
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstituent directed cellular imaging in the 800–850 nm range with BF2-azadipyrromethene fluorophores†
Cathal Caulfield ,
Dan Wu,
Massimiliano Garre and
Donal F. O'Shea*
,
Dan Wu,
Massimiliano Garre and
Donal F. O'Shea*
Department of Chemistry, RCSI, 123 St. Stephen's Green, Dublin 2, Ireland. E-mail: donalfoshea@rcsi.ie
First published on 16th May 2023
Abstract
Three bis(anilino)-substituted NIR-AZA fluorophores have been designed, synthesized and tested to bridge the availability gap of molecular fluorophores for live-cell microscopy imaging in the 800–850 nm spectral range. The concise synthetic route allows for the later stage introduction of three tailored peripheral substituents which guides the sub-cellular localization and imaging. Live-cell fluorescence imaging of lipid droplets, plasma membrane and cytosolic vacuoles was successfully achieved. Photophysical and internal charge transfer (ICT) properties of each fluorophore were examined through solvent studies and analyte responses.
Introduction
Instrumentation for fluorescence microscopy capable of imaging in the 800–850 nm range is becoming increasingly available providing access to the near-infrared spectral region beyond the more commonly used 550–700 nm wavelengths.1 At present, there are a limited number of molecular fluorophores displaying the necessary optical and physicochemical properties for intracellular live-cell tracking in this spectral region. Advantageously, fluorescence imaging at low energy near infrared wavelengths can reduce phototoxicity and background fluorescence allowing for prolonged live-cell imaging, and expand the spectral window for multi-colour imaging with lower wavelength probes.2,3 BF2-chelated tetraarylazadipyrromethenes (NIR-AZA) are a class of fluorophore with emission spectra in the near-infrared region.4,5 Numerous NIR-AZAs exhibiting emissions in the 700–750 nm spectral region have been reported owing to the versatile four-step synthesis of the core fluorophore scaffold 1b (Fig. 1).6 Careful optimization of peripheral substituents on 1 has garnered real-time continuous live cell and in vivo imaging applications. These include off to on bio-responsive imaging of cellular processes,7,8 bioconjugations for in vivo targeted tumour imaging9–11 and an emerging potential for clinical use in fluorescence-guided surgery.12,13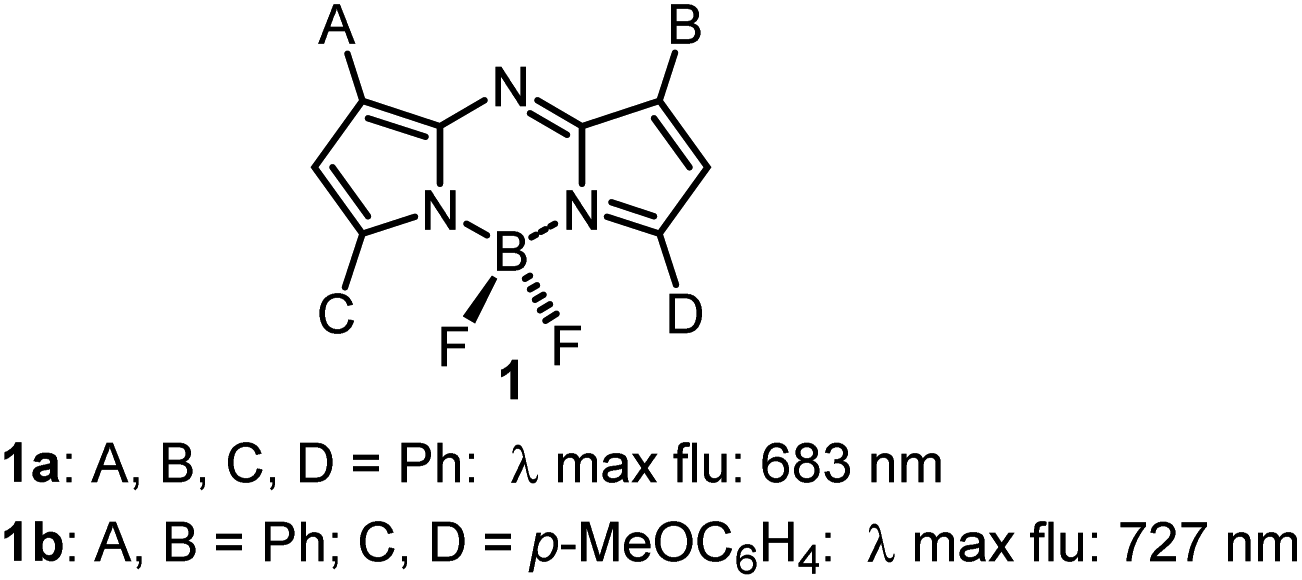 | ||
| Fig. 1 General structure of NIR-AZA fluorophores 1. | ||
Currently, our focus is on extending the emission wavelengths of this fluorophore class beyond the 700–750 nm region for the purpose of fluorescence microscopy imaging of specific subcellular compartments. There are a limited number of cyanine based fluorophores in the literature with emission in the 800–850 nm wavelength range, the majority having been engineered for the imaging of tissues within whole organisms.14 Cell microscopy imaging studies with clinically approved indocyanine green (ICG) 2 have been reported with the primary aim being to further understand in vivo cellular uptake, though some subcellular localizations were identified (Fig. 2).15 Cyanine fluorophore 3, which contains a protonatable primary amine substituent to concentrate it within acidic lysosomes, has been developed as a tool for understanding the fate of cell-based therapeutics (Fig. 2).16 Lysosomal staining with 3 was first tested in vitro where live-cell fluorescence imaging of human prostate cancer cells was achieved, while the major goals completed were cell tracking in tissue and whole organisms. Therefore, with such a limited range of fluorophores, a versatile synthesis of NIR-AZA fluorophores for imaging of specific cellular compartments in the 800–850 nm region would be of high value.
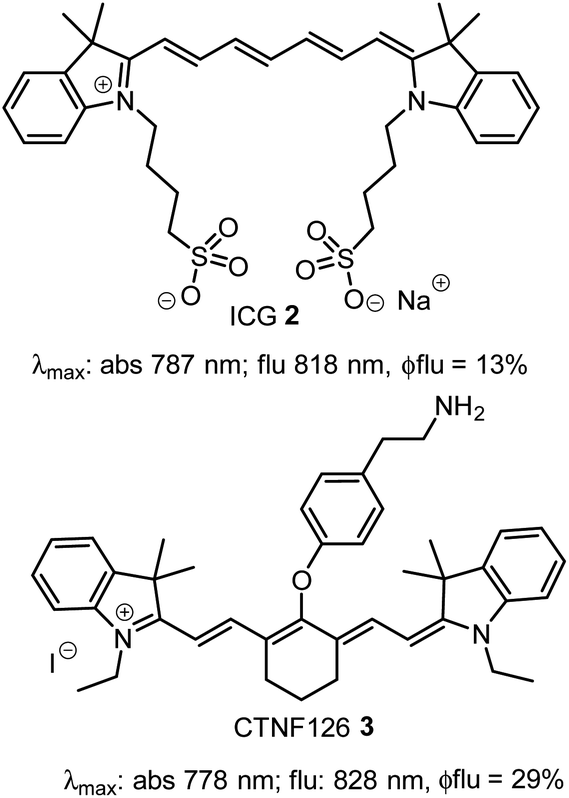 | ||
| Fig. 2 Structure and photophysical properties of cyanine fluorophores, indocyanine green 2 and CTNF126 3. | ||
The previously reported bis(anilino)-substituted NIR-AZA fluorophore 4 with a donor–acceptor–donor (D–A–D) design was identified as a good starting point for this study (Fig. 3).17 The strong electron donating dimethylamino groups of 4 coupled with the BF2-5–6–5 fused heterocyclic acceptor yields emission centred at 826 nm in aqueous solutions. The inclusion of these groups resulted in a substantial bathochromic shift in emission maximum of 154 nm relative to tetraphenyl-substituted 1a. This large red-shift is attributable to the internal charge transfer (ICT) established by this substituent pattern. The use of substituted anilino groups such as triphenylamine and julolidinyl moieties has recently been employed to extend the wavelengths of this fluorophore class but are overly lipophilic making them unsuitable for our requirements.18,19 It was envisaged that replacement of one methyl group of the dimethylamino substituents on 4 could be achieved while maintaining the desired wavelengths, and that different substituents could be selected to specifically direct the fluorophore to a sub-cellular localization and allow it to be imaged.
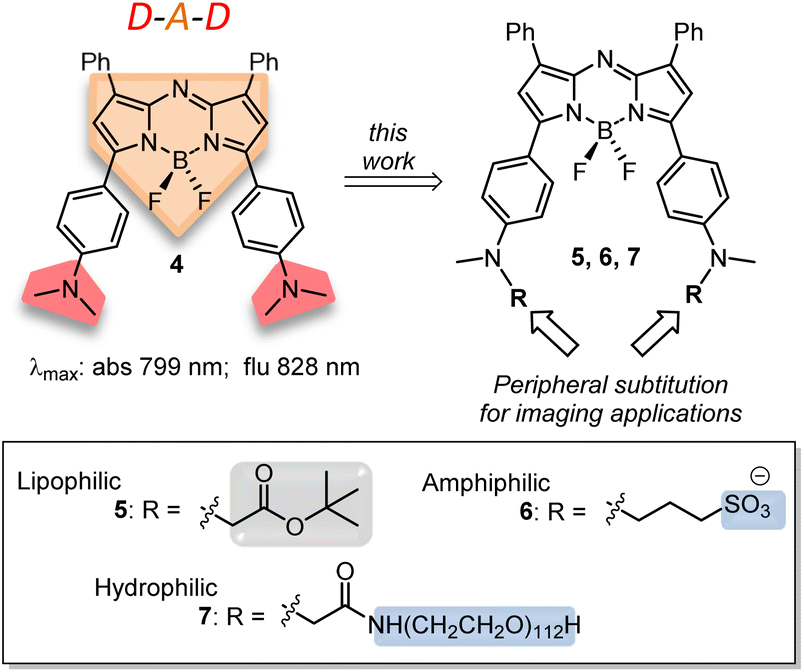 | ||
| Fig. 3 Structure and photophysical properties of NIR-AZA fluorophore 4, with donor–acceptor–donor (red-orange-red) design highlighted, and outline of current synthetic goals towards lipophilic, amphiphilic and hydrophilic 5, 6 and 7, respectively, with lipophilic (grey) and hydrophilic (blue) groups highlighted. | ||
Substituents were selected in order to impart lipophilic, anionic hydrophilic and charge neutral hydrophilic properties onto the fluorophore scaffold. Structurally this equated to the bis-inclusion of tert-butyl ester (CH2CO2tBu), alkyl sulfonic ((CH2)3SO3−) and polyethylene glycol (PEG) groups providing fluorophores 5, 6 and 7 respectively (Fig. 3). It was anticipated that the physicochemical properties imparted by each of these substituents would predetermine their sites of subcellular accumulation (Fig. 4). Specifically, 5 may favour accumulation in highly lipophilic intracellular vesicles such as lipid droplets (LD) which act as a reservoir of triacylglycerols and sterol esters.20 It was reasoned that the double hydrophilic sulfonic acid substitution on one face of 6 would impart both aqueous solubility and amphiphilic features due to the lipophilic phenyl groups on the opposing side of the molecule. These structural characteristics could favour accumulation within lipid bilayers such as the plasma membrane, with the ionic association of sulfonic acids with the ammonium component of membrane phospholipids assisting as a temporary anchoring mechanism.21 Over time internalization into the cell cytosol would occur as part of the normal processes giving rise to a secondary accumulation site within intracellular trafficking vesicles. The covalent attachment of polyethylene glycols (PEG) to drug molecules for enhanced selectivity is well established though its use with fluorescent agents is quite limited. In the case of 7 it was hoped that imparting complete aqueous solubility by PEGylation would promote intracellular accumulation in aqueous compartments within the cytosol without membrane staining (Fig. 4).22 Once synthesized, the impact of the tailored substitutions on each of the fluorophores ICT properties would be investigated via solvatochromic studies and ICT suppression through protonation of amino donors.23 With this knowledge in hand the temporal and spatial imaging characteristics of 5–7 would be investigated with an additional goal of identifying a common set of microscope excitation and emission parameters for general use.
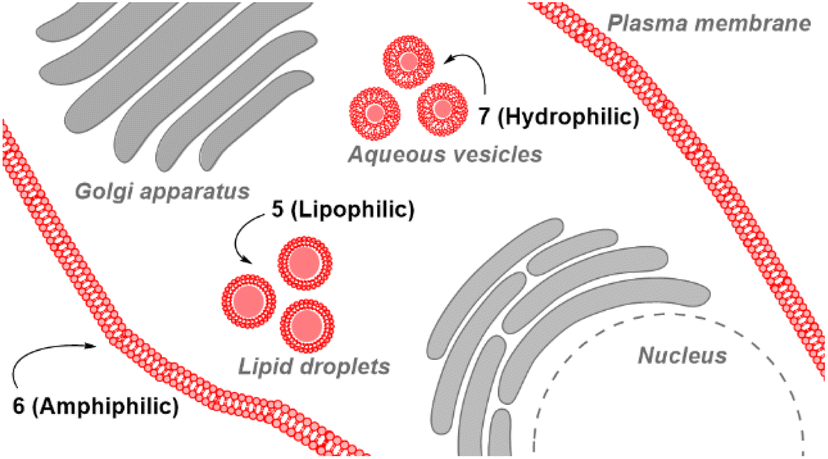 | ||
| Fig. 4 Anticipated subcellular localizations of fluorophores 5–7. Sub-cellular accumulation sites highlighted in red for 5 (lipid droplets), 6 (plasma membrane) and 7 (aqueous media containing vesicles). | ||
Results and discussion
Synthesis
A four-step synthesis to an advanced bis-methylanilino intermediate 11 commenced with an aldol condensation of commercially available 4-acetyl-N-methylaniline and benzaldehyde giving 4-methylaminochalcone 8 (Scheme 1). Michael addition of nitromethane to 8 under basic conditions gave 1,3-diaryl-4-nitrobutan-1-one 9 in 89% yield. Tetraarylazadipyrromethene 10 was reached through a mechanistically complex condensation of 9 with excess ammonium acetate at reflux for 24 h.24 Both chalcone 8 and azadipyrromethene 10 were isolated by direct precipitation out of the reaction solution using aqueous acid or water respectively and used in subsequent steps without further purification. However, the 1,3-diaryl-4-nitrobutan-1-one 9 was obtained as an oil after first diluting the reaction mixture with H2O and subsequently extracting with EtOAc. Transformation of 10 to the BF2 chelate 11 was achieved through reaction with boron trifluoride diethyl etherate in the presence of diisopropylethylamine (DIPEA) in CH2Cl2 under nitrogen for 24 h (Scheme 1).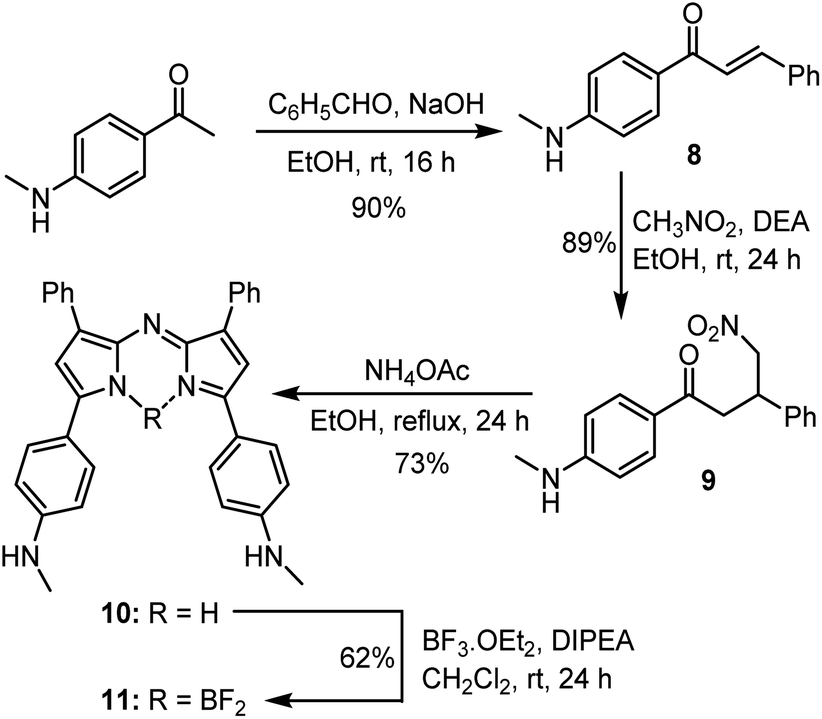 | ||
| Scheme 1 Synthetic route to BF2 chelated N-methyl-4-((methylamino)phenyl)-3-phenyl-1H-pyrrol-2-yl)imino)-3-phenyl-2H-pyrrol-5-yl)aniline 11. | ||
With the key intermediate 11 in hand, further nitrogen derivatization of the NH-methyl anilino groups via double nucleophilic substitution was explored (Scheme 2). Reaction of 11 with t-butyl bromoacetate under basic conditions at 60 °C for 16 h successfully introduced two tBu ester groups to provide the lipophilic fluorophore 5 following silica gel chromatography, in a 66% yield.25 1H NMR analysis was consistent with the structure of 5 with the occurrence of a singlet peak at 4.04 ppm in the spectrum corresponding to the N-methylene protons (Scheme 3B, ESI†). Next, reaction of 11 with 1,3-propane sultone in CH3CN with potassium carbonate at 60 °C for 16 h gave fluorophore 6 following reverse phase preparative chromatography.26 Product structure was confirmed by HRMS and NMR analysis with three new methylene peaks for the alkyl sulfonic acid component recorded at 3.6, 2.5 and 1.9 ppm (ESI†).
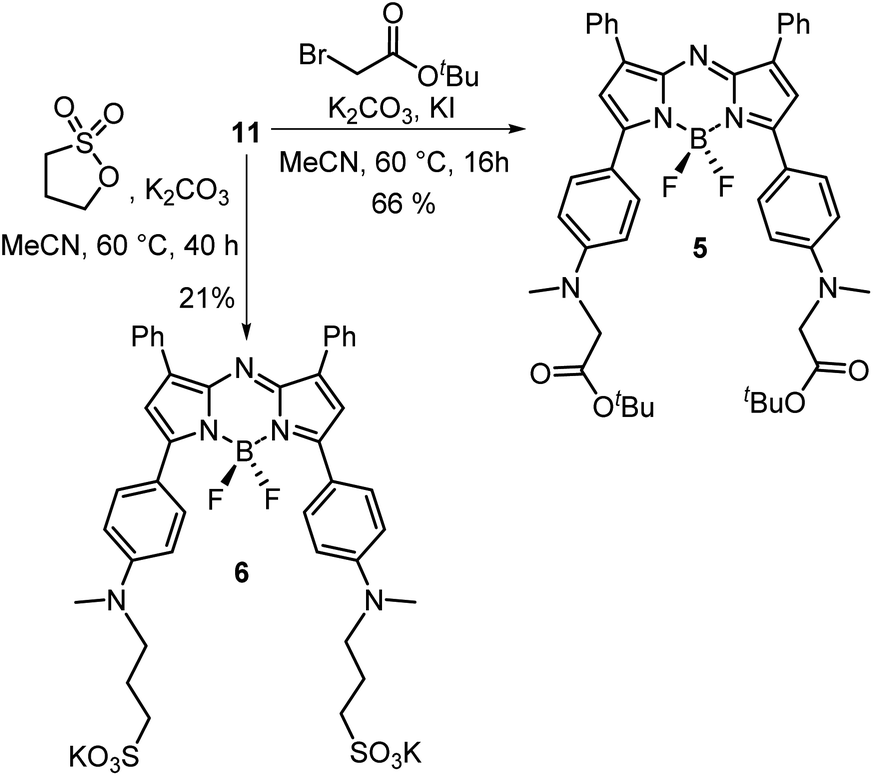 | ||
| Scheme 2 Transformation of intermediate 11 into fluorophores 5 and 6. | ||
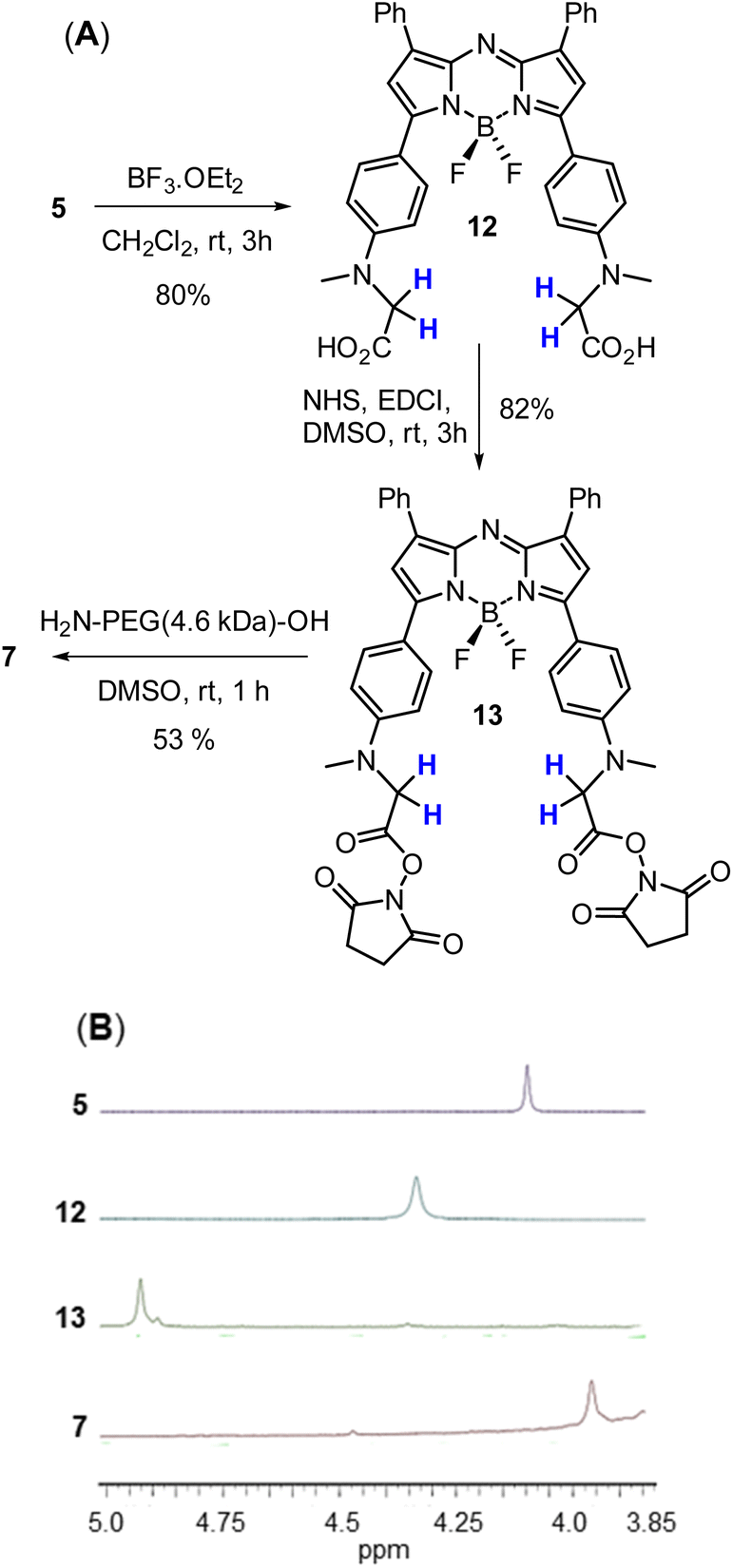 | ||
| Scheme 3 Generation and analysis of PEGylated NIR-AZA 7. (A) Three step route used for the synthesis of 7 from 5. (B) Partial 1H NMR spectra of 5, 12, 13 and 7 showing the chemical shift change in methylene protons (coloured blue). | ||
Access to fluorophore 7 was achieved through further derivatization of 5 starting with its ester saponification which was achieved in 80% yield with boron trifluoride diethyl etherate in DCM (Scheme 3A). The bis-acid 12 precipitated from the reaction, allowing isolation by filtration. The bis(succinimidyl ester) functionality of 13 was introduced by reacting 12 with N-hydroxysuccinimide in the presence of the coupling reagent N-ethyl-N-(3-dimethylaminopropyl)carbodiimide (EDCI) in dry DMSO for 3 h.
Reaction monitoring by analytical HPLC allowed the end point to be identified with product 13 isolated as a red solid in sufficient purity to proceed with the PEGylation via amide bond formation. Coupling of the activated ester groups of 13 with H2N-PEG(5kDa)-OH in dry DMSO, gave fluorophore 7 which was purified by reverse phase semi prep chromatography. The sequence of conversions of 5 to 12 to 13 to 7 could be followed by observing the change in chemical shift of the methylene protons in the 1H NMR spectrum going from 4.04 to 4.31 to 4.95 to 3.89 ppm respectively (Scheme 3B).
Photophysical properties of fluorophores 5–7
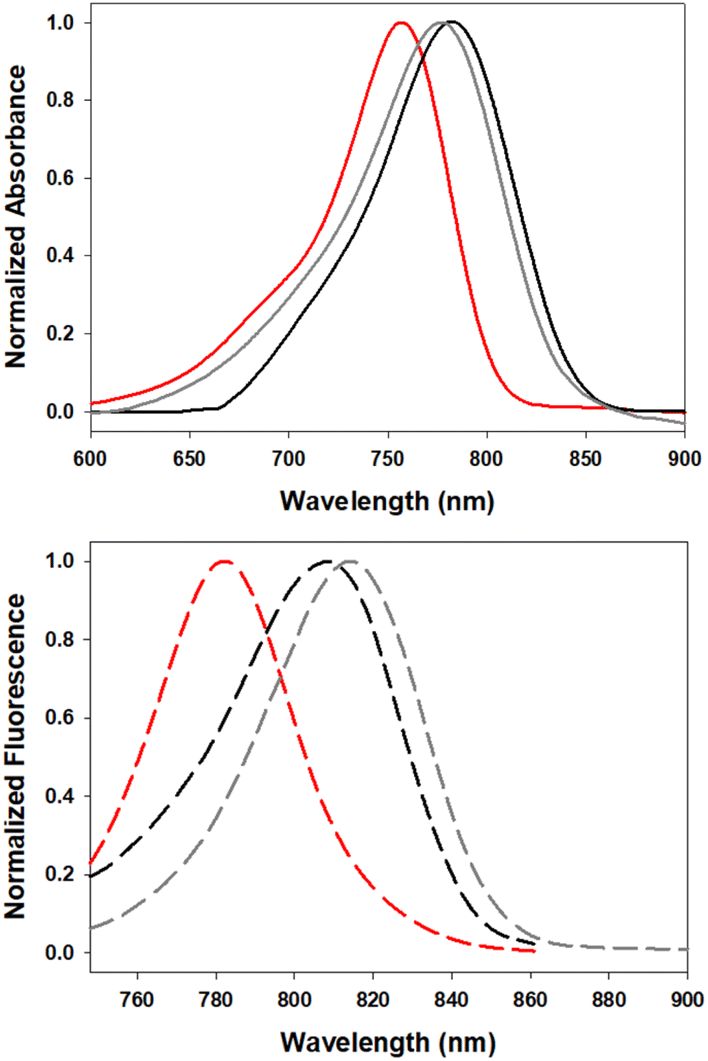
In order to investigate the impact of differing N-substituents on the photophysical properties of 5–7 they were cross compared using EtOH as solvent. Encouragingly, the long wavelength absorbance and emission maxima of 5 (780, 819 nm), 6 (806, 827 nm) and 7 (790, 820 nm) showed little variance, being within 26 nm for absorbance and 8 nm for emission (Fig. 5, Table 1).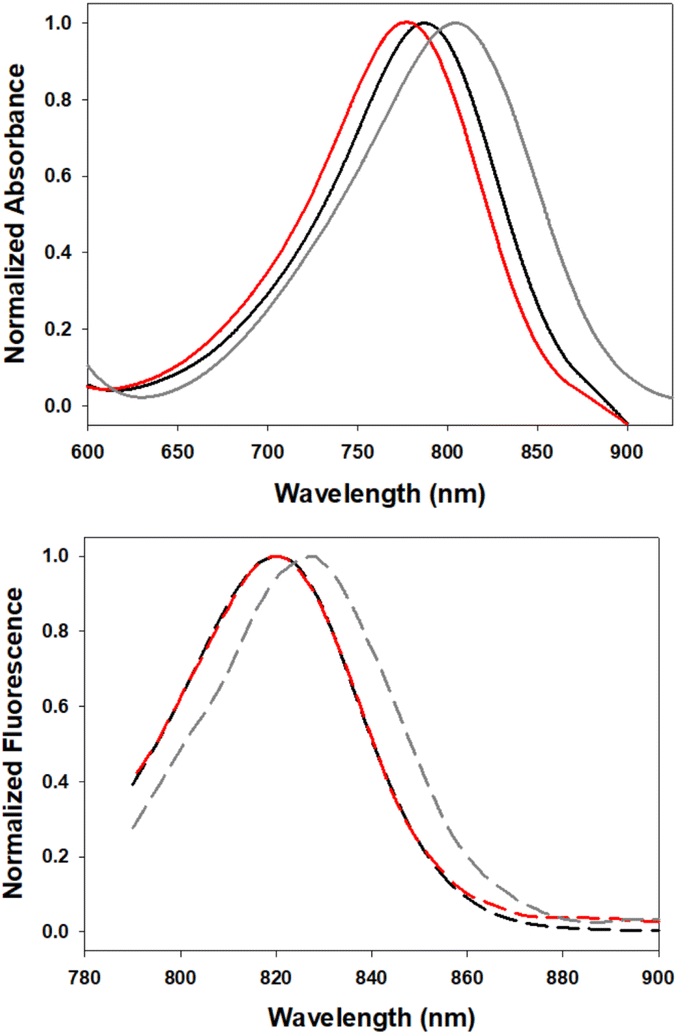 | ||
| Fig. 5 Normalized absorption (top) and emission (bottom) spectra of 5 (red), 6 (gray) and 7 (black) in EtOH (4 μM). | ||
The blue shift in absorbance wavelengths of 5 and 7 versus 6 could be rationalised in terms of the inductive electron withdrawing effect of the β-carbonyl substituents reducing the electronegativity at the amino donor groups.27 The absence of this functionality in 6 resulted in it being the most red-shifted absorbance of the three fluorophores, centred at 806 nm. It was positive to find that the different nitrogen substituents had only minor effects on the emission maxima values, with a small 7–8 nm bathochromic shift for 6 relative to 5 and 7. As would be expected, the quantum yields in ethanol were modest with tBu-ester-substituted 5 being the highest, PEG-amide-substituted 7 lower and the bis-sulfonic acid-substituted 6 lowest (Table 1).
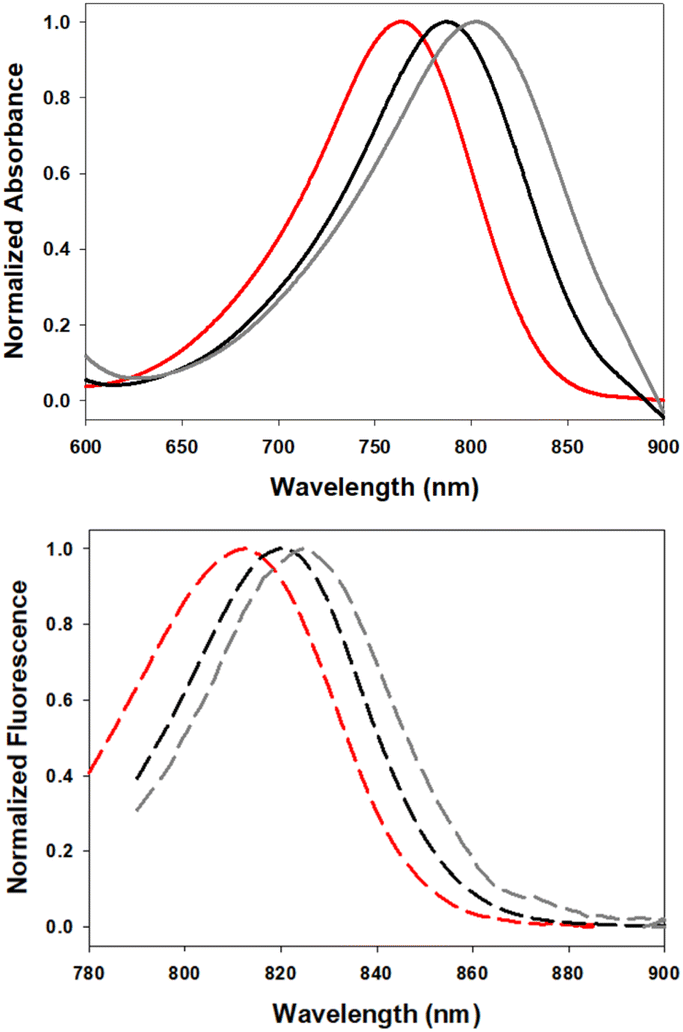
The solvatochromic ICT properties of the fluorophores were next examined in solvents of varying polarity (c-hexane, chloroform and ethanol). Additionally, their photophysical properties were assessed in solutions reflecting the micro-environment of their anticipated subcellular localisation; 5 in the triglyceride triolein, 6 in aqueous cetyltrimethylammonium bromide (CTAB) and 7 in water at varying pH. As expected for an ICT system such as 5, the wavelength trend for absorbance and emission maxima red shifted with increasing solvent polarity (Fig. 6, Table 2). Absorbance maxima values in c-hexane, CHCl3, and EtOH were recorded at 757, 776 and 780 nm respectively. A corresponding trend was seen for emission maxima (782, 815 and 820 nm) with quantum yields for 5 highest in c-hexane at 23.5% and decreased with increasing solvent polarity, such as CHCl3 (6.5%) and EtOH (1.6%), which is also consistent with an ICT system. In comparison the synthetic precursor compound 11 had lower quantum yields than 5 (c-hexane 8.4%, CHCl3 2.6%) which could be attributed to the NH group (ESI Fig. S1†). As LDs were the anticipated intracellular accumulation site for 5, its quantum yield was determined in triolein, a triglyceride of oleic acid which forms a major component of lipid droplet contents.29 Promisingly, a quantum yield of 11% was observed for 5 in triolein along with emission centred at 810 nm (Table 2).
 | ||
| Fig. 6 Normalized absorbance (top) and emission (bottom) spectra of 5 in c-hexane (red), CHCl3 (gray) and triolein (black) (2 μM). | ||
While the bis-anionic nature of 6 prohibited acquiring spectra in hexane and chloroform, its properties in aqueous CTAB were obtained. CTAB is a trimethyl ammonium salt of hexadecane comprising an anionic ammonium head group with an aliphatic hydrocarbon tail which can simulate an amphiphilic microenvironment in aqueous solutions.30 Encouragingly, a solution of 6 in aqueous CTAB (1%) exhibited an emission profile centred at 827 nm indicating its potential for its targeted imaging use (ESI Fig. S2†).
Examination of the solvent polarity effects on absorbance and emission profiles of 7 showed similar trends to those recorded for 5 with increasing solvent polarity again providing bathochromic shifts in absorbance and emission maxima (Fig. 7, Table 3). Fluorescence spectral analysis in chloroform, ethanol and water gave λmax values of 807, 820 and 826 nm respectively, with quantum yields being higher in less polar solvents.
 | ||
| Fig. 7 Normalized absorbance (top) and emission (bottom) spectra of 7 in CHCl3 (red), EtOH (black) and H2O (gray) (2 μM). | ||
When considering the use of 7 for imaging hydrophilic cellular vesicles two considerations needed to be investigated. These vesicles would not contain pure water but rather an aqueous mixture of more lipophilic materials, and fluctuating acidities can be expected with pH varying from 7 to 4. As such the fluorescence changes of 7 in mixed aqueous/EtOH solutions and at differing acidities were studied. Encouragingly, from an imaging standpoint, the emission intensity from a pure water solution of 7 increased sequentially as an increasing amount of EtOH was added, reaching a 5-fold enhancement for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/EtOH mixture when compared to water alone (Fig. 8).
:1 water/EtOH mixture when compared to water alone (Fig. 8).
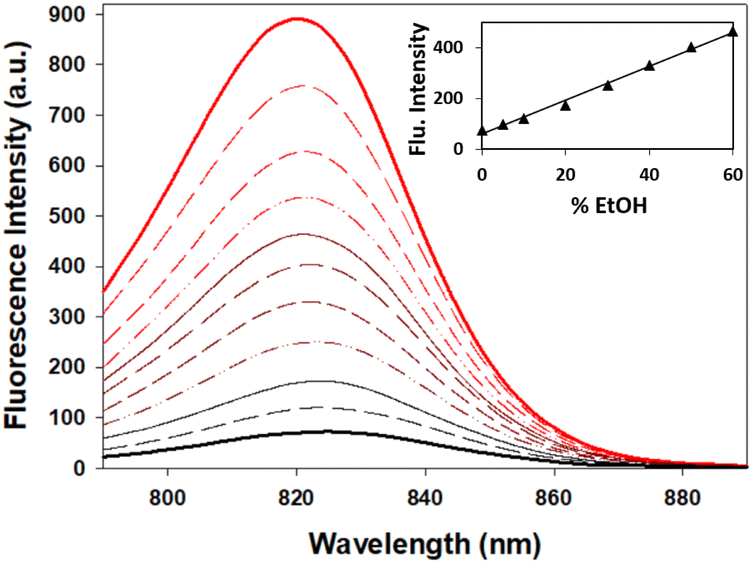 | ||
| Fig. 8 Fluorescence spectra of 7 in water/EtOH solutions (4 μM). Fluorescence spectra in pure H2O (solid black line) and EtOH (solid red line) with differing mixture compositions in between (excit. at 776 nm, 10 nm slit widths). Inset: graph of fluorescence intensity versus % EtOH in the solution mixture. | ||
As the desired emission wavelengths between 800 and 850 nm for 5–7 were as a result of the ICT from the bis-(anilino) substituents, it was necessary to test the suppression of this through nitrogen protonation and the likelihood of this occurring in a cellular imaging context. Using 7 as a representative example, changes to spectral features were recorded during an aqueous hydrochloric acid titration (Fig. 9). Importantly, no change in absorbance or emission was observed for the pH range of 7 to 2, confirming that emission properties would not be effected during cell imaging even in the most acidic intracellular vesicles (ESI Fig. S3†). The titration was continued below the pH scale to illustrate the impact of the D–A–D fluorophore design and the ICT role in delivering their extended wavelengths. The first protonation of an anilino nitrogen occurred across a 0.1 to 2.8 M HCl concentration with a stepwise disappearance of the 806 nm absorption band. In tandem, formation of a new band at 744 nm with a sharp isosbestic point at 768 nm occurred, indicating the formation of the monoprotonated A–A–D compound 7-H+. Continuation of the titration from 3 to 10 M HCl resulted in a decrease in the 744 nm band, with the subsequent formation of a new absorption at 654 nm for 7-2H+ with a second clear isosbestic point at 682 nm. A related sequence of changes was mirrored for the emission spectra with 7, 7-H+ and 7-2H+ having maxima at 826, 784 and 675 nm respectively (ESI, Table S1, Fig. S3†). It is of interest to note that the doubly protonated species 7-2H+ has absorption and emission properties very similar to that of 1a (Fig. 1). The robustness of 7 to extreme acidities may offer sensing applications beyond the scope of this current work.
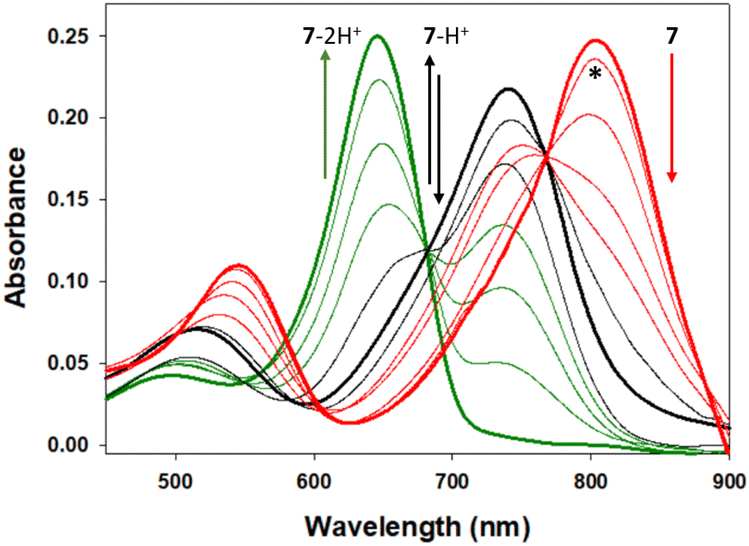 | ||
| Fig. 9 Absorbance spectral changes to 7 upon sequential anilino protonations. Absorbance maxima at pH 7, (bold red, 806 nm), 2.8 M HCl (bold black, 744 nm) and 10 M HCl (bold green, 654 nm) for 7, 7-H+ and 7-2H+ respectively with isosbestic points at 768 and 682 nm. *Spectrum at pH 1. See ESI Fig. S2† for corresponding emission changes. | ||
MDA-MB 231 cell imaging
Review of the absorption and emission spectra of 5, 6 and 7 in their cell model solutions of triolein, aqueous CTAB and water allowed a unified set of microscope imaging parameters to be selected as excitation at 777 nm with emission filter set from 800–850 nm (ESI Fig. S4†). Of the three fluorophores to be tested, the most lipophilic 5 was not aqueous soluble so the delivery vector poloxamer 188 (P188) was used to make stable aqueous solutions for cell imaging. P188 is a nonionic linear block copolymer consisting of poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO) segments structurally organised as (PEO)79-(PPO)28-(PEO)79.31 It has been investigated for transporting insoluble drug molecules such as doxorubicin and ibuprofen across cell membranes.32,33 It has also been previously shown to be highly effective in delivering molecular and nanoparticle fluorophores to cells.34,35 As LDs are the most concentrated lipophilic cellular regions, it was reasoned that these would be the preferred site of accumulation of 5. LDs are abundant intracellular organelles that are storage vessels for neutral lipids such as triacylglycerols and sterol esters. They play roles in both normal and abnormal cells beyond lipid storage including energy generation, membrane synthesis and protein degradation.36 It has been demonstrated that an increased number of LDs in breast cancer stem cells is connected with dysregulation of lipid metabolism in those cells.37 Previously reported LD staining fluorophores include the BF2 azadipyrromethene 1b with emission centred at 720 nm (ref. 35) with other classes including coumarin, statomerocyanine and naphthalimide based probes with emissions within the 600–700 nm range.38Imaging experiments used epithelial breast cancer MDA-MB 231 cells which are a highly aggressive triple-negative human cell line making imaging of its subcellular features of fundamental research importance. In practice, a chamber slide of seeded cells was positioned under the objective of a confocal laser scanning microscope (CLSM) surrounded by an incubator to maintain the temperature at 37 °C and CO2 at 5%. An imaging field of view containing viable cells was chosen and cells were treated with aqueous solution of 5 to a 5 μM concentration. Fluorescence images were acquired for 120 min using excitation wavelength of 777 nm with emitted light collected between 800 and 850 nm. During this time intracellular fluorescence was observed to grow in intensity from specifically localized vesicles throughout the cytoplasm (Fig. 10A and B, ESI Fig. S5† for repeat experimental images). Next, dual fluorophore experiments were carried out using the known LD stain 1b and 5 to confirm that the observed vesicles were LDs.35 Following co-incubation of cells with both fluorophores for 1 h, images were acquired in two channels using 630 and 777 nm excitations with collection filters set to 670–740 nm and 800–850 nm for 1a and 5 respectively. Overlaying of images generated from both channels showed the fluorophores were confined to the same subcellular vesicles (Fig. 10C, ESI Fig. S6† for repeat experimental images). Statistical image analysis of the colocalizations gave a Pearson correlation coefficient of 0.94 and Manders' coefficients values of 0.84 and 0.93 for tM1b and tM5 respectively, confirming a high level of co-localisation to LDs.
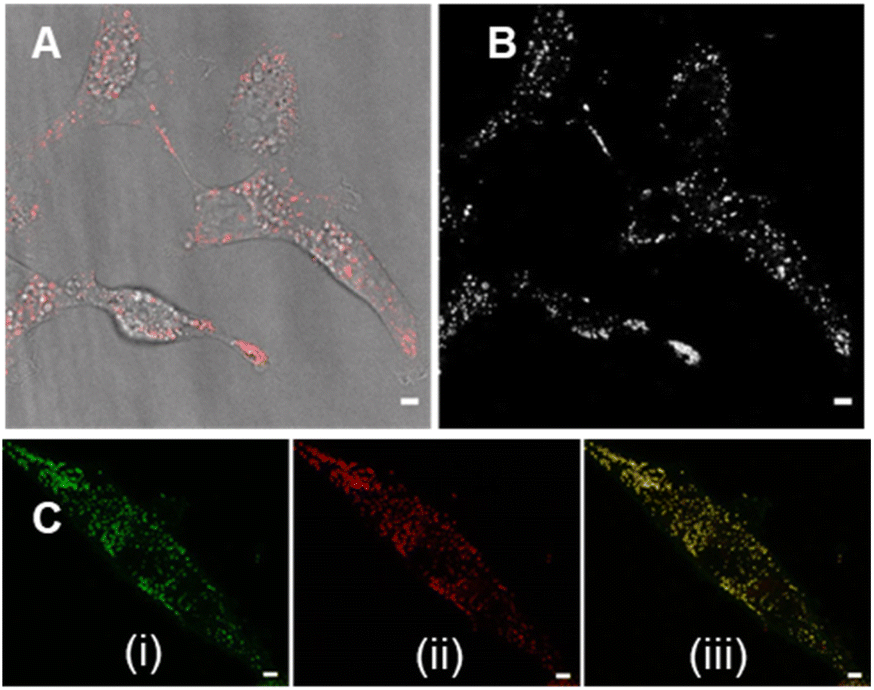 | ||
| Fig. 10 CLSM imaging of MDA MB-231 live cells following 120 min incubation with 5 (5 μM). (A) CLSM image (fluorescence in red) with bright field overlay. (B) Fluorescence shown in black and white for clarity. (C) Images of cells co-incubated with (i) 1b and (ii) 5 for 1 h with (iii) showing overlaid images with co-localisation (yellow) of both fluorophores in LDs. Scale bars 5 μm. | ||
Next, cell associations with the amphiphilic probe 6 were investigated using CLSM over 60 min. Pleasingly, imaging using the 777/800–850 nm microscope parameters the plasma membrane was distinguishable as framing the cell outline within 20 min following its addition (Fig. 11, panels A, B, ESI Fig. S7† for repeat experimental images). As would be expected in live cells, active transport through the plasma membrane occurred with 6 being internalized in vesicles within the cytosol by 1 h (Fig. 11, panels C, D, ESI Fig. S8, S9 for repeat experimental images and S10† for 4 h incubation images).39 These results confirmed that the design feature imparted by the alkyl sulfonic acid substituents could control the temporal and spatial accumulation of 6 in specific cellular microenvironments, allowing for them to be imaged.
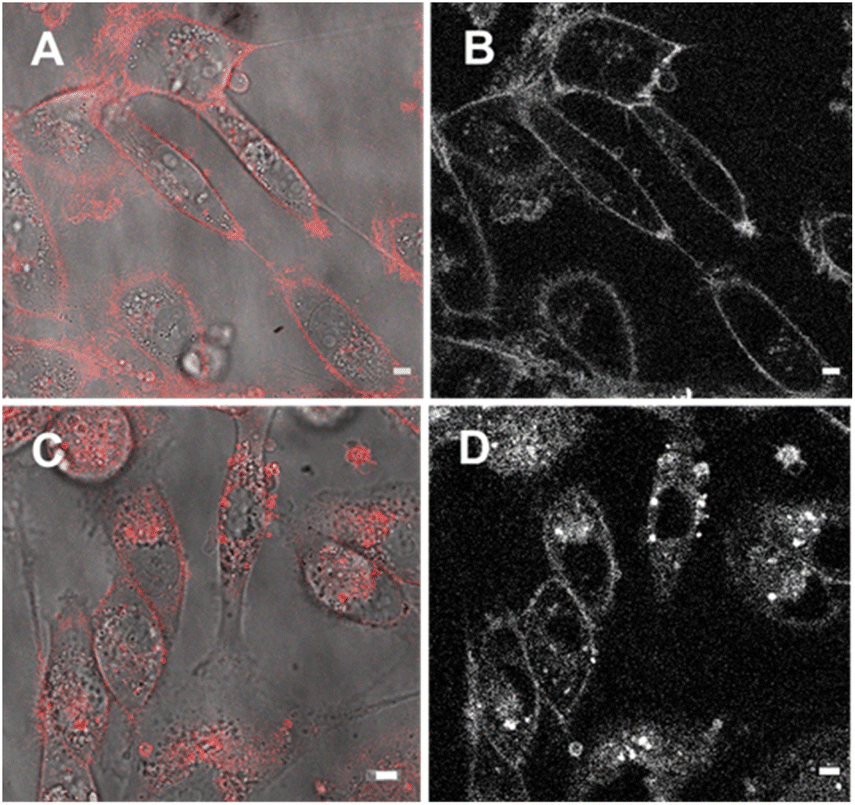 | ||
| Fig. 11 CLSM imaging of MDA MB-231 live cells over 60 min following incubation with 6 (5 μM). (A) CLSM image (fluorescence in red) with bright field overlay taken 20 min following the addition of 6. (B) Fluorescence shown in black and white for clarity. (C) CLSM image (fluorescence in red) with bright field overlay taken 60 min following the addition of 6. (D) Fluorescence shown in black and white for clarity. Scale bars 5 μm. | ||
The third fluorophore to be image tested was the aqueous soluble pegylated derivative 7. Unlike 6, it does not contain any structural feature that would promote a build-up within the plasma membrane but an effective cell uptake would be anticipated. Preliminary cell experiments showed that internalization of 7 was slower than that of 6, with no staining of the plasma membrane observable, and the optimal imaging time point identified as 24 h. Treatment of cells to a 5 μM concentration of 7 with incubation for 24 h gave an emission distribution from vesicles of varying size throughout the cytosol (Fig. 12A and B, ESI Fig. S9† for repeat experimental images).
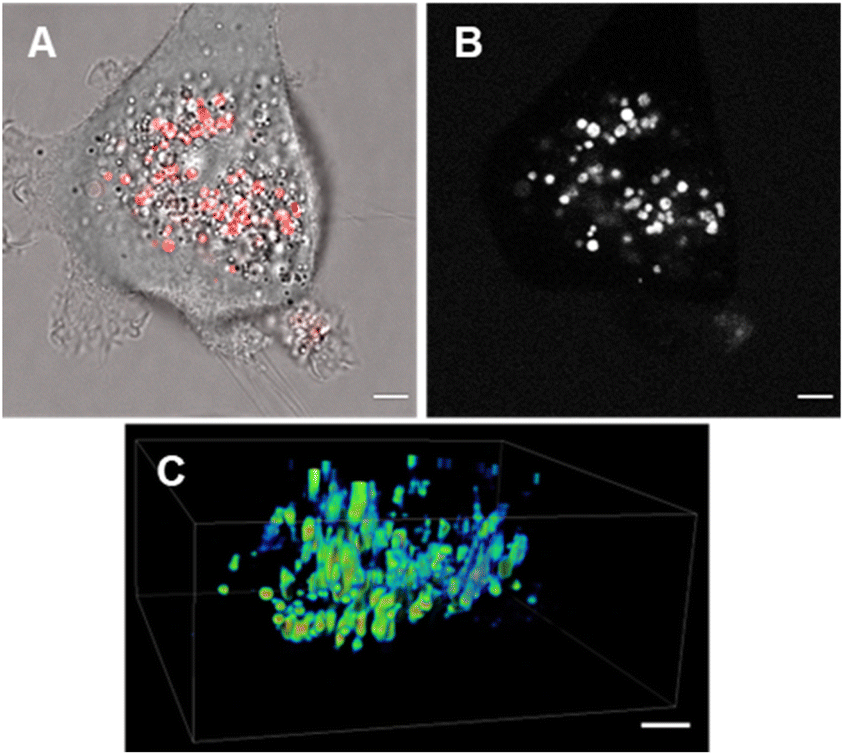 | ||
| Fig. 12 CLSM imaging of MDA MB-231 live cells following 24 h incubation with 7 (5 μM). (A) CLSM image (fluorescence in red) with bright field overlay. (B) Fluorescence shown in black and white for clarity. (C) Z-Stack analysis of an individual cell showing intracellular vesicles as intensity heatmap. Scale bars 5 μm. | ||
The intracellular staining pattern was attributable to cellular lysosomes and large acidic vacuoles which related to the metastatic nature of MDA-MB 231 cells.40 Z-Stack analysis of cells confirmed that regions of fluorescence were within the cytoplasm and their motion could be tracked over time (Fig. 12C and ESI Movie S1, S2†). Finally, as a comparison control imaging of 11 incubated with cells as an aqueous P188 solution showed both general cytosol staining and LDs (ESI Fig. S10†).
Conclusion
The design and synthesis of three NIR-AZA fluorophores with near infrared emissions in the 800–850 nm range has been realized. Photophysical studies showed that the long wavelengths were governed by an ICT which was achieved by the judicious placing of anilino substituents on the BF2-chelated tetraarylazadipyrromethene chromophore. The three fluorophores were differentiated from each other through a late stage introduction of key N-anilino substituents via alkylation reactions. The role of each substituent was to direct intracellular accumulation of each fluorophore to different sites within cells. The sub-cellular accumulation sites being LDs (tBu ester substituent), the plasma membrane (alkyl sulfonic acid substituent) and aqueous filled vesicles (PEG substituent) was confirmed for each using CLSM. With the knowledge gained from this study, improvement of quantum yields and other subcellular directing substituents are to be explored.Experimental
General
All reagents were used as received without further purification. Air-sensitive reagents were reacted in oven-dried glassware under nitrogen using syringe-septum cap techniques. All solvents were purified and degassed prior to use. Reactions were monitored using (i) thin-layer chromatography techniques (0.25 mm silica gel coated aluminium plates (60 Merck F254) using 254 nm UV light for visualisation), (ii) reverse phase chromatography on a HPLC (Shimadzu) equipped with analytical (YMC-Triart Phenyl, 4.6 × 150 mm I.D. S-5 μm) column, eluting with acetonitrile/water, and (iii) 1H NMR monitoring. If required, products were further purified using (i) flash-column chromatography techniques with Merck silica gel 60 under pressure or (ii) reverse phase semi-preparative HPLC using semi-preparative (YMC-Triart Phenyl, 10 × 150 mm I.D. S-5 μm, 12nm) column. Products were analysed by 1H NMR, 13C NMR and 19F NMR spectra recorded at 400 MHz, 101 MHz and 376 MHz, respectively, at rt and calibrated using residual non-deuterated solvent as an internal reference. Chemical shifts are reported in parts-per-million (ppm). Further analysis by reverse phase chromatography was carried out for final products using a HPLC (Shimadzu) as described above for reaction monitoring. ESI mass spectra were acquired using Waters LCT Classic in positive and negative modes as required. MALDI-TOF spectra were acquired using Bruker ‘autoflex max’ system with MALDI Imaging Platform. All absorbance spectra were recorded with a Varian Cary 50 scan UV-visible spectrophotometer and fluorescence spectra were recorded with a Varian Cary eclipse fluorescence spectrophotometer. Organic solvents for absorbance and fluorescence experiments were of HPLC quality and millipore filter HPLC grade water was used. Confocal microscope images were acquired using Leica Stellaris 8 (Leica objective 100×/1.49 HC PL APO CS2) and were processed using the Leica Lightning deconvolution wizard. Analysis of raw files using ImageJ 1.53q. MDA-MB 231 cells obtained from Merck Life Science Limited.:1, 125 mL) at rt until dissolved. 4-Acetyl-N-methylaniline (2.5 g, 0.0168 mol) and benzaldehyde (2.4 mL, 0.0235 mol) were added and the reaction stirred for 20 h at rt. The reaction was acidified to pH 2 using 5 M HCl (50 mL) and the product precipitated by adding H2O (300 mL). Product was isolated by filtration and dried under vacuum yielding (2E)-1-[4-(methylamino)phenyl]-3-phenyl-2-propen-1-one 8 as a yellow solid (3.6 g, 90%), mp = 120–125 °C. 1H NMR (400 MHz, CDCl3) δ: 7.97 (d, J = 8.7 Hz, 2H), 7.78 (d, J = 15.6 Hz, 1H), 7.63 (d, J = 6.2 Hz, 2H), 7.56 (d, J = 15.6 Hz, 1H), 7.43–7.36 (m, 3H), 6.62 (d, J = 8.7 Hz, 2H), 4.44 (br. s, 1H) 2.92 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 188.0, 153.2, 142.9, 135.6, 131.2, 130.1, 129.0, 128.4, 127.5, 122.3, 111.5, 30.3. HRMS (ESI−): calcd for C16H14NO [M − H]− 236.1081; found 236.1082.:1) to CH2Cl2 (100%). 1H NMR (400 MHz, DMSO-d6) δ: 8.18–8.11 (dd, J = 8.6 Hz, 8H), 7.58 (s, 2H), 7.51 (t, J = 7.5 Hz, 4H), 7.42 (t, J = 7.5 Hz, 2H), 6.95 (d, J = 5.0 Hz, 2H), 6.71 (d, J = 8.6 Hz, 4H), 2.84 (d, J = 5.0 Hz, 6H). 13C NMR (101 MHz, DMSO-d6): δ 154.9, 152.5, 143.9, 138.9, 132.5, 132.0, 128.7, 128.5, 118.5, 117.6, 111.8, 29.3 ppm. 19F NMR (376 MHz, DMSO-d6): −130.83 ppm. Abs (λmax): CHCl3 768 nm; c-hex 752 nm; EtOH 796 nm. Flu (λmax): CHCl3 814 nm Φflu 2.6%; c-hex 785 nm Φflu 8.4%; EtOH 827 nm. HRMS (ESI−): calcd for C34H27BF2N5 [M − H]− 554.2338 found 554.2343.:1) going to 100% CH2Cl2 yielding the product 5 as a red solid (140 mg, 66%), mp > 250 °C. 1H NMR (400 MHz, CDCl3) δ: 8.16–8.10 (dd, J = 20.5, 8.1 Hz, 8H), 7.45 (t, J = 7.3 Hz, 4H), 7.41–7.35 (m, 2H), 7.09 (s, 2H), 6.75 (d, J = 8.7 Hz, 4H), 4.04 (s, 4H), 3.16 (s, 6H, N(CH3)), 1.45 (s, 18H) ppm. 13C NMR (101 MHz, CDCl3): δ 169.5, 156.8, 150.9, 145.3, 141.4, 133.1, 131.8, 129.3, 128.8, 128.5, 120.2, 118.2, 112.1, 82.2, 55.2, 39.8, 28.2 ppm. 19F NMR (376 MHz, CDCl3): −32.49 (q, J = 32.7 Hz) ppm. HRMS (ESI+): calcd for C46H48BF2N5NaO4 [M + Na]+ 806.3665; found 806.3689.:55, 1.0 mL), filtered and the solution purified by reverse phase semi prep chromatography (eluent MeCN/H2O 45:55 going to 70:30, flow rate 4 mL min−1). Pure fractions were combined, concentrated by lyophilization yielding 6 as a dark red solid (6.2 mg, 21%), mp > 250 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.17 (d, J = 7.5 Hz, 8H), 7.62 (s, 2H), 7.51 (t, J = 7.5 Hz, 4H), 7.42 (t, J = 7.3 Hz, 2H), 6.92 (m, 4H), 3.61 (m, 4H), 3.09 (s, 6H, NH), 2.58–2.49 (m, 4H), 1.90 (dd, J = 7.7, 5.5 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ 154.6, 151.0, 144.1, 132.5, 131.8, 128.8, 128.8, 128.5, 118.7, 117.4, 111.9, 50.7, 48.6, 38.1, 23.0 ppm; 19F NMR (376 MHz, DMSO-d6) δ −130.95 ppm. HRMS (ESI−): calcd for C40H38BF2N5O6S2 [M − 2H]2− 398.6171; found 398.6174.:60 going to 70:30, flow rate 4 mL min−1) with pure fractions combined and aqueous solvent removed by lyophilization yielding 7 as a dark red solid product (64.5 mg, 53%) of greater than 99% purity, mp > 250 °C, 1H NMR (400 MHz, CDCl3) δ 7.95 (dd, J = 7.5 Hz, 8H), 7.34–7.27 (m, 6H), 6.98 (s, 2H), 6.93 (s, 2H), 6.66 (d, J = 8.3 Hz, 4H), 3.89 (s, 4H), 3.04 (s, 6H); 19F NMR (376 MHz, CDCl3): δ −132.06 (q) ppm. MS MALDI-TOF: calcd for C486H930BF2N7O226 10531.1556; found 10519.6354 (dist. 8713.9583–11064.6955).Preparation of triolein solution of 5
Fluorophore 5 (3.92 mg, 0.005 mmol) was diluted in triolein (8 mL), sonicated for 2 min, heated at 30 °C for 2 min and made up to the mark in a 10 mL volumetric flask to yield a 500 μM stock solution for use in quantum yield determination.Preparation of aqueous CTAB (1% w/v) solution of 6
CTAB (100 mg, 0.274 mmol) was dissolved in MeOH (10 mL) in a RBF. Stock solution of fluorophore 6 in MeOH (80.0 μL of a 500 μM stock) was added to the RBF and the mixture sonicated for 2 min to ensure solvation of CTAB. The solvent was removed by rotary evaporation and the RBF was dried for 30 min under high vacuum. The resulting solid was dissolved in DIW (8 mL), sonicated for 2 min, syringe filtered (PTFE/millipore) into a volumetric flask (10 mL), made up to the mark with DIW yielding a 4 μM sample of 6 and left at rt for 1 h to equilibrate.Preparation of aqueous P188 (1% w/v) solution of 5
Poloxamer-188 (100 mg, 0.055 mmol) was dissolved in dry THF (10 mL) in a RBF. Stock solution of fluorophore 5 in THF (80.0 μL of a 500 μM stock) was added and the mixture sonicated for 2 min to ensure solvation of P188. The solvent was removed by rotary evaporation and the RBF was dried for 30 min under high vacuum. The resulting solid was dissolved in DIW (8 mL), sonicated for 2 min, syringe filtered (PTFE/millipore) into a volumetric flask (10 mL), made up to the mark with DIW yielding a 4 μM sample of 5 and left at rt for 1 h to equilibrate.Quantum yield determination of fluorophores 5–7
Quantum yields were calculated using the comparative method and measured against a known standard (ICG 2; Φ = 0.13 in DMSO).28 A triplicate of five absorption/emission spectra were recorded respectively for each fluorophore. Graphs of absorbance at excitation wavelength versus integrated fluorescence were generated to determine Gradx and Gradstd which were inputted in the formula below to give quantum yield values.| ɸx = ɸstd (Gradx ÷ Gradstd) (ƞx2 ÷ ƞstd2) |
Cell culture and microscopy
MDA-MB 231 human breast cancer cells were seeded on to an eight well chamber slide (Ibidi) at a density of 1 × 104 cells per well 24 h before imaging. Cells were cultured in Dulbecco's Modified Eagles Media supplemented (DMEM) with 10% fetal bovine serum (FBS), 1% L-glutamine, and penicillin-streptomycin (1000 U mL−1), and incubated at 37 °C and 5% CO2. The slide was place on the microscope stage surrounded by an incubator to maintain the temperature at 37 °C and CO2 at 5%. Bright field imaging was used to choose a field of view and focus on a group of cells. CLSM and bright field images were acquired on a Leica Stellaris 8 Falcon microscope fitted with NKT Photonics White Light Laser (440–790 nm) and controlled by LAS X software (version. 4.4.0.24861). Live cell imaging experiments were carried out under Okolab incubation system. Excitation wavelength was using White Light Laser (WLL) tuned at 777 nm. Detection was using HyD S detector with collection range 800–850 nm, analogue mode, gain value 10. Images were acquired using a Leica HC PL APO CS2 100×/1.40 oil immersion objective. Image processing was completed by using software ImageJ 1.53f51 (National Institutes of Health, USA).Author contributions
DOS: Conceptualization, funding acquisition, supervision. CC and DOS: Writing original draft. CC: Synthesised, analysed and photophysical characterization of all compounds. DW: co-supervision. DW, MG: Carried out cell imaging studies. DW: Imaging data analysis.Conflicts of interest
DOS has a financial interest in patents filed and granted relating to NIR-fluorophores and processes for visual determination of tissue biology.Acknowledgements
DOS gratefully acknowledges funding support from the Irish Government Department of Business, Enterprise and Innovation's Disruptive Technology Innovation Fund and the Synthesis and Solid State Pharmaceutical Centre (SSPC) and Science Foundation Ireland for funding support, grant number 12/RC/2275_P2. CC acknowledges the RCSI Apjohn Fellowship for PhD funding. Images acquired in the RCSI Super Resolution Imaging Consortium funded by Science Foundation Ireland (18/RI/5723).References
- L. Ma and B. Fei, J. Biomed. Opt., 2021, 26, 010901–010911 CAS.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC.
- Y. Ge and D. F. O'Shea, Chem. Soc. Rev., 2016, 45, 3846–3864 RSC.
- Z. Shi, H. Xu, W. Hu, H. Bai, B. Peng, L. Ji, Q. Fan, L. Li and W. Huang, Chem. Soc. Rev., 2020, 49, 7533–7567 RSC.
- A. Gorman, J. Killoran and D. F. O'Shea, J. Am. Chem. Soc., 2004, 126, 10619–10631 CrossRef CAS PubMed.
- H. C. Daly, E. Conroy and D. F. O'Shea, Theranostics, 2020, 10, 3064–3082 CrossRef CAS PubMed.
- D. Wu, H. C. Daly, M. Grossi, E. Conroy, B. Li, W. M. Gallagher, R. Elmes and D. F. O'Shea, Chem. Sci., 2019, 10, 6944–6956 RSC.
- M. H. Y. Cheng, A. Maruani, H. Savoie, V. Chudasama and R. W. Boyle, Org. Biomol. Chem., 2018, 16, 1144–1149 RSC.
- S. Cheung, D. Wu, H. C. Daly, N. Busschaert, M. Morgunova, J. C. Simpson, D. Scholz, P. A. Gale and D. F. O'Shea, Chem, 2018, 4, 879–895 CAS.
- D. Wu, S. Cheung, M. Devocelle, L. J. Zhang, Z. L. Chen and D. F. O'Shea, Chem. Commun., 2015, 51, 16667–16670 RSC.
- N. Curtin, D. Wu, R. Cahill, A. Sarkar, P. M. Aonghusa, S. Zhuk, M. Barberio, M. A. Taher, J. Marescaux, M. Diana and D. F. O'Shea, Int. J. Med. Sci., 2021, 18, 1541 CrossRef CAS PubMed.
- H. C. Daly, G. Sampedro, C. Bon, D. Wu, G. Ismail, R. A. Cahill and D. F. O'Shea, Eur. J. Med. Chem., 2017, 135, 392–400 CrossRef CAS PubMed.
- H. Wada, H. Hyun, H. Kang, J. Gravier, M. Henary, M. W. Bordo, H. S. Choi and J. V. Frangioni, cAnn. Thorac. Surg., 2017, 103, 1132–1141 CrossRef PubMed.
- N. Onda, R. Mizutani-Morita, S. Yamashita, R. Nagahara, S. Matsumoto, T. Yoshida and M. Shibutani, Oncotarget, 2017, 8, 90278–90290 CrossRef PubMed.
- G. K. Park, J. H. Lee, A. Levitz, G. El Fakhri, N. S. Hwang, M. Henary and H. S. Choi, Adv. Mat., 2019, 31, 1806216 CrossRef PubMed.
- S. O. McDonnell and D. F. O'Shea, Org. Lett., 2006, 8, 3493–3496 CrossRef CAS PubMed.
- Y. Tian, H. Z. H. Zhou, Q. Cheng, H. Dang, H. Qian, C. Teng, K. Xiea and L. Yan, J. Mater. Chem. B, 2022, 10, 707–716 RSC.
- L. Bai, P. Sun, Y. Liu, H. Zhang, W. Hu, W. Zhang, Z. Liu, Q. Fan, L. Li and W. Huan, Chem. Commun., 2019, 55, 10920 RSC.
- J. A. Olzmann and P. Carvalho, Nat. Rev. Mol. Cell Biol., 2019, 20, 137–155 CrossRef CAS PubMed.
- C. W. Cairo, J. A. Key and C. M. Sadek, Curr. Opin. Chem. Biol., 2010, 14, 57–63 CrossRef CAS PubMed.
- A. V. Shubin, I. V. Demidyuk, A. A. Komissarov, L. M. Rafieva and S. V. Kostrov, Oncotarget, 2016, 7, 55863 CrossRef PubMed.
- S. Sasaki, G. P. Drummen and G. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC.
- M. Grossi, A. Palma, S. O. McDonnell, M. J. Hall, D. K. Rai, J. Muldoon and D. F. O'Shea, J. Org. Chem., 2012, 77, 9304–9312 CrossRef CAS PubMed.
- R. S. Mulla, M. T. Walden, D. S. Yufit, T. D. Lurie-Luke and J. A. Williams, Tetrahedron, 2017, 73, 6410–6420 CrossRef CAS.
- S. Zheng, Y. Yamashiro, I. Kitahara, W. Lin, J. Ericson, O. Siddiqui, W. Hsieh, P. Wang, C. R. Bartels, M. Kobuke, S. Noumi and A. Mochizuki, PatentPak, WO2017201482, 2017, pp. 41–44.
- C. A. Hoelzel, H. Hu, C. H. Wolstenholme, B. A. Karim, K. T. Munson, K. H. Jung, H. Zhang, Y. Liu, H. P. Yennawar, J. B. Asbury, X. Li and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 4785–4792 CrossRef CAS PubMed.
- R. C. Benson and H. A. Kues, J. Chem. Eng. Data, 1977, 22, 379 CrossRef CAS.
- R. V. Farese Jr and T. C. Walther, Cell, 2009, 139, 855–860 CrossRef PubMed.
- C. Peetla and V. Labhasetwar, Langmuir, 2009, 25, 2369–2377 CrossRef CAS PubMed.
- Q. T. H. Shubhra, J. Tóth, J. Gyenis and T. Feczkó, Polym. Rev., 2014, 54, 112–138 CrossRef CAS.
- M. Newa, K. H. Bhandari, D. X. Li, T.-H. Kwon, J. A. Kim, B. K. Yoo, J. S. Woo, W. S. Lyoo, C. S. Yong and H. G. Choi, Int. J. Pharm., 2007, 343, 228–237 CrossRef CAS PubMed.
- T. Tagami, Y. Ando and T. Ozeki, Int. J. Pharm., 2017, 517, 35–41 CrossRef CAS PubMed.
- B. Saremi, V. Bandi, S. Kazemi, Y. Hong, F. D'Souza and B. Yuan, Polymers, 2020, 12, 540 CrossRef CAS PubMed.
- N. Curtin, M. Garre, J.-B. Bodin, N. Solem, R. Méallet-Renault and D. F. O'Shea, RSC Adv., 2022, 12, 35655 RSC.
- J. A. Olzmann and P. Carvalho, Nat. Rev. Mol. Cell Biol., 2019, 20, 137–155 CrossRef CAS PubMed.
- L. Tirinato, F. Pagliari, T. Limongi, M. Marini, A. Falqui, J. Seco, P. Candeloro, C. Liberale and E. D. Fabrizio, Stem Cell. Int., 2017, 1656053 CAS.
- Y. Zhao, W. Shi, X. Li and H. Ma, Chem. Commun., 2022, 58, 1495–1509 RSC.
- D. Wu, S. Cheung and D. F. O'Shea, Biochim. Biophys. Acta Biomembr., 2018, 1860, 2272–2280 CrossRef CAS PubMed.
- W. Zhu, H. Qu, K. Xu, B. Jia, H. Li, Y. Du, G. Liu, H.-J. Wei and H.-Y. Zhao, Anim. Cell Syst., 2017, 21, 190–198 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures, tables, cell images, analytical spectra and HPLC traces available. See DOI: https://doi.org/10.1039/d2ra07942a |
| This journal is © The Royal Society of Chemistry 2023 |