DOI:
10.1039/D3RA00245D
(Paper)
RSC Adv., 2023,
13, 5925-5932
Efficient O-demethylation of lignin-derived aromatic compounds under moderate conditions†
Received
12th January 2023
, Accepted 9th February 2023
First published on 17th February 2023
Abstract
Lignin is a potential feedstock to produce renewable aromatic chemicals. However, lignin-derived aromatics are heavily methoxylated, which affects their reactivity in some downstream valorization attempts. Herein, we report an efficient method for the demethylation of the aromatics derived from lignin depolymerization using acidic concentrated lithium bromide (ACLB) under moderate conditions (e.g., 1.5 M HCl, 110 °C, and 2 h). Aromatics with one or two methoxy groups (G-type and S-type), alkyl hydroxyl and carbonyl groups, and electron-donating and electron-withdrawing substituents were used to investigate the demethylation mechanisms. S-type aromatics were demethylated faster than their G-type analogs. Alkyl hydroxyl groups were brominated under the conditions. Carbonyl groups (aldehydes and ketones) promoted unwelcome condensation. Electron-donating substituents promoted demethylation, whereas electron-withdrawing substituents retarded the demethylation. An ortho-carboxylic group enhanced the demethylation because of the formation of a stable intermediate.
Introduction
Due to environmental pollution and climate change caused by the overuse of fossil resources, the concept of a biorefinery in which lignocellulosic biomass is converted into a variety of products including fuels, chemicals, and materials, has attracted increasing interest.1 Lignin, one of the three main constituents of lignocellulose along with cellulose and hemicelluloses, is the most abundant renewable aromatic resource and has considerable potential to serve as a starting material for the production of aromatic compounds via thermochemical depolymerization to replace petroleum-based chemicals.2,3 Lignin contains three main types of aromatic units: p-hydroxyphenyl (H), coniferyl (G), and syringyl (S) units derived, respectively, from p-coumaryl, coniferyl, and sinapyl alcohols.4,5 These building units are interlinked through C–C (5–5, β–β, β–1, and β–5) and C–O (β–O–4 and 4–O–5) bonds, and the percentages of the monolignols and the linkages in lignin vary with plant species; bonds also formed beyond the radical coupling step of lignification in rearomatization reactions (and therefore not constituting inter-unit bonding per se), include α–O–4 (in β–5-coupled units) and α–O–γ (in β–β-coupled units).6 Many lignin depolymerization methods have been developed for aromatics production. For example, catalytic hydrogenolysis (or reductive catalytic fractionation, RCF) of lignin using hydrogen gas or a hydrogen donor is a popular and efficient method for breaking C–O linkages and producing aromatic and phenolic compounds in good yields.7,8 Oxidative depolymerization of lignin with oxygen, hydrogen peroxide, or peroxyacids under moderate conditions produces oxygenated aromatic compounds, such as vanillin, acetovanillone, and vanillic acid.9–11 Most ether bonds of lignin can be cleaved during hydrogenolysis and oxidation, but the aryl methyl ether structure (methoxy group) is often preserved in these lignin depolymerization processes.12 Chemical reactivity in some applications that need more phenolic hydroxy groups is consequently blocked. Developing efficient demethylation methods for lignin-derived aromatics under mild conditions is therefore of significant interest and importance for upgrading the aromatics into specific chemicals, such as catechol and pyrogallol derivatives.
Many methods have been reported for the cleavage of aryl methyl ethers, but most of them require harsh conditions (e.g., high temperature and long reaction time) and/or special reagents (Table S1†). For example, concentrated HI and HBr are the oldest reagents for the demethylation of lignin and lignin model compounds, and glacial acetic acid or phase-transfer catalysts are often used to improve the solubility of the substrates and/or the reaction efficiency.13–16 As a weaker acid than HI and HBr, HCl alone is ineffective for demethylation, but a salt-based reagent pyridinium chloride (Py·HCl) at 200 °C has been used for the demethylation of 4-methoxyphenylbutyric acid on a pilot scale.17 Some Lewis acids (e.g., BBr3 and AlCl3) are effective ether-cleaving reagents for demethylation. Their ether-cleavage efficiency is highly dependent on the reaction conditions. They are sensitive to air and water, so reactions must be conducted under dry conditions and an inert atmosphere.18–20 LiI-mediated aryl methyl ether cleavage was reported to promote the utilization of methoxy groups in lignin to produce acetic acid, methylated anilines, and ethanol. The ether cleavage function of LiI as a promoter requires the assistance of BF4− and other catalysts in the system.21–23 In recent years, ionic liquids (ILs) have been used as reaction media or catalysts for the demethylation of aryl methyl ethers and lignin. As ILs are non-volatile, non-flammable, and recyclable, they are considered to be green solvents (although their production and effects in the environment are not always benign), and reactions are usually conducted under mild conditions (<100 °C).24–26 However, ILs have some drawbacks including toxicity, preparation difficulty, and high cost; their mechanisms of action often remain unclear and complex depending on the IL system.27 Pressured hot water with Au/Nb2O5 catalyst or acid catalyst was also reported for demethylation of 4-propylguaiacol (PG) and related compounds. These systems require high temperature (e.g., 250 °C) and high pressure (e.g., 50 bar).12,28,29
It was observed in our previous studies that both native lignin in biomass and isolated industrial lignin (e.g., kraft lignin) can be extensively depolymerized in acidic concentrated lithium bromide (ACLB) solution by the cleavage of aryl ether (β–O–4) bonds under mild conditions (e.g., with 0.3 M HCl at 110 °C for 2 h).30,31 A follow-up investigation revealed that the ACLB at a higher acid concentration (e.g., 1.3 M HBr) can cleave all (linear, cyclic, and aromatic) ethers via nucleophilic substitution (SN2) mechanisms, except for biphenyl ethers with their characteristic 4–O–5 interunit bonding in lignin.32 It was demonstrated that the ACLB method can efficiently demethylate kraft lignin and convert it into an excellent radical and metal scavenger.33 Compared with other demethylation methods (Table S1†), the ACLB method required milder conditions (e.g., lower acid concentration, moderate temperature, and reaction under atmospheric pressure) and/or had better yield/selectivity. In addition, the chemicals used in the ACLB system are recyclable, less hazardous, and competitive in cost. Inspired by our previous studies, we hypothesized that the ACLB system could be an efficient approach to demethylate lignin-derived aromatics for their upgrading. We therefore carefully studied the cleavage of aryl methyl ethers in the ACLB system in this work using PG, one of the main products from the reductive catalytic fractionation of softwoods, as a test compound.34–36 Its demethylation product 4-propylcatechol (PC) can be used as a precursor in many applications, such as adhesives, pesticides, fragrances, and pharmaceuticals.37–39 The versatility of the ACLB system was also tested on other lignin-derived aromatic compounds (from both reductive and oxidative depolymerization of lignin) and substituted anisole derivatives. The effect of different functional groups/substituents on the cleavage of the aryl methyl ether bond was also investigated.
Experimental
Materials
PG, LiBr, N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA), anhydrous Na2SO4, and NaCl were purchased from Sigma-Aldrich. 4-Methylcatechol (MC) and ethyl acetate were purchased from Frontier Scientific. PC was purchased from Arctom. HCl was purchased from Macron. Vanillin, acetovanillone, vanillic acid, syringaldehyde, acetosyringone, syringic acid, anisole, guaiacol, 4-methoxyphenol, 2-methoxytoluene, 4-methoxytoluene, 2-nitroanisole, 4-nitroanisole, 2-methoxybenzoic acid, 4-methoxybenzoic acid, 3,4-dihydroxybenzaldehyde, 3,4-dihydroxyacetophenone, 3,4-dihydroxybenzoic acid, 3,4,5-trihydroxybenzaldehyde, 3,4,5-trihydroxybenzoic acid, phenol, catechol, hydroquinone, 2-methylphenol, 4-methylphenol, 2-nitrophenol phenol, 4-nitrophenol, 2-hydroxybenzoic acid, and 4-hydroxybenzoic acid were purchased from TCI. 4-Propylsyringol (PS), 4-propanolguaiacol (PGOH, guaiacylpropanol), and 4-propanolsyingol (PSOH, syringylpropanol) were synthesized with ∼99% purity (by NMR), as described in Section 1 in ESI (Fig. S1†).
Demethylation of lignin compounds in ACLB
Demethylation of lignin model compounds was carried out in a 20 mL pressure glass vial, stirred via a magnetic stirring bar. In a typical reaction, 0.1–1.0 mmol of the compound was mixed with a 2.5 mL acidic LiBr aqueous solution (32.5–65.9% w/w) with an HCl concentration of 0.25–1.50 M and then heated up to a preset temperature in an oil-bath. After the preset reaction time, the reaction was quenched by placing the vial in iced water. The mixture was diluted with brine (5 mL) and extracted with ethyl acetate (3 × 5 mL). The ethyl acetate phase was pipetted from the water phase and dried over anhydrous Na2SO4 before further analysis. All experiments were conducted in duplicate, and the average is reported.
GC-MS analysis
Qualitative analysis of demethylation products was conducted using a GC-MS system (GCMS-QP 2010S, Shimadzu Co., Addison, IL) with an Rtx-5MS column (30 m × 0.25 mm, L × ID, with 0.25 μm film thickness). The injection port was maintained at 280 °C, and the carrier gas was helium at a flow rate of 1.27 mL min−1 in a split mode (split ratio, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The GC oven temperature was programmed at a heating rate of 10 °C min−1 from 50 °C to 150 °C, 5 °C min−1 from 150 °C to 220 °C, and 10 °C min−1 from 220 °C to 300 °C. The MS detector was operated in electron-impact (EI) ionization mode and the scanning range was 35–600 m/z. The products from demethylation were silylated to improve the volatility and thermostability. In brief, a 200 μL aliquot of the ethyl acetate extract was loaded into a small screw-threaded vial and dried under a gentle stream of nitrogen. BSTFA (100 μL) was mixed and reacted with the dried products at room temperature for 3 h. The products were identified based on the NIST mass spectral library and/or by their molecular ion peaks.
:1). The GC oven temperature was programmed at a heating rate of 10 °C min−1 from 50 °C to 150 °C, 5 °C min−1 from 150 °C to 220 °C, and 10 °C min−1 from 220 °C to 300 °C. The MS detector was operated in electron-impact (EI) ionization mode and the scanning range was 35–600 m/z. The products from demethylation were silylated to improve the volatility and thermostability. In brief, a 200 μL aliquot of the ethyl acetate extract was loaded into a small screw-threaded vial and dried under a gentle stream of nitrogen. BSTFA (100 μL) was mixed and reacted with the dried products at room temperature for 3 h. The products were identified based on the NIST mass spectral library and/or by their molecular ion peaks.
GC-FID analysis
Quantitative analysis of demethylation products was conducted using a GC-FID system (GC-2014, Shimadzu Co., Canby, OR) with an Rtx-5MS column (30 m × 0.25 mm, L × ID, with 0.25 μm film thickness). The injector was maintained at 280 °C and the detector was set at 310 °C. The GC oven temperature and sample preparation were the same as for the GC-MS analysis above. Quantification was conducted based on the ratio of the peak area of the silylated products over the peak area of silylated MC as an internal standard. For accurate quantification, the relative response factor (RRF) of each aromatic compound was either determined using pure standard under the same conditions or estimated according to the model proposed by de Saint Laumer et al.40 More information about the determination and estimation of RRFs can be found in Section 2 in ESI.† Substrate conversion, product yield, and product selectivity were calculated using eqn (1)–(3):| | |
Conversion (%) = (nsubstrate,0 − nsubstrate)/nsubstrate,0 × 100%
| (1) |
| | |
Yield (%) = nproduct/nsubstrate,0 × 100%
| (2) |
| | |
Selectivity (%) = Yield/Conversion × 100%
| (3) |
where, nsubstrate,0 is the quantity of starting substrate, mol; nsubstrate is the quantity of residual (unreacted) substrate, mol; and nproduct is the quantity of product, mol.
Results and discussion
Demethylation of PG (G-type aromatic) in ACLB
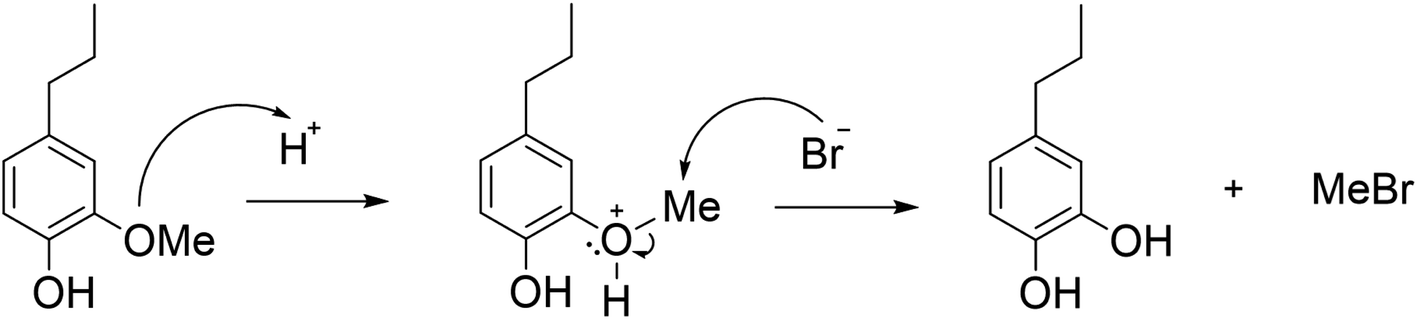
The effect of reaction conditions on the demethylation of PG in ACLB was first investigated. As shown in Scheme 1, the protonation of the ether oxygen, which results in a better leaving group (oxonium ion), is the prerequisite for the acid-catalyzed ether cleavage in ACLB. Br− then attacks the α-carbon (methyl carbon) to complete the cleavage of the ether bond following a typical SN2 mechanism.
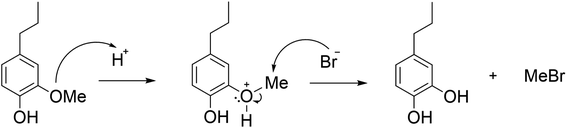 |
| | Scheme 1 Reaction mechanism for the demethylation of PG. | |
It was found that acid (as catalyst) concentration was a crucial parameter in PG demethylation. As shown in Fig. 1A, demethylation did not occur in 61.7% LiBr without HCl. When the HCl concentration was increased to 0.25 M, demethylation was observed, but the yield of PC was only 5%. The demethylation was further improved with increasing HCl concentration. PG was completely demethylated at 1.5 M HCl. Our previous studies demonstrated that the depolymerization and demethylation of lignin were efficient only at higher LiBr concentrations (40–62%).30–33 Similarly, the demethylation of PG increased with LiBr concentrations from 40.8% to 61.7% (Fig. 1B). At 61.7% LiBr, PG was completely converted in 2 h, yielding 96% PC. However, further increasing LiBr concentration to 65.9% showed no further positive effect on demethylation. ACLB solutions at a higher (>62%) concentration of LiBr are difficult to prepare due to the limited solubility of LiBr. These observations are consistent with the SN2 reaction mechanism and the unique properties of the LiBr trihydrate system. Theoretically, LiBr forms a molten salt hydrate system at 61.7% concentration (LiBr·3H2O). Li+ has an octahedral geometry (six coordination orbits), and thus each Li+ ion can coordinate with three water molecules (two Li+ ions share six water molecules), leaving Br−, which acts as a good nucleophile, naked and free in the solution at a high concentration.41 In addition, as Li+ has a great affinity to water molecules (oxygen), the H+ is more active (loosely hydrated) in the LiBr solution than in pure water, which increases the acidity of the acid.42 A relatively low concentration of acid (e.g., 1.5 M HCl) is therefore sufficient to protonate the ether oxygen, compared to traditional HI (57%, 7.6 M) and HBr (47–49%, 9.0 M) systems.43
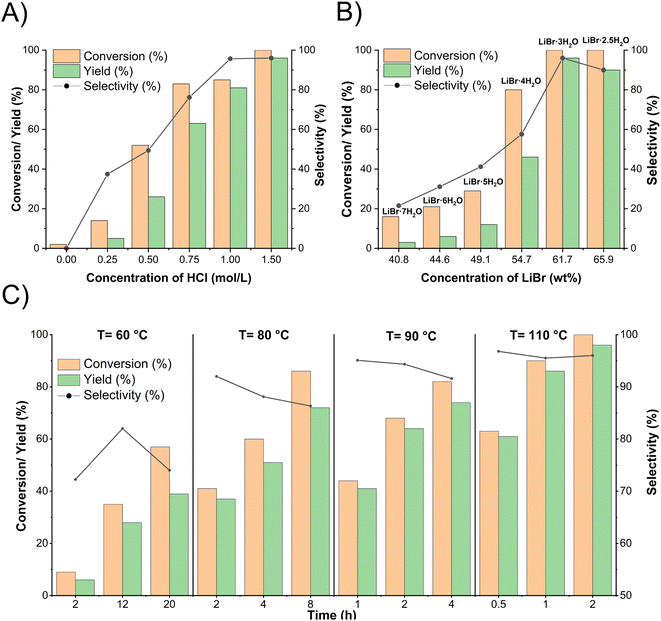 |
| | Fig. 1 Effect of reaction conditions (A) concentration of HCl, (B) concentration of LiBr, and (C) temperature and time on demethylation of PG to PC in ACLB. Note: reaction conditions: (A) 0.1 mmol PG, 2.5 mL ACLB with 61.7% LiBr, 110 °C, and 2 h; (B) 0.1 mmol PG, 2.5 mL ACLB with 1.5 M HCl, 110 °C, and 2 h; (C) 0.1 mmol PG, 2.5 mL ACLB with 61.7% LiBr and 1.5 M HCl. | |
The reaction temperature and time also play important roles in the demethylation of PG. As shown in Fig. 1C, only 39% PC was obtained after a 20 h treatment at 60 °C, whereas the yield of PC increased to 72% at 80 °C in 8 h. The demethylation of PG increased with time at the same temperature. For example, the PC yield jumped from 61 to 96% when the reaction time was extended from 0.5 to 2 h with other conditions fixed (61.7% LiBr, 1.50 M HCl, and 110 °C). However, a lower selectivity was observed at higher reaction times due to the side reaction of electrophilic aromatic bromination. A trace amount of brominated PC isomers was detected as side-products by GC-MS analysis (Fig. S2 in ESI†), indicating the existence of bromine in the system because Br− cannot brominate benzene rings; electrophilic aromatic substitution requires Br+; the methoxy and hydroxy groups in PG increase the electron density of the conjugated aromatic, facilitating electrophilic aromatic substitution (bromination). Bromine was most likely formed from the oxidation of Br− in the presence of air and acid (4H+ + 2Br− + O2 → Br2 + 2H2O), as was verified experimentally (Fig. S3 in ESI†).
Demethylation of PS (S-type aromatic) in ACLB
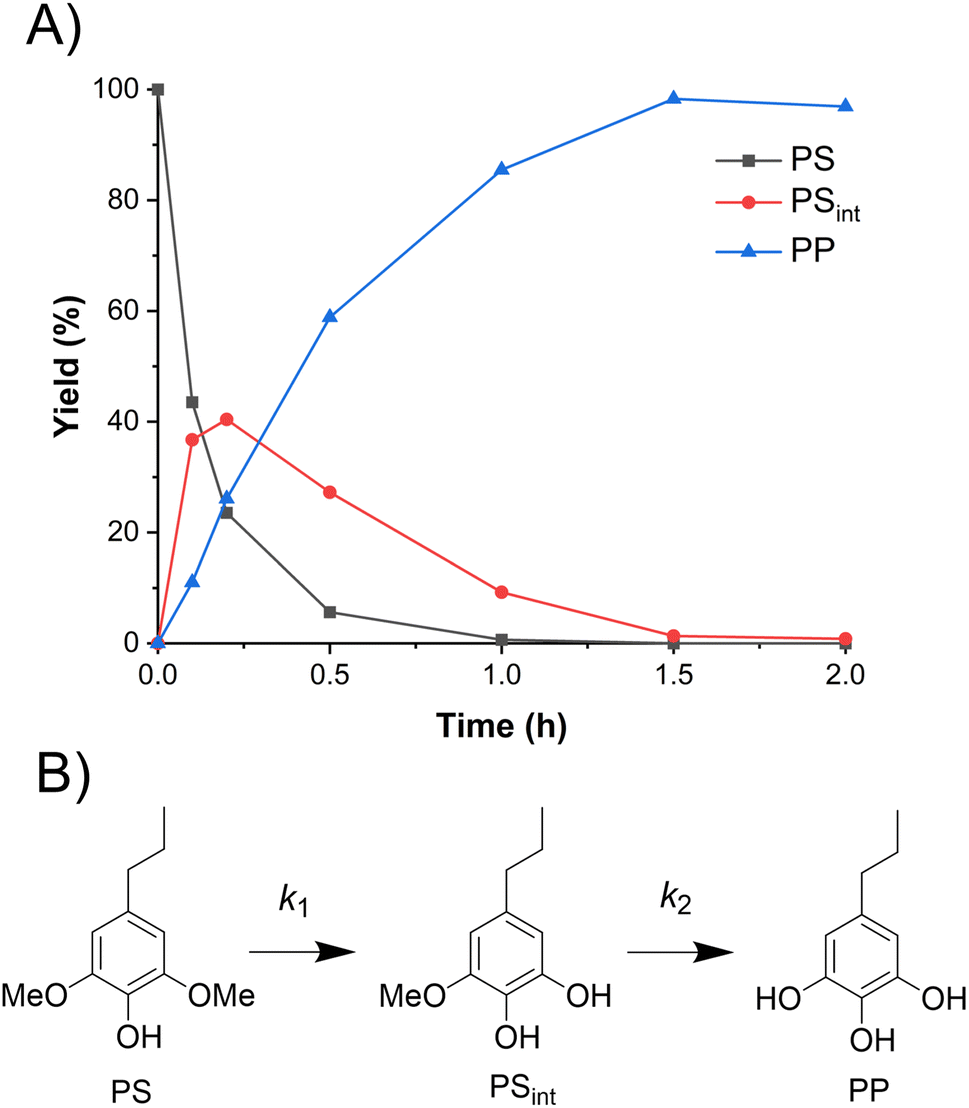
To compare the demethylation of S-type lignin compounds with that of the G-type above, PS was demethylated in ACLB. As shown in Fig. 2A, PS was completely demethylated to 5-propylpyrogallol (PP) with a yield of 98% in 2 h. PS undergoes demethylation via an intermediate (PSint), which is the product after one of the two methoxy groups of PS is demethylated (Fig. 2B). The yield of PSint increased quickly with the demethylation of the first methoxy group and then gradually decreased with the demethylation of the second methoxy group.
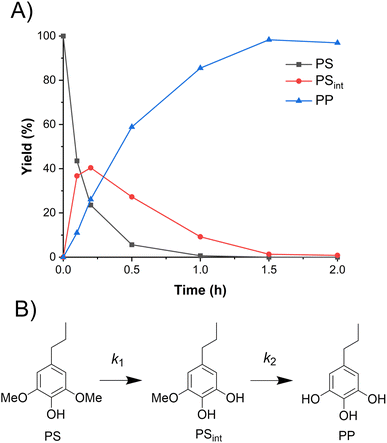 |
| | Fig. 2 Demethylation of PS in ACLB. (A) Reactant and product distribution of PS demethylation with time; (B) reaction pathways. Note: reaction conditions: 0.1 mmol substrate in 2.5 mL ACLB with 61.7% LiBr and 1.5 M HCl, and 110 °C. | |
It was observed that the demethylation of the same quantity of PS was faster than PG under the same conditions. To quantitatively compare their demethylation rates, their rate constants were calculated (Section 3 in ESI, Fig. S4 and S5†). As Br− was present in the reaction system in large excess, the demethylation reaction can be considered to be a pseudo-first-order reaction. As summarized in Table 1, the k1 of PS was more than twice that of PG at both 90 and 110 °C. The increased reaction rate of PS is mainly attributed to its two methoxy groups because both can react with Br− for the demethylation; the additional methoxy group also increases the electron density on the benzene ring, which eases the protonation of the methoxy oxygen for the following SN2 substitution.
Table 1 Rate constants for demethylation of PG and PS at different temperaturesa
| Substrate |
Temperature (°C) |
k1 (M−1 min−1) |
k2 (M−1 min−1) |
| Reaction conditions: 0.1 mmol substrate, 2.5 mL ACLB with 61.7% LiBr, and 1.5 M HCl. |
| PG |
110 |
0.041 |
— |
| PS |
110 |
0.087 |
0.068 |
| PG |
90 |
0.010 |
— |
| PS |
90 |
0.024 |
0.015 |
Demethylation of lignin-derived aromatics with sidechain functional groups (hydroxyl and carbonyl)
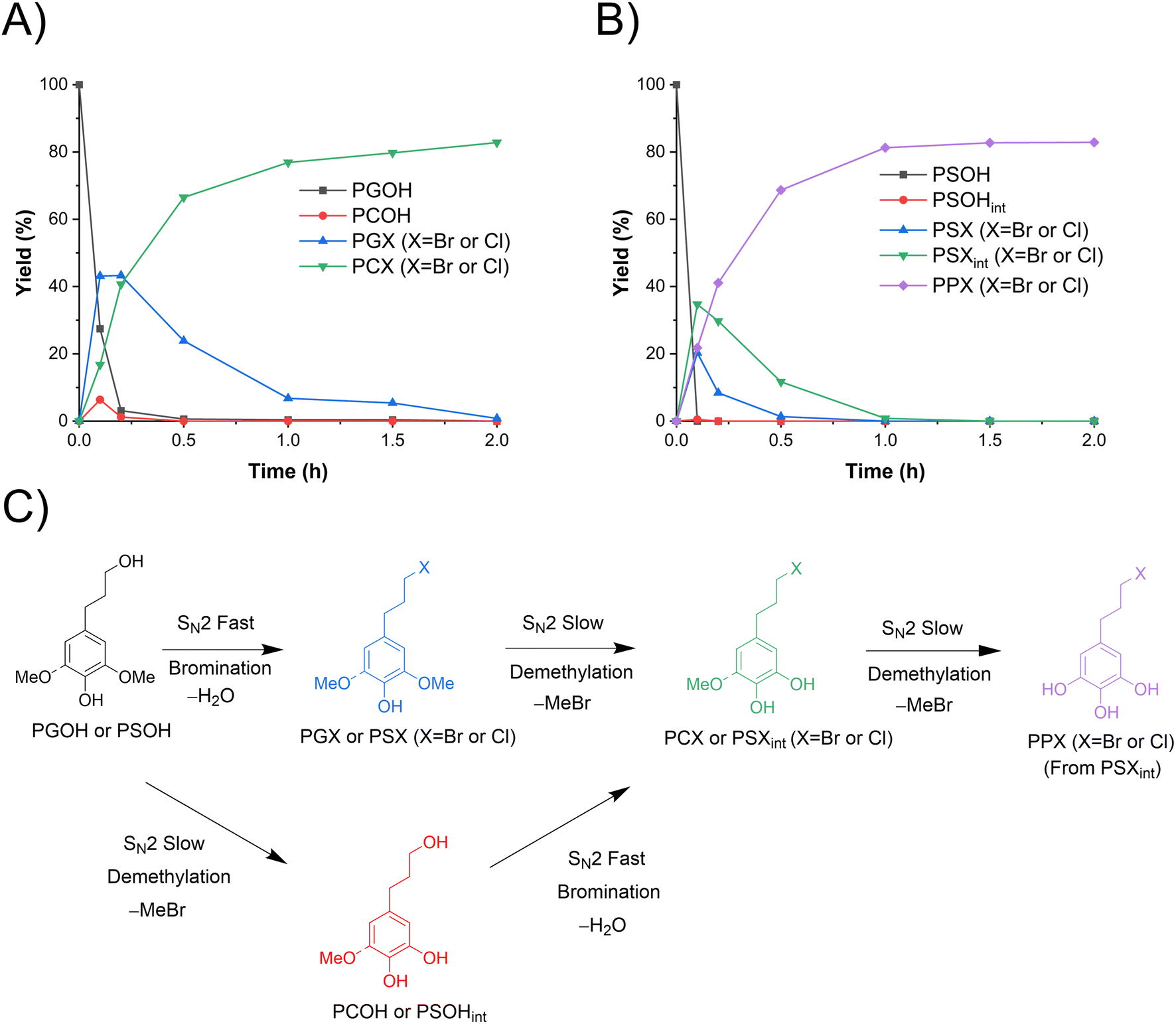
PGOH and PSOH, which have a propanol sidechain, are the products obtained from lignin hydrogenolysis, particularly with Pd as the catalyst.34–36 Their demethylation in ACLB was investigated. It was found that, in addition to demethylation, the aliphatic hydroxyl group was substituted by halogens. Both brominated and chlorinated products were identified, i.e., 4-(3-bromopropyl)-catechol (77%) and 4-(3-chloropropyl)-catechol (6%) from PGOH, and 5-(3-bromopropyl)-pyrogallol (75%) and 5-(3-chloropropyl)-pyrogallol (8%) from PSOH. It is apparent that the chlorinated products can be attributed to the HCl used as a catalyst; if HBr was used as the catalyst, no chlorinated products were detected. Due to the much lower concentration and lower nucleophilicity of Cl− than Br−, the brominated products dominated in both cases of PGOH and PSOH. The bromination (chlorination) of the aliphatic alcohol and the demethylation in ACLB follow analogous SN2 mechanisms (Fig. 3C), but it appears that the former is much easier than the latter. As shown in Fig. 3A, the primary hydroxy group of PGOH was brominated (chlorinated) quickly in 0.2 h at 110 °C. However, the demethylation was completed by only about 18% at the same time, and the complete demethylation of PGOH needed 2 h. These results and observations could be attributed to the more facile protonation of the aliphatic primary alcohol vs. the methoxyl.44 In addition, water in the former case is a better leaving group than phenol in the latter case. Similar results were observed in the demethylation of PSOH in ACLB (Fig. 3B), but the reactions and products of PSOH were more complicated than those of PGOH as multiple steps are required to deliver the final product (PPX).
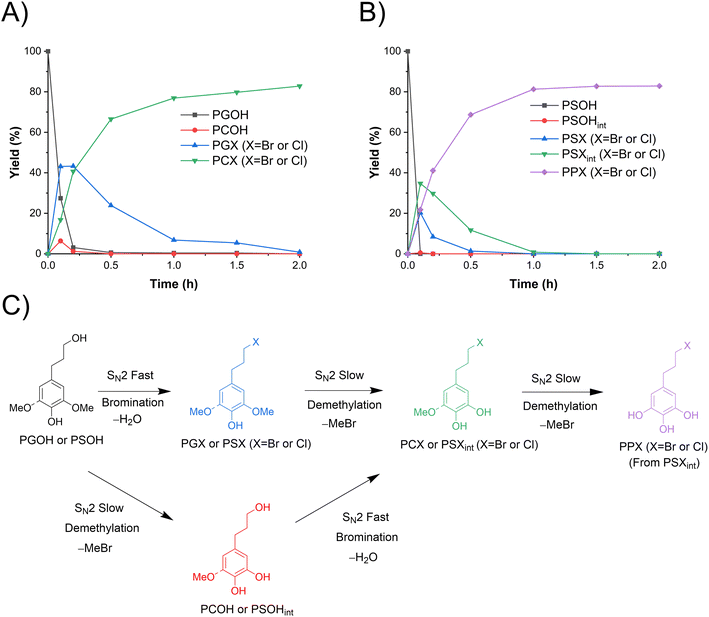 |
| | Fig. 3 Demethylation of PGOH and PSOH in ACLB. (A) Reactant and product distribution of PGOH demethylation with time; (B) reactant and product distribution of PSOH demethylation with time; and (C) reaction pathways. Note: reaction conditions: 0.1 mmol substrate, 2.5 mL ACLB with 61.7% LiBr and 1.5 M HCl, and 110 °C. | |
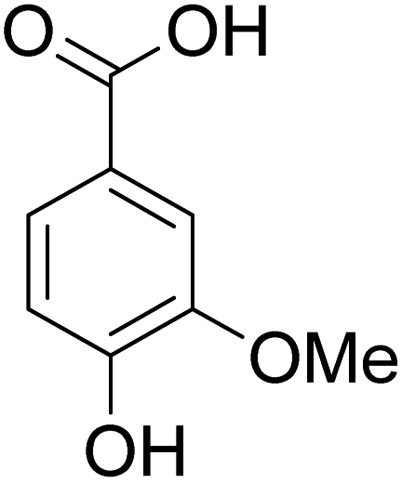
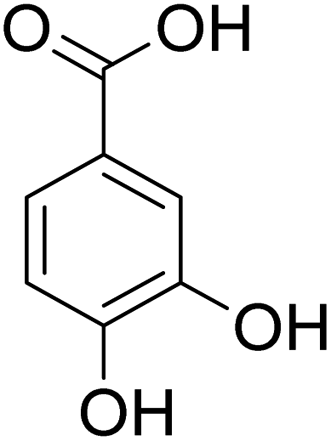
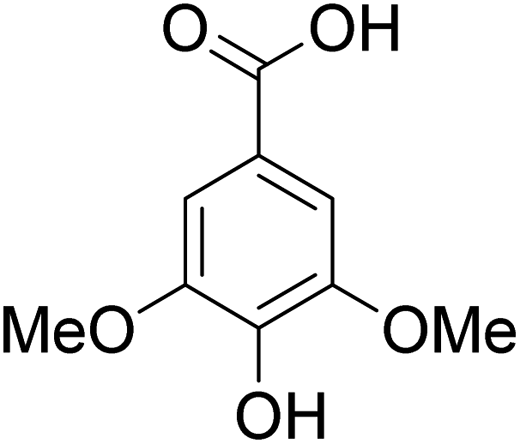
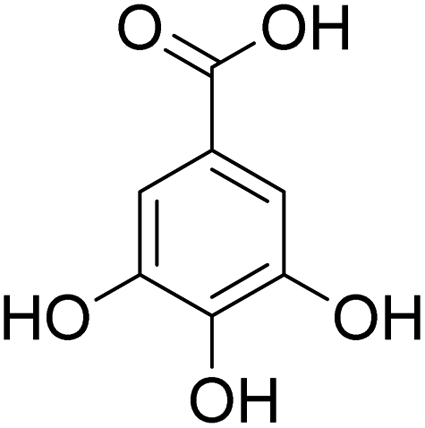
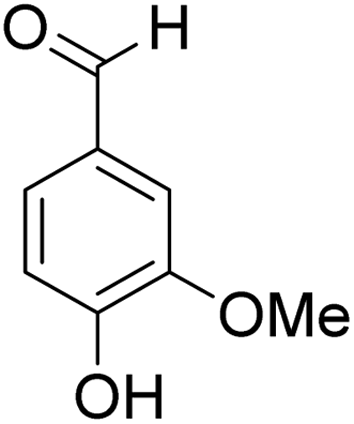
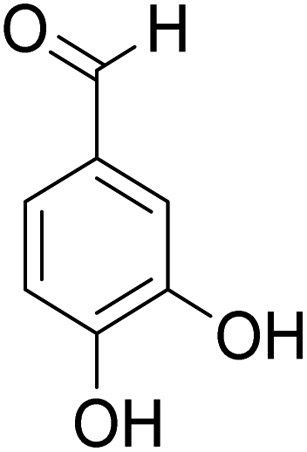
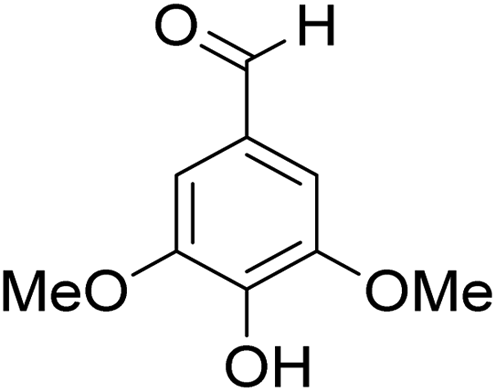
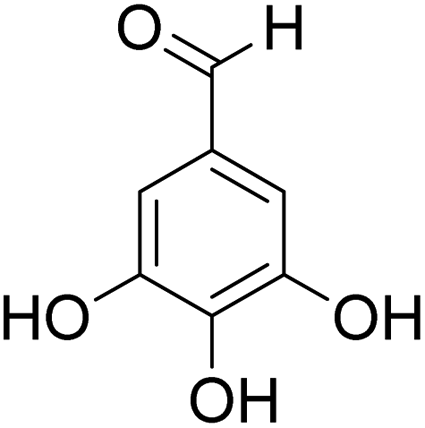
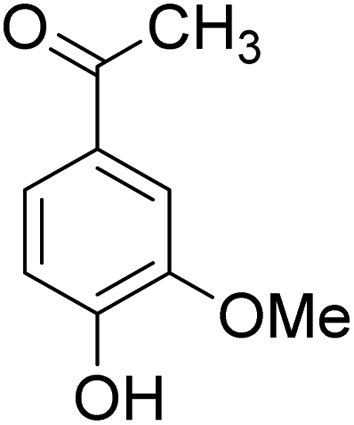
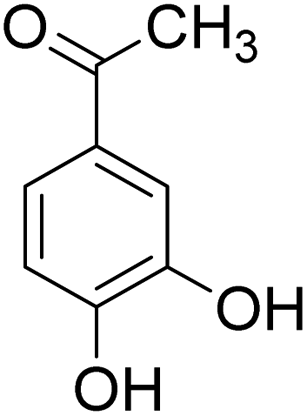
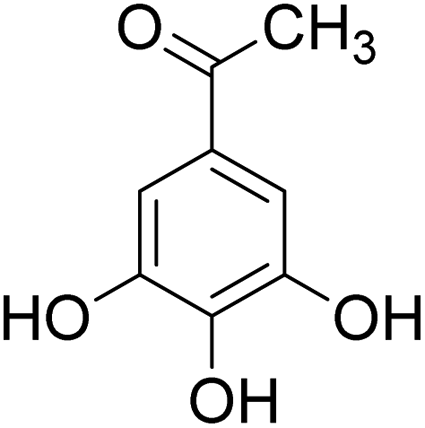
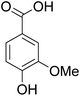
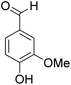
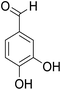
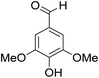
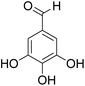
The behaviors of other functional groups (carboxyl, carbonyl, and aldehyde) in the aromatic compounds from oxidative depolymerization of lignin were investigated in ACLB. As summarized in Table 2, both vanillic acid and syringic acid (entries 1 and 2) were effectively demethylated with a high yield. However, vanillin, syringaldehyde, acetovanillone, and 3,5-dimethoxy-4-hydroxyacetophenone (entries 3–6) were completely converted but resulted in much lower yields of their demethylated products than the corresponding carboxylic acids. Black or purple solids were observed, and extending the reaction led to more solids and lower demethylation product yields because of condensation reactions on the compounds (both reactants and products). The same solid formation phenomenon was observed when heating 3,4-dihydroxybenzaldehyde and 3,4-dihydroxyacetophenone (the demethylation products of vanillin and acetovanillone) in ACLB. These observations suggested that the ketones and aldehydes and/or their demethylation products were unstable in ACLB and underwent condensation initiated by the carbonyl group. The carbonyl group can be protonated under the acidic conditions in ACLB, and the positively charged carbonyl carbon is vulnerable to attack by electron-rich aromatics (the nucleophile), resulting in the formation of new C–C bonds, i.e., condensation.45 With two methoxy groups, S-type compounds (syringaldehyde and 3,5-dimethoxy-4-hydroxyacetophenone) are more electron-rich than G-type compounds (vanillin and acetovanillone) and thus led to more condensation. Acetovanillone and 3,5-dimethoxy-4-hydroxyacetophenone could also undergo aldol condensation.
Table 2 Demethylation of compounds derived from oxidative depolymerization of lignina
| Entry |
Substrate |
Product |
Conversion (%) |
Yield (%) |
| Conditions: 0.1 mmol substrate, 2.5 mL, 61.7% LiBr with 1.5 M HCl, 110 °C, and 1 h. Time, 2 h. |
| 1 |
 |
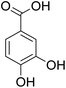 |
96 |
92 |
| 100b |
87b |
| 2 |
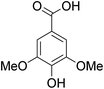 |
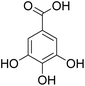 |
100 |
97 |
| 3 |
 |
 |
99 |
87 |
| 100b |
54b |
| 4 |
 |
 |
100 |
36 |
| 5 |
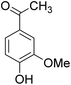 |
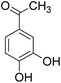 |
98 |
50 |
| 100b |
31b |
| 6 |
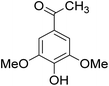 |
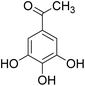 |
100 |
32 |
Effect of ring substituents on demethylation of aromatics
It was found in our previous study that the demethylation of creosol was much faster than that of anisole in ACLB.32 In this work, it was observed that S-type lignin compounds were more reactive than G-type ones. These observations imply that the electronic effects on the aromatic ring likely affects the protonation of the methoxy oxygen and thus the demethylation rate. To understand the effect of substituents on the demethylation of aromatics, anisole and its derivatives with different electron-donating and electron-withdrawing substituents were treated in ACLB.
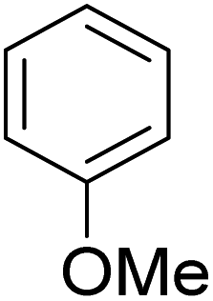
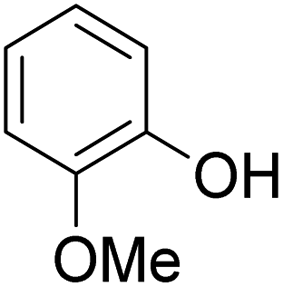
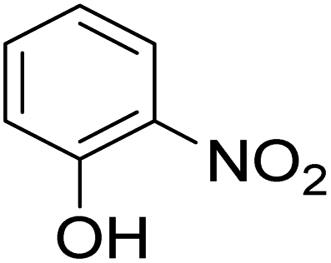
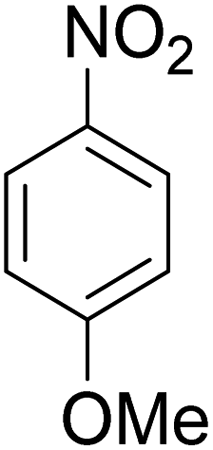
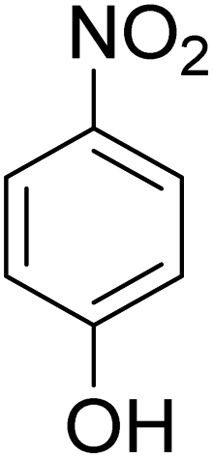
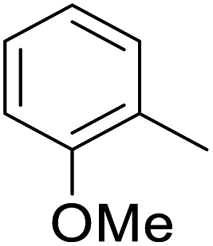
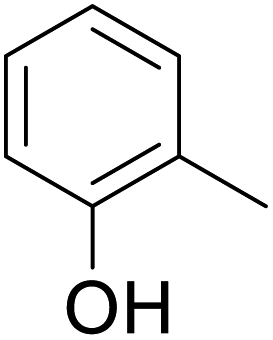
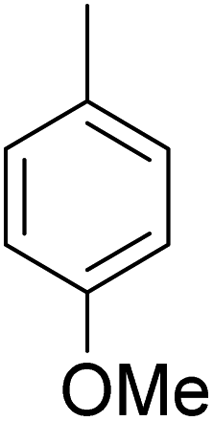
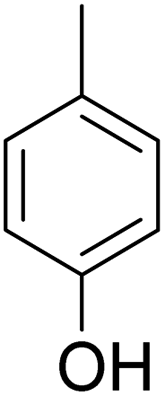
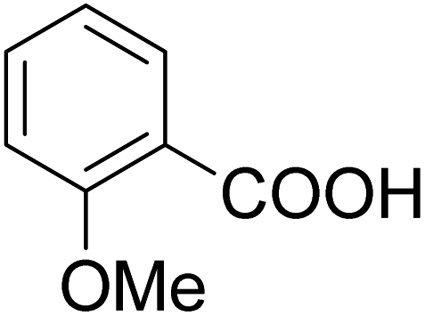
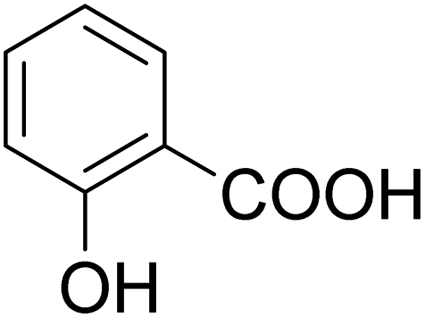
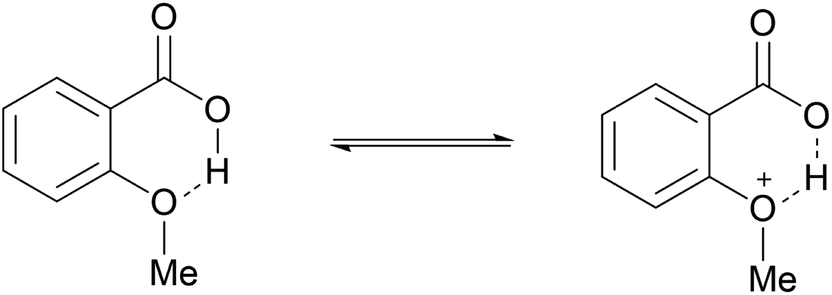
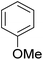
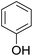
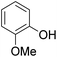
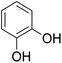
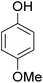
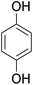
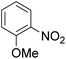
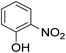
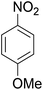
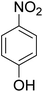
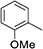
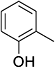
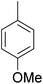
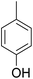
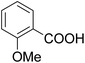
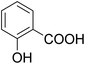
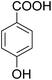
As shown in Table 3, the demethylation of guaiacol and 4-methoxyphenol (entries 2 and 3) was much faster than that of anisole (entry 1), and they were completely converted within 1 h with a high yield of their demethylation products. On the other hand, the demethylation of 2-nitroanisole and 4-nitroanisole was slower than anisole (entries 4 and 5). However, although with an electron-donating group (methyl), 2-methoxytoluene and 4-methoxytoluene cannot be demethylated effectively in ACLB (entries 6 and 7), which was mainly due to their poor solubility in ACLB. The effect of substrate solubility on demethylation efficiency was also supported by the difference between guaiacol and PG. Although having one more mildly electron-donating group (the propyl group) on the para position, the demethylation of PG was slower than that of guaiacol due to its low solubility in ACLB. 2-Methoxybenzoic acid was effectively demethylated to give salicylic acid in ACLB (entry 8) and had a much higher demethylation rate than 4-methoxybenzoic acid (entry 9) and anisole. The enhanced demethylation is most likely attributable to the intramolecular hydrogen bonding structure of 2-methoxybenzoic acid.46 As shown in Scheme 2, 2-methoxybenzoic acid can undergo an intramolecular protonation of its methoxy oxygen via a stable six-membered ring intermediate to accelerate the demethylation reaction; 4-methoxybenzoic acid obviously cannot undergo such intramolecular protonation.
Table 3 Effect of substituents on the demethylation of aromatic compoundsa
| Entry |
Substrate |
Product |
Conversion (%) |
Yield (%) |
| Conditions: 1 mmol substrate, 2.5 mL 61.7% LiBr with 1.5 M HCl, 110 °C, and 1 h. |
| 1 |
 |
 |
35 |
17 |
| 2 |
 |
 |
100 |
94 |
| 3 |
 |
 |
100 |
94 |
| 4 |
 |
 |
24 |
11 |
| 5 |
 |
 |
8 |
5 |
| 6 |
 |
 |
21 |
3 |
| 7 |
 |
 |
27 |
6 |
| 8 |
 |
 |
99 |
92 |
| 9 |
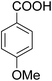 |
 |
33 |
25 |
 |
| | Scheme 2 Intramolecular protonation of methoxy oxygen in 2-methoxybenzoic acid. | |
Conclusions
This study has demonstrated that acidic concentrated lithium bromide (ACLB) can demethylate different lignin-derived aromatic compounds under moderate conditions (e.g., 1.5 M HCl, 110 °C, and 2 h). The demethylation was completed by the protonation of ether oxygen followed by the SN2 substitution with bromide. The versatility of the method was tested using different lignin-derived compounds with one or two methoxy groups, alkyl hydroxy, and carbonyl functional groups, and anisole derivatives with different substituents. It was found that compounds with two methoxy groups demethylated faster in ACLB. The sidechain functional groups led to side reactions (halogenation of alkyl alcohol and condensation due to the carbonyl groups) that affect the demethylation efficiency and selectivity. The substituents on the aromatic rings not only have electronic effects but also affect the solubility in ACLB, both of which impact the demethylation rate. Because of the moderate reaction conditions, the unique properties of the LiBr system (high thermal stability, low vapor pressure, low viscosity), its environment friendly nature, and the ease of separation of the products from the system by extraction, the ACLB method provides an efficient approach to upgrading lignin-derived aromatics. Phenolics produced by such demethylation allow enhanced downstream application opportunities, such as for lignin-based polymeric materials synthesis and platform chemicals production.
Author contributions
YW, JR, and XP conceived the idea and designed the research. YW and MC performed the research. YW, MC, YY, JR, and X.P analyzed the data. YW and XP wrote the original manuscript. YW, MC, YY, JR, and XP reviewed the manuscript and suggested improvements.
Conflicts of interest
There are no conflicts of interest to declare.
Note added after first publication
This article replaces the version published on 17 February 2023, which featured an incorrect product structure for entry 6 of Table 3 .
Acknowledgements
This project was supported by a USDA NIFA grant (2018-67009-27902) for XP and by the Swiss National Science Foundation (Sinergia) grant (CRS115_180258) for MC, WY, and JR. JR and the GLBRC NMR facility were funded by the DOE Great Lakes Bioenergy Research Center (DOE Office of Science, DE-SC0018409).
References
- B. Kumar and P. Verma, Fuel, 2021, 288, 119622 CrossRef CAS.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- C. Cai, Z. Xu, H. Zhou, S. Chen and M. Jin, Sci. Adv., 2021, 7, eabg4585 CrossRef CAS PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- M. Zaheer and R. Kempe, ACS Catal., 2015, 5, 1675–1684 CrossRef CAS.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- G. Lyu, C. G. Yoo and X. Pan, Biomass Bioenergy, 2018, 108, 7–14 CrossRef CAS.
- J. Bomon, M. Bal, T. K. Achar, S. Sergeyev, X. Wu, B. Wambacq, F. Lemière, B. F. Sels and B. U. W. Maes, Green Chem., 2021, 23, 1995–2009 RSC.
- M. Jablonksy, M. Botkova and J. Adamovska, Cellul. Chem. Technol., 2013, 49, 165–168 Search PubMed.
- H. Wang, T. L. Eberhardt, C. Wang, S. Gao and H. Pan, Polymers, 2019, 11, 1771 CrossRef CAS PubMed.
- K. Hwang and S. Park, Synth. Commun., 1993, 23, 2845–2849 CrossRef CAS.
- S. B. Waghmode, G. Mahale, V. P. Patil, K. Renalson and D. Singh, Synth. Commun., 2013, 43, 3272–3280 CrossRef CAS.
- C. R. Schmid, C. A. Beck, J. S. Cronin and M. A. Staszak, Org. Process Res. Dev., 2004, 8, 670–673 CrossRef CAS.
- D. Sang, X. Tu, J. Tian, Z. He and M. Yao, ChemistrySelect, 2018, 3, 10103–10107 CrossRef CAS.
- R. G. Lange, J. Org. Chem., 1962, 27, 2037–2039 CrossRef CAS.
- X. Li, J. He and Y. Zhang, J. Org. Chem., 2018, 83, 11019–11027 CrossRef CAS PubMed.
- Q. Mei, H. Liu, X. Shen, Q. Meng, H. Liu, J. Xiang and B. Han, Angew. Chem., Int. Ed. Engl., 2017, 56, 14868–14872 CrossRef CAS PubMed.
- Q. Mei, X. Shen, H. Liu, H. Liu, J. Xiang and B. Han, Chem. Sci., 2019, 10, 1082–1088 RSC.
- J. Zhang, Q. Qian, Y. Wang, B. B. Asare Bediako, J. Yan and B. Han, Chem. Sci., 2019, 10, 10640–10646 RSC.
- K. Lee and K. Kim, Bull. Korean Chem. Soc., 2010, 31, 3842–3843 CrossRef CAS.
- W. E. S. Hart, L. Aldous and J. B. Harper, Org. Biomol. Chem., 2017, 15, 5556–5563 RSC.
- W. Zhao, C. Wei, Y. Cui, J. Ye, B. He, X. Liu and J. Sun, Chem. Eng. J., 2022, 443, 136486 CrossRef CAS.
- K. Ghandi, Green Sustainable Chem., 2014, 4, 44–53 CrossRef CAS.
- L. Dong, Y. Xin, X. Liu, Y. Guo, C.-W. Pao, J.-L. Chen and Y. Wang, Green Chem., 2019, 21, 3081–3090 RSC.
- X. Wu, Y. Liao, J. Bomon, G. Tian, S. T. Bai, K. Van Aelst, Q. Zhang, W. Vermandel, B. Wambacq, B. U. W. Maes, J. Yu and B. F. Sels, ChemSusChem, 2022, 15, e202102248 CAS.
- X. Yang, N. Li, X. Lin, X. Pan and Y. Zhou, J. Agric. Food Chem., 2016, 64, 8379–8387 CrossRef CAS PubMed.
- N. Li, Y. Li, C. G. Yoo, X. Yang, X. Lin, J. Ralph and X. Pan, Green Chem., 2018, 20, 4224–4235 RSC.
- Z. Li, E. Sutandar, T. Goihl, X. Zhang and X. Pan, Green Chem., 2020, 22, 7989–8001 RSC.
- X. Yang, Z. Li, L. Li, N. Li, F. Jing, L. Hu, Q. Shang, X. Zhang, Y. Zhou and X. Pan, J. Agric. Food Chem., 2021, 69, 13568–13577 CrossRef CAS PubMed.
- J. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, ACS Catal., 2014, 4, 1574–1583 CrossRef CAS.
- C. Zhang and F. Wang, Acc. Chem. Res., 2020, 53, 470–484 CrossRef CAS PubMed.
- S. Van den Bosch, W. Schutyser, S. F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC.
- S. Zhao and M. M. Abu-Omar, Biomacromolecules, 2015, 16, 2025–2031 CrossRef CAS PubMed.
- E. Blondiaux, J. Bomon, M. Smoleń, N. Kaval, F. Lemière, S. Sergeyev, L. Diels, B. Sels and B. U. W. Maes, ACS Sustainable Chem. Eng., 2019, 7, 6906–6916 CrossRef CAS.
- Z. Dobi, B. N. Reddy, E. Renders, L. Van Raemdonck, C. Mensch, G. De Smet, C. Chen, C. Bheeter, S. Sergeyev, W. A. Herrebout and B. U. W. Maes, ChemSusChem, 2019, 12, 3103–3114 CrossRef CAS PubMed.
- J. Y. de Saint Laumer, S. Leocata, E. Tissot, L. Baroux, D. M. Kampf, P. Merle, A. Boschung, M. Seyfried and A. Chaintreau, J. Sep. Sci., 2015, 38, 3209–3217 CrossRef CAS PubMed.
- N. Li, X. Pan and J. Alexander, Green Chem., 2016, 18, 5367–5376 RSC.
- S. K. Franzyshen, Master thesis, The College of William and Mary in Virginia, 1985 DOI:10.21220/s2-cvb1-hh82.
- M. V. Bhatt and S. U. Kulkarni, Synthesis, 1983, 4, 249–282 CrossRef.
- M. Node, K. Ohta, T. Kajimoto, K. Nishide, E. Fujita and K. Fuji, Chem. Pharm. Bull., 1983, 31, 4178–4180 CrossRef CAS.
- R. Retnosari, I. B. Rachman, S. Sutrisno, M. E. F. Sari, D. Sukarianingsih and Y. Rukayadi, presented in part at the 4th International Seminar on Chemistry, 2021 Search PubMed.
- D. H. Buchanan, N. Takemura and J. N. O. Sy, J. Org. Chem., 1986, 51, 4291–4294 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00245d |
| ‡ Present address: Institute of Microbiology, Guangdong Academy of Sciences, 100 Xianlie Central Rd, Guangzhou, 510070, China. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Mingjie Chen‡
a,
Mingjie Chen‡