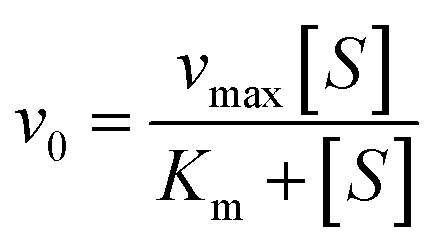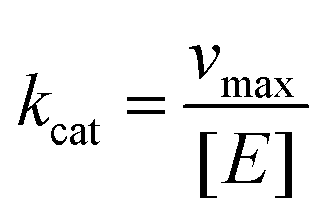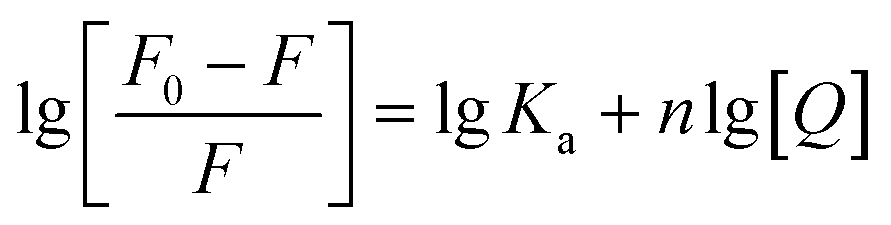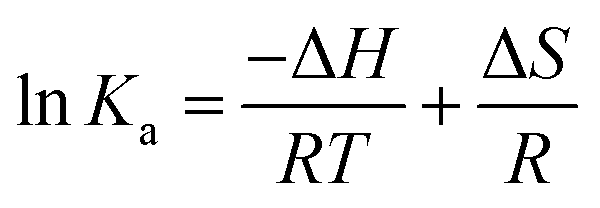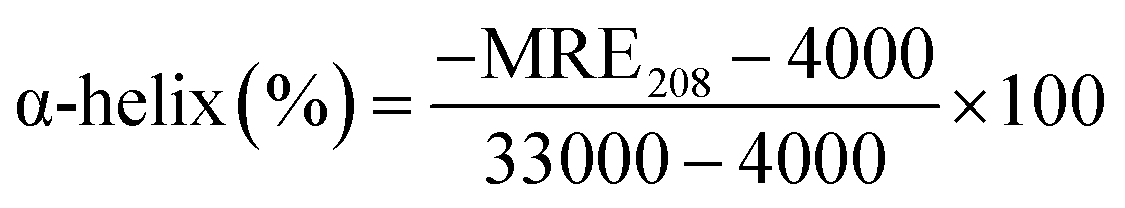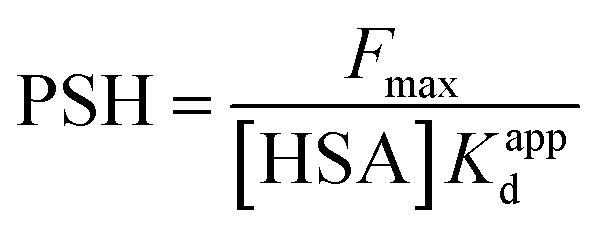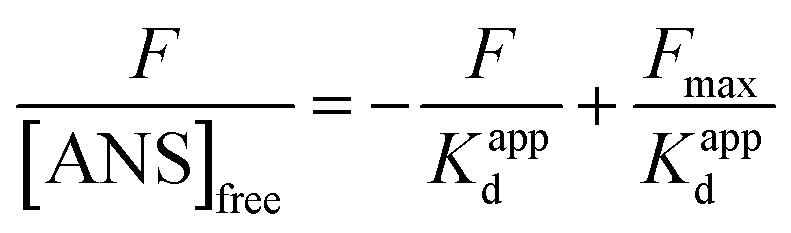DOI:
10.1039/D2RA07377C
(Paper)
RSC Adv., 2023,
13, 8281-8290
Unveiling the binding details and esterase-like activity effect of methyl yellow on human serum albumin: spectroscopic and simulation study
Received
20th November 2022
, Accepted 4th March 2023
First published on 14th March 2023
Abstract
The food sector uses methyl yellow (MY) extensively as a colorant. The primary transporter in vivo that influences MY absorption, metabolism, distribution, and excretion is human serum albumin (HSA). Exploring the binding process and looking at how HSA and MY work physiologically at the molecular level is therefore very important. Experiments using steady-state fluorescence and fluorescence lifetimes proved that HSA and MY's quenching mechanisms were static. The HSA–MY complex's binding constant was estimated using thermodynamic parameters to be around 104 M−1. The hydrophobic forces were a major factor in the binding process, as evidenced by the negative ΔG, positive ΔH, and ΔS, which suggested that this contact was spontaneous. Site tests showed that MY linked to HSA's site I. Circular dichroism and three-dimensional fluorescence analysis revealed that the 1.33% α-helix content dropped and the amino acid microenvironment altered. While HSA's protein surface hydrophobicity decreased when engaging MY, the binding of MY to HSA reduced in the presence of urea. The stability of the system was assessed using molecular modeling. Additionally, HSA's esterase-like activity decreased when MY was present, and Ibf/Phz affected the inhibition mechanism of MY on HSA. These findings offer a distinctive perspective for comprehending the structure and functioning of HSA and evaluating the safety of MY.
1. Introduction
Colorants are extensively used in the food industry and this is inseparable from the fact that they can directly affect organ perception and stimulate appetite.1–3 Methyl yellow (MY) is a common azo-dye used as food coloring in many regions.4 Azo dyes and their degradation byproducts may have potential carcinogenicity.5–8 In relevant microbial related experiments, MY has been proven to be a mutagen, whereas pharmaceutical experiments show carcinogenicity in mice injected with MY.9 Given the above-mentioned potential harm of azo compounds, relevant research and exploration on azo dye compounds is necessary to elucidate the relationship between azo dyes and human health.
A considerable part of plasma protein is human serum albumin (HSA),10 the HSA content in normal people is about 40 g L−1.11 The distribution and eradication of food additives are both handled by it.12 Additionally, HSA is crucial for preserving the blood's colloidal osmotic pressure.13 HSA is very special because of its internal hydrophobic cavity and special affinity, enabling its easy binding to small molecules.14–16 Most small molecules that have been shown to attach to HSA are mostly at Sudlow's site I (in subdomain IIA) and site II (in subdomain IIIA).17,18 At the same time, azo dyes can bind reversibly and effectively with HSA, which can regulate the effective concentration of limiting toxicity.19,20 The molecular recognition between the two molecules also affects the active function of biomolecules.3,21 Therefore, a careful investigation of the HSA and MY binding is essential to better clarify the colorant–protein interaction.
The interaction between azo dyes (such as citrus red 2 and tartrazine) and HSA has been thoroughly studied, providing another idea for the safety evaluation of azo dyes.14,22 The purpose of the current study was to look at how HSA and MY interact under simulated physiological conditions in vitro through multispectral, computer simulation, and other methods. The impact of this interaction on the structure and function of physiologically important proteins was also evaluated through fluorescence spectroscopy to identify the binding mechanism, the basic thermodynamic parameters, and binding related force.23–26 The quenching mechanism of the system was further discussed by fluorescence-lifetime measurements. The binding site of MY to HSA was identified by site experiment. Monitoring the changes of HSA conformation and amino acid residue microenvironment during the binding process by three-dimensional (3D) fluorescence and circular dichroism (CD). Fluorescence analysis was used to investigate anti-denaturation capacity and protein surface hydrophobicity (PSH) of HSA–MY system. Dynamical simulations (MD) and molecular docking were applied to theoretically evaluate the mode of binding and stability of the combination. In addition, the effect of MY on esterase-like activity of HSA was examined to understand its potential physiological safety. This paper explored the distribution and transport mechanism of MY at the molecular level and provided ideas to understand its toxicological evaluation.
2. Experimental
2.1. Chemicals and reagents
HSA stock solution was prepared by phosphate buffer solution (PBS, pH 7.2–7.4, 10 mM, 0.1 M NaCl) and stored in a 4 °C refrigerator, and HSA (>99%) was purchased from Sigma Chemical Company (St. Louis, MO, USA). Urea, ibuprofen (Ibf), p-nitrophenyl acetate (p-NPA), 8-anilino-1-naphthalenesulfonic acid (ANS) ammonium salt hydrate, phenylbutazone (Phz), phosphate buffer salt dry powder, and MY were obtained from J&K Scientific LLC (Beijing, China). Ibf, MY and Phz are directly dissolved in absolute ethanol separately to make their concentration 2 mM. The water used for all dilution operations is buffer solution. PBS dry powder was dissolved in Milli-Q water, which was purchased from Solarbio® (Beijing, China). Ensure that the ethanol content is small enough (2%) to maintain HSA activity. Other reagents used are analytical grade, which can be used directly without further treatment.
2.2. Fluorescence spectroscopy experiment
The steady-state fluorescence quenching experiment was conducted at three different temperatures, 298 K, 304 K, and 310 K, respectively, while the fluorescence spectrometer (Varian, USA) collected the pertinent data. The protein and small molecule concentrations were set at 2 μM and 0–6 μM, respectively. The excitation and emission wavelengths are 280 nm and 300 nm, respectively, and the excitation and emission slit widths are 5 and 10 nm.
In the site competing experiment, the concentrations of Ibf/Phz are 0, 1, 2, 3, 4, 5, 6 μM, the concentration of HSA and MY are 2 and 3 μM, respectively. The conditions of instrument are consistent with the steady-state fluorescence experiment.
In the three-dimensional fluorescence experiment, the concentration ratio of HSA to MY was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 and 1:1.5, and the excitation (emission) wavelength and slit width were both 200 nm and 5 nm.
:0 and 1:1.5, and the excitation (emission) wavelength and slit width were both 200 nm and 5 nm.
2.3. Fluorescence lifetime
The fluorescence lifetime of HSA combined with different concentrations of MY was measured by Jobin Yvon Fluorolog-3 spectrofluorometer (Horiba, LesUlis, FRA), and the proportions were 1:0, 1:1.5, 1:3, respectively.
2.4. Far-UV CD spectroscopy
The circular dichroism of HSA–MY supramolecular system was determined by spectrophotometer (Model 400, AVIV, USA), and the ratio of concentration was consistent with that of three-dimensional spectroscopy. The spectral wavelength ranges from 190 to 250 nm.
2.5. Molecular docking
Molecular docking has great advantages in predicting the conformation of binding processes in interaction system. The process of analyzing the docking conformation of HSA–MY can be realized by YASARA v17.4.17. The respective 3D correlation models were obtained from the protein database (PDB ID: 1H9Z) and PubChem (PubChem CID: 6053). The energy-minimization module of YASARA platform was used to optimize receptors and ligands. In the simulation, the algorithm adopted by the software program is based on semi-flexible docking, so as to obtain the ideal docking model. The algorithm can get 25 docking results, among which the optimal solution is used for later simulation analysis. The pH was set at 7.4. The temperature is room temperature 298 K. The two dimensional action diagram is treated with LIGPLOT program.
2.6. Molecular dynamics (MD) simulation
On the basis of the docking results, the same software is used to further evaluate the stability of the system. The relevant setting conditions include the following parameters, temperature set at 298 K, periodic boundary conditions and TIP3P model. The box is a cube with a side length of 100.52 Å, containing one MY and one HSA, and the total number of atoms is 102693. The charge status of MY is calibrated using the AM1-BCC model. The statistical ensemble is NPT, and the simulated force field is AMBER14 force field with constant atmospheric pressure (1 bar). In addition, the system is neutralized with Cl− or Na+ and simulated annealing with minimum steepness drop. Data were continuously recorded every 10 ps throughout the simulation.
2.7. PSH determination
A ternary system of HSA–MY–ANS was created by adding exogenous substance ANS to the HSA–MY complex solution. The concentration of ANS is 0, 0.5, 1, 1.5, 2, 2.3, 2.6, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 μM. The relevant test conditions were as follows: excitation and emission wavelengths were 380 nm and 480 nm, respectively, at room temperature. The experiment was tested by a Thermo Varioskan LUX.
2.8. Urea-induced unfolding studies
High concentration of urea was added to induce HSA unfolding to evaluate the stability of HSA–MY system binding at 298 K. For the unfolding experiments, different concentrations of MY (0–6 μM) were prepared while keeping constant the HSA (2 μM) and urea (6 M) concentrations. Incubated HSA–MY–Urea complex at room temperature for 8 hours, and the rest experimental parameters of urea denaturation are consistent with the fluorescence spectrum experiment.
2.9. Esterase-like activity measurements
Using Thermo Varioskan LUX at 405 nm to measure the absorbance value of p-nitrophenol, we analysed the impact of MY on HSA catalytic p-NPA degradation activity at 298 K, and the Phz/Ibf as control, HSA concentration was 10 μM. Determine the initial reaction rate (v0) by analysing the slope of the absorbance curve of p-nitrophenol. The parameters were set as follows: The number of absorbance readings for each plate hole during the dynamic cycle was 10 times with an interval of 15 s. Use Michaelis–Menten equation to substitute v0 and different concentrations of p-NPA into fitting to obtain kinetic parameters:| |
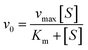 | (1) |
| |
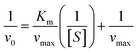 | (2) |
where vmax is the maximum degradation rate, [S] is the concentration of degraded substrate, Km is the Michaelis constant, and kcat is calculated as follows:| |
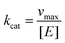 | (3) |
where [E] is the concentration of HSA.
3. Results and discussion
3.1. Study of molecular-recognition mechanism
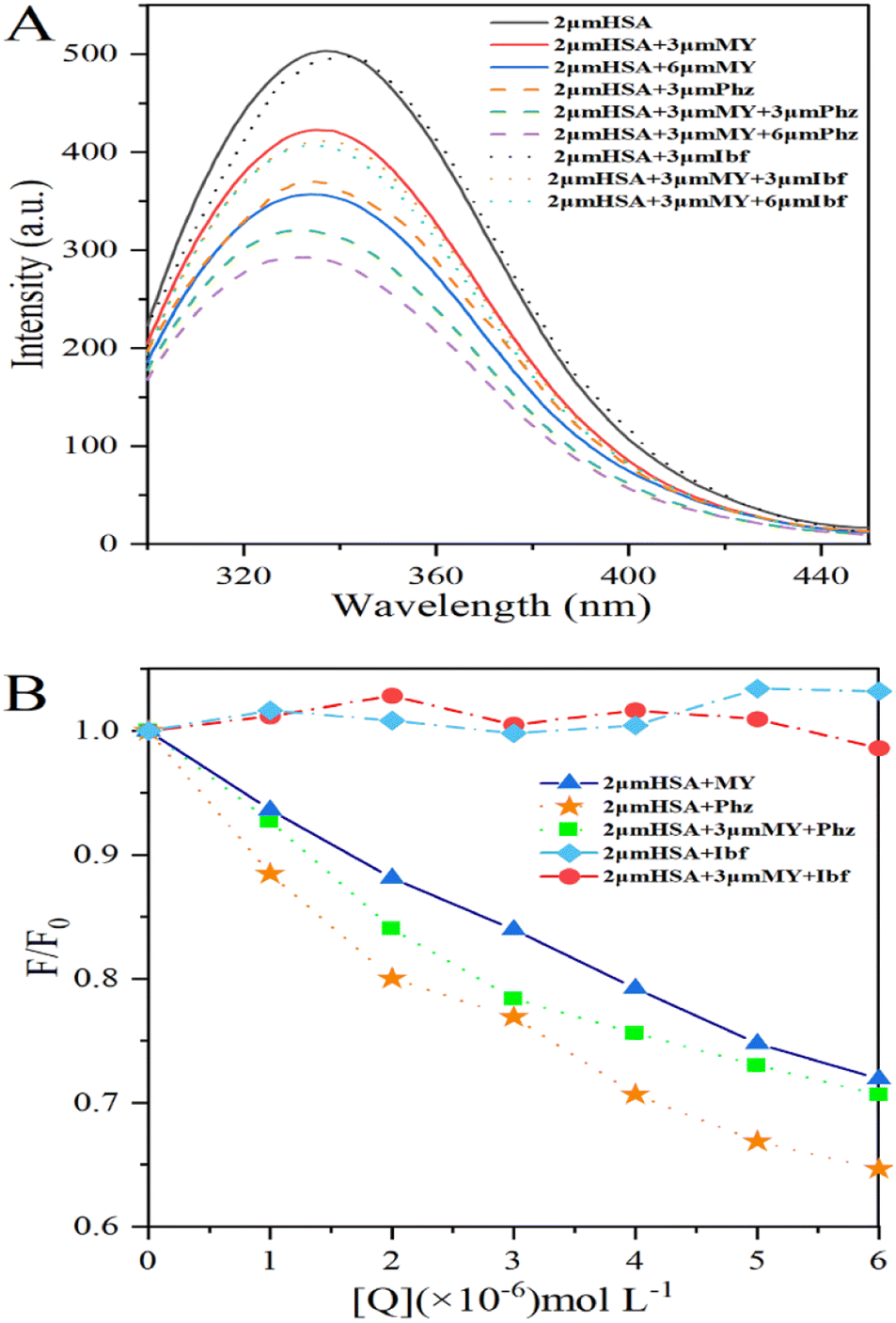
3.1.1. Analysis of fluorescence quenching and mechanism of MY on HSA. To investigate how various small-molecule ligands and proteins interact, molecular fluorescence spectroscopy may be employed efficiently and dependably. This method can be used to obtain specific information about the changes in biophysical properties caused by binding proteins, such as the type of quenching and binding ability. Fluorescence quenching is caused by changes in the microenvironment of chromophores, and results can be obtained by changes in protein fluorescence intensity. Quenching mechanisms may be static or dynamic. The shift of a fluorophore from its excited state to its ground state is known as dynamic quenching caused by short-term intermolecular collision between protein and quench agent. Static quenching is the generation of a new binding quench compound. With rising temperature, the static quenching constant (Ksv) decreases and is inversely proportional to the diffusion frequency.27Based on the above, the analysis is made as follows. After adding MY, the fluorescence was quenched (Fig. 1A). Increasing MY concentration caused a noticeable blue shift and a steady decrease in the emission intensity of HSA, that could be caused by the environment around Trp and Tyr becoming more hydrophobic. These findings demonstrated that MY interacted with HSA and reduced its inherent fluorescence. The connection between the quenching constant and temperature, which was inversely proportional to temperature for static quenching, may be used to distinguish between the two forms of quenching. The mathematical link between fluorescence-quenching findings and small-molecule quenchers can be expressed using the Stern–Volmer equation (eqn (4)):28
| |
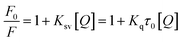 | (4) |
where
F is the fluorescence intensity of HSA–MY complex, and
F0 is that of HSA.
Ksv is the quenching constant,
Kq is rate constant of the fluorescence extinction. [
Q] is the MY concentration, and
τ0 is the of HSA's fluorescence lifetime.
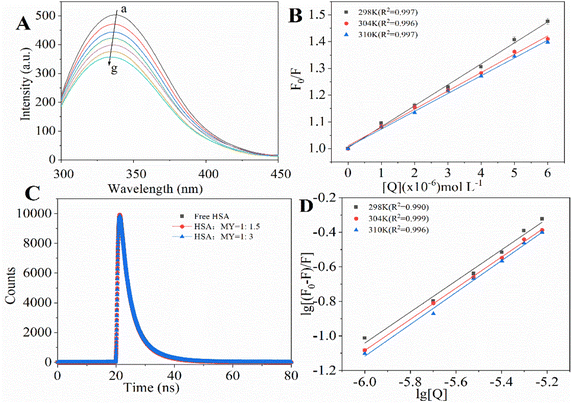 |
| | Fig. 1 (A) Fluorescence spectra of HSA–MY system; (B) Stern–Volmer curves of HSA–MY system at different temperatures; (C) fluorescence lifetime curve of the HSA–MY system; (D) double logarithmic plot of fluorescence quenching for the HSA–MY system at 298 K, 304 K and 310 K. | |
A linear dependence between relative fluorescence intensity (F0/F) and quench agent was obtained at 298 K, 304 K, and 310 K as shown in Fig. 1B. The slope of eqn (4) was represented by Ksv. The pertinent charts and figures showed that temperature climbed as Ksv values steadily fell (Table 1), and Kq values were in the order of 1012, which was significantly greater than the maximum quenching rate constant of HSA (2 × 1012). This discovery supported a static quenching process involving the formation of an equilibrium ground state HSA–MY complex.
Table 1 Binding and thermodynamic parameters of MY–HSA system at different temperature conditions. The results in the table represents the means ± SDs (n = 3)a
| T (K) |
Ksv (×104 M−1) |
Kq (×1012 M−1) |
n |
Ka (×104 M−1) |
ΔG (kJ mol−1) |
ΔH (kJ mol−1) |
ΔS (J mol−1 K−1) |
| As shown in the combination of HSA-MY system with relevant parameters, this indicates a static quenching mechanism. |
| 298 |
7.83 ± 0.19 |
7.83 ± 0.19 |
0.90 ± 0.04 |
4.32 ± 0.25 |
−26.46 |
|
|
| 304 |
6.84 ± 0.17 |
6.84 ± 0.17 |
0.91 ± 0.01 |
4.35 ± 0.07 |
−26.98 |
0.88 |
91.67 |
| 310 |
6.65 ± 0.14 |
6.65 ± 0.14 |
0.92 ± 0.03 |
4.41 ± 0.16 |
−27.56 |
|
|
In order to further verify the accuracy of the static quenching results obtained from the mathematical model, the time-resolved fluorescence spectroscopy method is used, which is based on measuring the lifetime of the chromophore. If the quenching mechanism of HSA–MY was dynamic, the fluorescence life would be reduced due to molecular collision; if the quenching was static, the fluorescence life would not change.29 Fig. 1C displayed the time-resolved fluorescence curves of samples with varying MY concentrations and constant HSA concentrations. HSA's decay curve was not significantly altered by the addition of various MY concentrations, demonstrating that HSA's fluorescence lifespan was unaffected by the MY addition.
Triexponential iterative fitting was used to evaluate the curves and get the pertinent parameters, namely decay times (τi), and pre-exponential factors (αi). And αi represents the relative fraction of τi (the lifetime of different components), whose sum is 1. Using the following equation, the average fluorescence lifespan (τ) of HSA was determined (eqn (5)):14
| | |
〈τ〉 = α1τ1 + α2τ2 + α3τ3
| (5) |
The factors and average fluorescence lifetime obtained by fitting a function are listed in Table 2. The accuracy of curve-fitting results can be evaluated by analyzing the chi-square value (χ2). A low χ2 means a good fit.15 The fitting results showed that the value was about 1, indicating that the curve fitting was accurate and the result was reliable. The fluorescence lifespan of HSA was unaffected by the addition of MY, which was negligible (<0.1 ns). These findings supported the findings of molecular-fluorescence spectroscopy by demonstrating the formation of a ground-state complex between MY and HSA and the static nature of the quenching process.
Table 2 Fluorescence lifetime of HSA at different MY concentrations
| System |
τ1 (ns) |
α1 |
τ2 (ns) |
α2 |
τ3 (ns) |
α3 |
τ (ns) |
χ2 |
| Free HSA |
3.093 |
0.36 |
0.610 |
0.40 |
6.931 |
0.24 |
3.024 |
1.073 |
| HSA:MY = 1:1.5 |
2.911 |
0.39 |
0.523 |
0.34 |
6.723 |
0.27 |
3.128 |
1.035 |
| HSA:MY = 1:3 |
2.837 |
0.41 |
0.554 |
0.34 |
6.685 |
0.25 |
3.014 |
1.059 |
3.1.2. Binding capability and forces. Based on the static quenching result and literatures,30–33 the relevant parameters of the binding process can be obtained by the following assumptions: If the quenching agent (Q) interacts independently with a group of equivalent sites (n) on the protein and closely combines to form (QnP), the equilibrium should be reached:34–36Then the binding equilibrium constant is derived:
| | |
Ka = [QnP]/([P]free × [Q]nfree)
| (7) |
If a binding ligand can alter a protein's intrinsic fluorescence and no intermediate is created, the binding of n solutes occurs in a sole elementary step, the modified Stern–Volmer equation can be obtained as:
| |
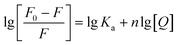 | (8) |
where [
Q] is the quencher concentration,
F0 is the fluorescence intensity of the protein without quencher and
F is that with quencher.
Ka is the binding constant.
For HSA–MY system, it is static quenching, so eqn (8) is applicable and can be used to further calculate relevant parameters. The Ka and n were calculated as indicated in Table 1. Ka had a magnitude of 104, and n was not far from 1. These results imply that MY attached to HSA with a single affinity site and a moderate strength (103 to 106 L mol−1).37 Additionally, heating promoted the binding of HSA and MY, thereby enhancing the stability of the HSA–MY supermolecule complex.
For interaction analysis, multiple forces including hydrophobic, hydrogen-bonding, electrostatic, and van der Waal interactions may all play a role in the binding of ligands to biomacromolecules.38 The sorts of molecular forces involved in molecule-binding processes (ΔG, ΔH, and ΔS) can be reflected in the quantities and symbols of relevant parameters. These parameters were calculated using the eqn (9) that represents Gibbs–Helmholtz equation and eqn (10) that represents the Van't Hoff equation, respectively:
| | |
ΔG = ΔH − TΔS = −RTlnKa
| (9) |
| |
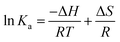 | (10) |
where the gas constant is in terms of
R. Using
Fig. 1D and above formula, Δ
H and Δ
S were measured. Δ
G was obtained by a simple calculation of
eqn (9).
39 According to
Table 1, a positive Δ
H meant that the binding of MY to HSA was an endothermic process and that the complex's formation was aided by the rise in temperature. On the other hand, Δ
H > 0 and Δ
S > 0 indicated that the molecular-force type was hydrophobic force. Additionally, the HSA–MY complex was formed spontaneously as evidenced by the negative Δ
G values, which again showed the spontaneity of the binding process.
3.1.3. Judgment of binding site of MY. The sites I or II of HSA are often occupied by small molecules during the binding and transport process.40 To determine the binding position of MY, the probes of Phz and Ibf were used in a competition experiment at 298 K as their binding sites were identified as sites I and II, respectively.41,42 By examining the shift in fluorescence intensity, the information about the MY binding site was discovered. The following eqn (11) was utilized to investigate MY binding sites:| |
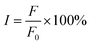 | (11) |
where F0 and F are the fluorescence intensity of the system before and after adding 0–6 μM MY/Phz/Ibf, respectively.As shown in Fig. 2A, the addition of Phz causes a greater fluorescence quenching degree of HSA than MY at the same concentration (3 μM), the result indicates that the quenching ability of Phz is stronger than that of MY, that is, Phz has bigger binding ability to HSA than MY. However, the fluorescence intensity of HSA is almost unchanged after adding Ibf, which means that Ibf has no quenching effect on protein intrinsic fluorescence. This phenomenon can be clearly illustrated by Fig. 2B, and these results are consistent with the previous reports.43,44 As shown in Fig. 2B, the F/F0 value of the HSA–MY–Phz system is higher than HSA–Phz system and lower than HSA–MY system. Since the binding constant of Phz/Ibf (105 M−1) is higher than MY (104 M−1),45,46 Phz competes with MY with the adding of Phz, resulting in a complex system of HSA–MY and HSA–Phz. Thus, the F/F0 value of HSA–MY–Phz complex system sits between the two blank systems when it reaches dynamic equilibrium. Also, the fluorescence intensity of the system will not change with the addition of Ibf. When system equilibrium reaches, Ibf does not compete with MY, HSA–MY–Ibf system that two ligands bind to a protein at the same time forms. Because Ibf is not a quencher, the F/F0 value remains the same as that of HSA–MY before adding Ibf. Thus, in this work, MY competes with only Phz on site I of HSA. In addition, the structural characteristics of MY can also provide information about binding sites. Conjugated molecules such as benzene rings are more likely to site I, while others, such as long chain fatty acids are more easily bound to site II,14,47–49 MY is azobenzene derivative molecule, which is more likely to bind at the site I. In a word, MY and Phz have a competitive binding in the same region on HSA, namely site I.
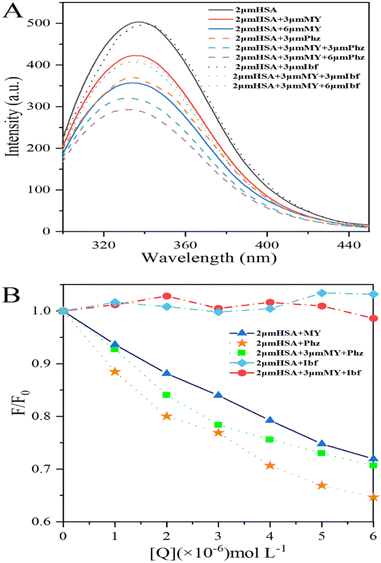 |
| | Fig. 2 (A) Fluorescence spectra of the HSA–MY-Probe system; (B) percentage conversion diagram of fluorescence intensity of HSA–MY-Probe system. The concentration of MY/Phz/Ibf is all 0–6 μM with the increment of 1 μM. | |
3.2. Analysis of HSA conformational changes caused by MY
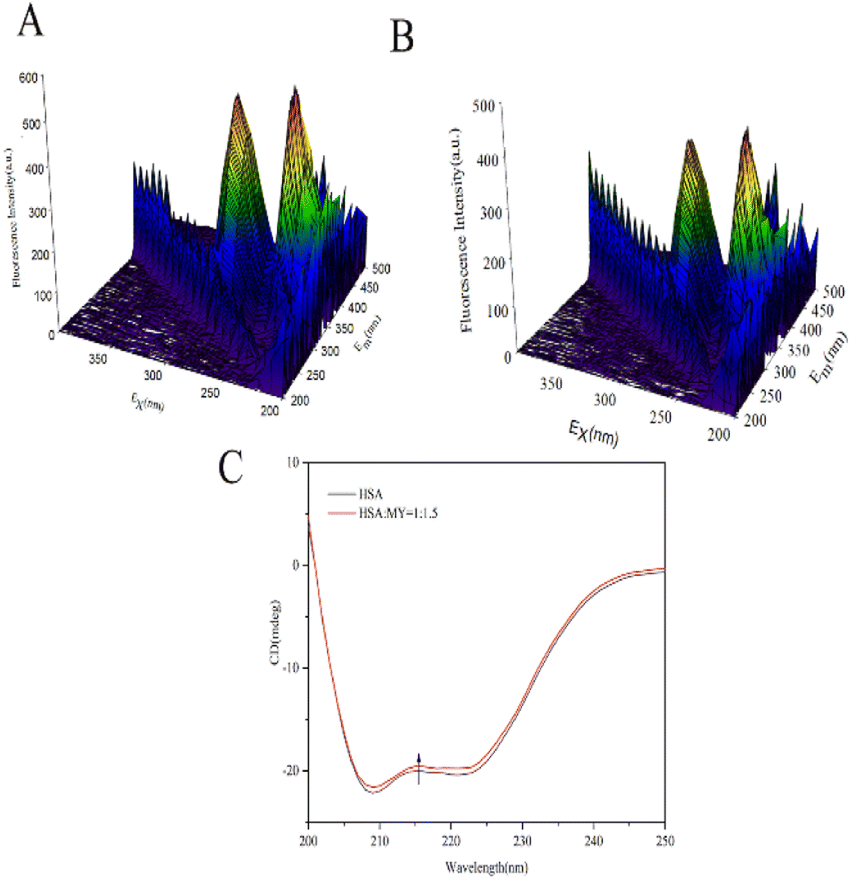
3.2.1. 3D fluorescence spectroscopy. 3D fluorescence spectra can comprehensively display sensitive information related to the protein skeleton, such as overall HSA particle size and the surrounding environment near the site.50 Fig. 3A and B show the spectra of HSA–MY at 298 K. The location and intensity of each peak are shown in the Table 3. Peak A was called the Rayleigh scattering peak, and the homogeneity and size of the HSA molecule were reflected in its fluorescence intensity. Peak I is connected to the n–Π* transition of Trp and Tyr residues, while peak II is related to the Π–Π* transition of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in the polypeptide skeleton structure, those can represent changes in the microenvironment of amino acid residues.51 After adding MY, the intensity of peak I and II decreased, which was due to the fluorescence quenching around Tyr and Trp caused by the combination of MY and HSA. In addition, there was a blue shift in the locations of peaks I and II in the two spectrograms (Table 3), indicating a rise in hydrophobicity, which was consistent with the effects of quenching experiment. As a result, MY binding had an impact on the local environment of aromatic residues in HSA.
O group in the polypeptide skeleton structure, those can represent changes in the microenvironment of amino acid residues.51 After adding MY, the intensity of peak I and II decreased, which was due to the fluorescence quenching around Tyr and Trp caused by the combination of MY and HSA. In addition, there was a blue shift in the locations of peaks I and II in the two spectrograms (Table 3), indicating a rise in hydrophobicity, which was consistent with the effects of quenching experiment. As a result, MY binding had an impact on the local environment of aromatic residues in HSA.
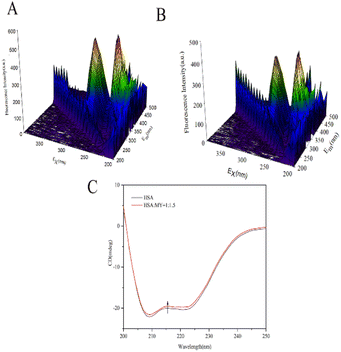 |
| | Fig. 3 (A) 3D fluorescence spectra of HSA; (B) 3D fluorescence spectra of HSA–MY; (C) CD spectral of HSA–MY system. | |
Table 3 3D fluorescence spectral parameters of HSA alone in the presence of MY
| System |
Peak |
Peak position λex/λem (nm/nm) |
Stokes shift Δλ (nm) |
Intensity |
| HSA |
A |
280/280 → 350/350 |
0 |
165.09 → 236.21 |
| 1 |
280/339 |
59 |
522.31 |
| 2 |
230/330 |
100 |
571.03 |
| HSA:MY = 1:1.5 |
A |
280/280 → 350/350 |
0 |
166.26 → 272.20 |
| 1 |
280/327 |
47 |
403.87 |
| 2 |
230/334 |
104 |
448.24 |
3.2.2. CD spectroscopy. The conformational helical variations of HSA may be simply and swiftly analysed by CD.52 The Fig. 3C depicted the spectrum, which had two negative bands that represent HSA-specific transition peaks. The eqn (12) and (13) may be used to compute and examine the effect of MY on the spiral based on pertinent data:| |
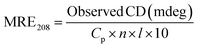 | (12) |
| |
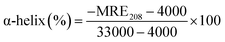 | (13) |
where Cp is the concentration of HSA, l is the route length of the cell, and n is the number of HSA's (585) amino acid residues. MRE208 is the mean residue ellipticity at 208 nm. Moreover, the MRE value of the pure α-helix is 33000 at 208 nm, whereas that of random curls and β form is 4000.29 MY addition increased the characteristic peak intensity of HSA but did not significantly change the peak shape and location. Using the above formula, it was determined that HSA has a α-helix (%) of 50.79%, and it decreased to 49.46% after adding 3 μM MY. A decrease of the α-helix illustrated that MY induced a change in the HSA conformation. However, the information about peak shape and location did not dramatically alter, the situation indicated that the combination of MY was mild and ensured the overall stability of HSA.
3.2.3. PSH determination. The transporter HSA's hydrophobicity on the surface affects the interaction of ligands. ANS can monitor changes in surface hydrophobicity, because its fluorescence is significantly enhanced when it combines with the corresponding hydrophobic region of HSA that relevant hydrophobic characteristic information can be measured by the numerical value of PSH.53 The following equation might be used to calculate PSH:| |
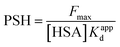 | (14) |
where Fmax is the maximum fluorescence intensity when ANS is combined with HSA to saturation state, and 1/Kappd is the affinity degree of ANS in HSA–MY system, and these two parameters can be obtained by using the eqn (15):| |
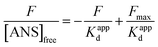 | (15) |
where [ANS]free is the concentration of free ANS. Similarly, the other related parameters are related as follows: ([ANS]bound = F/B in which F is fluorescence intensity, B is a dimensionless factor coefficient. In other words, F and bound ANS concentration exhibit a linear connection when the concentration of ANS is very low. Based on the correlation between total ANS concentration and bound ANS concentration, free ANS concentration is calculated. [ANS]free = [ANS]total − [ANS]bound). Fig. 4A's curve illustrated the increase in fluorescence intensity when ANS and HSA–MY bind to saturation. The Fmax was 288.06 ± 0.105 with MY and 211.82 ± 0.149 without MY respectively (Table 4). Notably, the values of 1/Kappd fell when MY was present, indicating that the competitive combination of MY and ANS in the drainage area leaded to the decrease of the combination amount of ANS and HSA, resulting in the above changes. A 68% decrease was also found in ΔPSH in the presence of MY. This result may be due to the tight binding between MY and HSA, made the hydrophobic cavity of the site shrink spatially. This finding also corresponded with the typical hydrophobic forces and the aforementioned decrease.
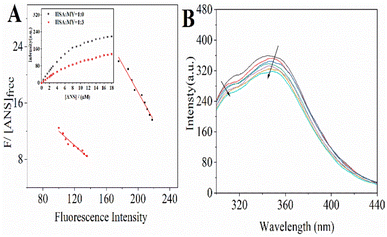 |
| | Fig. 4 (A) The Scatchard plots for the titration with increasing concentrations (0–18 μM) of ANS to HSA–MY. (Inset) Fluorescence intensity with binding of ANS to HSA–MY; (B) the fluorescence spectra when urea (6 M) is added in HSA–MY complex solution. (CHSA = 2 μM, CMY = 0–6 μM) at T = 298 K. | |
Table 4 Surface hydrophobicity parameters for HSA–MY system
| System |
Fmax |
1/Kappd |
PSH index |
| HSA–ANS |
288.06 ± 0.105 |
0.210 ± 0.0124 |
2.89 × 107 |
| HSA–MY–ANS |
211.82 ± 0.149 |
0.101 ± 0.0098 |
1.11 × 107 |
3.2.4. Urea effect on HSA–MY complex formation. By using urea-induced denaturation, the impact of MY binding on the stability and unfolding characteristics of HSA was investigated. Denaturation increased the exposure of phenylalanine, tyrosine, or tryptophan in protein molecules to water, thereby changing the emission characteristics of fluorescence properties.54,55 Compared with HSA, a new tyrosine peak with a slight red shift was found at 308 nm (Fig. 4B), indicating a change in the polar environment, which may be due to the increased distance between Trp and Tyr leading to a decrease or stop of energy transfer. Addition of exogenous urea reduced the fluorescence intensity characteristics of HSA (Fig. 4B), which may be due to its interaction with HSA and structural changes. Meanwhile, the change of fluorescence intensity was very weak, indicating that MY could not effectively quench the fluorescence of denatured HSA. Adding urea, the hydrophobicity around the interaction residues of MY and protein decreased, and Ka decreased significantly, that is, the hydrophobic force was more important in the binding process.
3.3. Analysis of computer simulation
3.3.1. Molecular docking. Introduce MY to form a supramolecular system, and obtain information about the docking mode of the system, this could be done using docking techniques, which can also validate fluorescence probe experimental findings. When analysing the advantages and disadvantages of the combination mode, a higher energy corresponded with better bonding. The one with the highest binding energy (8.29 kJ mol−1) was selected for analysis, and the constitutive model was shown in Fig. 5A and B, that MY binds to the hydrophobic cavity at site I of HSA. Docking results showed that the bonding between MY and HSA depended on hydrogen bond and hydrophobic forces. A hydrogen bond with a length of 3.07 Å and an energy of 10.90 kJ mol−1 existed between MY and the amino acid residue Asp324 close to the docking site. Meanwhile, there were hydrophobic forces between MY and the amino acid residues of Ala213, Leu331, Ala350, Leu327, Gly328, Phe228, Ser232, Lys212, Val235, Val216, Arg209. MY was entirely encased in site I of HSA as a result of the two forces. These results well agreed with the in vitro ones (Fig. 2), revealing that MY definitely bound with Sudlow's site I of HSA.
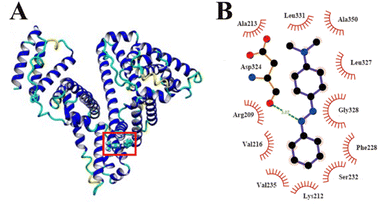 |
| | Fig. 5 (A) Molecular docking results of HSA–MY; (B) a schematic representation used by Ligplot software, where hydrogen bond is in green with distance. | |
3.3.2. MD simulation. To examine the HSA–MY system's natural dynamics, MD simulation was carried out under artificially realistic physiological settings that the optimal conformation comes from the docking experiment. The purpose was to obtain details about the information about the dynamic stability of the HSA–MY system (Fig. 5).In MD simulations, the deviation of protein skeleton atoms in the HSA–MY system is measured by root-mean-square deviation (RMSD), and it is also used to assess if the system is in equilibrium.56 The RMSD values of Cα atoms for the HSA–MY systems were shown during the simulation in Fig. 6A. In the process of 0–86 ns, the overall trend of RMSD of HSA–MY and HSA system is increasing, and the maximum values of RMSD are 4.99 Å and 3.94 Å respectively, which means that the skeleton structure of the two systems has experienced some dynamic changes. Meanwhile, the RMSD of HSA–MY system decreases during the process of 80–86 ns, contrary to that of HSA system, which shows the influence of MY on the skeleton structure of HSA. In the process of 86–100 ns, the RMSDs of the two systems are lower than the maximum value of the RMSD of each system, and the fluctuation range of RMSD of the two systems is within 1.8 Å, then the RMSD curves tend to be flat. According to the literature, the fluctuation range of the RMSD of the system was lower than 2 Å, indicating that the system was well equilibrated.57 Thus, the two systems reach dynamic equilibrium after 86 ns. Meanwhile, the structure of HSA was marginally more stable than that of HSA–MY, as evidenced by the fact that compared to HSA the RMSD of HSA–MY was lower. In other words, the binding of MY influences the amino acids microenvironment and the stability of protein structure.
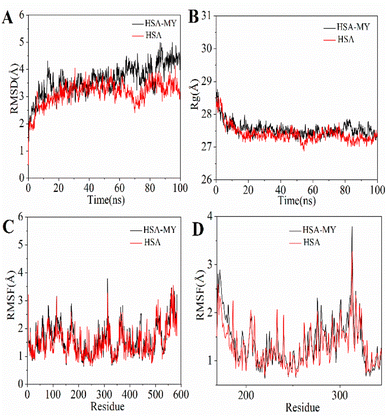 |
| | Fig. 6 (A) RMSD of HSA–MY system within 100 ns; (B) Rg curves of HSA–MY system during simulation procedure; (C) RMSF values of HSA–MY system versus residue numbers; (D) enlarged view of RMSF values from residue number 150 to 350. | |
Analysis of radius of gyration (Rg) can obtain information about the stability of the skeleton molecules of supramolecular systems.58 Rg was negatively correlated with the degree of protein volume contraction, i.e., a greater Rg corresponded with looser contraction. The skeleton of HSA–MY was looser during the simulation process, keeping the Rg of HSA high throughout (Fig. 6B), indicating that the addition of MY altered the amino acid environment surrounding the binding site, leading to the results mentioned above. This result was in line with those from 3D fluorescence experiments. After 90 ns, there is a narrow range of variation in the Rg of the two systems, indicating that the steady-state equilibrium has been reached.
Root-mean-square fluctuation (RMSF) could further reflect the degree of deviation freedom of skeleton atoms in supramolecular system. In the simulation procedure, a bigger RMSF was associated with a higher divergence of the corresponding amino acid residues compared to the initial state. Before and after the introduction of MY, RMSF has great similarity of continuous change (Fig. 6C). However, in the terminal region, i.e., the region with amino acid sequence over 500, RMSF produced a large fluctuation, indicating that the amino acids of the terminal region had a higher degree of freedom. Further analysis of the RMSF of amino acid residues near the binding site of MY on HSA, as shown in Fig. 6D, indicated that the RMSF value of HSA–MY was slightly higher than that of HSA, which increased the degree of freedom of related amino acid residues. For molecular dynamics simulation, the change in the system simulation process is small, because it can indicate the stability change of the system, as simulated by HSA–MY system. In other words, the RMSF of HSA–MY was larger when MY molecules were added to the simulation to create the supramolecular system, demonstrating that the binding of MY increased the amino acid freedom close to the binding site and loosened the skeleton.
3.4. MY's impact on HSA's esterase-like activities
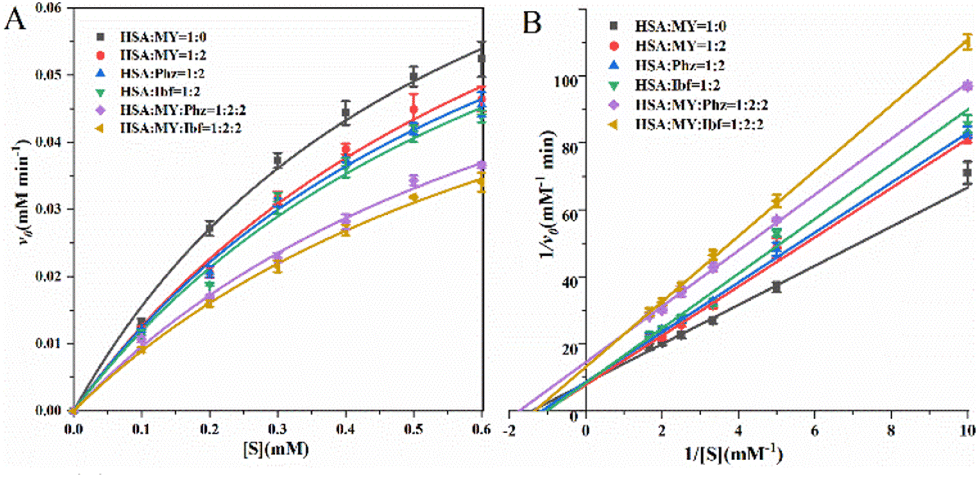
HSA serves as a transport protein in the majority, but it also acts as a catalyst for esterase-like activity,59 which means that it can be effectively affected by small molecules such as drugs. For example, the binding of noscapine to proteins resulted in decreased esterase-like activity of HSA. Among the process of HSA exerting esterase activity, Arg410 and Tyr411 play an important role that Tyr411 is closely related to p-NPA acetylation.60 For the HSA–MY system, v0 and [S] are substituted into the Michaelis equation for fitting under different molar ratios (Fig. 7A), and Phz/Ibf as control, relevant kinetic parameters are listed in Table 5. When MY/Phz/Ibf is added individually, the value of Km increases in three systems. Kmis related to the nature of HSA, the result indicates that the affinity of HSA decreases. And the value of vmax remains almost unchanged, as shown in Fig. 7B, the intercept (1/vmax) of the straight line is basically unchanged, but the slope (Km/vmax) gradually increases (Ibf > Phz > MY). This shows that when adding MY/Phz/Ibf, their inhibition mode is competitive inhibition.61 Meanwhile, higher Km means lower affinity, which shows that Ibf has stronger inhibitory effect than Phz or MY group. Because Ibf interacts with Tyr411 and Arg410, which affects the esterase-like activity.62,63 Thus, Phz/Ibf will reduce the enzyme activity of HSA by binding. Compared with MY/Phz binding to site I, Ibf binding to site II has the strongest inhibitory effect. In addition, the kcat/Km decreases with the addition of MY/Phz/Ibf, which implies that the binding ability of HSA and substrate p-NPA decreases with the order of Ibf > Phz > MY. Interestingly, when MY–Phz or MY–Ibf mixed, the values of v0 and vmax decrease, the values of Km are basically unchanged, which is a non-competitive inhibition mechanism.64 When multiple inhibitors interact with HSA, the structure of HSA will be more flexible, which may interfere with the binding process of substrate and cause the change of inhibition mechanism. Therefore, the results can be concluded that the conformational and hydrophobic changes caused by the combination of MY and HSA will interfere with the specific environment of substrate catalytic degradation, thus affecting the expression of HSA esterase-like activity.
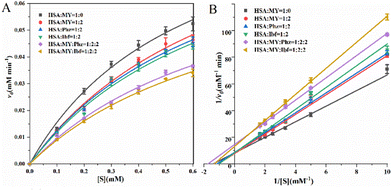 |
| | Fig. 7 (A) Michaelis–Menten diagram of MY affecting HSA esterase-like activity, and add probe Phz/Ibf as control; (B) Lineweaver–Burk diagram of the esterase-like activity of HSA–MY system, add probe Phz/Ibf as control. | |
Table 5 Kinetic parameters of esterase-like activity of HSA–MY system, and add Phz/Ibf as the control
| System |
vmax × 10−3 (mM min−1) |
Km × 10−2 (mM) |
kcat (min−1) |
kcat/Km (min−1 mM−1) |
| HSA:MY = 1:0 |
109.51 ± 5.36 |
61.54 ± 4.87 |
10.95 ± 0.54 |
17.79 ± 0.13 |
| HSA:MY = 1:2 |
107.55 ± 3.06 |
73.96 ± 3.88 |
10.76 ± 0.31 |
14.55 ± 0.10 |
| HSA:Phz = 1:2 |
108.46 ± 4.68 |
76.39 ± 5.53 |
10.85 ± 0.47 |
14.20 ± 0.12 |
| HSA:Ibf = 1:2 |
106.39 ± 3.76 |
78.82 ± 4.64 |
10.64 ± 0.38 |
13.49 ± 0.10 |
| HSA:MY:Phz = 1:2:2 |
86.35 ± 7.45 |
80.35 ± 6.54 |
8.65 ± 0.74 |
10.76 ± 0.16 |
| HSA:MY:Ibf = 1:2:2 |
81.53 ± 5.73 |
81.54 ± 7.22 |
8.15 ± 0.57 |
10.00 ± 0.15 |
4. Conclusion
In this paper, the molecular recognition mechanism, the structural changes, the system stability, and the disturbance of physiological functions between HSA and MY were studied in detail and completely. MY can quench HSA fluorescence through static quenching mechanism, which is a spontaneous, medium strength binding process driven by hydrophobic force. HSA's hydrophobicity and amounts of α-helices dropped, which caused the protein's shape to shift. PSH and urea denaturation experiment investigate the protein conformational stability, and it is verified that hydrophobic force is involved in the combination of MY on HSA. Binding site I was verified by molecular docking and MD, which also examined the stability of the HSA–MY system. HSA's esterase-like activity was suppressed by MY, the inhibitory effect of MY/Phz/Ibf on HSA is Ibf > Phz > MY. The study is anticipated to offer some crucial data for comprehending the physiological traits of MY.
Conflicts of interest
The relevant work of this article does not involve financial interest disputes without competition.
Acknowledgements
This work was supported by the Foundation the Fundamental Research Funds for the Central Universities (2022SCU12069), the Sichuan University postdoctoral interdisciplinary Innovation Fund, the National Natural Science Foundation of China (NNSFC Grant 22108180), and the Project funded by China Postdoctoral Science (2022M712232). Thanks to the Engineering Experimental Teaching Center, School of Chemical Engineering, Sichuan University for the technical assistance.
References
- M. R. Almeida, R. Stephani, H. F. Dos Santos and L. F. C. de Oliveira, J. Phys. Chem. A, 2010, 114, 526–534 CrossRef CAS.
- G. R. Ferreira, H. C. Garcia, M. R. C. Couri, H. F. Dos Santos and L. F. C. de Oliveira, J. Phys. Chem. A, 2013, 117, 642–649 CrossRef CAS PubMed.
- S. Kobylewski and M. F. Jacobson, Int. J. Occup. Environ. Health, 2012, 18, 220–246 CrossRef CAS PubMed.
- M. Oplatowska and C. T. Elliott, Analyst, 2011, 136, 2403–2410 RSC.
- A. Pielesz, I. Baranowska, A. Rybak and A. Wlochowicz, Ecotoxicol. Environ. Saf., 2002, 53, 42–47 CrossRef CAS.
- H. M. Pinheiro, E. Touraud and O. Thomas, Dyes Pigm., 2004, 61, 121–139 CrossRef CAS.
- F. Rafii, J. D. Hall and C. E. Cerniglia, Food Chem. Toxicol., 1997, 35, 897–901 CrossRef CAS.
- K. Yamjala, M. S. Nainar and N. R. Ramisetti, Food Chem., 2016, 192, 813–824 CrossRef CAS PubMed.
- K. T. Chung, G. E. Fulk and A. W. Andrews, Appl. Environ. Microbiol., 1981, 42, 641–648 CrossRef CAS.
- T. W. Evans, Aliment. Pharmacol. Ther., 2002, 16, 6–11 CrossRef CAS PubMed.
- S. Lamichhane and S. Lee, Arch. Pharmacal Res., 2020, 43, 118–133 CrossRef PubMed.
- H. Mohammadzadeh-Aghdash, N. Akbari, K. Esazadeh and J. Ezzati Nazhad Dolatabadi, Food Chem., 2019, 293, 491–498 CrossRef CAS PubMed.
- S. Prasanth and C. Sudarsanakumar, New J. Chem., 2017, 41, 9521–9530 RSC.
- D. Wu, J. Wang, D. Liu, Y. Zhang and X. Hu, Sci. Rep., 2019, 9, 1615 CrossRef PubMed.
- H. Xiang, Q. Sun, W. Wang, S. Li, X. Xiang, Z. Li, X. Liao and H. Li, Chemosphere, 2021, 270, 129431 CrossRef CAS PubMed.
- Q. Sun, H. Yang, P. Tang, J. Liu, W. Wang and H. Li, Food Chem., 2018, 243, 74–81 CrossRef CAS PubMed.
- Q. Sun, N. Gan, S. Zhang, L. Zhao, P. Tang, H. Pu, Y. Zhai, R. Gan and H. Li, Food Chem., 2019, 278, 127–135 CrossRef CAS PubMed.
- W. Wang, N. Gan, Q. Sun, D. Wu, R. Gan, M. Zhang, P. Tang and H. Li, Spectrochim. Acta, Part A, 2019, 219, 83–90 CrossRef CAS PubMed.
- X. Pan, P. Qin, R. Liu and J. Wang, J. Agric. Food Chem., 2011, 59, 6650–6656 CrossRef CAS PubMed.
- X. Pan, R. Liu, P. Qin, L. Wang and X. Zhao, J. Lumin., 2010, 130, 611–617 CrossRef CAS.
- C.-a. Hsu, T.-N. Wen, Y.-C. Su, Z.-B. Jiang, C.-W. Chen and L.-F. Shyur, Environ. Sci. Technol., 2012, 46, 5109–5117 CrossRef CAS.
- A. Basu and G. S. Kumar, Food Chem., 2015, 175, 137–142 CrossRef CAS PubMed.
- S. Khatun, Riyazuddeen and G. Rabbani, J. Chem. Thermodyn., 2019, 131, 9–20 CrossRef CAS.
- M. Manjushree and D.
H. Revanasiddappa, Spectrochim. Acta, Part A, 2019, 209, 264–273 CrossRef PubMed.
- M. Saeidifar, R. Sabbaghzadeh, H. M. Torshizi and A. A. Saboury, J. Iran. Chem. Soc., 2020, 18, 61–74 CrossRef.
- S. Zargar and T. A. Wani, Chem.-Biol. Interact., 2021, 350, 109707 CrossRef CAS.
- Y. Yue, J. Liu, R. Liu, Q. Dong and J. Fan, Spectrochim. Acta, Part A, 2014, 124, 46–51 CrossRef CAS PubMed.
- N. Bijari, Y. Shokoohinia, M. R. Ashrafi-Kooshk, S. Ranjbar, S. Parvaneh, M. Moieni-Arya and R. Khodarahmi, J. Lumin., 2013, 143, 328–336 CrossRef CAS.
- L. Zhao, J. Liu, R. Guo, Q. Sun, H. Yang and H. Li, RSC Adv., 2017, 7, 27796–27806 RSC.
- L. Yang, J. Lv, X. Wang, J. Zhang, Q. Li, T. Zhang, Z. Zhang and L. Zhang, J. Mol. Recognit., 2015, 28, 459–466 CrossRef CAS.
- E. Lissi, C. Calderon and A. Campos, Photochem. Photobiol., 2013, 89, 1413–1416 CrossRef CAS PubMed.
- D. M. Chipman, V. Grisaro and N. Sharon, J. Biol. Chem., 1967, 242, 4388 CrossRef CAS.
- H. Yan, S. Zhao, J. Yang, X. Zhu, G. Dai, H. Liang, F. Pan and L. Weng, J. Solution Chem., 2009, 38, 1183–1192 CrossRef CAS.
- M. Hu, X. Wang, H. Wang, Y. Chai, Y. He and G. Song, Luminescence, 2012, 27, 204–210 CrossRef CAS PubMed.
- R. G. Machicote, M. E. Pacheco and L. Bruzzone, Spectrochim. Acta, Part A, 2010, 77, 466–472 CrossRef PubMed.
- S. Hamdani, D. Joly, R. Carpentier and H. A. Tajmir-Riahi, J. Mol. Struct., 2009, 936, 80–86 CrossRef CAS.
- P. Zhao, G. Gao, L. Zhang, Q. Cai, N. Lu, L. Cheng, S. Li and X. Hou, J. Pharm. Biomed. Anal., 2017, 141, 262–269 CrossRef CAS PubMed.
- F. Wu, Q. Su, L. Zhou, P. Xu, A. Dong and W. Qian, Nano, 2021, 16, 2150052 CrossRef CAS.
- D. Wu, R. Duan, L. Tang, D. Zhou, Z. Zeng, W. Wu, J. Hu and Q. Sun, Lwt, 2022, 154, 112674 CrossRef CAS.
- Z. Suo, Q. Sun, H. Yang, P. Tang, R. Gan, X. Xiong and H. Li, RSC Adv., 2018, 8, 4742–4749 RSC.
- G. De Simone, A. di Masi and P. Ascenzi, Int. J. Mol. Sci., 2021, 22, 10086 CrossRef CAS PubMed.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1976, 12, 1052–1061 CAS.
- A. A. Sabour, A. Khan and M. R. Alhuzani, Molecules, 2022, 27, 7858 CrossRef CAS PubMed.
- S. Zargar, T. A. Wani, N. A. Alsaif and A. I. A. Khayyat, Molecules, 2022, 27, 8405 CrossRef PubMed.
- S. Yang, W. Zhang, Z. Liu, Z. Zhai, X. Hou, P. Wang, G. Ge and F. Wang, Anal. Bioanal. Chem., 2021, 413, 7431–7440 CrossRef CAS PubMed.
- N. E. Basken, C. J. Mathias and M. A. Green, J. Pharm. Sci., 2009, 98, 2170–2179 CrossRef CAS PubMed.
- S. Linciano, G. Moro, A. Zorzi and A. Angelini, J. Controlled Release, 2022, 348, 115–126 CrossRef CAS PubMed.
- W. Peng, F. Ding and Y. Xie, J. Photochem. Photobiol., B, 2016, 154, 40–50 CrossRef CAS PubMed.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed.
- S. Kandandapani, N. F. W. Ridzwan, S. B. Mohamad and S. Tayyab, J. Biomol. Struct. Dyn., 2020, 38, 4134–4142 CrossRef CAS PubMed.
- D. Li, D. Hong, H. Guo, J. Chen and B. Ji, J. Photochem. Photobiol., B, 2012, 117, 126–131 CrossRef CAS PubMed.
- M. Raza, Y. Jiang, B. Ahmad, A. U. Rahman, S. Raza, A. Khan, K. Tahir, S. Hassan, S. Khan and Q. Yuan, New J. Chem., 2021, 45, 7682–7693 RSC.
- L. Wang, G. Zhang and Y. Wang, Mol. Biol. Rep., 2014, 41, 3381–3391 CrossRef CAS PubMed.
- D. Patra, C. Barakat and R. M. Tafech, Colloids Surf., B, 2012, 94, 354–361 CrossRef CAS PubMed.
- S. A. Farsad, H. Haghaei, M. Shaban, M. Zakariazadeh and S. Soltani, J. Biomol. Struct. Dyn., 2022, 40, 6868–6879 CrossRef CAS PubMed.
- T. Sharifi, Y. Ghayeb and T. Mohammadi, Monatsh. Chem., 2017, 148, 781–791 CrossRef CAS.
- H. H. Ni, C. A. Sotriffer and J. A. McCammon, J. Med. Chem., 2001, 44, 3043–3047 CrossRef CAS PubMed.
- T. Mohammadi, Y. Ghayeb, T. Sharifi and T. Khayamian, Monatsh. Chem., 2017, 148, 1141–1151 CrossRef CAS.
- G. Rabbani and S. N. Ahn, Int. J. Biol. Macromol., 2019, 123, 979–990 CrossRef CAS PubMed.
- M. T. Rehman, H. Shamsi and A. U. Khan, Mol. Pharm., 2014, 11, 1785–1797 CrossRef CAS PubMed.
- N. Moradi, M. R. Ashrafi-Kooshk, S. Ghobadi, M. Shahlaei and R. Khodarahmi, J. Lumin., 2015, 160, 351–361 CrossRef CAS.
- G. Rabbani and S. N. Ahn, Int. J. Biol. Macromol., 2019, 123, 979–990 CrossRef CAS PubMed.
- Y. Kurono and K. Ikeda, Chem. Pharm. Bull., 1981, 29, 2993–3002 CrossRef CAS PubMed.
- P. Baruah, M. A. Rohman, S. O. Yesylevskyy and S. Mitra, Bioimpacts, 2019, 9, 79–88 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Na Gan,
Pu Ai,
Hui Li
*,
Na Gan,
Pu Ai,
Hui Li