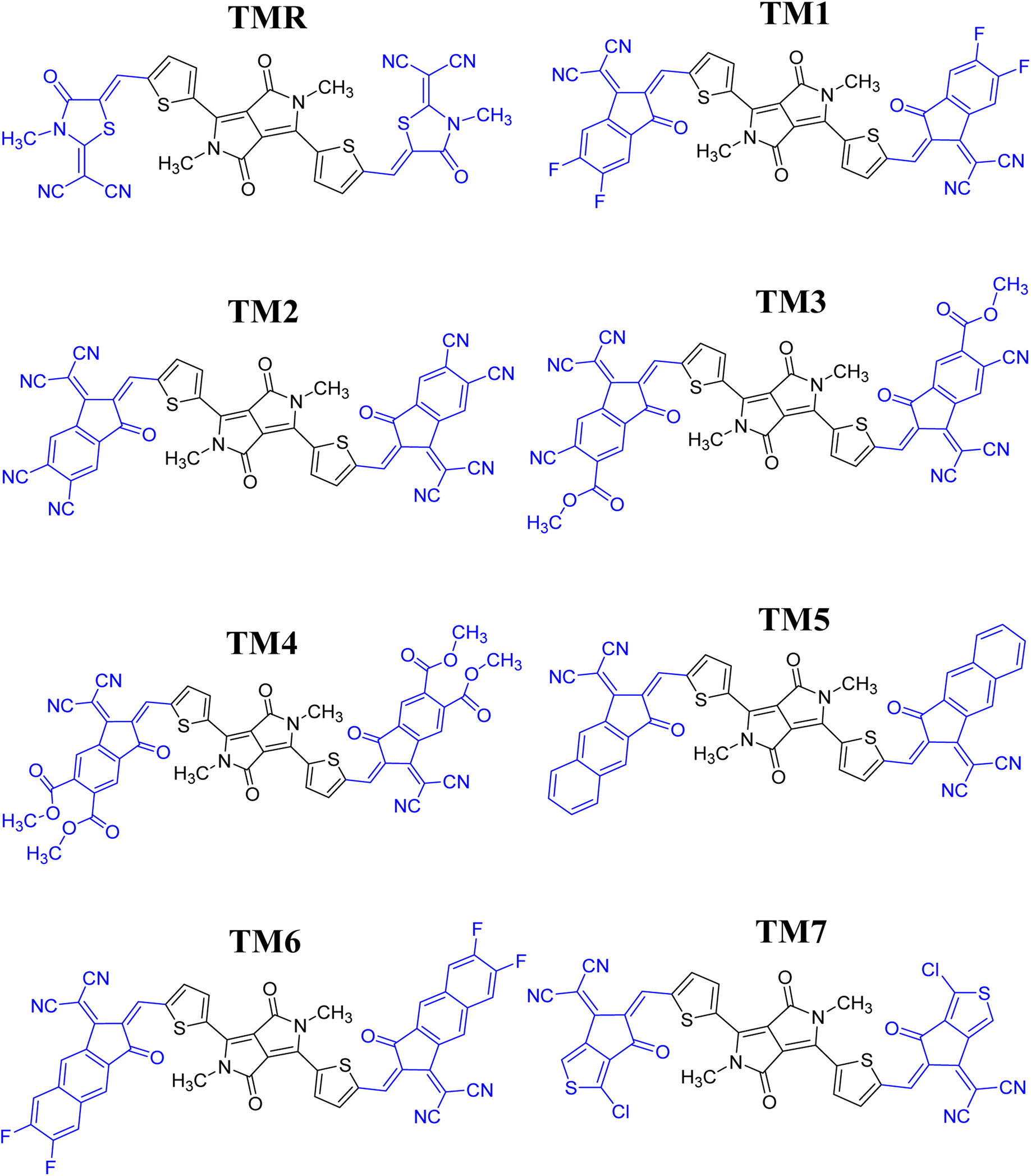
Seven photovoltaic compounds, designated TM1–TM7, were formulated by performing side-chain engineering of different acceptor groups at the peripheries of the A-π-D-π-A type23 reference TMR. These acceptor groups were selected for their prominent attributes in various computational studies and are known to have high electron-withdrawing abilities, along with significant conjugation in them, which aids in effective transfer of charge density towards them.24 The acceptors utilized were 2-(5,6-difluoro-2-methylene-3-oxo-indan-1-ylidene)malononitrile (TM1), 1-dicyanomethylene-2-methylene-3-oxo-indan-5,6-dicarbonitrile (TM2), 6-cyano-1-dicyanomethylene-2-methylene-3-oxo-indan-5-carboxylic acid methyl ester (TM3), 1-dicyanomethylene-2-methylene-3-oxo-indan-5,6-dicarboxylic acid dimethyl ester (TM4), 2-(2-methylene-3-oxo-2,3-dihydro-cyclopenta[b]naphthalen-1-ylidene)malononitrile (TM5), 2-(6,7-difluoro-2-methylene-3-oxo-2,3-dihydrocyclopenta[b]naphthalen-1-ylidene)malononitrile (TM6), and 2-(1-chloro-5-methylene-6-oxo-5,6-dihydrocyclopenta[c]thiophen-4-ylidene)malononitrile (TM7). When contrasted to TMR, these newly developed structures showed marked improvements in charge mobility, band gap reduction, and maximum absorption. Utilizing these acceptor moieties broadened the spectrum of absorption. Thiophene spacers also facilitated the transfer of charge from the central core to terminals (electron-accepting moieties). This suggests the active layer of molecules that have been formulated to be more efficient than the reference molecule TMR which could be beneficial to OSCs.
2. Computational details
All of the molecular structures under consideration were drawn using ChemDraw. Quantum mechanical computations of all the designed molecules and the TMR reference compound were carried out using Gaussian 09 software.25 To build, as well as to display, the 3D assemblies of all optimized compounds, we utilized GaussView 6.0.16.26 The in-depth investigation of TMR was done with DFT computing with its sub-method TD-DFT (time-dependent DFT), which was engaged to study the excited state properties. To execute the λmax (wavelength of maximum absorption), we used multiple approaches: B3LYP,27 CAM-B3LYP,28 MPW1PW91,29 and ωB97XD25 with 6-31G(d,p) basis set, in solvent phase. Spectral depiction of λmax was achieved through Origin 6.0 software.30 UV-visible absorption31,32 of TMR was contrasted with the cited (710 nm in chlorobenzene)33 value to determine the best functional. Closeness to the experimentally determined maximum absorption of TMR was achieved by MPW1PW91/6-31G(d,p). Using the IEFPCM (integral equation formalism polarizable continuum model), the maximum absorption characteristics adjoining the solvent were analyzed.34 Multiwfn 3.7 (ref. 35) and PyMolyze 1.1 (ref. 36) were used to obtain the spectra for the transition density matrix (TDM) analysis and the density of states (DOS), respectively.
The most notable factor, i.e. reorganization energy (RE), which influences the working efficiency of solar cells is also needed to be measured. The overall RE is distributed into two forms: internal (λint) and external (λext) RE. λext is related to the external deformations in the molecule, and is excluded because of the constant external environment of chloroform,37 whereas λint is related to changes that take place in the internal environment, i.e. variations in geometry/structure.38 We solely looked at the energy required for internal rearrangement in order to analyze charge transfer through electron–hole mobility. For the molecules, the Marcus rate equation39 has been employed to quantify the electron and hole reorganization energy:
| | |
λe = [E−0 − E−] + [E0− − E0]
| (1) |
| | |
λh = [E+0 − E+] + [E0+ − E0]
| (2) |
E−0 and
E+0 are anionic and cationic energies used in these equations to describe the ground state energy of a neutral compound. At a single-point ground state,
E0 represents the energy of a neutral molecule, whereas
E− and
E+ reflect the anion and cation single-point ground states energies after optimization.
E0− and
E0+ are the ground-level energies for anions and cations, correspondingly, after optimization.
3. Results and discussion
3.1. Method selection and geometry optimization
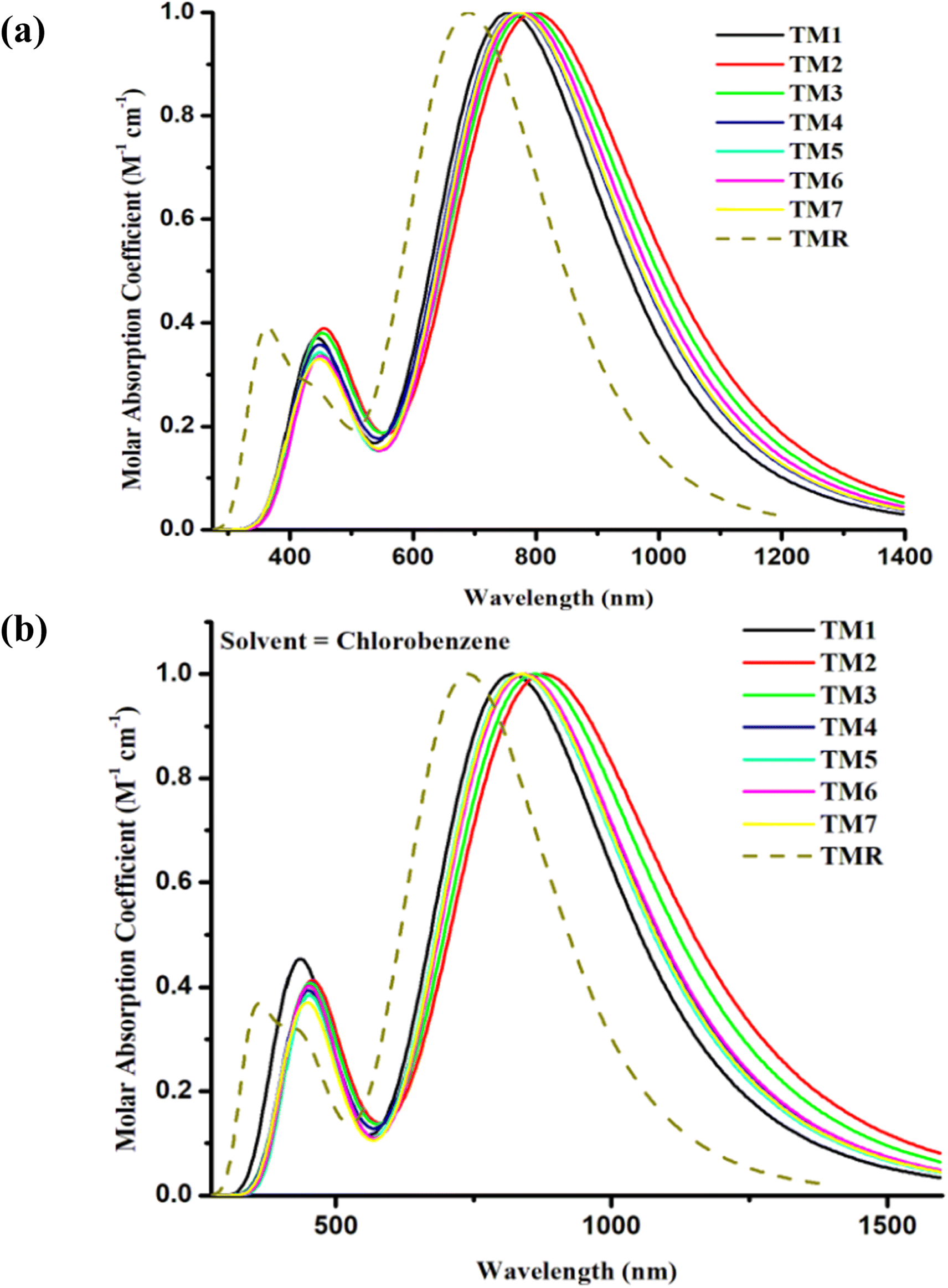
The aforementioned four alternative and valuable computational approaches with a 6-31G(d,p) basis set were used to optimize the ground state geometry of TMR. The reason for using these functional is to investigate the functional that could lead to desirable results in terms of λmax. Graphs depicting this behavior can be seen in Fig. 1(a) and (b). The MPW1PW91/6-13G(d,p) method provided an absorption maximum at 739 nm that was in close correlation with the experimental value (710 nm in chlorobenzene) as mentioned in the literature.33 Therefore, this approach was chosen as the optimum method for further research of all the formulated compounds. In Fig. 2 and 3, the chemical structures and optimized geometries of the reference TMR and designed compounds are illustrated.
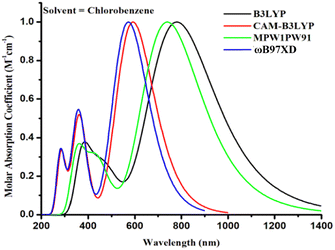 |
| | Fig. 1 UV-visible absorption of TMR for four different methods in chlorobenzene. | |
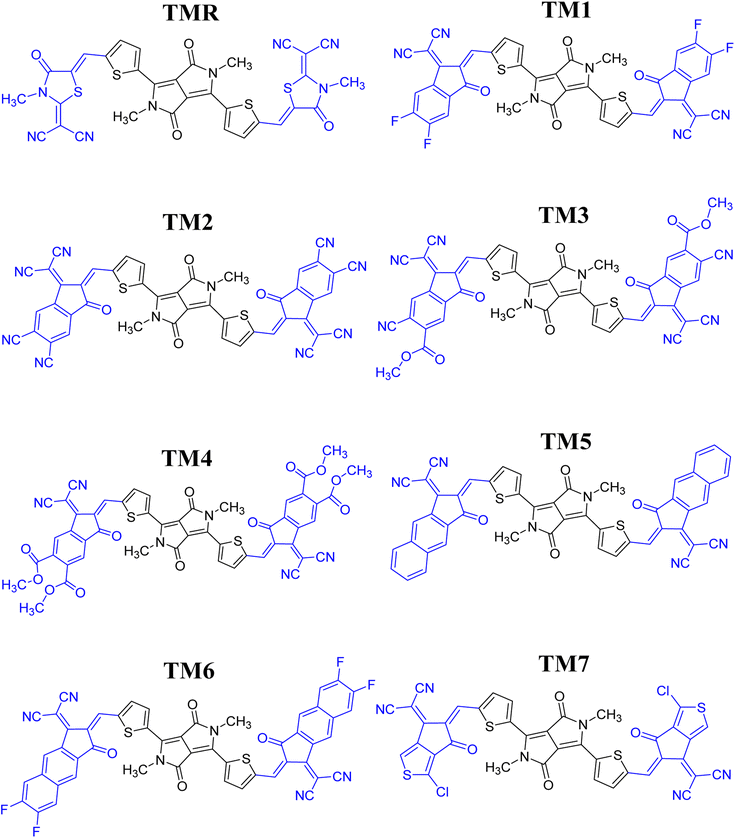 |
| | Fig. 2 ChemDraw structures of reference TMR and TM1–TM7 chromophores. | |
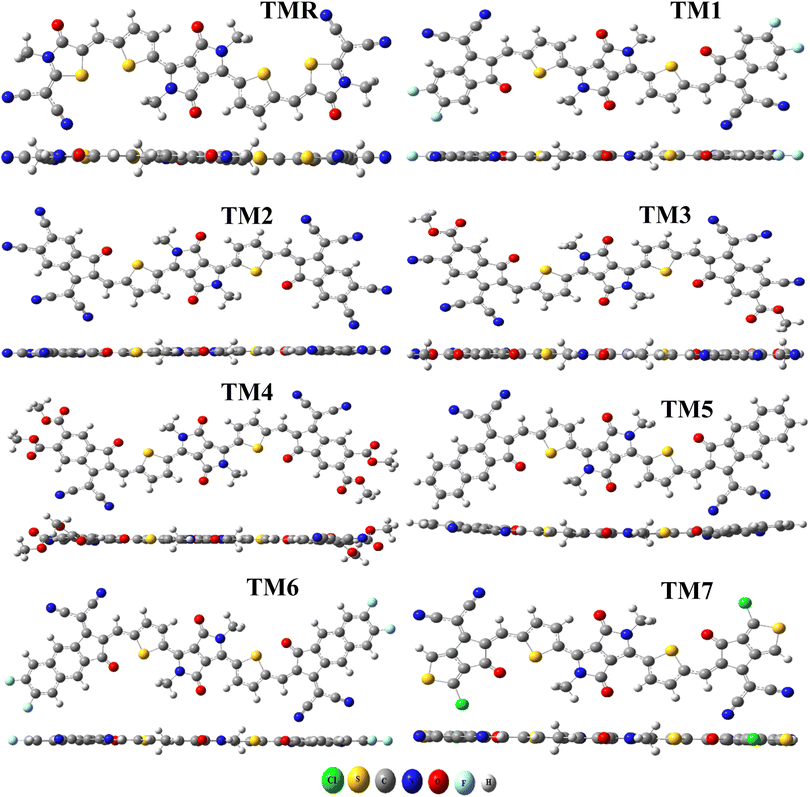 |
| | Fig. 3 Structural representation after optimization of TMR and tailored compounds TM1–TM7. | |
The bond lengths and dihedral angles (θ) of TMR and TM1–TM7 are presented in Table 1. All of the optimal structures of the investigated molecules (TMR and TM1–TM7) were found to have comparable geometries, with bond lengths and dihedral angles within a reasonable range. All the molecules studied had bond lengths between 1.418 Å and 1.424 Å, with the C–C single bond being 1.54 Å and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond being 1.33 Å, which exhibited enhanced charge transfer properties and also enabled conjugation through delocalization of π-electrons. All of the investigated molecules had dihedral angles within the range of −0.653° to 0.007°, indicating the absence of any twisting between the non-fused thiophene bridges and the peripheral acceptors, thereby indicating that all structures have planar geometry.
C bond being 1.33 Å, which exhibited enhanced charge transfer properties and also enabled conjugation through delocalization of π-electrons. All of the investigated molecules had dihedral angles within the range of −0.653° to 0.007°, indicating the absence of any twisting between the non-fused thiophene bridges and the peripheral acceptors, thereby indicating that all structures have planar geometry.
Table 1 Computed bond length and dihedral angle values for TMR and the modified chromophores TM1–TM7
| Molecule |
Bond length (Å) |
Dihedral angle (θ) |
| TMR |
1.424 |
−0.007 |
| TM1 |
1.422 |
−0.003 |
| TM2 |
1.418 |
0.007 |
| TM3 |
1.420 |
−0.002 |
| TM4 |
1.421 |
−0.428 |
| TM5 |
1.422 |
−0.653 |
| TM6 |
1.421 |
0.001 |
| TM7 |
1.422 |
0.004 |
3.2. Quantum chemical descriptor
Solar cells can be evaluated in general by analyzing the HOMOs and LUMOs derived from the optimal structures of the molecules under investigation. The collective name for these orbitals is “frontier molecular orbitals” (FMOs).40 As we are familiar with transfer of electrons from HOMO to LUMO, this gives a clear indication that HOMO resides on the electron-donating portion of the molecule, whereas LUMO resides on the electron-accepting portion.41,42 In other words, the LUMO is in the vicinity of the electrophilic part of the molecule, while the HOMO is close to the nucleophilic part.
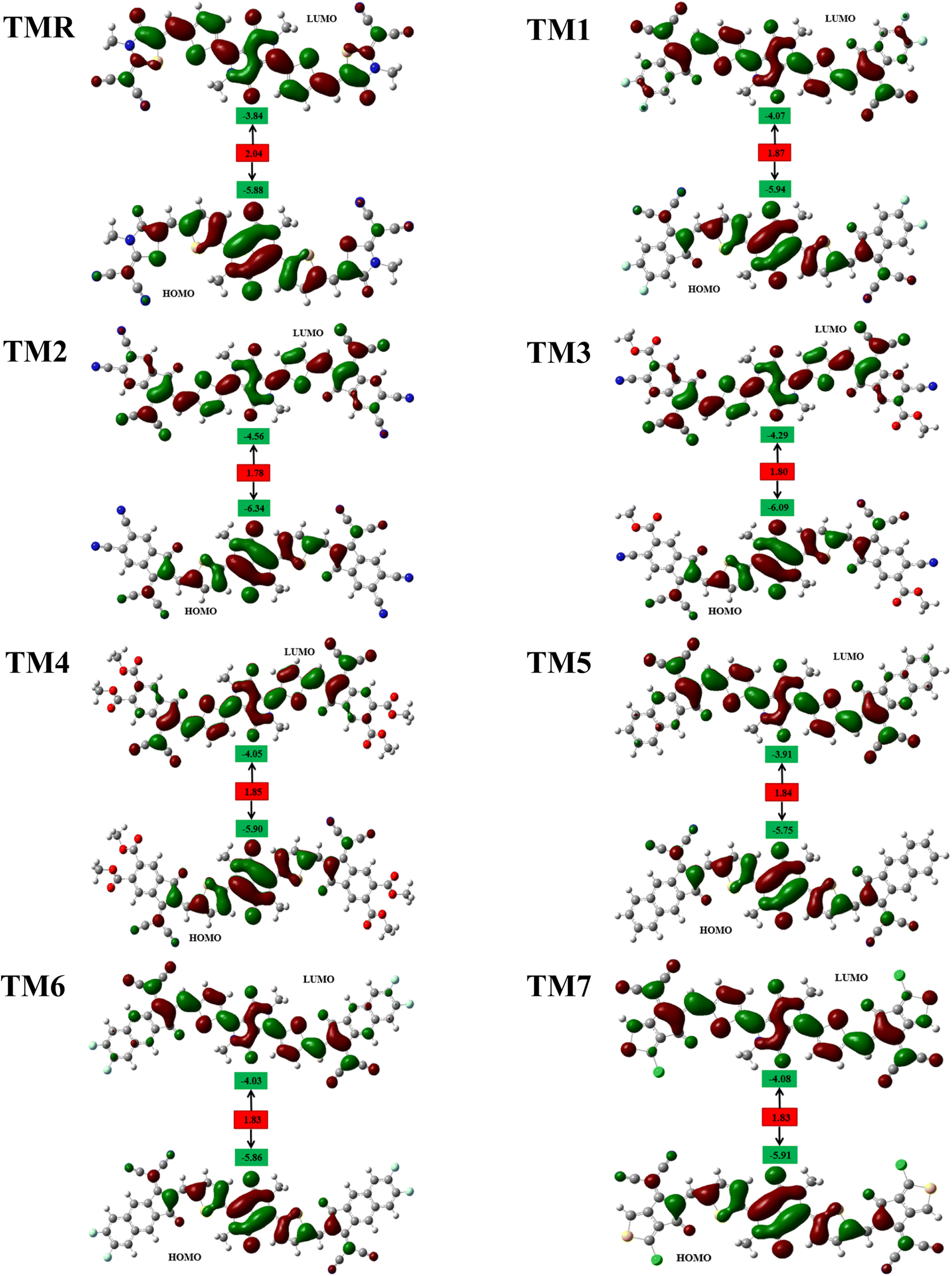
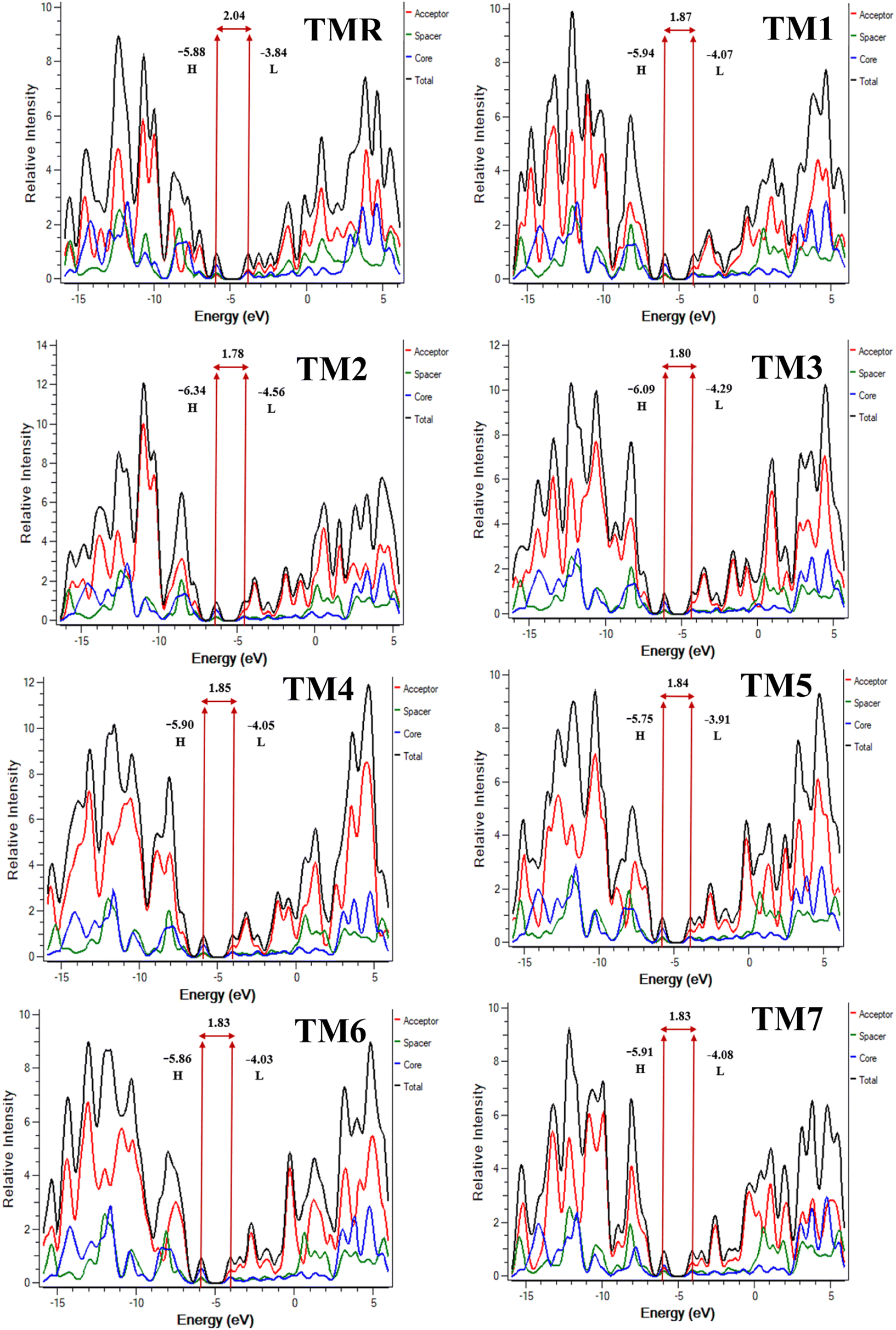
Fig. 4 displays the FMO charge distribution. Here, the red color is an indicator of a constructive (positive) phase, while green color shows a destructive (negative) one. Our reference TMR and TM1–TM7 molecules exhibited LUMO energies of −3.84 eV, −4.07 eV, −4.56 eV, −4.29 eV, −4.05 eV, −3.91 eV, −4.03 eV, and −4.08 eV, correspondingly. Whereas HOMO energies were −5.88 eV, −5.94 eV, −6.34 eV, −6.09 eV, −5.90 eV, −5.75 eV, −5.86 eV, and −5.91 eV, correspondingly (Table 2). Almost all the proposed compounds have lower HOMO and LUMO energies than the reference (TMR) due to the presence of strong peripheral electron-accepting entities. The FMO values of TM2 are shown to be the lowest among all the molecules in both the ground and excited states compared to those of the TMR molecule. TM5 > TM6 > TMR > TM7 > TM1 > TM3 > TM2 and TMR > TM5 > TM6 > TM4 > TM1 > TM7 > TM3 > TM2 are the orders of energy levels of HOMO and LUMO, respectively. The band gap (eqn (3)) plays a decisive role in maximizing charge transmission over the whole molecule and the molecule's overall efficiency. Band gap and power conversion efficiency (PCE) of OSCs are inversely correlated, as a molecule with a lower energy gap results in higher PCE and vice versa.
Here
EH and
EL are HOMO and LUMO energies, respectively. The minimum band gap can be achieved by increasing the HOMO of the donor, while decreasing the LUMO. The energy-gap values for the reference compound
TMR along with
TM1 to
TM7 chromophores were 2.04 eV, 1.87 eV, 1.78 eV, 1.80 eV, 1.85 eV, 1.84 eV, 1.83 eV, and 1.83 eV, respectively (
Table 2). All the planned structures have narrower band gaps than the standard compound
TMR. The
Eg values for all compounds fall in the range of 2.04–1.78 eV, as shown in
Fig. 4. The compounds have band gaps in the following order:
TMR >
TM1 >
TM4 >
TM5 >
TM6 =
TM7 >
TM3 >
TM2. The results show that the presence of the most electron-deficient units (acceptors) enhances charge mobility by decreasing the energy-gap values between the FMOs.
43,44
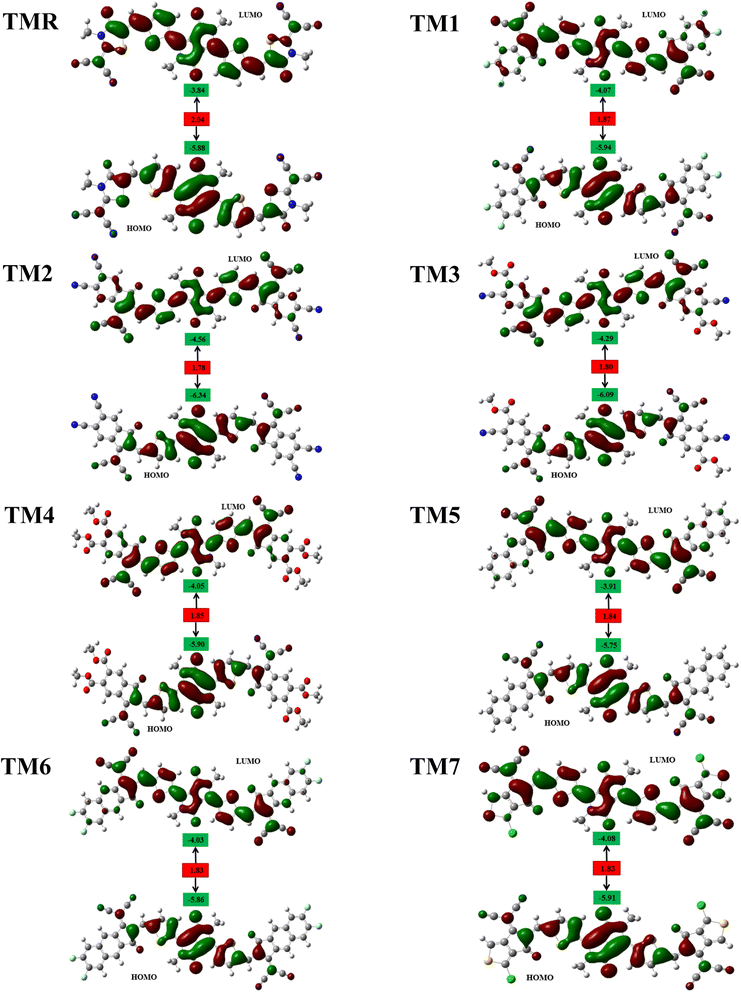 |
| | Fig. 4 FMO representation of TMR and tailored compounds TM1–TM7. | |
Table 2 Computed values for HOMO, LUMO, Eg, IP, and EA of TMR along with TM1–TM7 in eV
| Molecule |
HOMO |
LUMO |
Eg |
IP |
EA |
| TMR |
−5.88 |
−3.84 |
2.04 |
6.65 |
3.15 |
| TM1 |
−5.94 |
−4.07 |
1.87 |
6.67 |
3.42 |
| TM2 |
−6.34 |
−4.56 |
1.78 |
7.04 |
3.93 |
| TM3 |
−6.09 |
−4.29 |
1.80 |
6.80 |
3.67 |
| TM4 |
−5.90 |
−4.05 |
1.85 |
6.60 |
3.43 |
| TM5 |
−5.75 |
−3.91 |
1.84 |
6.45 |
3.29 |
| TM6 |
−5.86 |
−4.03 |
1.83 |
6.56 |
3.42 |
| TM7 |
−5.91 |
−4.08 |
1.83 |
6.64 |
3.43 |
The HOMO states of TMR and TM1–TM7 have almost the same degree of charge consistency in both the spacer and donor segments. Acceptor moieties had a small amount of charge consistency, and the substituted methyl groups in the central donor fragment did not experience electron density dispersion, due to their vertical alignment with respect to the molecules' horizontal plane. While the electron density is uniformly distributed in the donor, spacer as well as acceptor regarding the LUMOs of TMR, TM1, TM2, and TM7, with the exception of the methyl substituents in the acceptors and donor (core) section, the LUMOs of TM3, TM4, TM5, and TM6 revealed greater charge consistency over the donor core and spacer fragments but reduced charge consistency over the electron-accepting entities at the end-capped portions of the molecules. TMR, in contrast, shows almost equivalent electronic transitions between FMOs for all atoms excluding those in the methyl substituents. Because of the high density of charges in the LUMO of the acceptor and the HOMO of the donor, TM1 and TM7 are the most efficient in the interface of the OSC active layer. Also, the minute degree of charge density in the LUMO of donor and HOMO of acceptor could be due to the fairly planar geometry of the molecules.
3.3. Ionization potential and electron affinity
Ionization potential (IP) and electron affinity (EA) are useful tools for analyzing the charge-transfer efficiency of solar cells. The former is the energy necessary to eject an electron from an atom's outermost orbit, while the latter is the energy given by an atom when an electron is introduced to its outermost shell.45 But why we are discussing both of these in solar cells? This is for the reason that these variables work together in alternative ways to enhance the PCE of OSCs. Here IP values correspond to the energy needed by the HOMO in order to transfer its electron to the LUMO, hence they should be minimized so that electron will be removed with greater ease. EA, on the other hand, is linked to the LUMO values and so has to be greater. As groups that tend to donate electrons destabilize the HOMO by promoting electron transport, molecules with low IPs and high EAs are considered as good for passing charges. In contrast, the IPs of compounds with strong electron-withdrawing groups are higher, as the HOMO is more stable and electron removal is rather difficult. In accordance with Koopman's theorem46,47 the IP and EA of all the molecules under consideration were determined through eqn (4) and (5),48 and the outcomes are presented in Table 2.Here, IP denotes ionization potential and EA symbolizes the electron affinity of the system. The neutral molecule has cation with energy that is characterized via E+0 and anion with energy which is characterized via E−0. The energy of a neutral molecule is denoted by the symbol E0.40 Table 2 displays the calculated IP as well as EA values for the compounds under study. Almost all of the optimized molecules were found to have higher charge transfer efficiency than TMR. This was determined by the findings that almost all IP values for the optimized molecules were lower than those for TMR. TM5 has the lowest IP (6.45 eV) and highest charge extraction from HOMO. The IP value of the TM2 molecule is high indicating the poor charge transfer. Each of the synthesized molecules TM1–TM7 also had higher electron affinity than the standard reference molecule TMR.
3.4. Photovoltaic properties
The absorptive characteristics of the molecules in the gaseous as well as solution states were estimated using the pre-set functional and basis set at TD-DFT. The absorption spectra49 of molecules in both phases under investigation were acquired with the help of Origin 6.0, allowing us to clearly distinguish between the values of maximum absorption of the various compounds under research30 as shown in Fig. 5(a) and (b). The molar extinction coefficient was standardized on the y-axis with wavelength on the x-axis. Additionally, bar graphs comparing the experimental and the calculated values of λmax are shown in Fig. 5(a) and (b) to visualize the discrepancy between the two sets of results. Maximum absorption, oscillator strengths, excitation energies, light-harvesting efficiencies, etc. are shown in Tables 3 and 4.
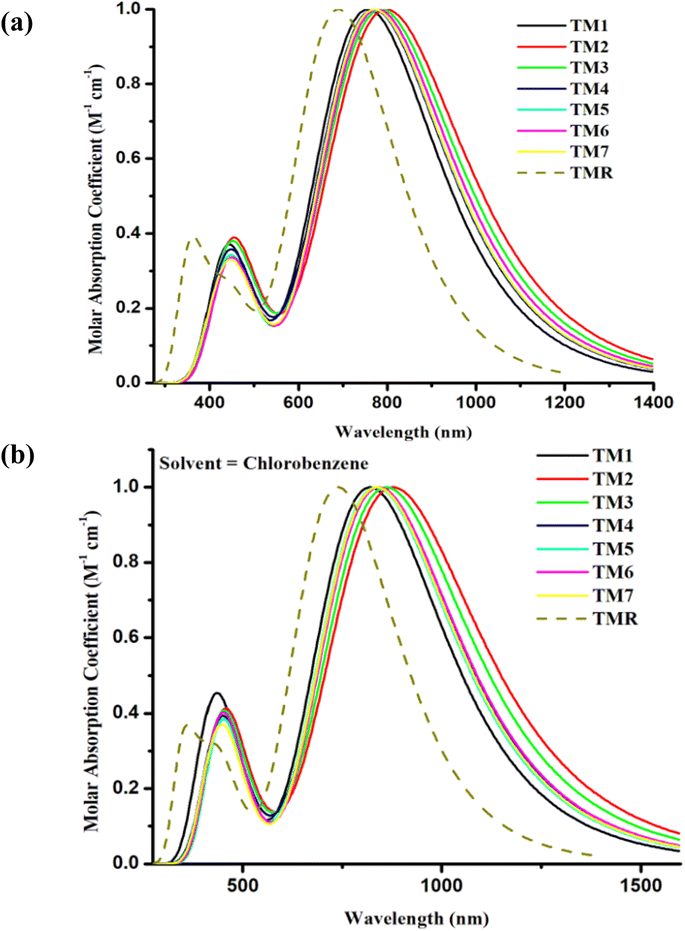 |
| | Fig. 5 Comparison plots for UV-visible absorption of TMR and tailored compounds TM1–TM7 in (a) gas phase and (b) chlorobenzene. | |
Table 3 Estimated results of λmax, LHE, excitation energy, oscillator strength, and transition character of TMR together with TM1–TM7 in the gas phase
| Molecule |
Calculated λmax (nm) |
Ex (eV) |
fos |
LHE |
Assignment |
Transition character (H → L) |
| TMR |
690 |
1.80 |
1.67 |
0.79 |
H > L |
71% |
| TM1 |
757 |
1.64 |
1.76 |
0.83 |
H > L |
71% |
| TM2 |
799 |
1.55 |
1.81 |
0.85 |
H > L |
71% |
| TM3 |
787 |
1.57 |
1.83 |
0.85 |
H > L |
71% |
| TM4 |
770 |
1.61 |
1.86 |
0.86 |
H > L |
71% |
| TM5 |
771 |
1.61 |
1.97 |
0.89 |
H > L |
71% |
| TM6 |
778 |
1.59 |
1.97 |
0.89 |
H > L |
71% |
| TM7 |
771 |
1.61 |
1.78 |
0.83 |
H > L |
71% |
Table 4 Estimated results of λmax, LHE, oscillator strength, excitation energy, and transition character of TMR together with TM1–TM7 in chlorobenzene along with exp. λmax
| Molecule |
Calculated λmax (nm) |
Exp. λmax (nm) |
Ex (eV) |
fos |
LHE |
Assignment |
Transition character (H → L) |
| TMR |
739 |
710 |
1.68 |
1.87 |
0.87 |
H > L |
71% |
| TM1 |
820 |
— |
1.51 |
1.98 |
0.90 |
H > L |
71% |
| TM2 |
878 |
— |
1.41 |
2.00 |
0.90 |
H > L |
71% |
| TM3 |
861 |
— |
1.44 |
2.02 |
0.90 |
H > L |
71% |
| TM4 |
840 |
— |
1.47 |
2.03 |
0.91 |
H > L |
71% |
| TM5 |
835 |
— |
1.48 |
2.18 |
0.93 |
H > L |
71% |
| TM6 |
842 |
— |
1.47 |
2.18 |
0.93 |
H > L |
71% |
| TM7 |
837 |
— |
1.48 |
2.01 |
0.90 |
H > L |
71% |
The absorption maxima of TMR and the tailored compounds TM1–TM7 were 690 nm, 757 nm, 799 nm, 787 nm, 770 nm, 771 nm, 778 nm, and 771 nm in gas phase and in the presence of the chlorobenzene solvent these were 739 nm, 820 nm, 878 nm, 861 nm, 840 nm, 835 nm, 842 nm, and 837 nm, respectively. All of the tailored compounds showed a clear red shift in absorption relative to TMR in gas phase, and significantly in solvent (near IR) as the polar excited state was stabilized by the polar solvent. In Fig. 5(a) and (b), even if the calculated absorption value of all the designed molecules has shifted to a greater extent in both gas phase and solvent, maximum wavelength is given by solvent, which is owing to higher conjugation in the solvent phase. This increased conjugation may result from the compounds' increased solubility in the solvent. The tightly conjugated and electron-accepting component of TM2 is responsible for its highest λmax (799 nm and 878 nm in both examined phases). In the gaseous state, the sequence of absorption is TM2 > TM3 > TM6 > TM5 = TM7 > TM4 > TM1 > TMR, while in the solvent (chlorobenzene) phase, the sequence is TM2 > TM3 > TM6 > TM4 > TM7 > TM5 > TM1 > TMR.
The excitation energy (Ex) is a crucial determinant of whether or not a compound can be used in OPVs. The energy utilized by an electron to jump from its ground state (S0) to its excited state (S1) is denoted by Ex.50 When Ex is small, it becomes easier for an electron to be excited, and for charge to move smoothly.51 The ability of the proposed molecules to efficiently transport electrons is revealed by a decrease in the Ex values of TM1–TM7 in both examined phases in contrast to TMR as presented in Tables 3 and 4. The smallest value for TM2 of first excitation energy, narrower band gap (1.78 eV), and greatest absorption maximum of 799 nm in gas phase and 878 nm in solvent indicated that the molecule has a higher charge transport ability. This is because of the existence of a potent end-capped electron-withdrawing acceptor entity. Because of its low Ex (1.54 eV in the gas phase and 1.41 eV in the solvent phase), TM2 has the potential to serve as an active layer in efficient OSCs.
Oscillator strength (fos)52 is the probability of electronic transition from the ground HOMO levels to the excited LUMO levels in photovoltaic cells. Higher absorption in the UV-visible zone means a higher fos, which in turn shows a greater charge transfer rate and hence a stronger oscillator strength. TMR and the constructed chromophores TM1–TM7 had oscillator strengths in the range of 1.97–1.67 in gas phase and 2.18–1.87 in solvent (Tables 3 and 4). The TM5 and TM6 structures with their strongest electron-deficient acceptor units at the terminal position have highest fos and as a result enhanced absorption and charge transformations.
Light harvesting efficiency (LHE) measures the ability of a molecule to absorb photons. According to the following eqn (6),53 its value is exactly proportional to the oscillator strength (fos):
Here, LHE represents light harvesting efficiency, and
f represents the oscillator strength, describing the relationship between the two variables. We make use of solvent-phase oscillator strength to evaluate LHE. High levels of LHE need a robust oscillator. The
fos has no units. Due to their more planar conformation,
TM5 and
TM6 displayed the greatest
fos values in both phases (0.89 and 0.93 respectively), as seen in
Tables 3 and
4.
3.5. Dipole moment
Dipole moment (μ) is another valuable parameter for assessing the effectiveness of solar devices. It arises in a molecule due to the difference in electronegativity of atoms contained in it. It is directly linked to the molecule's polarity.54 According to the fundamental tenet that “like dissolve like,” this increase in polarity results in better solubility in a polar solvent (for example, chlorobenzene). In contrast, symmetry results in a reduced μ, which in turn decreases solubility.55 An increase in dipole moment also has the benefit of increasing the electron mobility amongst HOMO and LUMO. Thus in this way the reactivity of a molecule increases which in turn increases the PCE of solar cells. Furthermore, the acceptors used in this research work are known to have a prominent effect on the dipole moment. The two fluoro-substituted malononitrile (TM1) groups significantly enhance the dipole moment due to enhanced π–π stacking found in them.56 Similarly, the acceptors of TM3 due to their polar character tend to increase the dipole moment as well.57 However, it should be kept in mind that the dipole moment of any symmetrical molecule despite a strongly electron-withdrawing group can reduce significantly due to the cancellation of opposite poles.58 However, regardless of their prominent symmetry, all the molecules except TM5 demonstrate better dipole moment than TMR. Table 5 displays the dipole moment of the reference TMR and modified molecules TM1–TM7 in the gaseous state μg, in the solvent (chlorobenzene) μe, and their difference Δμ. Both of the examined states (solvent and gas) have almost comparable order of dipole moments: TM5 > TM2 > TM6 > TM3 > TM4 > TM1 > TM7 > TMR and TM5 > TM3 > TM6 > TM2 = TM4 > TM1 > TM7 > TMR, correspondingly. The highest dihedral angle and the somewhat out-of-plane geometry of TM5 are responsible for its enhanced dipole moment. Because it is seen that dipole moment greatly depends on asymmetry, the lower the symmetry the greater the dipole moment.59 Similarly, TM2 has a nitrogen along with oxygen and TM6 has fluorine and oxygen, which increase their μ. These findings suggest that the modification of molecules with strong acceptor fragments may be advantageous in the future for more efficient OSCs.
Table 5 Computed values for gas-phase μg as well as solvent-phase μe dipole moment along with their difference Δμ in Debyes, for TMR and TM1–TM7
| Molecule |
μg |
μe |
Δμ |
| TMR |
0.0005 |
0.0005 |
0.0000 |
| TM1 |
0.0009 |
0.0011 |
0.0002 |
| TM2 |
0.0011 |
0.0039 |
0.0028 |
| TM3 |
0.0018 |
0.0019 |
0.0001 |
| TM4 |
0.0011 |
0.0013 |
0.0001 |
| TM5 |
0.8102 |
0.9807 |
0.1705 |
| TM6 |
0.0017 |
0.0020 |
0.0003 |
| TM7 |
0.0008 |
0.0009 |
0.0001 |
3.6. Density of states
The DOS for each of the compounds under study were calculated and analyzed to verify the results of FMO analysis. Donor cores, acceptors, and spacers are represented in the DOS plots by blue, red, and green lines, correspondingly (Fig. 6). These lines indicate the extent of involvement of various fragments in FMO enhancement. The entire DOS is shown as a black line. The HOMO is represented by the peak to the left of the central planar area, while the LUMO is represented by the peak to the right.60 This core planar empty area is essentially the band gap between the FMOs.24
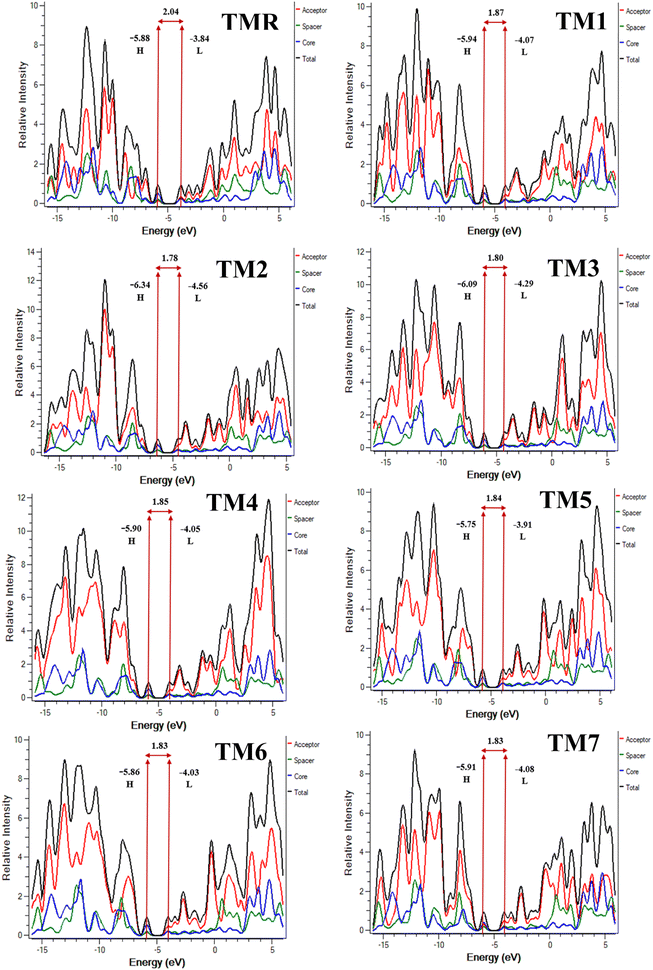 |
| | Fig. 6 DOS plots of TMR along with those of tailored compounds TM1–TM7. | |
Table 6 displays the results of a quantitative analysis of the three examined parts. It was discovered that the donor core made significant contributions towards the HOMO's improvement, spacers showed moderate and acceptors least involvement in HOMO elevation in the case of reference TMR and constructed structures TM1–TM7. For all our designed molecules (TM1–TM7) the percentage of donor involvement in the HOMO is greater than that for TMR. The results of FMO and the DOS are highly consistent with one another; the percentage of acceptors participating in the LUMO of TM1–TM7 is higher than in TMR, and it is lower in the case of the donor core. So, TM2 is the best because its donor moieties contribute the most (59.6%) to the HOMO. Similarly, TM7's acceptor fraction contributes 55.3% to the LUMO. Therefore, TM2 and TM7 stand out as the most notable molecules.
Table 6 Estimated values for percentage contributions of acceptor, donor core as well as spacer of TMR and TM1–TM7 in FMO formation
| Molecule |
FMO |
Acceptor (%) |
Spacer (%) |
Core (%) |
| TMR |
HOMO |
19.0 |
24.1 |
56.9 |
| LUMO |
38.2 |
31.1 |
30.7 |
| TM1 |
HOMO |
18.1 |
22.5 |
59.4 |
| LUMO |
49.0 |
26.1 |
24.9 |
| TM2 |
HOMO |
19.2 |
21.3 |
59.6 |
| LUMO |
53.3 |
24.5 |
22.2 |
| TM3 |
HOMO |
19.0 |
21.7 |
59.3 |
| LUMO |
52.5 |
24.7 |
22.9 |
| TM4 |
HOMO |
18.6 |
22.3 |
59.2 |
| LUMO |
50.5 |
25.4 |
24.1 |
| TM5 |
HOMO |
19.3 |
22.7 |
58.0 |
| LUMO |
50.8 |
25.0 |
24.1 |
| TM6 |
HOMO |
19.4 |
22.5 |
58.2 |
| LUMO |
51.5 |
24.8 |
23.7 |
| TM7 |
HOMO |
33.8 |
22.2 |
44.0 |
| LUMO |
55.3 |
25.4 |
19.3 |
3.7. Reorganization energy
Rearrangement energy analysis is a key factor in elucidating OSC performance. The RE of a molecule is the energy necessary to rearrange its constituent atoms and bonds followed by the transport of electrons and holes inside the molecule. Competent materials for OSCs are driven by the appropriate RE, which corresponds to the charge content which has to be transferred easily from donating to accepting fragments and is concerned with the hole–electron mobility.61 Thus this energy density (RE) is inversely linked to charge mobility. The more efficiently the charge transfers, the lower will be the RE value.62 The shape of cations and anions is just one of several important parameters that have an impact on RE. Table 7 displays the computed electron and hole RE values for all of the investigated compounds using eqn (1) and (2). RE of all of the molecules (TM1–TM7) including the reference (TMR) for positive (hole) and negative (electron) charges was examined through MPW1PW91/6-31G(d,p). Almost all of the seven altered molecules (TM1–TM7) had lower REs when compared with TMR proving that they are efficient electron transporters.
Table 7 Estimated values for hole RE λh along with electron RE λe of TMR and TM1–TM7
| Molecule |
λe (electron) |
λh (hole) |
| TMR |
0.01144 |
0.00967 |
| TM1 |
0.01060 |
0.00958 |
| TM2 |
0.00912 |
0.00917 |
| TM3 |
0.00977 |
0.00946 |
| TM4 |
0.01060 |
0.00981 |
| TM5 |
0.00977 |
0.00914 |
| TM6 |
0.00985 |
0.00930 |
| TM7 |
0.01001 |
0.00921 |
The RE (λh) values for holes of TMR and all modified compounds TM1–TM7 were 0.00967 eV, 0.00958 eV, 0.00917 eV, 0.00946 eV, 0.00981 eV, 0.00914 eV, 0.00930 eV, and 0.00921 eV, correspondingly. Similarly, the RE values for electrons of all the studied molecules were 0.01144 eV, 0.01060 eV, 0.00912 eV, 0.00977 eV, 0.01060 eV, 0.00977 eV, 0.00985 eV, and 0.01001 eV, correspondingly. These results for RE showed that all the designed molecules (with the exception of TM4) have lower hole RE compared to TMR, while the electronic RE (λe) for all the tailored structures TM1–TM7 are less than that of the reference TMR. Hole RE (λh) values determined via calculation are according to the order: TM4 > TMR > TM1 > TM3 > TM6 > TM7 > TM2 > TM5, while λe are in the order: TMR > TM1 = TM4 > TM7 > TM6 > TM3 = TM5 > TM2. TM2 has the lowest electron RE and TM5 has lowest hole RE of all the designed molecules. Thus TM2 is a good electron transporter, while TM5 is a good transporter of holes as they both provide the lowest RE values for both types of charges, respectively. The results show that the introduction of additional acceptor groups to the TM2 and TM5 molecules has improved the molecular charge transfer properties.
3.8. Molecular electrostatic potential
Another parameter which has proved effective to evaluate the degree of charge transfer between donor and acceptor sites is molecular electrostatic potential (MEP).63 MEP is the 3-D visualization of charge density at different positions of a molecule of interest. The MEP correlates with a molecule's reactive potential by highlighting its electrophilic and nucleophilic centers. Herein we make use of green, blue, and red shades to represent the location of different charges (positive, negative or neutral) over the molecule. Here the red color represents negative charge density which is certainly over the acceptor region where there may be the electronegative atoms present. Alternatively the blue color represents positive charge which is mainly over the donor portion (central core) and the neutral area with zero charge is revealed by a green shade.
Fig. 7 illustrates that the thiophene spacers are neutral parts of the molecules revealed by green color and the terminal acceptor groups are red indicating that they have a higher negative charge due to the presence of unsaturated electronegative atoms: oxygen, nitrogen, and sulphur. The tailored TM2 compound expresses more of a blue color over the acceptors and methyl groups of the donor part which reveals that these parts have more positive IP. Because electronegative atoms lie at a peripheral site the proposed compound TM3 showed more of a red color (negative charge) than any of the others, which shows that this molecule has greater negative IP. These results show that all the designed compounds promise an effective PCE of OSC systems compared with the reference TMR.
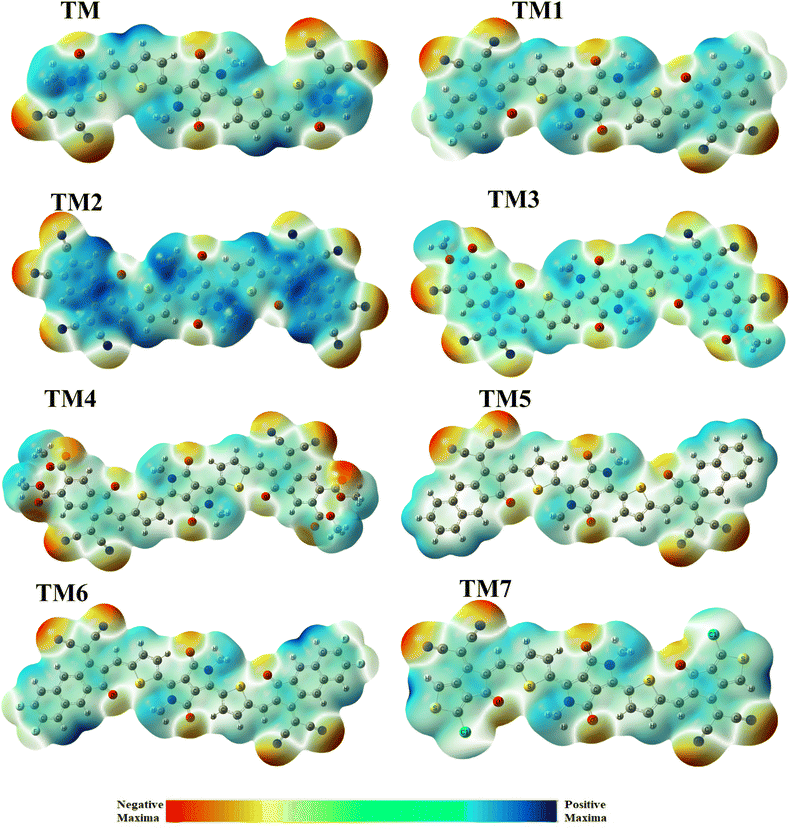 |
| | Fig. 7 Maps of MEPs of TMR along with those of tailored compounds TM1–TM7. | |
3.9. Exciton binding energy (Eb) TDM
The exciton binding energy (Eb) is a key parameter in the study and evaluation of OSCs.64 It represents the power required to bring the charge carriers apart once they have been created as a linked couple (exciton) This Eb is proportional to the coulombic interaction strength between the hole and the electron. Therefore, charge carriers with reduced Eb can travel conveniently towards their respective electrodes. Using eqn (7), we can hypothetically determine the binding energy of all the compounds that were studied:65Here, the excitation energy of the changed molecules is designated as Ex and the band gap as Eg, which is derived from eqn (3). The determined values for Eb of TMR and all other formulated structures TM1–TM7 are 0.24 eV, 0.23 eV, 0.23 eV, 0.23 eV, 0.24 eV, 0.23 eV, 0.24 eV, and 0.22 eV in the gas phase, while 0.36 eV, 0.36 eV, 0.37 eV, 0.36 eV, 0.38 eV, 0.36 eV, 0.36 eV, and 0.35 eV in solution phase (chlorobenzene), correspondingly. Based on the data shown in Table 8, the binding energies of all the compounds follow the sequence: TMR = TM4 = TM6 > TM1 = TM2 = TM3 = TM5 > TM7 in gas phase and TM4 > TM2 > TMR = TM1 = TM3 = TM5 = TM6 > TM7 in chlorobenzene. It is clear from this sequence that both TM4 and TM6 have the same binding energy as TMR in gaseous state, while TM1, TM3, TM5, and TM6 show this similar behavior in solution phase (chlorobenzene).
Table 8 Computed values for gas-phase as well as solvent (chlorobenzene)-phase exciton binding energy Eb of TMR and TM1–TM7
| Molecule |
Eb (eV) (gas phase) |
Eb (eV) (solvent phase) |
| TMR |
0.24 |
0.36 |
| TM1 |
0.23 |
0.36 |
| TM2 |
0.23 |
0.37 |
| TM3 |
0.23 |
0.36 |
| TM4 |
0.24 |
0.38 |
| TM5 |
0.23 |
0.36 |
| TM6 |
0.24 |
0.36 |
| TM7 |
0.22 |
0.35 |
TM7, the molecule with the lowest Eb values, has the greatest potential to enhance the working of OSCs in terms of charge transfer and PCE.
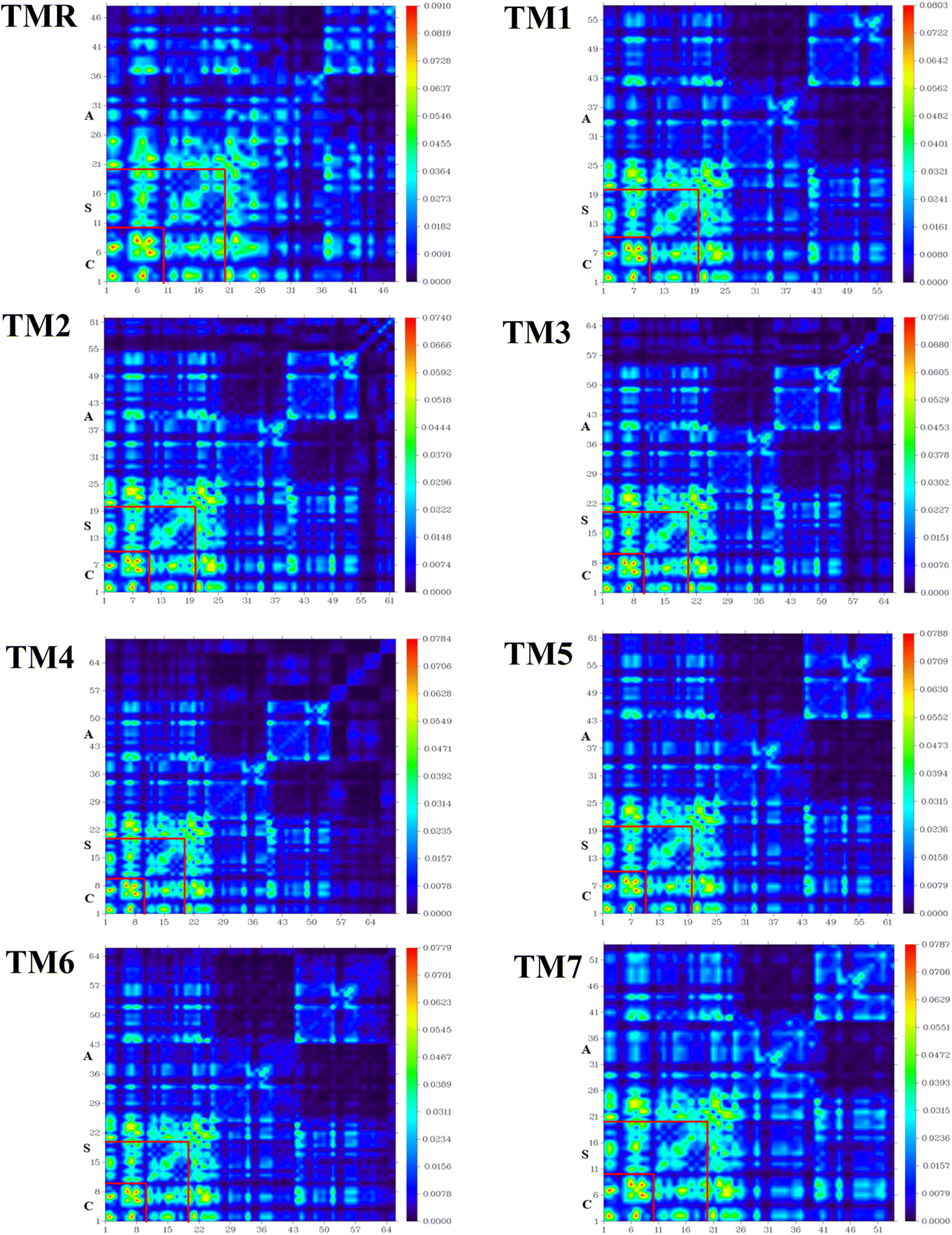
TDM has been shown to be the most convincing tool among all others when a variety of processes, including electronic excitation, charge production, separation, recombination, and diffusion, are analyzed sequentially.66 Through TDM, we can see how the quantum geometry of excited molecules works in practice. TDM determines the connection between excited donors and acceptors of molecules.67,68 TDM study of TMR and the designed molecules (TM1–TM7) was carried out using the desired hybrid DFT functional. Fig. 8 shows the TDM plots of TMR and the targeted molecules (TM1–TM7). Donor core (C), spacer (S) and acceptor (A) were the three divided slots for both the model molecule (TMR) as well as manufactured molecules (TM1–TM7). TDM plots depict atoms along the left and baseline axes, and electron density along the right axis. Hydrogen atoms have negligible impact, so they have been ignored.
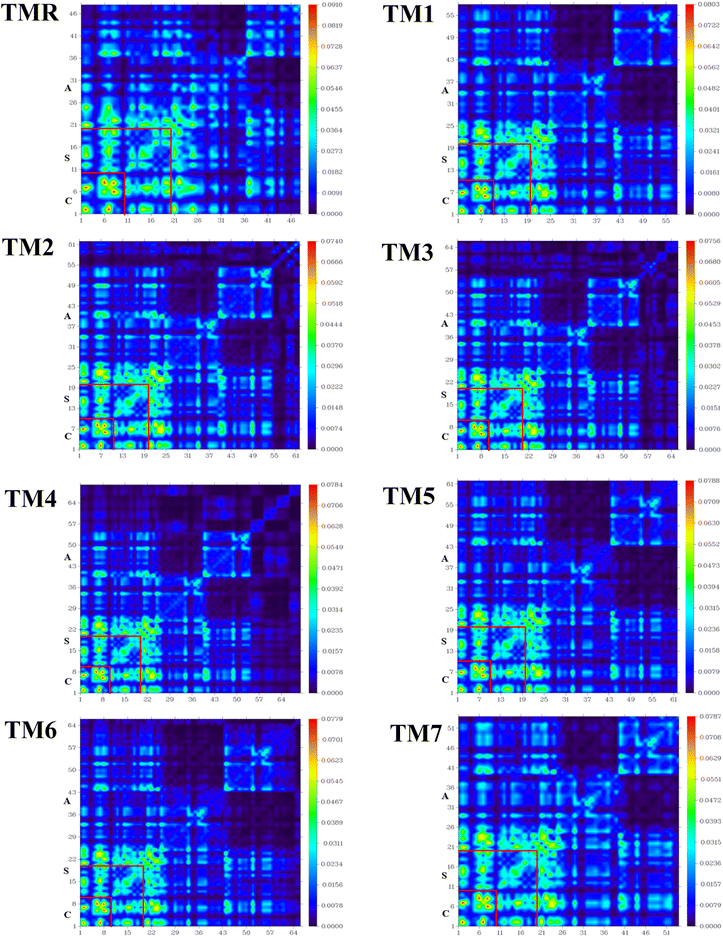 |
| | Fig. 8 TDM plots of TMR along with those of tailored compounds TM1–TM7. | |
A more stable and localized charge is found in the donor region (core) of the model (TMR) along with all the constructed TM1–TM7 compounds. Advantageous localized electron density (diagonal) and charge transfer (off-diagonal) properties are present in the newly formulated molecules (TM1–TM7), which also have a more uniform charge on the acceptor section. In the case of molecules TM3 and TM4, the donor core and spacer mainly reveal diagonal as well as off-diagonal behavior, while the acceptor fragment mainly demonstrated off-diagonal behavior. Both the localized electron density and charge transfer were shown by all the segments (acceptor, spacer, and core) of the TM5, TM6, and TM7 chromophore. As faster exciton dissociation is necessary for OSCs to be built, the TDM plots demonstrated significant diagonal as well as off-diagonal charge transfer from electron-rich center (donor) to electron-poor center (acceptor). This indicates that coupling of electrons and holes is weaker in the modified molecules. As shown in Fig. 8, we see a visual depiction of the interaction coefficient of the studied molecules in chlorobenzene. TDM analysis and a declining trend in the interaction coefficient show that the tailored compounds are superior at transferring charge from the donor moiety to the acceptor fragment through thiophene spacers. As a result, compared to TMR the formulated structures TM1–TM7 show remarkable charge dissociation capacity.
3.10. Open circuit voltage (VOC)
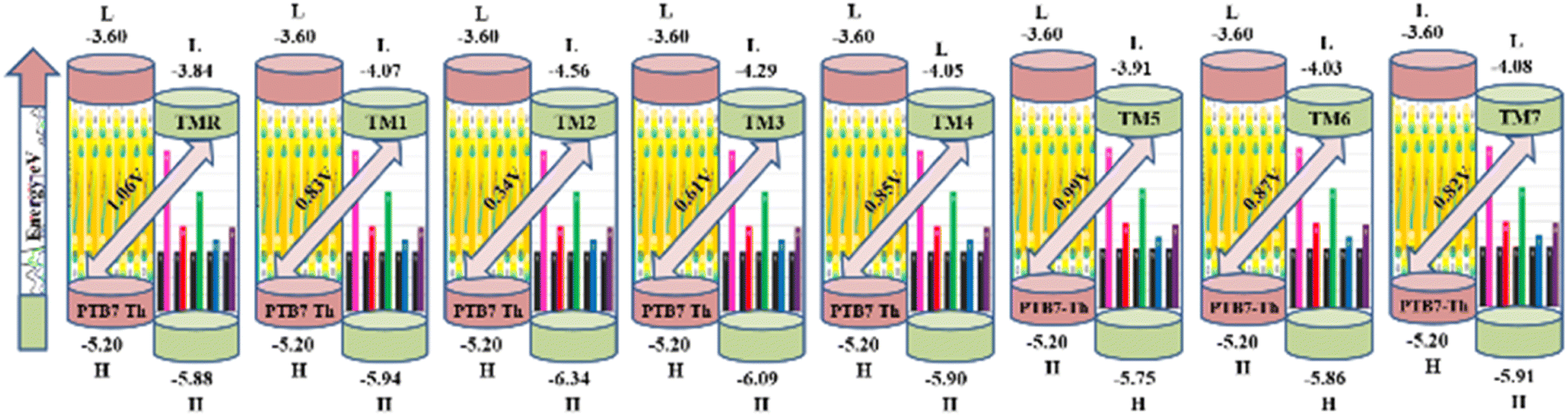
Open circuit voltage (VOC) provides a significant contribution in determination of the photovoltaic features of NFA-based organic photovoltaic devices. It is the greatest voltage that can be drawn from a photovoltaic device at zero current level.69–71 The open circuit voltage is influenced by several factors such as device temperature, molecule energy levels, light intensity, and charge-carrier recombination.72 The maximum value for VOC is calculated by comparing the HOMO value with LUMO value. To attain a value of VOC, the value of energy of HOMO of the donor must be smaller and the LUMO of the acceptor should be higher, which results in reduced band gap which is the desired condition for solar cells from any aspect. Here smaller value of HOMO corresponds to negative values which when becoming smaller, the actual HOMO value becomes higher and vice versa. Similar is the case with LUMO. A smaller band gap maximizes the absorption, which in return enhances the VOC and light harvesting efficiency. The maximum value for VOC is associated with success of NFA-based OSCs in terms of improved PCE. The VOC has direct relation with the effectiveness of OSCs: the higher the VOC the greater will be the efficiency.73 Visual representation of a comparison between the LUMO of the acceptor molecule with the HOMO of the donor polymer PTB7-Th to ascertain VOC is shown in Fig. 9. The VOC is determined mathematically through the use of the following eqn (8):74| |
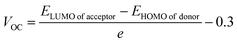 | (8) |
Here, e represents molecular charge having constant value of 1, 0.3 corresponds to empirical factors, which is also constant for every molecule, and E denotes energy of the utilized FMOs. PTB7-Th has values of −5.20 eV and −2.60 eV for HOMO and LUMO, respectively.44,75 The calculated values of open circuit voltage for reference molecule TMR and constructed compounds (TM1–TM7) are 1.06 V, 0.83 V, 0.34 V, 0.61 V, 0.85 V, 0.99 V, 0.87 V, and 0.82 V, respectively, as presented in Table 9. Amongst all the newly developed structures, TM5 (0.99 V) and TM6 (0.87 V) have been proved as molecules with highest VOC due to the involvement of the end-capped electron-withdrawing acceptor entities, from which it is concluded that TM5 and TM6 exhibit more charge delocalization, conjugation, and higher PCE than TM1, TM2, TM3, TM4, and TM7 molecules.
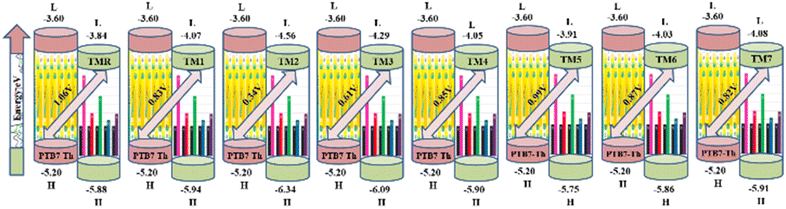 |
| | Fig. 9 Open circuit voltage of TMR and TM1–TM7 molecules with PTB7-Th. | |
Table 9 Computed values for VOC, normalized VOC along with the fill factor of TMR and TM1–TM7
| Molecule |
VOC (V) |
Normalized VOC |
Fill factor |
| TMR |
1.06 |
41.01 |
0.89 |
| TM1 |
0.83 |
32.11 |
0.86 |
| TM2 |
0.34 |
13.15 |
0.74 |
| TM3 |
0.61 |
23.60 |
0.83 |
| TM4 |
0.85 |
32.88 |
0.87 |
| TM5 |
0.99 |
38.30 |
0.88 |
| TM6 |
0.87 |
33.66 |
0.87 |
| TM7 |
0.82 |
31.72 |
0.86 |
3.11. Fill factor (FF)
The PCE and other photovoltaic properties of NFA-based OSCs can also be estimated through the utilization of a convincing tool, i.e. fill factor. The fill factor of reference TMR and other modified compounds TM1–TM7 can be measured with the application of eqn (9):76| |
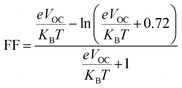 | (9) |
In this equation, open circuit voltage is represented by VOC, e denotes the charge of each compound, T characterizes the temperature of the system that was 300 K, Boltzmann constant is symbolized by KB and the normalized VOC is represented by 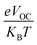 . The calculated fill factor values for the reference molecule and the designed chromophores (TM1–TM7) are summarized in Table 9, these values being 0.89, 0.86, 0.74, 0.83, 0.87, 0.88, 0.87 and 0.86, respectively. The measured results displayed that the molecules TMR, TM4, TM5 and TM6 have almost the similar value of fill factor, which is why they exhibit admirable photovoltaic properties compared with TM1, TM2, TM3, and TM7 having lower FF value. The descending pattern of FF of all compounds follows the sequence: TMR > TM5 > TM4 = TM6 > TM1 = TM7 > TM3 > TM2.
. The calculated fill factor values for the reference molecule and the designed chromophores (TM1–TM7) are summarized in Table 9, these values being 0.89, 0.86, 0.74, 0.83, 0.87, 0.88, 0.87 and 0.86, respectively. The measured results displayed that the molecules TMR, TM4, TM5 and TM6 have almost the similar value of fill factor, which is why they exhibit admirable photovoltaic properties compared with TM1, TM2, TM3, and TM7 having lower FF value. The descending pattern of FF of all compounds follows the sequence: TMR > TM5 > TM4 = TM6 > TM1 = TM7 > TM3 > TM2.
Jsc (short circuit current), VOC, power of incident light and FF contribute collectively to impart a positive effect on the PCE of NFA-based OSCs as represented by eqn (10):77,78
| |
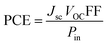 | (10) |
From the mentioned equation, it is clear that VOC, Jsc, and FF directly affect the PCE of OSC materials: by increasing the values of these parameters PCE will also be increased.79 On the other hand, power of incident light is inversely related to PCE. By knowing the numerical values of these parameters, we can judge effectively the value for PCE.
3.12. Iso-surface and reduced density gradient (RDG) analysis
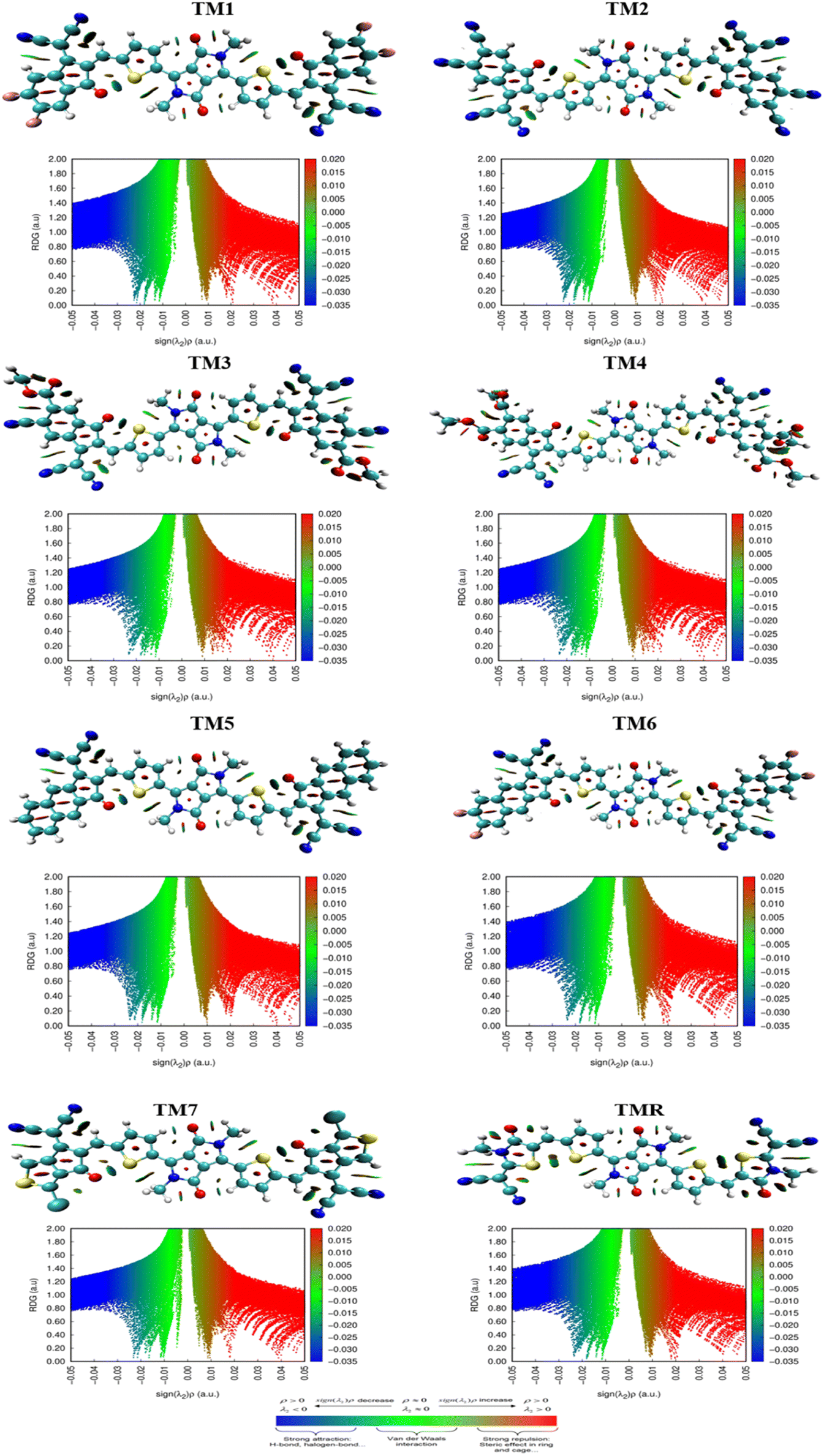
With an emphasis on non-covalent interactions between molecules, the non-covalent analysis is a 2D RDG graph that can discriminate among weak van der Waals forces, hydrogen bonds and repulsive steric forces. Molecular scatter plots are developed by the Multiwfn software tool. λ2 values are taken into account while assessing the type of NCIs. Graphical illustration of iso-surfaces and their respective NCIs for all derived molecules (TM1–TM7) along with TMR are shown in Fig. 10. Blue, green and red colours in graphs indicate strong interactions, van der Waal forces and weak interactions respectively as indicted in the coloured bar in Fig. 10.
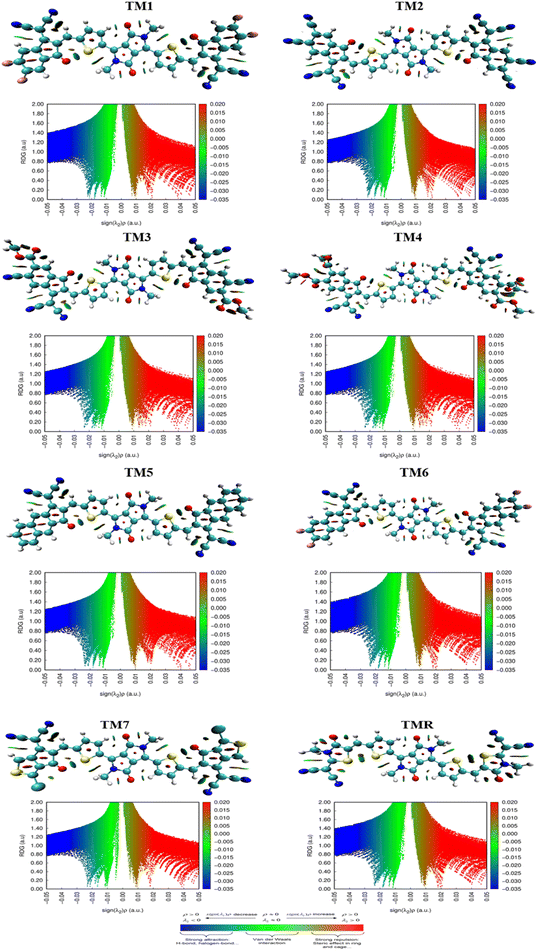 |
| | Fig. 10 NCIs and iso-surfaces of TMR and TM1–TM7 molecules. | |
4. Conclusion
The effects of end-capped acceptors on the charge transfer ability as well as on structural and photochemical activities of TMR have been investigated using state-of-the-art quantum chemical and quantum mechanical methods. Seven different acceptor molecules (TM1–TM7) were formulated via the end-cap engineering of the reference TMR. These compounds were designed with the purpose of investigating their optoelectronic characteristics for use in OSCs with enhanced efficiency. Light absorption, electron–hole RE, nucleophilic as well as electrophilic sections inside the molecule (MEP), band gap between FMOs, involvement of acceptor and donor in FMOs (DOS), and TDM were all investigated. All seven developed structures (TM1–TM7) when compared to the published reference TMR molecule exhibited notable improvement regarding all the above mentioned parameters, i.e. a reduced band gap in the range of 1.87–1.78 eV, a dipole moment that is larger than that of TMR in both the examined phases (gas and solvent), lower hole as well as electron REs, and maximum photon absorption (799 nm in gas phase and 878 nm in chlorobenzene solvent), which were very impressive, thanks to modifications by end-capped acceptor moieties. Therefore, it is suggested that all formulated candidates are promising for use in OSCs in order to obtain high PCE in the near future, since almost all of the unique acceptor molecules TM1–TM7 have better optical and electrical characteristics compared to the reference TMR.
Data availability
Data will be made available on request.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors acknowledge the technical support from the Department of Chemistry, University of Agriculture, Faisalabad, Pakistan. The authors would like to thank the Deanship of Scientific Research at Umm Al-Qura University for supporting this work by Grant Code: 22UQU4331174DSR48. We also thank Dr Khurshid Ayub, COMSATS University, Islamabad for additional resources.
References
- K. Kranthiraja and A. Saeki, Machine Learning-Assisted Polymer Design for Improving the Performance of Non-Fullerene Organic Solar Cells, ACS Appl. Mater. Interfaces, 2022, 14(25), 28936–28944 CrossRef CAS PubMed.
- Y. Yang, B. Xu and J. Hou, Solution-Processed Silver Nanowire as Flexible Transparent Electrodes in Organic Solar Cells, Chin. J. Chem., 2021, 39(8), 2315–2329 CrossRef CAS.
- Y. Sun, et al., Flexible organic solar cells: progress and challenges, Small Sci., 2021, 1(5), 2100001 CrossRef CAS.
- D. Hu, et al., 15.34% efficiency all-small-molecule organic solar cells with an improved fill factor enabled by a fullerene additive, Energy Environ. Sci., 2020, 13(7), 2134–2141 RSC.
- Y. Liu, et al., Recent progress in organic solar cells (Part I material science), Sci. China: Chem., 2021, 1–45 Search PubMed.
- Y. Liu, et al., Managing Challenges in Organic Photovoltaics: Properties and Roles of Donor/Acceptor Interfaces, Adv. Funct. Mater., 2022, 32(43), 2206707 CrossRef CAS.
- S. Sumaiya, K. Kardel and A. El-Shahat, Organic solar cell by inkjet printing—an overview, Technologies, 2017, 5(3), 53 CrossRef.
- C. Zhang, et al., Understanding the correlation and balance between the miscibility and optoelectronic properties of polymer–fullerene solar cells, J. Mater. Chem. A, 2017, 5(33), 17570–17579 RSC.
- W. U. Khan, et al., Robust thermal performance of red-emitting phosphor composites for white light-emitting diodes: energy transfer and oxygen-vacancy induced electronic localization, J. Colloid Interface Sci., 2021, 600, 219–228 CrossRef PubMed.
- W. U. Khan, et al., Robust thermal performance and color purity of samarium based deep red emitting phosphor for near-UV chip-based high-color-rendering, J. Alloys Compd., 2021, 888, 161538 CrossRef.
- W. U. Khan, et al., Bifunctional samarium activated red-emitting and thermal resistant phosphor for simultaneous field emission displays and white light-emitting diodes, Ceram. Int., 2022, 48(4), 5689–5697 CrossRef CAS.
- K. Vijayan, et al., A review on advancements, challenges, and prospective of copper and non-copper based thin-film solar cells using facile spray pyrolysis technique, Sol. Energy, 2022, 234, 81–102 CrossRef CAS.
- Y. M. Yang, et al., High-performance multiple-donor bulk heterojunction solar cells, Nat. Photonics, 2015, 9(3), 190–198 CrossRef CAS.
- I. C. Ghosekar and G. C. Patil, Review on performance analysis of P3HT:PCBM-based bulk heterojunction organic solar cells, Semicond. Sci. Technol., 2021, 36(4), 045005 CrossRef CAS.
- Y. Li, et al., Solution processable D–A small molecules for bulk-heterojunction solar cells, Energy Environ. Sci., 2010, 3(10), 1427–1436 RSC.
- S. F. Hoefler, et al., The effect of polymer molecular weight on the performance of PTB7-Th:O-IDTBR non-fullerene organic solar cells, J. Mater. Chem. A, 2018, 6(20), 9506–9516 RSC.
- J. Zhang, et al., Material insights and challenges for non-fullerene organic solar cells based on small molecular acceptors, Nat. Energy, 2018, 3(9), 720–731 CrossRef CAS.
- S. J. Akram, et al., Designing of the indacenodithiophene core-based small molecules for optoelectronic applications: a DFT approach, Sol. Energy, 2022, 237, 108–121 CrossRef CAS.
- B. L. Greenstein, D. C. Hiener and G. R. Hutchison, Computational evolution of high-performing unfused non-fullerene acceptors for organic solar cells, J. Chem. Phys., 2022, 156(17), 174107 CrossRef CAS PubMed.
- A. Mishra, N. Chaudhary, D. Bhardwaj, P. Pahari and A. Patra, et al., Poly[3,6-bis(2-thienyl)diketopyrrolopyrrole-selenophene]: effect of polymerization method on optical, electrochemical, and photovoltaic properties, Synth. Met., 2022, 291, 117170 CrossRef.
- Q. He, et al., A versatile star-shaped organic semiconductor based on benzodithiophene and diketopyrrolopyrrole, J. Mater. Chem. C, 2019, 7(22), 6622–6629 RSC.
- D. W. Polak, et al., Probing the electronic structure and photophysics of thiophene–diketopyrrolopyrrole derivatives in solution, Phys. Chem. Chem. Phys., 2022, 24(34), 20138–20151 RSC.
- Y. Zhang, et al., High-Performance Non-fullerene organic solar cells enabled by noncovalent Conformational locks and Side-Chain engineering, Chem. Eng. J., 2022, 446, 137206 CrossRef.
- A. Naveed, et al., Impact of end capped modification on BT-CIC molecule for high-performance photovoltaic attributes: a DFT approach, J. Mol. Model., 2022, 28(8), 1–16 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10(44), 6615–6620 RSC.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView 6.0.16, Semichem Inc., Shawnee Mission, KS, USA, 2016 Search PubMed.
- B. Civalleri, et al., B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals, CrystEngComm, 2008, 10(4), 405–410 RSC.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393(1–3), 51–57 CrossRef CAS.
- A. L. Macedo, L. H. Martorano, A. C. F. D. Albuquerque, R. G. Fiorot, J. W. Carneiro, V. R. Campos and F. M. D. Santos Jr, et al. Absolute configuration of (−)-cubebin, a classical lignan with pharmacological potential, defined by means of chiroptical spectroscopy, J. Braz. Chem. Soc., 2020, 31, 2030–2037 CAS.
- L. A. Deschenes and D. A. Vanden Bout, Origin 6.0: Scientific Data Analysis and Graphing Software Origin Lab Corporation (formerly Microcal Software, Inc.), University of Texas, ACS Publications, Web site: https://www.originlab.com, Commercial price: 595. Academicprice: 446, 2000 Search PubMed.
- S. Ullah, et al., Investigation of stability and rheological properties of silver nanoparticles stabilized by polyethylene glycol, J. Mater. Sci.: Mater. Electron., 2020, 31(13), 10470–10477 CrossRef CAS.
- M. Ans, A. Ayub, N. Alwadai, A. Rasool, M. Zahid, J. Iqbal and M. S. Al-Buriahi, Simultaneously enhanced efficiency of eco-friendly structural characterization of the dithienocyclopentacarbazole donor based acceptors with narrow bandgap for high-performance organic solar cells, J. Phys. D: Appl. Phys., 2022, 55(23), 235501 CrossRef.
- G. Y. Ge, et al., Unveiling the Interplay among End Group, Molecular Packing, Doping Level, and Charge Transport in N-Doped Small-Molecule Organic Semiconductors, Adv. Funct. Mater., 2022, 32(7), 2108289 CrossRef CAS.
- E. Cances, B. Mennucci and J. Tomasi, A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics, J. Chem. Phys., 1997, 107(8), 3032–3041 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
- A. Tenderholt, PyMOlyze, Version 1.1, Stanford University, CA Stanford, 2006 Search PubMed.
- S. J. Akram, et al., Impact of various heterocyclic π-linkers and their substitution position on the opto-electronic attributes of the A–π–D–π–A type IECIO-4F molecule: a comparative analysis, RSC Adv., 2022, 12(32), 20792–20806 RSC.
- H.-x. Li, R.-h. Zheng and Q. Shi, Theoretical study on charge carrier mobilities of tetrathiafulvalene derivatives, Phys. Chem. Chem. Phys., 2011, 13(13), 5642–5650 RSC.
- Z. Shuai, et al., Applying Marcus theory to describe the carrier transports in organic semiconductors: limitations and beyond, J. Chem. Phys., 2020, 153(8), 080902 CrossRef CAS PubMed.
- O. Adeniran and Z.-F. Liu, Quasiparticle electronic structure of phthalocyanine: TMD interfaces from first-principles GW, J. Chem. Phys., 2021, 155(21), 214702 CrossRef CAS PubMed.
- A. Eşme, Structural, spectral characterization, and topological study of (E)-5-(diethylamino)-2-((3,5-dinitrophenylimino)methyl)phenol, Struct. Chem., 2022, 1–12 Search PubMed.
- M. I. Khan, et al., Novel A-π-D-π-A-type non-fullerene acceptors for solution-processed organic photovoltaic cells: a DFT study, J. Solid State Chem., 2022, 123714 Search PubMed.
- Z. He, et al., Side chain engineering of indacenodithieno[3,2-b]thiophene (IDTT)-based wide bandgap polymers for non-fullerene organic photovoltaics, J. Mater. Chem. C, 2022, 10(39), 14633–14642 RSC.
- K. Zuo, et al., PTB7-Th-Based Organic Photovoltaic Cells with a High VOC of over 1.0 V via Fluorination and Side Chain Engineering of Benzotriazole-Containing Nonfullerene Acceptors, ACS Appl. Mater. Interfaces, 2022, 14(16), 18764–18772 CrossRef CAS PubMed.
- M. Rani, et al., Engineering of A-π-D-π-A system based non-fullerene acceptors to enhance the photovoltaic properties of organic solar cells; a DFT approach, Chem. Phys. Lett., 2022, 139750 CrossRef CAS.
- R. Costa, et al., Designed complexes based on betanidin and L0 Dyes for DSSCs: thermodynamic and optoelectronic properties from DFT study, Mol. Simul., 2022, 48(8), 647–661 CrossRef CAS.
- M. Khalid, et al., Exploration of nonlinear optical enhancement and interesting optical behavior with pyrene moiety as the conjugated donor and efficient modification in acceptor moieties, Opt. Quantum Electron., 2022, 54(7), 1–21 CrossRef.
- M. Waqas, et al., Impact of end-capped modification of MO-IDT based non-fullerene small molecule acceptors to improve the photovoltaic properties of organic solar cells, J. Mol. Graphics Modell., 2022, 108255 CrossRef CAS.
- W. U. Khan, et al., Single phase white LED phosphor Ca3YAl3B4O15: Ce3+, Tb3+, Sm3+ with superior performance: color-tunable and energy transfer study, Chem. Eng. J., 2021, 410, 128455 CrossRef.
- L. Palomo, L. Favereau, K. Senthilkumar, M. Stępień, J. Casado and F. J. Ramírez, Simultaneous Detection of Circularly Polarized Luminescence and Raman Optical Activity in an Organic Molecular Lemniscate, Angew. Chem., Int. Ed., 2022, 61(34), e202206976 CrossRef CAS PubMed.
- A. Naveed, et al., Tuning the optoelectronic properties of benzodithiophene based donor materials and their photovoltaic applications, Mater. Sci. Semicond. Process., 2022, 137, 106150 CrossRef CAS.
- E. U. Rashid, et al., Synergistic end-capped engineering on non-fused thiophene ring-based acceptors to enhance the photovoltaic properties of organic solar cells, RSC Adv., 2022, 12(20), 12321–12334 RSC.
- M. Rani, et al., Strategies toward the end-group modifications of indacenodithiophene based non-fullerene small molecule acceptor to improve the efficiency of organic solar cells; a DFT study, Comput. Theor. Chem., 2022, 113747 CrossRef CAS.
- I. Zubair, et al., Designing the optoelectronic properties of BODIPY and their photovoltaic applications for high performance of organic solar cells by using computational approach, Mater. Sci. Semicond. Process., 2022, 148, 106812 CrossRef CAS.
- Y. Chen, et al., Asymmetric alkoxy and alkyl substitution on nonfullerene acceptors enabling high-performance organic solar cells, Adv. Energy Mater., 2021, 11(3), 2003141 CrossRef CAS.
- R. Hussain, et al., Molecular engineering of A–D–C–D–A configured small molecular acceptors (SMAs) with promising photovoltaic properties for high-efficiency fullerene-free organic solar cells, Opt. Quantum Electron., 2020, 52(8), 1–20 CrossRef.
- D. Li, et al., Asymmetric Non-Fullerene Small-Molecule Acceptors toward High-Performance Organic Solar Cells, ACS Cent. Sci., 2021, 7(11), 1787–1797 CrossRef CAS PubMed.
- P. Gopikrishna, et al., Impact of symmetry-breaking of non-fullerene acceptors for efficient and stable organic solar cells, Chem. Sci., 2021, 12(42), 14083–14097 RSC.
- J. Zhang, et al., High-efficiency thermal-annealing-free organic solar cells based on an asymmetric acceptor with improved thermal and air stability, ACS Appl. Mater. Interfaces, 2020, 12(51), 57271–57280 CrossRef CAS PubMed.
- E. U. Rashid, et al., Depicting the role of end-capped acceptors to amplify the photovoltaic properties of benzothiadiazole core-based molecules for high-performance organic solar cell applications, Comput. Theor. Chem., 2022, 1211, 113669 CrossRef.
- J. Xiao, Z. Xiao, J. Hu, X. Gao, M. Asim, L. Pan and J. J. Zou, et al., Rational Design of Alkynyl-Based Linear Donor− π–Acceptor Conjugated Polymers with Accelerated Exciton Dissociation for Photocatalysis, Macromolecules, 2022, 55(13), 5412–5421 CrossRef CAS.
- M. Rafiq, et al., End-capped modification of dithienosilole based small donor molecules for high performance organic solar cells using DFT approach, J. Mol. Liq., 2022, 345, 118138 CrossRef CAS.
- R. M. Alghanmi, et al., Electronic spectral studies and DFT computational analysis of hydrogen bonded charge transfer complexes between chloranilic acid and 2,5-dihydroxy-p-benzoquinone with 2-amino-4-methylbenzothiazole in methanol, J. Mol. Liq., 2018, 256, 433–444 CrossRef CAS.
- Q.-Q. Pan, et al., A comparative study of a fluorene-based non-fullerene electron acceptor and PC61BM in an organic solar cell at a quantum chemical level, RSC Adv., 2016, 6(84), 81164–81173 RSC.
- M. R. Narayan and J. Singh, Roles of binding energy and diffusion length of singlet and triplet excitons in organic heterojunction solar cells, Phys. Status Solidi C, 2012, 9(12), 2386–2389 CrossRef CAS.
- M. Premont-Schwarz, Elementary solute-solvent interactions and the photophysical properties of photoacids, 2013 Search PubMed.
- C. Lu, et al., Linear and Nonlinear Photon-Induced Cross Bridge/Space Charge Transfer in STC Molecular Crystals, Nanomaterials, 2022, 12(3), 535 CrossRef CAS PubMed.
- R. A. Klaasen, et al., A fully automated method for the determination of serum belatacept and its application in a pharmacokinetic investigation in renal transplant recipients, Ther. Drug Monit., 2019, 41(1), 11–18 CrossRef CAS PubMed.
- T. Rakia, et al., Optimal design of dual-hop VLC/RF communication system with energy harvesting, IEEE Commun. Lett., 2016, 20(10), 1979–1982 Search PubMed.
- M. Ans, et al., Development of fullerene free acceptors molecules for organic solar cells: a step way forward toward efficient organic solar cells, Comput. Theor. Chem., 2019, 1161, 26–38 CrossRef CAS.
- A. Tang, et al., Benzotriazole-Based Acceptor and Donors, Coupled with Chlorination, Achieve a High VOC of 1.24 V and an Efficiency of 10.5% in Fullerene-Free Organic Solar Cells, Chem. Mater., 2019, 31(11), 3941–3947 CrossRef CAS.
- A. Tang, et al., ∼1.2 V open-circuit voltage from organic solar cells, J. Semicond., 2021, 42, 070202 CrossRef.
- M. U. Saeed, et al., End-capped modification of Y-Shaped dithienothiophen [3,2-b]-pyrrolobenzothiadiazole (TPBT) based non-fullerene acceptors for high performance organic solar cells by using DFT approach, Surf. Interfaces, 2022, 101875 CrossRef CAS.
- N. K. Elumalai and A. Uddin, Open circuit voltage of organic solar cells: an in-depth review, Energy Environ. Sci., 2016, 9(2), 391–410 RSC.
- M. I. Khan, et al., End-capped group modification on cyclopentadithiophene based non-fullerene small molecule acceptors for efficient organic solar cells; a DFT approach, J. Mol. Graphics Modell., 2022, 113, 108162 CrossRef CAS PubMed.
- L. Zhang, et al., Fine structural tuning of diketopyrrolopyrrole-cored donor materials for small molecule-fullerene organic solar cells: a theoretical study, Org. Electron., 2016, 32, 134–144 CrossRef CAS.
- U. Yaqoob, et al., Structural, optical and photovoltaic properties of unfused non-fullerene acceptors for efficient solution processable organic solar cell (Estimated PCE greater than 12.4%): a DFT approach, J. Mol. Liq., 2021, 341, 117428 CrossRef CAS.
- Z. Wang, et al., A–π–D–π–A Electron-Donating Small Molecules for Solution-Processed Organic Solar Cells: A Review, Macromol. Rapid Commun., 2017, 38(22), 1700470 CrossRef PubMed.
- M. Du, et al., Application of indacenodiselenophene central core and modulation of terminal group interaction for high-efficient P3HT-based organic solar cells, J. Mater. Chem. C, 2022, 10(27), 10114–10123 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Ahmed M. Shawky
*b,
Ahmed M. Shawky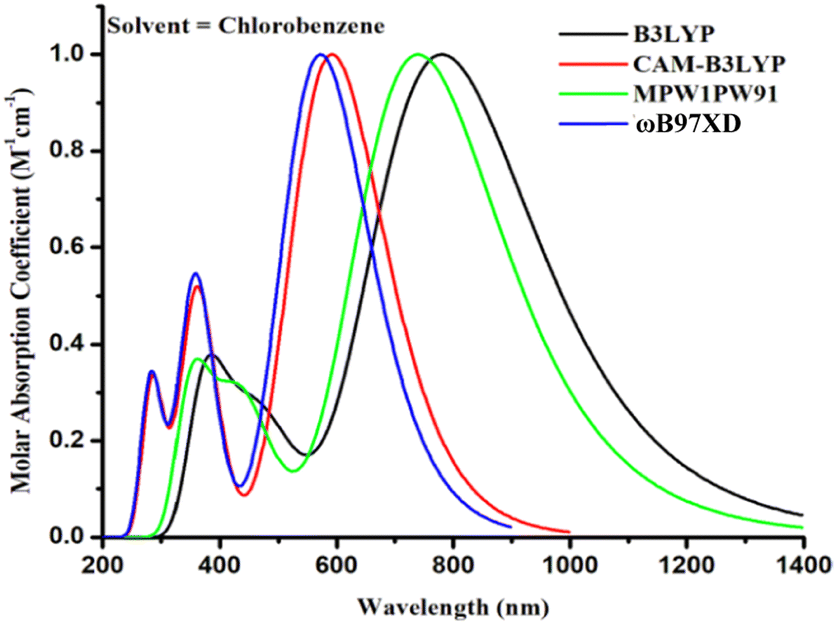

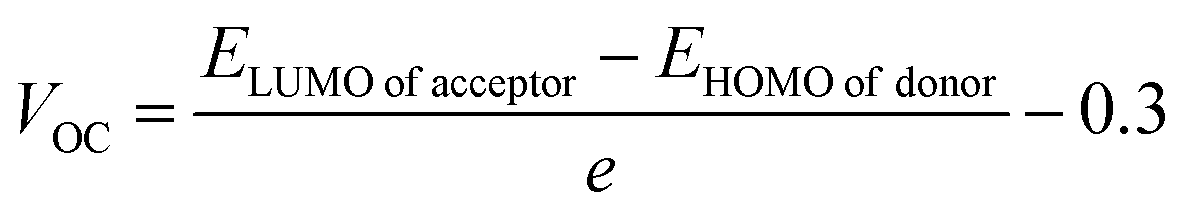
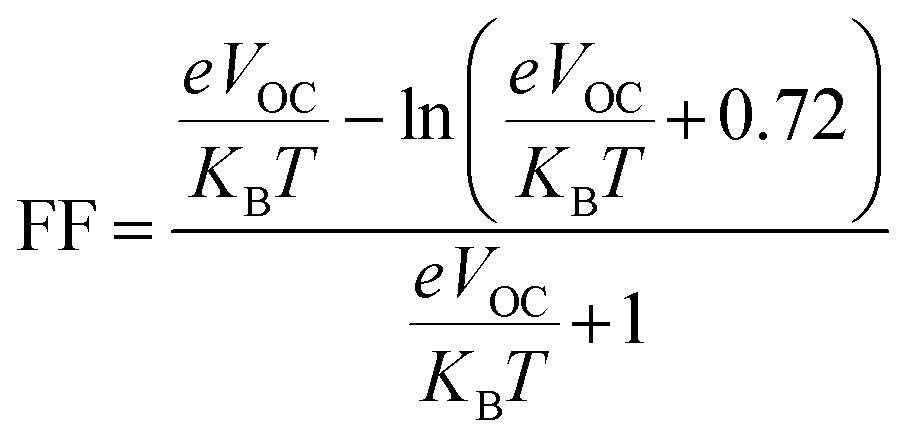
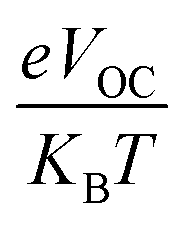 . The calculated fill factor values for the reference molecule and the designed chromophores (TM1–TM7) are summarized in Table 9, these values being 0.89, 0.86, 0.74, 0.83, 0.87, 0.88, 0.87 and 0.86, respectively. The measured results displayed that the molecules TMR, TM4, TM5 and TM6 have almost the similar value of fill factor, which is why they exhibit admirable photovoltaic properties compared with TM1, TM2, TM3, and TM7 having lower FF value. The descending pattern of FF of all compounds follows the sequence: TMR > TM5 > TM4 = TM6 > TM1 = TM7 > TM3 > TM2.
. The calculated fill factor values for the reference molecule and the designed chromophores (TM1–TM7) are summarized in Table 9, these values being 0.89, 0.86, 0.74, 0.83, 0.87, 0.88, 0.87 and 0.86, respectively. The measured results displayed that the molecules TMR, TM4, TM5 and TM6 have almost the similar value of fill factor, which is why they exhibit admirable photovoltaic properties compared with TM1, TM2, TM3, and TM7 having lower FF value. The descending pattern of FF of all compounds follows the sequence: TMR > TM5 > TM4 = TM6 > TM1 = TM7 > TM3 > TM2.