
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDistinct morphology-dependent behaviors for Au/γ-Al2O3 catalysts: enhanced thermal stabilization in CO oxidation reaction
Weimeng Cai†
,
Xinyu Sun†,
Yaojie Bao,
Jianhua Guo,
Anqiu Liu,
Kunhong Hu* and
Lipin Feng *
*
School of Energy Materials and Chemical Engineering, Hefei University, Hefei, 99 Jinxiu Rd, Hefei 230601, Anhui, People's Republic of China. E-mail: chemhu@hfuu.edu.cn; fenglipin2022@163.com; Tel: +86-551-62158315
First published on 20th March 2023
Abstract
The durability of supported metal catalysts usually suffers from sintering, the metal nanoparticles aggregating into larger sizes and subsequent loss of reactive surface, resulting in catalysts deactivation when heated at elevated temperatures. Herein, we investigate the evolution of Au species on different morphologies of γ-Al2O3 and surprisingly found vastly different behavior for the dispersion of surface Au nanoparticles. A nanorod-shaped γ-Al2O3 is prepared by the hydrothermal method resulting in an extraordinary catalyst support that can stabilize Au nanoparticles at annealing temperatures up to 700 °C. In contrast, the Au-supported catalyst prepared using commercial γ-Al2O3 shows a greater degree of inactivation under the same conditions. Remarkably, the unique morphology of such nanorod-shaped γ-Al2O3 is beneficial in preventing Au nanoparticles from sintering. The γ-Al2O3 nanorods are more effective than the commercial γ-Al2O3 at anchoring the Au nanoparticles. The results of X-ray photoelectron spectroscopy (XPS), in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and H2-TPR, reveal the interfacial interactions between Au nanoparticles and γ-Al2O3 nanorods, yielding a sinter-stability of the obtained Au/γ-Al2O3 nanorods catalyst. This synthetic strategy is simple and amenable to the large-scale manufacture of thermally stable γ-Al2O3 for industrial applications. Here, we investigate the morphology-dependent behavior of Au nanoparticles dispersed on different morphologies of γ-Al2O3. The result of X-ray photoelectron spectroscopy (XPS), in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and H2-TPR, reveal the interfacial interactions between Au nanoparticles and gamma alumina nanorods. Au nanoparticles on γ-Al2O3 nanorods exhibit higher sinter-resistant performance than those on commercial γ-Al2O3.
1 Introduction
Currently, supported metal catalysts perform a vital role in both the petrochemical and environmental field, such as catalytic reforming,1 catalytic hydrogenation,2 cleaning of automobile exhausts,3 and activation of nitrogen and methane.4 Of these, carbon monoxide is one of the most widespread and deadly pollutants in industrial and vehicle exhaust emissions.5 CO oxidation is of great importance in both basic research and practical applications. Oxide-loaded noble metal catalysts are also recognized as excellent CO oxidation catalysts.6 From there, such catalysts have been extensively applied in industrial or pollutant treatment processes.7–10 In most cases, due to the comparatively low Tammann temperature (Kelvin degrees, half of the bulk melting point) of the metal,11 the metal on the surface of the supported metal catalyst has liquid-like characteristics, exhibits a strong migration ability, and is easy to aggregate and sintering at high temperatures, thereby altering the catalytic performance.12–14 The regeneration of such catalysts requires a lot of material and financial resources, especially for the supported noble metal catalysts. Therefore, understanding the sintering process of metal and the preparation of durable supported metal catalysts has significant research and application value.In recent years, much effort has been devoted to developing new strategies for stabilizing metal nanoparticles. Recent developments in the preparation of sinter-resistant supported metal nanoparticle catalysts can be roughly divided into the following two categories: (i) strong metal-support interactions (SMSI). The intrinsic reason for sintering is ascribed to the weak interaction between metal particles and supports;15 hence, the SMSI effect can improve the anti-sintering performance of the catalyst. After the SMSI formation, the geometric and electronic effects of metal nanoparticles can be modified, which involves encapsulating the metal nanoparticles with oxide overlayers, electronic interaction between metal nanoparticles and oxide, etc. TiO2 (ref. 16–18) and CeO2 (ref. 19–21) are two of the most extensively investigated supports for the SMSI, due to their redox behavior and oxygen storage capacity.22–24 Notably, many industrially supported metal catalysts are on inert supports such as porous carbon or alumina. The SMSI strategy seems to be ineffective for these catalysts. Therefore, strategies to construct physical barriers to stabilize metal nanoparticles are still in great demand. (ii) Encapsulation with porous material such as oxide;12,25–27 carbon layers28–30 and zeolite crystals.31,32 The catalyst prepared by Arnal et al. encapsulating Au in ZrO2 hollow spheres can achieve 100% CO conversion at 260–280 °C even after a high-temperature treatment of 900 °C.27 Corma et al. reported that the nano-scale Pt species is confined in the pores of the MCM-22 molecular sieve, which can withstand heat treatment up to 540 °C in an air atmosphere. It has better stability performance in the propane dehydrogenation reaction when compared with the catalyst prepared by the usual impregnation method.31 Compared to the SMSI, encapsulation of metal nanoparticles with porous material indeed enhances the stability of metal nanoparticles due to the construction of physical barriers, which unfortunately reduces the accessible active sites simultaneously. In addition, the preparation process of this type of catalyst is relatively complicated, particularly for the fabrication of nanocage structures, and thus limits its large-scale industrial production. Therefore, it is of considerable interest to develop a simple and effective method to synthesize novel structured support to stabilize metal nanoparticles against sintering.
Gamma alumina is widely used in a variety of industrial processes, including ceramics, petroleum refinement, automobile emission control, etc. The γ-Al2O3 supported catalysts are widely used in the industrial catalyst field due to their essential properties of high surface area, good mechanical strength, and chemical and especially thermal stability.33 However, until now, it has been difficult to find a successful method using γ-Al2O3 to stabilize noble metals including Pt, Pd, and Au, particularly Au nanoparticles, which can anti-sintering at temperatures exceeding 700 °C. Thus, it is still a significant challenge to obtain gamma alumina with the properties mentioned above via a simple and effective strategy.
Au/Al2O3 catalysts show high performance in CO oxidation, catalytic purification of automotive exhausts, elimination of halogenated organic compounds and catalytic combustion of methane.34–37 However, the resistance to sintering and high dispersion of loaded gold nanocatalysts has been a difficult area of research in this field, particularly for the preparation of highly active gold catalysts using inert alumina as a carrier. Wang et al. added Ru as an accelerator to Au/Al2O3 to improve the stability of Au/Al2O3.38 Roberto Camposeco et al. synthesized Au/TiO2–Al2O3 catalysts with a high degree of dispersion, which showed good performance for the oxidation and anti-inactivity of CO.39 Su et al. prepared Au/LaFeO3/Al2O3 catalysts.40 The modification of LaFeO3 allowed the catalyst to increase its catalytic activity and form larger pores, and also prevented the aggregation and deactivation of gold nanoparticles. Therefore, the preparation of a catalyst with high thermal stability becomes very meaningful work.
Herein, we present a simple and reproducible hydrothermal synthesis method using ammonium oleate as the crystal face protector to synthesize nanorod-shaped porous gamma alumina. Subsequently, the Au/γ-Al2O3 catalyst is prepared by a simple equal-volume impregnation method. Then, we investigated the thermal stability of Au species on different morphology of γ-Al2O3 and surprisingly discovered vastly different sintering resistance behaviors in CO oxidation reaction. It was found that the nanorod-shaped Au/γ-Al2O3 catalyst can achieve 100% CO conversion at 300 °C even after high-temperature annealing at 700 °C, showing excellent thermal stability. It has better stability performance when compared with the catalyst prepared by the commercial gamma alumina under the same conditions. The interaction between the Au nanoparticles and the gamma alumina nanorods was confirmed by XPS, H2-TPR, and in situ DRIFTS. This study provides another solution for synthesizing catalyst supports capable of effectively stabilizing the metal nanoparticles on its surface. The main text of the article should appear here with headings as appropriate.
2 Experimental
2.1 Catalysts synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Then, the mixture solution was openly stirred for 1 h at 368 K to evaporate part of the isopropanol. Subsequently, a certain amount of deionized water was added to complement the lost solution, then an appropriate amount of nitric acid (2 mol L−1) was added to adjust the pH of the suspension to about 3–4. Finally, the mixture solutions were continuously stirred with a magnetic stirrer at 368 K under condensed reflux for 36 h in preparation for stable transparent aluminum sol.
:1. Then, the mixture solution was openly stirred for 1 h at 368 K to evaporate part of the isopropanol. Subsequently, a certain amount of deionized water was added to complement the lost solution, then an appropriate amount of nitric acid (2 mol L−1) was added to adjust the pH of the suspension to about 3–4. Finally, the mixture solutions were continuously stirred with a magnetic stirrer at 368 K under condensed reflux for 36 h in preparation for stable transparent aluminum sol.2.2 Catalyst characterization
X-ray diffraction (XRD) patterns were obtained using a Phillips X'Pro diffractometer with Cu Kα (λ = 1.5418 Å) radiation at 40 kV and 40 mA with 2θ values between 5 and 90°. The specific surface area was measured on an ASAP-2010 apparatus at liquid nitrogen temperature. The samples were degassed in vacuo at 573 K for 6 h before measurement. The Barrett–Joyner–Halenda (BJH) method was used to calculate the pore volume and the pore size distribution. The transmission electron microscopy (TEM) images were collected on a JEOL JEM-200CX instrument at an accelerating voltage of 200 kV. The surface elemental analysis was performed using a Thermo escalate 250XI X-ray photoelectron spectroscopy (XPS) equipped with Al Kα radiation. C 1s peak at 284.8 eV was used as a calibration peak.2.3 Evaluation of catalytic performance
In order to investigate the catalytic activity of the catalysts, the CO oxidation reaction was carried out in a laboratory-made fixed bed reactor. The specific steps were as follows: 20 mg of catalyst (20–40 mesh) was added to the reactor, first pretreat with N2 at 573 K for 1 h, and the reaction was started after cooling to room temperature. The mixed gas containing CO (2.0% CO, the rest was Ar) and O2 were introduced into the fixed bed to start the reaction. The reaction temperature was 323–673 K, and the mixed gas flow rate was 33 mL min−1. The reacted product was equipped with a Porapak-Q column gas chromatograph for online analysis.3 Results and discussion
3.1 Synthesis and characterization of Au/γ-Al2O3-r and Au/γ-Al2O3-c
Alumina was a polymorphic compound, γ-Al2O3 was obtained by calcining boehmite (γ-AlOOH) at 500–800 °C, and its structure was a defective spinel structure. From there, controlling the size and shape of the boehmite nanostructure was the key to alumina's performance and potential applications. In this study, the γ-Al2O3 nanorods were prepared by a two-step synthesis method, the schematic diagram of the preparation of γ-Al2O3 nanorods was shown in Scheme 1. The first step was to prepare aluminum sol by hydrolysis of aluminum isopropoxide under acidic conditions (using nitric acid as a pH adjuster). In the second step, the γ-Al2O3 nanorods were prepared by calcining boehmite precursor at 600 °C, while the boehmite precursor was produced through a hydrothermal synthesis using ammonia oleate as a topography inducing reagent.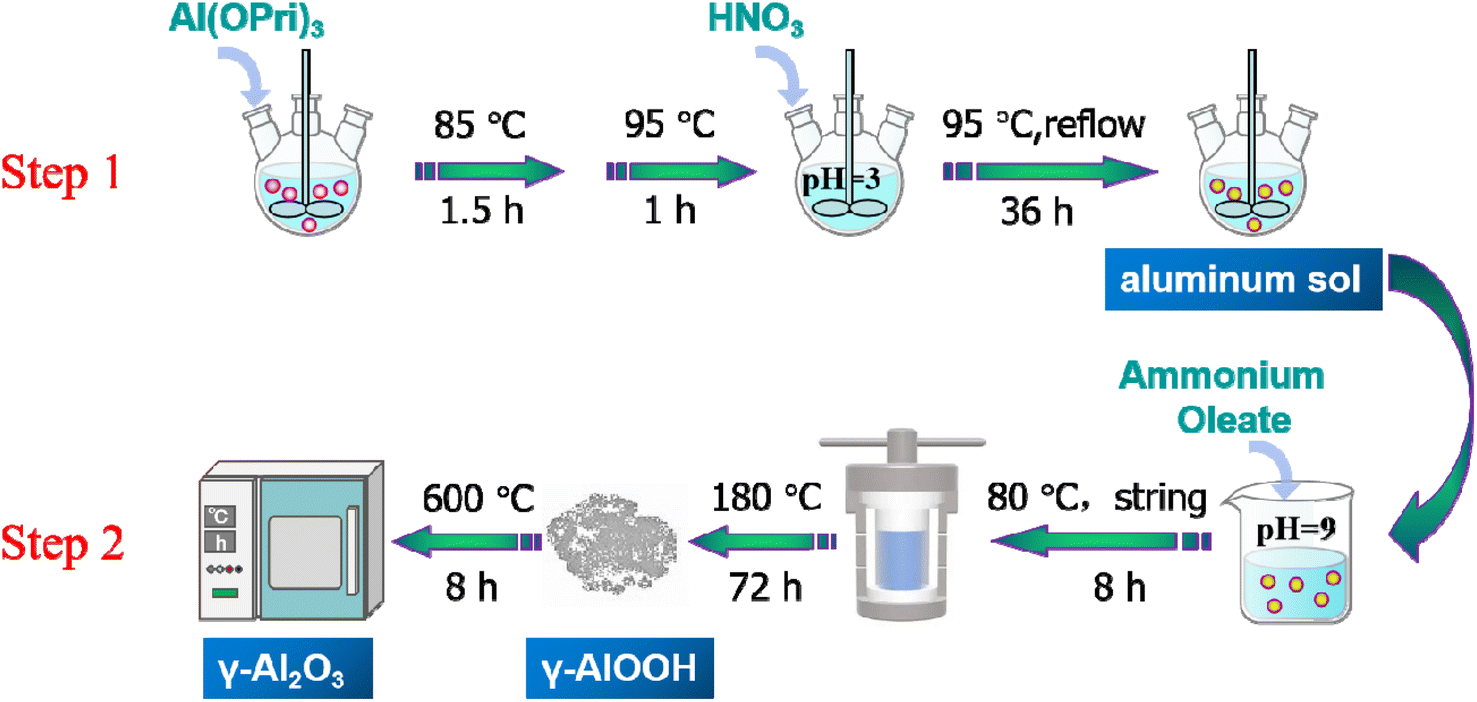 | ||
| Scheme 1 Schematic illustration of the preparation of the gamma-alumina nanorods. | ||
In order to preliminarily explore the formation mechanism of gamma alumina nanorods, we performed XRD characterization of the hydrated products, and the results were shown in Fig. 1(a). It can be seen that the 2θ angles at 14.5°, 28.2°, 38.3°, 48.9°, 55.2°, 64.03° and 71.9° were correspond to the (020), (120), (031), (051), (151), (231) and (251) crystal faces of γ-AlOOH (JCPDS# 21-1307). Boehmite (γ-AlOOH) can be converted to many different forms of alumina through thermal annealing. Therefore, controlling the size and shape of the boehmite precursor was key to improving the properties of the calcined product gamma-alumina. The XRD profiles of the two different alumina and 0.5% Au-loaded catalysts were displayed in Fig. 1(b). It can be seen from the figure that, for all alumina, the 2θ angles at 37.4°, 45.9° and 66.8° correspond to the (311), (400) and (440) crystal faces of γ-Al2O3 (JCPDS#29-0063). It can also be seen from the Fig. 1(b) that the prepared alumina has a narrower half-value width and stronger diffraction peak intensity, which indicates that the self-made alumina has better crystallinity than commercial alumina, and also confirms the regularity of its morphology. The XRD of the catalyst loaded with a mass fraction of 0.5% was also shown in Fig. 1(b). It can be seen from the figure that when the Au content is low (0.5%), the XRD diffraction peaks of the sample and the XRD characteristic diffraction peaks of the carrier Al2O3 are consistent, there is no characteristic diffraction peak of Au. This is due to the low loading of Au, which is lower than the detection limit of XRD, and no large particles of Au agglomerated during the roasting process (300 °C). Fig. 1(c) shows the N2 adsorption–desorption isotherm and pore distribution of γ-Al2O3-r and γ-Al2O3-c. The adsorption curves of γ-Al2O3-r and γ-Al2O3-c are of a typical type IV isotherm with an H3 hysteresis loop, indicating that the two types of alumina have a pore structure. The BET specific surface area of γ-Al2O3-r and γ-Al2O3-c estimated from this curve was 103 m2 g−1 and 240 m2 g−1 respectively. Furthermore, the pore size distribution curve of the two γ-Al2O3 obtained via BJH calculations (inset of Fig. 1(c)) reveals that the γ-Al2O3-r sample possesses a wide range of pore sizes from 2 to 50 nm with a peaked value at ∼14.5 nm. While the γ-Al2O3-c sample possesses a wide range of pore sizes from 2 to 25 nm with a peaked value at ∼5.2 nm.
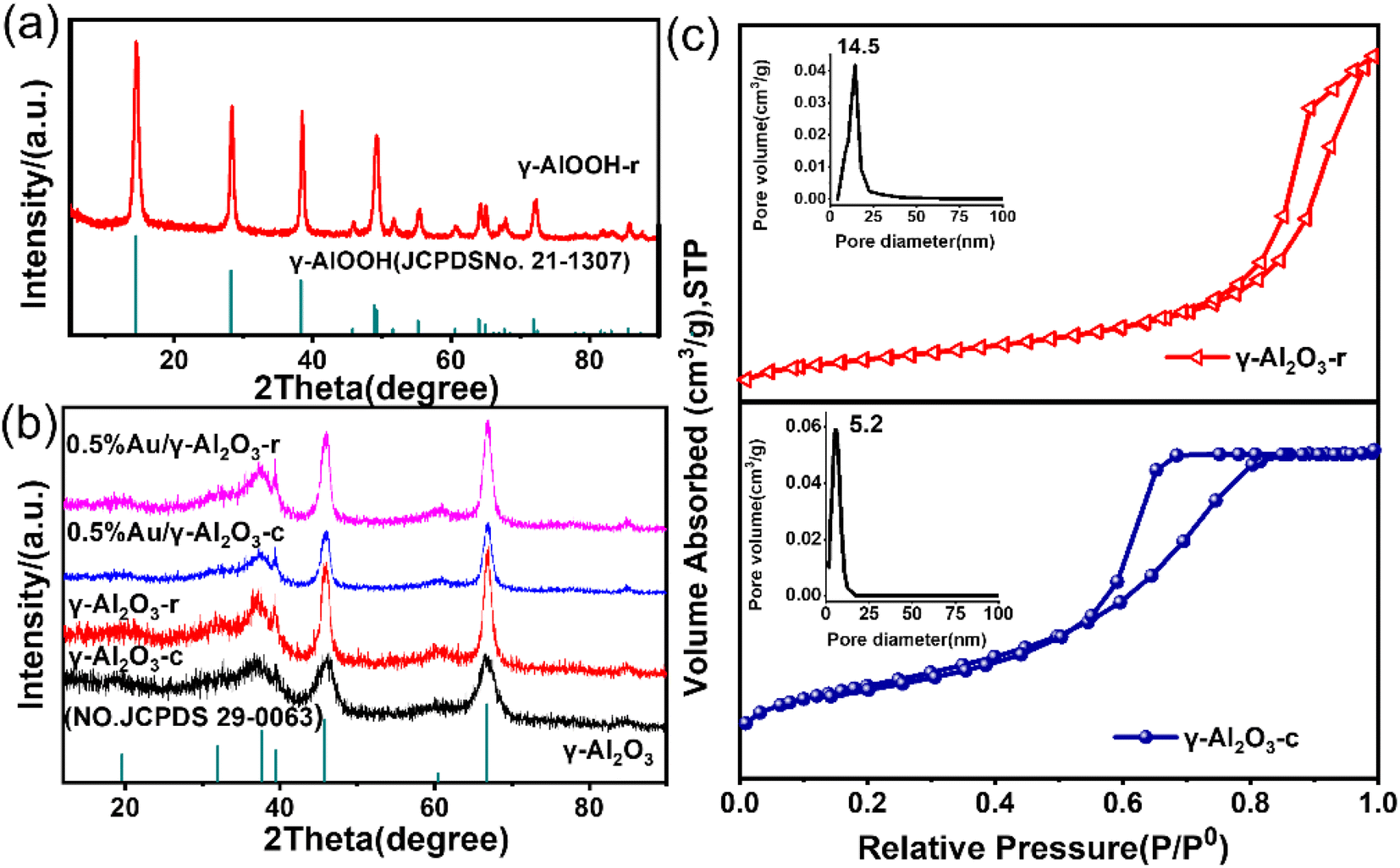 | ||
| Fig. 1 (a) XRD patterns of γ-AlOOH-r (b) XRD patterns of as-prepared γ-Al2O3-r and γ-Al2O3-c; 0.5%Au/γ-Al2O3-r and 0.5%Au/γ-Al2O3-c. (c) N2 sorption isotherm of γ-Al2O3-r and γ-Al2O3-c. | ||
The morphology of the γ-Al2O3-r and γ-Al2O3-c are shown in Fig. 2(a) and (b) respectively. The alumina prepared by the hydrothermal method shows a morphology different from that of commercial alumina, as shown in Fig. 2(a). The γ-Al2O3-r possesses a nanorod structure with a width of 8–10 nm and a length of 40–120 nm, while the commercial alumina presents the appearance of a porous structure formed by stacking many small spherical nanoparticles, as shown in Fig. 2(b). The source of the difference in the morphology of the two aluminas is the difference in their synthesis methods, which have undergone a hydrothermal synthesis process. The Au nanoparticles are loaded onto γ-Al2O3-r and γ-Al2O3-c via equal volume impregnation with a theoretical loading amount of 0.5 wt% and subsequent calcined under air atmosphere at 300 °C for 4 h. Fig. 2(c) and (d) show the TEM images of the 0.5%Au/γ-Al2O3-r and 0.5%Au/γ-Al2O3-c catalysts. As shown in Fig. 2(c), it can be seen that the Au nanoparticles on the surface of the γ-Al2O3-r catalyst are evenly distributed, and the aggregation of metal nanoparticles is not apparent. The size of most Au nanoparticles is below 5 nm (inset of Fig. 2(c)), indicating that the Au nanoparticles are highly dispersed on γ-Al2O3-r. The dispersibility of Au on γ-Al2O3-c can be clearly seen in Fig. 2(d), the size of Au nanoparticles is relatively large, with an average size of 8–9 nm (inset of Fig. 2(d)), indicating that using commercial γ-Al2O3 as support, the Au nanoparticles will agglomerate and grow during the calcination process. Therefore, the γ-Al2O3-r support has better dispersibility for Au nanoparticles than γ-Al2O3-c support.
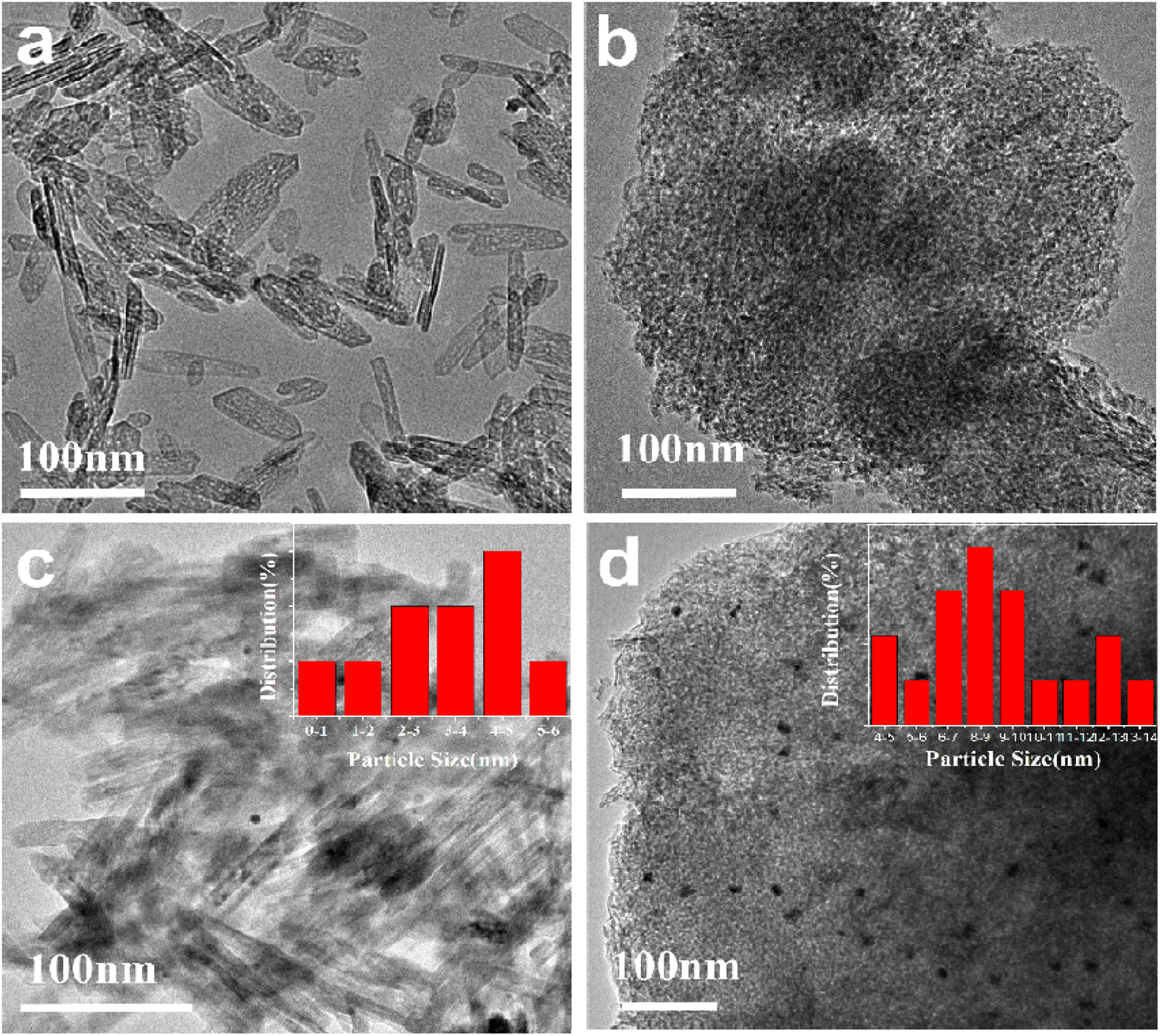 | ||
| Fig. 2 HRTEM image of the (a) γ-Al2O3-r; (b) γ-Al2O3-c; (c) 0.5%Au/γ-Al2O3-r; (d) 0.5%Au/γ-Al2O3-c. | ||
3.2 Evaluation of CO oxidation performance
As a typical and size-dependent reaction, the oxidation of carbon monoxide (CO) to carbon dioxide (CO2) is chosen as a simple test reaction to study the activity and thermal stability of Au/γ-Al2O3 catalysts, and explore the electron interactions between metal and support due to its special nanorod structure. Fig. 3 shows the CO oxidation performance of the Au/γ-Al2O3 catalysts calcined at different temperatures. It can be seen from Fig. 3 that whether the catalysts are calcined at 300 °C or 700 °C, the CO oxidation activity of the catalyst with nanorod-shaped γ-Al2O3 as the support was better than that of the catalyst with commercial γ-Al2O3 as the support. As shown in Fig. 3, the 0.5%Au/γ-Al2O3-r catalyst calcined at 300 °C can completely oxidize CO at 175 °C, while the complete conversion temperature of the 0.5%Au/γ-Al2O3-c catalyst was 350 °C. Previous studies have shown that gold particles with small size and high dispersibility have high CO oxidation activity,41 and catalysts with small gold particles have high oxidation activity and can completely convert carbon monoxide into carbon dioxide at low temperatures. Therefore, the results of CO oxidation performance are also consistent with the TEM results in Fig. 2(c) and (d). The catalyst with nanorod-shaped γ-Al2O3 as the support has smaller Au particles on the surface, and most of the gold particles are below 5 nm in size, in comparison, the Au nanoparticles on the surface of commercial γ-Al2O3 are larger, with an average size of 8–9 nm.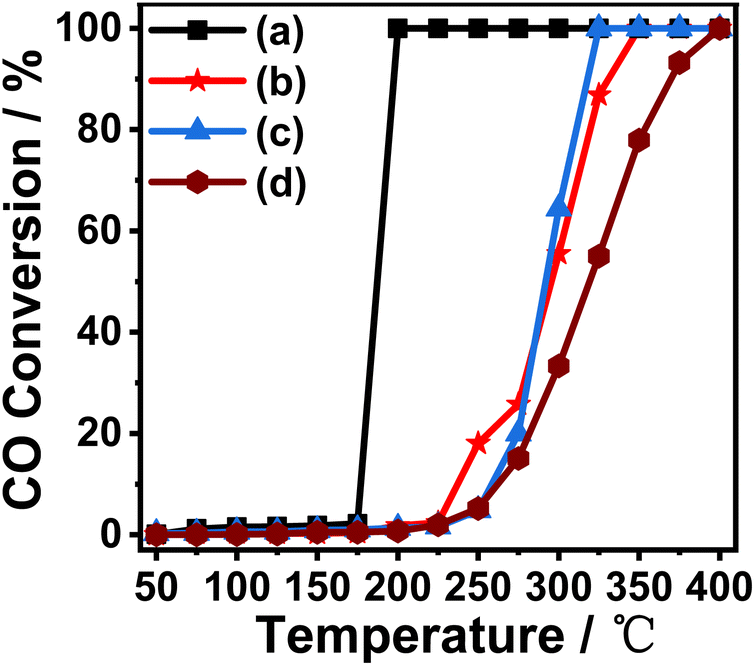 | ||
| Fig. 3 Catalytic activity of (a) 0.5%Au/γ-Al2O3-r-300 °C; (b) 0.5%Au/γ-Al2O3-c-300 °C; (c) 0.5%Au/γ-Al2O3-r-700 °C; (d) 0.5%Au/γ-Al2O3-c-700 °C in CO oxidation reaction. | ||
In order to investigate the thermal stability of the prepared catalysts, we calcined the two prepared catalysts (0.5%Au/γ-Al2O3-r, 0.5%Au/γ-Al2O3-c) under air atmosphere at 700 °C for 4 h. The performance of CO oxidation is tested and the results are also shown in Fig. 3. It can be seen from the figure that the catalyst with nanorod-shaped γ-Al2O3 as the support has good thermal stability. The temperature for complete CO conversion was 300 °C, which was much lower than the catalyst with commercial γ-Al2O3 as the support (400 °C). This experimental result shows that the supported Au catalyst prepared with nanorod-shaped γ-Al2O3 as the support has good anti-sintering performance, and even if it was calcined at a high temperature of 700 °C, its surface still has small-sized Au nanoparticles. The supported catalyst with commercial γ-Al2O3 as the support has poor thermal stability. After high-temperature roasting, the Au nanoparticles on the surface will aggregate and sinter, so the catalytic activity was poor.
3.3 Characterization of samples after roasting
To further explore the thermal stability of the prepared catalysts, the morphology of the samples after calcined at 700 °C is characterized by TEM, as shown in Fig. 4. Fig. 4(a) shows the TEM image of the 0.5%Au/γ-Al2O3-r catalyst after treatment at 700 °C. It can be seen from the figure that its morphology maintains the morphology of γ-Al2O3-r with an outer diameter of about 8–10 nm and a length of about 40–120 nm, and no obvious agglomerates of the Au nanoparticle can be found. The high-resolution TEM (Fig. 4(b)) shows that the size of the Au particles is about 5 nm, indicating that the Au nanoparticles still have a good dispersion on the surface of the alumina nanorods even after being calcined at a high temperature of 700 °C. Fig. 4(c) shows the TEM image of the 0.5%Au/γ-Al2O3-c catalyst after calcined at 700 °C. It can clearly see that many agglomerates with darker colors and a small number of large particles are present. The high-resolution TEM (Fig. 4(d)) shows more clearly that the size of the Au particles is about 10 nm, indicating that the Au nanoparticles agglomerated or even partially sintered on the surface of commercial γ-Al2O3 after calcined at 700 °C, which is also the reason for its low CO oxidation performance.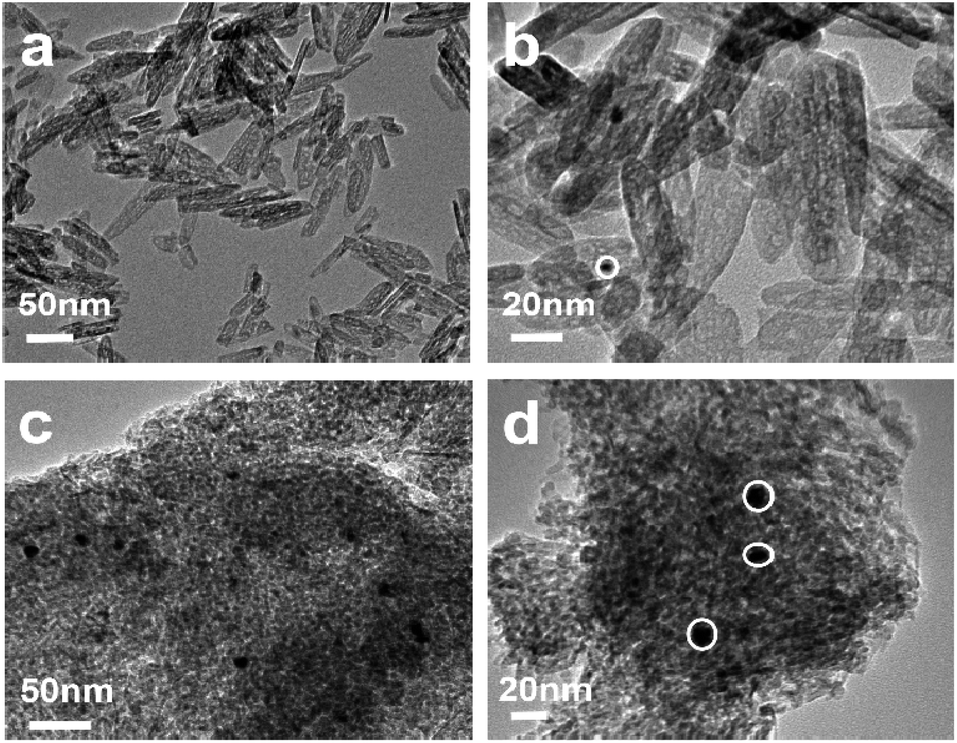 | ||
| Fig. 4 TEM image of the (a); (b) 0.5%Au/γ-Al2O3-r (c); (d) 0.5%Au/γ-Al2O3-c after calcined at 700 °C under air atmosphere. | ||
3.4 Investigation of the morphology-dependent behaviors of support
High resolution X-ray photoelectron spectroscopy (XPS) was used to explore the effect of the morphology of the γ-Al2O3 support on the metal nanoparticles as well as the chemical states of the Au nanoparticles present in the 0.5%Au/γ-Al2O3-r and 0.5%Au/γ-Al2O3-c catalysts after calcined at 700 °C under air atmosphere. As shown in Fig. 5(a), the full scan spectra of the two catalysts clearly show the presence of five peaks assigned to O 1s, C 1s, Al 2s, Au 4f and Al 2p on the surface of both the 0.5%Au/γ-Al2O3-r and the 0.5% Au/γ-Al2O3-c catalyst, which indicated that the elemental composition of the surfaces of the two catalysts was the same. To explore the possibility of electronic interactions between Au and γ-Al2O3 support, the spectrum of Au 4f of the two catalysts was shown in Fig. 5(b). For the 0.5%Au/γ-Al2O3-c catalyst (Fig. 5(b), blue line), the Au 4f spectrum shows two main peaks, and the binding energy (BE) at around 86.8 eV and 83.1 eV following the literature values for 4f 5/2 and Au 4f 7/2 of Au0, respectively,42 suggesting that Au exists as a metallic phase in the 0.5%Au/γ-Al2O3-c catalyst. In the case of 0.5%Au/γ-Al2O3-r catalyst (Fig. 5(b), red line), the Au 4f 5/2 and 4f 7/2 peaks shift to higher BE by approximately 0.2 eV with the binding energy (BE) at around 87.0 eV and 83.3 eV compared to the 0.5%Au/γ-Al2O3-c catalyst. In this study, whether the composition, the preparation method or the treatment conditions of the two catalysts were the same, the only difference was the support morphology of the two catalysts. Therefore, the positive shift of the 0.5%Au/γ-Al2O3-r catalyst was due to the interactions between the Au and the special nanorod alumina support. These results clearly show that after calcined at high temperature, there was an electronic interaction between Au and γ-Al2O3-r support, and Au transfers part of the electrons to the γ-Al2O3-r support, producing the Auδ+ species on the surface of the 0.5%Au/γ-Al2O3-r catalyst calcined at 700 °C. Furthermore, the experimental results were consistent with this literature.43 Gaurav Kumar et al. using Hammett and DFT studies evaluating differences in the active-site electronics of supported Au nanoparticle catalysts, and the logical conclusion was that the less active catalysts have more negatively charged Au active sites while the more active catalysts are less negatively (or perhaps more positively) charged. Tus the better CO oxidation activity of the 0.5%Au/γ-Al2O3-r catalyst maybe due to the more positively charged Au species as certified by XPS. Fig. 5(c) shows that the C 1s peak positions of the two catalysts were almost the same. As shown in Fig. 5(d), the O 1s peaks of the 0.5% Au/γ-Al2O3-c and 0.5% Au/γ-Al2O3-r catalysts were at about 531.08 eV and 531.2 eV, respectively, and the O 1s peaks could be attributed to the lattice oxygen for γ-Al2O3 that have appeared in the literature.44 The negative shift of the 0.5%Au/γ-Al2O3-r catalyst maybe due to Au transferring part of the electrons to the γ-Al2O3-r, the surface of the γ-Al2O3-r was partially negatively charged. According to the above XPS results, due to the special morphological effect of γ-Al2O3-r, after calcined at high temperature, there was an electronic interaction between the Au and the γ-Al2O3-r support, and the Au transfers part of the electrons to the γ-Al2O3-r support, making it partially positively charged. The partially positively charged Auδ+ species has higher activity in the CO oxidation reaction, so the Au/γ-Al2O3-r catalysts show enhanced thermal stabilization.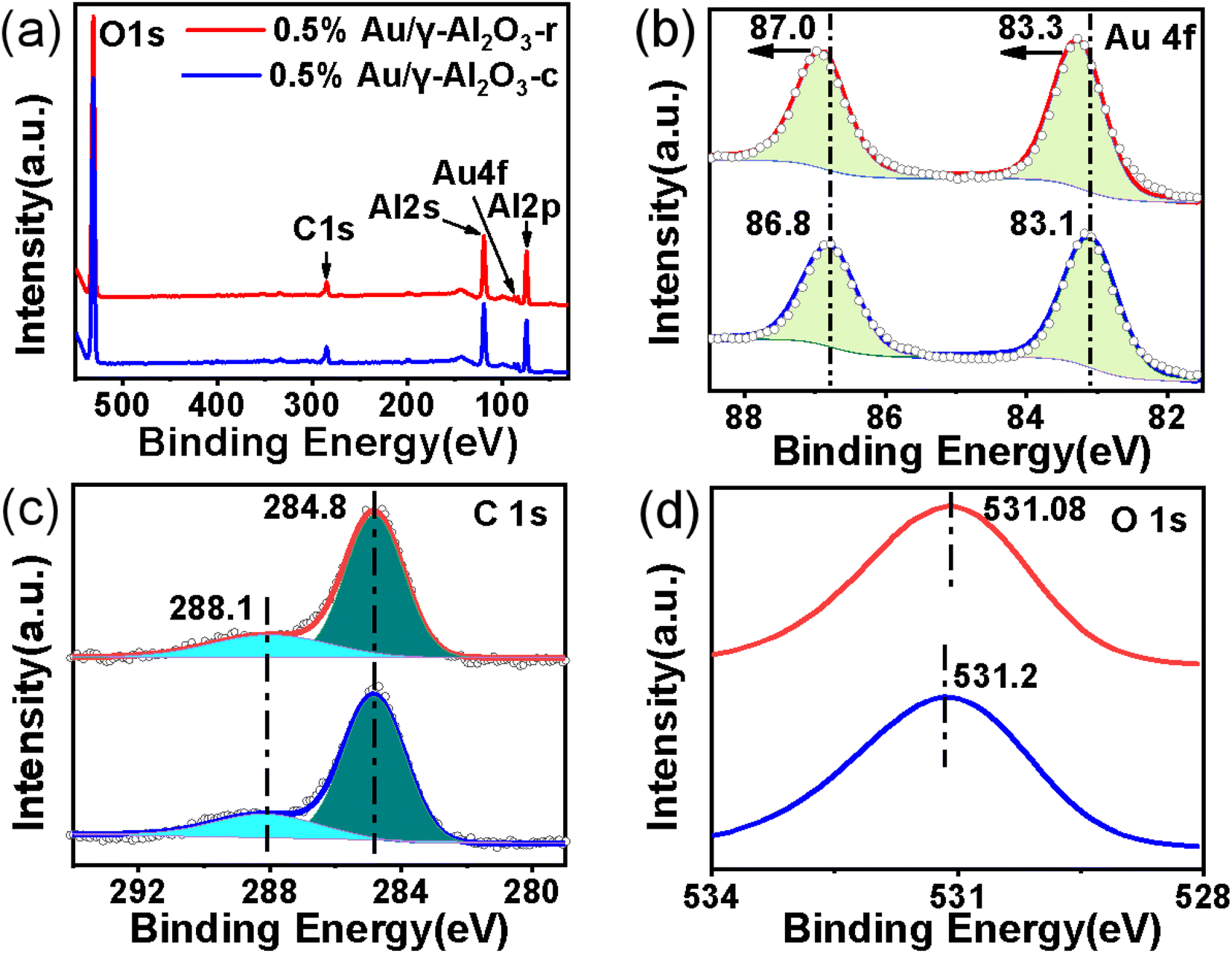 | ||
| Fig. 5 The XPS spectra of (a) the full scan spectra of 0.5%Au/γ-Al2O3-r and 0.5%Au/γ-Al2O3-c after calcined at 700 °C under air atmosphere; (b) Au 4f; (c) C 1s; (d) O 1s. | ||
To further explore the surface chemical properties of the Au on the two γ-Al2O3 supports with different morphology under high calcining temperatures (700 °C), in situ DRIFTS was used to measure the adsorbed CO. Fig. 6 compares IR spectra of CO adsorbed on the 0.5%Au/γ-Al2O3-r and 0.5%Au/γ-Al2O3-c catalysts. As shown in Fig. 6 (blue line), two bands were detected on the 0.5%Au/γ-Al2O3-c catalyst at 2200 and 2088 cm−1, respectively. The former was attributed to CO adsorbed on cationic gold (Au(I) or Au(III)).45 The latter belonged to two linear CO species adsorbed on the metallic Au sites.46 In comparison with the Au-supported catalyst on commercial γ-Al2O3, adsorptions of CO on the 0.5%Au/γ-Al2O3-r catalyst (Fig. 6, red line) result in the appearance of two bands at 2206 and 2100 cm−1, which can also be assigned to CO adsorbed on cationic gold (Au(I) or Au(III)) and the metallic Au sites respectively. In addition, there was a blue shift (ca. 12 cm−1) on the 0.5%Au/γ-Al2O3-r catalysts, which was a result of the electronic modification, and corresponds to a formally positively charged Au, the blue shift of the CO adsorption band may be attributed to electron withdrawing from Au NPs to γ-Al2O3-r support. The DRIFTS measurements show the presence of Auδ+ species on the surface of 0.5%Au/γ-Al2O3-r catalyst after calcined at 700 °C, which was consistent with the results of XPS (Fig. 5). These characterization results further confirm that there was electronic interaction between the Au nanoparticles and the special nanorod-like gamma alumina support after being calcined under high temperature, which resulted in better anti-sintering performance.
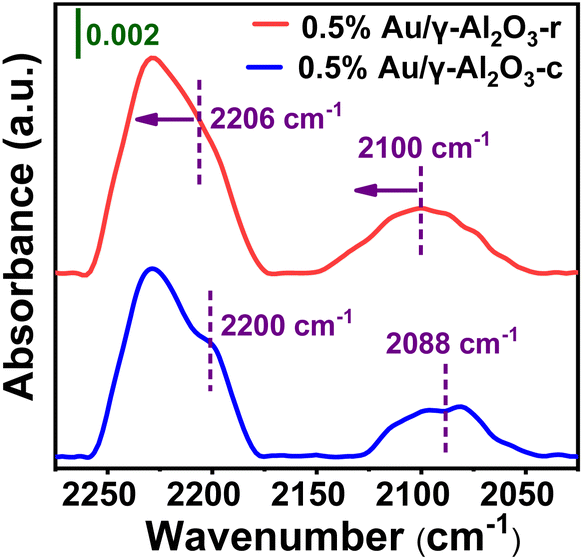 | ||
| Fig. 6 In situ DRIFT spectra of CO adsorption on 0.5%Au/γ-Al2O3-r (red line) and 0.5%Au/γ-Al2O3-c (blue line) after calcined at 700 °C under air atmosphere. | ||
The redox performance was a critical parameter during deep oxidation reactions such as CO oxidation reaction, which was usually investigated by H2-TPR. In order to determine the reduction characteristics of Au/γ-Al2O3 catalysts at different morphology and calcination temperatures, we performed H2-TPR experiments. The 100 mg sample was pretreated with a stream of argon gas (50 mL min−1) at 353 K for 2 h. The H2 reduction curve was recorded with a thermal conductivity detector using a 5% H2/N2 mixture as a reference stream introduced from room temperature to 823 K (10 K min−1) at a 50 mL min−1 rate. As shown in Fig. 7, the H2-TPR plots show the characteristic peaks of the Au/γ-Al2O3 catalysts over the temperature range from 297 K to 823 K. In both Au/γ-Al2O3 catalysts, similar H2 consumption profiles were showed observed, in which a single peak existed at about 334 K for the 0.5%Au/γ-Al2O3-r catalyst and around 361 K for the 0.5%Au/γ-Al2O3-c catalyst. The peak was attributed to oxygen included in gold oxide (AuOx) on γ-Al2O3-r and γ-Al2O3-c.47,48 Considering the low-temperature peaks (334 K and 361 K) of the Au/γ-Al2O3 catalysts, the Au nanoparticles on the γ-Al2O3 support contained some oxygen content that can be used for the oxidation of CO. Furthermore, whether calcined at 300 °C or 700 °C, the peak of the Au/γ-Al2O3-r catalyst appeared at a temperature around 30 K lower than that of the Au/γ-Al2O3-c catalyst, confirming that the oxygen species of gold oxide on γ-Al2O3 nanorod were more active concerning oxidation of reductant than the oxygen content of gold oxide on commercial γ-Al2O3. Combined with the results of activity evaluation, the lower the reduction peak temperature, the better the activity, which can be attributed to the reduction peak of active gold oxide species. The result of H2-TPR was also consistent with the results of CO catalytic oxidation, with the gold supported on γ-Al2O3 nanorods having better catalytic activity. The H2-TPR results of the catalysts calcined at different temperatures showed that the area of the low-temperature reduction peak became smaller after high-temperature calcination (700 °C). Combined with the activity evaluation results, the larger the reduction peak area, the better the activity. Take the 0.5%Au/γ-Al2O3-r catalyst as an example, the 0.5%Au/γ-Al2O3-r-300 °C catalyst has a larger reduction peak area than that of 0.5%Au/γ-Al2O3-r-700 °C, which means that there were more active oxide species, so its activity was better. In addition, the difference between the peak types of the two catalysts can also be clearly seen in Fig. 7. The reduction peak of the 0.5%Au/γ-Al2O3-r catalyst was narrow, while the reduction peak of the 0.5%Au/γ-Al2O3-c catalyst was broader. According to the literature,49 the particle size distribution of metal particles on the catalyst surface with a narrow reduction peak was uniform. The reason was that the diffusion and adsorption properties of hydrogen were similar, and the reduction process will proceed simultaneously, so the peak was narrow. Whereas the species corresponding to broad reduction peaks have different properties. This result was consistent with our previous TEM characterization results (Fig. 2 and 4). The Au dispersion on the surface of the gamma alumina nanorods was more uniform, which may be related to the regular morphology of the alumina support. To sum up, the results of H2-TPR further confirmed the morphology-dependent effects of the gamma alumina support. The active gold species of the catalyst supported by γ-Al2O3 nanorods were more easily reduced and thus have higher activity in CO oxidation reaction. The interaction between the γ-Al2O3 nanorods support and Au can prevent the aggregation and growth of the Au nanoparticles at high temperature, so it has better thermal stability.
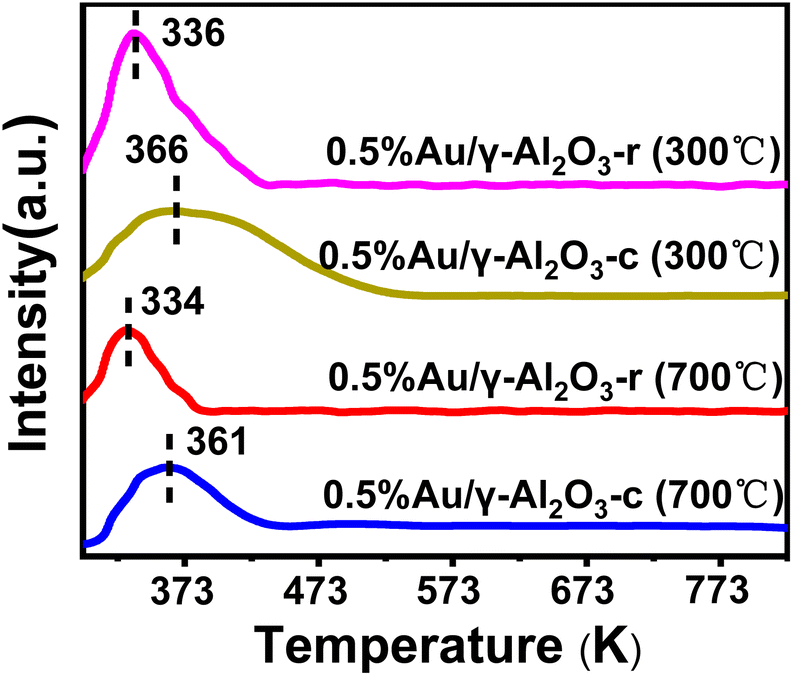 | ||
| Fig. 7 H2-TPR profiles of 0.5%Au/γ-Al2O3 catalysts with different shapes and calcination temperatures. | ||
4 Conclusions
The nanorods gamma alumina with regular and uniform morphology has been synthesized via a simple hydrothermal synthesis method. The special morphology of the alumina support and interfacial interactions are responsible for stabilizing Au nanoparticles at high temperatures. The morphology of the support, that is, the exposed crystal facets, can influence the catalytic performance through metal-support interactions (MSI). A charge transfer from the Au nanoparticles to the γ-Al2O3 nanorods was observed by XPS, CO-DRIFTS and H2-TPR characterization and higher thermal stability in CO oxidation was observed as compared to Au on commercial γ-Al2O3. Even after calcination at 700 °C, higher stability and excellent catalytic activity can be achieved, which further confirm the small size and high dispersion of Au nanoparticles. Therefore, this finding reveals the distinct morphology-dependent behaviors of γ-Al2O3. The nanorod-shaped gamma alumina can stabilize the Au nanoparticles against sintering through the MSI, and such a strategy of tuning the catalyst performance by modulating the morphology of the support can be used to guide the synthesis of other thermally stable catalysts.Author contributions
W. C. and L. F conceived the work. W. C. and X. S. designed the experiments and wrote the paper. W. C., X. S. and Y. B. performed the experiments. A. L. and J. G. helped perform the analysis with constructive discussions. K. H. guided the work. All authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Natural Science Foundation of China (52075144); University Natural Sciences Research Project of Anhui Province (Project Number: 2022AH051802) and the Anhui Province Key Research and Development Program (202104a05020065).Notes and references
- P. Biloen, J. N. Helle, H. Verbeek, F. M. Dautzenberg and W. Sachtler, J. Catal., 1980, 63, 112–118 CrossRef CAS.
- G. X. Pei, X. Y. Liu, X. Yang, L. Zhang, A. Wang, L. Li, H. Wang, X. Wang and T. Zhang, ACS Catal., 2017, 7, 1491–1500 CrossRef CAS.
- H. Tang, J. Wei, F. Liu, B. Qiao, X. Pan, L. Li, J. Liu, J. Wang and T. Zhang, J. Am. Chem. Soc., 2015, jacs.5b11306 Search PubMed.
- K. An, S. Alayoglu, N. Musselwhite, K. Na and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 6830–6833 CrossRef CAS PubMed.
- H. Guan, Y. Chen, C. Ruan, J. Lin, Y. Su, X. Wang and L. Qu, Chin. J. Catal., 2020, 41, 613–621 CrossRef CAS.
- B. Qiao, J. Lin, A. Wang, Y. Chen, T. Zhang and J. Liu, Chin. J. Catal., 2015, 36, 1505–1511 CrossRef CAS.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346, 1498–1501 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- S. Liu, W. Xu, Y. Niu, B. Zhang, L. Zheng, W. Liu, L. Li and J. Wang, Nat. Commun., 2019, 10, 5790 CrossRef CAS PubMed.
- J. Lu, B. Fu, M. C. Kung, G. Xiao, J. W. Elam, H. H. Kung and P. C. Stair, Science, 2012, 335, 1205–1208 CrossRef CAS PubMed.
- E. Wolf and F. Alfani, Catal. Rev., 1982, 24, 329–371 CrossRef CAS.
- J. Radnik, A. Benhmid, V. N. Kalevaru, M. M. Pohl, A. Martin, B. Lücke and U. Dingerdissen, Angew. Chem., Int. Ed., 2005, 44, 6771–6774 CrossRef CAS PubMed.
- J. Zhang, H. Wang, L. Wang, S. Ali, C. Wang, L. Wang, X. Meng, B. Li, D. S. Su and F.-S. Xiao, J. Am. Chem. Soc., 2019, 141, 2975–2983 CrossRef CAS PubMed.
- Y. Yao, Z. Huang, P. Xie, L. Wu, L. Ma, T. Li, Z. Pang, M. Jiao, Z. Liang and J. Gao, Nat. Nanotechnol., 2019, 14, 851–857 CrossRef CAS PubMed.
- S. Zhang, P. N. Plessow, J. J. Willis, S. Dai, M. Xu, G. W. Graham, M. Cargnello, F. Abild-Pedersen and X. Pan, Nano Lett., 2016, 16, 4528–4534 CrossRef CAS PubMed.
- M. Xu, S. He, H. Chen, G. Cui, L. Zheng, B. Wang and M. Wei, ACS Catal., 2017, 7, 7600–7609 CrossRef CAS.
- H. Y. Kim, H. M. Lee and G. Henkelman, J. Am. Chem. Soc., 2012, 134, 1560–1570 CrossRef CAS PubMed.
- H. Ha, S. Yoon, K. An and H. Y. Kim, ACS Catal., 2018, 8, 11491–11501 CrossRef CAS.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga and X. I. Pereira Hernández, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- J. Kašpar, P. Fornasiero and M. Graziani, Catal. Today, 1999, 50, 285–298 CrossRef.
- S. Carrettin, P. Concepción, A. Corma, J. M. López Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538–2540 CrossRef CAS PubMed.
- C. T. Campbell and C. H. Peden, Science, 2005, 309, 713–714 CrossRef CAS PubMed.
- Z. Gao and Y. Qin, Acc. Chem. Res., 2017, 50, 2309–2316 CrossRef CAS PubMed.
- J. Huo, J.-P. Tessonnier and B. H. Shanks, ACS Catal., 2021, 11, 5248–5270 CrossRef CAS.
- P. M. Arnal, M. Comotti and F. Schüth, Angew. Chem., Int. Ed., 2006, 45, 8224–8227 CrossRef CAS PubMed.
- W. Zhan, Y. Shu, Y. Sheng, H. Zhu, Y. Guo, L. Wang, Y. Guo, J. Zhang, G. Lu and S. Dai, Angew. Chem., Int. Ed., 2017, 56, 4494–4498 CrossRef CAS PubMed.
- W. Chen, Z. Fan, X. Pan and X. Bao, J. Am. Chem. Soc., 2008, 130, 9414–9419 CrossRef CAS PubMed.
- Y. Wu, Angew. Chem., Int. Ed., 2019, 58, 18388–18393 CrossRef PubMed.
- A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS PubMed.
- P. Munnik, P. E. De Jongh and K. P. De Jong, J. Am. Chem. Soc., 2014, 136, 7333–7340 CrossRef CAS PubMed.
- K. Murata, Y. Mahara, J. Ohyama, Y. Yamamoto, S. Arai and A. Satsuma, Angew. Chem., 2017, 129, 16209–16213 CrossRef.
- B. Qiao, J. Lin, A. Wang and Y. Chen, Chin. J. Catal., 2015, 36(9), 1505–1511 CrossRef CAS.
- T. Chaieb, L. Delannoy, S. Casale, C. Louis and C. Thomas, Chem. Commun., 2015, 51, 796–799 RSC.
- F. Cárdenas-Lizana, S. Gómez-Quero, A. Hugon, L. Delannoy, C. Louis and M. A. Keane, J. Catal., 2009, 262, 235–243 CrossRef.
- S. Yao, Z. Chen, S. Weng, L. Mao, X. Zhang, J. Han, Z. Wu, H. Lu, X. Tang and B. Jiang, J. Hazard. Mater., 2019, 373, 698–704 CrossRef CAS PubMed.
- X. Wang, G. Lu, Y. Guo, Z. Zhang and Y. Guo, Environ. Chem. Lett., 2011, 9, 185–189 CrossRef CAS.
- R. Camposeco and R. Zanella, Environ. Sci. Pollut. Res., 2022, 29, 76992–77006 CrossRef CAS PubMed.
- H. Su, Y. Zheng, X. Sun, L. Sun, X. Xu and C. Qi, Kinet. Catal., 2020, 61, 304–309 CrossRef CAS.
- R. Grisel, C. Weststrate, A. Goossens, M. Crajé, A. Van der Kraan and B. Nieuwenhuys, Catal. Today, 2002, 72, 123–132 CrossRef CAS.
- Z. Huo, C.-k. Tsung, W. Huang, X. Zhang and P. Yang, Nano Lett., 2008, 8, 2041–2044 CrossRef CAS PubMed.
- G. Kumar, L. Tibbitts, J. Newell, B. Panthi, A. Mukhopadhyay, R. M. Rioux, C. J. Pursell, M. Janik and B. D. Chandler, Nat. Chem., 2018, 10, 268–274 CrossRef CAS PubMed.
- B. R. Strohmeier, Surf. Sci. Spectra, 1994, 3, 135–140 CrossRef CAS.
- N. Parkyns, J. Phys. Chem., 1971, 75, 526–531 CrossRef CAS.
- E. Roze, P. Gravejat, E. Quinet, J. L. Rousset and D. Bianchi, J. Phys. Chem. C, 2009, 113, 1037–1045 CrossRef CAS.
- S.-Y. Lai, Y. Qiu and S. Wang, J. Catal., 2006, 237, 303–313 CrossRef CAS.
- M. J. Kim, G.-H. Han, S. H. Lee, H. W. Jung, J. W. Choung, C. H. Kim and K.-Y. Lee, J. Hazard. Mater., 2020, 384, 121341 CrossRef CAS PubMed.
- X. Tang, B. Zhang, Y. Li, Y. Xu, Q. Xin and W. Shen, Catal. Lett., 2004, 97, 163–169 CrossRef CAS.
Footnote |
| † These authors contribute equally to this research. |
| This journal is © The Royal Society of Chemistry 2023 |