DOI:
10.1039/D3RA00139C
(Review Article)
RSC Adv., 2023,
13, 10715-10756
Ring forming transformations of ynamides via cycloaddition
Received
8th January 2023
, Accepted 29th March 2023
First published on 4th April 2023
Abstract
Ynamides are N-alkyne compounds bearing an electron withdrawing group at the nitrogen atom. They offer unique pathways for the construction of versatile building blocks owing to their exceptional balance between reactivity and stability. Recently several studies have been reported that explore and illustrate the synthetic potential of ynamides and ynamide-derived advanced intermediates in cycloadditions with different reaction partners to yield heterocyclic cycloadducts of synthetic and pharmaceutical value. Cycloaddition reactions of ynamides are the facile and preferable routes for the construction of structural motifs having striking importance in synthetic, medicinal chemistry, and advanced materials. In this systematic review, we highlighted the recently reported novel transformations and synthetic applications that involved the cycloaddition reaction of ynamides. The scope along with the limitations of the transformations are discussed in detail.
1. Introduction
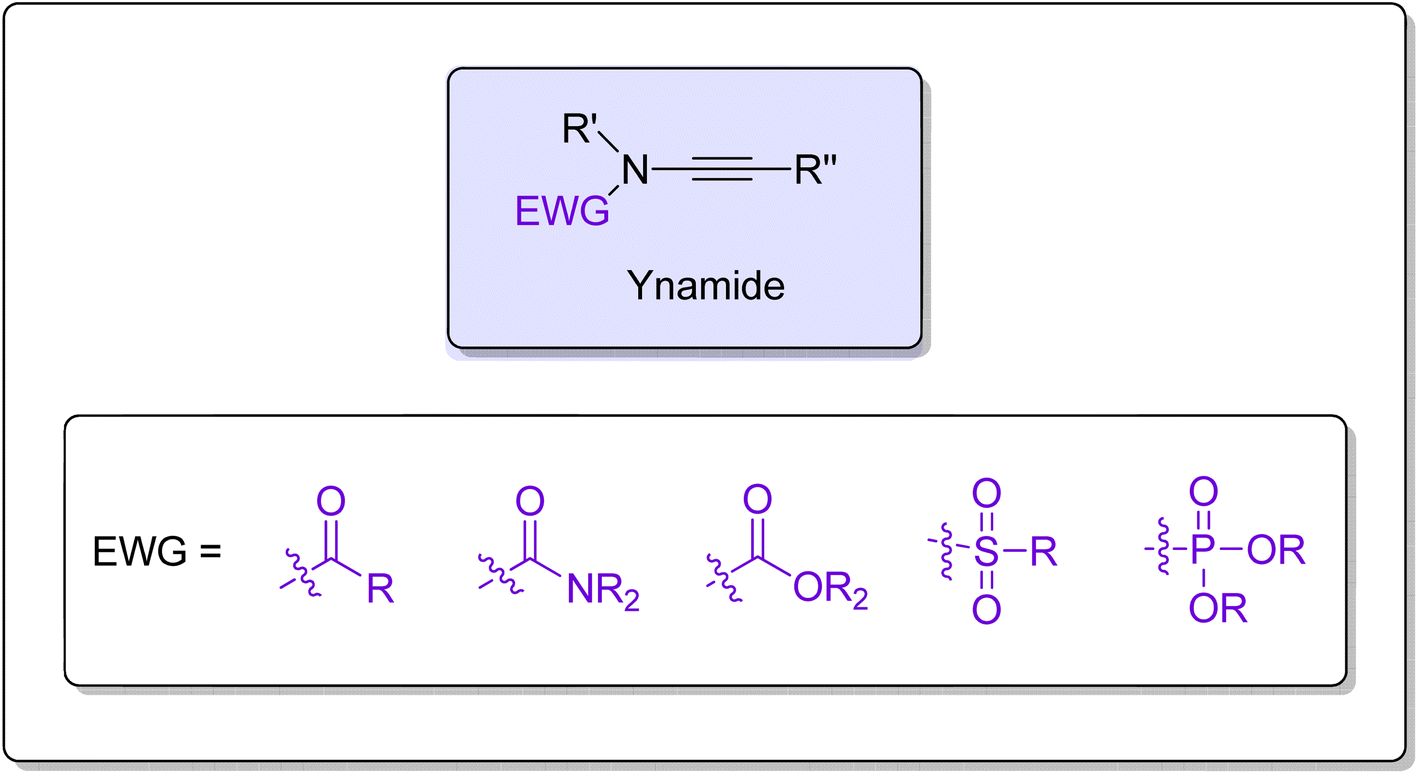
Ynamides are scaffolds that have N-alkyne as an active core, with the nitrogen atom further linked to an electron withdrawing group (EWG) (Fig. 1). Stimulated by the atom-economical and facile synthesis of ynamides, the scope, and reactivity of ynamides were explored and expanded in recent years.1 In the past decade, exceptional reactivity and stability of ynamides have been featured persistently and employed in a wide array of synthetic manifestations to realize the synthesis of natural as well as synthetic products. Ynamide is a modern functional group that can be employed as a versatile tool in organic synthesis to realize the synthesis of a diverse range of cyclic/polycyclic building blocks containing nitrogen within or close to the ring.2–5 The nitrogen atom in the ynamides accounts for its unusual reactivity and regioselectivity as it induces strong polarization in the alkyne triple bond.6 Owing to polarization, alkyne's α-carbon end serves as an electrophilic center while the β-carbon serves as a nucleophilic center. The electrophilic and nucleophilic centers generated within the same molecule render the alkyne with exceptional reactivity. Moreover, by varying the EWG attached to the nitrogen atom, the electronic properties could be modulated, which ultimately controls the reactivity of ynamides.7,8
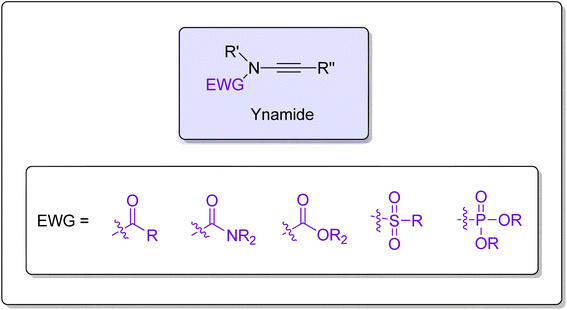 |
| | Fig. 1 The general structure of ynamide. | |
With the advent of ynamide's reactivity and novel methodologies for more efficient and regioselective transformations, there is a surge of ongoing interest in the development and synthetic applications of cycloaddition reactions.5,9–12 Among the several conceivable synthetic transformations of ynamides, cycloaddition reactions are one of the preferable routes that offer rapid assembly of novel complex frameworks with structural as well as molecular diversity.5 An impressive number of ynamide's cycloaddition reactions realizing the C–C and C–X (X = heteroatom) bond synthesis with excellent regioselectivity, stereoselectivity, and chemoselectivity expanded the asymmetric synthesis toolbox.10
The catalyst-activated ynamides afford the generation of advanced reaction intermediates that readily underwent cycloaddition (either intermolecular or intramolecular) to yield cycloadduct. This review aims to highlight the most prominent progress by discussing the novel and improved reported methodologies of ynamides and ynamide derived intermediates, along with a brief discussion on product diversity and selectivity during cycloaddition reactions (from 2014 to 2021). Moreover, in this review applicability, limitations, and mechanistic rationale of the synthetic transformations for the construction of structural motifs having high synthetic value is discussed. We anticipate that this review will expand the research spectrum by providing the reader with a broad and thorough overview of the expanding area of ynamide's chemistry.
2. Cycloaddition reactions of ynamides
The cycloaddition reactions of ynamides have been reported extensively and furnished a diverse range of synthetically and biologically important cores such as carbocycles and heterocycles. The transformation of ynamides provided efficient and direct access to a wide array of cyclic products.13,14 The following section comprises a brief overview of the prominent progress in the cycloaddition reactions.
2.1. [2 + 2]-cycloaddition
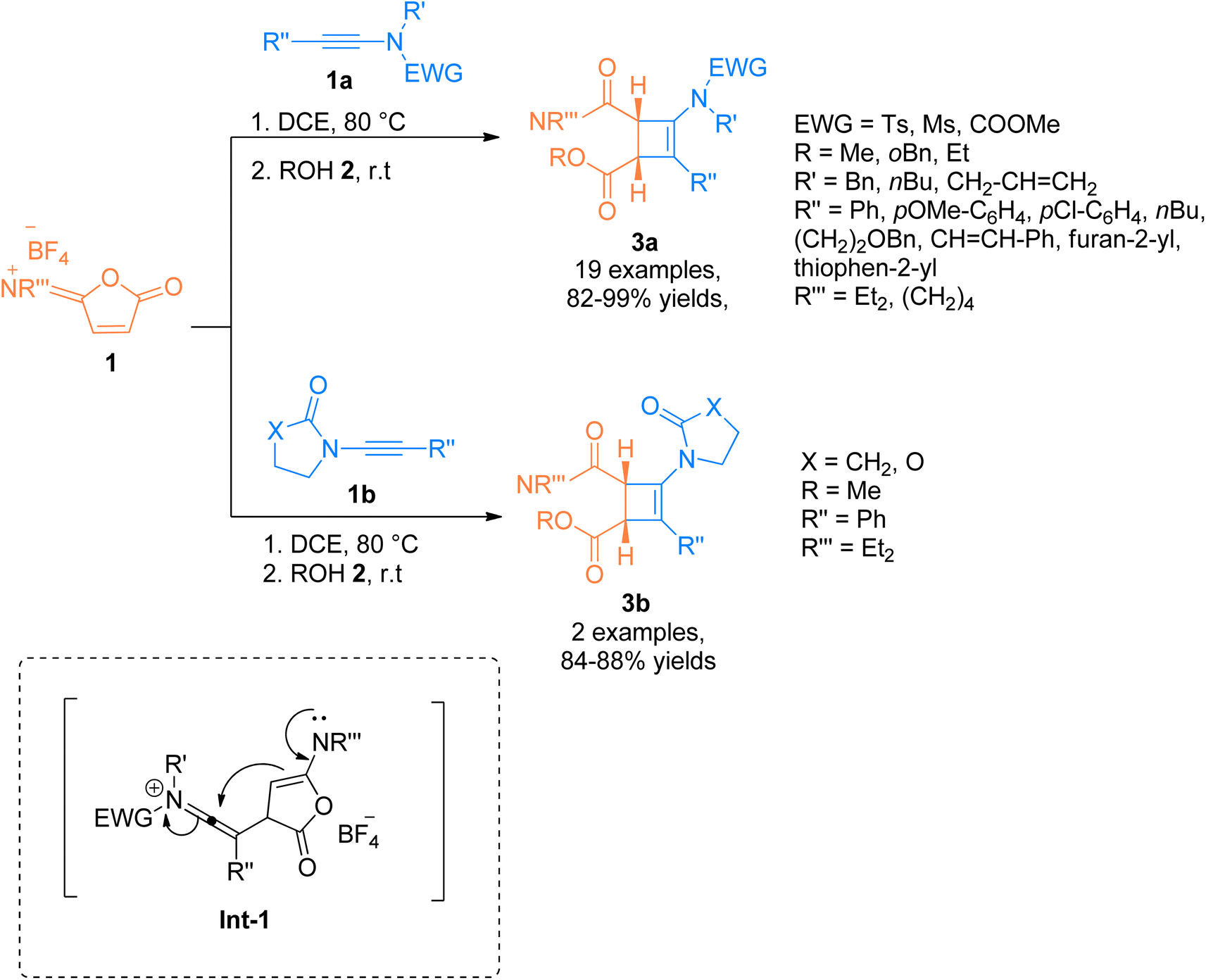
2.1.1. Synthesis of cyclobutenes via [2 + 2]-cycloaddition. There is a surge of interest to develop atom economical and facile methodologies to afford functionalized cyclobutenes via cycloaddition of ynamides.15 Yuan et al. (2014), established a unique and efficient catalyst-free synthetic route for catalyst-free, [2 + 2]-cycloaddition of ynamides. The ynamides 1a/1b reacted with cyclic isoimidium salt (α,β-unsaturated carbonyl compound) 1 (serving as electron-deficient alkene partners), and alcohol 2 via thermally driven regiospecific Ficini [2 + 2]-cycloaddition reaction via in situ generation of carbene intermediate Int-1. The reaction sequence happened using dichloroethane at 80 °C followed by the reaction with alcohol 2 at room temperature to yield stable cyclobutenamides 3a (19 examples, 82–99% yields) and 3b (2 examples, in 84–88% yields) (Scheme 1).16
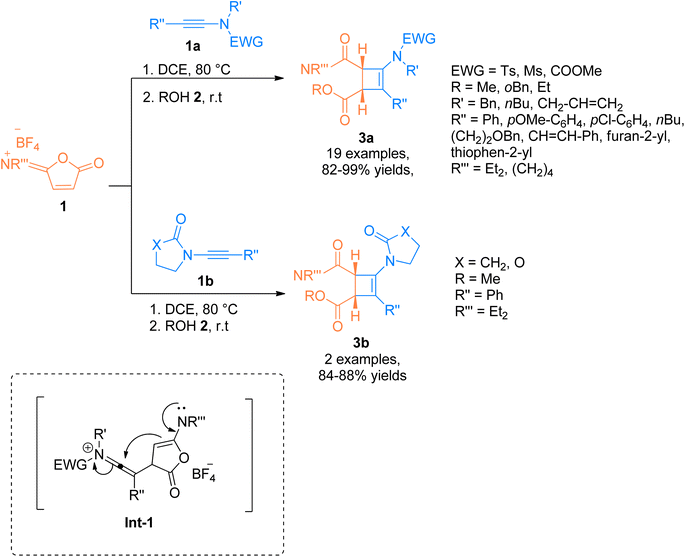 |
| | Scheme 1 Synthesis of substituted aminocyclobutenes 3a and 3b via [2 + 2]-cycloaddition of ynamides 1a and 1b and reaction partner isoimidium salt 1 and alcohol 2, respectively. | |
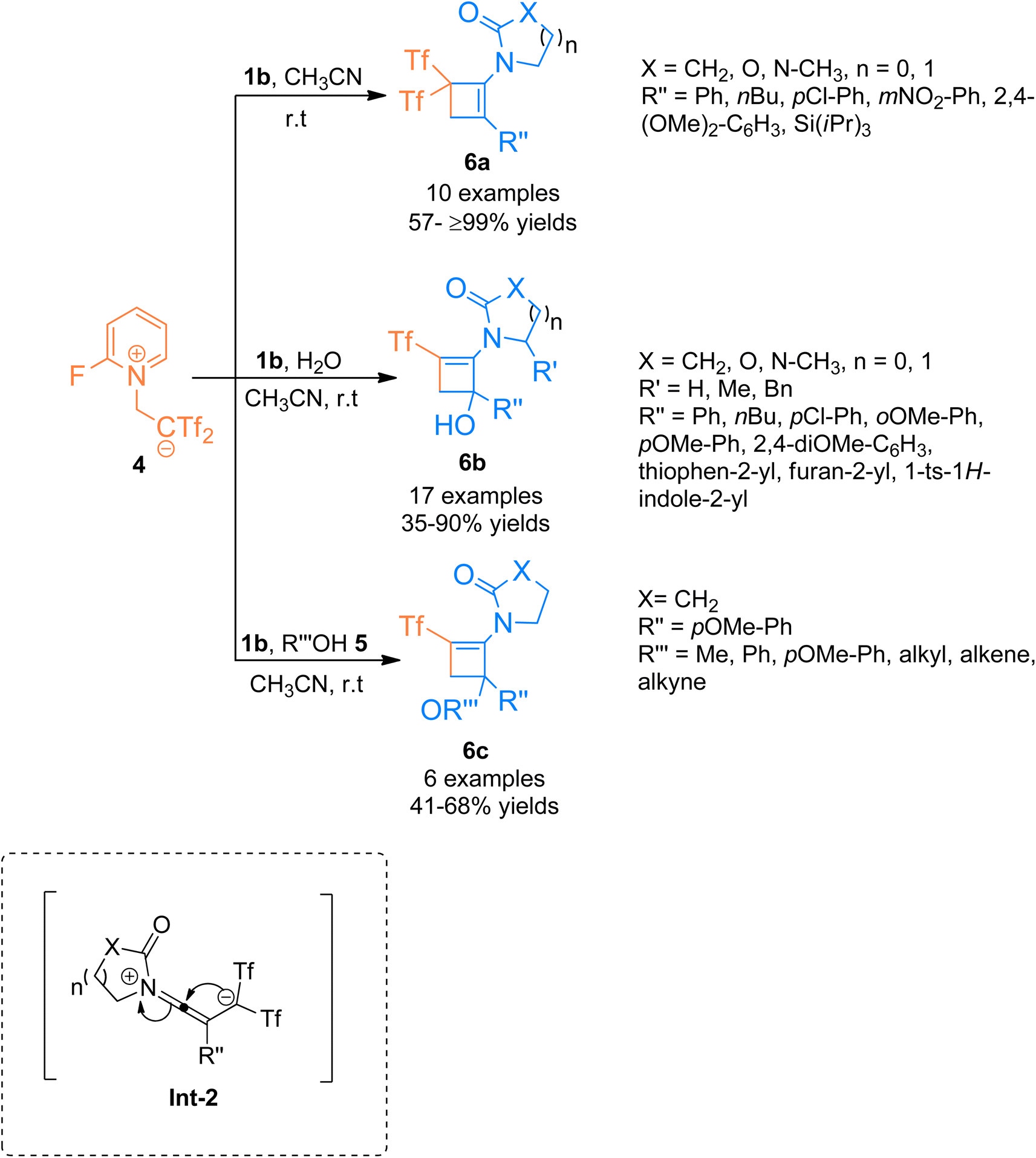
In addition to the above, the catalyst-free regiospecific [2 + 2]-cycloaddition of the ynamide 1b and zwitterion 4 as a source of bis((trifluoromethyl)sulfonyl)ethane was described by Alcaide et al. (2016), under mild conditions to afford aminocyclobutenes 6a–c. Aminocyclobutene 6a was afforded using acetonitrile at room temperature within a short reaction time of two minutes (10 examples, 57- ≥99% yields) (Scheme 2). Electron-rich aromatic scaffolds tethered at the carbon terminal of ynamide 1b switched the reactivity with the zwitterion 4 using water/alcohol 5. The synthetic route proceeded via a cyclization reaction of the intermediate Int-2 followed by a hydroalkoxylation reaction within a single step. Using acetonitrile and water solvents the cycloaddition reaction furnished aminocyclobutenyl alcohols 6b at room temperature (17 examples, 35–90% yields). While the three-component cycloaddition reaction between ynamides 1b, zwitterion 4, and alcohols 5 yielded aminocyclobutenyl ethers 6c (6 examples, 41–68% yields) using acetonitrile (anhydrous) at room temperature. This synthetic manifestation offers the advantage of catalyst-free transformation at room temperature with short reaction times.17
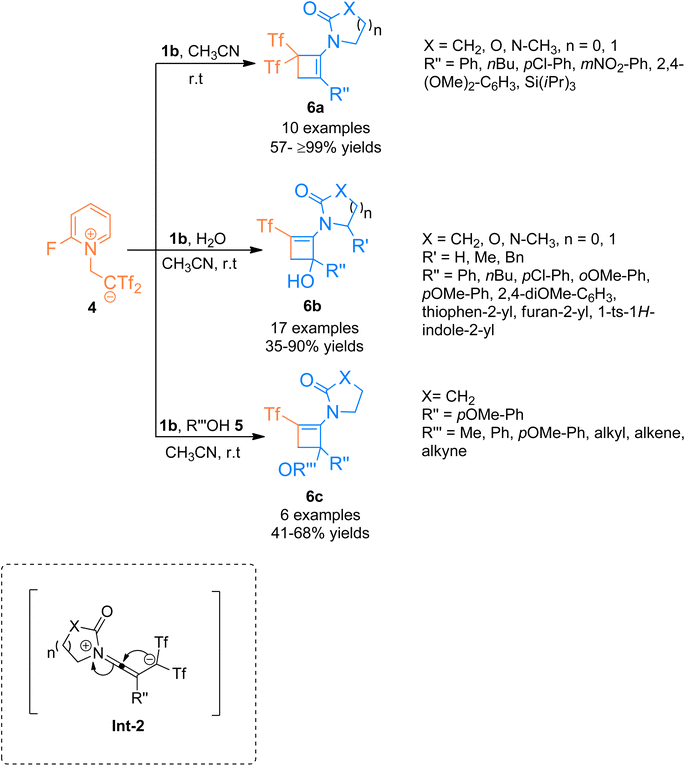 |
| | Scheme 2 Synthesis of substituted aminocyclobutenes 6a–c via [2 + 2]-cycloaddition of ynamides 1b. | |
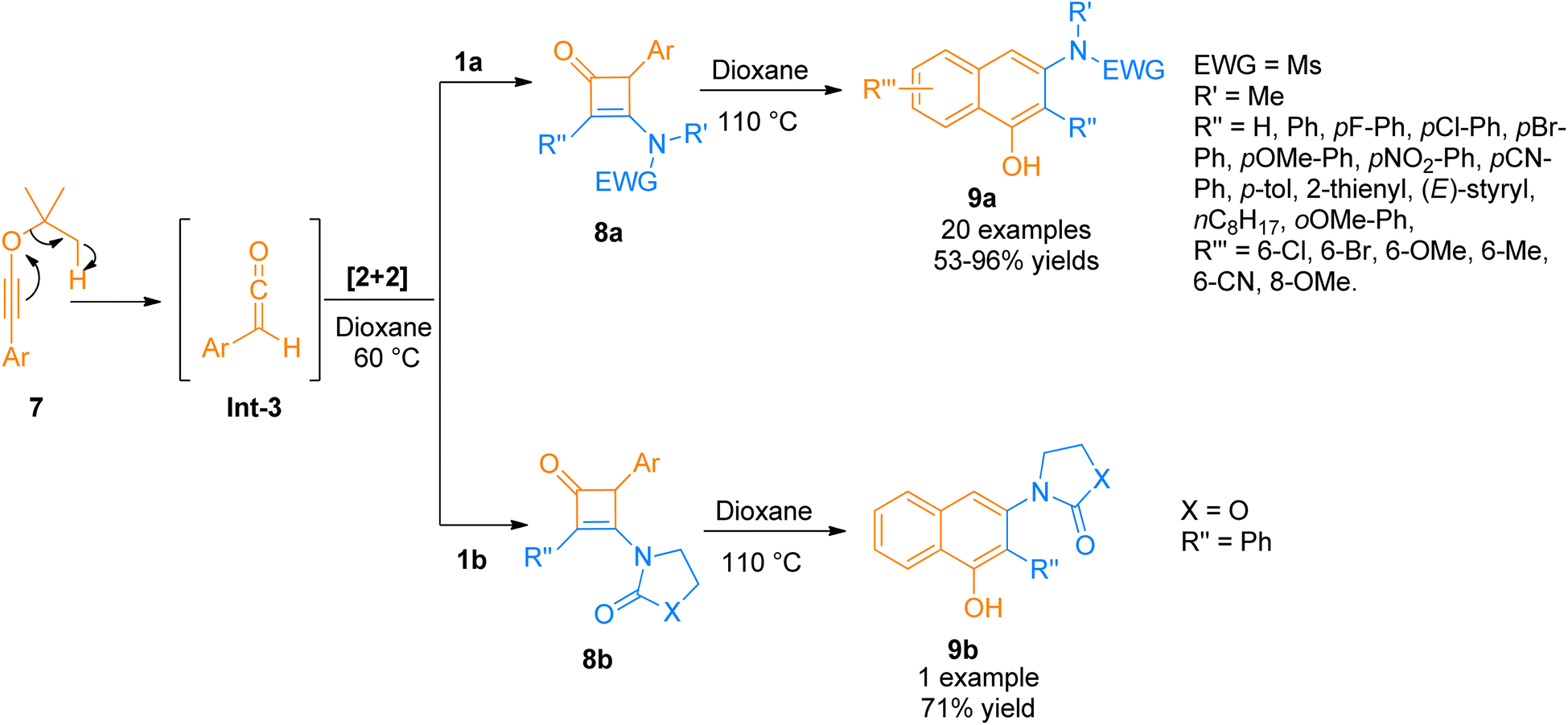
2.1.2. Synthesis of cyclobutenones via [2 + 2]-cycloaddition. Cyclobutenone is an important class of synthetic intermediates owing to their high reactivity. The ring strain associated with high reactivity makes them suitable for a variety of imperative synthetic transformations. Strained cyclobutenones having unsaturation and a carbonyl group undergo facile carbon–carbon bond cleavage.18 One of the most convenient approaches for synthesizing cyclobutenones is the [2 + 2]-cycloaddition reaction of ketene with alkynes. Hence, a novel protocol for benzannulation was developed by Bai et al. (2015), using readily available aromatic tBu-ynol ethers 7 (as a precursor of aryl ketene intermediate Int-3 via [1,5] H-shift), and the aryl ketene intermediate Int-3 react with ynamides 1a/1b to afford substituted cyclobutanone 8a/8b as key intermediate via [2 + 2]-cycloaddition using dioxane at 60 °C. Further, cyclobutanone 8a/8b underwent 4π electron electrocyclic cleavage/6 electron electrocyclic ring closure followed by a tautomerization process in dioxane solvent at 110 °C to yield 3-amino-1-naphthols 9a/9b (21 examples, 53–96% yields and 1 example, 71% yield) (Scheme 3). The synthetic route supported a wide array of ynamides functionalized with different moieties but the imidazole tethered ynamide did not undergo reaction at all under the given conditions.19
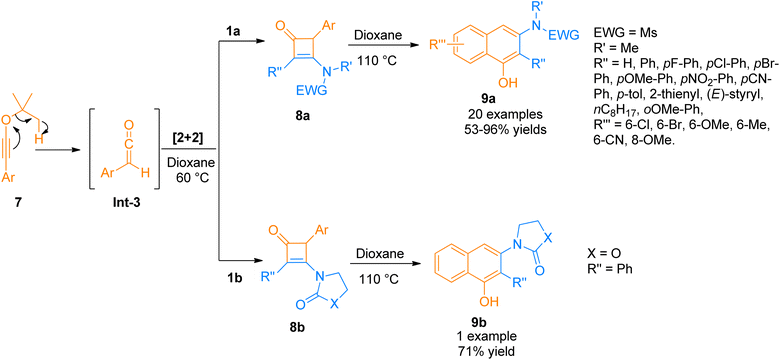 |
| | Scheme 3 Synthesis of 3-amino-1-naphthols 9a/9b via cycloaddition and benzannulation of ynamide 1a/1b and ynol-ethers 7. | |
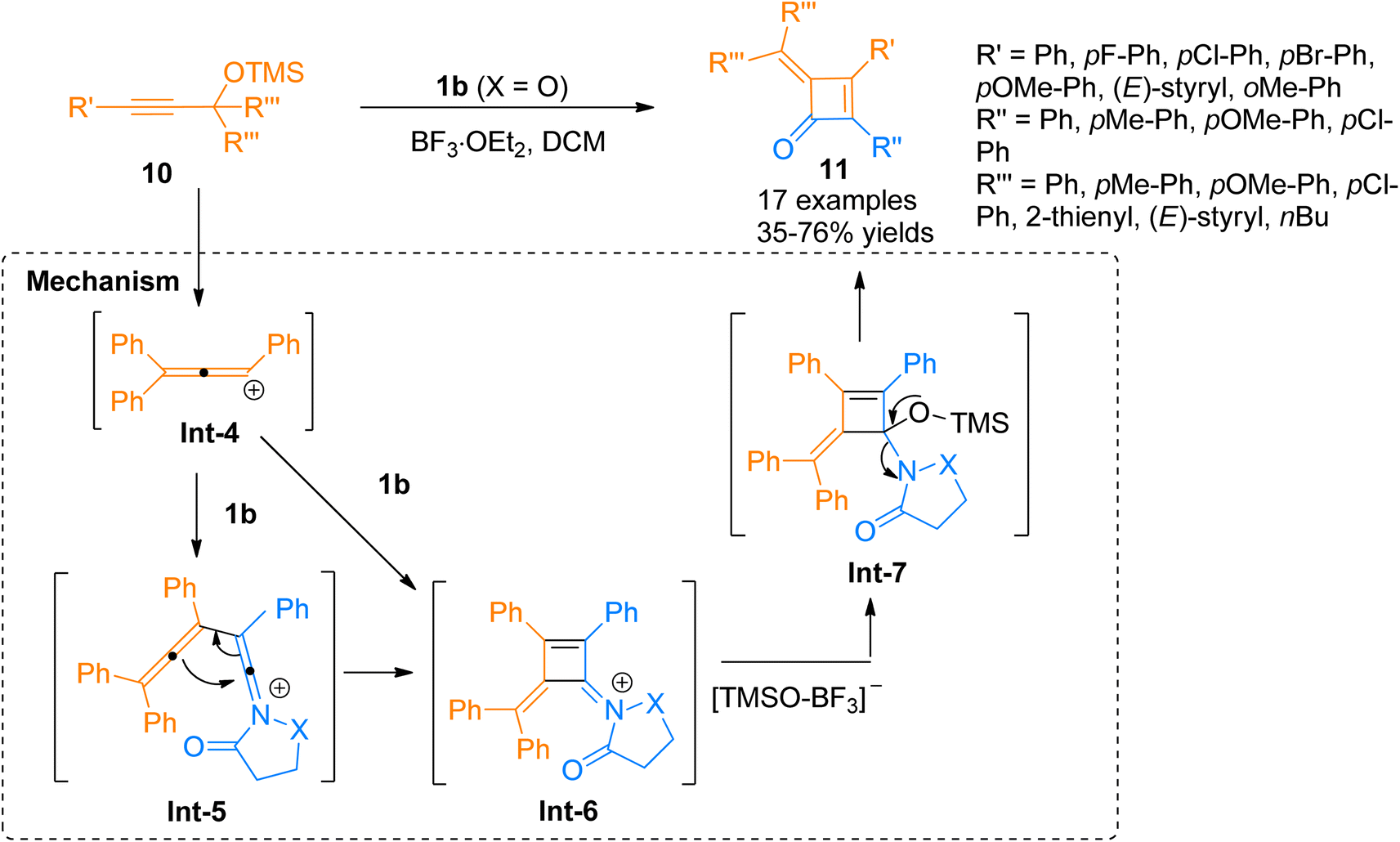
Chen et al. (2016), developed a Lewis acid-catalyzed novel, and facile protocol for regioselective [2 + 2]-cycloaddition to afford polysubstituted cyclobutenones. The developed protocol used readily available ynamides 1b and propargyl silyl ethers 10 as substrates to afford four-membered cyclic enones 16 in BF3·OEt2 catalyzed [2 + 2]-cycloaddition in dichloromethane solvent at room temperature. The reaction involved the reaction of in situ generated carbocation intermediate Int-4 (from propargyl silyl ether 10), with the ynamide 1b to either access the intermediate Int-6 via concerted [2 + 2] cycloaddition or afford keteniminium ion Int-5 which further underwent 4π electron electrocyclization to yield highly unstable intermediate Int-6. In the next step, the addition of the [TMSO-BF3]− and elimination of OTMS in subsequent steps yielded intermediate Int-7 and product 11, respectively. The synthesized alkylidene cyclobutenones 11 can further undergo valuable transformations by reacting with primary amines, water, and organometallic compounds such as organolithium compounds and Grignard reagents to yield structurally diverse products of significant importance via ring-opening and ring expansion reactions. However, the reaction was found with certain limitations as the styryl substituted ynamide afforded the product in lower yield (35%). Moreover, the reaction did not afford the title product when the R′′′ group in propargyl silyl ether was methyl or hydrogen (Scheme 4).20
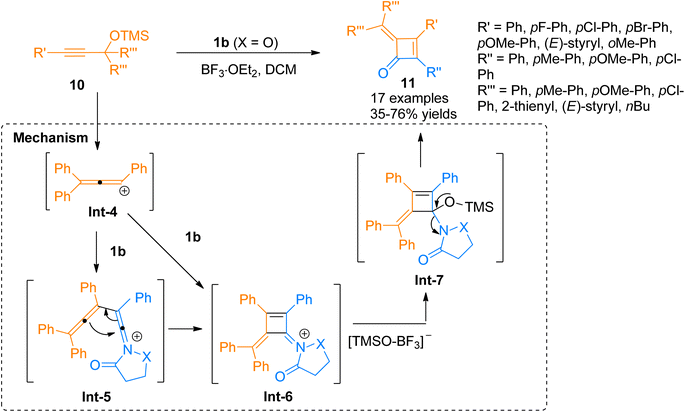 |
| | Scheme 4 Synthesis of cyclobutenones 11 via [2 + 2]-cycloaddition between ynamide 1b and propargyl silyl ether 10. | |
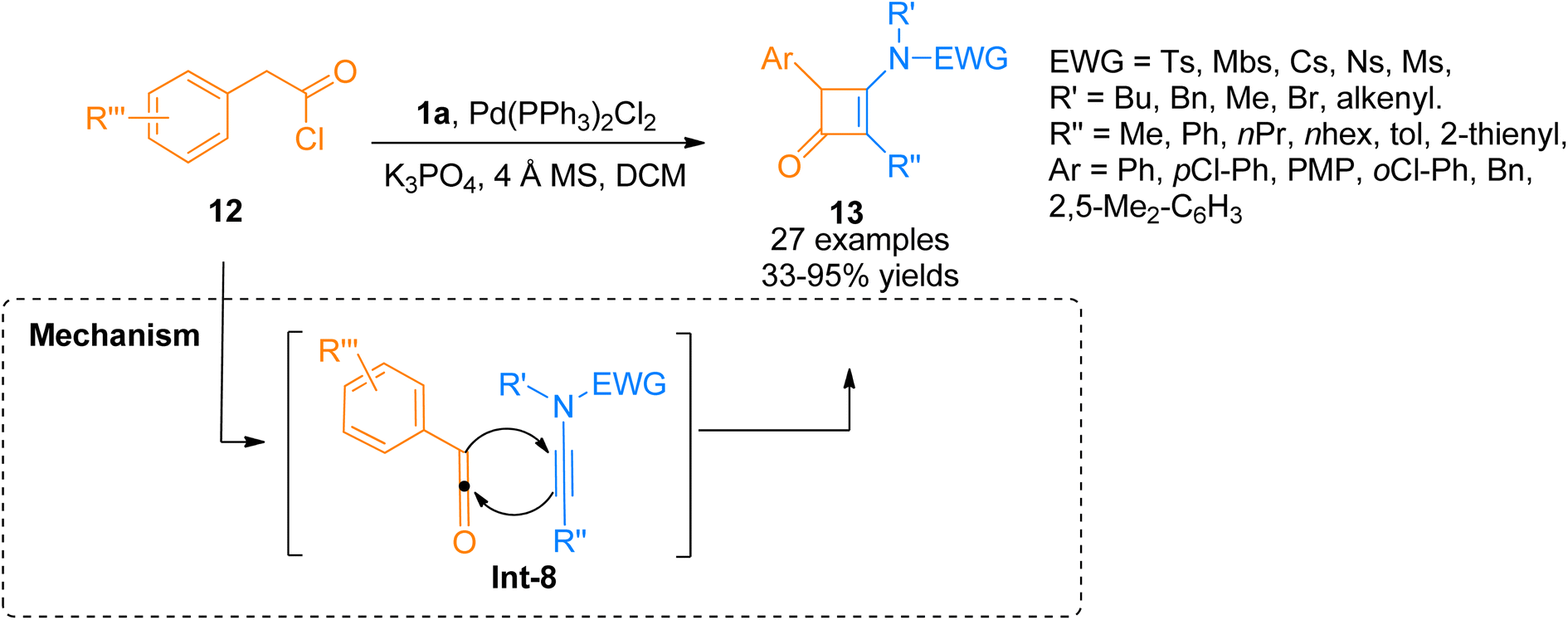
In 2018, Peng et al. used acyl chlorides in a palladium-catalyzed cyclization reaction taking ynamides as reaction partners to realize the synthesis of 3-aminocyclobutenones. Acyl chloride 12 generated monosubstituted ketenes in situ via dehydrohalogenation reaction. The in situ generated monosubstituted ketenes react with ynamides 1a via a [2 + 2]-cycloaddition in a concerted step to yield 3-aminocyclobutenones 13 via intermediate Int-8 (Scheme 5). The reaction was carried out in dichloromethane solvent by maintaining the temperature at 60 °C under a nitrogen atmosphere (27 examples, 33–95% yield range). The generated 3-aminocyclobutenones could be subsequently transformed via a pericyclic ring-opening/ring-closure reaction catalyzed by heat to generate synthetically important scaffolds 3-amino-1-naphthols in promising yields.21
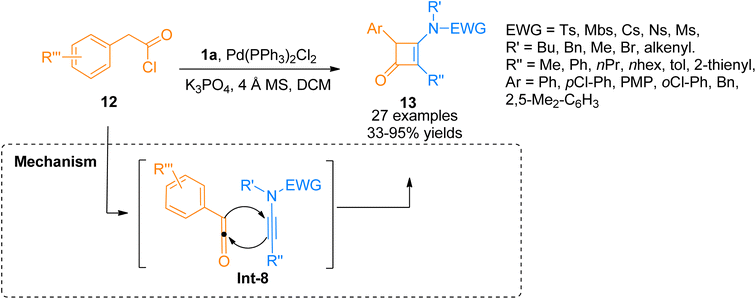 |
| | Scheme 5 Synthesis of cyclobutenones 13 via [2 + 2]-cycloaddition of ynamide 1a and acyl chlorides 12. | |
2.1.3. Synthesis of quinolines via [2 + 2]-cycloaddition. A tandem regiocontrolled benzannulation strategy for the construction of highly substituted quinolones 23 via a single-step reaction of N-propargyl functionalized ynamide 1c and cyclobutenones 14/diaoketones 15 was described by Willumstad et al. (2015). The benzannulation initiates with the thermolysis or irradiation of the cyclobutenone and the reaction proceeds via a cascade strategy involving four types of pericyclic transformations. Cascade reaction involved the 4π electron electrocyclic ring-opening reaction of cyclobutenone derivative 14 or photochemical Wolff rearrangement of α-diazo ketone derivative 15 to generate vinyl ketene intermediate Int-9, followed by [2 + 2]-cycloaddition to afford new cyclobutenone intermediate Int-10. Cyclobutanone intermediate Int-10 readily underwent a reversible 4π electron electrocyclic ring-opening reaction to generate dienyl ketene intermediate Int-11 that cyclize via 6π electron electrocyclic ring closure to form highly substituted aniline derivatives 16 in a regiocontrolled manner. The substituted aniline derivatives 16 (4 examples, 55–95% yields) with a suitable electrophile such as iodine underwent Larock cyclization to afford highly functionalized quinolones 17 (4 examples, 40–87% yields) (Scheme 6).22
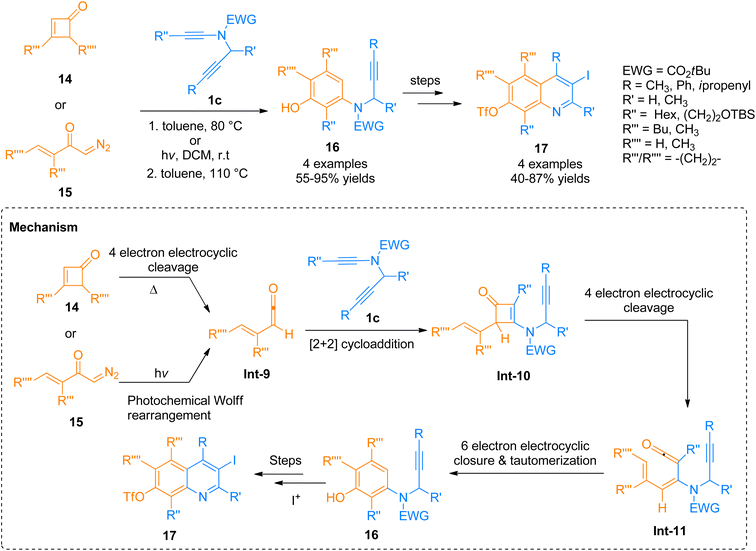 |
| | Scheme 6 Synthesis of substituted quinolones 17 via [2 + 2]-cycloaddition of ynamide 1c with cyclobutenones 14 or diaoketones 15. | |
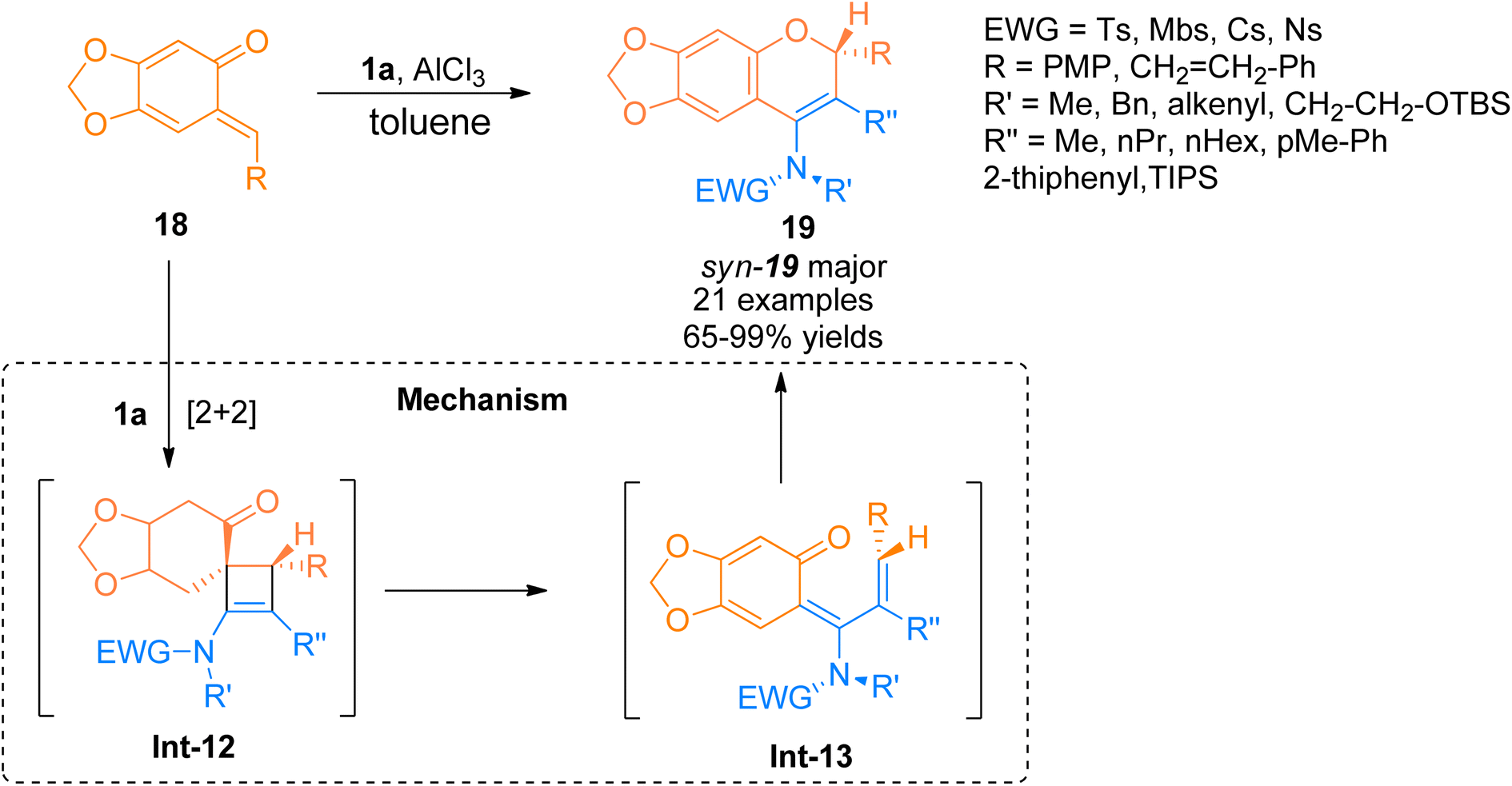
2.1.4. Synthesis of 4-amino-2H chromenes via [2 + 2]-cycloaddition. For the construction of these amino substituted chromene scaffolds, only a few studies have been reported taking ynamides as starting materials. In this respect, Yang et al. (2016) reported the facile route for the annulation reaction of terminally substituted ynamide 1a with o-quinone methides 18 with excellent control over central as well as axial chirality. The reaction sequence followed the AlCl3 promoted [2 + 2]-cycloaddition reaction to yield spiro cyclic intermediate Int-12, which underwent 4π electron electrocyclic torquoselective ring-opening reaction to yield Int-13. Further, the 6π-electrocyclic ring closure of Int-13 afforded the diastereomeric 4-amino-2H-chromenes 19 (21 examples, 65–99% yield range) with high stereoselectivity in toluene solvent at −40 °C (Scheme 7). Terminal substitution at ynamides plays a significant role in determining the [2 + 2]-cycloaddition or [4 + 2]-cycloaddition pathway for the construction of 4-amino-2H chromenes and 2-amino-4H-chromenes, respectively. The terminally substituted ynamides selectively underwent [2 + 2] cycloaddition while terminally unsubstituted ynamides underwent [4 + 2] cycloaddition. The reaction provided a facile route to construct non-biaryl atropimers which can be further transformed into novel atropisomers.23
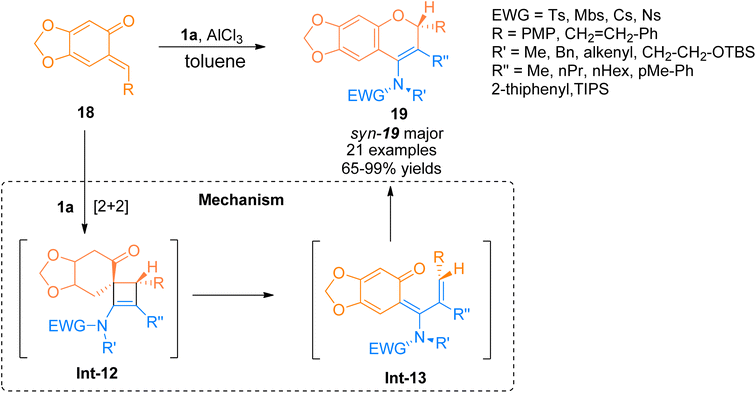 |
| | Scheme 7 Synthesis of 4-amino-2H-chromenes 19 via [2 + 2]-cycloaddition of ynamide 1a and o-quinone methides 18. | |
2.2. [3 + 2]-cycloaddition
2.2.1. Synthesis of indole derivatives via [3 + 2]-cycloaddition.
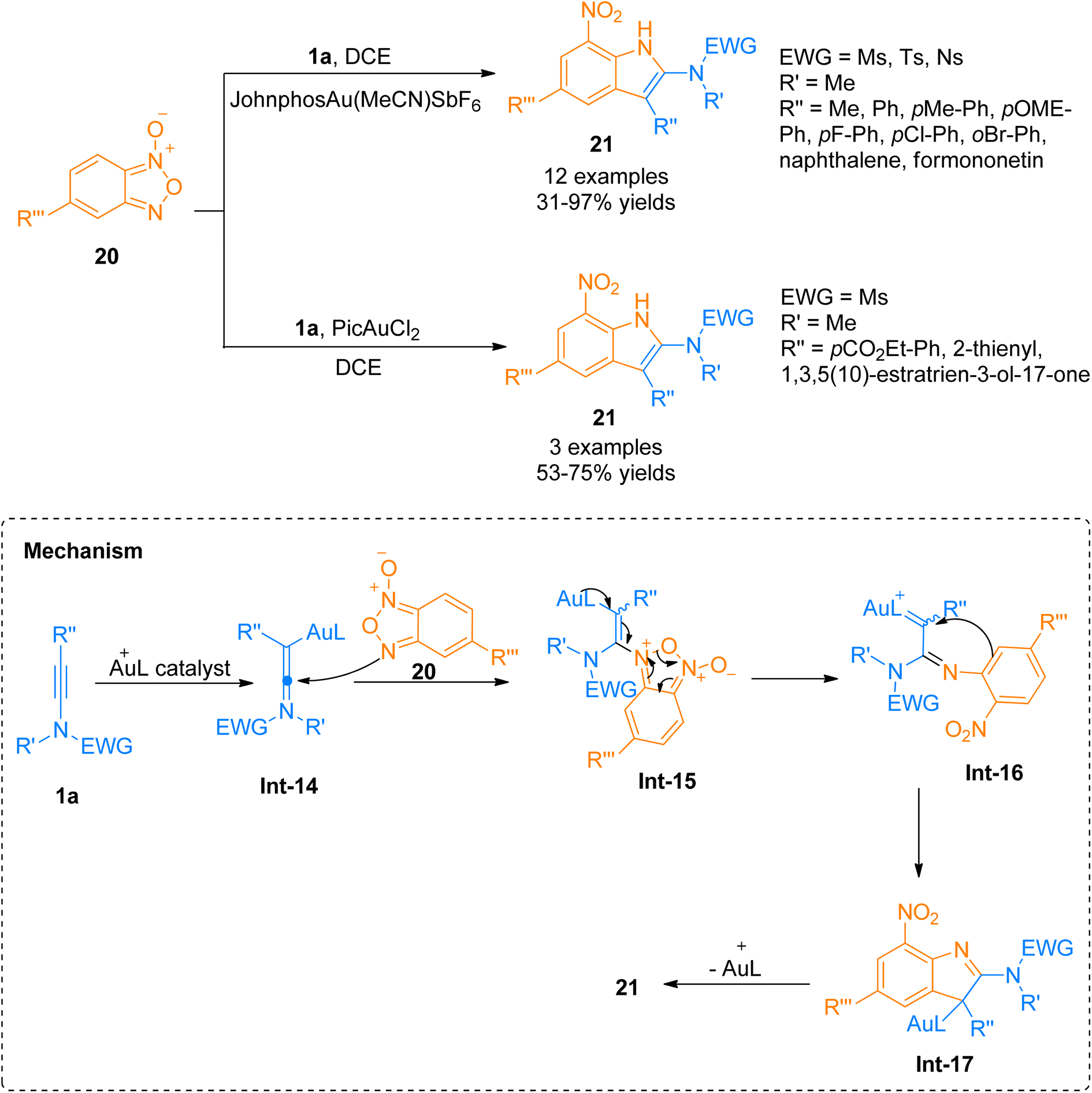
2.2.1.1. Synthesis of 2-aminoindoles. Among the N-heterocycles, synthesis of 7-nitroindoles has been previously reported by limited synthetic routes that have serious drawbacks such as harsh reaction conditions and narrow substrate scope. Moreover, the achievement of higher regioselectivity is one of the major challenges.24–26 In 2019, Xu et al. the gold(I) complex as catalyst was found to be an efficient catalyst to afford α-imino-Au-carbene intermediate taking benzofuran N-oxide 20 as a precursor which further undergoes [3 + 2]-cycloaddition with ynamide 1a to achieve functionalized 7-nitroindoles 21 as reported by Xu et al. The optimized parameters involved the use of (acetonitrile)[(2-biphenyl)di-tert-butylphosphine]gold(I)hexafluoroantimonate (Johnphos Au(MeCN)SbF6) catalyst (5 mol%), dichloroethane, and 25 °C temperature. The proposed reaction mechanism (based on the controlled reactions) involves the formation of ynamide derived advanced intermediate, keteniminium ion Int-14 that reacts with the N-oxide 20 to afford Int-15. Int-15 transforms to α-imino-gold-carbene intermediate Int-16 via ring fragmentation reaction. Nucleophilic attack of the phenyl ring on gold carbene Int-16 furnished Int-17. It was followed by subsequent aromatization and denaturation of Int-17 which provided access to 7-nitroindole 21. This reaction with moderate substrate scope afforded required substituted 7-nitroindoles 21 (12 examples, 31–97%) (Scheme 8). However, some of the ynamides i.e. heteroaryl substituted ynamide, under standard conditions, afforded several side products, but catalyst switching to PicAuCl2 (5 mol%) obtained the products in promising yields (3 examples, 53–75% yields).27
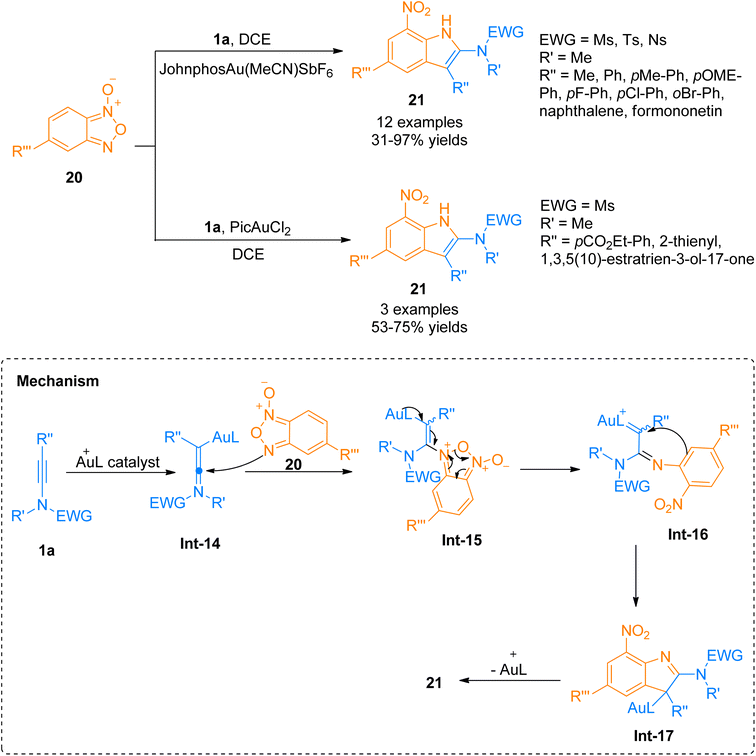 |
| | Scheme 8 Synthesis of 7-nitroindoles 21 via [3 + 2]-cycloaddition of ynamides 1a with benzofuran N-oxide 20. | |
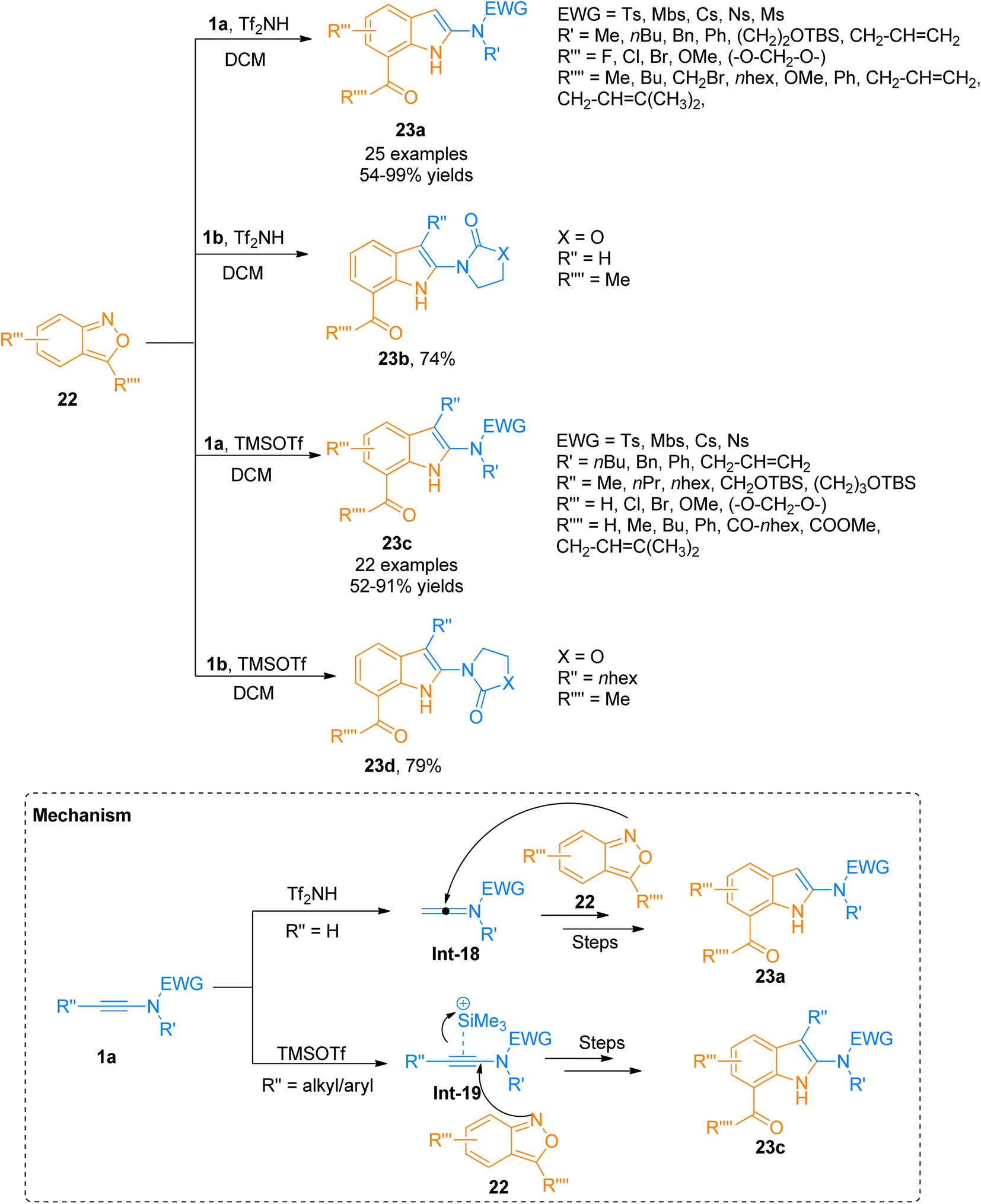
Although many powerful synthetic routes to realize the synthesis of indole structural motifs have been developed, approaches for the efficient synthesis of 2-aminoindoles are limited. Zhang et al. (2021), recently developed a metal-free protocol for [3 + 2]-cycloaddition of the ynamide 1a/1b with anthranils 22 that provided a flexible and facile route for the synthesis of functionalized 2-aminoindoles 23a–d (Scheme 9). The terminally unsubstituted ynamides 1a/1b underwent this transformation using Tf2NH (0.5 equiv.) in dichloromethane as solvent at room temperature to afford 23a (25 examples, 54–99% yields)/23b (1 example, 74% yield). While terminally substituted ynamide 1a/1b afforded the products 23c (23 examples, 52–91% yields)/23d (1 example, 79% yield) via TMSOTf (0.5 equiv.) catalyzed cycloaddition reaction using dichloromethane. The reaction afforded the products in excellent regioselectivity under mild conditions (temperature −20 to −50 °C). According to the postulated mechanism, the reaction initiates with the electrophilic addition of a proton to the ynamide 1a that generates keteniminium ion intermediate Int-18, while in the case of terminally substituted ynamide, the silicon π-alkyne species Int-19 generates as a result of coordination of TMSOTf catalyst with ynamide alkyne core. Further, in the next step anthranil 22 traps both intermediates which are followed by the same reaction sequence of ring fragmentation, aromatization, and hydride shift to afford their respective final products. The metal-free protocol for annulation provides an efficient route for the interesting synthesis of 2-aminoindoles 23a/23c, which are building blocks for the construction of diverse 2-aminoindolyl frameworks (Scheme 9).28
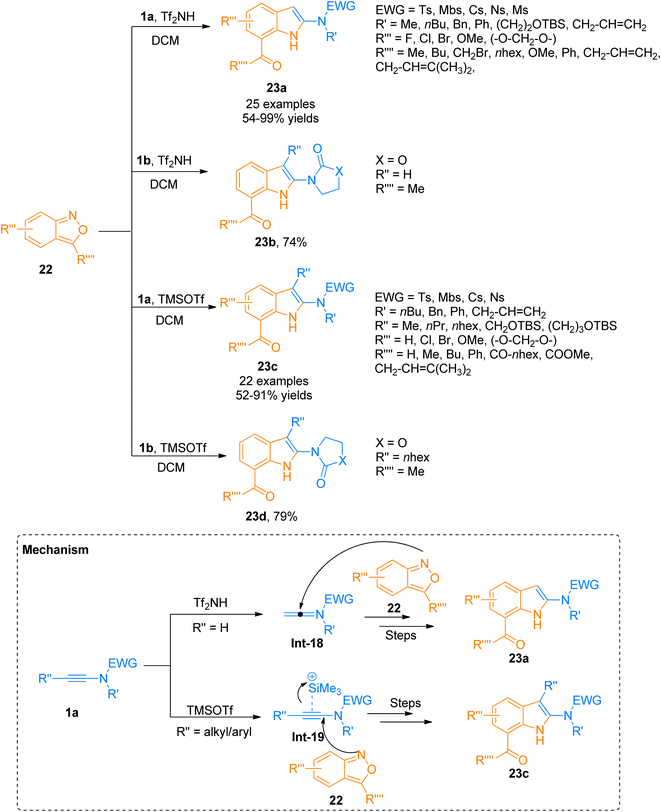 |
| | Scheme 9 Synthesis of 2-aminoindoles 23a–d via [3 + 2]-cycloaddition of ynamide 1a,b with anthranils 22. | |
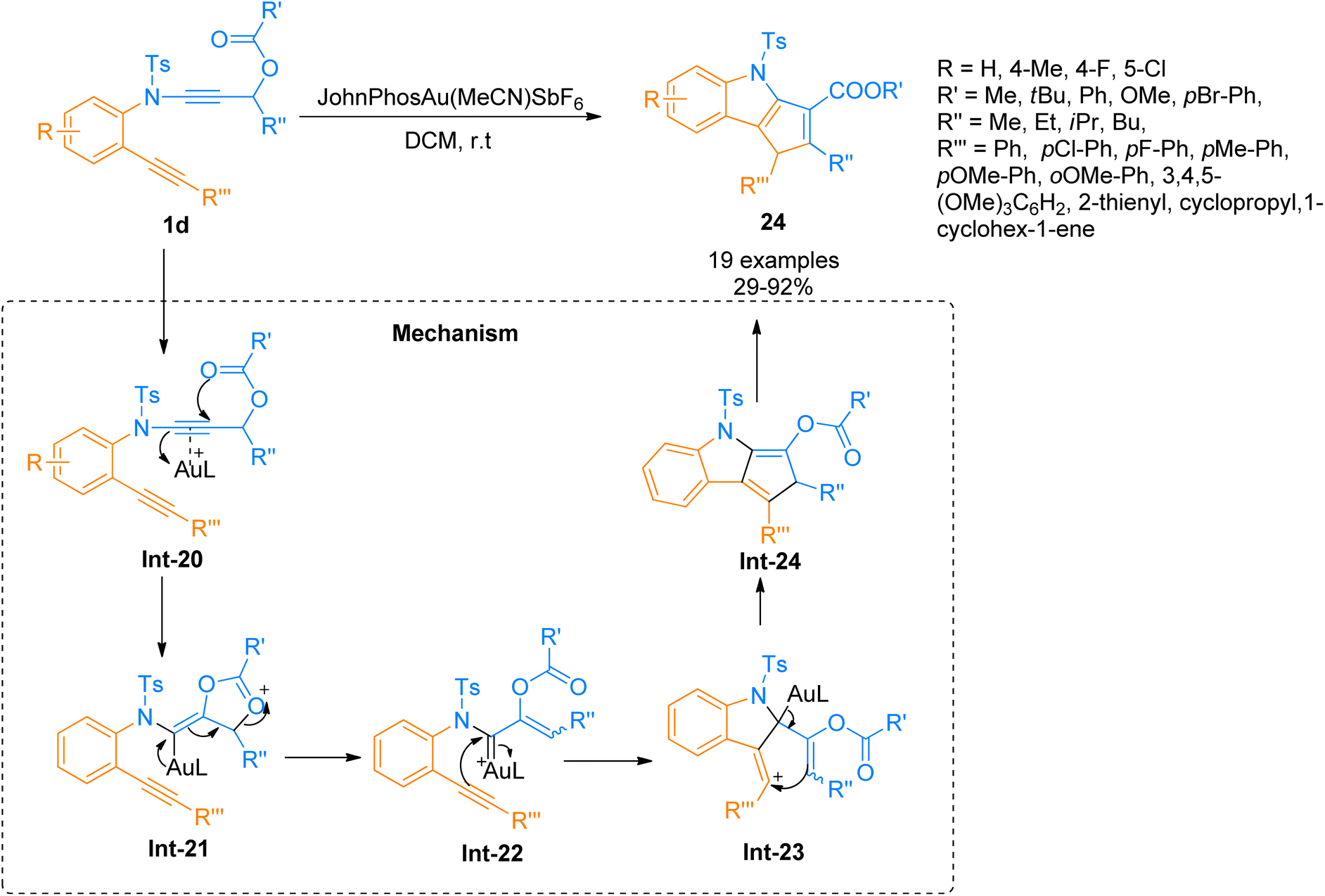
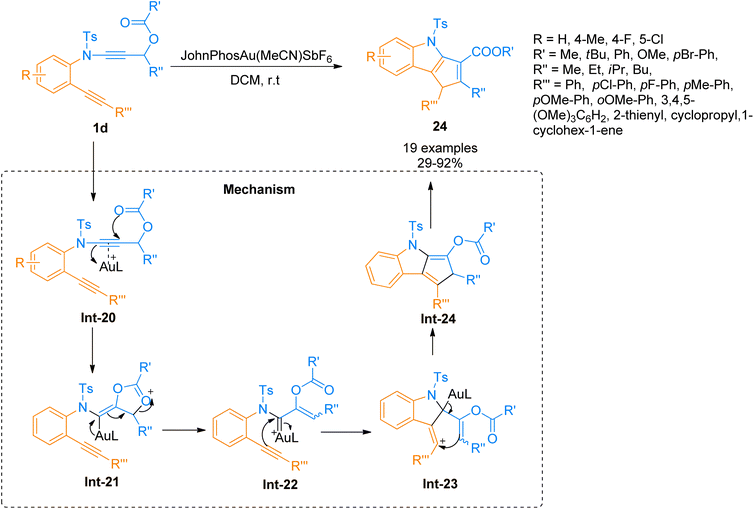 |
| | Scheme 10 Synthesis of cyclopenta[b]indoles 24 via intramolecular [3 + 2]-cycloaddition of ynamide 1d. | |
2.2.2. Synthesis of pyrroles via [3 + 2]-cycloaddition. The conventional multistep methods such as Paal Knorr synthesis and Hantzch reaction for the construction of pyrrole rings have various drawbacks such as poor step economy, low yields, and limited functional group compatibility. In this regard, the cycloaddition reactions of the ynamides and advanced intermediates derived from ynamides are preferable alternatives for the construction of functionalized pyrrole scaffolds.30–33
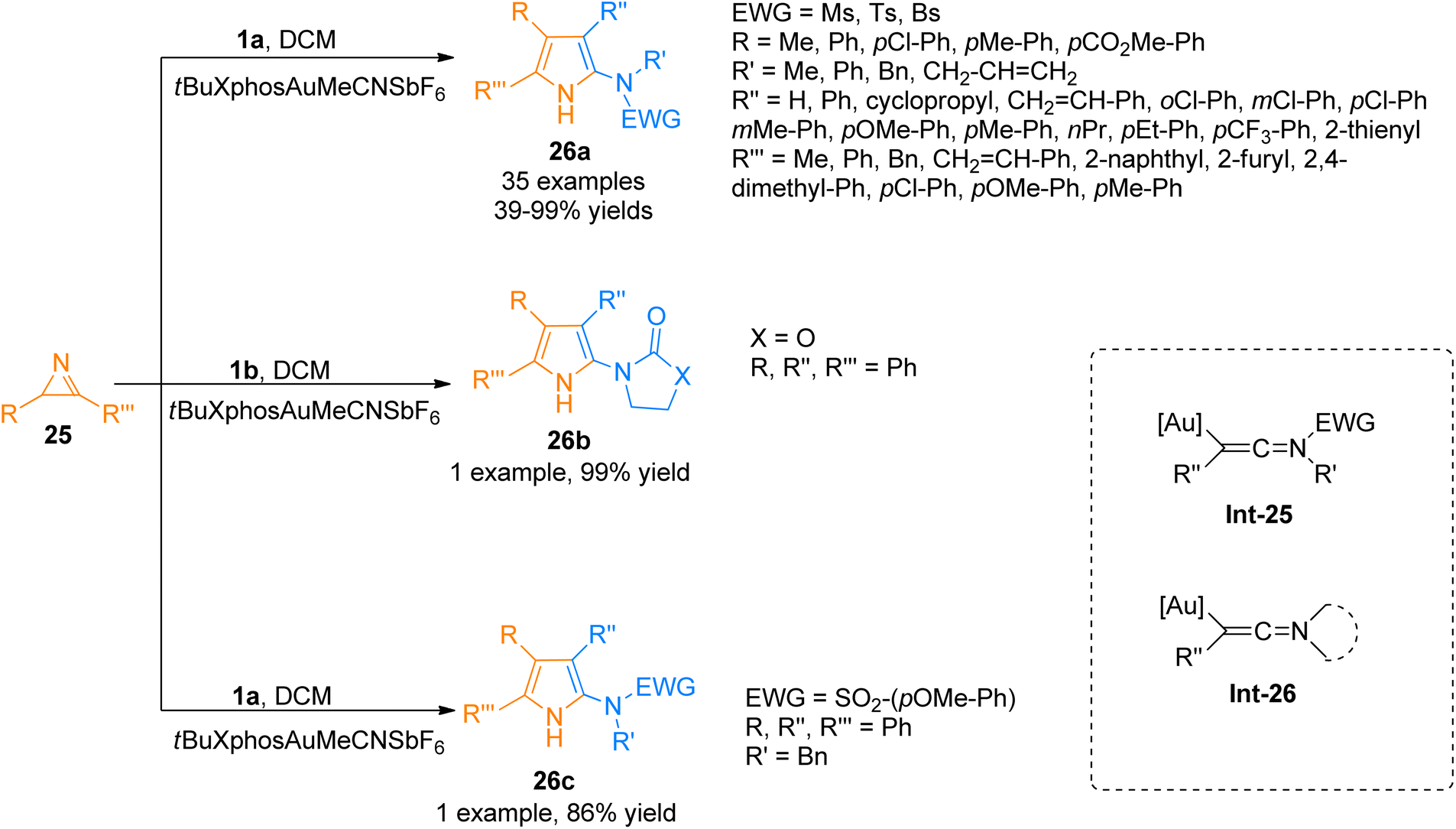
2.2.2.1. Synthesis of polysubstituted pyrroles. Pyrroles are considered versatile building blocks and are used as precursors for the construction of pyrrolepyrimidine, pyrrolotriazine, and pyrrolopyridine analogs in organic synthesis.34,35 Zhu et al. (2015), developed an efficient method of [3 + 2]-cycloaddition reaction of ynamides 1a with 2H-azirines 25 that involved intermolecular nitrene transfer using the gold catalyst for the synthesis of highly substituted pyrroles 26a (35 examples, 39–99% yields). The ynamide containing cyclic electron withdrawing group 1b and substituted sulfonyl group 1a reacted with 2H-azirines 25 to afford 26b (99% yield) and 26c (86% yield). The reaction provides a facile route through which the ynamides 1a/1b activation by gold catalyst yield keteniminium intermediate Int-25/Int-26 which is highly electrophilic and reacts with 32 to realize the transformation into respective products 26a–c (Scheme 11). The reaction was realized using mild reaction conditions, and low catalyst loading. It afforded a broad range of polysubstituted pyrroles 26a–c with higher atom economy. However, the ynamide bearing Ns electron withdrawing group was not found compatible with the said cycloaddition reaction.36
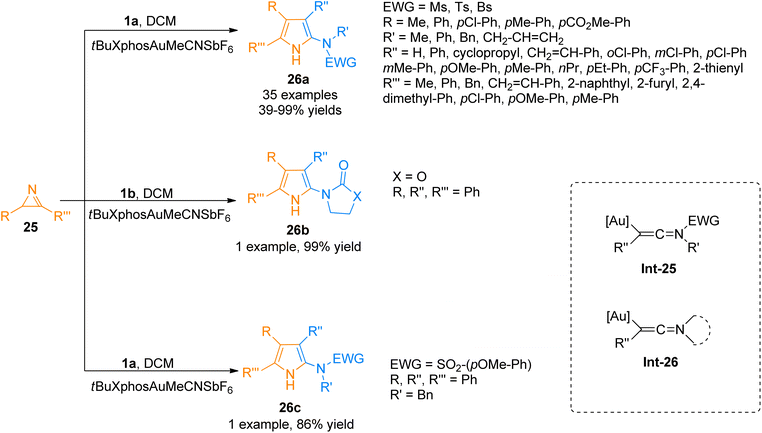 |
| | Scheme 11 Synthesis of polysubstituted pyrroles 26a–c via [3 + 2]-cycloaddition of ynamides 1a/1b with 2H-azirines 25. | |
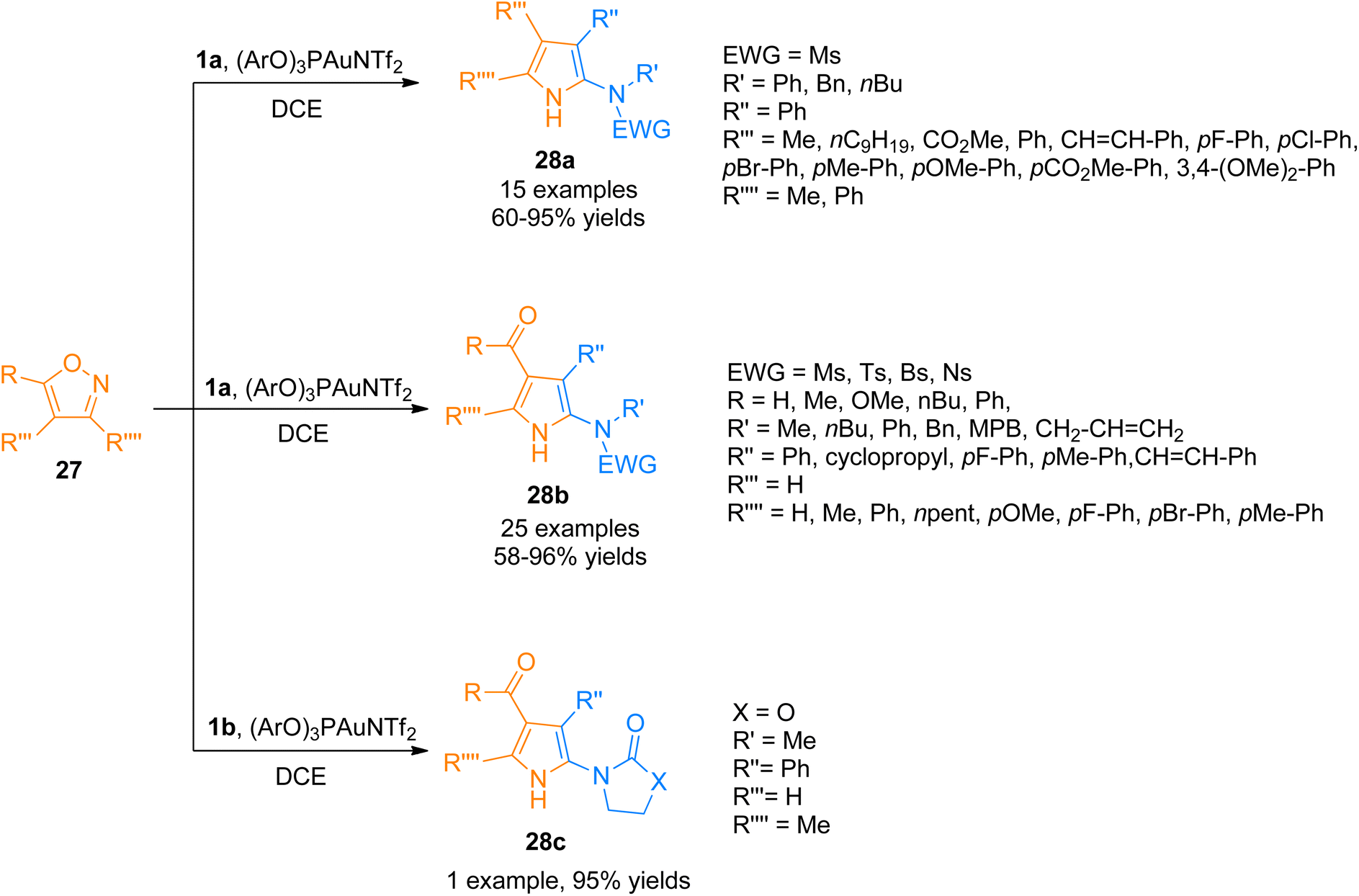
Moreover, the generation of the gold-carbene intermediate in the gold catalyzed intermolecular reaction of alkynes and nucleophiles has been of considerable interest but the developed protocols have the disadvantage of low atom economy.37–40 Zhou et al. (2015), developed the atom economical approach to construct highly functionalized 2-aminopyrroles 28a–c by formal [3 + 2]-cycloaddition reaction of ynamides 1a/1b with isoxazoles 27 using a gold(I) catalyst (Scheme 12). The reaction proceeded by the generation of α-amino-gold-carbene intermediate providing a novel and atom-economical route to access gold carbene intermediates using (ArO)3PAuNTf2 (5 mol%) (Ar = 2,4-di-tert-butylphenyl) catalyst, dichloroethane solvent at 80 °C. The developed protocol can introduce up to four substituents to the pyrrole ring 28a (15 examples, 60–95% yields), 28b (25 examples, 58–96% yields), 28c (1 example, 95% yields).41
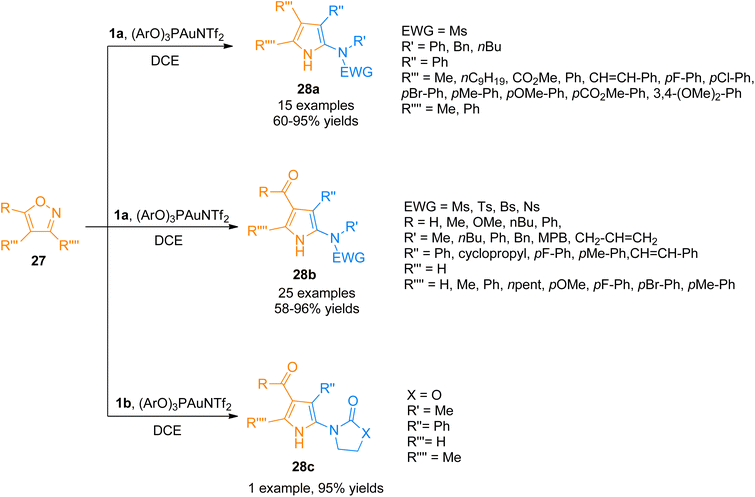 |
| | Scheme 12 Synthesis of polysubstituted 2-aminopyrroles 28a–c via [3 + 2]-cycloaddition of ynamides 1a/1b with isoxazoles 27. | |
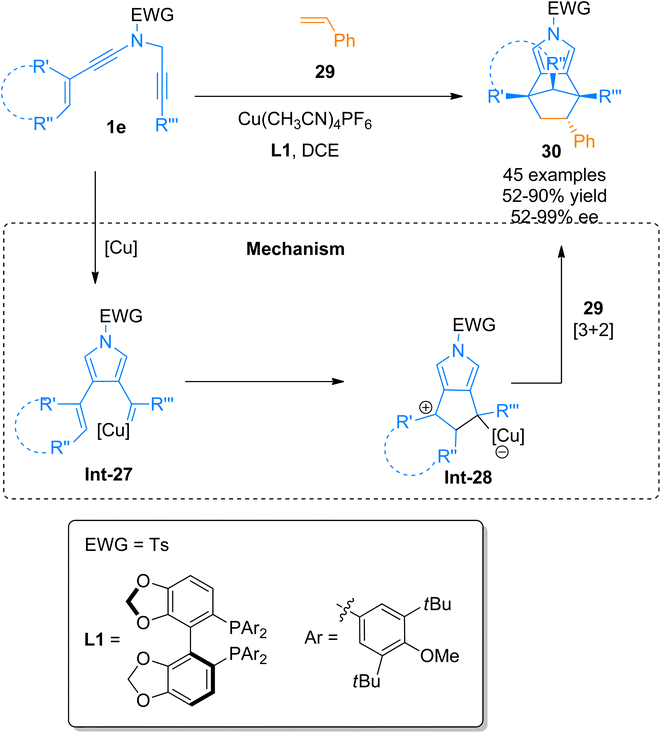 |
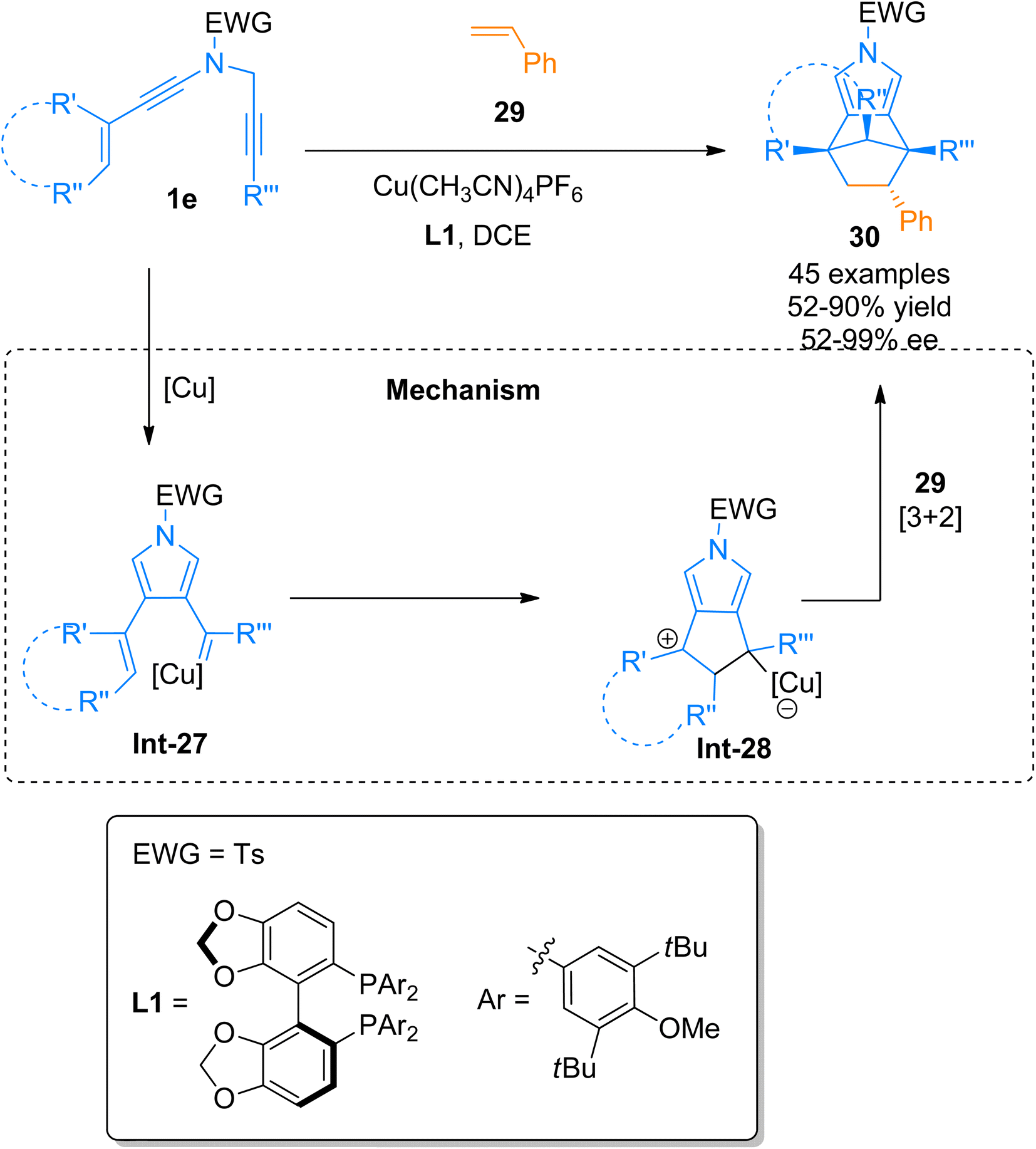
| | Scheme 13 Synthesis of pyrrole-fused scaffolds 30 via [3 + 2]-cycloaddition of ynamides 1e and alkenes 29. | |
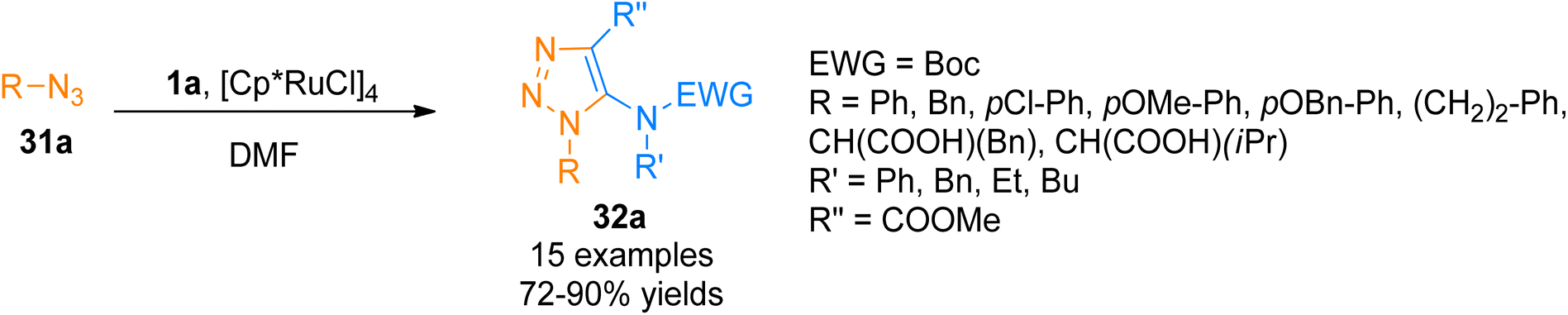
2.2.3. Synthesis of triazoles via [3 + 2]-cycloaddition. Ferrini et al. (2015), devised a protocol for [3 + 2]-cycloaddition reaction of ynamides 1a and azides 31a using ruthenium metal catalyst to yield protected triazoles 32a containing amino acid in a regio-controlled manner (Scheme 14).45 Further the work on Rhodium catalyzed azide–alkyne 1,3-dipolar cycloaddition was extended and modified by Liao et al. (2017).46 The reaction afforded functionalized 5-aminotriazoles via regioselective 1,3-dipolar cycloaddition reaction of ynamide and azides in acetonitrile solvent at room temperature or 100 °C in an open flask. The transformation also involves the [Rh(CO2)Cl]2 catalyzed cycloaddition of internal ynamides that yielded aminotriazoles in excellent yield and regioselectivity. The developed methodology expanded the substrate scope of the methodology reported by Ferrini et al. (2015)45 by employing a range of substituted azides with diversely functionalized ynamides bearing Ms, Ts, and Bs electron withdrawing groups (31 examples, 17–98% yield range).46
 |
| | Scheme 14 Synthesis of 1,2,3-triazoles 32a via [3 + 2]-cycloaddition of ynamides 1a with azide 31a. | |
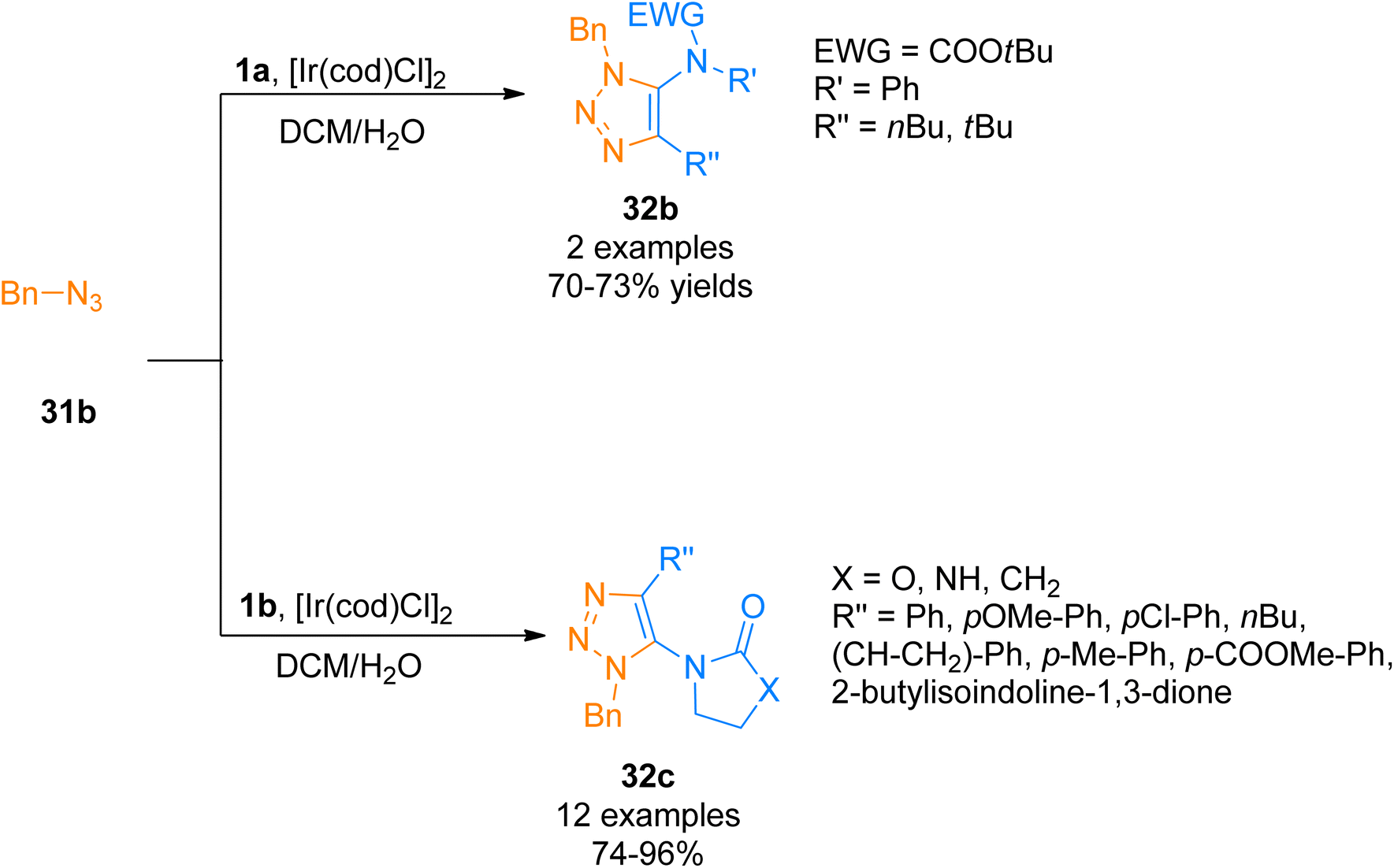
Similarly, Song and Zheng, (2017), developed an iridium-catalyzed regioselective route to access functionalized 5-amido-1,2,3-triazoles 32b–c via [3 + 2]-cycloaddition of azide 31b and ynamide 1a/1b by addition of [Ir(cod)Cl]2 catalysts in dichloromethane or water as a solvent in open air flask at room temperature. The methodology achieved excellent regioselectivity (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0) via iridium-ynamide coordination complex that reverses the regioselectivities of Cu-catalyzed azide–alkyne cycloaddition while enhancing the regioselectivities of Ru-catalyzed azide–alkyne cycloaddition. The advantages of the developed methodology involve low catalyst toxicity, room temperature, insensitivity to oxygen and water, compatibility with organic, aqueous as well as biological reaction environments, and higher yields (14 examples, 70–96% yield range) (Scheme 15).47
:0) via iridium-ynamide coordination complex that reverses the regioselectivities of Cu-catalyzed azide–alkyne cycloaddition while enhancing the regioselectivities of Ru-catalyzed azide–alkyne cycloaddition. The advantages of the developed methodology involve low catalyst toxicity, room temperature, insensitivity to oxygen and water, compatibility with organic, aqueous as well as biological reaction environments, and higher yields (14 examples, 70–96% yield range) (Scheme 15).47
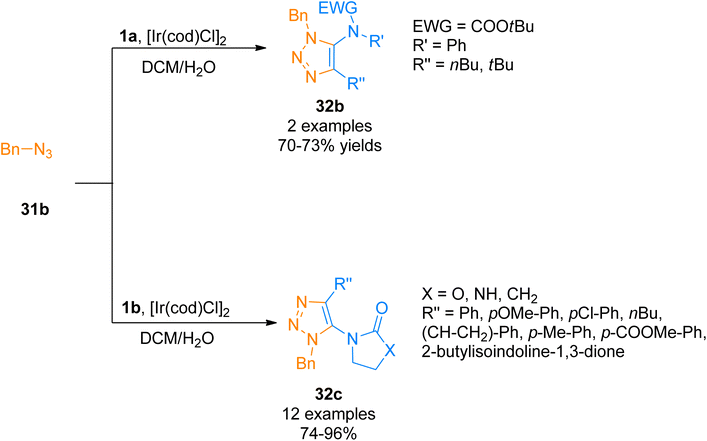 |
| | Scheme 15 Synthesis of 1,2,3-triazoles 32b/32c via [3 + 2]-cycloaddition of ynamides 1a/1b with azide 31b. | |
2.2.4. Synthesis of imidazole-fused oxo-substituted frameworks via [3 + 2]-cycloaddition.
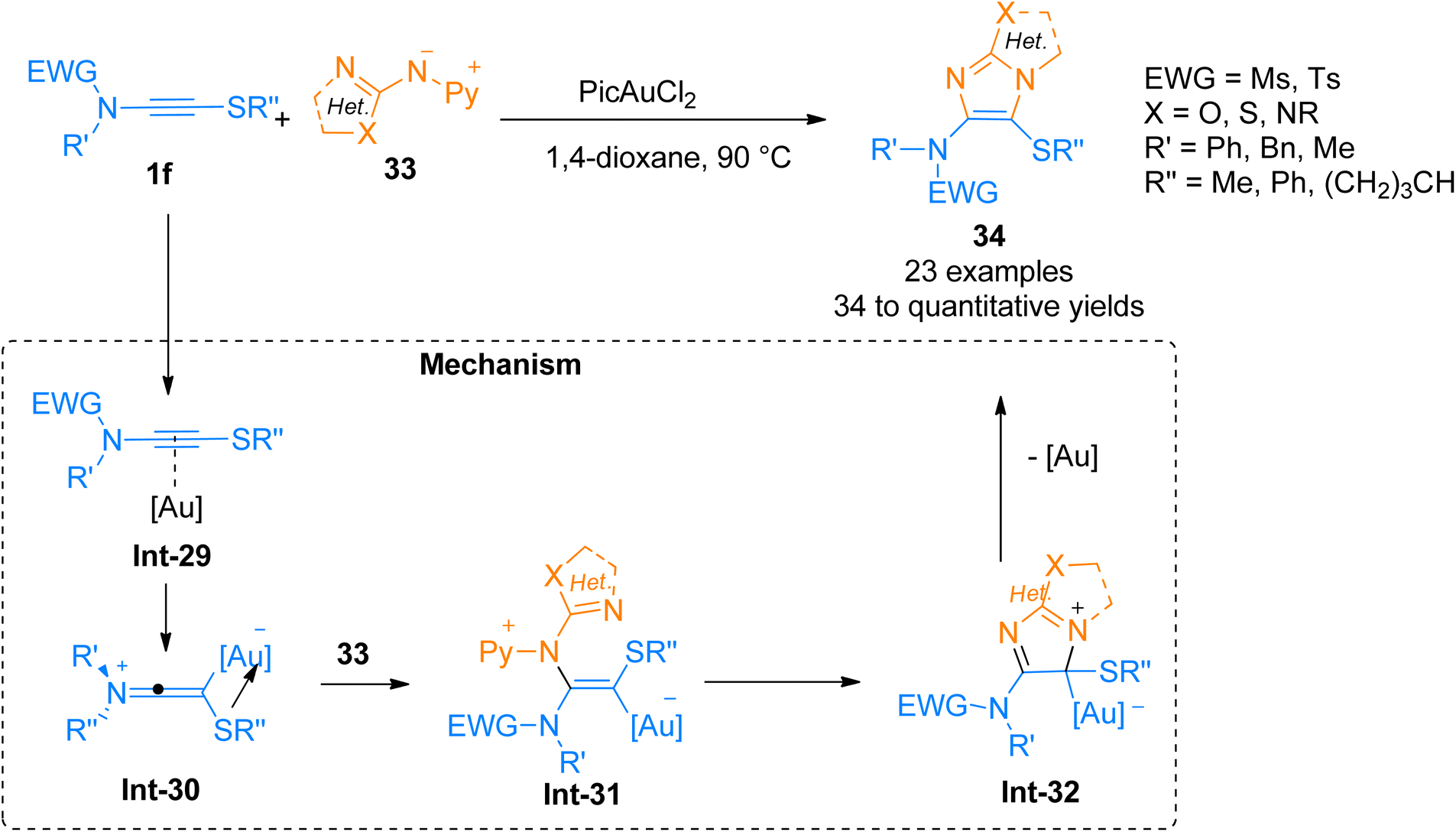
2.2.4.1. Synthesis of heteroaromatic ring-fused 4-aminoimidazoles. Oxo-substituted imidazole frameworks are attractive targets owing to their vital importance in medicinal and agrochemical industries.48–50 Arce, et al. (2020), developed a method to access oxo-substituted heteroaromatic-fused 4-aminoimidazoles 34 by the cycloaddition of nucleophilic nitrenoid 33 with sulfenyl substituted ynamides 1f. This protocol offered a proficient and regioselective synthetic route to access geminally substituted amino sulfenyl containing N-heterocycles by the addition of PicAuCl2 (5 mol%) catalyst in 1,4-dioxane at 90 °C, or 1,2-dichlorobenzene at 125 °C (23 examples, 34 to quantitative yields) (Scheme 16). The mechanism of the synthetic route starts with the gold-coordinated ynamide Int-29 derived through the coordination of gold catalyst with the alkyne core of ynamide. Int-29 converts to keteneiminium ion intermediate Int-30. The nitrenoid specie 33 reacts with Int-30 and afforded the final product 34 via cyclization and elimination of gold catalyst from Int-31 and Int-32, respectively. This transformation could also afford highly hetero-substituted heterocycles which can be transformed into desirable compounds such as heteroarylsulfoxide and heteroarylsulfone.49
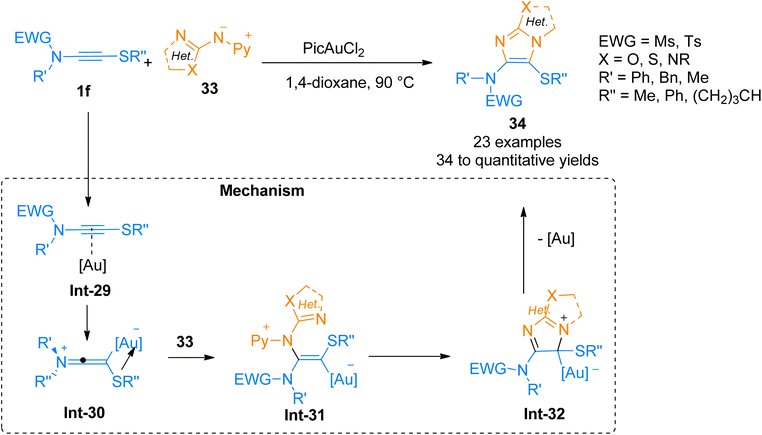 |
| | Scheme 16 Synthesis of o–o′-heteroatom-linked imidazole derivative 34 via [3 + 2]-cycloaddition of ynamide 1f and nitrenoid 33. | |
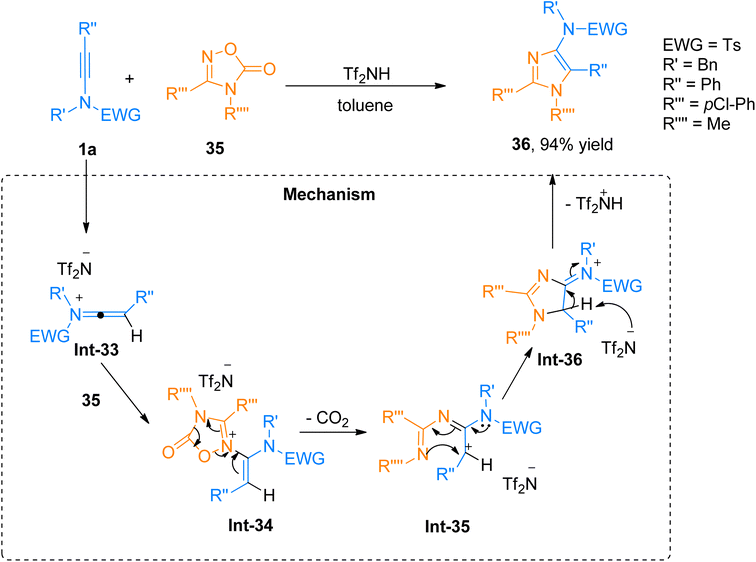 |
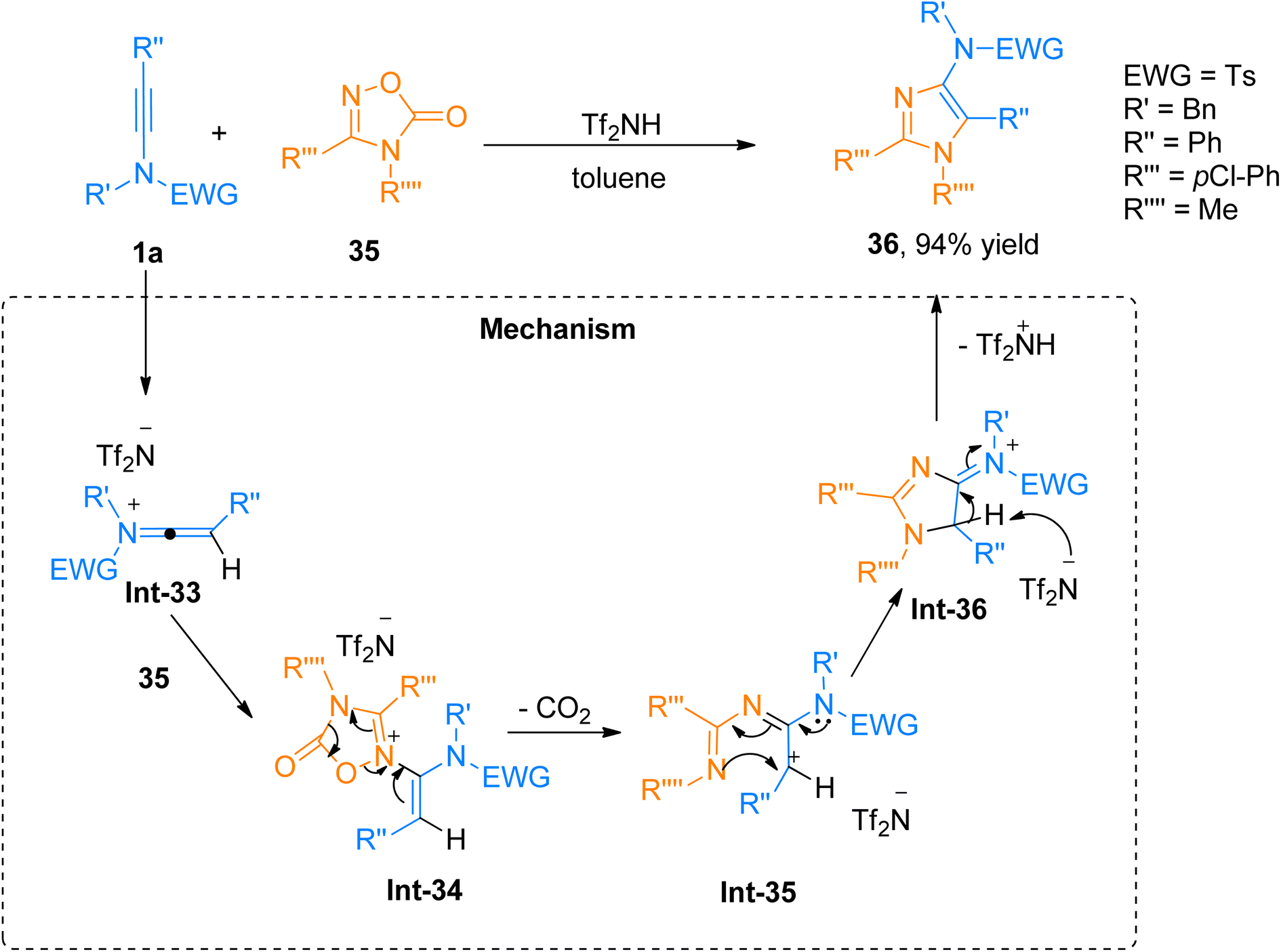
| | Scheme 17 Synthesis of 5-aminoimidazoles 36 via [3 + 2]-cycloaddition of ynamides 1a and oxadiazolones 35. | |
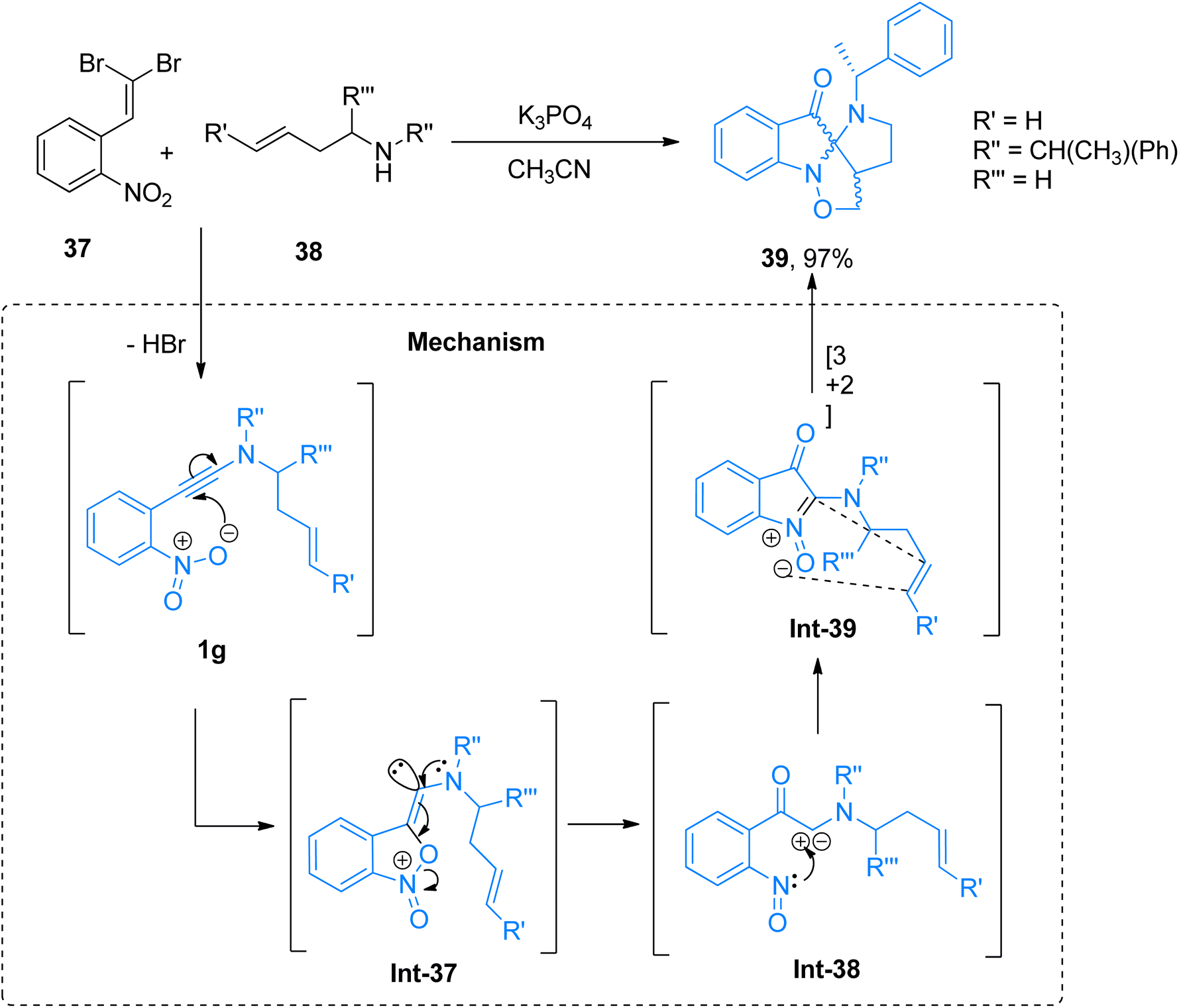
2.2.5. Synthesis of spiropseudoindoxyls via [3 + 2]-cycloaddition. Marien et al. (2018), described a domino reaction to afford polycyclic spiropseudoindoxyls in a metal-free, one-pot multicomponent reaction of 1-dibromovinyl-2-nitro-substituted arene and 2° amines. The reaction was afforded by the addition of K3PO4 in acetonitrile solvent at a reaction temperature of 70 °C. The reaction mechanism was predicted based on the control experiments and theoretical calculations based on topological analysis tools.52 The scaffold with the fused ring was achieved by the generation of ortho-nitroarylated ynamine/ynamide intermediate 1g (generated in situ) which further underwent cycloisomerization (involving the generation of Int-37 to Int-39) to yield N-alkenyl-tethered 2-aminoisatogens via a route that is different than standard electron pair donation. The developed methodology afforded spiro-compounds in poor to excellent yield range (28 examples, 11–97% yields). Spiropseudoindoxyl obtained through this methodology are novel scaffolds that find their practical applications in medicinal chemistry. A highlighted example of this methodology is presented in Scheme 18. The compound 39 was afforded in 97% yield via cycloaddition of the ynamide 1g generated in situ by the reaction of arene 37 and secondary amine 38.52
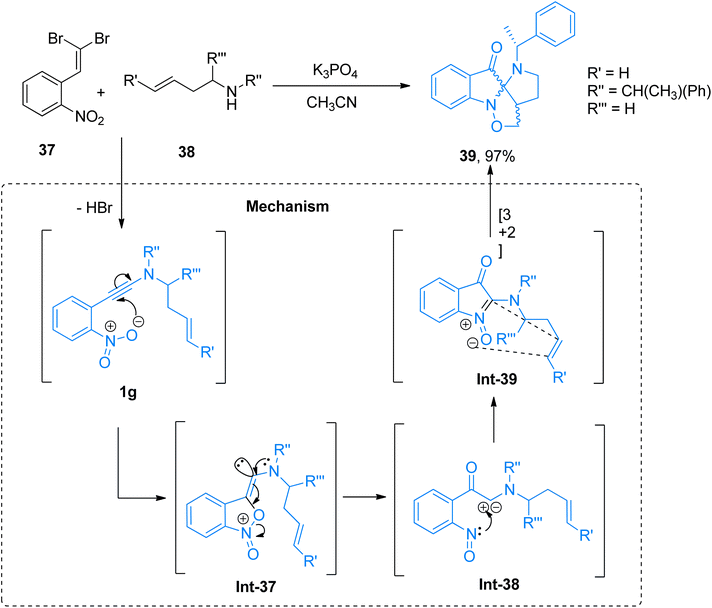 |
| | Scheme 18 Synthesis of spiropseudoindoxyls 39 via intramolecular [3 + 2]-cycloaddition of ynamide intermediate 1g. | |
2.2.6. Synthesis of oxazole derivatives via [3 + 2]-cycloaddition. Classical methods for the construction of oxazoles involve the ring derivatization or cyclization of acyclic precursors. But recent developments in organometallic reagents catalyzed and transition metal-catalyzed bimolecular cycloaddition reactions offer alternative approaches to furnish oxazole derivatives using mild reaction conditions.53,54
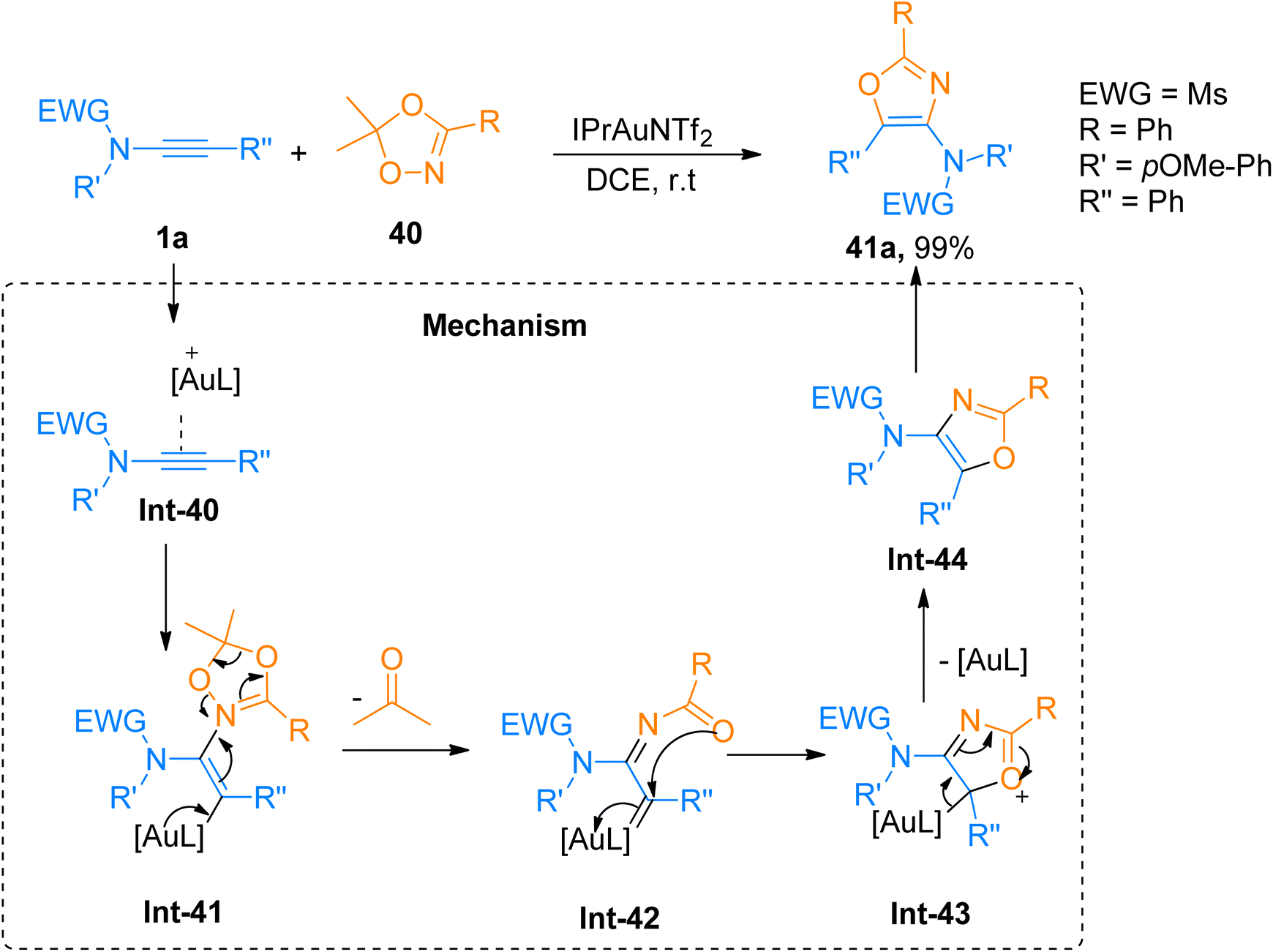
2.2.6.1. Synthesis of 4-aminooxazoles. Chen et al. (2016), developed a facile route for the construction of highly substituted oxazole based scaffolds via regioselective [3 + 2]-cycloaddition reaction between 1,4,2-dioxazoles and ynamides using a gold catalyst. In the reaction mechanism, the Int-40 reacts with 1,4,2-dioxazole that acts as N-acyl nitrene species, to generated iminium ion intermediate Int-41. Further Int-41 via the ring opening converters to α-imino-gold-carbene intermediate Int-42 by elimination of ketone using IPrAuNTf2 catalyst (5 mol%), in dichloroethane at room temperature or 80 °C. The α-imino-gold-carbene Int-42 further undergoes subsequent steps of cyclization and elimination of gold catalyst to afford the title product via the formation of Int-43 and Int-44, respectively. The product 41a was accessed in 99% yield via cycloaddition of 1a and 1,4,2-dioxazole 40 and the reaction was carried out using IPrAuNTf2 catalyst (5 mol%) and dichloroethane (Scheme 19). The selectivity of the cycloaddition reaction provided the access to functional oxazoles with biologically active chiral centers (36 examples, 30–99% yields).55
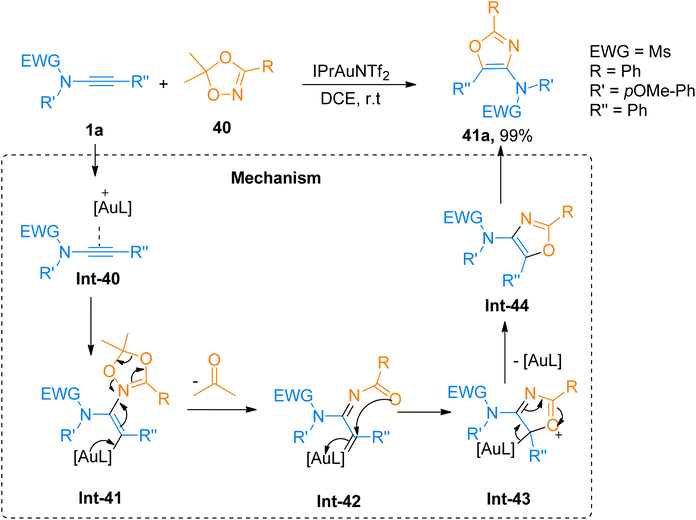 |
| | Scheme 19 Synthesis of 4-aminooxazoles 41a from [3 + 2]-cycloaddition of ynamides 1a with dioxazole 40. | |
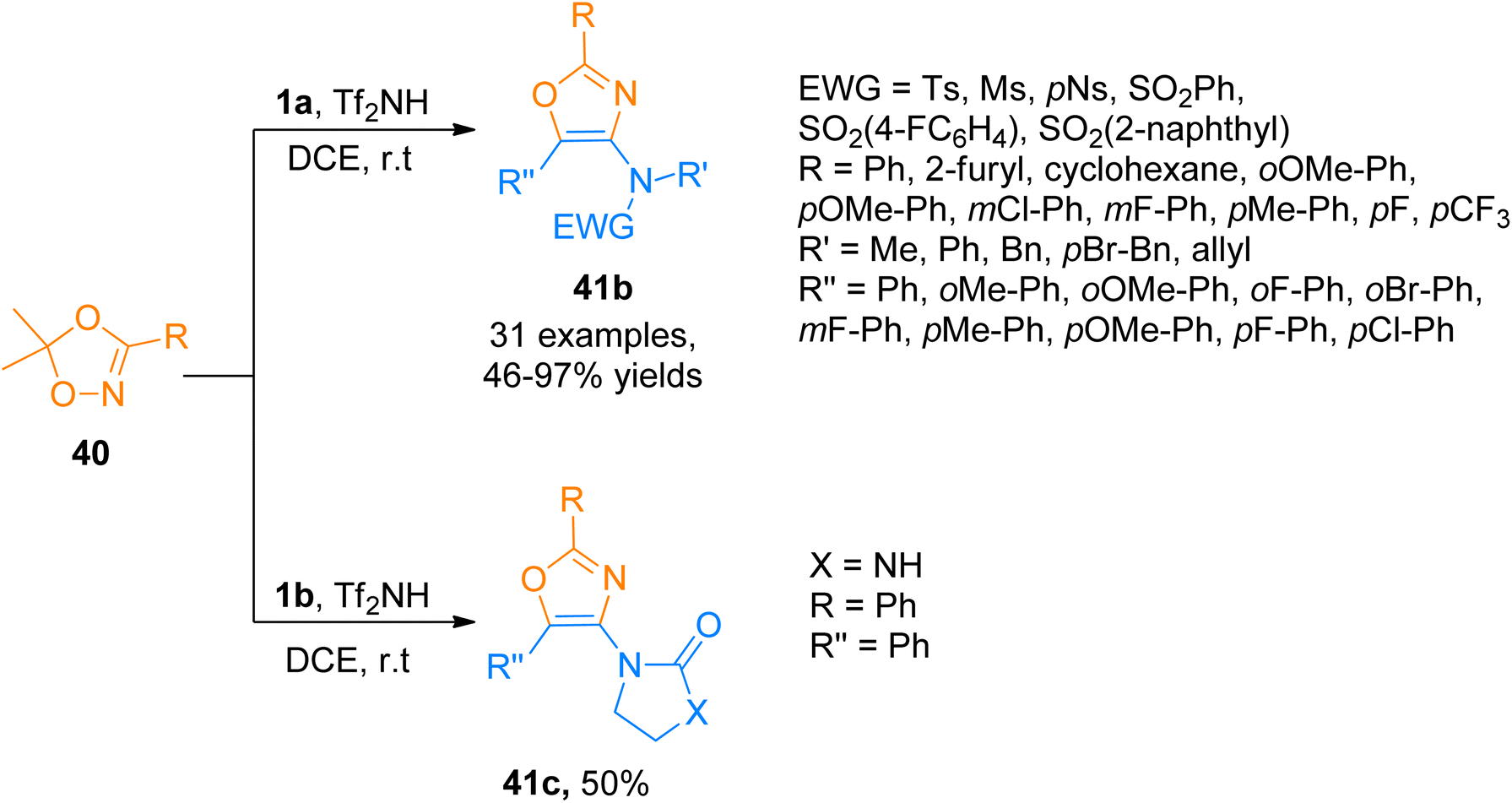
Similarly, Zhao et al. (2017), accessed 4-aminooxazoles 41b via Tf2NH-catalyzed, [3 + 2]-cycloaddition reaction of various ynamides 1a and dioxazoles 40 using dichloroethane solvent at room temperature. Along with the ynamide bearing aliphatic electron withdrawing group, ynamides 1b was also employed in the developed methodology, which underwent a cycloaddition reaction with dioxazoles 40 to afford 41c in 50% yield (Scheme 20). The predicted reaction mechanism suggested the role of the catalyst in the stabilization of the keteniminium ion and further, it facilitates the elimination of the H in the last step, in the same manner as depicted previously in Scheme 19. This protocol offers a metal-free approach under mild reaction conditions to carry out the transformation in short reaction times. The developed methodology offered the advantages of low cost and readily available catalyst, highly efficient transformation, and broad substrate scope to afford 4-aminooxazoles (32 examples, 46–97% yields). This metal-free approach suggests the potential application of the metal-free methodology for the synthesis of pharmaceutically important scaffolds.56
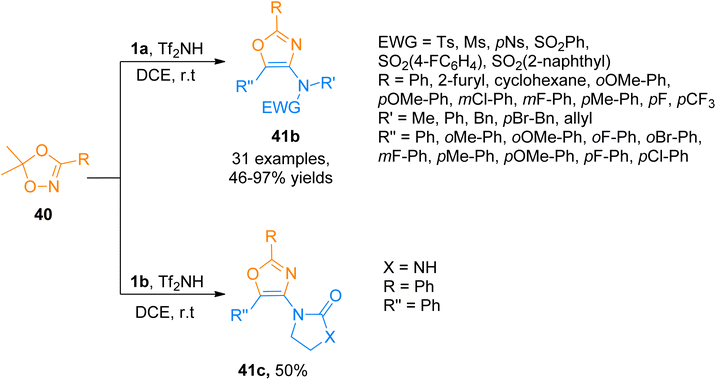 |
| | Scheme 20 Synthesis of 4-aminooxazoles 41b-c from [3 + 2]-cycloaddition of ynamides 1a/1b and dioxazoles 40. | |
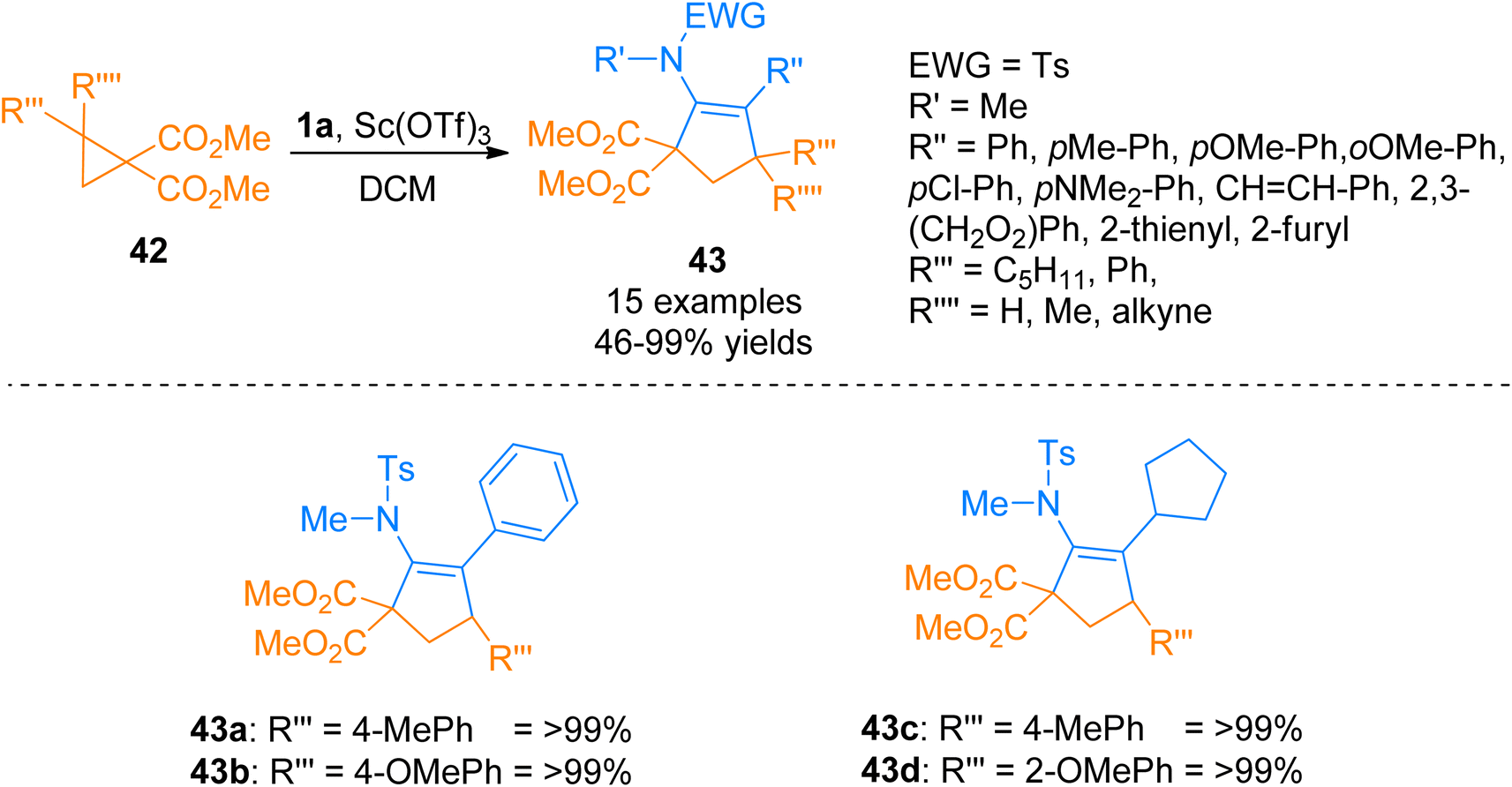
2.2.7. Synthesis of cyclopentenes via [3 + 2]-cycloaddition. Mackay et al. (2014), described the [3 + 2] cyclization of ynamides 1a and cyclopropanes 42 to attain corresponding cyclopentene sulfonamides 43 in excellent yields. Subsequent deprotection and hydrolysis of cyclopentene sulfonamides furnished 2,3-substituted cyclopentanones with high diastereoselectivities. Ynamides 1a underwent cycloaddition with cyclopropanes 42 by adding Sc(OTf)3 catalyst (10 mol%) and dichloromethane solvent at room temperature (Scheme 21). In this scandium-catalyzed annulation, cyclopropanes 42 acts as a donor while ynamides 1a as an acceptor. The developed synthetic route afforded the products with high diastereoselectivities in a moderate to quantitative yield range (15 examples, 46 to >99% yields).57
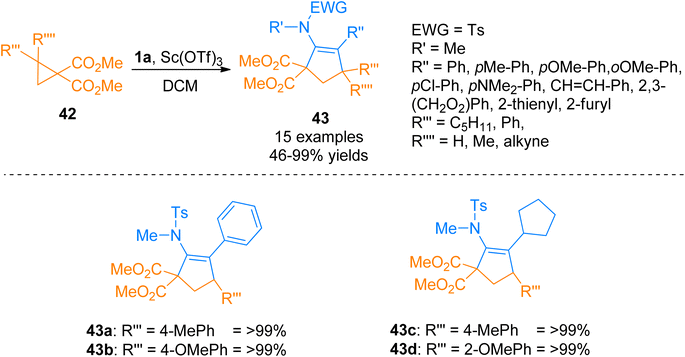 |
| | Scheme 21 Synthesis of cyclopentene sulfonamides 43 via [3 + 2]-cycloaddition of ynamides 1a with cyclopropanes 42. | |
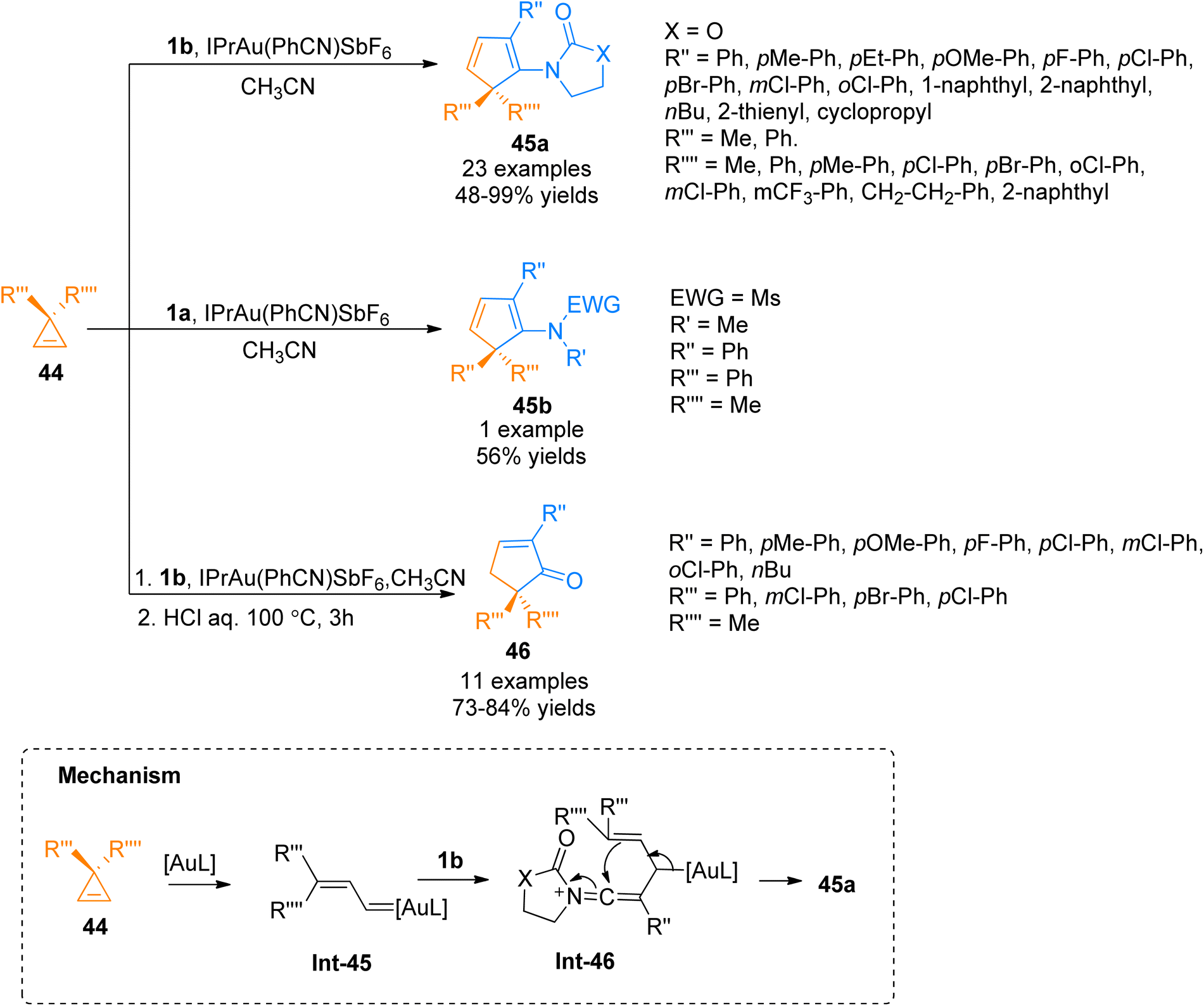
2.2.8. Synthesis of cyclopentadienes and cyclopentenones via [3 + 2]-cycloaddition. Cheng et al. (2017), investigated the gold-catalyzed intermolecular cycloisomerization of ynamides 1a/1b and cyclopropenes 44 to yield cyclopentadienes 45a–b and cyclopentenones 46. The transformation involves the activation of double bonds over triple bonds and achieving cyclopentadienes in high yields. In comparison with the already reported transformations involving gold-catalyzed enyne cycloisomerization, this novel methodology presented a rare example of the gold-catalyzed transformation with alkenes activation priority over alkynes (formation of Int-45 and Int-46). The reaction afforded the cyclopentadienes 45a and 45b via cycloaddition of cyclopropenes 44 with ynamides 1a and 1b, respectively with optimized conditions of IPrAu(PhCN)SbF6 catalyst (5 mol%) in acetonitrile solvent at 100 °C. Moreover, the developed protocol could be extended to realize the synthesis of functionalized cyclopentenone 46 via one-pot, formal [3 + 2]-cycloaddition between cyclopropenes 44 and ynamides 1b using optimized conditions followed by the treatment with concentrated HCl at 100 °C (Scheme 22). The reaction afforded cyclopentadienes 45a–b (24 examples, 48–99% yields) and cyclopentenones 46 (11 examples, 64–84% yields) in a moderate to excellent yield range.58
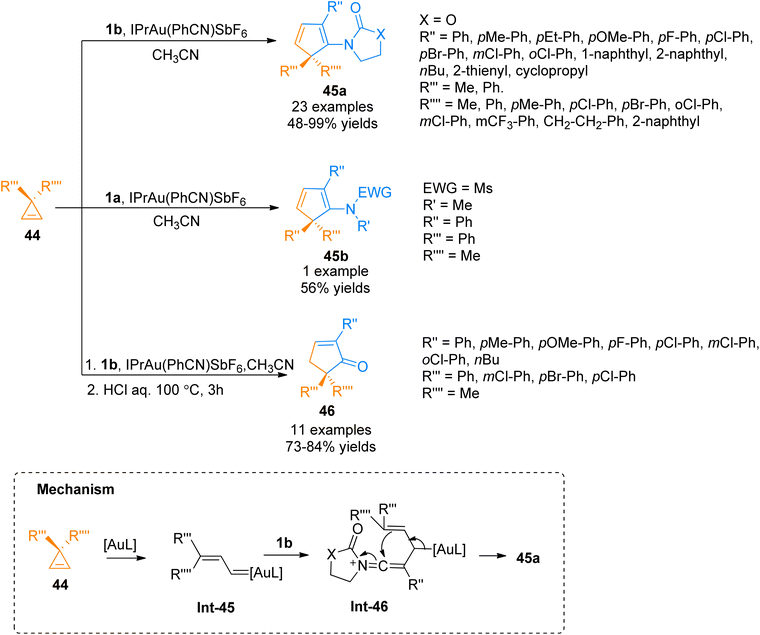 |
| | Scheme 22 Synthesis of cyclopentadienes 45a/45b and cyclopentenones 46 via [3 + 2]-cycloaddition between ynamides 1a/1b and cyclopropenes 44. | |
2.3. [4 + 1]-cycloaddition
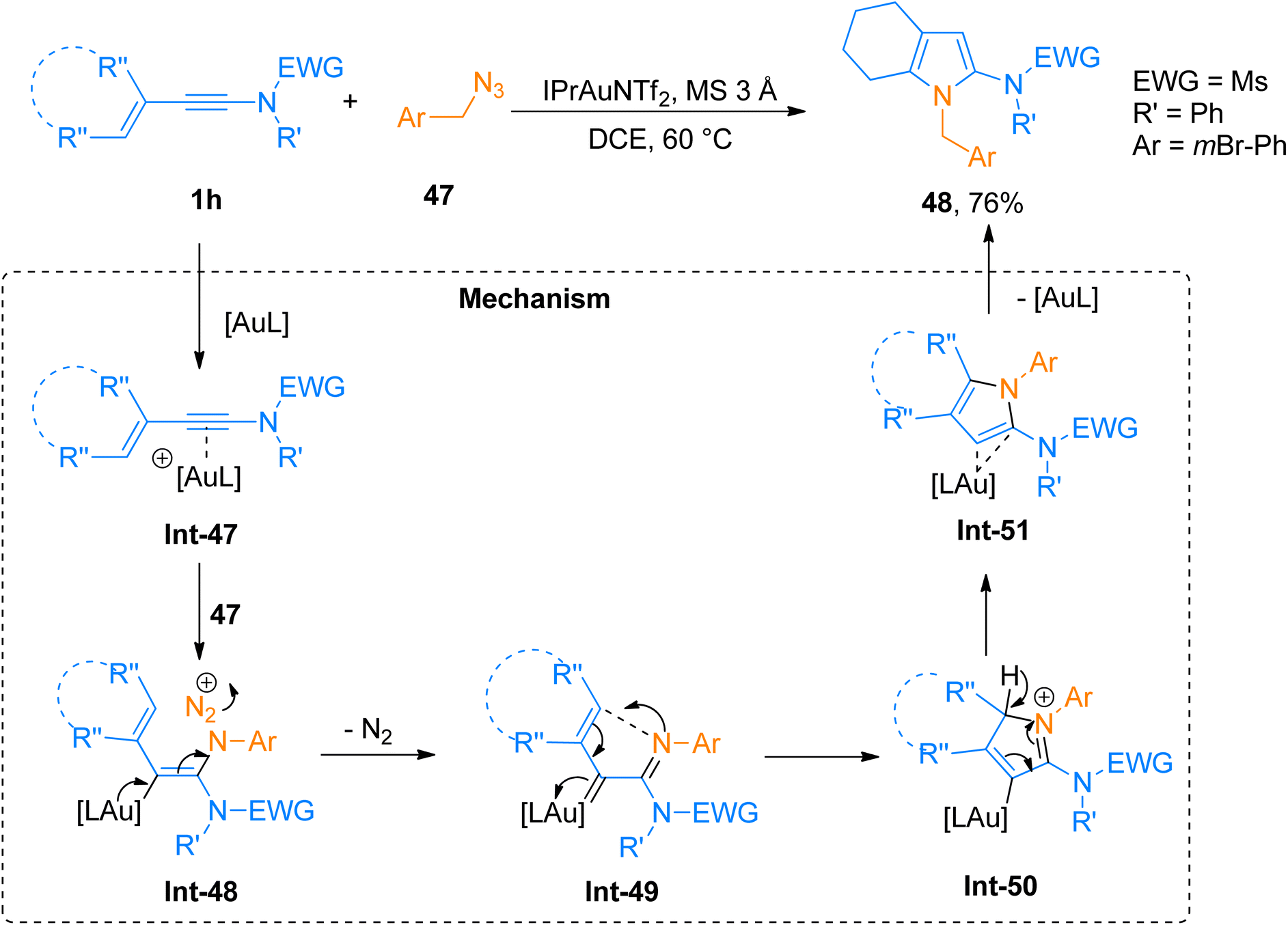
2.3.1. Synthesis of 2-aminopyrroles via [4 + 1]-cycloaddition. Shu et al. (2016), carried out a novel gold-catalyzed aza-Nazarov cycloaddition via intermolecular ynamide amination for facile synthesis of substituted 2-aminopyrroles in promising yields. The reaction generated highly functionalized aminopyrroles via formal [4 + 1]-cycloaddition of 3-en-1-ynamides with azides using the gold catalyst. This tandem reaction sequence afforded the title products (17 examples, 50–76% yields) in high regioselectivity by using IPrAuNTf2 catalyst (10 mol%) and MS 3 Å in dichloroethane solvent at 60 °C. Among the synthesized series compound 48 was afforded 76% yield via cycloaddition of ynamide 1h and azide 47 (Scheme 23). The reaction mechanism was proposed based on DFT calculations of the relative free energies of the intermediates and transition states.59 The gold activated ynamide intermediate Int-47 reacted with 47 to generate imine-gold-carbene intermediate Int-49. The Int-49 undergoes cyclization and proton transfer in subsequent steps affording Int-50 and Int-51, respectively. Finally, the elimination of gold catalyst from Int-51 yielded the final product. The facile and convenient synthetic route with wide substrate scope suggested this strategy as a preferable alternative for the construction of a diverse range of substituted 2-aminopyrroles.59
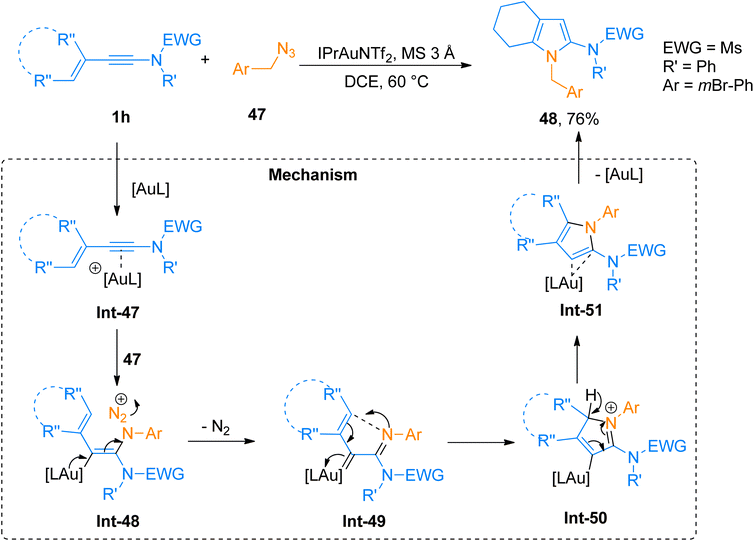 |
| | Scheme 23 Synthesis of 2-aminopyrrole 48 via [4 + 1]-cycloaddition of 3-en-1-ynamide 1h with azide 47. | |
2.4. [4 + 2]-cycloaddition
2.4.1. Synthesis of quinolines via [4 + 2]-cycloaddition.
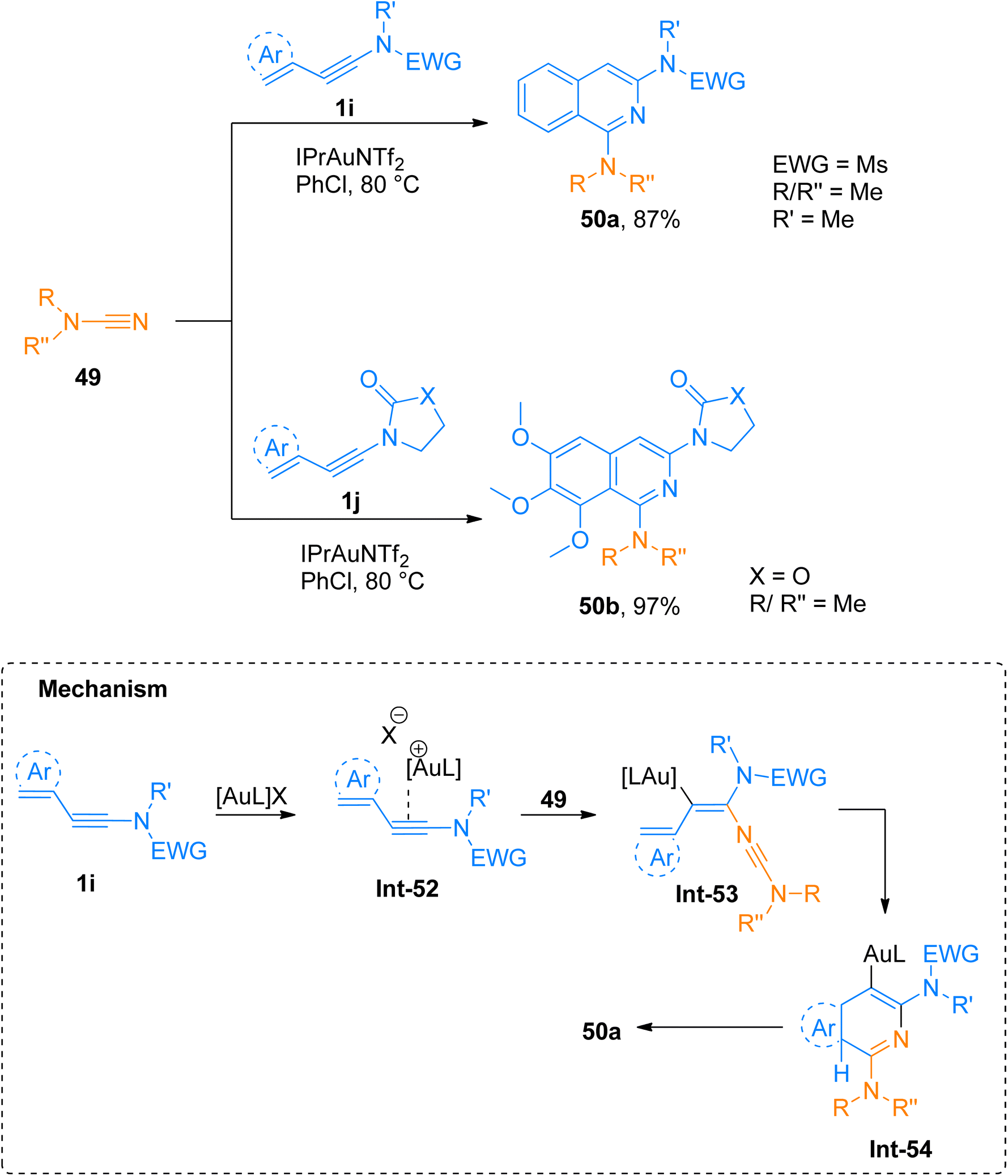
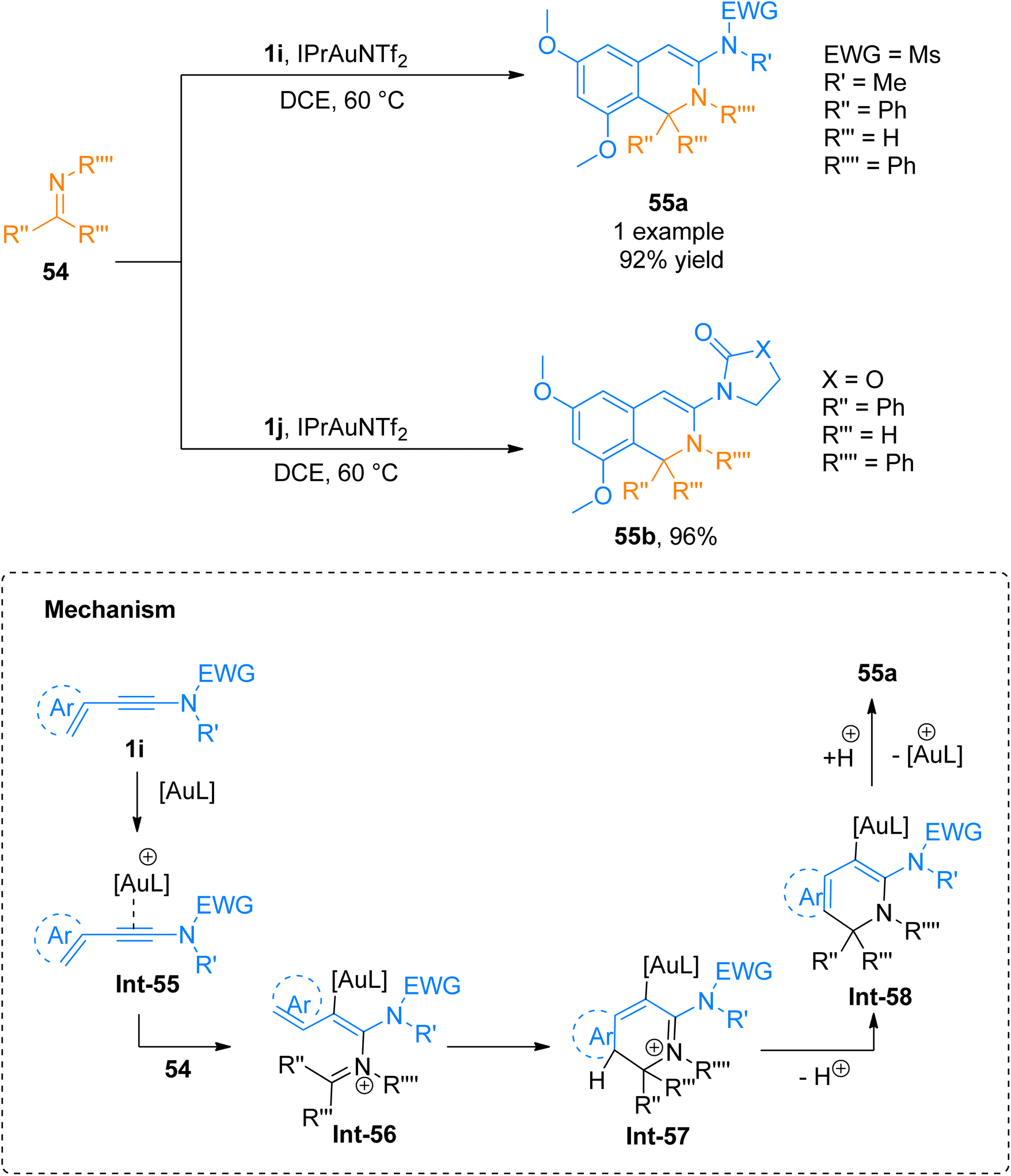
2.4.1.1. Synthesis of isoquinolines. Cyanamides differ in reactivity from conventional nitriles due to the dual nature of cyanamide scaffold bearing electrophilic and nucleophilic nitrogen atoms. Because of this dual reactivity cyanamides are called push–pull nitriles.60 Amino groups present in ynamide and cyanamides allow the one-step and facile introduction of valuable functional substituents in title products. Dubovtsev et al. (2020), described thermodynamically controlled conditions to modulate the reactivity switch either as [2 + 2 + 2] or [4 + 2]-cycloaddition resulting in 1,3-diaminoisoquinolines. The reaction underwent [4 + 2]-cycloaddition of ynamides and cynamides using IPrAuNTf2 catalyst (5 mol%) in chlorobenzene solvent at 80 °C affording diaminoisoquinolines with acyclic electron withdrawing groups (5 examples, 62–87%) and diaminoisoquinolines bearing cyclic electron withdrawing groups (13 examples, 41–97%) (Scheme 24). The reaction mechanism under thermodynamically controlled conditions involves the reaction of activated ynamide intermediate Int-52 with 49 to generate Int-53 that undergoes cyclization and quenching of proton to afford the final product via Int-54. The reaction is of remarkable importance as it proceeded efficiently using mild conditions and open the possibilities of introduction of dialkyl, diaryl, and amino substituents to isoquinoline cores with excellent regioselectivity. Under optimized reaction conditions ynamides, 1i and 1j reacted with cyanamides 49 to afford products 50a and 50b with 87% and 97% yields, respectively.61
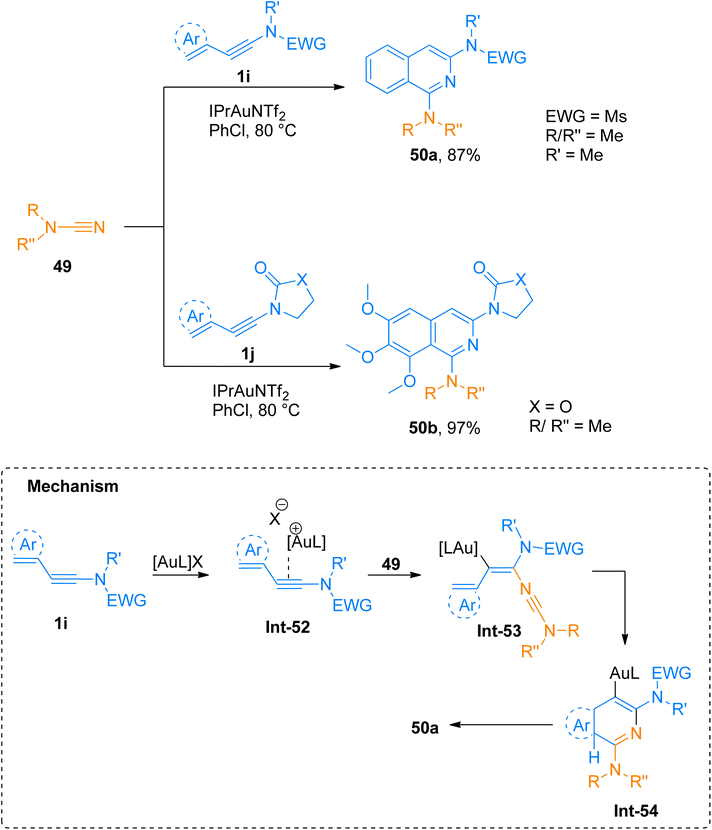 |
| | Scheme 24 Synthesis of 1,3-diaminoisoquinolines 50a and 50b via [4 + 2]-cycloaddition of cyanamides 49 with ynamides 1i and 1j, respectively. | |
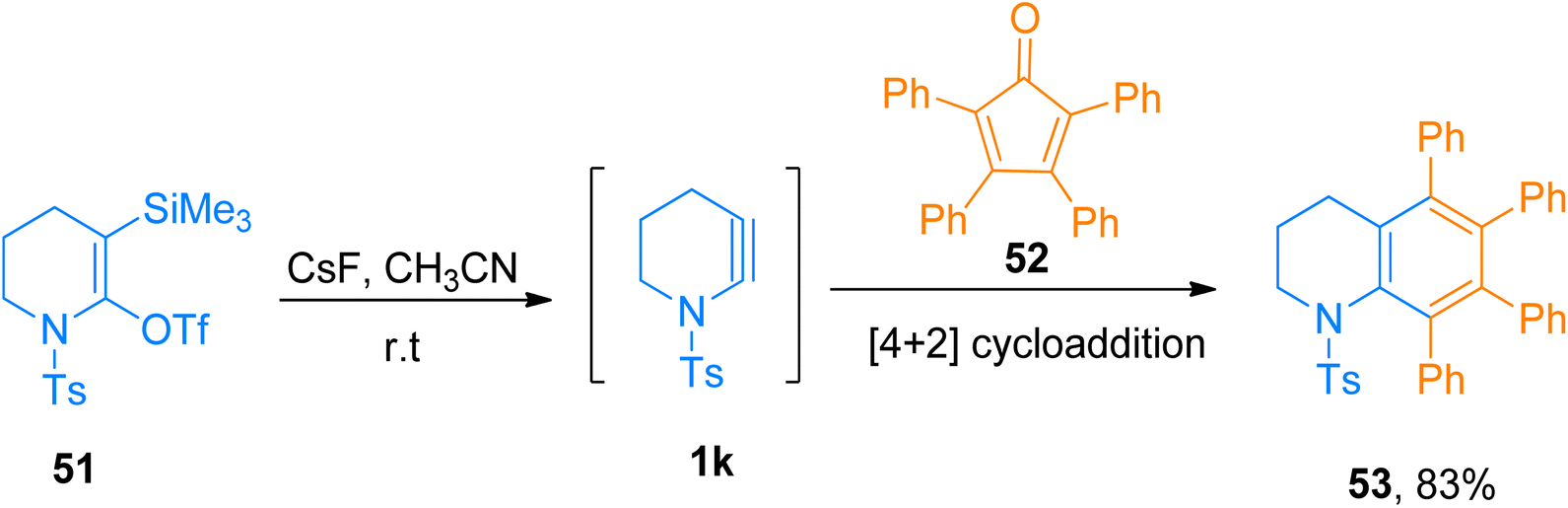
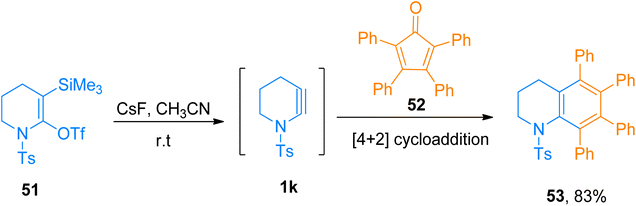 |
| | Scheme 25 Synthesis of tetrahydroquinoline 53 via [4 + 2]-cycloaddition of cyclic ynamide 1k with cyclopentadienone 52. | |
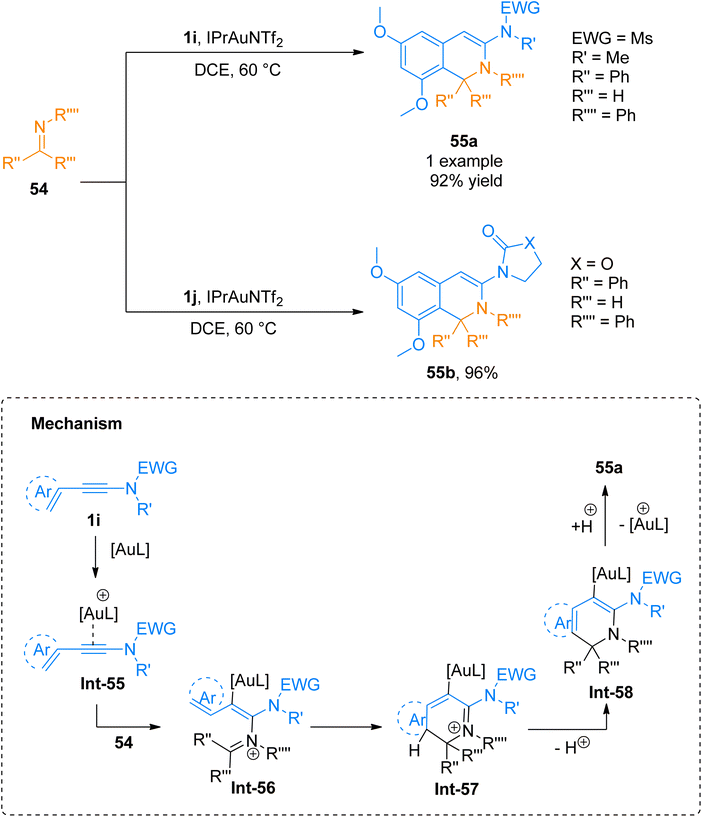 |
| | Scheme 26 Synthesis of 1,2-dihydroisoquinolines 55a/55b via [4 + 2]-cycloaddition of ynamides 1i/1j with imines 54. | |
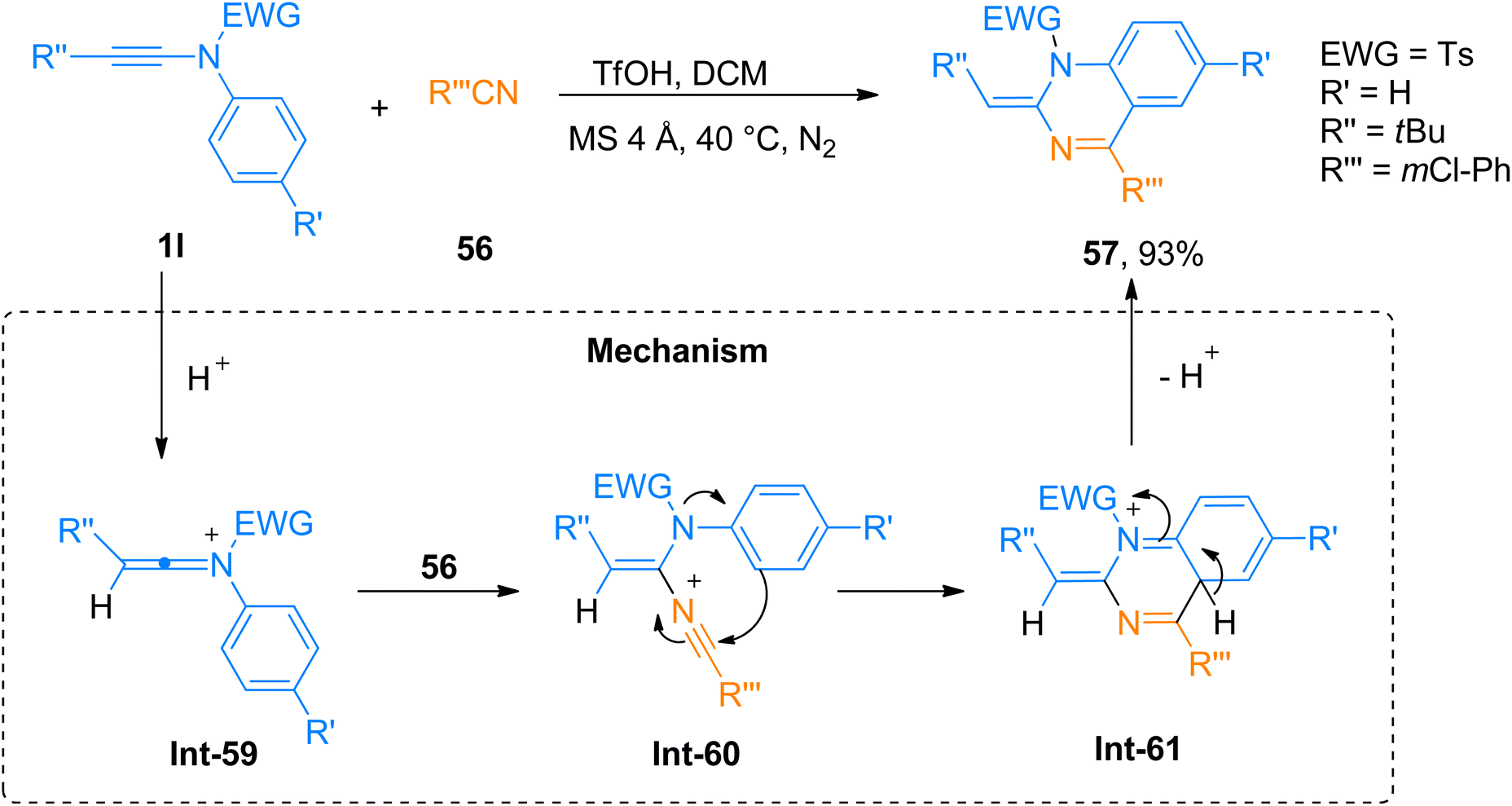
2.4.2. Synthesis of 1,2-dihydroquinazoline via [4 + 2]-cycloaddition. Quinazoline is an imperative core present in alkaloids and medicinal compounds. Quinazolines attracted great interest owing to their potent biological activities as antifungal, anticancer, and antihypertensive agents.64 Wu et al. (2019), described the efficient and straightforward TfOH catalyzed [4 + 2]-cycloaddition of ynamides and nitriles to yield 1,2-dihydroquinazoline derivatives in dichloromethane solvent, using MS 4 Å, inert environment (N2), at 40 °C (25 examples, 34–93% yields). The highest yield of 93% of product 57 was afforded via cycloaddition of ynamide 1l and nitrile 56 (Scheme 27). The proposed mechanism suggests the activation of ynamide 1l by TfOH to yield keteniminium ion Int-59. In the next step nitrile 56 attack the keteniminium ion Int-59 to generate Int-60. Subsequently, 6-endo-dig cyclization of the Int-60 realized the formation of Int-61 which via elimination reaction afforded the respective product. The straightforward and atom-economical synthesis yields the functionalized 1,2-dihydroquinazoline derivatives in a short reaction time with higher yields and excellent diastereoselectivity. Moreover, the use of non-toxic and inexpensive catalysts made it an auspicious pathway to realize pharmaceutically active constructs.64
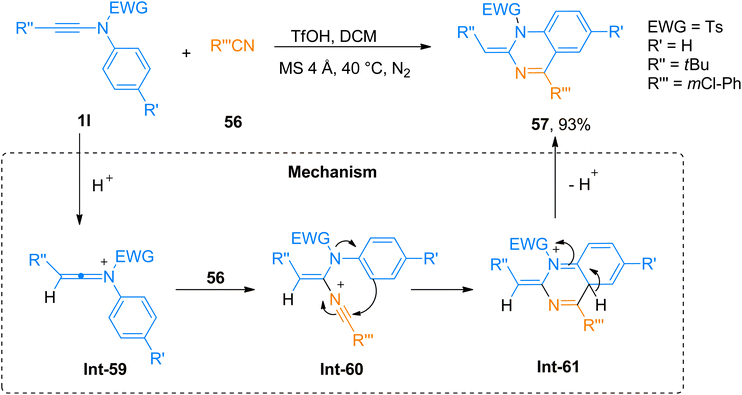 |
| | Scheme 27 Synthesis of 1,2-dihydroquinazoline 57 via [4 + 2]-cycloaddition of ynamides 1l with nitrile 56. | |
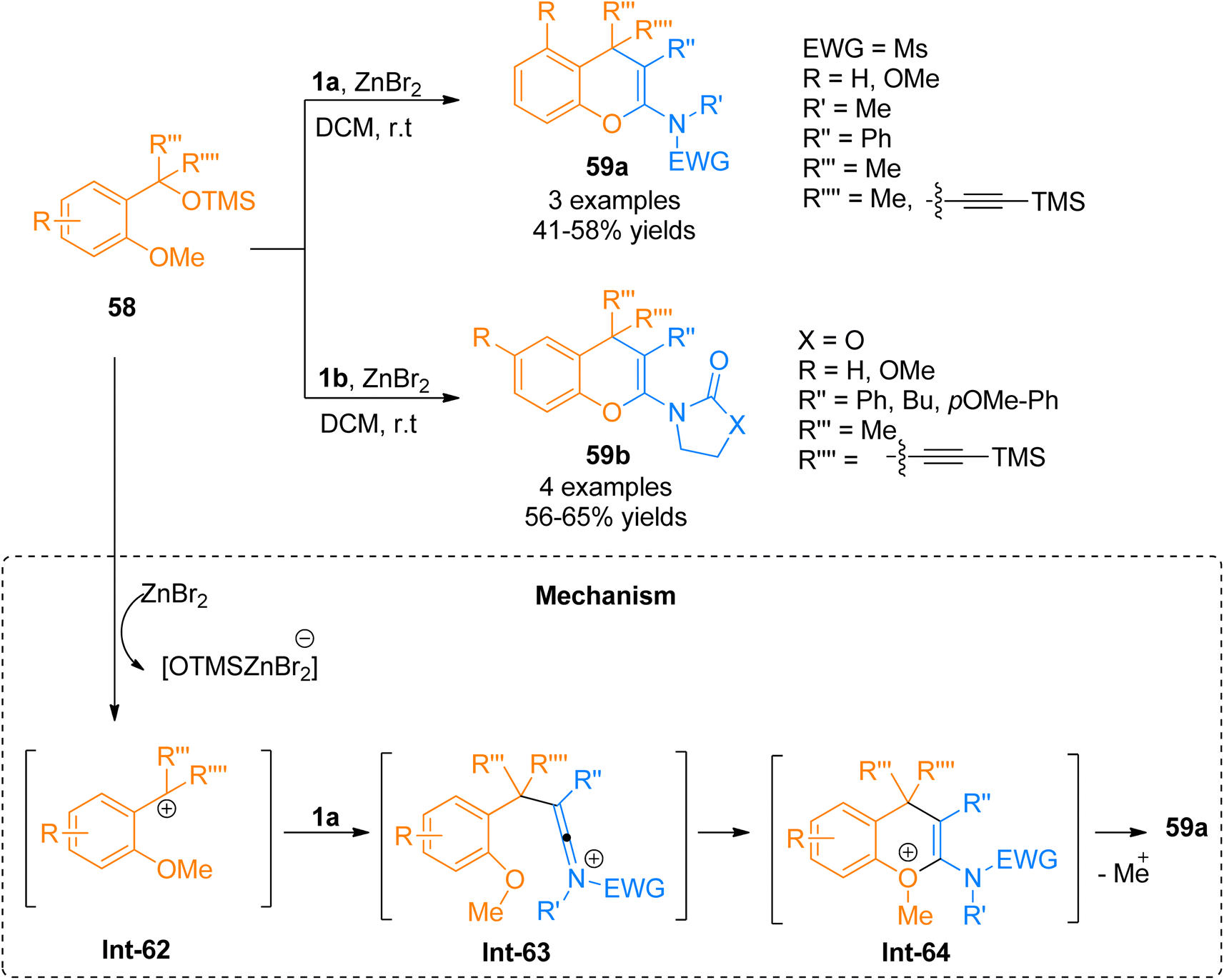
2.4.3. Synthesis of 2-amino-4H-chromenes via [4 + 2]-cycloaddition. Due to the notable and highly pronounced activities of 2-amino-4H-chromenes, a multitude of protocols has been developed for the convenient and facile synthesis of 2-amino-4H-chromenes.23,65 Chen et al. (2016), recently reported a Lewis acid-catalyzed cycloaddition reaction of 58 and ynamides 1a or 1b to construct title compounds 59a (3 examples, 41–58% yield) and 59b (4 examples, 56–65% examples), respectively, by ZnBr2 promoted transformation using N2 atmosphere at room temperature. The reaction mechanism proceeds by the ZnBr2 promoted the removal of the OTMS group from silyl ether 58 resulting in carbocation intermediate Int-62 which further reacts with ynamide 1a to yield Int-63. The final product was afforded by the cyclization followed by the elimination of the methyl group from Int-64 (Scheme 28). However, the developed methodology faced certain drawbacks such as low yields and limited substrate scope.65
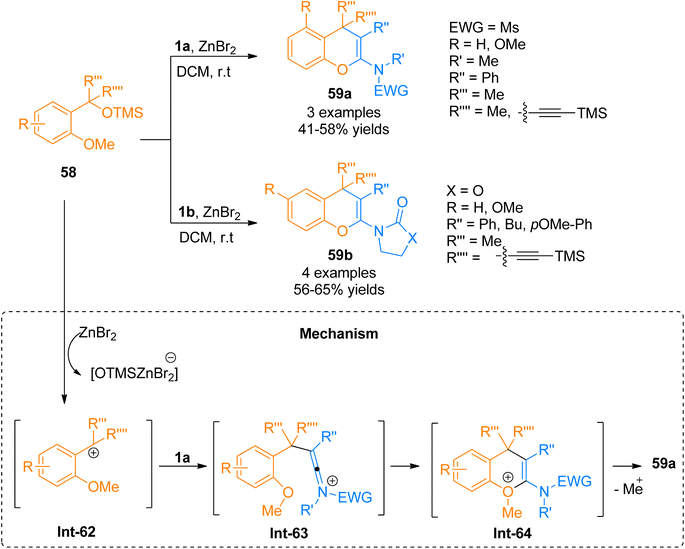 |
| | Scheme 28 Synthesis of 2-amino-4H-chromenes 59 via [4 + 2]-cycloaddition of ynamides 1a/1b with silyl ether 58. | |
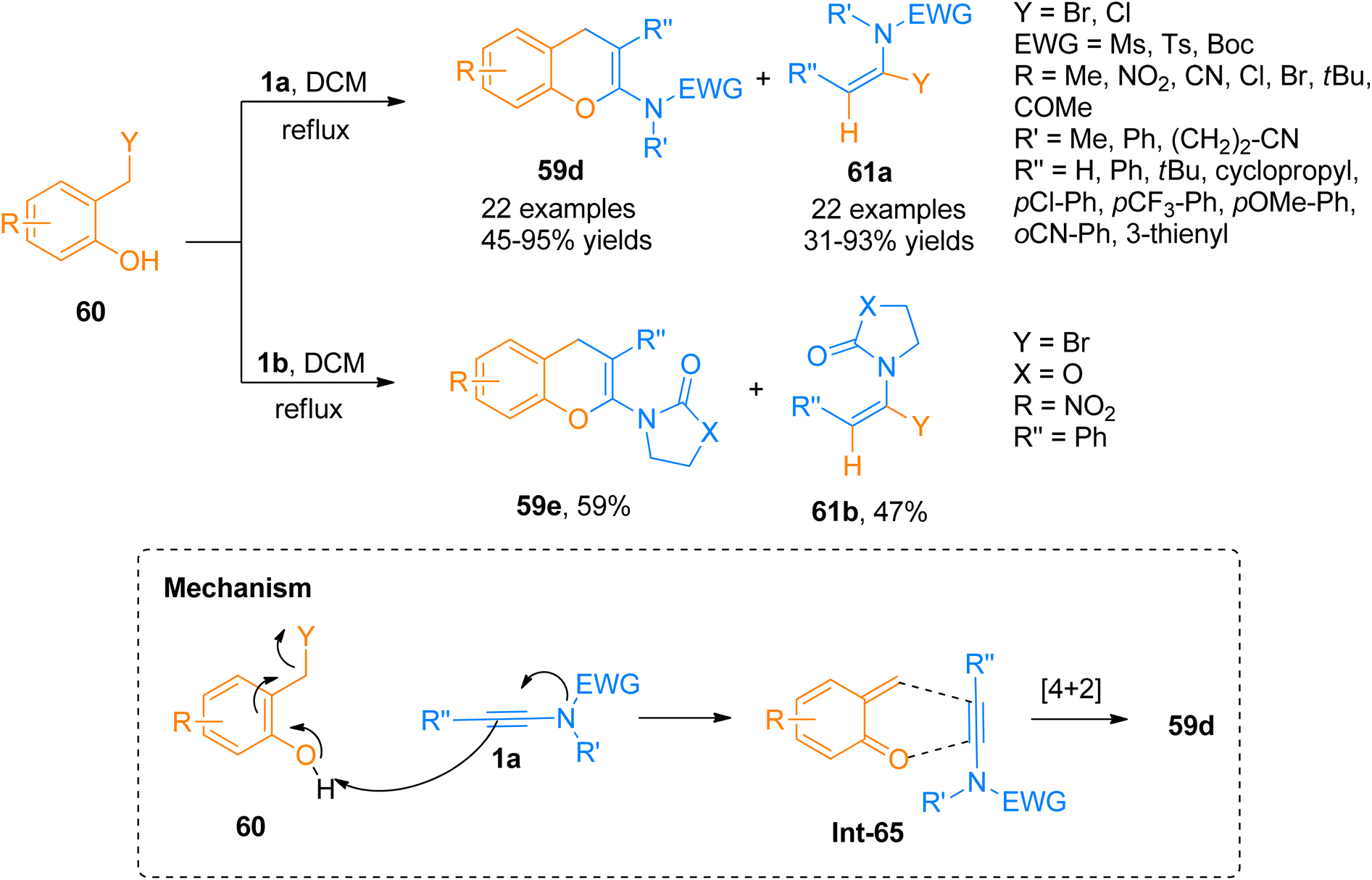
Wen et al. (2020), presented one of the well-developed methods for the synthesis of the 2-amino-4H-chromenes via catalyst-free cycloaddition of ynamides by overcoming the salient drawbacks of previously reported methods. The novel concise and flexible route for the construction of functionalized 2-amino-4H-chromenes 59d–e and α-halo enamides 61a–b involves [4 + 2]-cycloaddition reaction of ynamides 1a and 1b and 2-halomethyl phenols 60 via keteniminium intermediate formation under catalyst-free conditions (Scheme 29). Moreover, the facile protocol afforded higher yields of 59d (23 examples, 45–95% yields) and 59e (1 example, 59% yield) using mild conditions and reliable tolerance for a broad range of functionalities. Along with functionalized 2-amino-4H-chromenes, this reaction also afforded α-halo enamides 61a (22 examples, 31–93% yields) and 61b (1 example, 47% yield) in good to excellent yields which are key intermediates for versatile transformations and can be readily converted into a range of functional groups by halogen metal exchange reaction.66
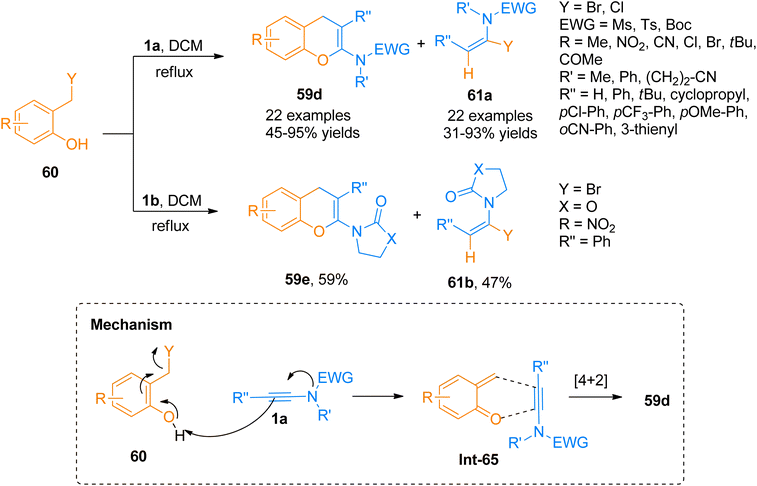 |
| | Scheme 29 Synthesis of 2-amino-4H-chromenes 59d–e via [4 + 2]-cycloaddition of ynamides 1a/1b. | |
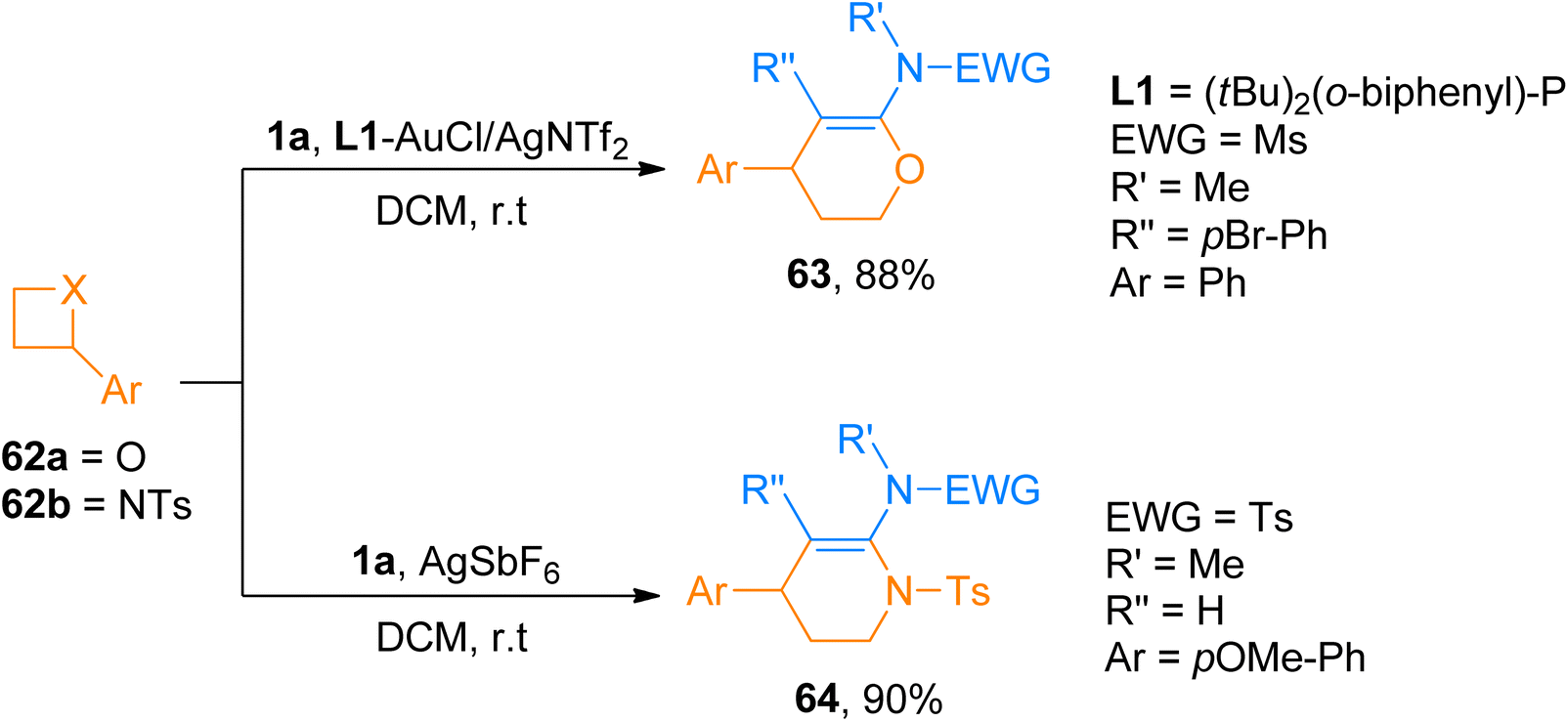
2.4.4. Synthesis of oxa/azacyclic six-membered heterocycles via [4 + 2]-cycloaddition. The use of oxetanes and azitidines for the construction of four-membered frameworks via metal-catalyzed [4 + n]-cycloaddition reactions is limited. The less strained ring is responsible for their lower reactivity due to which they require metal complexes in large quantities.67 Pawar et al. (2014), developed an alternative route to cover this drawback which provided a facile route to access oxa and aza cyclic six-membered heterocycles. The [4 + 2]-cycloaddition reaction between ynamides and oxetanes using gold catalyst afforded required oxa/azacyclic six-membered heterocycles (15 examples, 64–88% yields). In L1-AuCl/AgNTf2 catalyzed (5 mol%) cycloaddition, gold-p-ynamides act as electrophiles and oxetanes as nucleophiles affording the cycloadducts in a regioselective manner in dichloromethane solvent at room temperature. While for azetidines, [4 + 2]-cycloaddition reaction with ynamides occurred more efficiently using a silver catalyst, AgSbF6 (10 mol%), and dichloromethane solvent at room temperature affording nitrogen-containing 6-membered heterocycles (14 examples, 45–90% yields). Both gold and silver catalyst work similarly to activate ynamide which get attacked by heteroatom of oxetane or azetidine. Among the synthesized series of oxa cyclic heterocycles, compound 63 was afforded an 88% yield by the gold-catalyzed reaction of ynamide 1a and oxetane 62a. Similarly, in the aza cyclic heterocyclic series, compound 64 was afforded the highest yield of 90% by silver catalyzed cycloaddition of ynamide 1a and azetidine 62b (Scheme 30).68
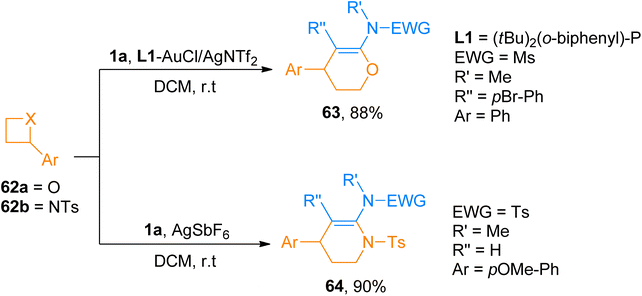 |
| | Scheme 30 Synthesis of six-membered oxa-heterocycle 63 and aza-heterocycle 64 via [4 + 2]-cycloaddition of ynamide 1a and oxetane 62a/azetidine 62b. | |
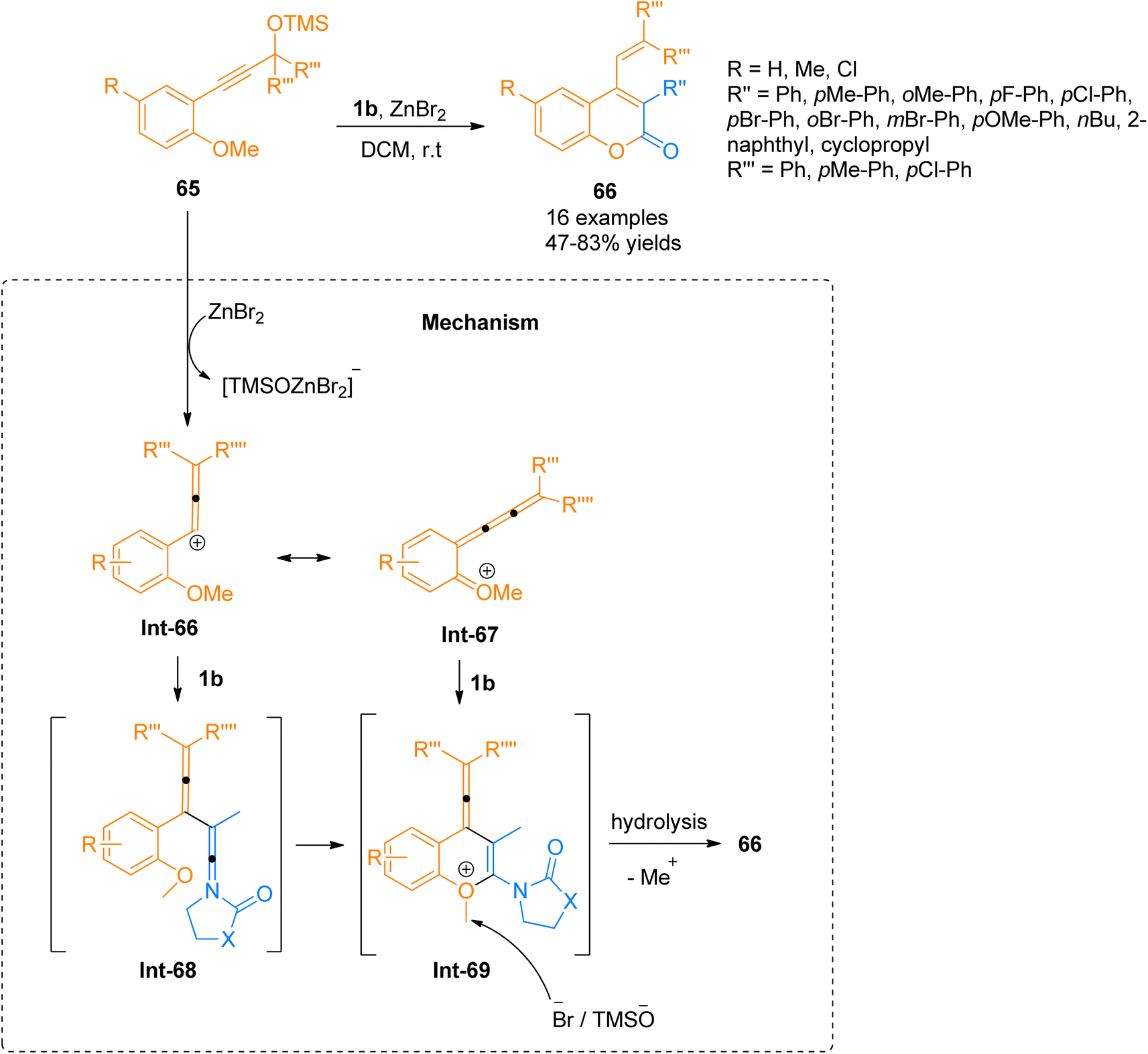
2.4.5. Synthesis of coumarins via [4 + 2]-cycloaddition. Chen et al. (2016), described a novel synthetic manifestation for the [4 + 2]-cycloaddition reaction of o-anisole functionalized propargyl silyl ethers 65 with ynamides 1b to afford polycyclic 4-vinylcoumarins 66 (16 examples, 47–83% yields). Fluorescence analysis was used for the screening and optimization of the reaction conditions for 6π-electrocyclization. The synthesis was carried out using ZnBr2 as a Lewis acid catalyst, in dichloromethane solvent at room temperature (Scheme 31). The proposed reaction suggests the initiation of reaction by ZnBr2 promoted propargyl silyl ether 65 conversions to carbocation Int-66 which is in conjugation with Int-67. There are two possible pathways, during the first pathway ynamide 1b reacts with Int-66 to yield highly unstable intermediate Int-68 which ultimately converts to Int-69 via nucleophilic attack and cyclization. While the alternative pathway involves the concerted [4 + 2]-cycloaddition of the Int-67 and ynamide 1b to directly afford Int-69. Elimination of the methyl group followed by hydrolysis yields the final product.69
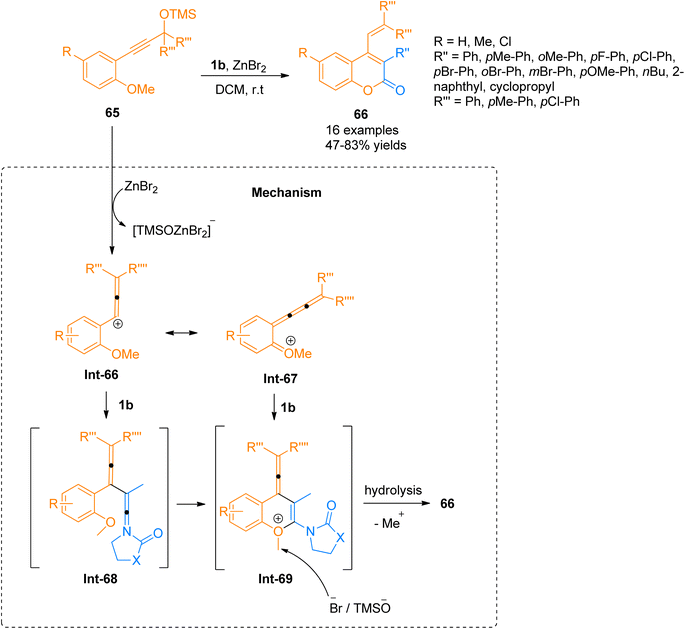 |
| | Scheme 31 Synthesis of substituted coumarins 66 via [4 + 2]-cycloaddition of ynamide 1b with 4-vinylcoumarins 65. | |
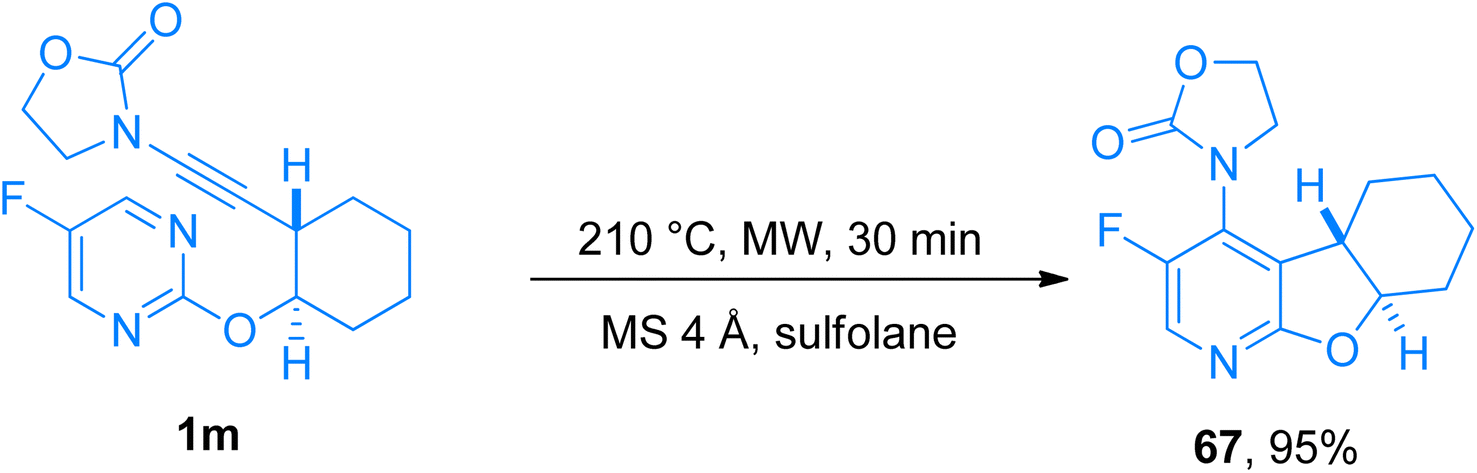
2.4.6. Synthesis of 4-aminopyridines via [4 + 2]-cycloaddition. 4-Aminopyridines are biologically as well as synthetically important scaffolds that attracted the attention of the pharmaceutical and agrochemical industry.70 Despite the skeletal diversity associated with the 4-aminopyridines, the synthetic manifestations of this class still have shortcomings and limitations. However, an intramolecular inverse electron demand hetero Diels–Alder reaction (IEDDA)/retro-Diels–Alder reaction (rDA) could provide direct access to this synthetically challenging, and valuable class of compounds. Duret et al. (2017), presented the synthetic manifestation to access diverse polycyclic fused and spiro 4-aminopyridines exhibiting considerable structural variability from commercially available pyrimidines. This methodology reported the use of ynamides in IEDDA for the first time to yield aminopyridines. The three-step transformation includes the retro [4 + 2]-cycloaddition between ynamide bearing cyclic group at the nitrogen terminal and pyrimidine (electron-deficient heterodiene) as a key step which set up the first example of ynamide behaving as an electron-rich dienophile in [4 + 2]-cycloaddition reaction. Furthermore, IEDDA in heated toluene overcomes the limited scalability of the reaction. Starting from C2-substituted pyrimidines, tetra or penta substituted 4-aminopyridines can be achieved in excellent yields (26 examples, 11–95% yields) using MS 4 Å, and sulfolane by microwave irradiation at 225 °C for 1 min and further at 210 °C, for 30 min. Compound 67 was furnished in 95% yield by the novel methodology via microwave-promoted cycloaddition of ynamide 1m at 210 °C (Scheme 32).71
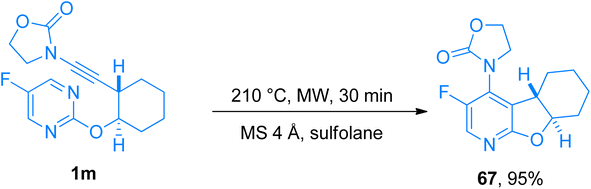 |
| | Scheme 32 Synthesis of 4-aminopyridines 67 via intramolecular inverse [4 + 2]-cycloaddition of ynamide 1m. | |
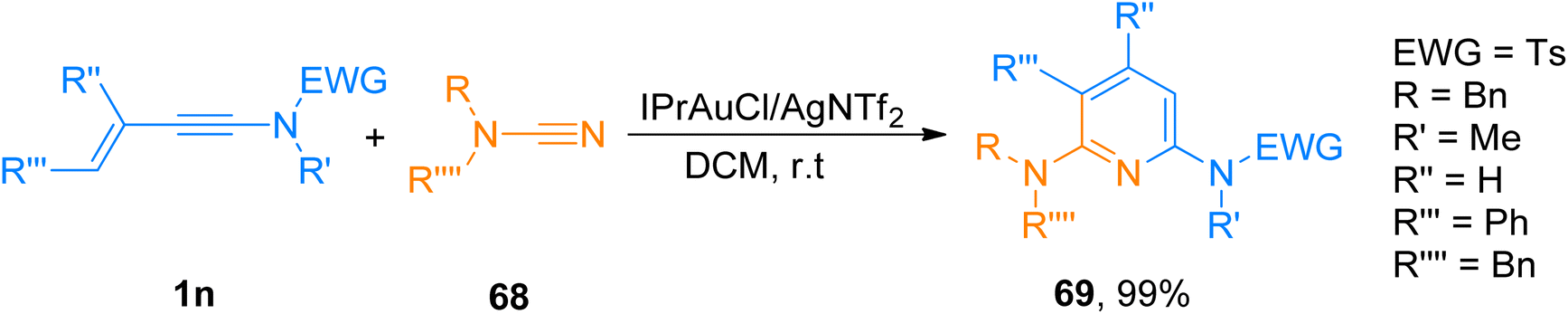
2.4.7. Synthesis of diaminopyridines via [4 + 2]-cycloaddition. Hetero-tetradehydro-Diels–Alder reaction (TDDA) for the construction of pyridines is unique and only a few examples have been reported yet.72,73 For TDDA reaction between enynamide and cyanamide reaction partners to afford diaminopyridines. Shcherbakov et al. (2021), developed a gold(I) catalyzed protocol. The novel protocol proceeds under mild reaction conditions such as IPrAuCl/AgNTf2 (5 mol%) catalyst, in dichloromethane solvent at room temperature, and presents an efficient route for diversely functionalized 2,6-diaminopyridines. In addition to the above, the developed methodology also possessed broad substrate scope as different substituents on cyanamide and ynamides were tolerated well (28 examples, 70–99% yields). Among the synthesized series compound 69 was afforded in 99% yield via hetero-TDDA between ynamide 1n and cyanamide 68 (Scheme 33).74
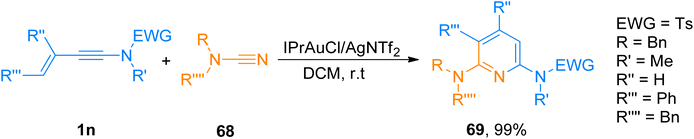 |
| | Scheme 33 Synthesis of 2,6-diaminopyridines 69 via hetero-TDDA reaction between enynamide 1n and cyanamide 68. | |
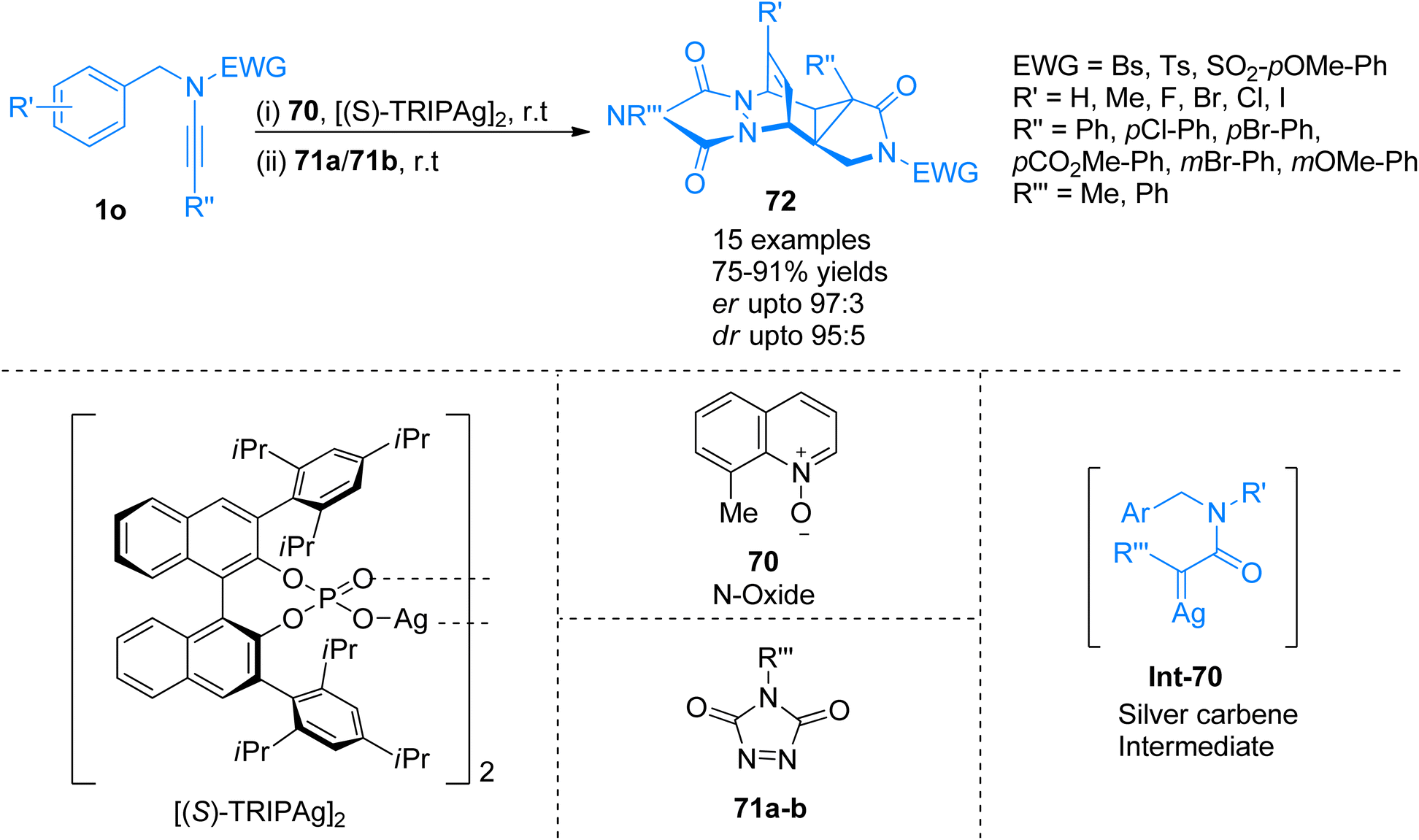
2.4.8. Synthesis of polycyclic aromatic compounds via [4 + 2]-cycloaddition. Dearomatization of arenes is a direct method to convert aromatic compounds to functionalized carbocycles, however, protocols for highly enantioselective dearomatization of arenes are limited as compared to indoles and phenols. Ito and coworkers (2021), developed a diazo-free novel protocol for the construction of silver carbene species using ynamide 1o to employ in the enantioselective dearomatization of nonactivated arenes. Norcaradiene species Int-70 generated as a transition state and trapped via intermolecular [4 + 2]-cycloaddition to afford polycycles 72 with five consecutive stereogenic centers (Scheme 34). The reaction mechanism involved the dearomatization reaction in the presence [(S)-TRIPAg]2 (2.5 mol%), N-oxide 70 (2 equiv.), and MS 3 Å, in PhCl solvent at room temperature. It was followed by pericyclic reactions and [4 + 2]-cycloaddition reaction using 71a/71b (1 eq.) and PhCl solvent at room temperature which serves as the basis for achieving chemo and enantioselectivity. The reaction tolerated a range of ynamides with different aryl groups at the terminal position. The reaction observed wide substrate scope as ynamide terminally with aryl groups having electron-rich, as well as electron withdrawing substituents, were employed to access a series of functionalized fused-cyclohexenes with excellent yields and stereoselectivity (15 examples, 75–91% yields). On the other hand, the developed methodology faced certain limitations as intermolecular reactions with benzene did not afford the target product.75
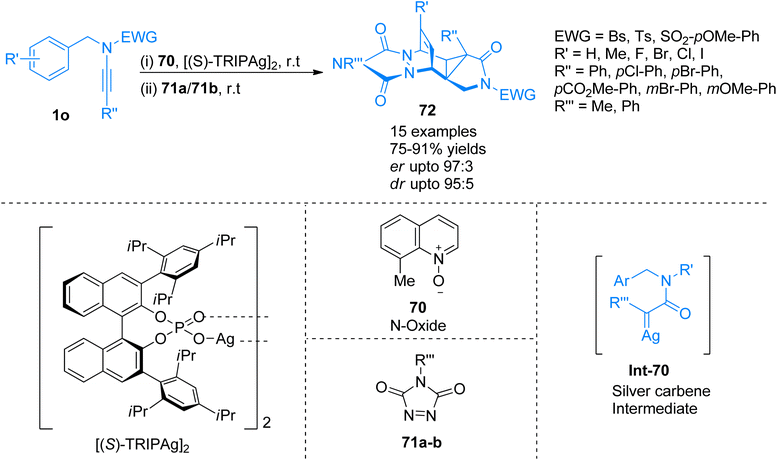 |
| | Scheme 34 Synthesis of polycycles 72 via [4 + 2]-cycloaddition of ynamide 1o. | |
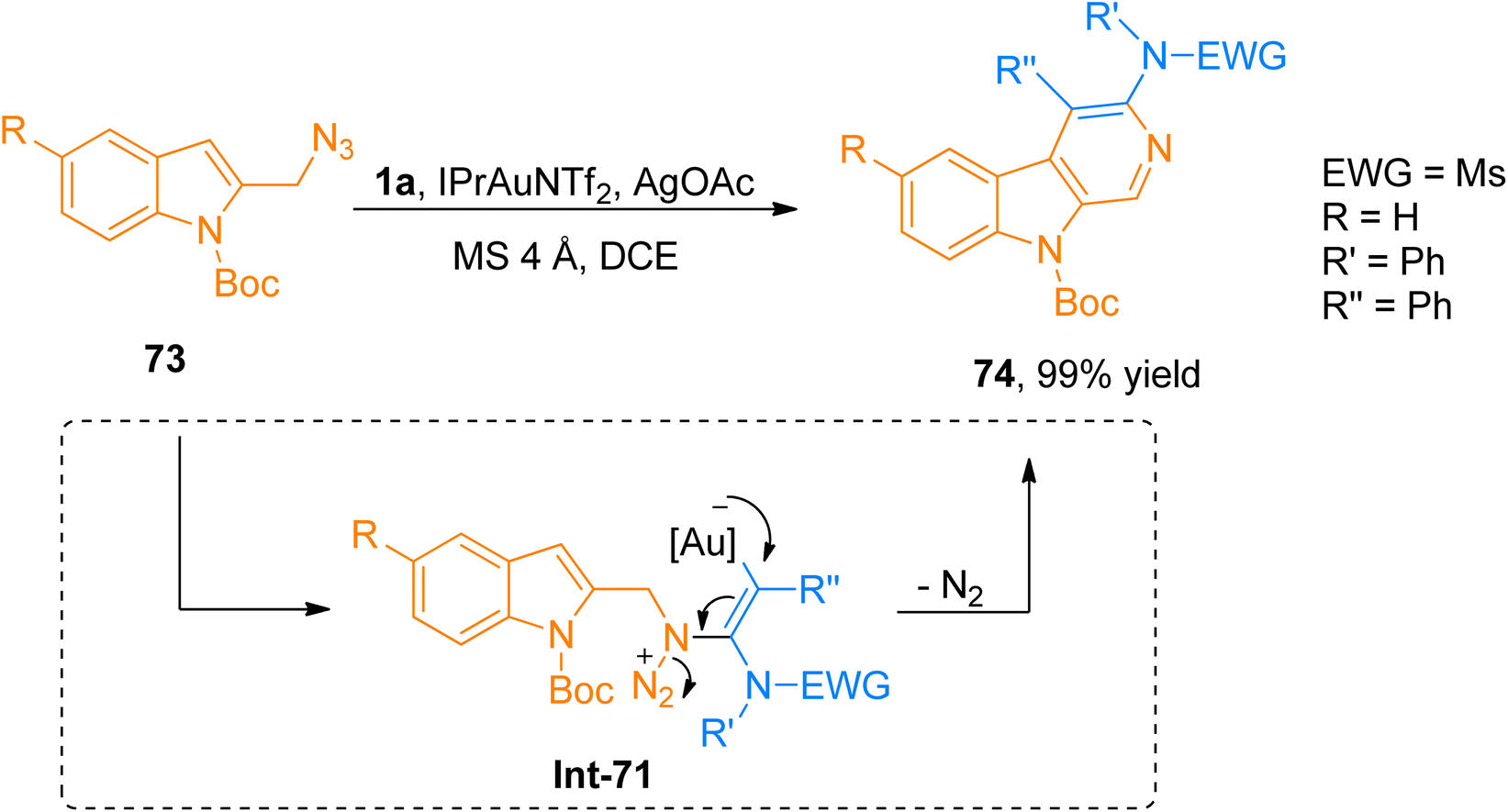
2.4.9. Synthesis of 3-amino-β-carboline via [4 + 2]-cycloaddition. Shu et al. (2015), disclosed the generation of α-imino-gold-carbenes during gold catalyst promoted intermolecular [4 + 2]-cycloaddition of ynamide and azide to provide regioselective access to the 3-amino-β-carboline product in promising yield range (17 examples, 65–99% yields). The synthesis of 3-amino-β-carboline 74 (99% yield) was afforded via the generation of α-imino carbene intermediate from ynamide 1a and azide 73 by the addition of IPrAuNTf2 (5 mol%), AgOAc (1.1 eq.), and MS 4 Å in dichloroethane solvent at 60 °C (Scheme 35). During the reaction, α-imino carbene intermediate got trapped by nucleophilic indolyl core to yield respective 1-H-pyrido[3,4-b]indole derivative which upon dehydrogenative oxidation afforded 3-amino-β-carboline species. When compared to previous methods reported for the synthesis of gold-carbene intermediate his protocol offers various advantages such as atom economical synthesis and flexible amino group introduction. A wide array of different ynamides and substituted azides were successfully engaged with the reaction. This transformation provided a straightforward route to access the biologically important 3-amino-β-carbolines having potent antitumor activities rather than tedious synthetic pathways, however, alkyl-substituted ynamide failed to afford the targeted product and furnished α,β-unsaturated imidamide instead of 3-amino-β-carboline.76
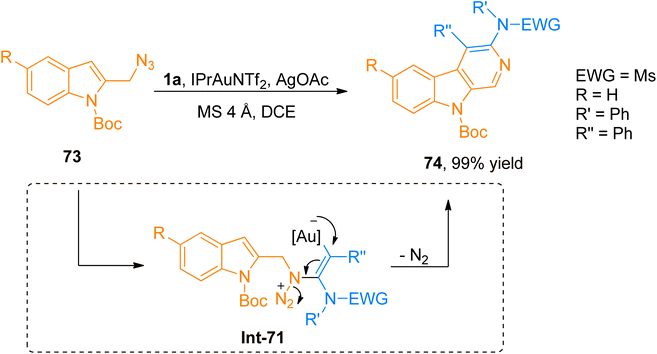 |
| | Scheme 35 Synthesis of 3-amino-β-carboline 74 via [4 + 2]-cycloaddition between ynamides 1a and azides 73. | |
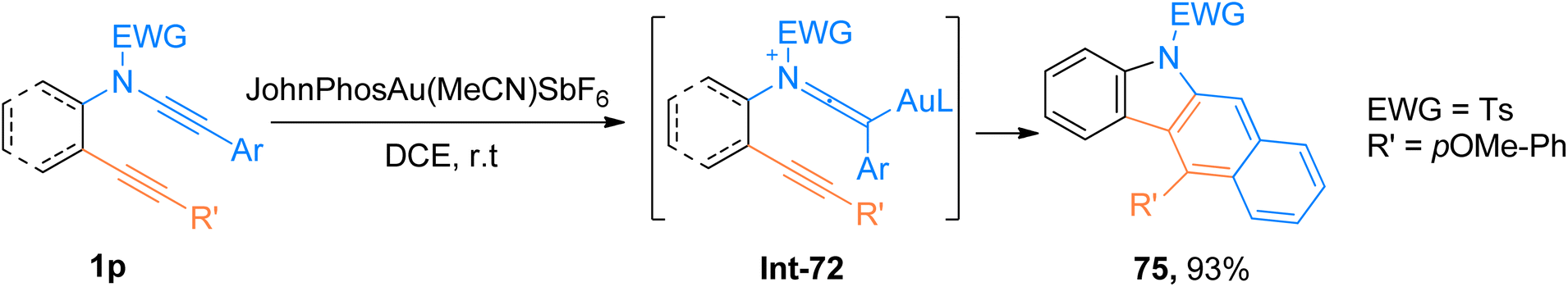
2.4.10. Synthesis of benzo[b]carbazoles via [4 + 2]-cycloaddition. Practical applications of tetradehydro Diels–Alder (TDDA) cycloaddition are limited due to the requirement of pre-functionalized substrates and harsh reaction conditions. As the TDDA allows the single-step construction of benzene rings, hence the demand for advancements in the facile and synthetically valuable TDDA is continuously increasing.77,78 Xu et al. (2018), carried out cycloisomerization of ynamide-ynes 1p following a gold catalyst-promoted dehydro-Diels–Alder reaction that provided facile access to benzo[b]carbazole derivatives. The cycloaddition involved the regioselective attack by alkyne scaffold to gold-ketenium intermediate which ultimately underwent benzannulation to yield the final product using JohnPhosAu(MeCN)SbF6 (5 mol%) catalyst and dichloroethane solvent at room temperature. Among synthesized series, compound 75 afforded the highest yield of 93% via the generation of gold-ketenium ion Int-72 as an intermediate during intramolecular cycloaddition of ynamide-ynes 1p (Scheme 36). This protocol offered a highly efficient route with mild reaction conditions and moderate substrate scope (15 examples, 18–93% yields), which also serves as a competitive alternative to the thermal dehydro-Diels–Alder reaction.79
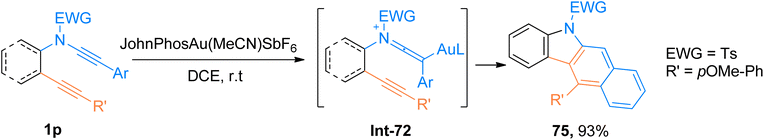 |
| | Scheme 36 Synthesis of benzo[b]carbazole derivatives 75 via intramolecular tetrahydro-Diels–Alder reaction of ynamides 1p. | |
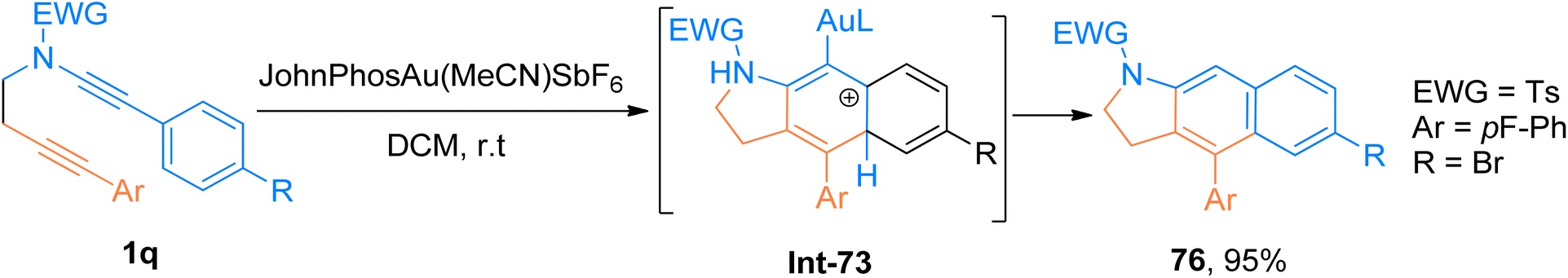
2.4.11. Synthesis of dihydrobenzo[f]indoles via [4 + 2]-cycloaddition. Prabagar et al. (2019), described gold-catalyzed intramolecular TDDA of aryl substituted ynamides 1q to access 2,3-dihydrobenzo[f]indole derivatives. Substrates having aryl groups at ynamide terminus are crucial for gold-catalyzed TDDA reaction using JohnPhosAu(MeCN)SbF6 (2 mol%), dichloromethane solvent. The reaction sequence involves the in situ construction of vinyl cation by following 5-exo-dig cyclization reaction of alkyne and Au-keteniminium ynamide species. The reaction was not possible if the vinyl cation do not get trapped by the aryl group substituted at the ynamide terminus. The protocol showed good tolerance for different substituents and afforded the products good to excellent yields (17 examples, 30–95% yields). The compound 76 was accessed in 95% yield via the formation of intermediate Int-73 during cycloaddition of 1q (Scheme 37).80
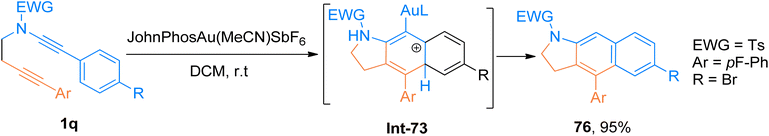 |
| | Scheme 37 Synthesis of dihydrobenzoindole derivatives 76 via intramolecular tetrahydro-Diels–Alder reaction of ynamides 1q. | |
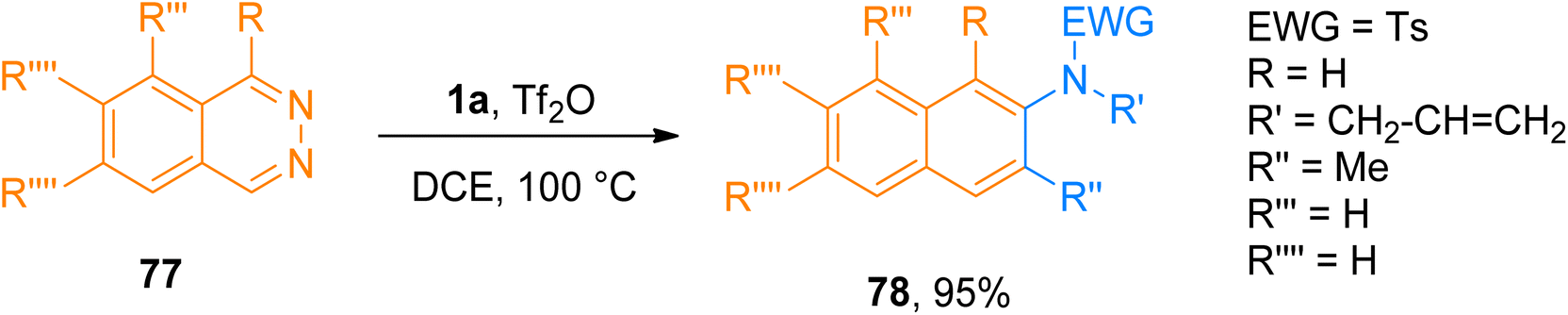
2.4.12. Synthesis of 2-aminonaphthalenes and 2-aminoanthracenes via [4 + 2]-cycloaddition. The cycloaddition reaction between diene (electron-rich) and dienophile (electron-deficient) species is called the normal electron demand Diels–Alder reaction. It has been well developed and has considerable applications in current organic synthesis.81–83 In contrast the Diels–Alder reaction of diene (electron-deficient) and dienophile (electron-rich) is known as inverse electron demand Diels–Alder reaction (IEDDA) which is relatively less developed.84,85 Xue et al. (2018), developed a novel metal-free IEDDA reaction between ynamides and 1,2-diazines to afford. Tf2O-catalyzed IEDDA allowed the efficient construction of functionalized 2-aminonaphthalene derivatives and 2-aminoanthracene derivatives in a promising yield range (24 examples, 56–95% yields). Moreover, the developed novel protocol offered a straightforward way to synthesize diversified products using a Tf2O catalyst and dichloroethane solvent at 100 °C. Product 78 was accessed using the developed methodology in 95% yield by IEDDA reaction of ynamide 1a and 1,2-diazine 77 (Scheme 38).86
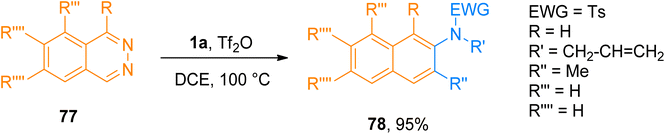 |
| | Scheme 38 Synthesis of 2-aminonaphthalenes 78 via IEDDA reaction between ynamides 1a and 1,2-diazines 77. | |
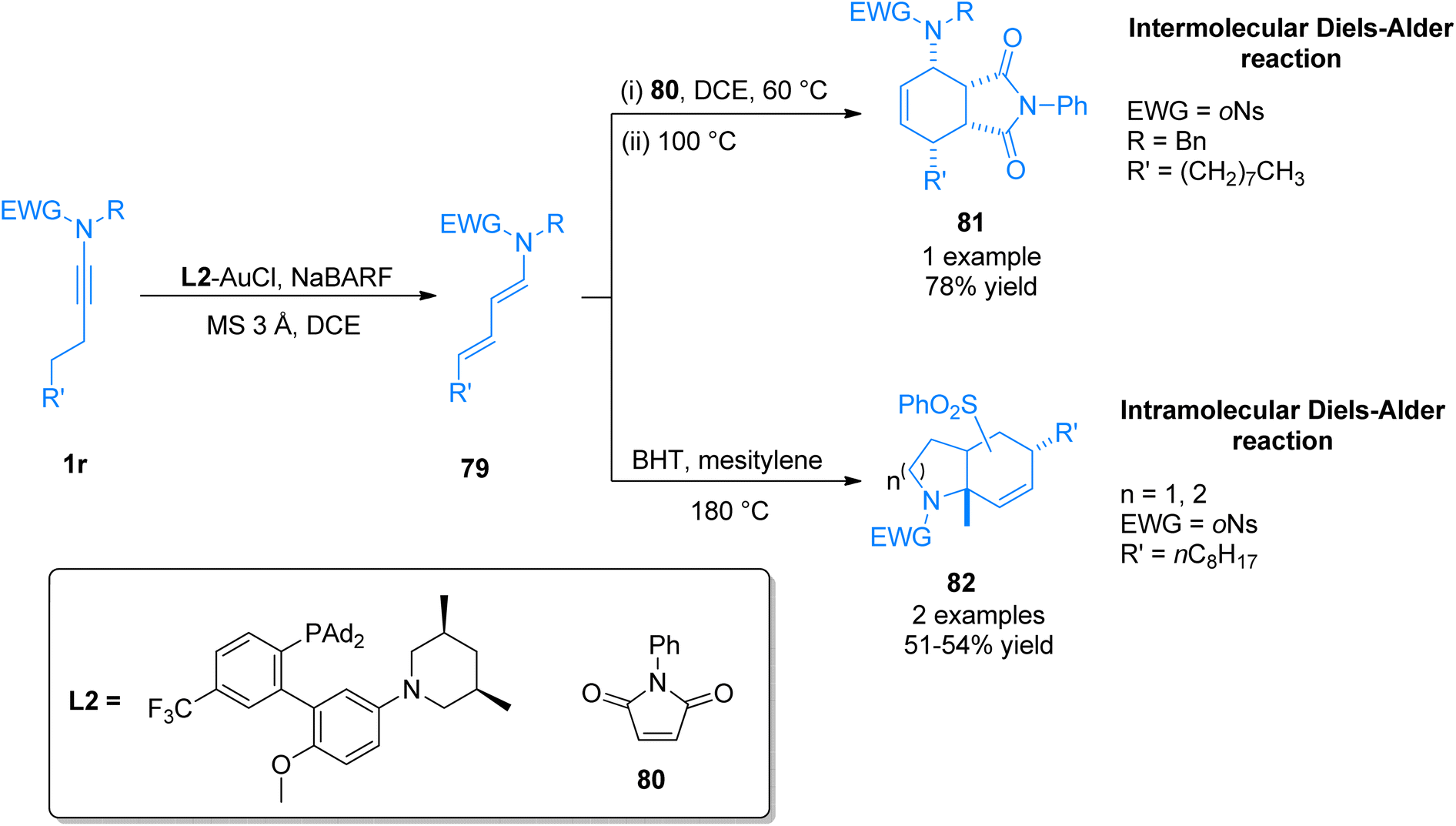
2.4.13. Synthesis of tetrahydroisoindoledione, hexahydroindole, octahydroquinoline via [4 + 2]-cycloaddition. Li et al. (2017), disclosed the synthetic pathway of gold catalyst promoted one-pot Diels–Alder reaction to furnish nitrogen-containing bicyclic scaffolds. The developed methodology directly converted N-alkynyl-o-nosylamides 1r to (1E,3E)-1-amido-1,3-dienes 79 in high diastereoselectivities and excellent yields. The reaction involves via gold-catalyzed ynamide isomerization reaction using L2-AuCl (5 mol%) catalyst, tetrakis[3,5-bis(trifluoromethyl)phenyl]boron sodium complex (NaBARF), MS 3 Å, and dichloroethane solvent at 60 or 80 °C. The isomerization reaction was followed by intermolecular or intramolecular one-pot Diels–Alder reaction to afford tetrahydroisoindoledione 81 (1 example, 78% yield) and hexahydroindole/octahydroquinoline 82 (2 examples, 51–54% yields). Intermolecular Diels–Alder reaction proceeded by treating allenamide 79 with N-phenylmaleimide 80 using dichloroethane solvent at 60 °C and the temperature was further raised to 100 °C to afford 81 in 78% yield (Scheme 56). While the intramolecular Diels–Alder reaction furnished 82 via the treatment of 79 with butylated hydroxytoluene (BHT) using mesitylene solvent at 180 °C (Scheme 39). The developed methodology afforded excellent (3E)-selectivity for distal (attached with β-carbon) carbon–carbon double bond along with good regioselectivity. In contrast, Brønsted acid-catalyzed reaction was associated with poor regio and stereo-selectivity.87
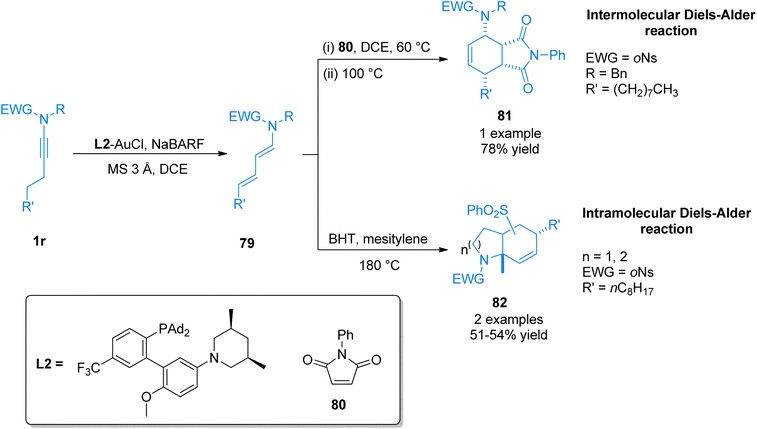 |
| | Scheme 39 Synthesis of bicyclic compounds 81 and 82 via one-pot Diels–Alder reaction of ynamide 1r. | |
2.5. [4 + 3]-cycloaddition
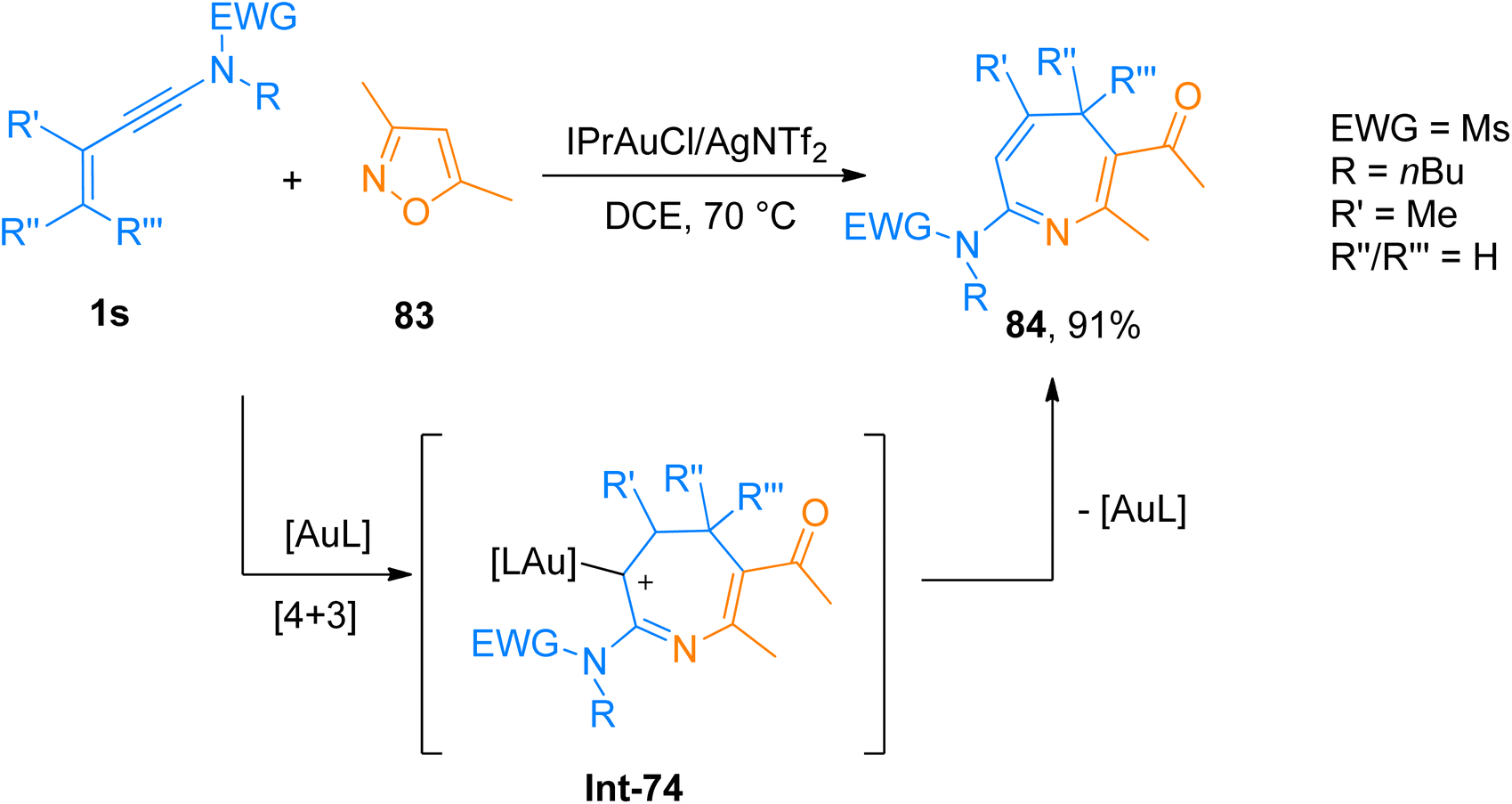
2.5.1. Synthesis of 4H-azepines via [4 + 3]-cycloaddition. Giri et al. (2018), developed [4 + 3]-cycloaddition reaction of 3-en-1-ynamides and isoxazoles to yield 4H-azepines using the gold catalyst. The reaction involved the 6π electrocyclization of in situ generated gold stabilized 3-azaheptatrienyl cation Int-74 to access 4H-azepines. The developed methodology employed a range of ynamides and substituted isoxazoles to afford the products in a favorable yield range using IPrAuCl/AgNTf2 (10 mol%) catalyst and dichloroethane solvent at 70 °C (23 examples, 16–91% yields). Among the synthesized series compound 84 was afforded in 90% yield by [4 + 3]-cycloaddition of ynamide 1s and functionalized isoxazole 83 (Scheme 40). The reaction could be extended to construct other valuable scaffolds such as highly substituted pyridine derivatives using Zn(OTf)2.70
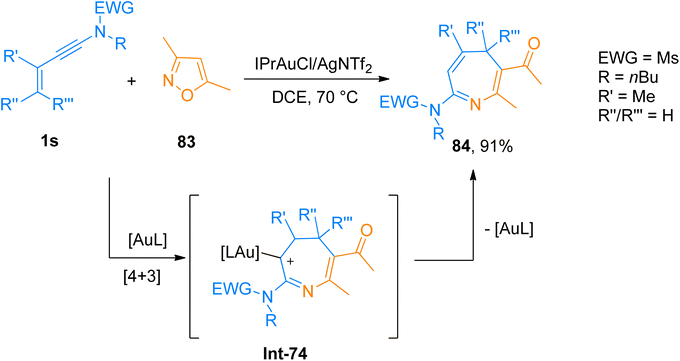 |
| | Scheme 40 Synthesis of 4H-azepines 84 via [4 + 3]-cycloaddition of ynamides 1s and substituted isoxazoles 83. | |
2.6. [8 + 2]-cycloaddition
2.6.1. Synthesis of cycloheptapyrroles via [8 + 2]-cycloaddition. High-order cyclization reactions are useful tools for the construction of medium sized rings. 8-Azaheptafulvenes is the most reliable partner to undergo the [8 + n]-cycloaddition reaction, more specifically the [8 + 2]-cycloaddition reaction provided an efficient route to access cycloheptapyrrole core that is pervasive in several bioactive compounds. [8 + 2]-cycloaddition of 8-azaheptafulvenes is of considerable interest to yield in situ generated intermediates88–90 and to achieve high enantioselectivity.91 Rodríguez et al. (2021), described [8 + 2]-cycloaddition reaction of ynamides 1a/1b and azaheptafulvenes 85 to furnish 2-amido-1,4-dihydrocycloheptapyrroles 86a/86b in a regioselective manner. This transformation was carried out using gold catalyst JohnPhos AuNTf2 (5 mol%), and toluene solvent at 110 °C (Scheme 41). The reaction mechanism involves the initiation with the construction of electrophilic intermediate Int-75 and Int-76 by the coordination of gold-cation intermediate to triple bond of ynamide 1a, followed by nucleophilic attack of 8-azaheptafulvene 85 to generate Int-77. The subsequent cyclization of Int-77 afforded Int-78 that undergo elimination of gold catalyst to realize the target products 86a (5 examples, 22–90% yields) and 86b (5 examples, 74–86% yields).92
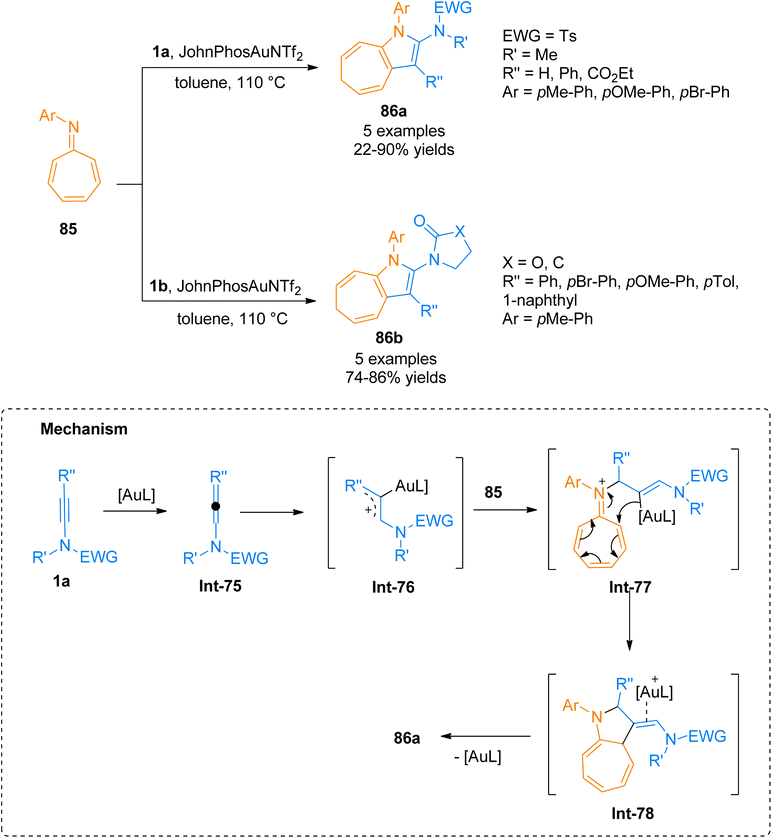 |
| | Scheme 41 Synthesis of 2-amido-1,4-dihydrocycloheptapyrroles 86a/86b via [8 + 2]-cycloaddition of ynamides 1a and 1b with 8-aryl-8-azaheptafulvenes 85, respectively. | |
2.7. [2 + 2 + 1]-cycloaddition
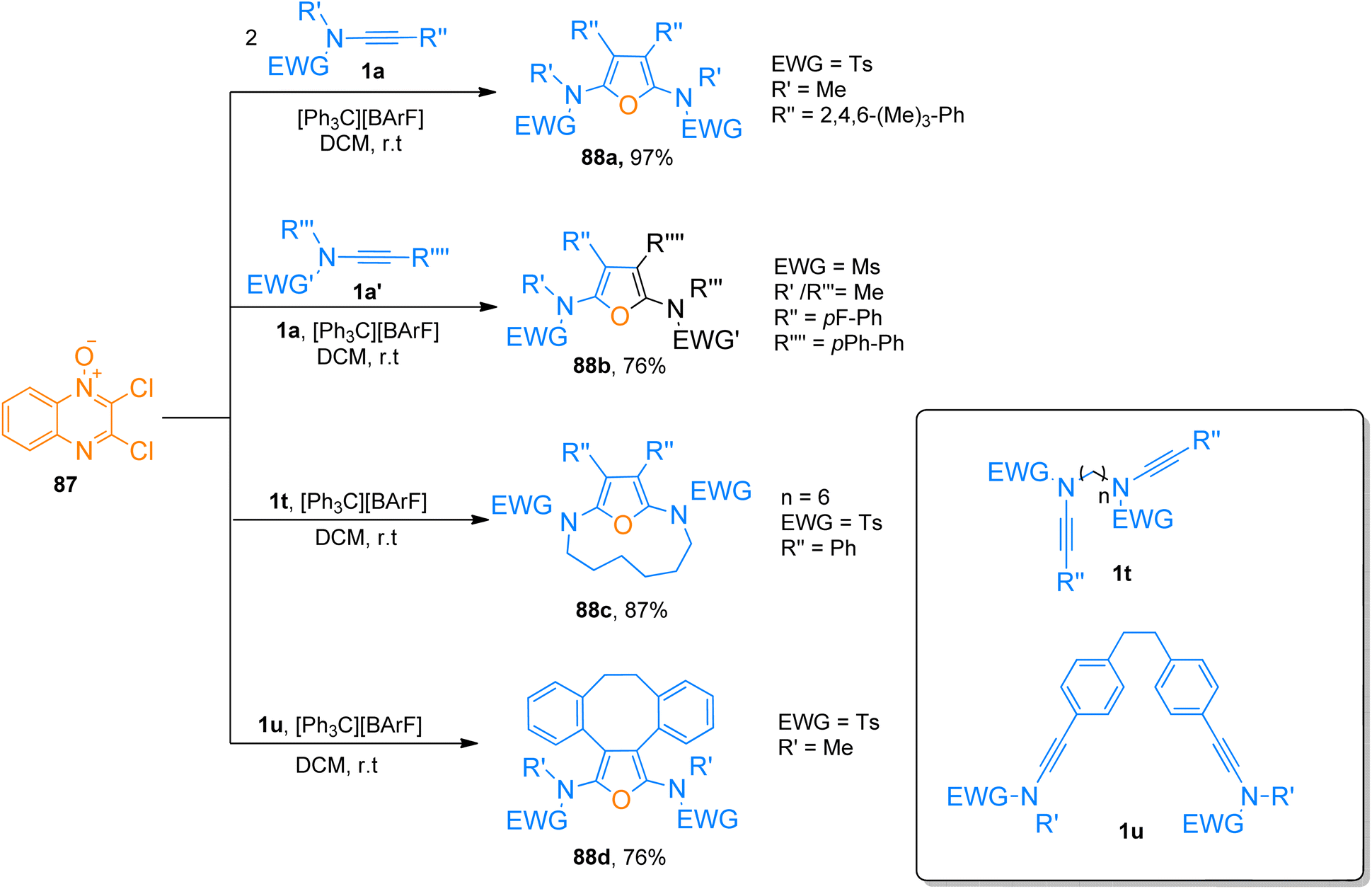
2.7.1. Synthesis of furans via [2 + 2 + 1]-cycloaddition. Jin et al. (2019), described trityl promoted oxidative [2 + 2 + 1]-cycloaddition of ynamides and quinoxaline N-oxide to construct highly substituted furan rings. This facile and convergent methodology with mild reaction conditions yielded symmetric as well as unsymmetric products (28 examples, 60–97% yields). The reaction mechanism involves the activation of the alkyne core using carbocation catalyst [Ph3C][BArF] (2 mol%) and dichloromethane solvent at room temperature. While the quinoxaline N-oxide prevented the over-oxidation of ynamides. The quinoxaline N-oxide 87 underwent cycloaddition with two equivalents of ynamide 1a to afford the furan derivative 88a with the highest yield of 97%. While to access 88b (76%) two different ynamides 1a and 1a′ were involved in cycloaddition with 87. The methodology also works for intramolecular macrocyclization of di-ynamides to realize macro-cyclic furan frameworks (9 examples, 20–87% yield). The macrocyclic products 88c (87%) and 88d (76%) were furnished by cycloaddition of 87 with di-ynamides 1t and 1u, respectively (Scheme 42).93
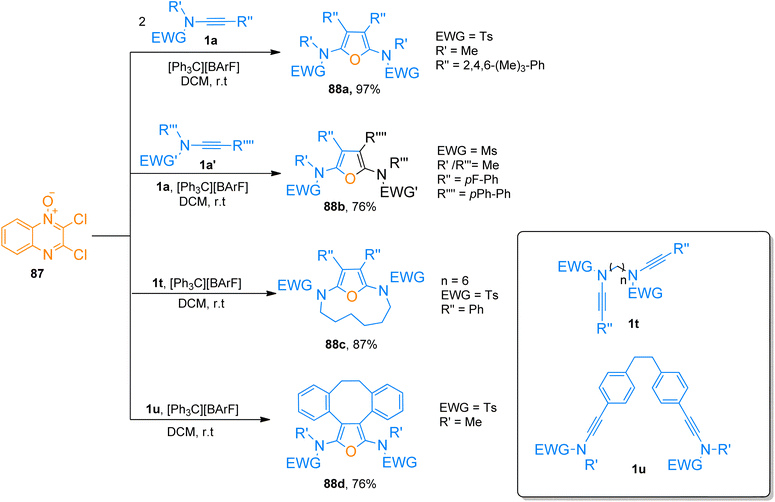 |
| | Scheme 42 Synthesis of highly substituted furan rings 88a–b and macro-cyclic furan derivatives 88c–d via [2 + 2 + 1]-cycloaddition of ynamides 1a, 1a′, 1t, and 1u with 2,3-dichloroquinoxaline-N-oxide 87. | |
2.8. [2 + 2 + 2]-cycloaddition
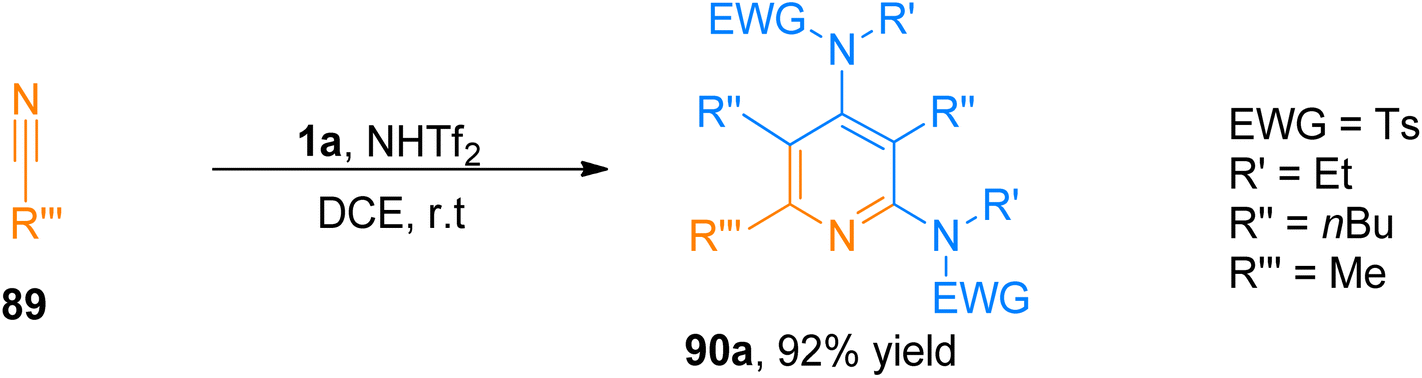
2.8.1. Synthesis of pyridines via [2 + 2 + 2]-cycloaddition. The construction of 2,4-diaminopyridine frameworks using a low-valent metal catalyst is a challenging task.94,95 Wang et al. (2016), disclosed a novel synthetic route to access highly substituted pyridines by following a metal-free, HNTf2 (10 mol%) promoted [2 + 2 + 2]-cycloaddition of ynamide and nitrile using dichloroethane solvent under nitrogen atmosphere at room temperature. Brønsted acid-catalyzed mild intermolecular cycloisomerization reaction sequence proceeded to achieve fully substituted pyridines because of the low nucleophilicity of the counter ion NTf2. The reaction sequence offered complementary excellent chemoselectivity as compared to gold-catalyzed synthesis and high regioselectivity (28 examples, 63–92% yields). Among the synthesized series the product, 90a was achieved with the highest yield of 92% by HNTf2-catalysed cycloaddition reaction of ynamide 1a and nitrile 89 (Scheme 43).96
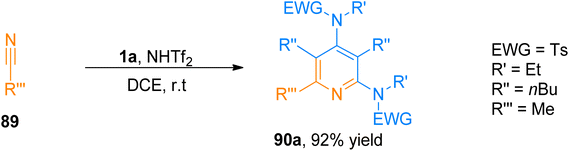 |
| | Scheme 43 Synthesis of 2,4-diaminopyridines 90a via [2 + 2 + 2]-cycloaddition of ynamide 1a with substituted nitrile 89. | |
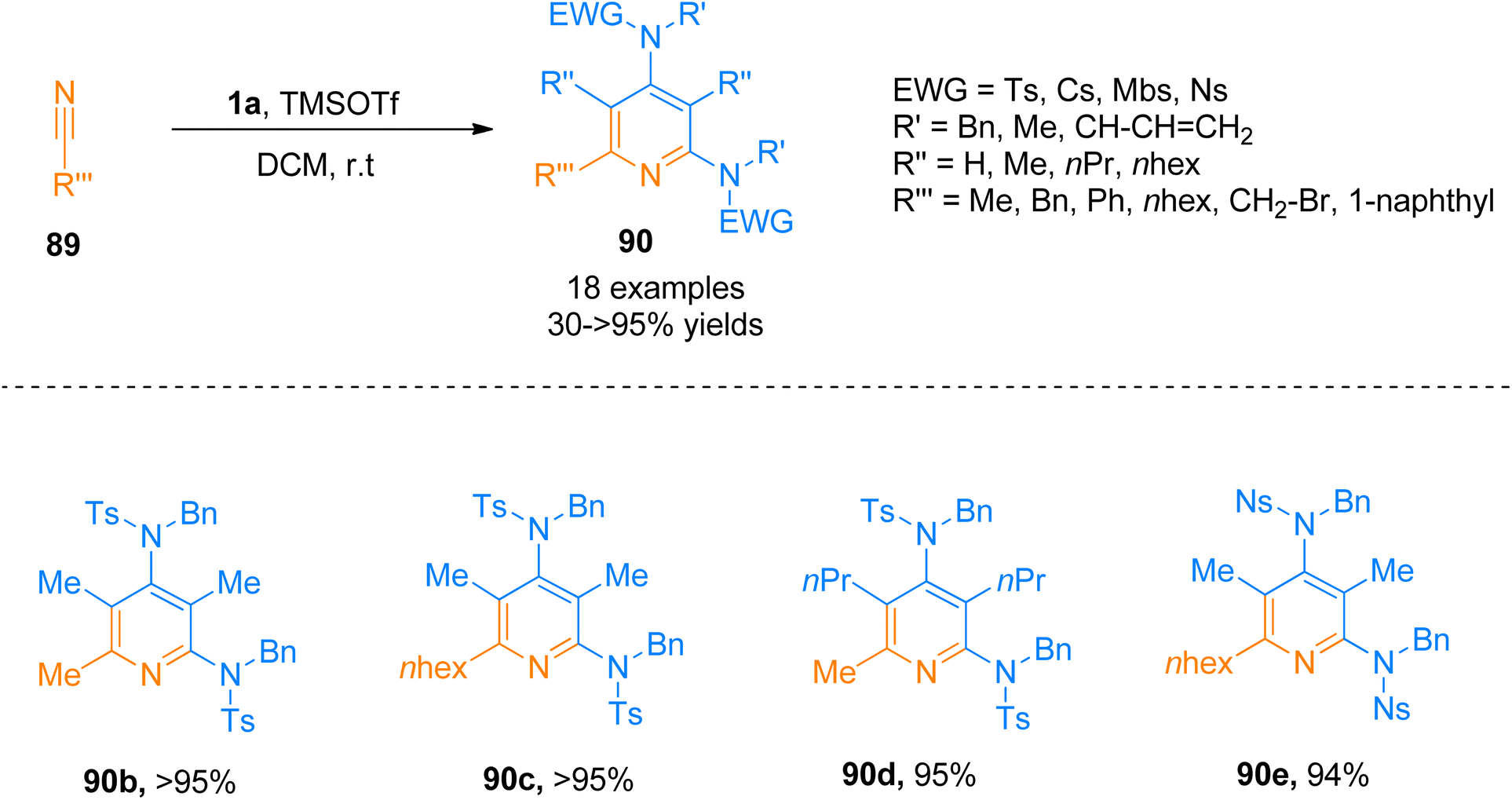
Similarly, Zhang et al. (2016), presented [2 + 2 + 2]-cycloaddition reaction of ynamides that provided a metal-free and straightforward route to afford 2,4-diaminopyridines. This novel protocol allowed the concomitant construction of carbon–carbon as well as the carbon–nitrogen bond between ynamide 1a and nitrile 89. The developed reaction exhibited moderate substrate scope, and afforded products with excellent chemoselectivity, and high regioselectivity (18 examples, 30 to >95% yields). Developed transformation offers facile access to 90 by TMSOTf (0.5 equiv.) catalyzed cycloaddition reaction of ynamide 1a and nitrile 89 using dichloromethane at room temperature (Scheme 44).97
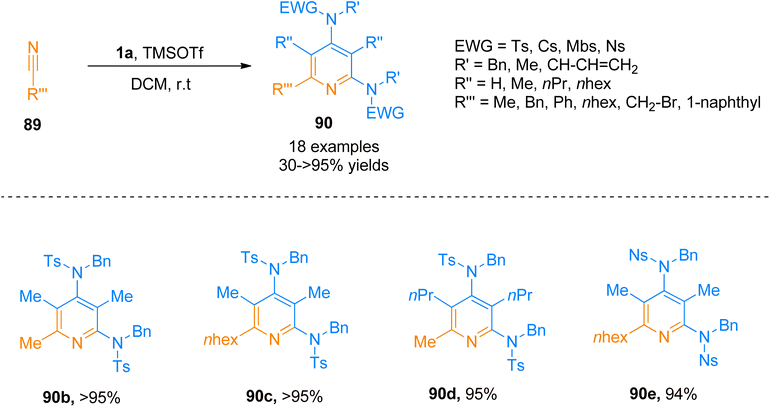 |
| | Scheme 44 Synthesis of 2,4-diaminopyridines 90 via [2 + 2 + 2]-cycloaddition of ynamides 1a with substituted nitrile 89. | |
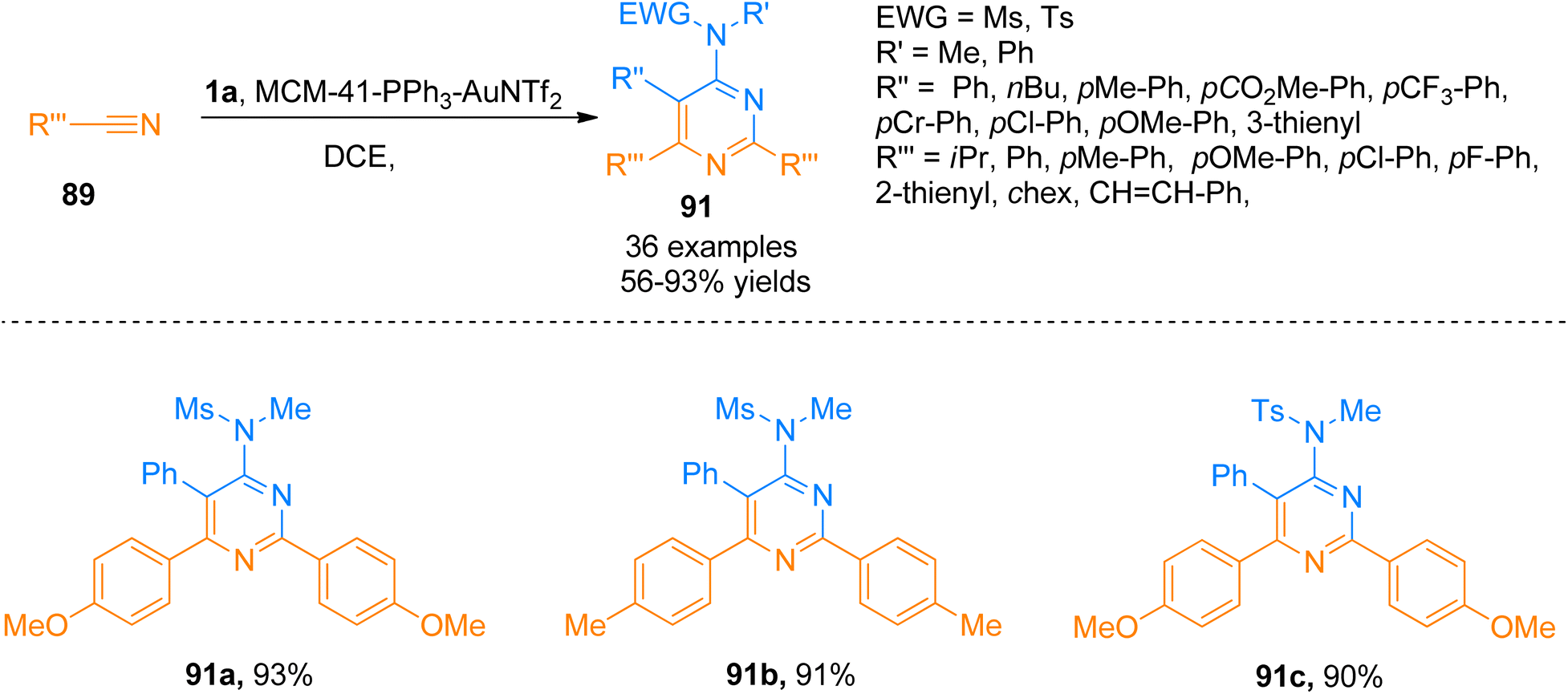
2.8.2. Synthesis of pyrimidines via [2 + 2 + 2]-cycloaddition. 4-Aminopyrimidines are biologically active scaffolds that have considerable importance in pharmaceutical chemistry. These scaffolds are biologically active cores in numerous natural, pharmaceutical and agrochemical products. Moreover, these cores also serve as precursors to realizing the construction of versatile products. Previously reported intermolecular [2 + 2 + 2]-cycloaddition reactions of alkyne with nitriles promoted by metal catalysts were limited to the synthesis of pyridines only rather than pyrimidines.98,99 Karad and Liu (2014), for the first time, described an intermolecular [2 + 2 + 2]-cycloaddition between ynamides and two discrete nitriles to construct monomeric 4-aminopyrimidines (27 products with 45–93% yields).98 One of the drawbacks of the developed transformation realizing the synthesis of 4-aminopyrimidines was the use of expensive catalysts. To overcome this problem, a metal-free TfOH-mediated [2 + 2 + 2]-cycloaddition was designed and realized by Chen et al. (2016) (32 examples, 36–97% yields).100Further, Liu et al. (2018), developed a highly efficient novel [2 + 2 + 2] annulation reaction between ynamide 1a and nitriles that provided a general and powerful tool to access a variety of tetra-substituted pyrimidines or monomeric aminopyrimidines. This reaction presented the first reported example of gold(I) promoted cycloaddition for the synthesis of pyrimidines 91 using mild conditions that presented an alternative to classical synthetic transformations. The presented atom economical protocol afforded the product 91 (36 examples, 56–93% yields) using MCM-41-immobilized phosphine gold(I) complex (MCM-41-PPh3-AuNTf2, 5 mol%) catalyst, mild reaction conditions (80 or 30 °C), and dichloroethane. The gold catalyst used in this annulation reaction is recoverable and reusable for up to 8 runs without any substantial loss of catalytic activity. The developed methodology is tolerant to a range of functional groups yielding a wide variety of tetrasubstituted pyrimidines (Scheme 45).101
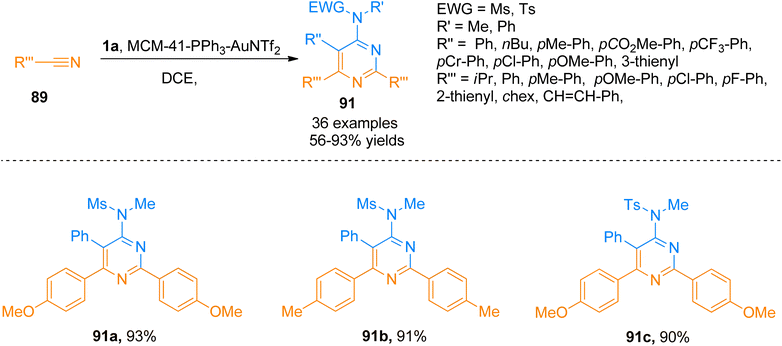 |
| | Scheme 45 Synthesis of substituted pyrimidines 91 by [2 + 2 + 2] cycloaddition of substituted nitrile 89 and ynamide 1a. | |
Theoretical and computational studies with the help of DFT calculations were undertaken by Liang et al. (2016), on the novel [2 + 2 + 2]-cycloadditions between ynamide and two nitrile species promoted by gold catalyst.102 The reaction performed by the Liu group101 was taken as a model reaction in which ynamide 1a and nitrile underwent cycloaddition to afford 4-aminopyridine using PPh3Au+ X− (5 mol%) (X = NTf2−, SbF6−, OTf−) catalysts in dichloroethane solvent at 75 °C. The study involved the explanation of the possible mechanism involved in transformation and the part that the electron withdrawing group attached to N of ynamide 1a plays in the reaction. The study confirmed that gold-ynamide species 1a induced dimerization of two nitriles to construct the target product while the catalyst alone was unable to do so. The EWG attached to the N of alkyne resulted in the stabilization of the electron pair on the nitrogen atom while the electron-donating group did not favor the reaction. The study provided a comprehensive understanding of the catalytic [2 + 2 + 2]-cycloaddition reaction that may be helpful for the design of further reactions.102
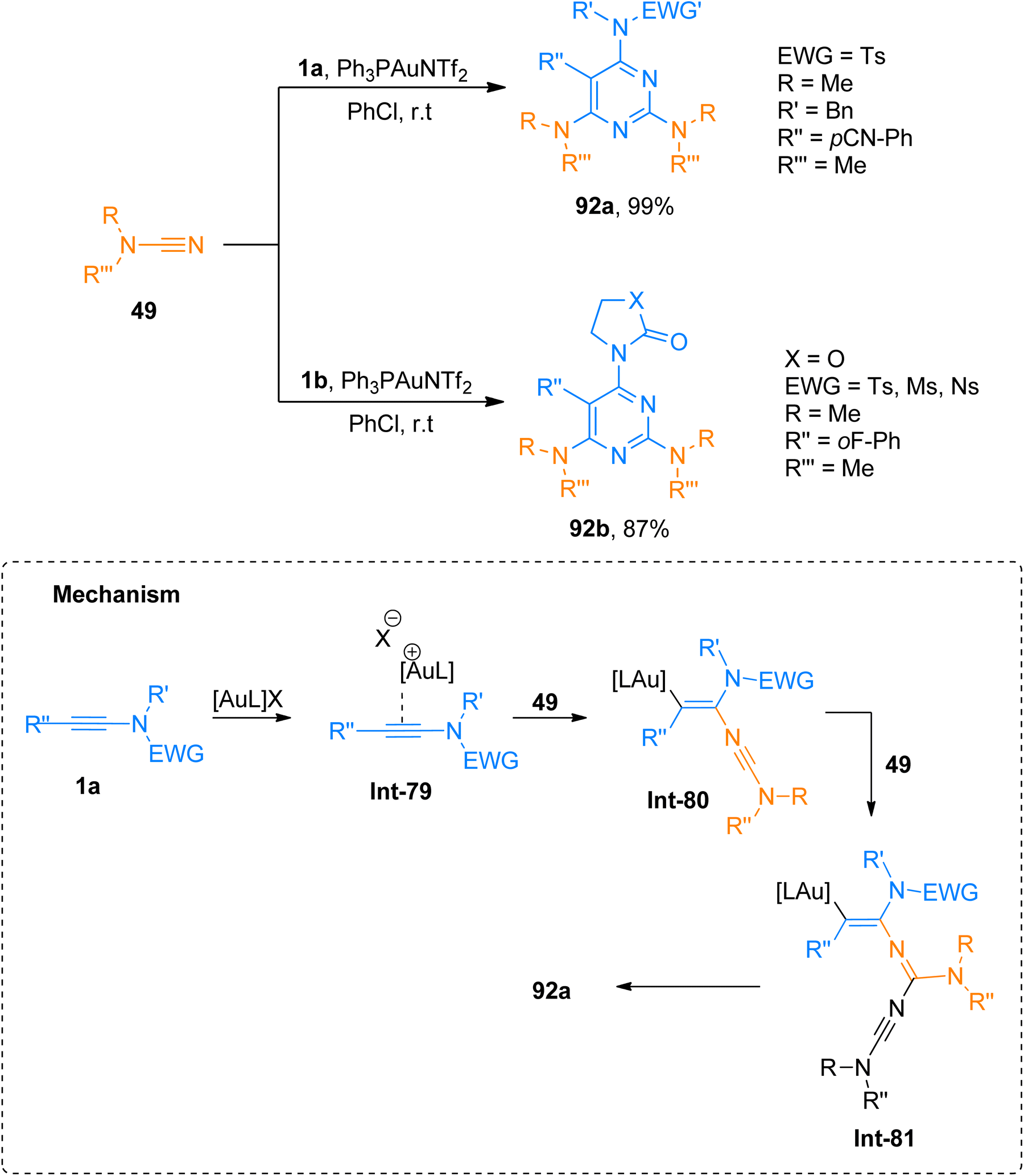
Dubovtsev et al. (2020), described the interaction between ynamide and cyanamide reaction during a gold catalyzed cycloaddition that demonstrated condition-dependent mechanistic selectivity. The [2 + 2 + 2]-cycloaddition proceeded via formation of Int-79 to Int-81 using kinetically controlled conditions of Ph3PAuNTf2 (5 mol%) catalyst and PhCl at room temperature to afford 2,4,6-triaminopyrimidines (19 examples, 35–99% yields). The methodology was well-tolerated by a range of substrates and dialkyl, diaryl, and heterocyclic amino-functional groups could be incorporated into the target pyrimidine framework. The developed methodology offers the advantages of mild reaction conditions, regioselectivities, and versatile products via post-functionalization. Among the synthesized series, the compounds 92a and 92b were obtained in the highest yields of 99% and 87% via the cycloaddition of cyanamide 49 with 1a and 1b, respectively (Scheme 46).61
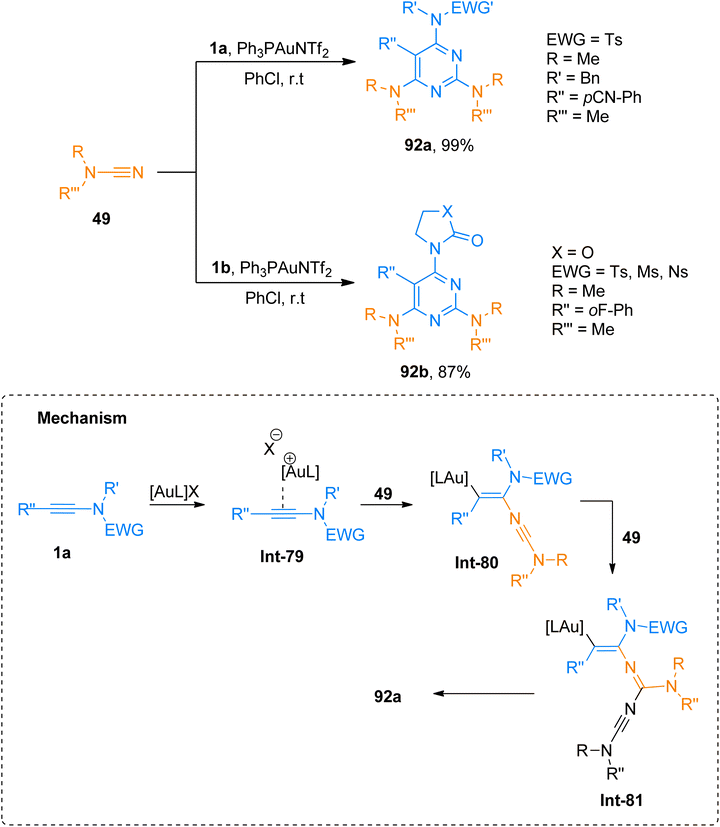 |
| | Scheme 46 Synthesis of 2,4,6-triaminopyrimidines 92a and 92b via [2 + 2 + 2]-cycloaddition of cyanamides 49 and ynamides 1a and 1b, respectively. | |
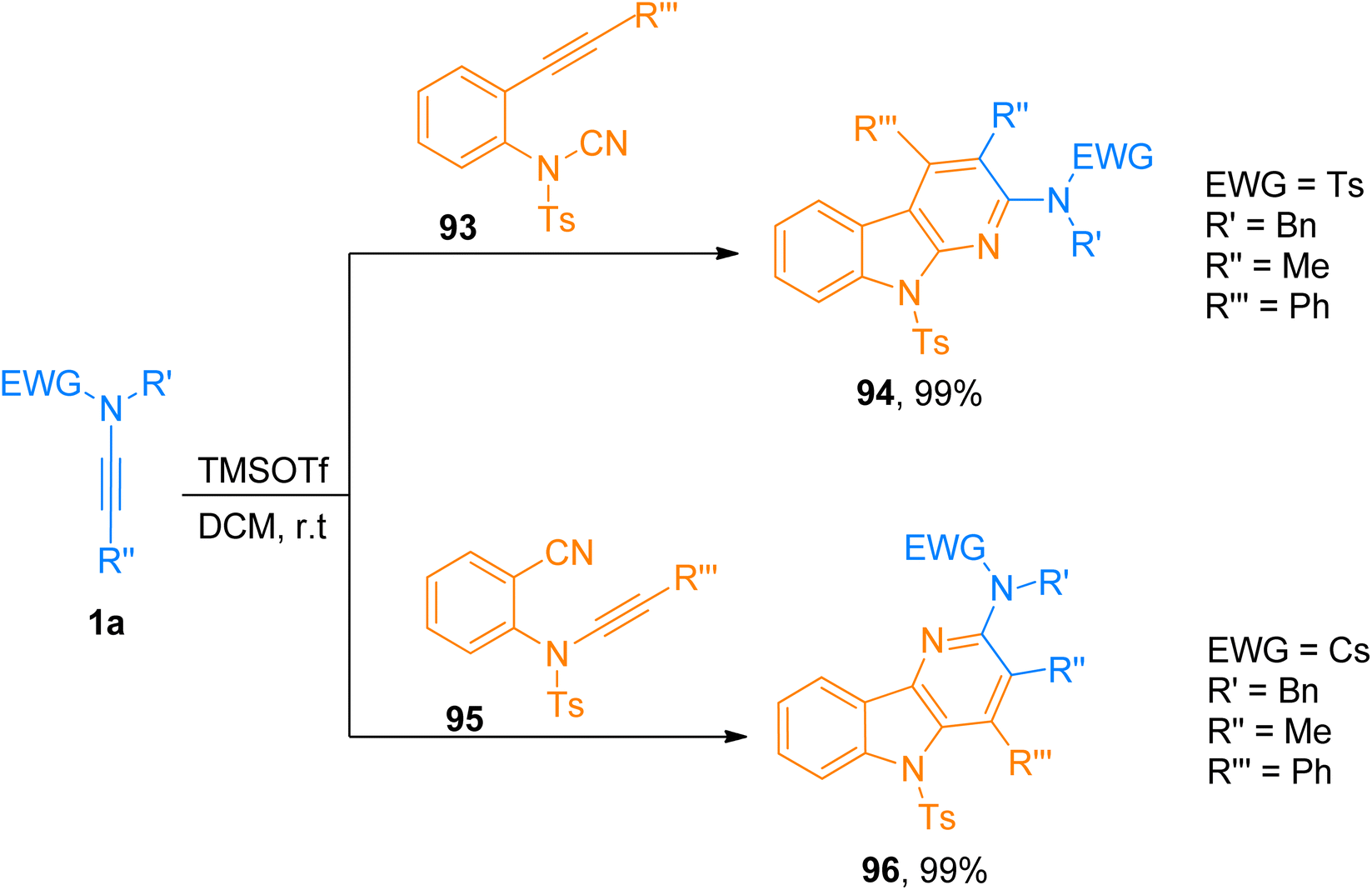
2.8.3. Synthesis of 2-amino-α-carbolines. Zhang et al. (2019), presented a [2 + 2 + 2]-cycloaddition of ynamides with alkyne-cyanamides and ynamide-nitriles in the metal-free environment to yield amino substituted α-carbolines and δ-carbolines, respectively. The developed efficient methodology provided an environment-friendly synthetic route with wide functional group compatibility for the highly regioselective construction of carboline derivatives in a promising yield range. This TMSOTf (0.2 equiv.) catalyzed cycloaddition reaction in dichloromethane at room temperature is the first method to realize the synthesis of 2-amino-α-carbolines (26 examples, 35–99% yields) and 3-amino-δ-carbolines (27 examples, 40–99% yields) via cycloaddition. Among the series of 2-amino-α-carbolines, compound 94 was accessed in 99% yield by cycloaddition of ynamide 1a and alkyne-cyanamide 93. While from the series of 3-amino-δ-carbolines, compound 96 obtained in 99% yield via cycloaddition of ynamide 1a and ynamide-nitrile 95 (Scheme 47).103
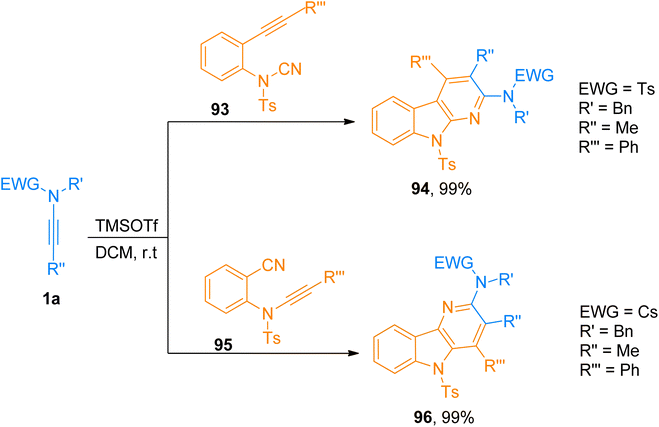 |
| | Scheme 47 Synthesis of 2-aminocarbolines 94 and 3-aminocarbolines 96 via [2 + 2 + 2]-cycloaddition of ynamide 1a with alkyne-cyanamide 93 or ynamide-nitrile 95. | |
2.9. [5 + 1] and [5 + 2]-cycloaddition
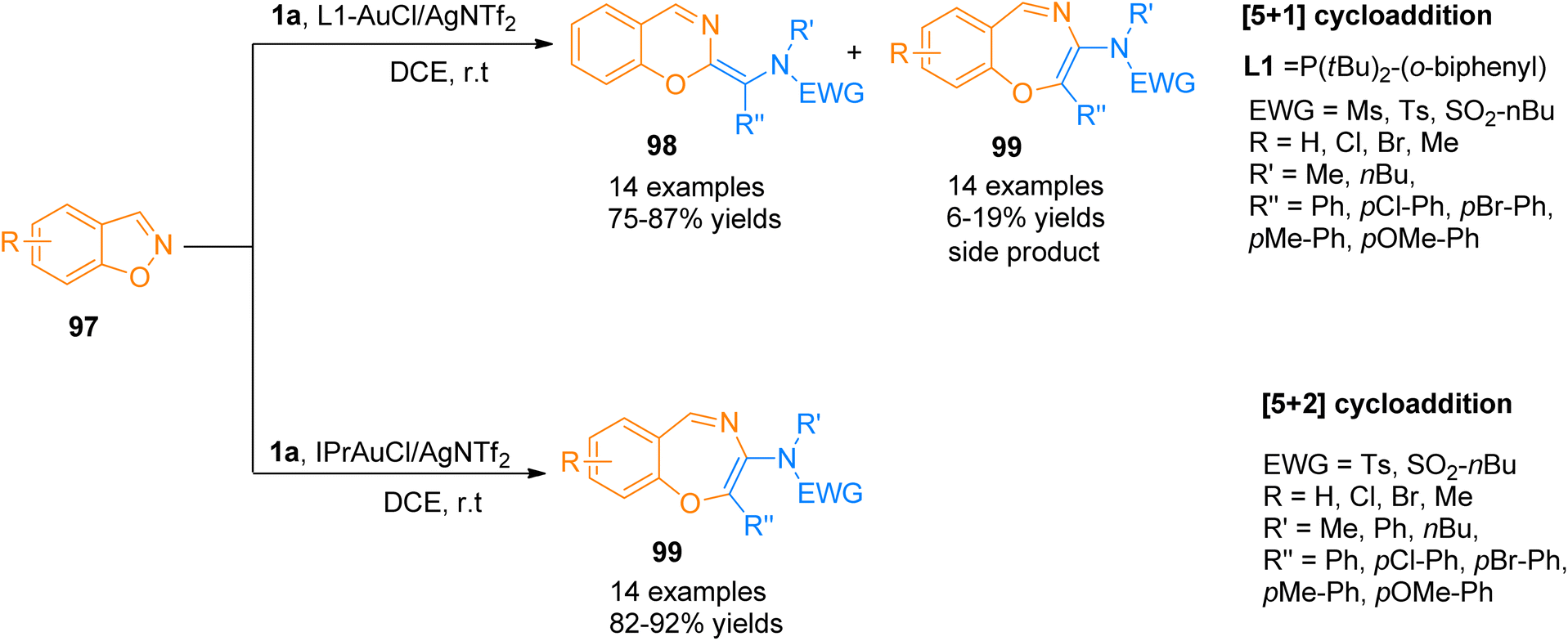
2.9.1. Synthesis of oxazines and oxazepines via [5 + 1] and [5 + 2]-cycloadditions. Jadhav et al. (2018), described gold-catalyzed ligand controlled chemoselective [5 + 1] and [5 + 2] annulation reactions between ynamides 1a and 1,2-benzisoxazoles 97. Using L1-AuCl/AgNTf2 catalyst (5 mol%) using dichloroethane solvent at room temperature, aryl-substituted ynamides 1a underwent [5 + 1]-cycloaddition reaction and yielded six-membered heterocycles 98 as the major products (14 examples, 75–87% yields) along with seven-membered heterocycles 99 as side products. While the chemoselectivity could be switched to [5 + 2]-cycloaddition reaction by using IPrAuCl/AgNTf2 (5 mol%) catalyst and dichloroethane solvent at room temperature to yield benzo[f][1,4]oxazepines 99 (14 examples, 82–92% yields) (Scheme 48). However, for some of the ynamides, chemoselectivity remained unaffected by the change in the gold catalyst. The suggested mechanism for the reaction involves the construction of a common intermediate that underwent two distinct types of N–O cleavage to afford gold carbenes that yielded final products either by [5 + 1] or [5 + 2]-cycloaddition reaction.104
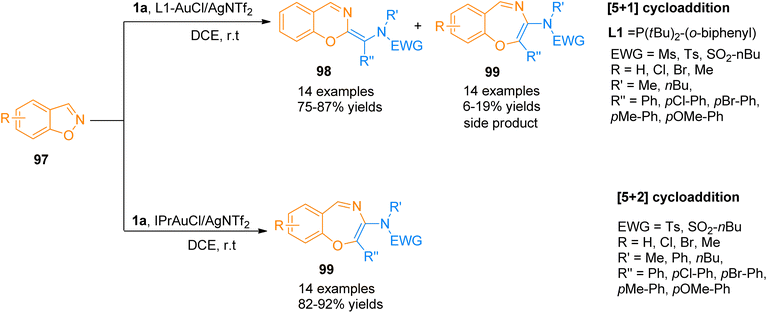 |
| | Scheme 48 Synthesis of six-membered heterocycles 98 and seven-membered heterocycles 99 via [5 + 1] and [5 + 2]-cycloaddition of ynamide 1a and 1,2-benzisoxazole 97. | |
2.10. [5 + 2 + 1]-cycloaddition
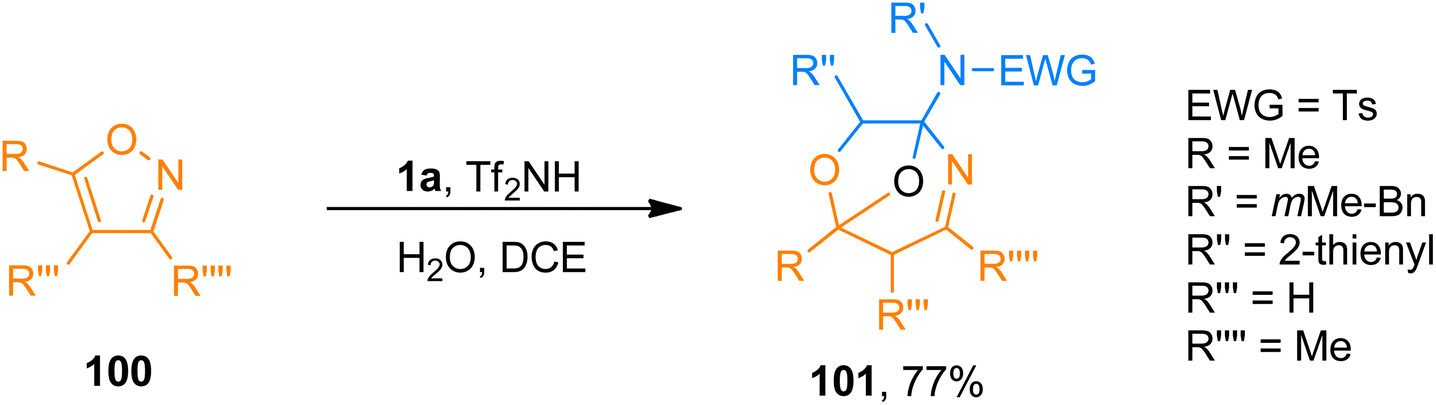
2.10.1. Synthesis of oxygen-bridged tetrahydro-1,4-oxazepines via [5 + 2 + 1]-cycloaddition. Zhao et al. (2018), presented [5 + 2 + 1]-cycloaddition of ynamides, isoxazoles, and water catalyzed by Brønsted acid that yielded oxygen-bridged tetrahydro-1,4-oxazepine derivatives. The Tf2NH (15 mol%) catalyzed transformation provides an atom economical synthesis of the oxygen-bridged cycloadducts with distinct selectivity than gold-catalyzed reactions using H2O and dichloroethane solvent at −10 °C or −20 °C. The oxygen of the bridge originates from water. The reaction exhibited good substrate scope with promising yields (27 examples, 40–77% yields). Among the series of oxygen-bridged tetrahydro-1,4-oxazepines, this novel protocol provided access to product 101 (77% yield) by [5 + 2 + 1]-cycloaddition of ynamide 1a, isoxazole 100, and water (Scheme 49).105
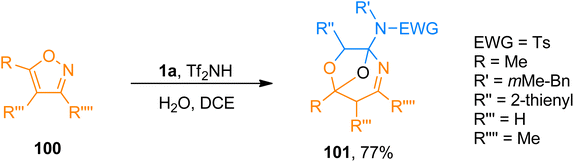 |
| | Scheme 49 Synthesis of tetrahydro-1,4-oxazepine derivatives 101 via [5 + 2 + 1]-cycloaddition of ynamide 1a, and isoxazole 100. | |
2.11. Sydnone alkyne cycloaddition
2.11.1. Synthesis of pyrazoles via sydnone alkyne cycloaddition. Sydnones are one of the most important scaffolds belonging to meso-ionic heteroaromatic compounds that take part as 1,3 dipoles during cycloaddition with alkynes.106,107 Such cycloaddition reactions require electron-deficient dienophiles, high temperature, and long reaction time for better yields, however, the regioselectivity of the pyrazole products usually depends on precursors.108 Wezeman et al. (2017) reported improved sydnone-alkyne cycloaddition with significant substrate scope and functional group tolerance. 4-Aminopyrazoles 103 (4 examples, 54–64% yield) were accessed via copper-mediated cycloaddition of aliphatic ynamides 1a and sydnones 102 using CuSO4·5H2O, 1,10-phenanthroline, sodium ascorbate, Et3N, and tBuOH:H2O (1:1). Moreover, a mixture of 4,5,6,7-tetrahydro-2H-pyrazolo[x,y-b]pyridine isomers was accessed from the reaction of sydnones with strained ynamide 1k via copper-free sydnone alkyne cycloaddition in acetonitrile solvent at room temperature (8 examples, 47–94% yields). Strained ynamides 1k generated in situ via reaction of N-Ts-azacyclohexyne with CsF. The isomers products 104a and 104b were afforded by strain-promoted in situ generated ynamide 1k and syndone 102 in 2:7 with an overall yield of 94% (Scheme 50).109
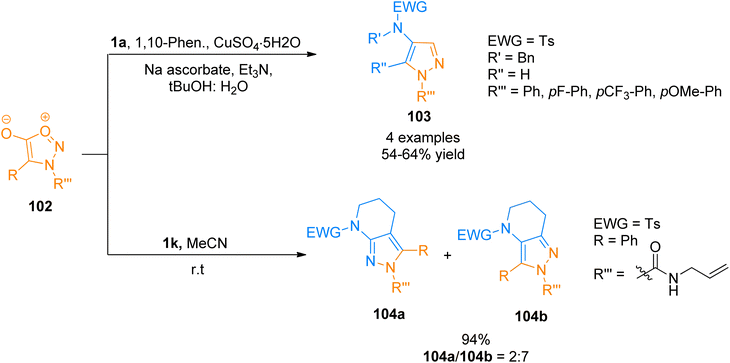 |
| | Scheme 50 Synthesis of 4-aminopyrazoles 103 and 4,5,6,7-tetrahydro-2H-pyrazolo[x,y-b]pyridine 104a and 104b via sydnone-alkyne cycloaddition of aliphatic ynamides 1a and 1k with sydnones 102. | |
2.12. Applications
2.12.1. Synthetic applications. During the last decade, extensive work on ynamides revealed their attractive synthetic features and broad application spectrum that considerably stimulated the development of the asymmetric synthesis of N-heterocycles.110–118 In addition to this, the synthetic applications of the developed methodologies for the total synthesis of natural products with biological and optoelectronic properties have been achieved.119–121 Further the application of the transformations could be extended for the synthesis of biologically active and pharmaceutical molecules via late-stage functionalization.52,56,64,98,100,130
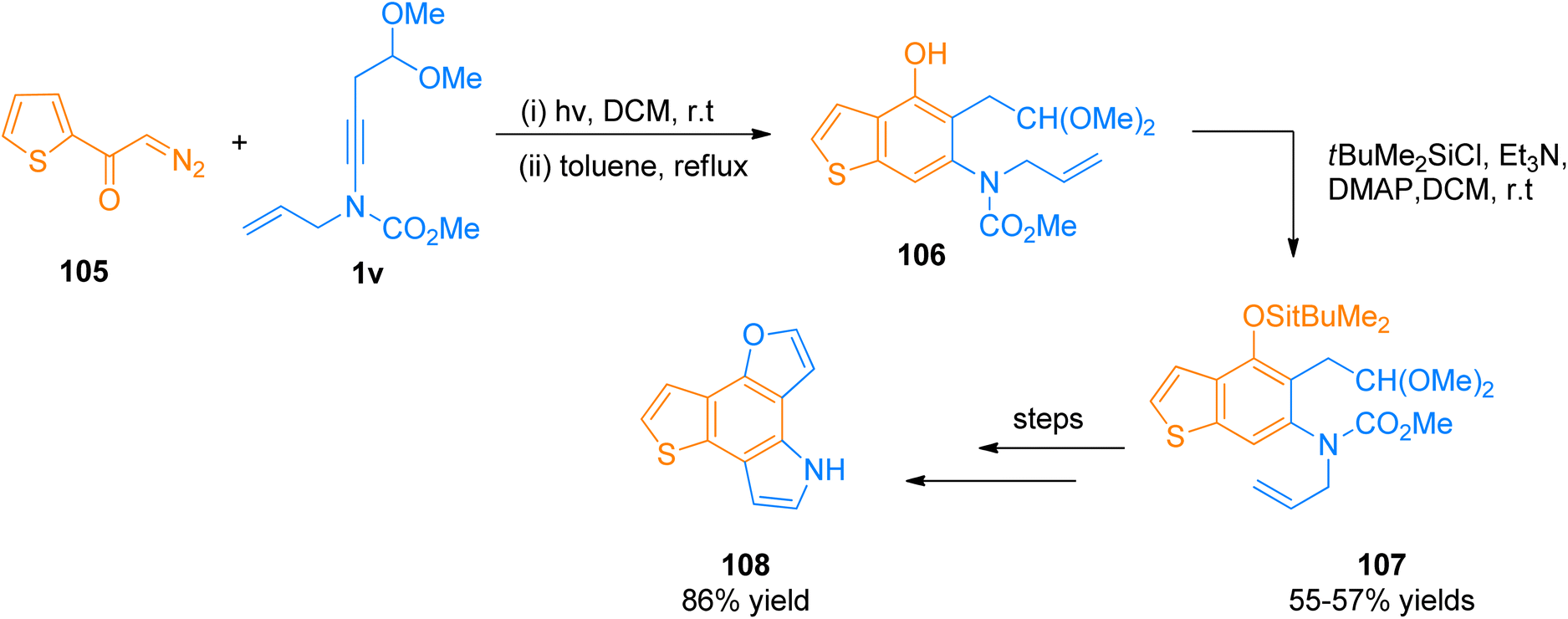
2.12.1.1. Total synthesis of furo[2,3-g]thieno[2,3-e]indole (FTI). Synthesis of FTI containing tetracyclic system was carried out by Forneris et al. (2018). Photochemically induced benzannulation of ynamide and α-diazo ketone afforded functionalized benzothiophene that acts as a building block for the construction of N and O-containing heterocyclic rings. The powerful methodology involved the in situ realization of vinyl ketene intermediate by the electrocyclic ring opening or photochemical Wolff rearrangement of cyclobutenone or α-diazo ketone precursor, respectively. The benzannulation sequence includes four discrete pericyclic reactions. Vinyl ketene intermediate reacted with highly substituted benzene in a regioselective [2 + 2]-cycloaddition, 4π-electron electrocyclic ring-opening, and 6π-electrocyclization manner to afford vinylcyclobutenone intermediate, dienylketene intermediate, and tetracyclic aromatic benzannulation product respectively.122 The synthetic transformation involves the benzannulation of ynamide 1v and diazo ketone 105 via regioselective [2 + 2]-cycloaddition, 4π-electron electrocyclic ring-opening, and 6π-electrocyclization reaction sequence in two steps. The reaction was carried out by irradiation of the degassed solution in dichloromethane at room temperature followed by reflux in toluene as solvent. The benzannulation product 106 was subjected to column chromatography for purification before the next reaction. In the next step, the phenolic hydroxyl group of compound 106 underwent protection by TBDMS using tBuMe2SiCl, Et3N, 4-DMAP, and dichloromethane solvent at room temperature to furnish silyl ether derivative 107 in 55–57% overall yield starting from 106. Further, the colorless FTI 108 was afforded by passing through several steps such as oxidative cleavage via modified Lemieux-Johnson conditions, deprotection of silyl ethers, and one-pot double cyclization (Scheme 51).122
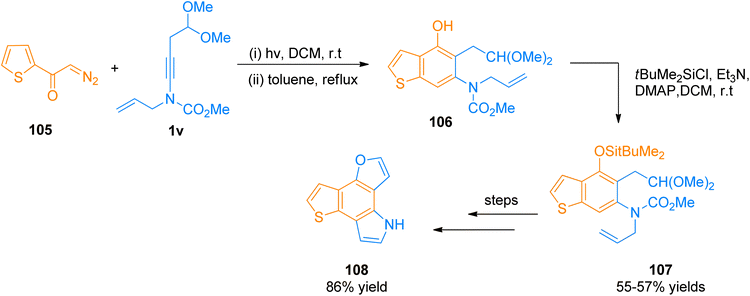 |
| | Scheme 51 Synthesis of FTI 108 via [2 + 2]-cycloaddition ynamide 1v with diazo ketone 105. | |
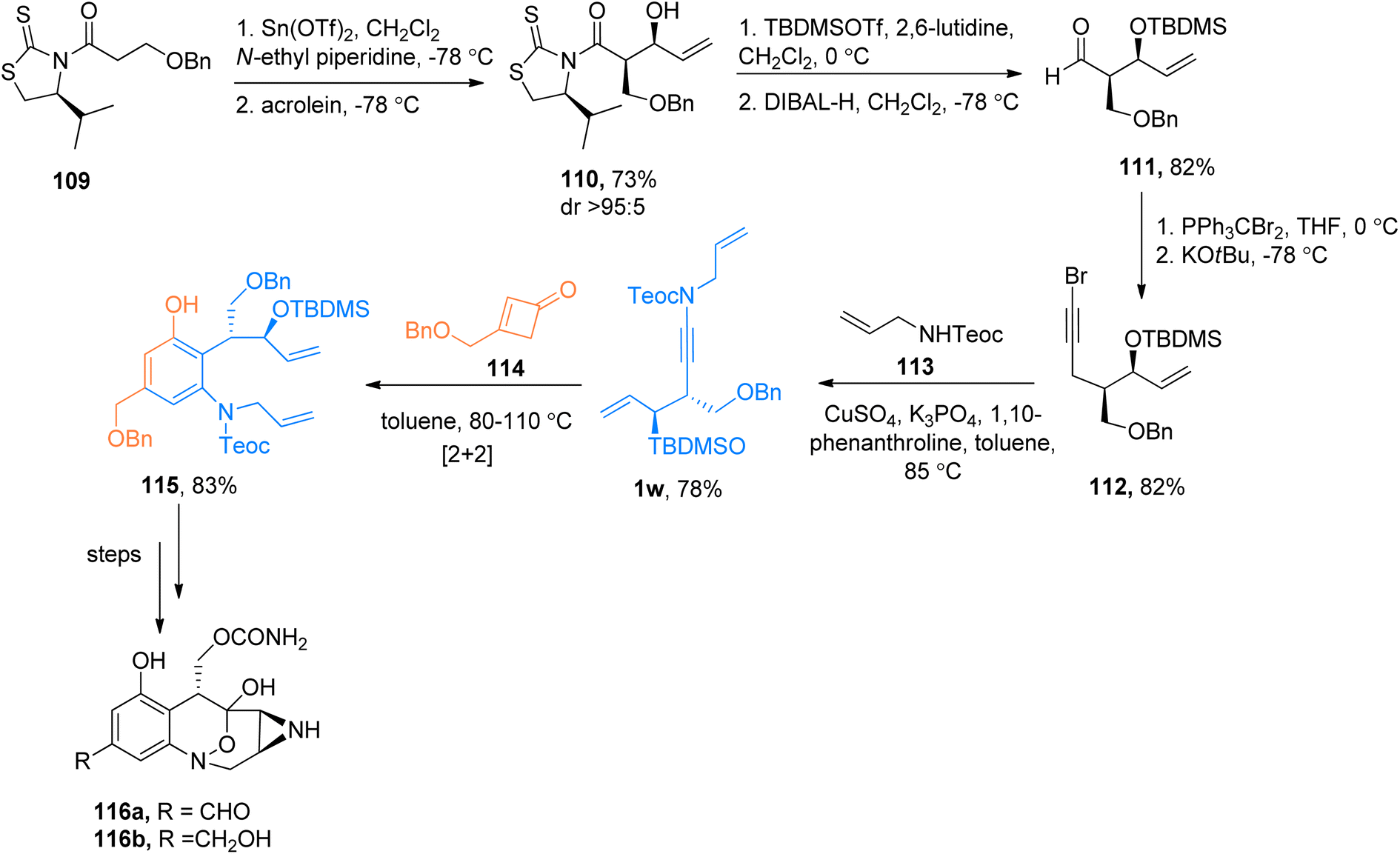
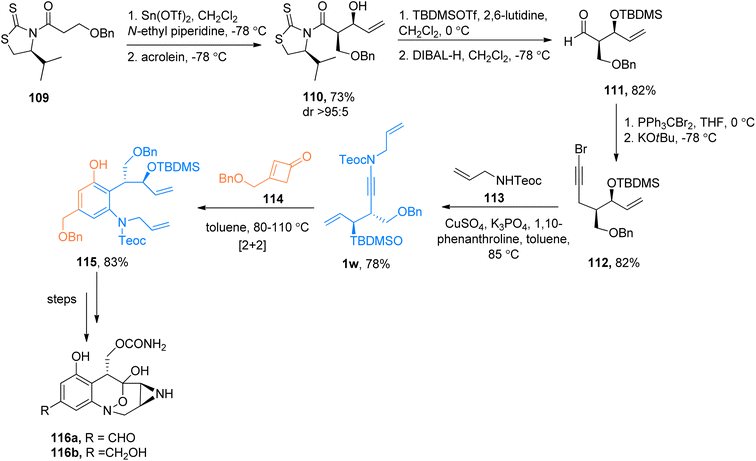 |
| | Scheme 52 The total synthesis of the anticancer agents 116a and 116b via [2 + 2]-cycloaddition of ynamide 1w and cyclobutenone 114. | |
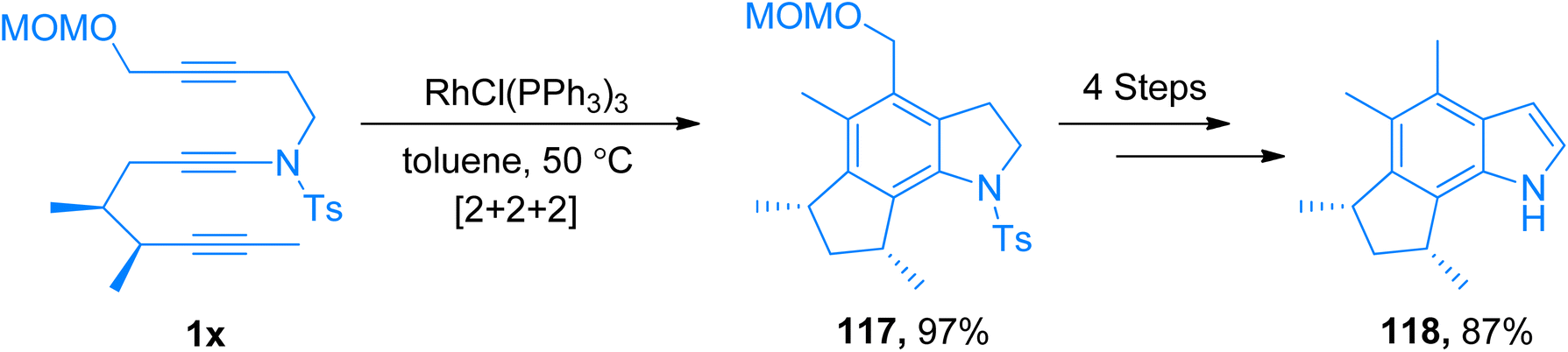
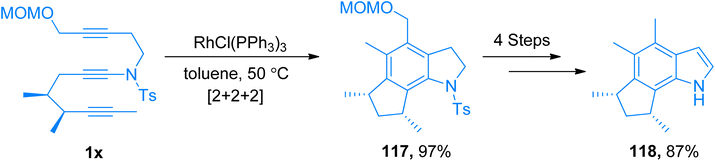 |
| | Scheme 53 Synthesis of (−)-herbindole A 118 via intramolecular [2 + 2 + 2]-cycloaddition of ynamide 1x. | |
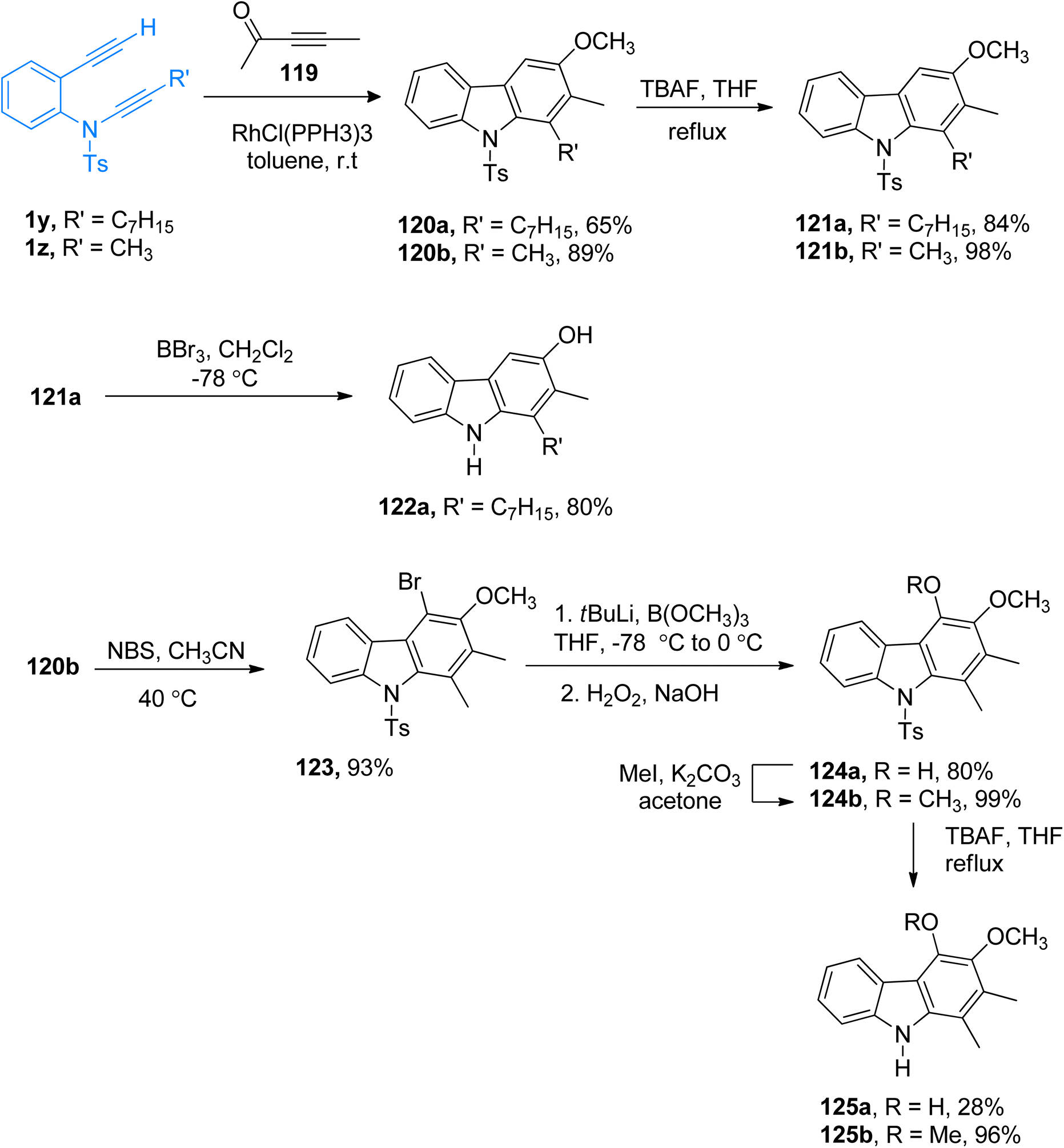
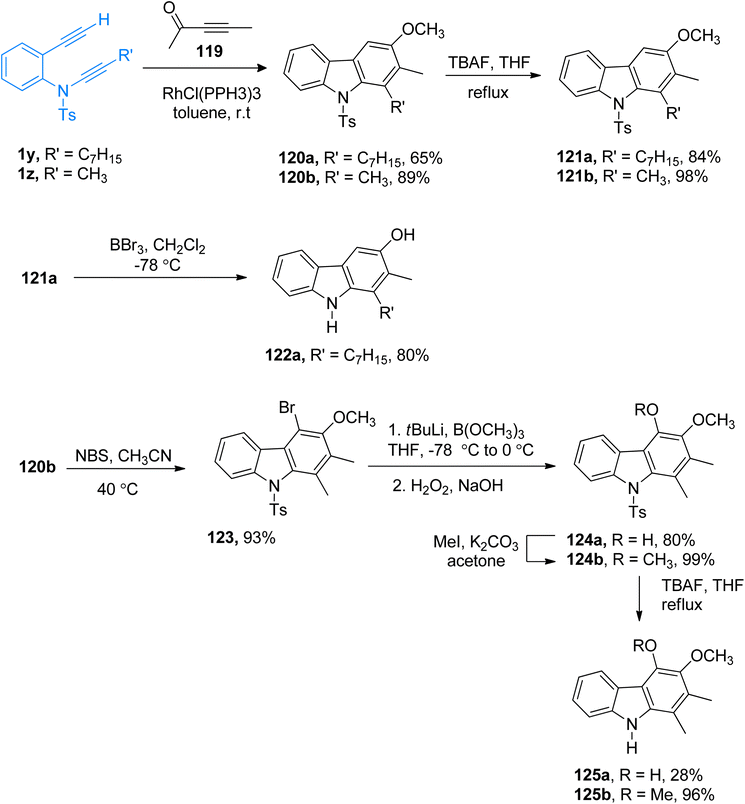 |
| | Scheme 54 Synthesis of 4-deoxycarbazomycin B 121b, carazostatin 122a, carbazomycin B 125a, and carbazomycin A 125b via [2 + 2 + 2] cycloaddition of ynamides 1y/1z. | |
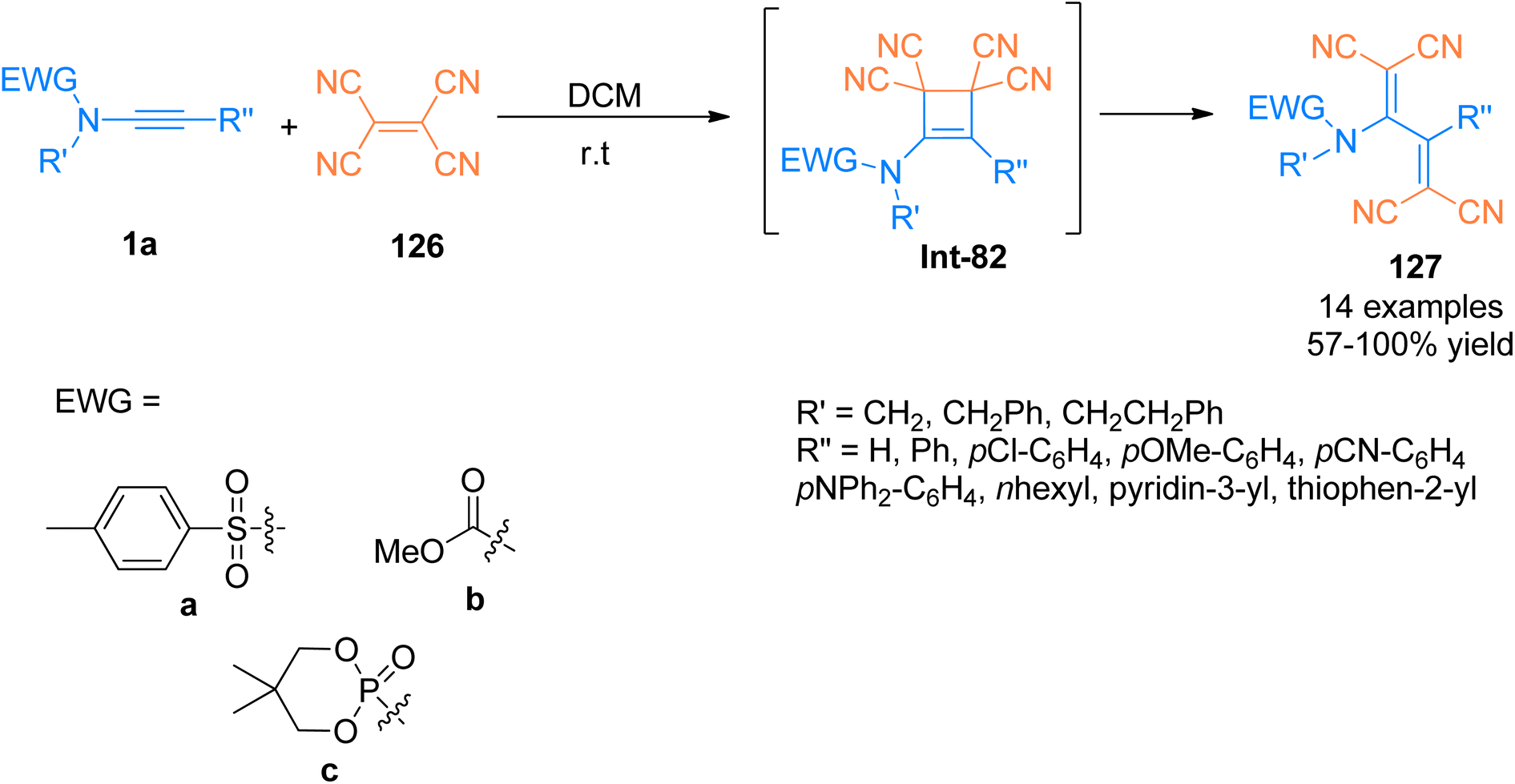
Previously, [2 + 2]-cycloaddition reaction between tetracyanoethylene (TCNE) and alkynyl-transition metal complexes for the synthesis of tetracyanobutadiene (TCNB) have been studied extensively.128,129 Betou et al. (2014), reported a novel and efficient [2 + 2]-cycloaddition-retro electrocyclization protocol for the reaction of ynamide 1a and tetracyanoethylene 126 to generate intermediate species Int-82. The species Int-82 readily underwent ring opening to afford TCNB species 127 with excellent optoelectronic properties. The metal-free transformation was realized under mild reaction conditions. Different types of ynamides were reacted with TCNE at room temperature using dichloromethane as a solvent to attain good to quantitative yields (57–100%) of novel TCNB derivatives. This methodology could further develop novel molecules exhibiting excellent properties for their potential use in optoelectronic devices (Scheme 55).130
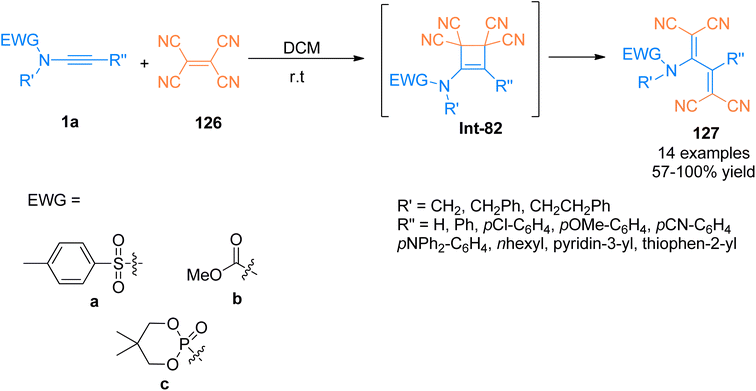 |
| | Scheme 55 Synthesis of tetracyanobutadiene 127 by [2 + 2]-cycloaddition of ynamide 1a and tetracyanoethylene 126. | |
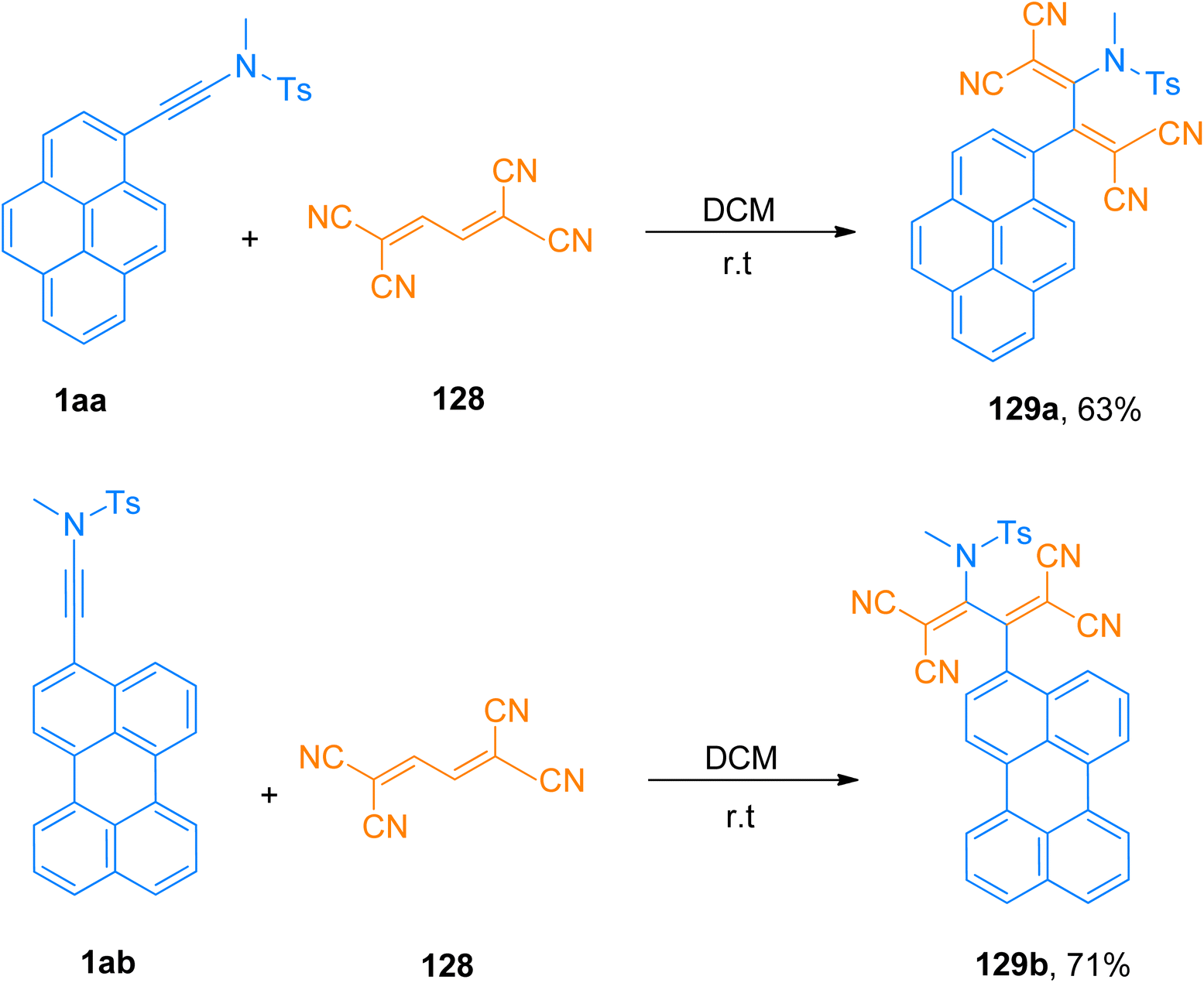
Bui et al. (2020), designed and constructed two tetracyanobutadinene derivatives 129a (63% yield) and 129b (71% yield) via [2 + 2]-cycloaddition followed by retrocyclization reaction of tetracyanobutadiene 128 with pyrene substituted ynamide 1aa and perylene substituted ynamide 1ab, respectively. The reaction proceeded using dichloromethane at room temperature (Scheme 56). Due to panchromatic absorptions, synthesized compounds exhibited Near Infrared (NIR) photoluminescence with a detectable limit of 1350 nm.131
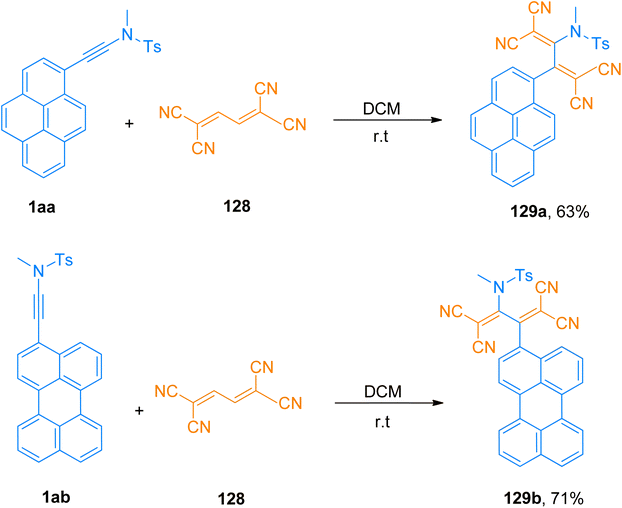 |
| | Scheme 56 Synthesis of tetracyanobutadiene derivatives 129a and 129b via [2 + 2]-cycloaddition of between pyrene/perylene substituted ynamide 1aa and 1ab and tetracyanobutadiene 128, respectively. | |
3. Conclusion and future outlooks
Ynamides pioneered the recent decade with extensive research, and still, there is a surge of studies reporting improved strategies and novel methodologies involving ynamides in cycloaddition reactions. Several studies were reported in the literature with efficient and facile metal-catalyzed as well as catalyst-free transformations to form complex nitrogen-containing frameworks with excellent yields from readily available substrates. The perfect balance of stability and reactivity makes ynamides ideal substrates to carry out a wide array of synthetic transformations via cycloaddition. Despite the realization of a wide variety of heterocyclic paradigms, still, certain limitations and challenges need to be considered in future research.
The gold catalysts as well as the Brønsted acid catalyst promote the cycloaddition of the ynamide with other reaction partners in a quite similar manner via α-addition (chemoselectivity). However, inverted regioselectivity to achieve cycloadducts of high synthetic and medicinal value is highly desirable. Hence, the catalytic systems need to be further explored to achieve the β-addition. Similarly, the area of enantioselective cycloaddition of the ynamides to realize the synthesis of asymmetric products is still under development for which the catalytic systems and chiral ligands should be employed for the enantioselective realization of heterocycles/carbocycles.132
In addition to the above, several studies proposed the mechanism of the transformation by isotope labeling experiments, and kinetic as well as theoretical mechanistic studies that help understand the mechanistic grounds of catalytic systems during the reactions. But still, there is great room for theoretical as well as practical investigation of the reaction mechanisms to better understand the reactivity and rational design of novel routes for ynamide cycloadditions. Because some of the studies suggested mechanisms of the transformations based on the previously reported mechanism without controlled experiments and theoretical grounds for the specific reaction.
This review recapitulates the recent state-of-the-art advances in the field of cycloaddition reactions unveil the synthetic potential of ynamides and we hope that it will help readers to further explore the reactivity of ynamides for facile and efficient future strategies.
Data availability
Data sharing does not apply to this article as no datasets were generated or analyzed during the current study.
Author contributions
Ramsha Iftikhar: conceptualization, data collection, writing – original draft, visualization.
Aqsa Mazhar, Muhammad Saqlain Iqbal: writing – original draft, writing – review, and editing.
Faiza Zahid Khan: english proofreading, writing – review, and editing.
Syed Hassan Askary, Hifza Sibtain: writing – review, and editing.
Conflicts of interest
The authors have no conflict of interest to declare.
Acknowledgements
The work was supported and facilitated by the Government College University Faisalabad, Faisalabad, Pakistan. Ramsha Iftikhar would like to thank Ameer Fawad Zahoor for his endless efforts and contribution to improving and proofreading the manuscript.
References
- K. DeKorver, H. Li, A. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. Hsung, Chem. Rev., 2010, 110, 5064–5106 CrossRef CAS PubMed.
- G. Evano, K. Jouvin and A. Coste, Synthesis, 2012, 45, 17–26 CrossRef.
- R. Hsung, J. Mulder and K. Kurtz, Synlett, 2003, 1379–1390 CrossRef.
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed.
- X. Wang, H. Yeom, L. Fang, S. He, Z. Ma, B. Kedrowski and R. Hsung, Acc. Chem. Res., 2013, 47, 560–578 CrossRef PubMed.
- B. Zhou, T. Tan, X. Zhu, M. Shang and L. Ye, ACS Catal., 2019, 9, 6393–6406 CrossRef CAS.
- C. Zificsak, J. Mulder, R. Hsung, C. Rameshkumar and L. Wei, Tetrahedron, 2001, 57, 7575–7606 CrossRef CAS.
- G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed.
- F. Hong and L. Ye, Acc. Chem. Res., 2020, 53, 2003–2019 CrossRef CAS PubMed.
- B. Prabagar, N. Ghosh and A. K. Sahoo, Synlett, 2017, 28, 2539–2555 CrossRef CAS.
- D. J. Campeau, A. Pommainville and F. Gagosz, J. Am. Chem. Soc., 2021, 143, 9601–9611 CrossRef CAS PubMed.
- F. Pan, C. Shu and L. W. Ye, Org. Biomol. Chem., 2016, 14, 9456–9465 RSC.
- Y. B. Chen, P. C. Qian and L. W. Ye, Chem. Soc. Rev., 2020, 49, 8897–8909 RSC.
- C. C. Lynch, A. Sripada and C. Wolf, Chem. Soc. Rev., 2020, 49, 8543–8583 RSC.
- K. Enomoto, H. Oyama and M. Nakada, Chem.–Eur. J., 2014, 21, 2798–2802 CrossRef PubMed.
- Y. Yuan, L. Bai, J. Nan, J. Liu and X. Luan, Org. Lett., 2014, 16, 4316–4319 CrossRef CAS.
- B. Alcaide, P. Almendros and C. Lázaro-Milla, Chem.–Eur. J., 2016, 22, 8998–9005 CrossRef CAS PubMed.
- J. C Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485–1538 CrossRef PubMed.
- Y. Bai, J. Yin, Z. Liu and G. Zhu, J. Org. Chem., 2015, 80, 10226–10233 CrossRef CAS PubMed.
- L. Chen, J. Cao, Z. Xu, Z. J Zheng, Y. M Cui and L. W. Xu, Chem. Commun., 2016, 52, 9574–9577 RSC.
- C. Peng, J. Zhang, J. Xue, S. Li, X. N. Wang and J. Chang, J. Org. Chem., 2018, 83, 9256–9266 CrossRef CAS PubMed.
- T. P. Willumstad, P. D Boudreau and R. L. Danheiser, J. Org. Chem., 2015, 80, 11794–11805 CrossRef CAS PubMed.
- Y. Yang, H. Liu, C. Peng, J. Wu, J. Zhang, Y. Qiao, X. N. Wang and J. Chang, Org. Lett., 2016, 18, 5022–5025 CrossRef CAS PubMed.
- Y. Miki, M. Umemoto, H. Maruyama, M. Kuromatsu and H. Hamamoto, Heterocycles, 2007, 71, 2457–2463 CrossRef CAS.
- B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 12898–12899 CrossRef CAS PubMed.
- T. Zieliński, P. Dydio and J. Jurczak, Tetrahedron, 2008, 64, 568–574 CrossRef.
- W. Xu, Y. Chen, A. Wang and Y. Liu, Org. Lett., 2019, 21, 7613–7618 CrossRef CAS PubMed.
- J. Zhang, Y. Li, C. Zhang, X. Wang and J. Chang, Org. Lett., 2021, 23, 2029–2035 CrossRef CAS PubMed.
- J. Liu, M. Chen, L. Zhang and Y. Liu, Chem.–Eur. J., 2014, 21, 1009–1013 CrossRef PubMed.
- R. G. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- V. Estevez, M. Villacampa and J. C. Menendez, Chem. Soc. Rev., 2014, 43, 4633–4657 RSC.
- A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS PubMed.
- A. Ghoshal, A. Yadav and A. K. Srivastava, J. Org. Chem., 2020, 85, 14890–14904 CrossRef CAS PubMed.
- S. K. Pawar, R. L. Sahani and R. S. Liu, Chem.–Eur. J., 2015, 21, 10843–10850 CrossRef CAS PubMed.
- A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS PubMed.
- L. Zhu, Y. Yu, Z. Mao and X. Huang, Org. Lett., 2014, 17, 30–33 CrossRef PubMed.
- X. Y. Xiao, A. H. Zhou, C. Shu, F. Pan, T. Li and L. W. Ye, Chem.–Asian J., 2015, 10, 1854–1858 CrossRef CAS PubMed.
- A. Mukherjee, R. B. Dateer, R. Chaudhuri, S. Bhunia, S. Karad and R. S. Liu, J. Am. Chem. Soc., 2011, 133, 15372–15375 CrossRef CAS PubMed.
- E. P. A. Talbot, M. Richardson, J. M. McKenna and F. D. Toste, Adv. Synth. Catal., 2014, 356, 687–691 CrossRef CAS PubMed.
- D. B. Huple, S. Ghorpade and R. S. Liu, Chem.–Eur. J., 2013, 19, 12965–12969 CrossRef CAS PubMed.
- A. H Zhou, Q. He, C. Shu, Y. F. Yu, S. Liu, T. Zhao, W. Zhang, X. Lu and L. W. Ye, Chem. Sci., 2015, 6, 1265–1271 RSC.
- S. I. Murahashi and Y. Imada, Chem. Rev., 2019, 119, 4684–4716 CrossRef CAS.
- N. De and E. J. Yoo, ACS Catal., 2017, 8, 48–58 CrossRef.
- F. L. Hong, Y. B. Chen, S. H. Ye, G. Y. Zhu, X. Q. Zhu, X. Lu, R. Liu and L. W. Ye, J. Am. Chem. Soc., 2020, 142, 7618–7626 CrossRef CAS PubMed.
- S. Ferrini, J. Z. Chandanshive, S. Lena, M. F. Comes Franchini, G. Giannini, A. Tafi and M. Taddei, J. Org. Chem., 2015, 80, 2562–2572 CrossRef CAS PubMed.
- Y. Liao, Q. Lu, G. Chen, Y. Yu, C. Li and X. Huang, ACS Catal., 2017, 7, 7529–7534 CrossRef CAS.
- W. Song and N. Zheng, Org. Lett., 2017, 19, 6200–6203 CrossRef CAS PubMed.
- M. Garzón, E. M. Arce, R. J. Reddy and P. W. Davies, Adv. Synth. Catal., 2017, 359, 1837–1843 CrossRef.
- E. M. Arce, S. G. Lamont and P. W. Davies, Adv. Synth. Catal., 2020, 362, 2503–2509 CrossRef CAS.
- W. Xu, Y. Chen, A. Wang and Y. Liu, Org. Lett., 2019, 21, 7613–7618 CrossRef CAS PubMed.
- Y. Zhao, Y. Hu, X. Li and B. Wan, Org. Biomol. Chem., 2017, 15, 3413–3417 RSC.
- N. Marien, B. N. Reddy, F. D. Vleeschouwer, S. Goderis, K. V. Hecke and G. Verniest, Angew. Chem., 2018, 130, 5762–5766 CrossRef.
- S. Bresciani and N. C. O. Tomkinson, Heterocycles, 2014, 89, 2479–2543 CrossRef CAS.
- A. D. Gillie, R. J. Reddy and P. W. Davies, Adv. Synth. Catal., 2016, 358, 226–239 CrossRef CAS.
- M. Chen, N. Sun, H. Chen and Y. Liu, Chem. Commun., 2016, 52, 6324–6327 RSC.
- Y. Zhao, Y. Hu, C. Wang, X. Li and B. Wan, J. Org. Chem., 2017, 82, 3935–3942 CrossRef CAS PubMed.
- W. D. Mackay, M. Fistikci, R. M. Carris and J. S. Johnson, Org. Lett., 2014, 16, 1626–1629 CrossRef CAS PubMed.
- X. Cheng, L. Zhu, M. Lin, J. Chen and X. Huang, Chem. Commun., 2017, 53, 3745–3748 RSC.
- C. Shu, Y. Wang, B. Zhou, X. Li, Y. Ping, X. Lu and L. Ye, Org. Lett., 2016, 47, 3254–3257 CrossRef PubMed.
- M. R. Prabhath, L. Williams, S. V. Bhat and P. Sharma, Molecules, 2017, 22, 615 CrossRef PubMed.
- A. Y. Dubovtsev, N. V. Shcherbakov, D. V. Dar'in and V. Y. Kukushkin, Adv. Synth. Catal., 2020, 362, 2672–2682 CrossRef CAS.
- S. F. Tlais and R. L. Danheiser, J. Am. Chem. Soc., 2014, 136, 15489–15492 CrossRef CAS PubMed.
- Z. Xin, S. Kramer, J. Overgaard and T. Skrydstrup, Chem.–Eur. J., 2014, 20, 7926–7930 CrossRef CAS PubMed.
- H. Wu, Y. Liu, M. He, H. Wen, W. Cao, P. Chen and Y. Tang, Org. Biomol. Chem., 2019, 17, 8408–8416 RSC.
- L. Chen, L. Yu, Y. Deng, Z. Zheng, Z. Xu, J. Cao and L. Xu, Adv. Synth. Catal., 2016, 358, 480–485 CrossRef CAS.
- H. Wen, W. Yan, P. Chen, Y. Li and Y. Tang, J. Org. Chem., 2020, 85, 12870–12881 CrossRef CAS PubMed.
- J. A. Burkhard, G. Wuitschik, M. R. Evans, K. Muller and E. M. Carreira, Angew. Chem., 2010 (Angew. Chem., Int. Ed., 2010, 49, 9053–9067) Search PubMed.
- S. K. Pawar, D. Vasu and R. S. Liu, Adv. Synth. Catal., 2014, 356, 2411–2416 CrossRef CAS.
- L. Chen, Y. M. Cui, Z. Xu, J. Cao, Z. J. Zheng and L. W. Xu, Chem. Commun., 2016, 52, 11131–11134 RSC.
- S. S. Giri and R. S. Liu, Chem. Sci., 2018, 9, 2991–2995 RSC.
- G. Duret, R. Quinlan, B. Yin, R. E. Martin, P. Bisseret, M. Neuburger, V. Gandon and N. Blanchard, J. Org. Chem., 2017, 82, 1726–1742 CrossRef CAS PubMed.
- S. R. Dubbaka, M. Kienle, H. Mayr and P. Knochel, Angew. Chem., Int. Ed., 2007, 46, 9093–9096 CrossRef CAS PubMed.
- L. G. Xie, S. Niyomchon, A. J. Mota, L. González and N. Maulide, Nat. Commun., 2016, 7, 10914 CrossRef PubMed.
- N. V. Shcherbakov, D. V. Dar’in, V. Y. Kukushkin and A. Y. Dubovtsev, J. Org. Chem., 2021, 86, 7218–7228 CrossRef CAS PubMed.
- T. Ito, S. Harada, H. Homma, H. Takenaka, S. Hirose and T. Nemoto, J. Am. Chem. Soc., 2020, 143, 604–611 CrossRef PubMed.
- C. Shu, Y. H. Wang, B. Zhou, X. L. Li, Y. F. Ping, X. Lu and L. W. Ye, J. Am. Chem. Soc., 2015, 137, 9567–9570 CrossRef CAS PubMed.
- P. Wessig and G. Müller, Chem. Rev., 2008, 108, 2051–2063 CrossRef CAS PubMed.
- W. Li, L. Zhou and J. Zhang, Chem.–Eur. J., 2015, 22, 1558–1571 CrossRef PubMed.
- W. Xu, G. Wang, X. Xie and Y. Liu, Org. Lett., 2018, 20, 3273–3277 CrossRef CAS PubMed.
- B. Prabagar, S. Dutta, V. Gandon and A. K. Sahoo, Asian J. Org. Chem., 2019, 8, 1128–1132 CrossRef CAS.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS PubMed.
- M. Juhl and D. Tanner, Chem. Soc. Rev., 2009, 38, 2983 RSC.
- E. Chirkin and F. H. Porée, Curr. Org. Chem., 2016, 20, 2284–2325 CrossRef CAS.
- X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515–5546 CrossRef CAS PubMed.
- G. Duret, V. L. Fouler, P. Bisseret, V. Bizet and N. Blanchard, Eur. J. Org. Chem., 2017, 2017, 6816–6830 CrossRef CAS.
- J. Xue, E. Gao, X. N. Wang and J. Chang, Org. Lett., 2018, 20, 6055–6058 CrossRef CAS PubMed.
- X. Li, Z. Wang, X. Ma, P. N. Liu and L. Zhang, Org. Lett., 2017, 19, 5744–5747 CrossRef CAS PubMed.
- S. Wang, C. R. Escrich and M. A. Pericàs, Angew. Chem., 2017, 129, 15264–15268 CrossRef.
- R. Manzano, A. Romaniega, L. Prieto, E. Díaz, E. Reyes, U. Uria, L. Carrillo and J. L. Vicario, Org. Lett., 2020, 22, 4721–4725 CrossRef CAS PubMed.
- C. He, Z. Li, H. Zhou and J. Xu, Org. Lett., 2019, 21, 8022–8026 CrossRef CAS PubMed.
- N. Abe and T. Gunji, Heterocycles, 2010, 82, 201–248 CrossRef CAS PubMed.
- T. S. Rodríguez, Á. L. S. Sobrino and A. Ballesteros, Chem.–Eur. J., 2021, 27, 7154–7159 CrossRef.
- H. Jin, M. Rudolph, F. Rominger and A. S. K. Hashmi, ACS Catal., 2019, 9, 11663–11668 CrossRef CAS.
- Y. L. Chen, P. Sharma and R. S. Liu, Chem. Commun., 2016, 52, 3187–3190 RSC.
- P. Garcia, Y. Evanno, P. George, M. Sevrin, G. Ricci, M. Malacria, C. Aubert and V. Gandon, Chem.–Eur. J., 2012, 18, 4337–4344 CrossRef CAS PubMed.
- G. Wang, X. You, Y. Gan and Y. Liu, Org. Lett., 2016, 19, 110–113 CrossRef PubMed.
- J. Zhang, Q. Zhang, B. Xia, J. Wu, X. N. Wang and J. Chang, Org. Lett., 2016, 18, 3390–3393 CrossRef CAS PubMed.
- S. N. Karad and R. S. Liu, Angew. Chem., Int. Ed., 2014, 53, 9072–9076 CrossRef CAS PubMed.
- Z. Zeng, H. Jin, X. Song, Q. Wang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem. Commun., 2017, 53, 4304–4307 RSC.
- P. Chen, C. X. Song, W. S. Wang, X. L. Yu and Y. Tang, RSC Adv., 2016, 6, 80055–80058 RSC.
- D. Liu, Q. Nie and M. Cai, Tetrahedron, 2018, 74, 3020–3029 CrossRef CAS.
- H. Liang, S. Bi, Y. Liu, Y. N. Tang and C. Liu, Org. Biomol. Chem., 2016, 14, 2637–2644 RSC.
- J. Zhang, M. Guo, Y. Chen, S. Zhang, X. N. Wang and J. Chang, Org. Lett., 2019, 21, 1331–1336 CrossRef CAS PubMed.
- P. D. Jadhav, X. Lu and R. S. Liu, ACS Catal., 2018, 8, 9697–9701 CrossRef CAS.
- Y. Zhao, C. Wang, Y. Hu and B. Wan, Chem. Commun., 2018, 54, 3963–3966 RSC.
- D. L. Browne and J. P. Harrity, Tetrahedron, 2010, 66, 553–568 CrossRef CAS.
- S. K. Bhosale, S. R. Deshpande and R. D. Wagh, J. Chem. Pharm. Res., 2012, 4, 1185–1199 CAS.
- S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS PubMed.
- T. Wezeman, J. C. Barceló, M. Nieger, J. P. A. Harrity and S. Bräse, Org. Biomol. Chem., 2017, 15, 1575–1579 RSC.
- Y. Yu, G. Chen, L. Zhu, Y. Liao, Y. Wu and X. Huang, J. Org. Chem., 2016, 81, 8142–8154 CrossRef CAS PubMed.
- X. L. Han, C. J. Zhou, X. G. Liu, S. S. Zhang, H. Wang and Q. Li, Org. Lett., 2017, 19, 6108–6111 CrossRef CAS PubMed.
- J. Brioche, C. Meyer and J. Cossy, Org. Lett., 2015, 17, 2800–2803 CrossRef CAS PubMed.
- M. Sugiura, H. Hagio, R. Hirabayashi and S. Kobayashi, J. Am. Chem. Soc., 2001, 123, 12510–12517 CrossRef CAS PubMed.
- X. L. Han, X. D Nie, Z. D. Chen, C. M. Si, B. G. Wei and G. Q. Lin, J. Org. Chem., 2020, 85, 13567–13578 CrossRef CAS PubMed.
- T. Kuzuguchi, Y. Yabuuchi, T. Yoshimura and J. I. Matsuo, Org. Biomol. Chem., 2017, 15, 5268–5271 RSC.
- Q. Zhao, D. F. L. Rayo, D. Campeau, M. Daenen and F. Gagosz, Angew. Chem., 2018, 130, 13791–13795 CrossRef.
- W. Xu, J. Zhao, X. Li and Y. Liu, J. Org. Chem., 2018, 83, 15470–15485 CrossRef CAS PubMed.
- X. L. Han, X. G. Liu, E. Lin, Y. Chen, Z. Chen, H. Wang and Q. Li, Chem. Commun., 2018, 54, 11562–11565 RSC.
- Y. Yamaoka, T. Yoshida, M. Shinozaki, K. I. Yamada and K. Takasu, J. Org. Chem., 2015, 80, 957–964 CrossRef CAS PubMed.
- S. Nonaka, K. Sugimoto, H. Ueda and H. Tokuyama, Adv. Synth. Catal., 2016, 358, 380–385 CrossRef CAS.
- G. Duret, V. Le Fouler, P. Bisseret, V. Bizet and N. Blanchard, Eur. J. Org. Chem., 2017, 46, 6816–6830 CrossRef.
- C. C. Forneris, Y. P. Wang, G. Mamaliga, T. P. Willumstad and R. L. Danheiser, Org. Lett., 2018, 20, 6318–6322 CrossRef CAS PubMed.
- X. Y. Mak, A. L. Crombie and R. L. Danheiser, J. Org. Chem., 2011, 76, 1852–1873 CrossRef CAS PubMed.
- N. Saito, T. Ichimaru and Y. Sato, Org. Lett., 2012, 14, 1914–1917 CrossRef CAS PubMed.
- C. Alayrac and B. Witulski, Heterocycles, 2021, 103, 205–217 CrossRef CAS PubMed.
- T. Michinobu, J. C. May, J. H. Lim, C. Boudon, J. P. Gisselbrecht, P. Seiler, M. Gross, I. Biaggio and F. Diederich, Chem. Commun., 2005, 737–739 RSC.
- T. Shoji and S. Ito, Chem.–Eur. J., 2017, 23, 16696–16709 CrossRef CAS PubMed.
- M. I. Bruce, Aust. J. Chem., 2011, 64, 77–103 CrossRef CAS.
- M. I. Bruce, J. R. Rodgers, M. R. Snow and A. G. Swincer, J. Chem. Soc., Chem. Commun., 1981, 6, 271–272 RSC.
- M. Betou, N. Kerisit, E. Meledje, Y. Leroux, C. Katan, J. Halet, J. Guillemin and Y. Trolez, Chem.–Eur. J., 2014, 20, 9553–9557 CrossRef CAS PubMed.
- A. T. Bui, C. Philippe, M. Beau, N. Richy, M. Cordier, T. Roisnel, L. Lemiègre, O. Mongin, F. Paul and Y. Trolez, Chem. Commun., 2020, 56, 3571–3574 RSC.
- B. Zhou, T. D. Tan, X. Q. Zhu, M. Shang and L. W. Ye, ACS Catal., 2019, 9, 6393–6406 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Aqsa Mazharb,
Muhammad Saqlain Iqbalc,
Faiza Zahid Khand,
Syed Hassan Askarye and
Hifza Sibtain
*a,
Aqsa Mazharb,
Muhammad Saqlain Iqbalc,
Faiza Zahid Khand,
Syed Hassan Askarye and
Hifza Sibtain