
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-dependent fluorescence behaviour of imide-fused [n]phenacenes (n = 3, 5, 7)†
Keito Nosea,
Kaito Yoshiokaa,
Minoru Yamaji b,
Fumito Tanic,
Kenta Gotoc and
Hideki Okamoto*ad
b,
Fumito Tanic,
Kenta Gotoc and
Hideki Okamoto*ad
aDivision of Molecular Sciences, Graduate School of Natural Science and Technology, Okayama University, Okayama 700-8530, Japan. E-mail: hokamoto@okayama-u.ac.jp
bDivision of Molecular Science, Graduate School of Science and Engineering, Gunma University, Ota 373-0057, Japan
cInstitute for Materials Chemistry and Engineering, Kyushu University, Fukuoka 819-0395, Japan
dDepartment of Chemistry, Faculty of Natural Science and Technology, Okayama University, Okayama 700-8530, Japan
First published on 30th January 2023
Abstract
Imide-fused [n]phenacenes (nPDIs, n = 3, 5, 7) were systematically synthesised and their electronic features were investigated by electrochemical and electronic spectral measurements. nPDIs showed two reduction waves attributed to formation of radical ions and dianions. 3PDI produced blue fluorescence independent of solvent polarity. In contrast, 5PDI and 7PDI displayed marked positive solvatofluorochromism due to intramolecular charge transfer characters between the imide moieties and phenacene π cores in the excited state. The spectral features were analyzed by the Lippert–Mataga relationship and theoretical calculations.
Introduction
[n]Phenacenes are polycyclic aromatic hydrocarbons (PAHs) consisting of a zigzag alignment of n benzene rings. These are referred to as one-dimensional graphene ribbons with armchair edges. Although phenacenes are chemically quite stable and robust, they have attracted less interest as functional materials because of their low solubility, low fluorescence efficiency and low light absorbing ability in the visible region.1–3 Conventionally, π-extended [n]phenacenes, i.e. n ≥ 5, have received little attention in functional chemistry and materials chemistry. Meanwhile, it was found that large phenacenes served as high-performance p-channel semiconductors and even as aromatic superconductors.4,5 The results have opened the door to the material sciences of phenacenes, namely in organic electronics.6,7Recently, the electronic properties and unprecedented optical characteristics of large phenacenes have been revealed; e.g., picene ([5]phenacene) displayed fluorescence from the second excited state (S2) in the vapour phase, a [2,2](5,8)picenophane derivative showed an exciplex fluorescence band and tetraester-functionalised [12]phenacene behaved as a unique nematogen, emitting polarised fluorescence from monomeric and dimeric forms.8–10 It is thus expected that phenacenes would be a promising platform for constructing novel functional luminophores.
Imide-fused PAHs, typified by rylene diimides and acene diimides, have been extensively utilised as n-type organic electronic molecules, efficient fluorophores and supramolecular components.11–13 In particular, synthesis and elucidation of electronic natures of rylene-diimide based functional molecules are one of the central topics in current fundamental and materials sciences.14–16 Additionally, diverse molecular designs of polycyclic aromatic molecules are proposed and their electronic and optical properties are extensively explored. An imide fused fluoranthene, symmetrically combined with anthracene core, was reported to be solvatfluorochromic.17 A naphthalimide derivative, substituted with two different functionalities on the imide moieties, showed fluorescence colour changes by interacting with aromatic solvents through exciplex formation.18 Helicene and the related helical diimides provide optical properties due to the intra-molecular conjugation between the imide functionalities.19,20
In contrast to such established aromatic imide compounds, there are only a few studies on phenacenes incorporating imide functionalities. Previously reported imide-fused phenacenes have imide functionalities in the branching direction of the molecular axis because of the limited synthetic protocols.21 A fulminene ([6]phenacene) diimide was reported to provide a fluorescence band with a maximum at 500 nm.22 Extremely π-extended phenacene diimides exhibit an absorption band in a 450–470 nm region that is little dependent on numbers of the benzene rings involved in the molecules.23 We recently synthesized picenes bearing imide moieties in the long molecular axis directions, Cn-PicDIs (see Fig. 1 for the chemical structure), and revealed that they served as n-channel organic semiconductors and displayed fluorescence band in the deep blue region.24
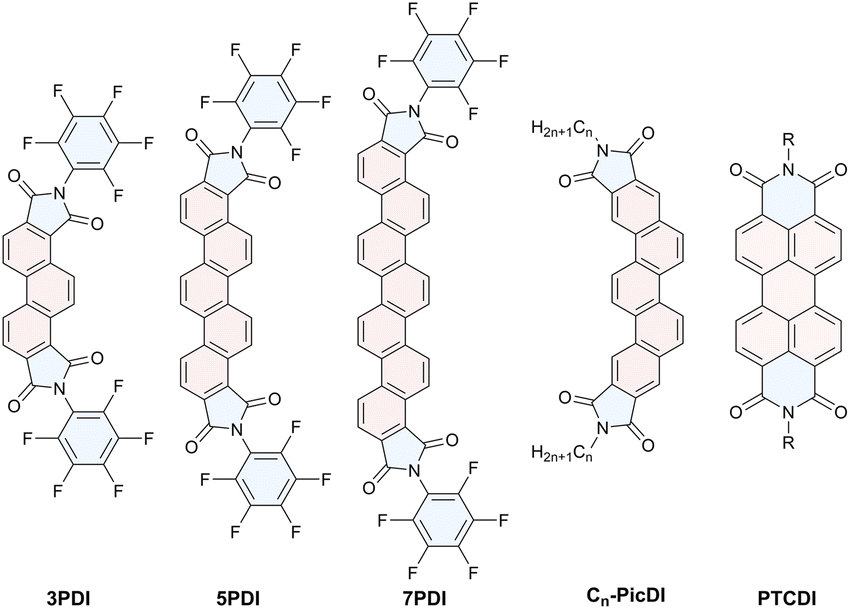 | ||
| Fig. 1 Chemical structures of nPDIs and the related aromatic diimides. | ||
Due to chemical stability and robustness, efficient functional dyes would be constructed employing the phenacene π core through an appropriate molecular design, e.g., proper imidation. However, little is known about active manipulation of the electronic and spectral features of functionalised phanacenes. Therefore, we aimed to produce a phenacene-based functional dye. In this study, we synthesised [n]phanacene derivatives, nPDIs (n = 3, 5 and 7, see Fig. 1 for the chemical structures), incorporating imide functionalities at both edges of the molecules. The electronic features of nPDIs investigated by electrochemical and photophysical measurements are described, namely, it has been revealed that fluorescence behaviour of nPDIs can be effectively manipulated by solvent polarity.
Synthesis of nPDIs
To construct phenacene frameworks, the Mallory photoreaction of diarylethenes is one of the most efficient and reliable methods.25 We have fully taken advantages of this strategy in the final step to effectively obtain nPDIs (Scheme 1). Pentafluorophenyl moieties have been introduced to enhance the electron-attracting ability of imide functionality.26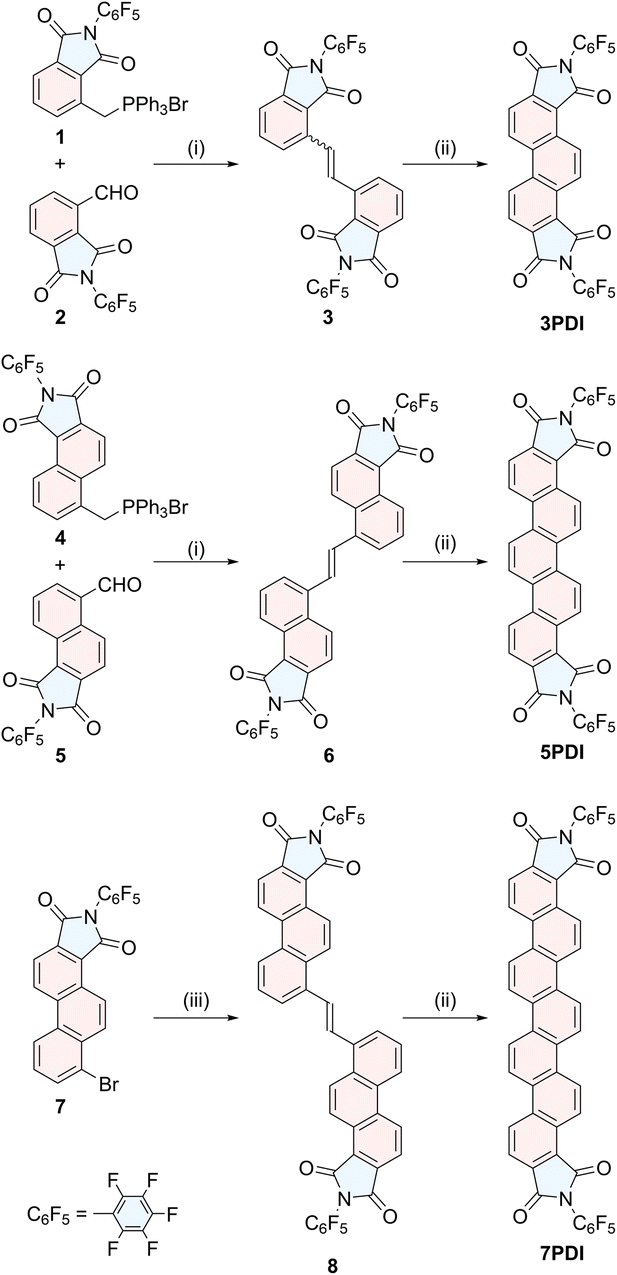 | ||
| Scheme 1 Synthetic routes to nPDIs. Reagents and conditions: (i) Bu4NF, CH2Cl2, (ii) hν, I2, air, toluene, (iii) K2CO3, Bu4NBr, CH2Cl2-THF then Bu4NF, (iv) (E)-1,2-bis(tetrabutylstannyl)ethene, Pd(PPh3)4. | ||
Diarylethene precursors 3, 6 and 8 were prepared by either Wittig alkene synthesis between the appropriate arylaldehyde and arymethylphosphonium salt (for precursors 3 and 6) or Migita–Kosugi–Stille coupling27 between distannylethene and bromoarene 7 (for precursor 8). These precursors were illuminated with black-light lamps (352 nm), in the presence of a catalytic amount of I2, producing the desired nPDIs in moderate to good yields, 3PDI (95%), 5PDI (76%) and 7PDI (40% from 7). The detailed synthetic procedures of nPDIs and the intermediate building blocks, and their compound data are deposited in the ESI.†
Electrochemical characterization of nPDIs
To reveal the electronic features of nPDIs, electrochemical measurements were carried out using cyclic voltammetry (CV) and square wave voltammetry (SWV) techniques (Fig. 2). 3PDI and 5PDI showed two reversible reduction peaks in CV owing to the formation of radical anion and dianion species, as in the case of the reported rylene diimides.28 7PDI showed a single reduction peak; the two reduction waves were considered to be merged into one reduction wave. Such electrochemical behaviour was similar to that reported for aromatic diimides.28,29 The first reduction potential (Ered1) was estimated to be −1.37, −1.49 and −1.54 V (vs. Fc/Fc+) for 3PDI, 5PDI and 7PDI, respectively. The electron affinity of the nPDI series was higher for the smaller homologue because the radical anion form of the smaller nPDI was more effectively stabilized by the two imide moieties in proximity. As for the second reduction potential (Ered2), the order is inverted: −1.66, −1.60 and −1.54 V (vs. Fc/Fc+) for 3PDI, 5PDI and 7PDI, respectively. It is considered that the negative charges distributed to the two imide moieties in the dianions of nPDIs, produced during two successive one-electron reduction processes, led to repulsive coulombic interactions in the smaller homologue, namely, 3PDI.28 The energy levels of the lowest unoccupied molecular orbitals (LUMO, ELUMO) were estimated to be −3.43, −3.31 and −3.26 eV for 3PDI, 5PDI and 7PDI, respectively [ELUMO = −(4.8 + Ered1) eV].30 The ELUMO values were slightly higher than those of related picene diimides (ELUMO = ca. −3.9 eV)24 indicating that the imide-fusing position affected the electron accepting ability.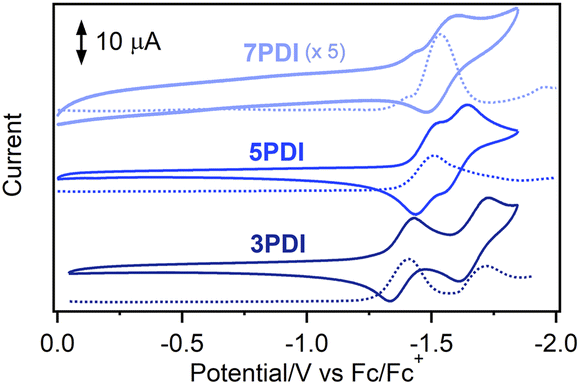 | ||
| Fig. 2 CV (full lines) and SWV (dotted lines) curves for nPDIs observed in benzonitrile, in the presence of Bu4NPF6 (0.1 M). | ||
Electronic spectra
The electronic spectral characteristics of nPDIs were investigated by absorption and fluorescence spectral measurements in various solvents (Fig. 3), and the photophysical parameters are summarised in Table 1. nPDIs showed a small absorption band in the longer wavelength region (400–450 nm). The absorption band slightly red shifted upon increasing the number of fused benzene rings from n = 3 to 5, and λabs values of 5PDI and 7PDI were similar to each other; λabs = 404–409 nm for 3PDI, 428–434 nm for 5PDI, 423–431 nm for 7PDI (cf. Fig. S6 in the ESI†). The entire absorption spectral profiles for each nPDIs were almost independent of the solvent polarity.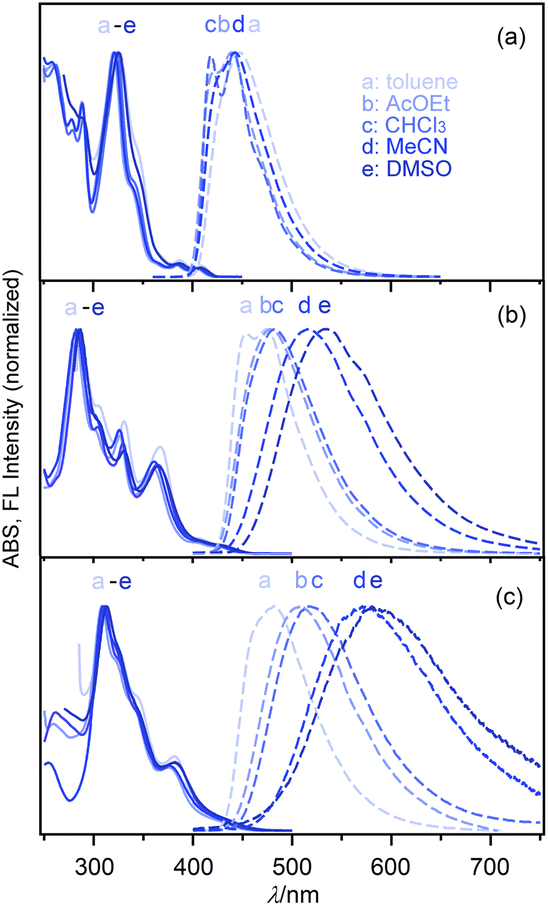 | ||
| Fig. 3 Electronic absorption (full lines) and fluorescence spectra (dashed lines) of nPDIs in various solvents: (a) 3PDI, (b) 5PDI and (c) 7PDI. | ||
| Compound | Solvent | λabs (nm) | λFLa (nm) | Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) b (cm−1) b (cm−1) |
ΦFc |
|---|---|---|---|---|---|
| a Fluorescence excitation spectra were consistent with absorption spectra (Fig. S1–S3 in ESI).b Stokes shift.c Fluorescence quantum yield determined under aerated conditions.d The positions of the 0–0 band were estimated from simulated spectra by peak fitting (Fig. S4 and S5, in ESI).e No detectable fluorescence emission was observed. | |||||
| 3PDI | Toluene | 409 | 429d | 863 | 0.17 |
| CHCl3 | 409 | 417d | 485 | 0.11 | |
| AcOEt | 404 | 415d | 677 | 0.15 | |
| MeCN | 405 | 419d | 836 | 0.17 | |
| DMSO | 408 | —e | —e | —e | |
| 5PDI | Toluene | 428 | 454 | 1340 | 0.32 |
| CHCl3 | 434 | 483 | 2340 | 0.26 | |
| AcOEt | 429 | 478 | 2390 | 0.15 | |
| MeCN | 430 | 516 | 3880 | 0.29 | |
| DMSO | 433 | 533 | 4330 | 0.07 | |
| 7PDI | Toluene | 429d | 482 | 2580 | 0.35 |
| CHCl3 | 431d | 515 | 3790 | 0.28 | |
| AcOEt | 423d | 510 | 4020 | 0.30 | |
| MeCN | 430d | 571 | 5740 | 0.10 | |
| DMSO | 430d | 581 | 6040 | 0.08 | |
In fluorescence spectra, 3PDI displayed an emission band in the 400–600 nm region. The emission band was essentially insensitive to solvent polarity (Fig. 3a). In DMSO, no fluorescence emission of 3PDI was detected. For 5PDI and 7PDI, the fluorescence band was significantly sensitive to solvent polarity and bathochromically shifted depending on solvent polarity (Fig. 3b and c). As a result, the fluorescence colour changed from blue (λFL = 454 nm in toluene) to yellow (λFL = 533 nm in DMSO) for 5PDI, and from sky blue (λFL = 482 nm in toluene) to orange (λFL = 581 nm in DMSO) for 7PDI.
Regardless of the solvent used, the fluorescence quantum yield of 3PDI (ΦF = ca. 0.15) was slightly higher than that of parent phenanthrene (ΦF = 0.049 in aerated CHCl3).3 The ΦF values of 5PDI and 7PDI were, respectively, determined to be 0.32 and 0.35 in toluene, which were about 4-fold larger than that for parent picene (ΦF = 0.088 in aerated CHCl3).3 Upon increasing solvent polarity, the ΦF values tended to decrease. These results suggest that the lowest excited states (S1) of 5PDI and 7PDI are of intramolecular charge transfer (ICT) nature.
The solvatofluorochromic behaviour of nPDIs was analysed using the Lippert–Mataga relationship (eqn (1)),31,32
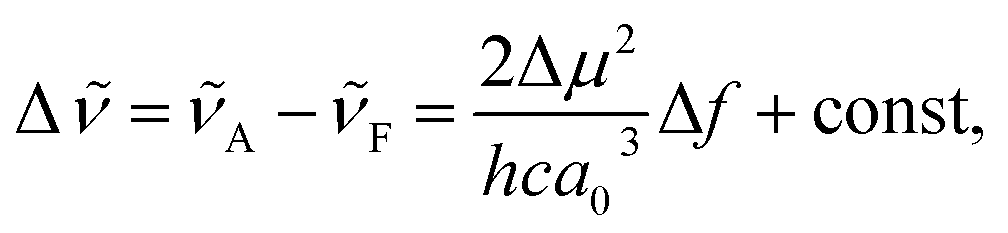 | (1) |
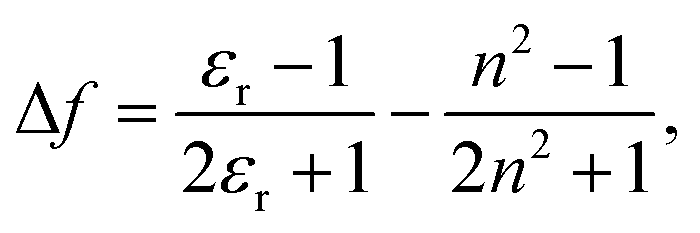 | (2) |
is the Stokes shift estimated by the difference in peak wavenumbers between the absorption and fluorescence emission bands (cf. Fig. S4 and S5 in the ESI†), h is the Planck constant, c is the velocity of light in vacuum, Δμ is the dipole moment change between the ground and excited states, a0 is the Onsagar cavity radius and Δf is the orientation polarizability of solvents expressed by dielectric constant (εr) and refractive index (n) of the solvents (eqn (2)). The Lippert–Mataga plots showed linear correlations (Fig. 4) and the Δμ values were estimated from the slopes; Δμ = 4.8 D for 3PDI, 11.3 D for 5PDI and 13.8 D for 7PDI. It was, thus, revealed that 3PDI showed little change in dipole moment upon excitation whereas 5PDI and 7PDI were significantly polarised in the S1 state.
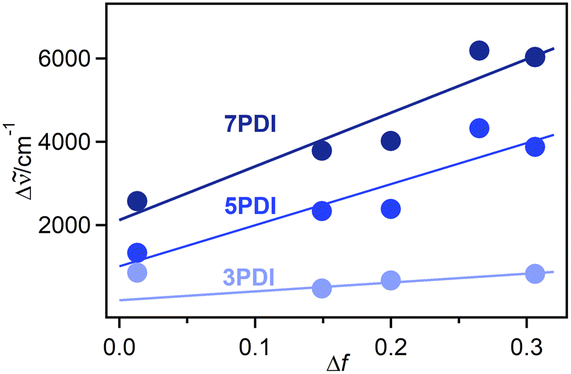 | ||
| Fig. 4 Lippert–Mataga plots for nPDIs. The point for 3PDI in toluene (Δf = 0.013) was omitted as it deviated from the linear correlation. The Δf values were calculated by eqn (2). The n and εr values were obtained from ref. 33. | ||
In the case of related picene diimide, C8-PicDI, the solvatofluorochromic shift was substancially less significant (λFL = 412 nm in toluene, λFL = 425 nm in DMSO, Fig. S7a in the ESI†) compared to that of 5PDI (λFL = 428 nm in toluene, λFL = 533 nm in DMSO, Table 1). It can be concluded that the imide-fusing positions play an important role for inducing the solvent-dependent fluorescence behaviour. Additionally, only slight solvent-dependent electronic spectral shift of PTCDI (R = octyl) was observed indicating that the perylene diimide, incorporating no substituent on the aromatic core, insignificantly responded to solvent environments (Fig. S7b in the ESI†).
It would be worth mentioning that 3PDI displayed fluorescence behaviour different form that of 5PDI and 7PDI. The fluorescence band observed in toluene was apparently broadened and its onset wavelength slightly red shifted compared to that observed in the other solvent. Additionally, in DMSO, the fluorescence of 3PDI was totally quenched, whereas that of 5PDI and 7PDI did not vanish. A Stern–Volmer plot for the fluorescence quenching of 3PDI with DMSO apparently showed a linear relationship in a low DMSO concentration region (<75 mM, Fig. S8 in the ESI†). It is considered that there is a specific interaction between 3PDI and DMSO molecules through a dynamic process. DMSO has been shown to cause fluorescence quenching of ICT fluorophores via specific mechanisms such as hydrogen bonding and improved twisted intramolecular charge transfer (TICT).34,35 However, in the case of 3PDI, such mechanisms could be excluded as 3PDI has no hydrogen donor and the pentafluorophenyl moieties would not contribute to the frontier orbitals (cf. Fig. 5). It has been mentioned that DMSO quenched fluorescence of Zn-bis(dipyrromethenate)s through coordination-mediated interactions and/or photo-induced electron transfer (PET).36 In the case of 3PDI-DMSO system, fluorescence quenching through a PET mechanism might be an alternative possibility. The detailed quenching mechanism is currently not clear and is being investigated.
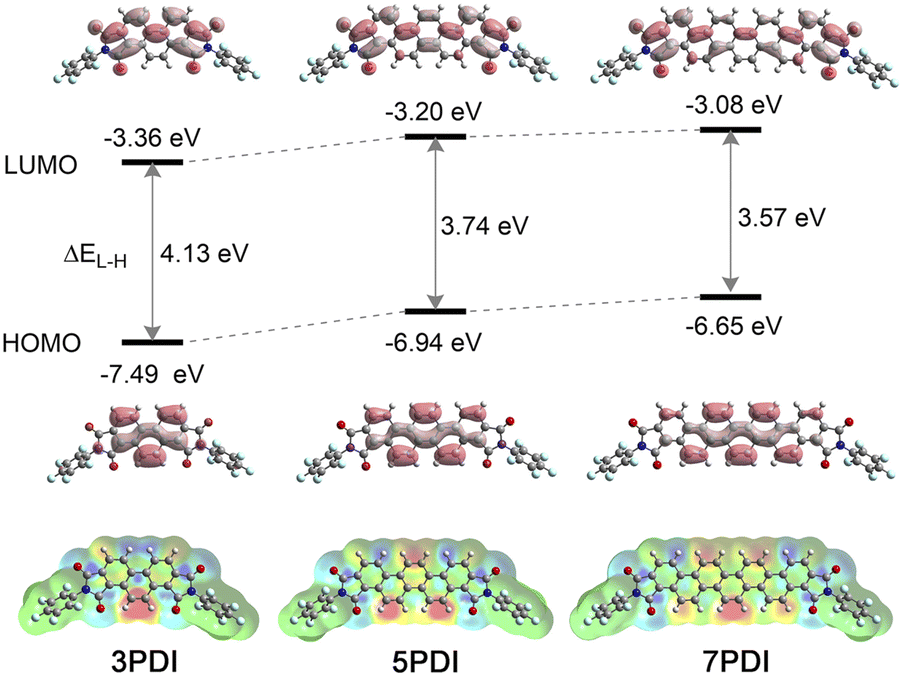 | ||
| Fig. 5 Upper: Frontier molecular orbital diagrams of nPDIs. The calculated energy levels of HOMO and LUMO, and HOMO–LUMO energy gap (ΔEL–H) are shown. Lower: Density difference mappings for S0–S1 electronic transition; the red regions indicate reduced electron density and the blue regions indicate increased electron density upon the photoexcitation. | ||
Theoretical analyses
To further obtain insights into the electronic features of the excited state of nPDIs, theoretical analyses were carried out at the (TD) PBE0/6-311+G(2df,2p) level of theory.37 Fig. 5 shows the calculated frontier molecular orbitals and energy levels, and density difference mapping corresponding to the electronic transition from the S0 to S1 states. The energy gap for the highest occupied molecular orbitals (HOMO) and LUMO (ΔEL–H) gradually decreases with increasing benzene ring numbers in nPDIs.The molecular orbital diagrams of the HOMO display similar features for the nPDI series, i.e., the HOMO is localised exactly on the phenacene cores. In contrast, the LUMO was delocalised over the entire molecules extending to the two imide moieties. The LUMO energy levels (ELUMO) increased with the increasing number of fused benzene rings in nPDIs. It is reasonable to consider that, for 3PDI, more efficient conjugation between the phenanthrene core and electron-accepting imide moieties more effectively stabilises the LUMO compared to larger nPDIs. The order of the calculated ELUMO values was consistent with that of the first reduction potentials estimated by electrochemical measurements (Fig. 2).
The excited state natures of nPDIs were investigated in vacuum using the TD-DFT method. The calculated transition wavelengths [λ(S0–S1)], oscillator strengths (f) and configurations of the electronic transitions are summarized in Table S1 in the ESI.† Additionally, the calculated absorption spectra were compared with the experimental absorption spectra recorded in CHCl3 (Fig. S9 in the ESI†). The calculation results slightly overestimated the transition energies compared with the experimentally observed absorption spectra. The S1 state is mainly attributed to the HOMO–LUMO transition for all nPDIs (Table S1 in the ESI†). Based on the results that the HOMO is located on the phenacene cores and the LUMO expands to the two imide moieties (Fig. 5), the S1 state of nPDIs is considered to possess ICT properties between the phenacene π core and imide functionality.
The density difference mappings for the S0–S1 electronic transition (Fig. 5, lower) provide additional insight into the ICT characteristics of nPDIs in the S1 state. Upon excitation, the electron density at the peripheral double bonds in the central phenacene cores decreased (red regions), whereas that at the imide carbonyl moieties increased (blue regions). Consequently, in the cases of 5PDI and 7PDI, an enhanced electronic dipole moment was induced in the S1 state resulting in appreciable positive solvatofluorochromism. In the case of 3PDI, the two negatively charged imide moieties opposed the positively charged double bond. They behave like quasi-quadrupoles to reduce the polarized character of the entire molecule in the S1 state. Thus, 3PDI showed minimal fluorescence response to solvent polarity.
Conclusions
A series of imide-fused phenacenes nPDIs was photochemically synthesized and their electronic features were electrochemically and photophysically investigated. The ELUMO of 5PDI estimated from the electrochemical measurements, −3.31 eV, was slightly higher than that of related Cn-PicDI, ca. −3.9 eV. The results suggested that electron accepting ability of imide-fused phenacenes can be manipulated by the position of the imide functionalities. Due to the ICT characters between the phenacene π-core and the imide moieties, 5PDI and 7PDI displayed marked positive solvatofluorochromism. 3PDI displayed negligible solvent effects on fluorescence and showed effective quenching in DMSO. The present results provide a novel strategy for developing fluorophores utilising [n]phenacenes, specifically n ≥ 5, which have rarely been used in development of luminescent functional materials. To our knowledge, 5PDI and 7PDI provide the first phanacene-based fluorophores responding to solvent environments, and their A–D(π)–A electronic alignment would be potentially applicable to non-linear optics.38Author contributions
Keito Nose: investigation, validation. Kaito Yoshioka: investigation, validation. Minoru Yamaji, investigation, validation, writing–original draft. Fumito Tani: investigation, validation. Kenta Goto: investigation, validation. Hideki Okamoto: conceptualization, validation, writing–original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present study was supported by Grants-in-Aid for Scientific Research, KAKENHI, from JSPS, Japan (JP18H02043 and JP20K05648), and by the Cooperative Research Program of the ‘Network Joint Research Centre for Materials and Devices.’Notes and references
- F. B. Mallory, K. E. Butler, A. C. Evans, E. J. Brondyke, C. W. Mallory, C. Yang and A. Ellenstein, J. Am. Chem. Soc., 1997, 119, 2119–2124 CrossRef CAS.
- F. B. Mallory, K. E. Butler, A. C. Evans and C. W. Mallory, Tetrahedron Lett., 1996, 37, 7173–7176 CrossRef CAS.
- H. Okamoto, M. Yamaji, S. Gohda, K. Sato, H. Sugino and K. Satake, Res. Chem. Intermed., 2013, 39, 147–159 CrossRef CAS.
- H. Okamoto, N. Kawasaki, Y. Kaji, Y. Kubozono, A. Fujiwara and M. Yamaji, J. Am. Chem. Soc., 2008, 130, 10470–10471 CrossRef CAS PubMed.
- R. Mitsuhashi, Y. Suzuki, Y. Yamanari, H. Mitamura, T. Kambe, N. Ikeda, H. Okamoto, A. Fujiwara, M. Yamaji, N. Kawasaki, Y. Maniwa and Y. Kubozono, Nature, 2010, 464, 76–79 CrossRef CAS PubMed.
- Y. Kubozono, X. He, S. Hamao, K. Teranishi, H. Goto, R. Eguchi, T. Kambe, S. Gohda and Y. Nishihara, Eur. J. Inorg. Chem., 2014, 3806–3819 CrossRef CAS.
- Y. Kubozono, H. Mitamura, X. Lee, X. He, Y. Yamanari, Y. Takahashi, Y. Suzuki, Y. Kaji, R. Eguchi, K. Akaike, T. Kambe, H. Okamoto, A. Fujiwara, T. Kato, T. Kosugi and H. Aoki, Phys. Chem. Chem. Phys., 2011, 13, 16476–16493 RSC.
- T. Itoh, M. Yamaji and H. Okamoto, Chem. Phys. Lett., 2013, 570, 26–28 CrossRef CAS.
- M.-C. Tang, Y.-C. Wei, Y.-C. Chu, C.-X. Jiang, Z.-X. Huang, C.-C. Wu, T.-H. Chao, P.-H. Hong, M.-J. Cheng, P.-T. Chou and Y.-T. Wu, J. Am. Chem. Soc., 2020, 142, 20351–20358 CrossRef CAS PubMed.
- G. Farias, D. S. Simeão, T. S. Moreira, P. L. dos Santos, A. Bentaleb, E. Girotto, A. P. Monkman, J. Eccher, F. Durola, H. Bock, B. de Souza and I. H. Bechtold, J. Mater. Chem. C, 2019, 7, 12080–12085 RSC.
- M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef PubMed.
- Y. Avlasevich, C. Li and K. Müllen, J. Mater. Chem., 2010, 20, 3814–3826 RSC.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS.
- W. Jiang and Z. Wang, J. Am. Chem. Soc., 2022, 144, 14976–14991 CrossRef CAS PubMed.
- S. Maniam, H. F. Higginbotham, T. D. M. Bell and S. J. Langford, Chem.–Eur. J., 2019, 25, 7044–7057 CrossRef CAS PubMed.
- A. Herrmann and K. Müllen, Chem. Lett., 2006, 35, 978–985 CrossRef CAS.
- H. Ishikawa, K. Katayama, J. Nishida, C. Kitamura and T. Kawase, Tetrahedron Lett., 2018, 59, 3782–3786 CrossRef CAS.
- M. Pandeeswar and T. Govindaraju, RSC Adv., 2013, 3, 11459–11462 RSC.
- F. Saal, F. Zhang, M. Holzapfel, M. Stolte, E. Michail, M. Moos, A. Schmiedel, A.-M. Krause, C. Lambert, F. Würthner and P. Ravat, J. Am. Chem. Soc., 2020, 142, 21298–21303 CrossRef CAS PubMed.
- G. Zhang, J. Tan, L. Zhou, C. Liu, J. Liu, Y. Zou, A. Narita and Y. Hu, Org. Lett., 2021, 23, 6183–6188 CrossRef CAS PubMed.
- L. Sturm, F. Aribot, L. Soliman, H. Bock and F. Durola, Eur. J. Org. Chem., 2022, e202200196 CAS.
- R. Wang, K. Shi, K. Cai, Y. Guo, X. Yang, J.-Y. Wang, J. Pei and D. Zhao, New J. Chem., 2016, 40, 113–121 RSC.
- T. S. Moreira, M. Ferreira, A. Dall'armellina, R. Cristiano, H. Gallardo, E. A. Hillard, H. Bock and F. Durola, Eur. J. Org. Chem., 2017, 4548–4551 CrossRef CAS.
- Y. Guo, K. Yoshioka, S. Hamao, Y. Kubozono, F. Tani, K. Goto and H. Okamoto, RSC Adv., 2020, 10, 31547–31552 RSC.
- F. B. Mallory and C. W. Mallory, Org. React., 1984, 30, 1–456 CAS.
- T. Korenaga, K. Kadowaki, T. Ema and T. Sakai, J. Org. Chem., 2004, 69, 7340–7343 CrossRef CAS PubMed.
- V. Farina, V. Krishnamurthy and W. J. Scott, Org. React., 1997, 50, 1–652 CAS.
- S. K. Lee, Y. Zu, A. Herrmann, Y. Geerts, K. Müllen and A. J. Bard, J. Am. Chem. Soc., 1999, 121, 3513–3520 CrossRef CAS.
- X. Zhao, Y. Xiong, J. Ma and Z. Yuan, J. Phys. Chem. A, 2016, 120, 7554–7560 CrossRef CAS.
- H. Qu, W. Cui, J. Li, J. Shao and C. Chi, Org. Lett., 2011, 13, 924–927 CrossRef CAS PubMed.
- E. Lippert, Z. Naturforsch., 1955, 10, 541–545 CrossRef.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1955, 28, 690–691 CrossRef CAS.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, 3rd edn, 2003 Search PubMed.
- A. Maliakal, G. Lem, N. J. Turro, R. Ravichandran, J. C. Suhadolnik, A. D. DeBellis, M. G. Wood and J. Lau, J. Phys. Chem. A, 2002, 106, 7680–7689 CrossRef CAS.
- D. Cho and W. L. Mattice, J. Phys. Chem., 1990, 94, 3847–3851 CrossRef CAS.
- A. A. Ksenofontov, G. B. Guseva, E. V. Antina, I. A. Khodov and A. I. Vyugin, Sens. Actuators, B, 2017, 251, 858–868 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- O. Mongin, L. Porrès, M. Charlot, C. Katan and M. Blanchard-Desce, Chem.–Eur. J., 2007, 13, 1481–1498 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra of the new compounds, theoretical calculation results. See DOI: https://doi.org/10.1039/d2ra07771j |
| This journal is © The Royal Society of Chemistry 2023 |