
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion of cellulose into valuable chemicals using sulfonated amorphous carbon in 1-ethyl-3-methylimidazolium chloride†
Thien-Hang Thi Nguyen‡
ab,
Ha Bich Phan‡abc,
Trinh Hao Nguyen ab,
Kim Nguyen Tranab,
Linh Ho Thuy Nguyenbd,
Tan Le Hoang Doanbd and
Phuong Hoang Tran*ab
ab,
Kim Nguyen Tranab,
Linh Ho Thuy Nguyenbd,
Tan Le Hoang Doanbd and
Phuong Hoang Tran*ab
aDepartment of Organic Chemistry, Faculty of Chemistry, University of Science, Vietnam National University, Ho Chi Minh City, Vietnam. E-mail: thphuong@hcmus.edu.vn; Tel: +84-903-706-762
bVietnam National University, Ho Chi Minh City, Vietnam
cInstitute of Public Health, Ho Chi Minh City, Vietnam
dCenter for Innovative Materials and Architectures (INOMAR), Ho Chi Minh City, Vietnam
First published on 6th March 2023
Abstract
In this study, three carbon-based solid acid catalysts were prepared via the one-step hydrothermal procedure using glucose and Brønsted acid, including sulfuric acid, p-toluenesulfonic acid, or hydrochloric acid. The as-synthesized catalysts were tested for their ability to convert cellulose into valuable chemicals. The effects of Brønsted acidic catalyst, catalyst loading, solvent, temperature, time, and reactor on the reaction were investigated. The as-synthesized C–H2SO4 catalyst containing Brønsted acid sites (–SO3H, –OH, and –COOH functional groups) demonstrated high activity in the transformation of cellulose into valuable chemicals with the yield of total products of 88.17% including 49.79% LA in 1-ethyl-3-methylimidazolium chloride ([EMIM]Cl) solvent at 120 °C in 24 h. The recyclability and stability of C–H2SO4 were also observed. A proposed mechanism of cellulose conversion into valuable chemicals in the presence of C–H2SO4 was presented. The current method could provide a feasible approach for the conversion of cellulose into valuable chemicals.
Introduction
Research on the transformation of available carbohydrates into valuable chemicals has received much attention over the past decade.1–5 The transformation of cellulose based on agricultural residues into useful chemicals has been studied extensively for practical applications.6–8 Facing the shortage of fossil fuels and fluctuating prices of gasoline, the development of clean, safe, and sustainable alternative energy sources has become an urgent mission.9–11 Among the new kinds of energy resources, biomass feedstocks are essential sources that are expected to bring many benefits in the future as it is the most widely abundant resource on earth.12–14 Substituting conventional energy sources with biomass is a potential solution to lower dependence on coal, oil, and gas and environmental pollution. The conversion of carbohydrates into valuable manufacturing chemicals such as 5-hydroxymethylfurfural (HMF), 2,5-diformylfuran (DFF), levulinic acid (LA), formic acid (FA), 2,5-furandicarboxylic acid (FDCA), furfural (Fur) has been investigated extensively in the past decade.4,15–26 Formic acid is a chemical that is used to make formaldehyde, rubber, plasticizers, pharmaceuticals, and textiles, among other things. Furthermore, using FA as a hydrogen storage material for automotive applications or as an internal hydrogen source for bio-based processes is gaining popularity.27,28 One of the most interesting products is LA, which is the platform chemical for various high value-added products.29 LA is the starting point for several chemicals from fields as diverse as solvents and fuels, polymers and plasticizers, food additives, pharmaceuticals. In the pharmaceutical industry, calcium levulinate is a calcium supplement enhancing bone formation and muscular excitability. In agriculture, its derivate δ-aminolevulinic acid is a biodegradable herbicide,30 levulinate potash can be used as a highly effective fertilizer.31 LA can be converted from different biomass raw materials,32,33 especially from cellulose. Over the past decade, several studies have employed homogeneous acidic catalysts for the production of LA (e.g., hydrochloric acid, nitric acid, sulfuric acid, …).34–37 However, these acids suffer from disadvantages, such as difficulty in recovery, equipment corrosion, and environmental pollution. Consequently, a variety of solid acid catalysts have received much attention in LA production from different feedstocks, including acidic TiO2 nanoparticles,38 and sulfonated carbon.39 Based on LA synthesis processes, high temperatures and long reaction times are also required for LA generation from cellulose using solid catalysts.Cellulose has become the focus material in many studies thanks to its low cost and abundant quantity. Swatloski and coworkers40 reported an exciting result for cellulose dissolution with 1-ethyl-3-methylimidazolium chloride ([EMIM]Cl). Several studies have indicated the role of liquid acids in hydrolyzing cellulose in the industry.10 However, these liquid–acid catalysts demand a special method in the form of neutralization, which involves costly and inefficient catalyst extraction from products and leads to unrecyclable sulfate waste. Therefore, the use of recyclable strong solid acids has been encouraged in response to the criteria of green and sustainable chemistry. Comparative studies of different solid acid catalysts showed that carbon materials with acid sites could be considered the most promising cellulose depolymerization catalysts as their unique properties include high thermal and chemical stability, low cost, recyclability, and high surface area.41–44
Carbon-based solid acid catalysts containing Brønsted acidic sites (–SO3H, –COOH, and –OH) have been demonstrated as effective solid catalysts for the conversion of cellulose.45–47 The catalytic effect demonstrated the significant ability of hydrolysis of cellulose; the large effective area and acid groups supported the hydration of monosaccharides to furanic compounds.48 The cellulose hydrolysis reaction in the presence of carbon-based solid acid provided a glucose yield of 40% with high selectivity.49 Recently, the hydrolysis of cellulose was carried out in ionic liquids, in which cellulose was dissolved in ionic liquids and then hydrolyzed.50,51 The ionic liquids supported the dissolution of cellulose by disrupting the crystalline structure of cellulose and made the β-1,4-glycosidic bonds more favorable for hydrolysis. Ignatyev and coworkers reported Smopex-101 hydrolyzed cellulose in [BMIM]Cl.52 According to previous studies, [BMIM]Cl and [EMIM]Cl ionic liquids are two suitable solvents to dissolve cellulose as well as convert cellulose into some basic products.53,54 In addition, [BMIM]Cl ionic liquid is more expensive than [EMIM]Cl. When compared to economic efficiency, we choose [EMIM]Cl to conduct research. Besides, the carbonaceous catalysts were used for the hydrolysis of hemicellulose to monosaccharides and the dehydration of these compounds to furfural and HMF.55–57
In this study, we report a method for the conversion of cellulose into valuable chemicals in a one-pot reaction using Brønsted acidic carbon materials in ionic liquid 1-ethyl-3-methylimidazolium chloride ([EMIM]Cl) as catalytic systems. Among three Brønsted acidic carbon materials, C–H2SO4 was prepared via direct hydrothermal carbonization of glucose, citric acid, and sulfuric acid for 4 h at 180 °C providing the highest activity in the conversion of cellulose into organic compounds. These results motivated us to continue investigating the synthesis of organic products from cellulose and agricultural by-products.
Materials and methods
Materials
D-Glucose monohydrate (>99%), cellulose (>99%), HMF (>99%), DFF (>99%), FA (>99), LA (>99%), Fur (>99%), FDCA (>99%), choline chloride (>99%), citric acid monohydrate (>99%), oxalic acid dihydrate (>99%), malonic acid (>99%), fumaric acid (>99%), adipic acid (>99%), [EMIM]Cl (98%), p-toluenesulfonic acid (>99%), sulfuric acid (98%), hydrochloric acid (36%), THF (>99%) were obtained from Sigma-Aldrich. All chemical substances were used without any further purification.Preparation of Brønsted acidic carbon materials
Brønsted acidic carbon materials were synthesized by one-step hydrothermal carbonization using the procedure reported by Xiao and coworkers with a little modification.58 The mixture of the D-glucose (10 g), citric acid monohydrate (4 g, 20 mmol), Brønsted acid (p-toluenesulfonic acid (TsOH), sulfuric acid (H2SO4) or hydrochloric acid (HCl), 10 mmol), and 80 mL distilled water was charged in a 100 mL Teflon-lined stainless-steel autoclave. The mixture was heated at 180 °C for 4 h in an oven. Black carbonaceous materials were obtained by filtration and washed several times with distilled water and followed by methanol, and then dried at 80 °C for 6 h. Three samples were prepared through this procedure including C–TsOH, C–H2SO4, and C–HCl, which had synthesized yields per 100 g of D-glucose of 36.3, 38.1, and 34.5 wt%, respectively.Characterization techniques
The neutralization titration method of every functional group over carbonaceous solid acid catalysts prepared above was similar to the study of Wang et al.59 with a little modification of conditions, and the detailed method was as follows. To measure the total functional groups (–SO3H, –COOH, and –OH groups), NaOH (1 N, 40 mL) was added to a vessel with a catalyst (80 mg). The mixture was stirred at room temperature for 24 h, and the reaction mixture was centrifuged to separate the solid. The supernatant solution was titrated by HCl (0.1 N) aqueous solution using phenolphthalein as an indicator. The content of the –SO3H group was determined by adding NaCl (1 N, 40 mL) to the catalyst (80 mg), and the reaction mixture was stirred for 24 h at room temperature. After centrifugal separation, the supernatant solution was titrated by NaOH (0.01 N) using phenolphthalein as an indicator.Fourier-transform infrared spectroscopy (FT-IR) were recorded on a Bruker's VERTEX 70 series FT-IR spectrometers and Shimadzu IRPrestige-21, with a measuring range of 600–4000 cm−1. The crystallinity of the synthesized powders was analyzed using Bruker-D8 Advance powder X-ray diffractometer (P-XRD, CuKα radiation, λ = 0.15418 nm) with a step size and a time per step fixed at 0.01 and 0.25 s, respectively. Thermal gravimetric analysis (TGA) was recorded on a TGA Q500 V20.13 Build 39 instrument, heated to 800 °C (10 °C min−1) in an air atmosphere. Scanning electron microscopy (SEM) image and Energy dispersive X-ray (EDX) spectra were measured on a Hitachi S4800 (Japan) instrument, with magnification 500–5000 times, an acceleration voltage of 10 kV. The concentration of sulfur was determined by inductively coupled plasma mass spectrometry (Agilent ICP-MS 7700x instrument).
Preparation of deep eutectic solvents (DESs)
DESs were prepared based on the study of Abbott and coworkers.60 The choline chloride and HBD (citric acid (C), oxalic acid (O), malonic acid (M), fumaric acid (F), or adipic acid (A)) with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio were heated by a magnetic stirrer for 2 h at 110 °C until they formed a homogeneous colorless liquid. DESs were used directly as solvents for the conversion of cellulose without purification. The obtained DESs are denoted as DES-C, DES-O, DES-M, DES-F, and DES-A, respectively.
:1 molar ratio were heated by a magnetic stirrer for 2 h at 110 °C until they formed a homogeneous colorless liquid. DESs were used directly as solvents for the conversion of cellulose without purification. The obtained DESs are denoted as DES-C, DES-O, DES-M, DES-F, and DES-A, respectively.
Cellulose transformation in C–H2SO4/DESs
In a typical experiment, cellulose (162 mg), DES (4 g), and C–H2SO4 (10 mg) were put into the reaction vessel. The reaction mixture was stirred with a magnetic bar at a temperature of 120 °C in an oil bath for 18 h. After completion of the reaction, samples were diluted with distilled water to analyze by the HPLC method.Cellulose conversion by C–H2SO4/[EMIM]Cl
In a typical experiment, cellulose (162 mg), [EMIM]Cl (1 g, 7 mmol), and C–H2SO4 (0, 1, 5, 8, 10, and 20 mg) were put into the reaction tubes with a magnetic bar. The mixture was heated to 120 °C in an oil bath with a magnetic stirrer. Samples were taken from the reaction mixture at a period of time for HPLC analysis to study the effect of catalyst, reaction times, and reaction temperatures on the yield of the product.Recycling and reuse of C–H2SO4/[EMIM]Cl
After completion of the reaction, valuable products were extracted by tetrahydrofuran (THF). The remaining [EMIM]Cl and catalyst were dried at 100 °C for 3 h and used for subsequent recycling.Products analysis
The reaction products were quantified by Agilent Technologies 1260 Infinity HPLC with a PAD detector (285 nm, 210 nm). The column InertSustain C18 (5 μm, 4.6 × 150 mm) was used to separate the components of the reaction mixture. The column temperature was maintained at 30 °C. A mixture of methanol and 2.5 mM sulfuric acid was used as eluent with a flow rate of 0.7 mL min−1.According to HPLC analysis, the amounts of HMF, DFF, LA, FA, Fur, and FDCA were calculated as follows:
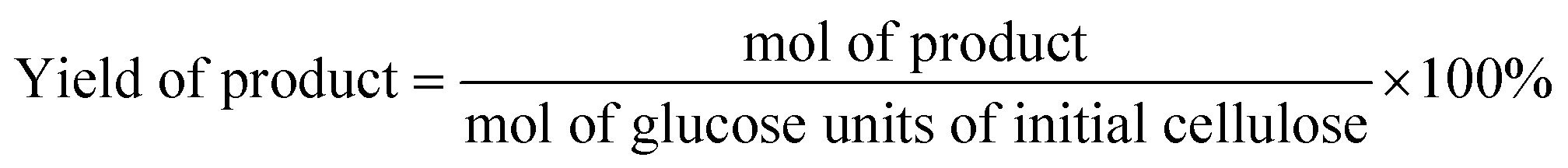
Results and discussion
Characterization of catalysts
Three different types of catalysts were synthesized by one-pot hydrothermal carbonization. To obtain strong acidity on the surface of catalysts, p-toluenesulfonic acid or sulfuric acid or hydrochloric acid were mixed in glucose solution to prepare C–TsOH, C–H2SO4, and C–HCl catalysts, respectively. The procedure for the carbonaceous solid acid catalysts was shown in Fig. 1. The colors of the three materials were obtained in black color after the hydrothermal reaction at 180 °C for 4 h.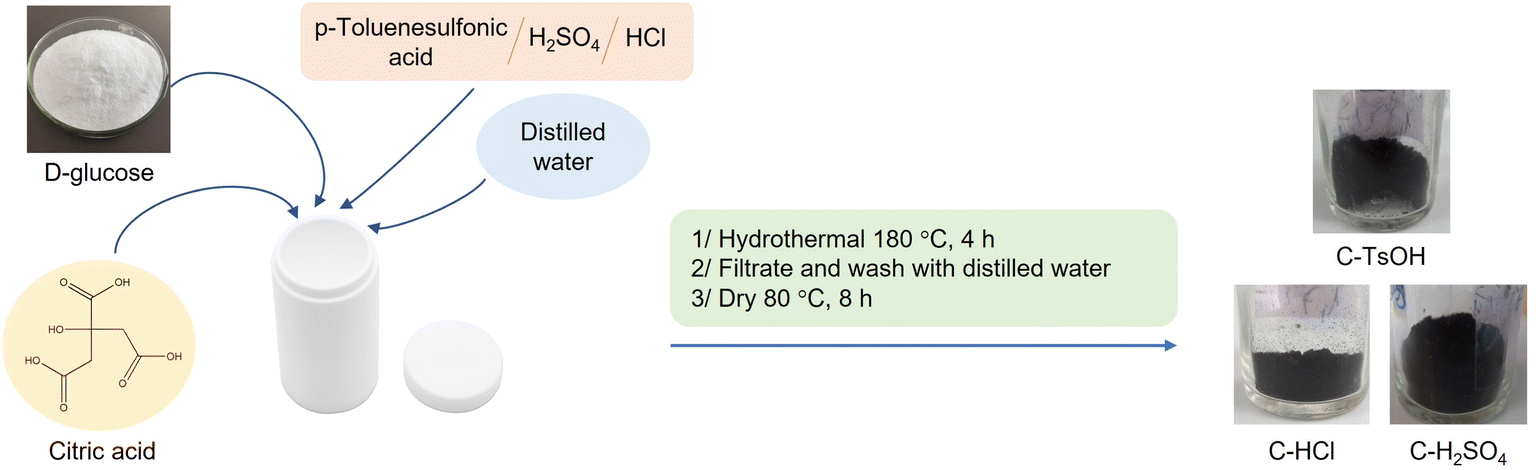 | ||
| Fig. 1 Preparation of carbonaceous acidic catalysts. | ||
The proposed mechanism for the formation of these catalysts is the conversion of glucose to organic substances such as furfural in the first step. These compounds can react with citric acid, TsOH, H2SO4, or HCl to form the corresponding acid functional groups. Then, polymerization reactions occur, forming soluble polymers. These polymers, after being aromatized, gradually aggregate into larger spheres by forming stable oxygen groups such as ethers or quinones.61 The presence of citric acid helps to provide the carboxyl acid group and improves the acidity of the catalyst. The sulfonic acid groups can also convert to another group, such as sulfonates during hydrothermal processes.
From the neutralization titration results, the total acid content of three materials was 2.2, 2.2, and 1.9 mmol g−1 for C–TsOH, C–H2SO4, and C–HCl particles, respectively. These total acid contents are similar to previous literature. Wang and coworkers developed carbon-based solid acid, which was synthesized from glucose and p-toluenesulfonic acid, and the acidity was 2.0 mmol g−1.59 Nata and coworkers reported the preparation of sulfonated carbonaceous material generated by hydrothermal carbonization of glucose at 180 °C for 4 h in the presence of acrylic acid and hydroxyethylsulfonic acid, and the acidity of this material was 2.10 mmol g−1.62
The structure of C–H2SO4 was determined by XRD, FT-IR, TGA, SEM, and EDX. The FT-IR spectra of catalysts are shown in Fig. 2a. Upon hydrothermal carbonization of glucose and acids, these rich carbon materials with functional groups including phenolic hydroxyl, carbonyl, sulfonic acid groups, and carboxylic groups were obtained. The characteristic band at ∼3414 cm−1 in three samples is attributed to the O–H stretching. The aliphatic C–H band is shown by the band 3000–2815 cm−1. The absorption band at approximately 1694 cm−1 is assigned to stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, indicating the existence of carboxylic acid groups. The absorption bands at approximately 1162 cm−1 and 1020 cm−1 are assigned to stretching SO3− and OSO, respectively. These signals confirmed that the –SO3H groups were successfully grafted onto the carbon surface.59 The band at approximately 1600 cm−1 belongs to CC groups.
O groups, indicating the existence of carboxylic acid groups. The absorption bands at approximately 1162 cm−1 and 1020 cm−1 are assigned to stretching SO3− and OSO, respectively. These signals confirmed that the –SO3H groups were successfully grafted onto the carbon surface.59 The band at approximately 1600 cm−1 belongs to CC groups.
 | ||
| Fig. 2 Characterization of C–H2SO4: (a) FT-IR spectrum, (b) XRD pattern, (c) TGA curve, (d) SEM image, and (e) EDX spectra. | ||
The XRD pattern of the catalyst was presented in Fig. 2b. The result showed no noticeable difference in the crystalline structure of the three samples. The XRD pattern exhibited a broad diffraction peak at 2θ angle of 15–30°, which was attributed to (002) planes of amorphous carbon. Besides, the weak diffraction peak (101) around 45° was also observed due to the axis of the graphite structure. These diffraction patterns were typical of amorphous carbon consisting of disordered polycyclic aromatic carbon sheets.63 TGA was measured to investigate the thermal stability of the prepared material. As shown in Fig. 2c, the sample presented a slight weight loss (about 5%) below 200 °C, which is mainly due to the loss of a small amount of bonded water. The thermogravimetric curve of the catalyst at higher temperatures (above 300 °C) showed more significant weight loss because of the gradual desorption and thermal decomposition process of organic functional groups. Thus, it could be concluded that the catalyst was stable up to 300 °C. The EDX spectra (Fig. 2d) showed the presence of C, O, and S elements. In addition, ICP-MS revealed that the obtained material had a calculated sulfur content of 620 mg kg−1. The SEM image of C–H2SO4, as depicted in Fig. 2e showed that the resulting particle was spherical in shape with 5–8 μm diameter size.
Catalytic performance of carbonaceous catalysts
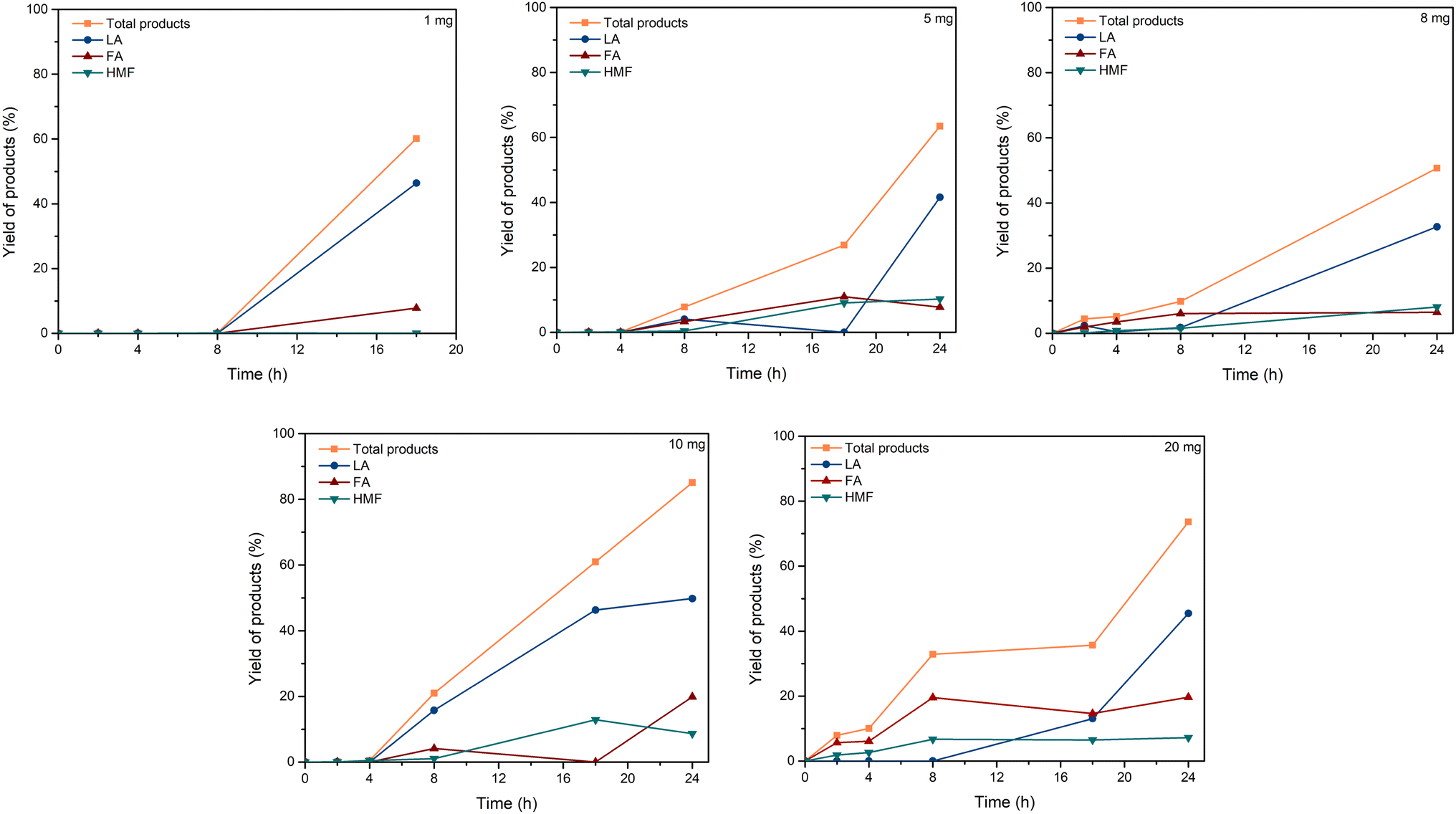 | ||
| Fig. 3 Effect of C–H2SO4 dosage. Reaction conditions: cellulose (162 mg), C–H2SO4 (1, 5, 8, 10, 20 mg), [EMIM]Cl (1 g, 7 mmol), 120 °C. | ||
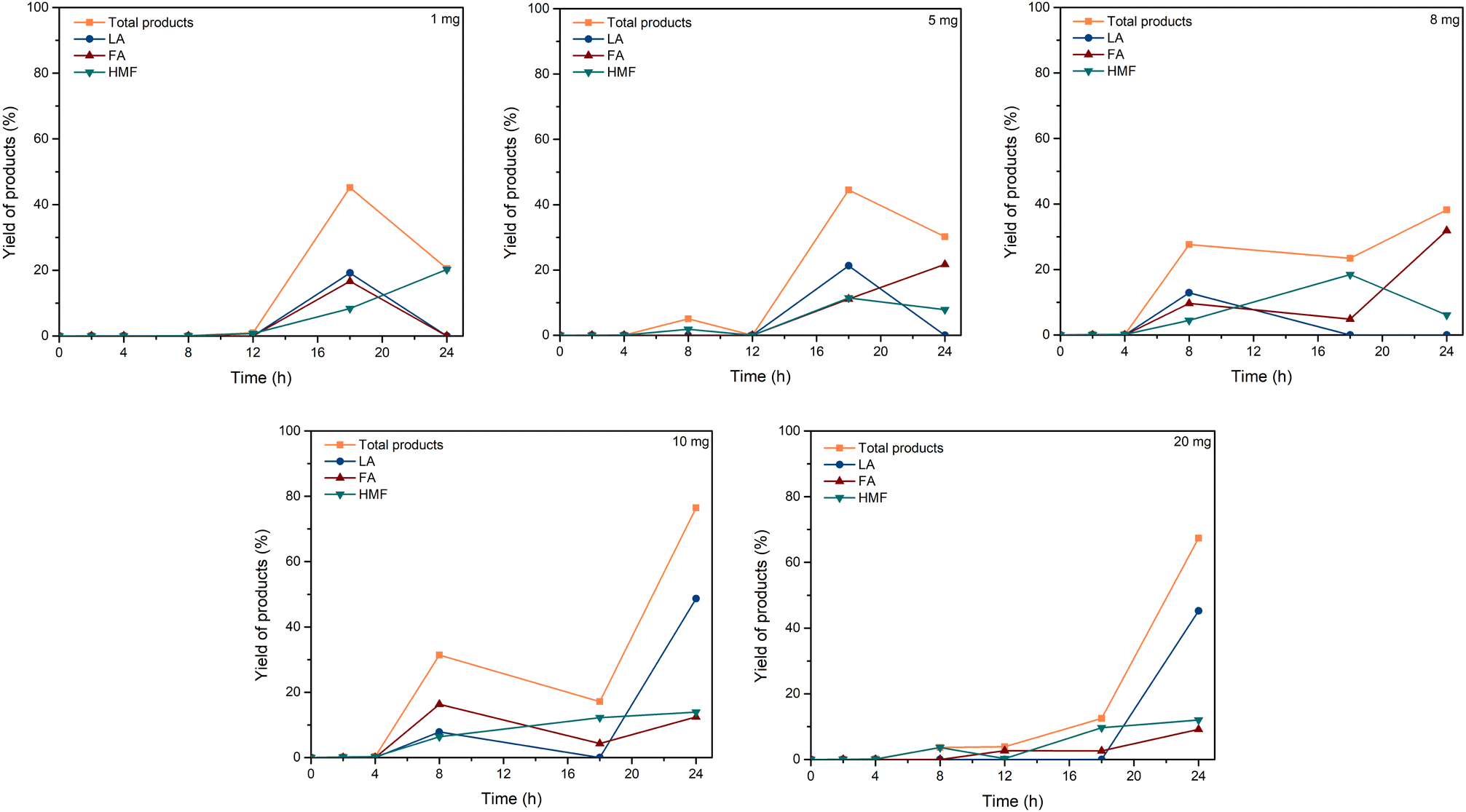 | ||
| Fig. 4 Effect of C–TsOH dosage. Reaction conditions: cellulose (162 mg), C–TsOH (1, 5, 8, 10, 20 mg), [EMIM]Cl (1 g, 7 mmol), 120 °C. | ||
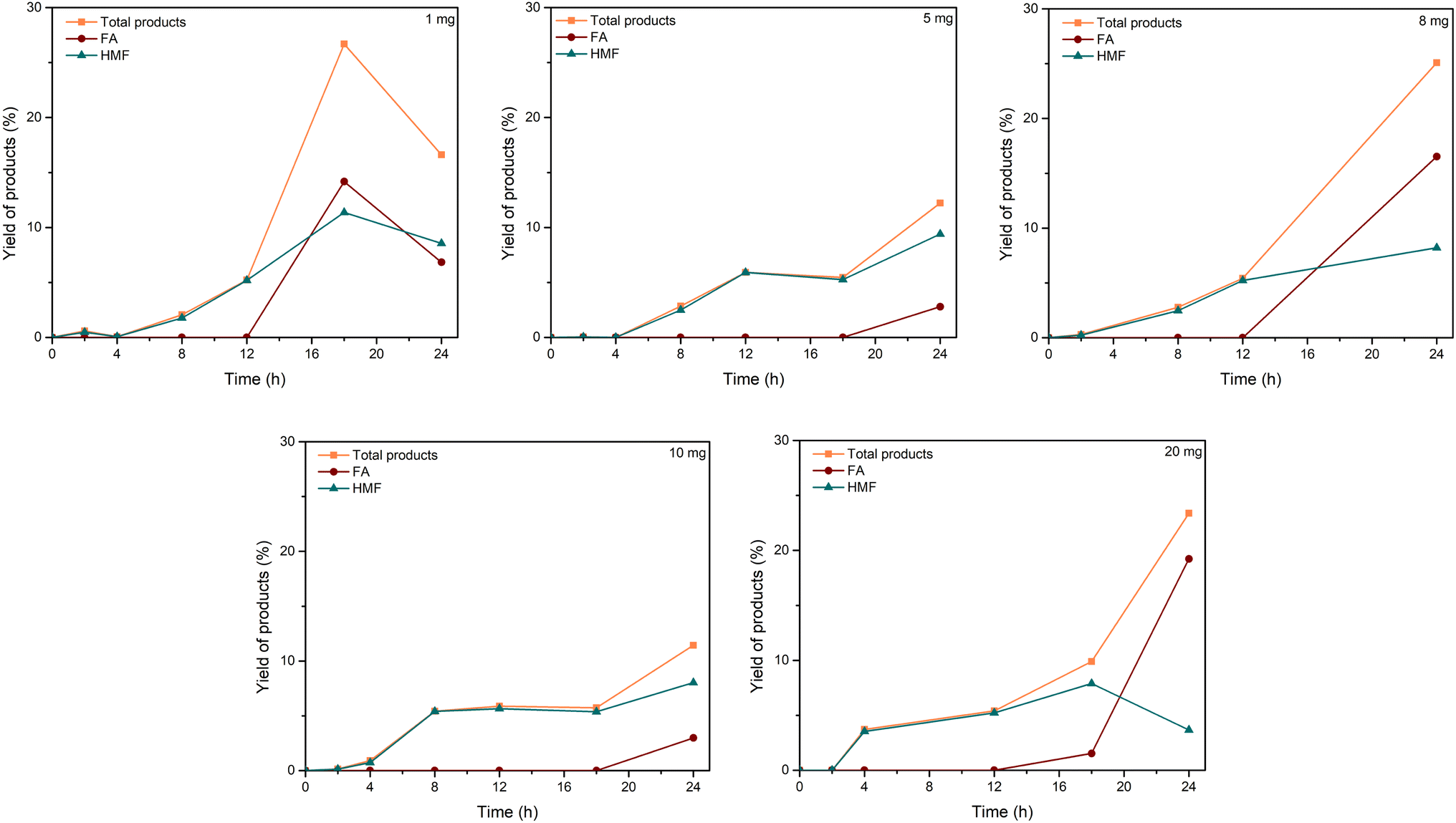 | ||
| Fig. 5 Effect of C–HCl dosage. Reaction conditions: cellulose (162 mg), C–HCl (1, 5, 8, 10, 20 mg), [EMIM]Cl (1 g, 7 mmol), 120 °C. | ||
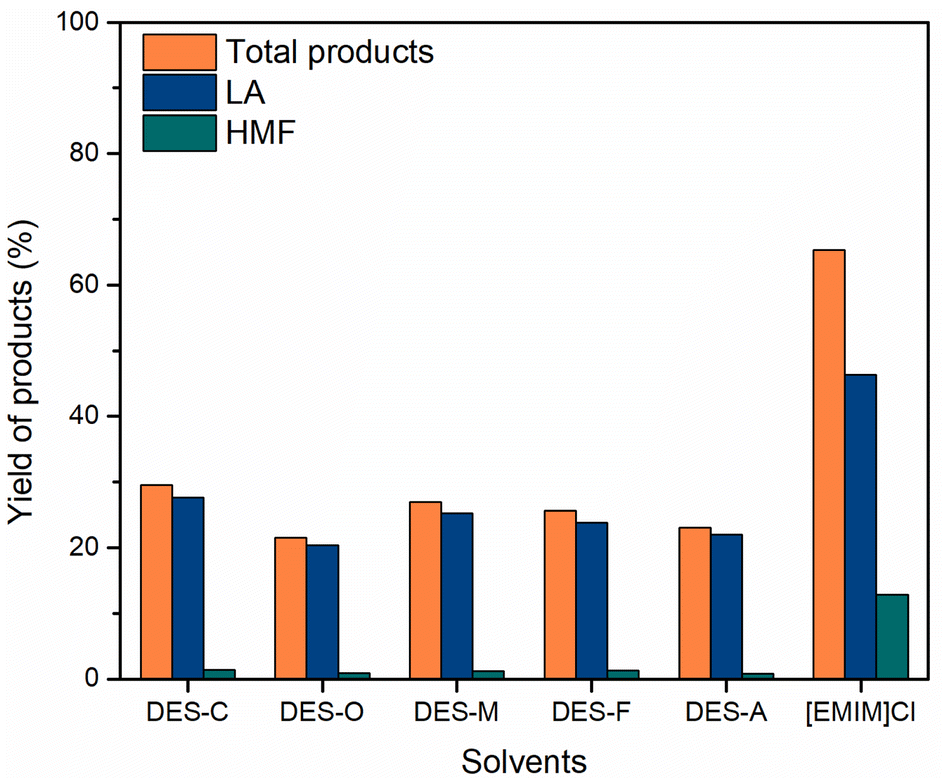 | ||
| Fig. 6 Effect of various solvents on cellulose conversion. Reaction conditions: cellulose (162 mg), C–H2SO4 (10 mg), [EMIM]Cl (1 g, 7 mmol), 120 °C, 18 h. | ||
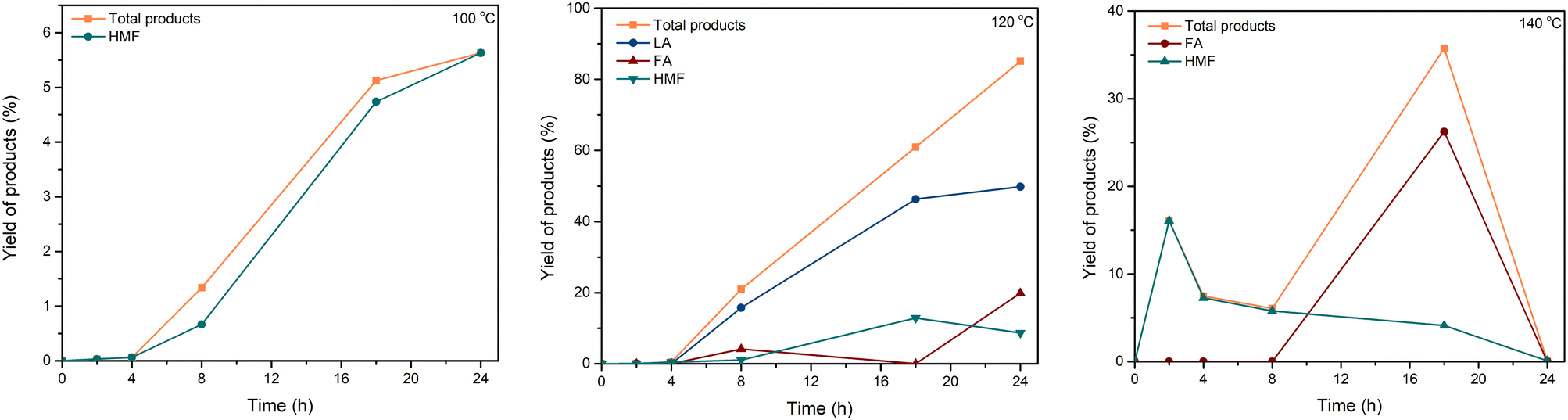 | ||
| Fig. 7 Effect of reaction time and temperature on total yield of products. Reaction conditions: cellulose (162 mg), C–H2SO4 (10 mg), [EMIM]Cl (1 g, 7 mmol). | ||
 | ||
| Fig. 8 Cellulose conversion towards valuable chemicals in the recycling test. Reaction conditions: cellulose (162 mg), C–H2SO4 (10 mg), [EMIM]Cl (1 g, 7 mmol), 120 °C, 24 h. | ||
The comparison of the current work with the reported literature on the conversion of cellulose into valuable substances was presented in Table 1. It can be seen that the catalytic activity of C–H2SO4 in [EMIM]Cl showed high efficiency in the conversion of cellulose to valuable chemicals. The previous literature revealed that the conversion of cellulose also afforded products in high yields, but those procedures still suffered drawbacks, including the requirement of expensive catalysts, organic solvents, or high-pressure conditions.
| Entry | Reaction condition | Yield (%) | Ref. | ||
|---|---|---|---|---|---|
| HMF | FA | LA | |||
| 1 | Microcrystalline cellulose (0.5 g), 1-(4-sulfonic acid) ethyl-3-methylimidazolium hydrogen sulfate (1.0 g), 0.2 M FeCl2 (1 mL), MIBK (8 mL), 150 °C, 300 min, 101325 Pa | 33.86 | 18.61 | 5.93 | 66 |
| 2 | Cellulose (50 mg), [C3SO3Hmim]HSO4 (1.000 g), H2O (2.000 g), 160 °C, 30 min, MW | — | — | 55.0 | 21 |
| 3 | Cellulose (0.05 g), [BSmim]HSO4 (1.5 g), H2O (0.3 g, 20 wt%), 120 °C, 120 min | 4.3 | — | 39.4 | 67 |
| 4 | Cellulose (162 mg), C–H2SO4 (10 mg), [EMIM]Cl (1 g, 7 mmol), 120 °C, 24 h | 8.67 | 19.89 | 46.31 | This work |
The mechanism of this conversion was proposed in Scheme 1. Kitano and coworkers reported the amorphous carbon could absorb β-1,4 glucan.41 Thus, we proposed that the mechanism began with the sorption of cellulose on the catalyst through the formation of hydrogen bonds between the oxygen of cellulose and OH groups in the carbonaceous catalyst. As previously reported, hydrogen bonds forming with the oxygen of glycosidic bond with the OH group of the carbonaceous materials were more favorable than the inter- and intramolecular hydrogen bonds of cellulose. Thus, the β-1,4-glycosidic bonds of cellulose were associated with priority with the acidic sites of the carbonaceous material for hydrolysis.41 Hence, the catalytic activity of the carbonaceous catalyst increased the synergistic effect of adsorption to cellulose on the surface and the strong –SO3H acidic sites attributed to the hydrolysis of cellulose.41 Based on the work of Cao et al.68 and our experiment, the mechanism of solid acid-catalyzed hydrolysis of cellulose in [EMIM]Cl was started with the Brønsted acid sites on the surface of the catalyst attacked to the oxygen of the β-1,4-glycosidic bond. Then, the C–O bond was cleavaged and released glucose molecules. Since Brønsted acid has a poor effect on the isomerization of glucose to fructose, the formation of HMF and LA can be explained by levoglucosan (LGA) intermediate formation.17,68,69 The long reaction time in the Brønsted acid condition favored the formation of LA and FA.
 | ||
| Scheme 1 Proposed reaction mechanism for cellulose hydrolysis-dehydration by C–H2SO4 in [EMIM]Cl. | ||
Conclusions
We have successfully developed the conversion of cellulose to valuable chemicals using the catalytic system C–H2SO4/[EMIM]Cl for the first time. The yield of total products could reach 88.17% (including 49.79% LA) when using C–H2SO4/[EMIM]Cl under optimal conditions. Meanwhile, C–HCl gave low yields of products, which could be explained by the activity of the chloride group being weaker than that of the –SO3H group in the hydrolysis/dehydration reaction. The current method showed that high reaction temperature accelerated the reaction to provide high yields of products. Nonetheless, excessive-high temperature resulted in a decrease in product yield due to the formation of humins. Besides, there was no considerable loss in catalytic activity in three consecutive cycles, indicating the reusability and stability of the catalytic system.Data availability
The data of this study are available from the corresponding author.Author's contributions
Hang Thien Thi Nguyen: methodology, investigation, writing-original draft-review and editing. Ha Bich Phan: methodology, investigation, data curation. Trinh Hao Nguyen and Kim Nguyen Tran: investigation, data curation. Linh Ho Thuy Nguyen and Tan Le Hoang Doan: data curation. Phuong Hoang Tran: methodology, writing-review and editing, project administration. All authors read and approved the final manuscript.Consent for publication
The authors give the publisher permission to publish the work.Conflicts of interest
The authors declare no competing interest.Acknowledgements
The authors acknowledge the financial support from Vietnam National University, Ho Chi Minh City (VNU-HCM) under grant number 562-2022-18-03. The authors would also to thank Dr Duy-Khiem Nguyen Chau for the valuable discussion. The authors express their sincere thanks to the Center for Innovative Materials & Architectures (INOMAR), Viet Nam National University, Ho Chi Minh City for their support in using the TGA and FT-IR measurements.References
- F. Chacón-Huete, C. Messina, B. Cigana and P. Forgione, ChemSusChem, 2022, e202200328 Search PubMed.
- L. Hu, L. Lin, Z. Wu, S. Zhou and S. Liu, Renewable Sustainable Energy Rev., 2017, 74, 230–257 CrossRef CAS.
- H. B. Phan, C. M. Luong, L. P. Nguyen, B. T. Bui, H. T. Nguyen, B. V. Mai, T. V.-T. Mai, L. K. Huynh and P. H. Tran, ACS Sustainable Chem. Eng., 2022, 10, 8673–8684 CrossRef CAS.
- P. H. Tran and P. V. Tran, Fuel, 2019, 246, 18–23 CrossRef CAS.
- R. Radhakrishnan, P. Patra, M. Das and A. Ghosh, Renewable Sustainable Energy Rev., 2021, 149, 111368 CrossRef CAS.
- J. B. Ocreto, W.-H. Chen, A. P. Rollon, H. C. Ong, A. Pétrissans, M. Pétrissans and M. D. G. De Luna, Chem. Eng. J., 2022, 136733 CrossRef CAS.
- V. Ashokkumar, R. Venkatkarthick, S. Jayashree, S. Chuetor, S. Dharmaraj, G. Kumar, W.-H. Chen and C. Ngamcharussrivichai, Bioresour. Technol., 2022, 344, 126195 CrossRef CAS PubMed.
- R. Kumar, T. H. Kim, B. Basak, S. M. Patil, H. H. Kim, Y. Ahn, K. K. Yadav, M. M. Cabral-Pinto and B.-H. Jeon, J. Cleaner Prod., 2021, 130180 Search PubMed.
- N. S. Ab Rasid, A. Shamjuddin, A. Z. A. Rahman and N. A. S. Amin, J. Cleaner Prod., 2021, 321, 129038 CrossRef CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr, J. P. Hallett, D. J. Leak and C. L. Liotta, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- A. M. Elgarahy, A. Hammad, D. M. El-Sherif, M. Abouzid, M. S. Gaballah and K. Z. Elwakeel, J. Environ. Chem. Eng., 2021, 9, 106503 CrossRef CAS.
- W. Deng, Y. Feng, J. Fu, H. Guo, Y. Guo, B. Han, Z. Jiang, L. Kong, C. Li, H. Liu, P. T. T. Nguyen, P. Ren, F. Wang, S. Wang, Y. Wang, Y. Wang, S. S. Wong, K. Yan, N. Yan, X. Yang, Y. Zhang, Z. Zhang, X. Zeng and H. Zhou, Green Energy Environ., 2023, 8, 10–114 CrossRef CAS.
- Y. Zhao, K. Lu, H. Xu, L. Zhu and S. Wang, Renewable Sustainable Energy Rev., 2021, 139, 110706 CrossRef CAS.
- B. Beig, M. Riaz, S. R. Naqvi, M. Hassan, Z. Zheng, K. Karimi, A. Pugazhendhi, A. Atabani and N. T. L. Chi, Fuel, 2021, 287, 119670 CrossRef CAS.
- M. J. Antal Jr, W. S. Mok and G. N. Richards, Carbohydr. Res., 1990, 199, 91–109 CrossRef CAS PubMed.
- D. B. Bevilaqua, M. K. Rambo, T. M. Rizzetti, A. L. Cardoso and A. F. Martins, J. Cleaner Prod., 2013, 47, 96–101 CrossRef CAS.
- C. E. Bounoukta, C. Megías-Sayago, F. Ammari, S. Ivanova, A. Monzon, M. A. Centeno and J. A. Odriozola, Appl. Catal., B, 2021, 286, 119938 CrossRef CAS.
- C. M. Lew, N. Rajabbeigi and M. Tsapatsis, Ind. Eng. Chem. Res., 2012, 51, 5364–5366 CrossRef CAS.
- J. Li, D. J. Ding, L. Deng, Q. X. Guo and Y. Fu, ChemSusChem, 2012, 5, 1313–1318 CrossRef CAS PubMed.
- C. Moreau, R. Durand, S. Razigade, J. Duhamet, P. Faugeras, P. Rivalier, P. Ros and G. Avignon, Appl. Catal., A, 1996, 145, 211–224 CrossRef CAS.
- H. Ren, Y. Zhou and L. Liu, Bioresour. Technol., 2013, 129, 616–619 CrossRef CAS PubMed.
- Y. Román-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef PubMed.
- X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS.
- P. Hoang Tran, ChemSusChem, 2022, 15, e202200220 CrossRef CAS PubMed.
- H. B. Phan, Q. B. T. Nguyen, C. M. Luong, K. N. Tran and P. H. Tran, Mol. Catal., 2021, 503, 111428 CrossRef CAS.
- X. Zhang, X. Zhang, N. Sun, S. Wang, X. Wang and Z. Jiang, Renewable Energy, 2019, 141, 802–813 CrossRef CAS.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC.
- W. Supronowicz, I. Ignatyev, G. Lolli, A. Wolf, L. Zhao and L. Mleczko, Green Chem., 2015, 17, 2904–2911 RSC.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. L. Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- M. Mascal and S. Dutta, Green Chem., 2011, 13, 40–41 RSC.
- J. Zhang, S. Wu, B. Li and H. Zhang, ChemCatChem, 2012, 4, 1230–1237 CrossRef CAS.
- S. Van de Vyver, J. Thomas, J. Geboers, S. Keyzer, M. Smet, W. Dehaen, P. A. Jacobs and B. F. Sels, Energy Environ. Sci., 2011, 4, 3601–3610 RSC.
- H. Anggorowati, S. Jamilatun, R. B. Cahyono and A. Budiman, 2018.
- B. Girisuta, L. Janssen and H. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef CAS.
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184–189 CrossRef CAS.
- C. Chang, M. Xiaojian and C. Peilin, Chin. J. Chem. Eng., 2006, 14, 708–712 CrossRef CAS.
- V. Tarabanko, M. Y. Chernyak, S. Aralova and B. Kuznetsov, React. Kinet. Catal. Lett., 2002, 75, 117–126 CrossRef CAS.
- C.-H. Kuo, A. S. Poyraz, L. Jin, Y. Meng, L. Pahalagedara, S.-Y. Chen, D. A. Kriz, C. Guild, A. Gudz and S. L. Suib, Green Chem., 2014, 16, 785–791 RSC.
- F. Chambon, F. Rataboul, C. Pinel, A. Cabiac, E. Guillon and N. Essayem, Appl. Catal., B, 2011, 105, 171–181 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- M. Kitano, D. Yamaguchi, S. Suganuma, K. Nakajima, H. Kato, S. Hayashi and M. Hara, Langmuir, 2009, 25, 5068–5075 CrossRef CAS PubMed.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033–1037 RSC.
- J. Pang, A. Wang, M. Zheng and T. Zhang, Chem. Commun., 2010, 46, 6935–6937 RSC.
- M. Hara, T. Yoshida, A. Takagaki, T. Takata, J. N. Kondo, S. Hayashi and K. Domen, Angew. Chem., 2004, 116, 3015–3018 CrossRef.
- P. H. Tran, V. H. Huynh, P. E. Hansen, D. K. N. Chau and T. N. Le, Asian J. Org. Chem., 2015, 4, 482–486 CrossRef CAS.
- A. Onda, T. Ochi and K. Yanagisawa, Top. Catal., 2009, 52, 801–807 CrossRef CAS.
- H. Guo, X. Qi, L. Li and R. L. Smith Jr, Bioresour. Technol., 2012, 116, 355–359 CrossRef CAS PubMed.
- S. Suganuma, K. Nakajima, M. Kitano, S. Hayashi and M. Hara, ChemSusChem, 2012, 5, 1841–1846 CrossRef CAS PubMed.
- L. Gan, J. Zhu and L. Lv, Cellulose, 2017, 24, 5327–5339 CrossRef CAS.
- J. H. Jordan, M. W. Easson and B. D. Condon, RSC Adv., 2020, 10, 39413–39424 RSC.
- S. Suzuki, Y. Takeoka, M. Rikukawa and M. Yoshizawa-Fujita, RSC Adv., 2018, 8, 14623–14632 RSC.
- I. A. Ignatyev, C. V. Doorslaer, P. G. Mertens, K. Binnemans and D. E. d. Vos, Holzforschung, 2012, 66, 417–425 CAS.
- B. Kosan, C. Michels and F. Meister, Cellulose, 2008, 15, 59–66 CrossRef CAS.
- O. M. Gazit and A. Katz, ChemSusChem, 2012, 5, 1542–1548 CrossRef CAS PubMed.
- M. Nahavandi, T. Kasanneni, Z. S. Yuan, C. C. Xu and S. Rohani, ACS Sustainable Chem. Eng., 2019, 7, 11970–11984 CAS.
- Z. Qi, Q. Wang, C. Liang, J. Yue, S. Liu, S. Ma, X. Wang, Z. Wang, Z. Li and W. Qi, Ind. Eng. Chem. Res., 2020, 59, 17046–17056 CrossRef CAS.
- R. Ormsby, J. R. Kastner and J. Miller, Catal. Today, 2012, 190, 89–97 CrossRef CAS.
- H. Xiao, Y. Guo, X. Liang and C. Qi, J. Solid State Chem., 2010, 183, 1721–1725 CrossRef CAS.
- J. Wang, W. Xu, J. Ren, X. Liu, G. Lu and Y. Wang, Green Chem., 2011, 13, 2678–2681 RSC.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Carbon, 2009, 47, 2281–2289 CrossRef CAS.
- I. F. Nata, C. Irawan, P. Mardina and C.-K. Lee, J. Solid State Chem., 2015, 230, 163–168 CrossRef CAS.
- X.-Y. Liu, M. Huang, H.-L. Ma, Z.-Q. Zhang, J.-M. Gao, Y.-L. Zhu, X.-J. Han and X.-Y. Guo, Molecules, 2010, 15, 7188–7196 CrossRef CAS PubMed.
- R. O. Almeida, A. Moreira, D. Moreira, M. E. Pina, M. G. Carvalho, M. G. Rasteiro and J. A. Gamelas, RSC Adv., 2022, 12, 3979–3989 RSC.
- Y.-L. Chen, X. Zhang, T.-T. You and F. Xu, Cellulose, 2019, 26, 205–213 CrossRef CAS.
- F. Tao, H. Song and L. Chou, ChemSusChem, 2010, 3, 1298–1303 CrossRef CAS PubMed.
- Y. Shen, J.-K. Sun, Y.-X. Yi, B. Wang, F. Xu and R.-C. Sun, Bioresour. Technol., 2015, 192, 812–816 CrossRef CAS PubMed.
- F. Cao, T. J. Schwartz, D. J. McClelland, S. H. Krishna, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2015, 8, 1808–1815 RSC.
- S. H. Krishna, T. W. Walker, J. A. Dumesic and G. W. Huber, ChemSusChem, 2017, 10, 129–138 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00177f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |