DOI:
10.1039/D3RA00144J
(Paper)
RSC Adv., 2023,
13, 4578-4583
Linear π-conjugated polycyclic compounds consisting of four-, five-, and six-membered rings: benzo[1′′,2′′:3,4;4′′,5′′:3′,4′]bis(cyclobuta[1,2-c]thiophene)†‡
Received
8th January 2023
, Accepted 27th January 2023
First published on 3rd February 2023
Abstract
Linear π-conjugated polycyclic compounds, BBCTs, containing linearly annulated 5-, 4-, 6-, 4-, and 5-membered rings were produced via copper-mediated double intramolecular coupling reactions. The absorption spectra and electrochemical results confirmed their moderate optical energy gaps and high HOMO energy levels, respectively. In a crystalline state, the BBCT molecules adopt a herringbone structure, while the methylated molecules form slipped one-dimensional columns. The local and global aromaticity of the new polycyclic compounds is discussed based on the experimental results and theoretical predictions. The present fundamental findings are useful for the further design and synthesis of novel π-conjugated polycyclic compounds containing four-membered rings with potential applications in electronic materials.
Introduction
Acene has attracted research interest for over a century, including investigations of its fundamental properties and aromaticity and its applications in organic electronic devices.1,2 The incorporation of heteroatom(s) into acene alters its stability, electronic properties, and intermolecular interaction modes. The incorporation of nitrogen or sulfur atoms modulates the energy levels of the frontier orbitals and crystal packing structures of acene, and some of these compounds are n- or p-type organic semiconducting materials, respectively.3–8 Another approach to tailoring the properties of acene is the incorporation of hydrocarbon rings of different sizes. The inclusion of a four-membered ring (4MR) is particularly interesting because of its planarity and inherent anti-aromatic character.9 Vollhardt and co-workers reported linear [n]phenylenes in which the 4MR bridges every benzene ring, resulting in unique local aromaticity and electronic properties compared to acene with similar sizes.10–12 Linear π-conjugated polycyclic compounds containing 4MRs have also been reported to show intriguing photophysical and electronic properties owing to the modulated local aromaticity and frontier orbital energies.13–16 Moreover, the incorporation of both nitrogen atoms and 4MRs into acene strongly affects these properties, with some of these compounds exhibiting distinguished semiconducting and luminescence properties.17–26
The incorporation of both sulfur atoms and 4MRs into acene affects its aromaticity and electronic properties. We previously investigated the geometries, electronic properties, and aromaticity of benzo[3,4]cyclobuta[1,2-c]thiophene (cBCT) and its extended homologues using density functional theory (DFT) calculations (Fig. 1).27 While the synthesis of cBCT was reported previously,25,28 its extended homologues have not been experimentally explored. Among the homologues, benzo[1′′,2′′:3,4;4′′,5′′:3′,4′]bis(cyclobuta[1,2-c]thiophene) (BBCT) was chosen as the first test molecule for experimental investigation for the following reasons. BBCT will be more stable than its b-type isomer.27,29,30 The energy gap (EGap) and HOMO energy level of BBCT predicted by the DFT calculations are moderate due to the production of a p-type organic semiconducting material. BBCT will show moderate global aromaticity; the two fused 4MRs will attenuate the aromaticity of the central benzene ring and the fused thiophene ring at the 3,4-bond (c-type) with low electron density because its inherent diene character will attenuate the antiaromatic character of the 4MRs.27,31–35 Finally, BBCT has a unique structural feature, i.e., the linear annulation of 5-, 4-, 6-, 4-, and 5-membered rings.36–43
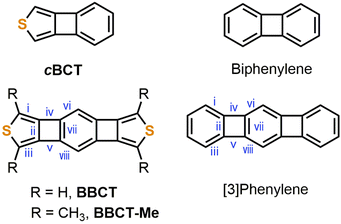 |
| | Fig. 1 Chemical structures of cBCT, biphenylene, BBCT, BBCT-Me, and [3]phenylene. Roman numerals on the BBCTs and [3]phenylene indicate bond positions. | |
In this context, we herein report the synthesis, electronic properties, crystal structures, and aromaticity of BBCT and its methylated compound BBCT-Me and compare them with the properties and structure of [3]phenylene as a reference compound,10 for future potential application of these molecules as p-type semiconducting materials.
Results and discussion
For the construction of the small, yet strained 4MRs, we chose a copper-mediated intramolecular coupling reaction of dibromobiaryls under ambient conditions.12,44 We first tested the synthesis of biphenylene and cBCT. Biphenylene and cBCT were isolated in 54% and 29% yields from 2,2′-dibromobiphenyl and 3-bromo-4-(2-bromophenyl)thiophene (2), respectively (Scheme 1). The lower yield obtained for cBCT is attributed to the larger strain associated with 4MR formation between the rings of different sizes, as supported by DFT calculations (Fig. S1, ESI‡). While the isolated yield of cBCT is not high, it is higher than the reported values.25,28,45 Thus, we employed this reaction for the synthesis of BBCT. Precursors 4a and 4b were synthesized via a palladium-catalyzed cross-coupling reaction between 3,4-dibromothiophenes and 1,4-dibromo-2,5-diiodobenzene. The double-fold 4MR formation in both 4a and 4b afforded BBCT and BBCT-Me in 4% and <1% (2% based on the NMR internal standard method) isolated yields, respectively. In the case of BBCT-Me, the addition of N,N,N′,N′-tetramethylethylenediamine (TMEDA) is needed due to the steric influence of the methyl groups. The BBCTs containing the strained 4MRs are stable under ambient conditions. As a reference compound, we synthesized [3]phenylene (see ESI‡).
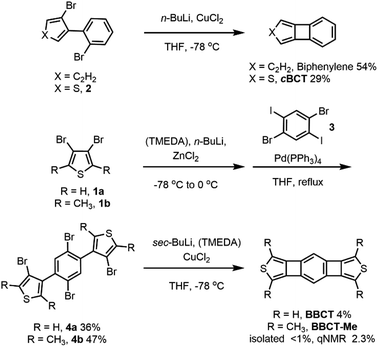 |
| | Scheme 1 Synthesis of biphenylene, cBCT, BBCT, and BBCT-Me. | |
Fig. 2 shows the absorption spectra of cBCT, BBCT, BBCT-Me, and [3]phenylene in CH2Cl2 at room temperature. The absorption spectrum of cBCT shows two strong bands with maxima at 348 and 249 nm. The absorption spectra of the BBCTs are similar, displaying strong bands with maxima at 423 and 285 nm for BBCT and 427 and 304 nm for BBCT-Me. The red shift in absorption for the BBCTs with respect to cBCT is due to the extension of π-electron conjugation. The absorption spectrum of [3]phenylene is similar to those of the BBCTs, although the very weak absorption band extends to 520 nm. The optical band gap (EOpt.Gap) values estimated from the absorption edges are 3.43, 2.68, 2.69, and 2.38 eV for cBCT, BBCT, BBCT-Me, and [3]phenylene, respectively (Table 1). The smallest excitation energy (calc. Eexcitation) values predicted by time-dependent (TD) DFT calculations at the B3LYP/6-311+G(d,p) level agree with the EOpt.Gap values. Moreover, TD-DFT calculations support that the lowest-energy transitions of these compounds involve the HOMO, LUMO, and LUMO+1 (Fig. S2, S3 and Tables 1, S2–S5, ESI‡). The blue shift of the adsorption edges of the BBCTs with respect to that of [3]phenylene is attributed to the lower LUMO level of [3]phenylene.
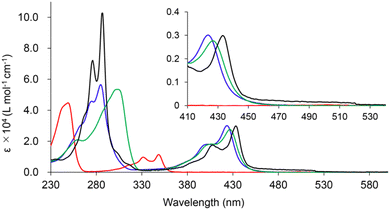 |
| | Fig. 2 UV-vis absorption spectra of cBCT (red), BBCT (blue), BBCT-Me (green) and [3]phenylene (black) at room temperature in CH2Cl2. | |
Table 1 Electronic properties of BBCT, BBCT-Me, cBCT, and [3]phenylene
| Compound |
λedge (nm) |
Exp. EOpt.Gapa (eV) |
Calc. Eexcitationb (eV) |
Calc. EGapc (eV) |
Eonset (V) |
Epa (V) |
Exp. EHOMOd (eV) |
Calc. EHOMOc (eV) |
| Determined from the absorption edges in the UV-vis absorption spectra. The smallest excitation energies estimated by TD-DFT calculations at the B3LYP/6-311+G(d,p) level of theory. Estimated from the optimized geometries at the B3LYP/6-311+G(d,p) level of theory. Derived from the onsets of the first oxidation waves in the cyclic voltammograms (Fig. 3). |
| BBCT |
463 |
2.68 |
2.78 |
3.47 |
0.39 |
0.59 |
−5.2 |
−5.35 |
| BBCT-Me |
461 |
2.69 |
2.87 |
3.53 |
0.21 |
0.38 |
−5.0 |
−5.09 |
| cBCT |
362 |
3.43 |
3.72 |
4.51 |
0.86 |
1.23 |
−5.7 |
−5.83 |
| [3]Phenylene |
520 |
2.38 |
2.25 |
2.97 |
0.26 |
0.47 |
−5.1 |
−5.13 |
Cyclic voltammetry measurements of cBCT, BBCT, BBCT-Me, and [3]phenylene were conducted at room temperature in CH2Cl2 using [n-Bu4N][ClO4] as the supporting electrolyte (Table 1 and Fig. 3). The cyclic voltammogram of cBCT shows one irreversible oxidation wave at Epa = 1.23 V (Fc/Fc+). Two irreversible oxidation waves were recorded in the voltammograms of BBCT and BBCT-Me, with the first oxidation waves peaking at Epa = 0.59 and 0.38 V, respectively. [3]Phenylene also shows two irreversible oxidation waves, with the first peaking at Epa = 0.47 V. The electrochemically derived HOMO energy levels (exp. EHOMO) of cBCT, BBCT, and BBCT-Me were −5.7, −5.2, and −5.0 eV, respectively, based on the onsets of each first oxidation wave in the voltammograms (Eonset). These values qualitatively agree with those predicted by DFT simulation (calc. EHOMO, Table 1). These results suggest that the size of the π-conjugated system and methyl groups affect the electronic properties.46 Based on comparison with the HOMO energy levels of thienoacenes with the same number of rings, the BBCTs are reasonable electron donors.47,48
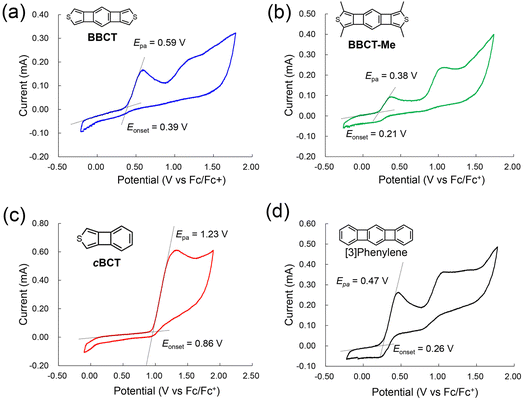 |
| | Fig. 3 Cyclic voltammograms of BBCT (a), BBCT-Me (b), cBCT (c), and [3]phenylene (d) in CH2Cl2 (concentration: 2 mM; supporting electrolyte: [n-Bu4N][ClO4]; scan rate: 100 mV s−1). Black dotted lines are the linear extrapolations which were used for the estimation of the onset potentials (Eonset). The potentials are against the ferrocene/ferrocenium (Fc/Fc+) couple. Epas are the peak top potentials of the first oxidation waves. | |
Platelet single crystals of BBCT and BBCT-Me suitable for X-ray diffraction analysis were grown from THF/EtOH and CH2Cl2/hexane, respectively. Both compounds adopt virtually planar geometries, confirming the linearly annulated 5-, 4-, 6-, 4-, and 5-membered rings (Fig. 4a and 5a). The BBCT and BBCT-Me molecules pack in the P21/c space group. The BBCT molecules adopt a herringbone structure (Fig. 4b and c).49 Within the column consisting of molecules with the same orientation, the adjacent molecules slip completely with limited intermolecular contacts. The nearest interatomic distances between the carbon atoms in the same and different columns are 3.46 Å. The interatomic distances between the hydrogen and carbon atoms range from 2.73 to 3.26 Å, suggesting (C–H)–π interactions between the columns.50 Moreover, there are close contacts between the sulfur and hydrogen atoms (3.09 Å).51 On the other hand, the BBCT-Me molecules form slipped one-dimensional columns with two different orientations (Fig. 5b and c). The nearest interatomic distances of the carbon atoms of adjacent molecules within the column are 3.43 Å. The methyl groups of BBCT-Me are located on the thiophene rings of the adjacent molecules in the different columns with distances of 2.86 Å between the hydrogen atoms of the methyl groups and the centroid of the thiophene rings, indicating (C–H)–π interactions. The methyl groups of BBCT-Me are also close to the 4MRs of adjacent molecules within the column. The nearest distances between the hydrogen atoms of the methyl groups and the carbon atoms of the 4MRs are 2.78 Å, again indicating (C–H)–π interactions. The methyl groups influence the crystal packing structure of the BBCTs. The non-covalent interaction (NCI) plots52 were calculated at the r2SCAN-3c/def2-mTZVPP level of theory53 to visualize these interactions (Fig. 4d–f, 5d–f, S10 and S11‡). Charge-transfer integral calculations predict that the BBCT molecules show molecular orbital interactions in the herringbone structure (Fig. S12, ESI‡), implying potential in the application of p-type semiconducting material.
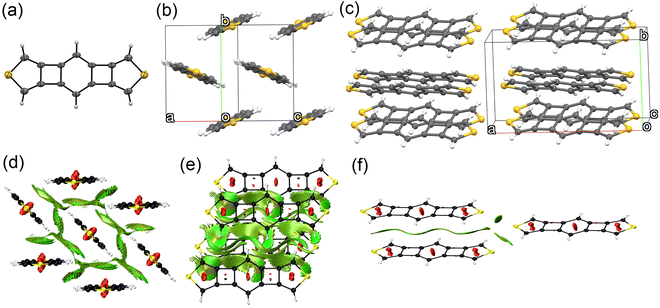 |
| | Fig. 4 Crystal structure of BBCT. (a) Molecular structure of BBCT with thermal ellipsoids at 30% probability. (b) Six molecules of BBCT viewed from the direction of the medium line of the unit cell vectors a and c. (c) Twelve molecules of BBCT viewed from the direction of the unit cell vector c. (d)–(f) NCI plots of the BBCT molecules in the crystal structure calculated at the r2SCAN-3c/def2-mTZVPP level (isovalue: 0.60 a.u.). 2D reduced density gradient vs. sign(λ2)ρ plots are shown in Fig. S10 (ESI‡). | |
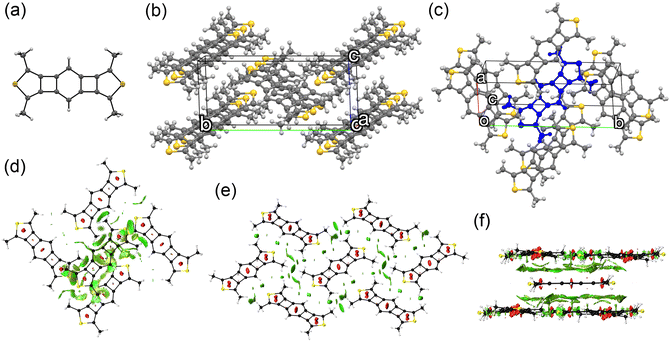 |
| | Fig. 5 Crystal structure of BBCT-Me. (a) Molecular structure of BBCT-Me with thermal ellipsoids at 30% probability. (b) Fifteen molecules of BBCT-Me viewed from the direction of the unit cell vector a showing slipped one-dimensional columns with two different orientations. (c) Eight molecules of BBCT-Me viewed from the unit cell vector c. The central molecule in which the methyl groups show short contacts with adjacent molecules is colored blue. (d)–(f) NCI plots of the BBCT-Me molecules in the crystal structure calculated at the r2SCAN-3c/def2-mTZVPP level (isovalue: 0.60 a.u.). 2D reduced density gradient vs. sign(λ2)ρ plots are shown in Fig. S11 (ESI‡). | |
The local and global aromaticity of the novel polycyclic compounds is an intriguing subject. We analyzed the bond lengths of BBCTs from the crystal structures. The two shared and four non-shared bonds of the central benzene ring of the BBCTs are similar in length (Table 2 and Fig. S7, S8, ESI‡), indicating negligible bond length alternation. The shared bonds show single-bond character to reduce the electron density in the 4MRs.27,35 As the structural criterion for the local aromaticity, we performed the harmonic oscillator model of aromaticity (HOMA).54 The HOMA value of the central benzene ring of BBCT is large (0.88, Fig. S9, ESI‡), which is attributed to the small degree of bond length alteration rather than cyclic π-electron delocalization.10 The thiophene rings retain diene character (Table 2). It should be noted that the lengths of the shared bonds in the 4MR differ by 0.018 Å between the benzene and thiophene sides. Moreover, the shared bonds of the central benzene rings are elongated in BBCT compared with those of [3]phenylene,55 indicating the larger strain of BBCT. This could be a factor in the low efficiency of 4MR construction in BBCT. As the measure of aromatic ring current effect, we compared the signals of the hydrogen atoms in the 1H NMR spectra recorded in acetone-d6 (Fig. 6). The signals of the hydrogen atoms (Ha) of BBCT and [3]phenylene resonated at 6.73 and 6.41 ppm, respectively, while those of the hydrogen atoms of the benzene ring in cBCT appeared at 7.02–6.90 ppm. The aromatic shielding effect from the diatropic ring currents of the central benzene ring is attenuated in BBCT, although it is still stronger than that in [3]phenylene. A similar trend was observed in the chemical shift of the hydrogen atoms attached to the thiophene ring (6.74 and 6.64 ppm for cBCT and BBCT, respectively). The nucleus-independent chemical shift (NICS) scans and calculated current paths and strengths also agree with the above observations (Fig. S4–S6, ESI‡).27,56 The local aromaticity of the central benzene ring and thiophene rings is attenuated in BBCT. Moreover, theoretically predicted paratropic character of the 4MRs in BBCT is weaker than those of [3]phenylene, indicating the limited anti-aromatic character of the 4MRs in BBCT.27 Overall, BBCT has moderate global aromaticity.
Table 2 Bond lengths (Å) of BBCT, BBCT-Me, and [3]phenylene determined by X-ray single-crystal analysis
| Compounda |
i |
ii |
iii |
iv |
v |
vi |
vii |
viii |
| Bond positions are described in Fig. 1. Bond lengths of [3]phenylene were referred from ref. 55. |
| BBCT |
1.341(4) |
1.443(3) |
1.342(4) |
1.505(3) |
1.496(3) |
1.384(3) |
1.425(3) |
1.387(3) |
| BBCT-Me |
1.349(2) |
1.443(2) |
1.353(2) |
1.502(2) |
1.501(2) |
1.393(2) |
1.430(2) |
1.389(2) |
| [3]Phenyleneb |
1.363(2) |
1.419(2) |
1.365(2) |
1.514(2) |
1.514(2) |
1.392(2) |
1.416(2) |
1.390(2) |
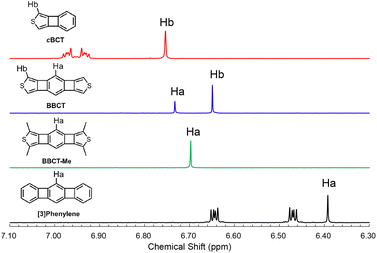 |
| | Fig. 6 1H NMR spectra of cBCT (red), BBCT (blue), BBCT-Me (green), and [3]phenylene (black) in acetone-d6 at 25 °C. | |
Conclusions
In conclusion, we synthesized a new family of linearly fused polycyclic compounds, BBCTs, containing the strained 4MRs that bridge thiophene and benzene rings. UV-vis absorption and electrochemical investigations confirmed that the BBCTs have moderate EOpt.Gap values and high HOMO energy levels. X-ray single-crystal analysis revealed that BBCT and BBCT-Me adopt herringbone and one-dimensional columnar structures, respectively. The structural features derived from the X-ray single crystal analysis and the aromatic ring current effect estimated by the 1H NMR spectra indicated moderate aromatic character in BBCT. This finding is useful for producing novel organic materials based on polycyclic compounds containing 4MRs that bridge thiophene and benzene rings.
Author contributions
Conceptualization: KT. Investigation, experimental design, and formal analysis: TK, DA, SH. Experimental data collection: TK, DA, SH. Validation: TK, DA, SH. Project administration: KT. Funding acquisition: KT. Resources: KT. Writing (original draft): TK, KT. Writing (review & editing): TK, SH, KT.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Tokuyama Science Foundation. We thank Prof. Noriharu Nagao (Meiji University) for his support with the single-crystal X-ray diffraction analyses.
References
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed.
- M. Randić, Chem. Rev., 2003, 103, 3449–3605 CrossRef PubMed.
- U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676–1686 CrossRef CAS PubMed.
- B. Djukic and D. F. Perepichka, Chem. Commun., 2011, 47, 12619–12621 RSC.
- X. Shi, T. Y. Gopalakrishna, Q. Wang and C. Chi, Chem.–Eur. J., 2017, 23, 8525–8531 CrossRef CAS PubMed.
- T. Yamamoto and K. Takimiya, J. Am. Chem. Soc., 2007, 129, 2224–2225 CrossRef CAS PubMed.
- M. Mamada and Y. Yamashita, in Polycyclic Arenes and Heteroarene, ed. Q. Miao, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016, pp. 277–308 Search PubMed.
- M. L. Tang, T. Okamoto and Z. Bao, J. Am. Chem. Soc., 2006, 128, 16002–16003 CrossRef CAS PubMed.
- W. C. Lothrop, J. Am. Chem. Soc., 1941, 63, 1187–1191 CrossRef CAS.
- B. C. Berris, G. H. Hovakeemian, T.-H. Lai, H. Mestdagh and K. P. C. Vollhardt, J. Am. Chem. Soc., 1985, 107, 5670–5687 CrossRef CAS.
- C. Dosche, H.-G. Löhmannsröben, A. Bieser, P. I. Dosa, S. Han, M. Iwamoto, A. Schleifenbaum and K. P. C. Vollhardt, Phys. Chem. Chem. Phys., 2002, 4, 2156–2161 RSC.
- M. Watanabe and T. Ohashi, Jpn. Kokai Tokkyo Koho, 2008, JP2008247853A Search PubMed.
- R. R. Parkhurst and T. M. Swager, J. Am. Chem. Soc., 2012, 134, 15351–15356 CrossRef CAS PubMed.
- Z. Jin, Y. C. Teo, S. J. Teat and Y. Xia, J. Am. Chem. Soc., 2017, 139, 15933–15939 CrossRef CAS PubMed.
- Z. Jin, Z.-F. Yao, K. P. Barker, J. Pei and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 2034–2039 CrossRef CAS PubMed.
- J. Wang, M. Chu, J.-X. Fan, T.-K. Lau, A.-M. Ren, X. Lu and Q. Miao, J. Am. Chem. Soc., 2019, 141, 3589–3596 CrossRef CAS PubMed.
- J. M. Blatchly, J. F. W. McOmie and S. D. Thatte, J. Chem. Soc., 1962, 5090–5095 RSC.
- M. P. Cava, D. R. Napier and R. J. Pohl, J. Am. Chem. Soc., 1963, 85, 2076–2080 CrossRef CAS.
- S. Hünig and H. Pütter, Chem. Ber., 1977, 110, 2532–2544 CrossRef.
- S. Yang, D. Liu, X. Xu and Q. Miao, Chem. Commun., 2015, 51, 4275–4278 RSC.
- S. Yang, M. Chu and Q. Miao, J. Mater. Chem. C, 2018, 6, 3651–3657 RSC.
- S. Yang, B. Shan, X. Xu and Q. Miao, Chem.–Eur. J., 2016, 22, 6637–6642 CrossRef CAS PubMed.
- P. Biegger, M. Schaffroth, C. Patze, O. Tverskoy, F. Rominger and U. H. F. Bunz, Chem.–Eur. J., 2015, 21, 7048–7052 CrossRef CAS PubMed.
- P. Biegger, M. Schaffroth, O. Tverskoy, F. Rominger and U. H. F. Bunz, Chem.–Eur. J., 2016, 22, 15896–15901 CrossRef CAS PubMed.
- V. Engelhardt, J. G. Garcia, A. A. Hubaud, K. A. Lyssenko, S. Spyroudis, T. V. Thimofeeva, P. Tongwa and K. P. C. Vollhadt, Synlett, 2011, 280–284 CAS.
- Y. C. Teo, Z. Jin and Y. Xia, Org. Lett., 2018, 20, 3300–3304 CrossRef CAS PubMed.
- S. Hashimoto and K. Tahara, J. Org. Chem., 2019, 84, 9850–9858 CrossRef CAS PubMed.
- P. J. Garratt and K. P. C. Vollhardt, J. Chem. Soc. D, 1970, 109 RSC.
- J. W. Barton and D. J. Lapham, Tetrahedron Lett., 1979, 20, 3571–3572 CrossRef.
- J. Nakayama, J. Synth. Org. Chem., Jpn., 1994, 52, 308–317 CrossRef CAS.
- F. Fringuelli, G. Marino, A. Taticchi and G. A. Grandolini, J. Chem. Soc., Perkin Trans., 1974, 2, 332–337 RSC.
- C. K. Frederickson, L. N. Zakharov and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 16827–16838 CrossRef CAS PubMed.
- Y. Ohtomo, K. Ishiwata, S. Hashimoto, T. Kuroiwa and K. Tahara, J. Org. Chem., 2021, 86, 13198–13211 CrossRef CAS PubMed.
- S. Hashimoto, R. Kishi and K. Tahara, New J. Chem., 2022, 46, 22703–22714 RSC.
- S. Hashimoto and K. Tahara, Chemistry, 2022, 4, 1546–1560 CrossRef CAS.
- D. Cagardová, M. Michakík, P. Poliak and V. Lukeš, J. Mol. Struct., 2019, 1175, 297–306 CrossRef.
- D. N. Nicolaides, Synthesis, 1977, 127–129 CrossRef CAS.
- M. L. Leow and J. A. H. MacBride, Tetrahedron Lett., 1984, 25, 4283–4284 CrossRef CAS.
- A. Fukazawa, H. Oshima, Y. Shiota, S. Takahashi, K. Yoshizawa and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 1731–1734 CrossRef CAS PubMed.
- A. Fukazawa, H. Oshima, S. Shimizu, N. Kobayashi and S. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 8738–8745 CrossRef CAS PubMed.
- P. J. Mayer, O. E. Bakouri, T. Holczbauer, G. F. Samu, C. Janáky, H. Ottosson and G. London, J. Org. Chem., 2020, 85, 5158–5172 CrossRef CAS PubMed.
- T. Gazdag, P. J. Mayer, P. P. Kalapos, T. Holczbauer, O. E. Bakouri and G. London, ACS Omega, 2022, 7, 8336–8349 CrossRef CAS PubMed.
- M. Nishijima, K. Mutoh, R. Shimada, A. Sakamoto and J. Abe, J. Am. Chem. Soc., 2022, 144, 17186–17197 CrossRef CAS PubMed.
- S. M. H. Kabir, M. Hasegawa, Y. Kuwatani, M. Yoshida, H. Matsuyama and M. Iyoda, J. Chem. Soc., Perkin Trans., 2001, 159–165 RSC.
- P. J. Garratt and K. P. C. Vollhardt, J. Am. Chem. Soc., 1972, 94, 7087–7092 CrossRef CAS.
- M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891–4945 CrossRef CAS PubMed.
- S. Shinamura, I. Osaka, E. Miyazaki, A. Nakao, M. Yamagishi, J. Takeya and K. Takimiya, J. Am. Chem. Soc., 2011, 133, 5024–5035 CrossRef CAS PubMed.
- M. L. Tang, A. D. Reichardt, T. Siegrist, S. C. B. Mannsfeld and Z. Bao, Chem. Mater., 2008, 20, 4669–4676 CrossRef CAS.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318–4328 CrossRef CAS PubMed.
- M. Nishio, M. Hirota and Y. Umezawa, The CH/π Interaction: Evidence, Nature, and Consequences, Wiley-VCH, New York, 1998 Search PubMed.
- H. A. Fargher, T. J. Sherbow, M. M. Haley, D. W. Johnson and M. D. Pluth, Chem. Soc. Rev., 2022, 51, 1454–1469 RSC.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed.
- J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef.
- A. Schleifenbaum, N. Feeder and K. P. C. Vollhardt, Tetrahedron Lett., 2001, 42, 7329–7332 CrossRef CAS.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Shigeru Yamago on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2223524 and 2223525. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra00144j |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*






