
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBrushite mineralised Scots pine (Pinus sylvestris L.) sapwood – revealing mineral crystallization within a wood matrix by in situ XRD†
Edita Garskaite *a,
Giedrius Balciunasb,
Marian Drienovskyc,
Denis Sokold,
Dick Sandberga,
Alexandre C. Bastose and
Andrei N. Salake
*a,
Giedrius Balciunasb,
Marian Drienovskyc,
Denis Sokold,
Dick Sandberga,
Alexandre C. Bastose and
Andrei N. Salake
aWood Science and Engineering, Department of Engineering Sciences and Mathematics, Luleå University of Technology, Forskargatan 1, SE-931 87 Skellefteå, Sweden. E-mail: Edita.garskaite@ltu.se
bLaboratory of Thermal Insulating Materials and Acoustics, Institute of Building Materials, Vilnius Gediminas Technical University, Linkmenu g. 28, Vilnius LT-08217, Lithuania
cInstitute of Materials Science, Faculty of Materials Science and Technology in Trnava, Slovak University of Technology in Bratislava, Ulica Jana Bottu 2781/25, 91724 Trnava, Slovakia
dInstitute of Chemistry, Faculty of Chemistry and Geosciences, Vilnius University, Naugarduko 24, Vilnius LT-03225, Lithuania
eDepartment of Materials and Ceramics Engineering and CICECO – Aveiro Institute of Materials, University of Aveiro, 3810-193 Aveiro, Portugal
First published on 16th February 2023
Abstract
Dicalcium phosphate dihydrate (CaHPO4·2H2O, DCPD, brushite) crystals were synthesised within Scots pine sapwood via a wet-chemistry route from aqueous solutions of Ca(CH3COO)2 and NH4H2PO4 salts. SEM/EDS analysis was used to assess the saturation of the wood cell lumina and cell wall as well as morphological features and elemental composition of the co-precipitated mineral. Brushite mineral crystallization and crystallite growth within the wood matrix was studied by in situ XRD. The chemical composition of the mineral before and after the dissolution was evaluated using FTIR spectroscopy. The overall impact of brushite on the thermal behaviour of wood was studied by TGA/DSC and TGA/DTA/MS analysis under oxidative and pyrolytic conditions. Bending and compression strength perpendicular and parallel to the fibre directions as well as bending strengths in longitudinal and transverse directions of the mineralised wood were also evaluated. Results indicate the viability of the wet-chemistry processing route for wood reinforcement with crystalline calcium phosphate (CaP)-based minerals, and imply a potential in producing hybrid bio-based materials that could be attractive in the construction sector as an environmentally friendly building material.
1. Introduction
Interest in employing timber in the construction sector is on the rise and using wood and wood-based materials instead of fossil-based materials to provide the same function can reduce the negative climate impact.1–3 Thus, the development of wood products by enhancing their physicochemical properties and extending their useful lifetime is an integral part of sustainable production and consumption.4,5 The wood-material properties required to be improved are in the first hand protection against photodegradation, thermal stability or fire retardancy, mechanical properties, resistance to biological degradation, and others.1,6–8Wood is a natural composite material, and the structural components of wood cell wall are mainly cellulose (approximately 40–45%), hemicelluloses (25–30%), and lignin (20–30%).7,9,10 The chemical composition and hierarchical porous structure of wood offers diverse possibilities for its modification.11 Conventionally, wood material is treated by several means, i.e. applying coatings, modifying wood cell wall (thermally or chemically), or impregnating.8,12,13 Chemical modification exploits hydroxyl (OH) groups of wood constituents which react with different chemical reagents resulting in formation of covalent bonding. During the impregnation, wood is treated passively, i.e. chemicals diffuse into either cell wall or lumen. The impregnation usually proceeded from solutions of water-soluble compounds. However, migration of such additives to the wood surface and leakage can occur when the wood is exposed to elevated moisture. As a result, an effective lifetime of the product is likely to be of shorter duration.
Lately, studies were conducted to enhance wood properties via “hybrid” modification pathway, namely when impregnation with or without vacuum-pressure is performed from aqueous solutions at ambient temperature and as a chemical reaction proceeds an insoluble solid is coprecipitated within wood matrix when the reactive ions combine.3 For instance, CaCO3 was successfully synthesised inside the cell-wall structure of beech and spruce wood utilising approach of alkaline hydrolysis of dimethyl carbonate precursors in the presence of calcium ions.14 In another study, BaSO4 was precipitated within wood scaffold by alternating infiltration with concentrated electrolyte solutions.15 Preparation of struvite (MgNH4PO4·6H2O)-wood composite with improved flame retardancy and transverse strength and stiffness under compression was also demonstrated.16,17 An incorporation of water-insoluble material within wood matrix, when leaching of intercalated material is prevented, can be a way to make wood firmer and more durable.
Calcium phosphates (CaPs) are a group of minerals widely used in the field of bone tissue engineering.18–20 They have a similar mineral structure to the main mineral of human bones and teeth. Among them, the most widely investigated are hydroxyapatite (Ca10(PO4)6(OH)2, HAP) and tricalcium phosphate (Ca3(PO4)2, TCP).21–23 Under certain conditions, some of these CaPs are able to transform from one to another, which in consequence determines the density and microstructure of material. Dicalcium phosphate dihydrate, CaHPO4·2H2O (DCPD), known as brushite (crystallises in the monoclinic space group Ia with unit cell parameters a = 5.812(2) Å, b = 15.180(3) Å, c = 6.239(2) Å and β = 116.42(3)°, Z = 4),24 is formed in dental and urinary calculus.25–27 It has been shown that brushite mineral (pKSP of 6.59 at 25 °C) is one of the precursors to prepare HAP (pKSP of 116.8 at 25 °C);28–31 under treatment with NaOH it immediately transforms into the HAP mineral.32 It was also reported that DCPD can be transformed into whitlockite (Ca9Mg(HPO4)(PO4)6 or Ca3(PO4)2·MgHPO4), which has a similar crystal structure to that of β-TCP.33 Extensive studies were performed to understand CaP formation in solutions (in vitro and in vivo) and variety of techniques were used to assess the nucleation and phase transformation.34–37 However, to our knowledge, no study on the formation of CaP-based mineral within the wood matrix has been reported.
CaP bioceramics are conventionally produced by chemical precipitation from aqueous solutions containing calcium and phosphate ions.38–40 Solution-based chemical synthesis is usually environment-friendly and proceeds at room temperature. Owing to a wide range of precursors, different types of solvents and organic additives (complexing agents, surfactants) that can be used, such a synthesis offers inexpensive processing way to phase-pure material with desired crystallinity and particle/crystal sizes, which are known to have a profound effect on the material properties, and thereby on the performance of the final product. The synthesis parameters, such as concentration, temperature, pH, reaction time can be also adjusted.
CaP minerals exhibit strong ionic and/or covalent bonding which result in their high hardness and compressive strength, high melting points, low thermal and electrical conductivity, and chemical inertness. These unique physicochemical properties make CaPs excellent candidates to be considered as reinforcement material for wood protection. Thus, exploring the synergistic effects of the wood material and CaP minerals, when an insoluble material is formed within wood matrix via wet-chemistry route, might be a worthwhile strategy aiming both low-environmental impact and extend the useful lifetime of the wood-based materials.
In the present work, the mineralization of Scots pine (Pinus sylvestris L.) sapwood with CaP-mineral was studied. The mineralization of wood matrix was achieved via wet-chemistry approach by performing cyclic impregnation and using vacuum setup. Morphological features of coprecipitated mineral as well as saturation of wood matrix were assessed by SEM/EDS analysis. Mineral crystallization within wood matrix was studied by in situ XRD analysis. The thermal stability under different conditions and gas evolution of the mineralised wood was studied via TGA/DSC and TGA/DTA/MS analyses. Chemical functionalities of mineralised wood before and after the leaching were evaluated using FTIR spectroscopy. Mechanical properties such as compression and bending strengths of produced composites were also measured. Proposed wet-chemistry route for wood reinforcement with CaP-based materials opens possibilities for the upscaling, and the developed hybrid wood composites exhibit potential for its use in different application, including construction sector.
2. Experimental
2.1. Wood specimens, solutions and wood mineralization
Specimens of Scots pine (Pinus sylvestris L.) sapwood with dimensions of 1 × 1 × 1 cm and 1 × 1 × 15 cm (tangential (T) × radial (R) × longitudinal (L)) were cut from sawn timber obtained from northern Sweden. Wood slabs of 100 μm thick were prepared by microtomy slicing layers of the wood block 1 × 1 × 1 cm wetted with a distilled water.Calcium acetate (Ca(CH3COO)2, 99%, Aldrich) and ammonium dihydrogen phosphate (NH4H2PO4, 99%, Aldrich) were used as the cations precursors. To study mineral crystallization in aqueous solutions and within 100 μm wood slabs, an aqueous solutions of 0.5 M Ca(CH3COO)2 and 0.3 M NH4H2PO4 were prepared by dissolving 0.395 g (0.0025 mol) of Ca(CH3COO)2 salt and a stoichiometric amount 0.1725 g (0.0015 mol) of NH4H2PO4 salt in separate beakers containing 5 mL of distilled H2O. Solutions were mixed for 5 min to ensure complete dissolution of salts. The Ca–P–O precipitate formation was performed by placing one drop of solution containing Ca ions on the glass plate and then adding the drop of solution containing phosphate ions. Additionally, solutions of 0.05 M Ca(CH3COO)2 and 0.03 M NH4H2PO4 were prepared and used to study Ca–P–O precipitate formation.
To mineralize the wood bocks, 0.5 M, 1.0 M and 1.5 M Ca(CH3COO)2 solutions (volume of 1 L) and the corresponding 0.3 M, 0.6 M and 0.9 M of NH4H2PO4 solutions (volume of 1 L) were prepared. The reactive solutions were used in the consecutive order that molar ratio of calcium to phosphate ions was maintained to be 1.67. Mineralization of wood blocks was performed in glass round-bottom flasks connected to the vacuum pump. First, wood specimens were vacuumed for 1 hour to remove the air from the matrix cavities. A 0.5 M Ca(CH3COO)2 solution was then injected into the flask and the wood blocks were left under vacuum for 2 hours to achieve complete saturation of wood matrix. In a following step, these impregnated specimens were removed from the solution and dried for 3 days at room temperature. Dried specimens were then placed additionally in a round bottom flask connected to the vacuum pump and vacuumed for 1 hour. After the air was removed, a 0.3 M NH4H2PO4 solution was injected into the flask. Soaking of wood blocks was performed for 2 hours to allow complete saturation of wood matrix. Magnetic stirrer was used to ensure homogeneous ion distribution within the solution. Turbidity of the solution was observed as well as precipitate formation on the surface of wood blocks. By the end of impregnation procedure, the wood blocks were settled on the bottom of the beaker. Saturated wood blocks were then removed from turbid solution and left for 3 days to dry at room temperature, i.e. to reduce the moisture content within the wood matrix. In all series of experiments, the same order of solution accession was maintained. The mineralised wood specimens prepared from the systems of 0.5 M Ca(CH3COO)2 and 0.3 M NH4H2PO4, 1.0 M Ca(CH3COO)2 and 0.6 M NH4H2PO4, and 1.5 M Ca(CH3COO)2 and 0.9 M NH4H2PO4 are designated as DCPD(1)-wood, DCPD(2)-wood and DCPD(3)-wood.
2.2. Mineral dissolution tests
Testing of the mineral dissolution from the wood matrix was conducted in water medium on five replicates. Mineralised wood slabs of 100 μm thick were immersed in polypropylene containers with 5 mL of distilled H2O and kept for 30 min. The wood slabs were then carefully transferred with tweezers into the other containers containing the same volume of distilled H2O. For each specimen water was changed three times, and the duration of submersion time was 30 min. After the final leaching step each wood slab was placed on the microscopy glass plate and dried for 48 hours at room temperature.2.3. Characterization
Morphological features of pure wood and brushite mineralised wood (microtome-cut slabs of 100 μm thick) were evaluated using a field emission scanning electron microscope (FE-SEM, SU70, Hitachi). A secondary electron imaging was used and the electron beam acceleration voltage was 2.0–5.0 kV. Before analysis, the samples were coated with ∼10 nm layer of Ag. The samples were also studied using a Hitachi TM3000 Tabletop SEM equipped with the energy dispersive X-ray spectrometer (EDS), and the spectrometer was controlled by the INCA software (Oxford Instruments). A backscattered electron detector was applied and the electron beam acceleration voltage was 15 kV. An X-ray acquisition time of 315 s was used to obtain the EDS spectra (number of independent measurements (n) = 3–5 for each feature of interest) and the elemental mapping images. Infrared spectra were recorded using a Fourier transform infrared (FT-IR) spectrometer (Frontier FT-IR, PerkinElmer, ZnSe/Diamond ATR crystal, DTGS detector, 4000 to 600 cm−1, 4 scans). Thermal behaviour of the untreated and mineralised wood was studied under oxidative conditions in air atmosphere (up to 600 °C) and pyrolysis conditions in N2 (up to 855 °C) and He (up to 1000 °C) atmospheres. Thermogravimetric analysis (TG) and differential scanning calorimetry (DSC) were performed using a PerkinElmer STA 6000 Simultaneous Thermal Analyzer. Dried samples of 5–10 mg were heated from 25 to 855 °C at a rate of 10 °C min−1 in N2 atmosphere (20 mL min−1). Simultaneous Thermal Analyser (Netzsch STA 409CD) equipped with Quadrupole Mass Spectrometer (MS) was used to study samples in synthetic air (80(O2)/20(N2)) and He atmospheres. For the analysis in air (60 mL min−1) the samples of 3.8 mg were used. The samples were placed in aluminium pans covered with pierced lid in order to enhance contact with synthetic air during the heating (at rate of 10 °C min−1). The measurements performed in He 6.0 atmosphere were with a gas flow rate of 55 mL min−1. The samples (2.5 mg) placed in Al2O3 pans were put inside the sample chamber, which was evacuated and purged prior each measurement. The measurements were conducted upon heating between room temperature and 1000 °C with a heating rate of 5 °C min−1. A measurement with empty pans (correction) was also made to eliminate background signals from TGA and MS. The MS detector was set to measure atomic mass units from 13 to 313. Videos of mineral precipitate formation in solutions were recorded using Colour CCD camera Panasonic WV-CP242EX, with OPTEM Zoom 70 optical lens.2.4. In situ XRD studies of brushite formation
Formation of the crystalline phases was monitored using a PANalytical X'Pert powder diffractometer (Ni-filtered Cu Kα radiation, step 0.02°, exposure time ∼1 s per step) over a 2-theta range at room temperature. One drop of each solution (calcium acetate and ammonium phosphate) were spilled on dry wooden slab placed on the horizontal platinum strip of the HTK-16N chamber. After this procedure, the specimen was fully wetted. The XRD data were collected at ambient pressure. The scans were recorded every 3.5 min. The water evaporation from the specimen was not controlled. Prior the measurements, the same solutions with the same amounts were dropped on a zero-background holder and kept in air until complete desiccation. The XRD patterns of resulting traces were recorded over the range of 10–80° and analysed using the PDF-4+ database (2022).2.5. Mechanical properties
The compressive strength tests were performed with a universal testing machine Hounsfield H10KS (Hounsfield Test Equipment Ltd, Redhill, UK) and a Qmat Professional software. The cell load was of 10 kN with a resolution of 0.1 N (load measurement accuracy: ±0.5% of indicated load from 2% to 100% capacity). The load speed was 20 mm min−1 (position measurement accuracy: 0.001 mm, speed accuracy: ±0.005% of set speed). To calculate density (ρ), the weight and the linear dimensions of prepared specimens were estimated using precision balance Mettler Toledo MS1003S (e = 0.01 g, d = 0.001 g) and DIGI-MET indicator (e = 0.01 mm, d = 0.001 mm) from Helios Preisser, respectively.3. Results and discussion
3.1. Surface morphology
The saturation of wood matrix, morphological features and elemental composition of the formed precipitate were assessed by performing SEM/EDS analysis of the cross-sectioned layers of the mineralised-wood blocks. The SEM/EDS images of the representative wood sample mineralised from the 0.5 M Ca(CH3COO)2/0.3 M NH4H2PO4 solution system are presented in Fig. 1. Examination of the internal cross-sectioned layers showed that the saturation of wood matrix is homogeneous with only a few filled wood cell lumina (Fig. 1(a)). The coprecipitated material exhibited plate-shaped morphology (Fig. 1(b) and (c)), indicating the crystallization of brushite mineral.19,32,41 | ||
| Fig. 1 SEM and EDS images showing (a) and (b) wood cell lumina filling with brushite mineral, (c) layered structure of brushite crystals, (d) EDS mapping (RGB colours assigned as Ca – red, P – green and C – blue; the examined area is marked in the image (a)), and (e) line EDS analysis and corresponding spectra showing the Ca and P presence within the cell walls (background of spectrum for element C is shadowed to emphasise cell wall position, and the green line at 20 μm shows position marked in the SEM image). | ||
SEM images with the EDS-based elemental mapping analysis of the same mineralised wood sample are shown in Fig. 1(d). Along with carbon, oxygen, phosphorus and calcium, aluminium and silver were also detected, and these last two were due to the sample holder and pre-sputtering, respectively. The average atomic percent (%) ratios of Ca to P of the precipitate within cell lumina was calculated to be around 1 (n = 5), which further suggests a formation of the mineral brushite. Various studies have repeatedly shown that phase composition of the CaP mineral strongly depends on the pH of solution where reaction takes place,42–44 and brushite is the most thermodynamically stable phase under acidic conditions (pH between 2 and 6.5).20,33,45 When 0.5 M Ca(CH3COO)2 (the measured pH = 8.47) and 0.3 M NH4H2PO4 (pH = 4.34) are mixed together, the CaP mineral formation resulted in a drop of the solution pH to 4.96, and allowing the precipitate maturation to proceed further, the pH value of the mixture after 30 min was 4.92. Furthermore, one shall also note that wood material has slightly acidic pH.46 As the pH of the solution 0.5 M Ca(CH3COO)2/0.3 M NH4H2PO4 system is acidic, the formation of the brushite mineral within wood matrix is expected. Furthermore, the EDS-based elemental mapping (Fig. 1(d)) showed homogeneous distribution of Ca and P over the entire wood matrix, i.e. precipitate formation within cell lumina and the cell wall. The EDS line-based analysis performed (Fig. 1(e)) also confirmed the Ca and P presence within the cell walls. A similar complete saturation of wood cell wall from aqueous solutions has recently been demonstrated.7 It was also observed that the Ca/P ratio measured from the cell wall was slightly higher compared to that of the mineral formed within the cell lumina, and this can be attributed to the excess of Ca ions that remain in the wood matrix unreacted. The minerals crystalized within wood cell lumina from the 1.0 M Ca(CH3COO)2/0.6 M NH4H2PO4, and 1.5 M Ca(CH3COO)2/0.9 M NH4H2PO4 systems exhibited similar morphology, only the saturation of wood matrix slightly increased with an increase in concentration of solutions (data not presented). The EDS elemental analysis of mineralised wood samples produced from the 1.0 M Ca(CH3COO)2/0.6 M NH4H2PO4, and 1.5 M Ca(CH3COO)2/0.9 M NH4H2PO4 systems showed that at% ratios of Ca/P of the mineral formed within cell lumina were approx. 1. Moreover, comparison of layers cut of the modified block surface with those that were cut from the middle of the block, revealed that the saturation of the outermost layers was slightly higher compared to the middle section. This can be ascribed to the higher concentration of reactive species on the wood surface. During the drying of impregnated wood (cycle-I) the migration of Ca(CH3COO)2 solution through the entire wood block occurs. Subsequently, when wood is treated with the solution containing phosphate ions (cycle-II impregnation) the reaction takes place instantly, and as more species are available to react, the higher saturation of the cell lumina at the surface of wood block is inevitable.
To visualise formation of precipitate, the reaction between 0.5 M Ca(CH3COO)2 and 0.3 M NH4H2PO4 solutions was recorded while observing the appearance of solution turbidity (ESI,† Video 1: reaction between 0.5 M Ca(CH3COO)2 and 0.3 M NH4H2PO4 solutions). The precipitation of CaP material was instantaneous after the drops of two solutions came into contact. Solution saturation was found to be strong. The precipitate formation was additionally inspected from the less concentrated solutions of 0.05 M Ca(CH3COO)2 and 0.03 M NH4H2PO4. For this system, however, no evidence of solution turbidity was observed (ESI†, Video 2: reaction between 0.05 M Ca(CH3COO)2 and 0.03 M NH4H2PO4 solutions). This indicates the presence of constituents in low abundance and insufficient activation energy to allow nucleation and mineral growth from the present environment. It is also known, that calcium ions precipitate with phosphate ions when the formed product exceeds the solubility product constants of calcium phosphate solid phases.47 The effects of the initial pH and the reactive ion concentrations on the mineralization and morphology of brushite particles during aqueous solution synthesis has been reported by Toshima et al.41 They showed that solution saturation has profound effect on crystal formation and its shape, i.e. no product was observed for at least 24 h from solutions of low concentrations and increasing the supersaturation ratio and/or initial pH value shifted the morphology from the liquid phase to the petal-like structure, and then to the plate-like structure.
An additional inspection of the precipitate formation from 0.5 M Ca(CH3COO)2/0.3 M NH4H2PO4 and 0.05 M Ca(CH3COO)2/0.03 M NH4H2PO4 systems was done under conditions when the reactive ions were coursed to react together within wood slabs of 100 μm thick. The SEM/EDS analysis of wood slabs mineralised from solutions of lower concentrations showed marginal precipitate formation within the wood cell lumina, and this can be attributed to the increased concentration of reactive species when solvent from wood matrix evaporates. The solid precipitate formed within cell lumina exhibited similar surface morphology, i.e. plate-shaped crystals, compared to that of precipitate formed from the solutions of higher concentrations (data not presented).
To evaluate the stability of the precipitated mineral and its intercalation within wood matrix, mineral dissolution tests were performed. The wood slabs mineralised from the 0.5 M Ca(CH3COO)2/0.3 M NH4H2PO4 and 0.05 M Ca(CH3COO)2/0.03 M NH4H2PO4 systems were examined. The SEM/EDS analysis revealed that precipitate remained within dried wood matrix after the leaching. The representative SEM/EDS analysis data of the “leached” wood are presented in Fig. S1 of ESI.† Further morphological examination of the samples (slabs derived from 0.5 M Ca(CH3COO)2/0.3 M NH4H2PO4 system) revealed regions of precipitate exhibiting slightly different morphology (Fig. 2). Though the overall morphology of plate-like crystals remained unchanged within wood matrix (Fig. 2(a) and (b)), the regions of smoothly filled cell lumina as well as cell walls having smoothened “cementitious” coverage were also observed (Fig. 2(c) and (d)). This might be explained by considering the motion of ions in the liquid. As demonstrated above, the instantaneous formation of the mineral take place when solutions with reactive species come into contact. This leads to the change in concentration in regions, as the flux of ions through the liquid to the slab boundaries is impeded. As a result, different rate of matrix mineralization is induced. Furthermore, the removal of excess ions during the leaching might invoke a partial dissolution of the mineral, that could lead to a spatial rearrangement of the formed material. As noted previously, an extensive number of studies have showed that concentration of reactive species, pH, temperature, and other variables affect the CaP phase and morphology.19,32,35,37,48–50
 | ||
| Fig. 2 FE-SEM images of the precipitate formed within Scots pine slabs (100 μm) showing different morphology after the leaching tests: (a and b) plate-like laminated crystals, and (c and d) “smoothened” mineral. | ||
3.2. In situ XRD studies of material formation
To monitor mineral crystallization and crystal growth the in situ XRD study was carried out. The XRD pattern of the powder precipitated within wood matrix from the mixture of a 0.5 M Ca(CH3COO)2 solution and a 0.3 M NH4H2PO4 solution is shown in Fig. 3. It was found from the analysis of the pattern that all the observed diffraction peaks belong to the crystalline phase of calcium phosphate hydroxide hydrate, CaHPO4 2H2O (or CaPO3(OH)·2H2O) (ICDD card no. 00-011-0293 (ref. 51)). No other crystalline phase has been detected. As seen from the inset in Fig. 3, the background in not really flat, which suggests amorphous phase(s) likely formed as a result of drying of the solution remained after precipitation of calcium phosphate.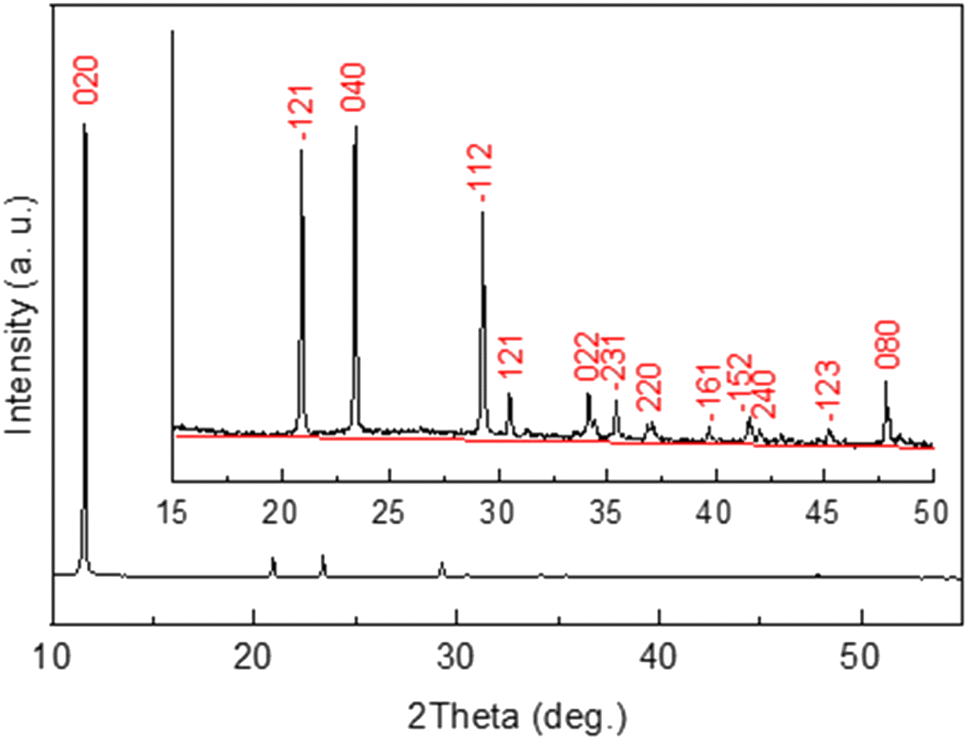 | ||
| Fig. 3 XRD pattern of CaHPO4·2H2O precipitated from the mixture of a 0.5 M Ca(CH3COO)2 solution and a 0.3 M NH4H2PO4 solution and dried at room temperature on a zero-background holder. Inset shows the magnified range of the pattern with the most intense diffraction reflections indexed based on the ICDD card no. 00-011-0293. | ||
The diffraction peak ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 12 was chosen to monitor crystallization of CaHPO4·2H2O since it was intense enough and less dependent on possible wooden slab deformation than the peaks in the lower 2-theta range of the XRD pattern.
12 was chosen to monitor crystallization of CaHPO4·2H2O since it was intense enough and less dependent on possible wooden slab deformation than the peaks in the lower 2-theta range of the XRD pattern.
Fig. 4 shows the time evolution of the diffraction peak 12. The peak is detectable in about 8 min after the precipitation experiment starts. It should be stressed here that a white precipitate is observed literally in few seconds after the solutions are mixed (Video 1 in ESI†). Although the amount of the precipitated phase looked sufficient to be detected by XRD already in the first scan, no peak was observed suggesting that the first-minutes precipitate is mainly amorphous. One can see from the figure that the intensity of the peak is not a monotonous function of time.
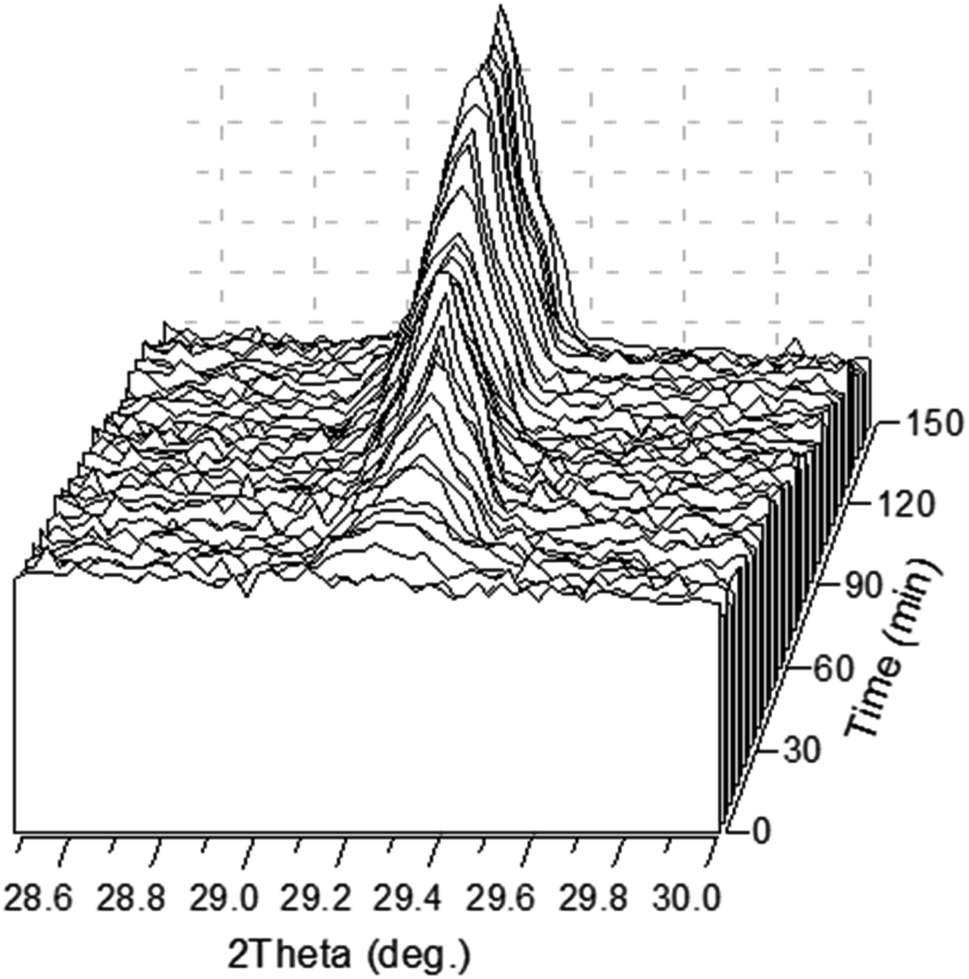 | ||
| Fig. 4 3D representation of the XRD pattern range that includes the 12 diffraction reflection of CaHPO4·2H2O precipitated from a mixture of 0.5 M Ca(CH3COO)2 solution and a 0.3 M NH4H2PO4 solution in a 100 μm wood slab as a function of crystallization time. | ||
The time dependence of the integral intensity of the diffraction peak 12 is depicted in Fig. 5. As seen from the figure, there are two ranges, which can be associated with an increase of the crystalline phase amount. To analyse this feature, one should take into account two parallel processes, namely precipitation followed by crystallization and evaporation of water from the reaction solution. After first 40 min, precipitation and crystallization considerably slow down because concentration of the calcium- and phosphate-containing species in the solution becomes lower. The process stagnates until concentration of the remaining species rises due to evaporation of the rest of solution. The last minutes (between about 100 and 120 min since the beginning), when the solution dries, is a time of active crystallization.
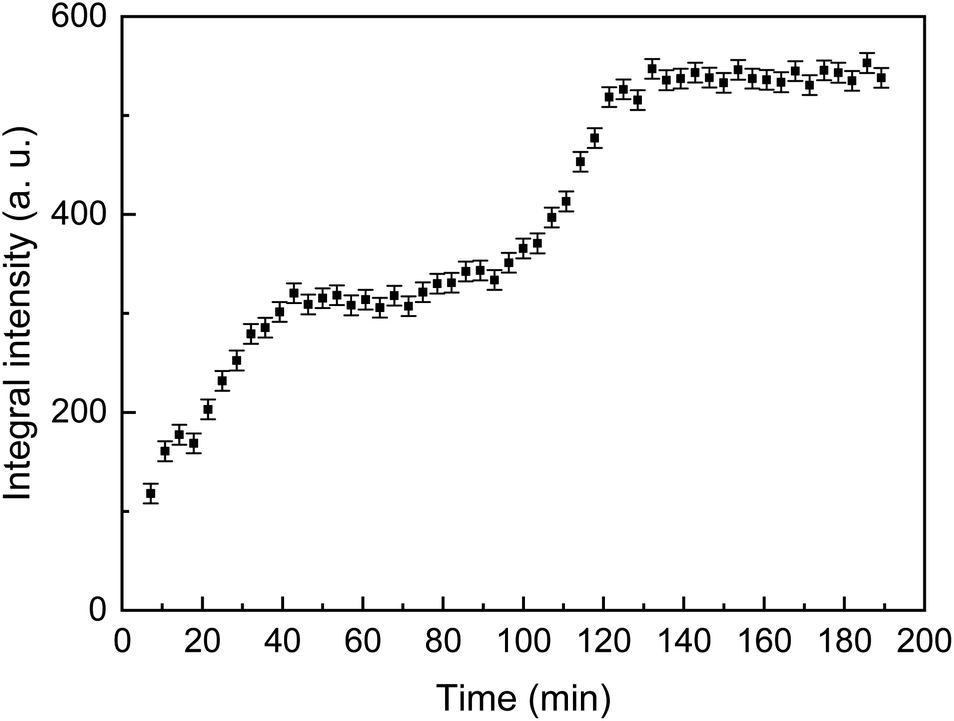 | ||
| Fig. 5 The integral intensity of the 12 diffraction reflection of CaHPO4·2H2O as a function of the crystallization time. | ||
After about 150 min, a considerable regular shift of the peak was observed without any change of intensity which indicated deformation (bending) of the wood slab due to a completing desiccation.
3.3. Structure evaluation via spectroscopy
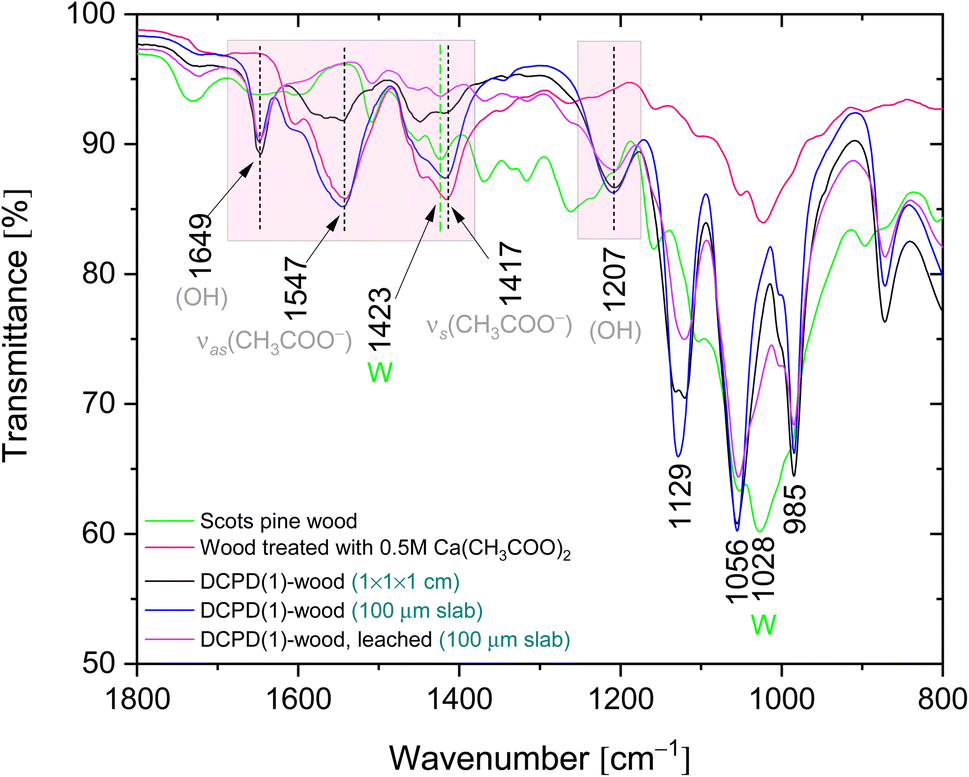
Chemical composition of the mineralised wood as well as brushite mineral integration within wood matrix was evaluated using FTIR analysis. IR spectra of the untreated wood and brushite-mineralised wood are presented in Fig. 6, and the assignment of the bands for modified wood is summarised in Table 1. The spectrum from the untreated Scots pine wood showed typical absorption bands that are assigned to the main chemical components of wood, i.e. cellulose, hemicellulose and lignin.7,52 The bands located in 2940–2840 cm−1 region originated from the C–H asymmetric stretching in methyl and methylene groups of aliphatic hydrocarbons.7 In the 1750–1540 cm−1 region, the lignin gives characteristic IR absorption bands. The carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching vibrations of unconjugated ketones and esters were present at 1740–1710 cm−1 (maximum at 1735 cm−1), and conjugated carbonyl structures of lignin absorbed radiation in 1690–1620 cm−1. At lower wavenumbers, such as 1508 cm−1 (the aromatic skeletal vibrations of lignin), 1450 cm−1 (CC, C–H, O–H in plane deformation, –CH3 asymmetric bending (lignin)) and 1419 cm−1 (C–H aromatic skeletal vibrations (lignin), –CH2 bending deformation (cellulose)) were also present. The fingerprint 1150–950 cm−1 region exhibited bands due to various polysaccharide vibrations.7,52,53
O) stretching vibrations of unconjugated ketones and esters were present at 1740–1710 cm−1 (maximum at 1735 cm−1), and conjugated carbonyl structures of lignin absorbed radiation in 1690–1620 cm−1. At lower wavenumbers, such as 1508 cm−1 (the aromatic skeletal vibrations of lignin), 1450 cm−1 (CC, C–H, O–H in plane deformation, –CH3 asymmetric bending (lignin)) and 1419 cm−1 (C–H aromatic skeletal vibrations (lignin), –CH2 bending deformation (cellulose)) were also present. The fingerprint 1150–950 cm−1 region exhibited bands due to various polysaccharide vibrations.7,52,53
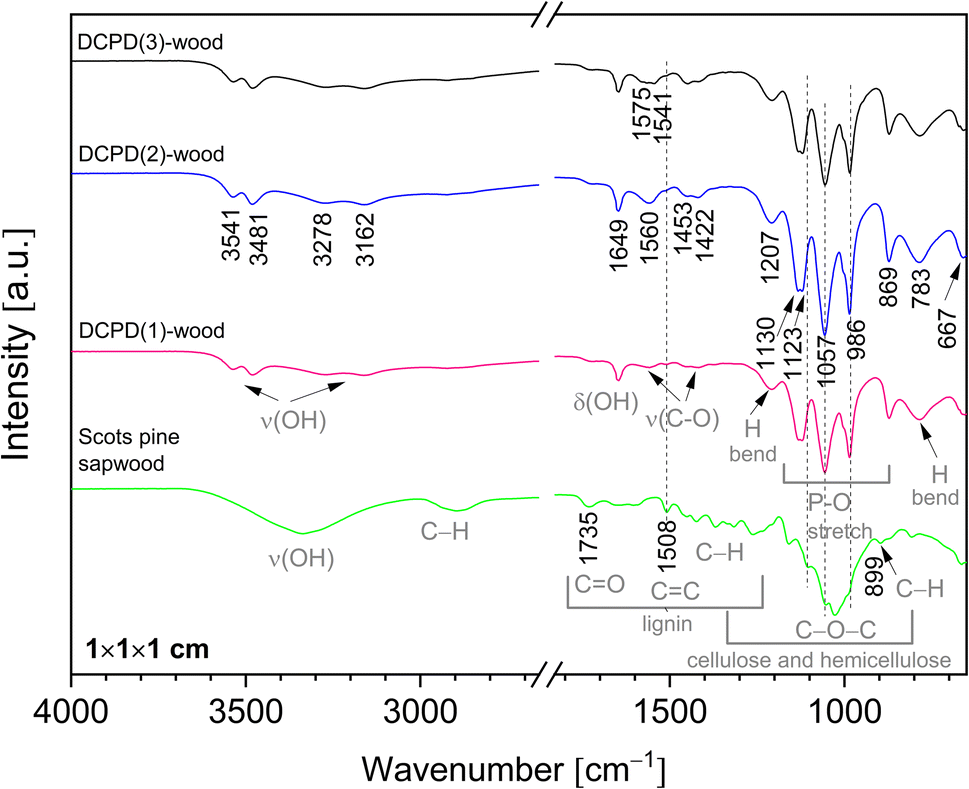 | ||
| Fig. 6 FTIR spectra of the untreated Scots pine sapwood and the brushite-mineralised wood (‘dash’ black line indicates position of the band maxima for the untreated wood). | ||
| Frequency (in cm−1) | Vibrating species | Reference |
|---|---|---|
| 3541, 3481 | H2O, stretch, (crystal water) | 27, 29, 56, 58 and 59 |
| 3278, 3162 | H2O, stretch, (crystal water) | 27, 29 and 56 |
| 1649 | H2O bending | 27, 29, 56, 58 and 59 |
| 1571, 1541 | COO− group, ν, antisymmetric C–O stretching mode (acetate) | 60 and 61 |
| 1453, 1422 | COO− group, ν, symmetric C–O stretching mode (acetate) | 60 and 61 |
| 1207 | H-in-plane bending (HPO4 group) | 27, 29, 56 and 57 |
| 1123 | P–O stretch (HPO4 group) | 27, 29, 56 and 57 |
| 1057 | P–O stretch (HPO4 group) | 27, 29, 56 and 57 |
| 986 | P–O stretch (HPO4 group) | 27, 29, 56 and 57 |
| 869 | P–O stretch (HPO4 group) | 27, 29, 56 and 57 |
| 783 | H-out-of-plane bend (HPO4 group), P–OH | 27, 29, 56 and 57 |
| 667 | H2O libration | 27, 56, 58 and 59 |
IR spectra of the brushite-mineralised wood exhibited different spectral features. The clearly distinguishable absorption peaks observed at ∼1207, 1125 (appearing as doublet at 1130, 1123 cm−1), 1057, 986 and 869 cm−1 are characteristic of brushite: the vibrations that occur due to the movement of H atom of the HPO4 group are present at 1207 cm−1, while the P–O stretching vibrations of HPO4 group gives bands in the 1150–850 cm−1 region.27,54–57 The broad band present at ∼783 cm−1 could be assigned to overlapped bands attributed to the lignin (C–H out of-plane bending vibrations) and HPO4 group (P–O–H out of plane bending).7,56 In the spectra of mineralised wood, water vibrations, i.e. internal vibrations and libration modes, were also observed. The broad absorption bands in the region of 3600–3150 cm−1 (appearing as doublets at 3541, 3481 cm−1 and 3278, 3162 cm−1) are assigned to the O–H stretching modes of the crystal water molecules.27,29,56,58,59 The band detected at 1649 cm−1 was assigned to the H–O–H bending vibrations, and the broad band at ∼667 cm−1 to the H2O librational mode.27,56,58,59 Furthermore, the bands located in the 1610–1520 cm−1 and 1480–1380 cm−1 regions could be assigned to the C–O stretching modes of carboxyl (COO−) groups of Ca(CH3COO)2 molecules.60,61 The FTIR spectrum of wood sample treated solely with the 0.5 M Ca(CH3COO)2 solution (Fig. 7, and ESI Fig. S2.†) shows distinctive bands at 1551 cm−1 (1604 cm−1 sh) and 1417 cm−1 (1449 cm−1 sh) that are due to the asymmetric ν(C–O) and symmetric ν(C–O) modes of acetate, respectively.60,61
 | ||
| Fig. 7 FTIR spectra of the Scots pine sapwood: untreated, impregnated from solution of 0.5 M Ca(CH3COO)2, and mineralised with brushite before and after the leaching (W – wood, ‘dash dot’ green line indicates position of the band maxima for the untreated wood, ‘short dash’ black line indicates position of the band maxima for the treated wood). | ||
Brushite-wood composites after the dissolution tests were also examined. The IR spectrum of leached DCPD(1)-wood sample (Fig. 7) shows that bands assigned to the C–O vibrations of acetate ions diminish, and only the bands belonging to the C–H groups of lignin skeletal vibrations as well as –CH2 groups of cellulose (at ∼1423 cm−1) are present. The absorption bands of the OH and PO4 groups remained in the IR spectrum, and no shift in wavelength or band shape changes was observed thereby confirming mineral brushite stability and firm intercalation within Scots pine wood matrix. This agrees well with the XRD and SEM/EDS data affirming that under current experimental conditions the formation of CaHPO4·2H2O phase within matrix was preferable. One can assume that the pH of solution likely was the defining factor of brushite coprecipitation. This can be corroborated with different studies that have repeatedly shown that calcium phosphate powder processing from solutions under acidic pH conditions leads to the brushite or monetite phase formation.62,63 The full region of the IR spectra recorded of the mineralised wood derived from the mixture of 0.5 M Ca(CH3COO)2 and 0.3 M NH4H2PO4 solutions before and after the leaching is shown in ESI Fig. 3.†
3.4. Thermal behaviour
The combustion and pyrolysis of the mineralised wood was also studied. The TG/DSC/DTG curves of the untreated wood and DCPD(1)-wood composite are presented in Fig. 8. The results show that thermal decomposition in oxidizing conditions for the untreated wood progress in three main steps leaving about 6% carbonaceous residue at ∼500 °C. The first weight loss of about 5% was observed at temperatures up to 100 °C (maximum at 67 °C, DSC curve) and was attributed to the evaporation of a surface-bound water. An additional weight loss of 55% occurred upon further heating up to 350 °C following strong exothermic peak at ∼340 °C. This loss could be attributed to the degradation of hemicelluloses and cellulose, and the removal of volatile components such as CO, CO2, H2O and low molecular weight hydrocarbons. Hemicellulose has an amorphous structure, whereas cellulose is semi-crystalline polymer and this leads to its higher thermal stability.64 Heating to 515 °C produced an additional 34% weight loss and simultaneously exothermic reaction (DSC curve) with a maximum at 470 °C which is mainly related to the decomposition of lignin. Lignin is more thermally stable than hemicellulose and cellulose due to its aromatic structure; the decomposition starts slowly over a wider temperature range (150–900 °C). The decomposition of the untreated wood was completed at ∼500 °C.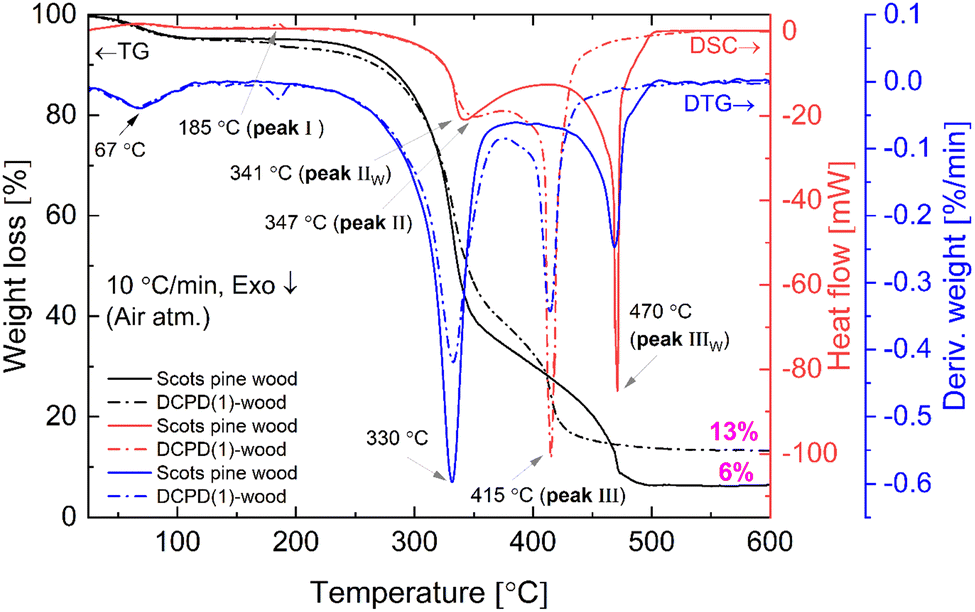 | ||
| Fig. 8 TG/DSC and DTG curves (oxidative atmosphere) of the untreated and DCPD(1)-mineralised Scots pine sapwood. | ||
Brushite-mineralised wood exhibited different thermal behaviour. The TG/DSC/DTG curves of the DCPD(1)-, DCPD(2)- and DCPD(3)-wood samples showed that thermal degradation of mineralised wood in an oxidative atmosphere proceeds in four steps (Fig. 8 and ESI Fig. S4†). One can observe that heating the samples up to 200 °C resulted in two weight-loss events. The first ∼5% weight loss (maximum at ∼67 °C, DTG curve) was observed up to 100 °C (release of adsorbed water) and the second small weight loss (up to 3%) appears in the temperature range of 170–200 °C with a small endothermic event (maximum at ∼186 °C, DSC curves). The latter event was attributed to the removal of water chemically-bonded to wood matrix as well as to initial dehydration of brushite molecules. In fact, literature shows that brushite starts to dehydrate at around 80 °C resulting into a gradual transformation of brushite to monetite (CaHPO4) and formation of an amorphous phase.65,66 The third weight loss (∼48%, 47% and 38% for the DCPD(1)-, DCPD(2) and DCPD(3)-wood, respectively) takes place up to 375 °C, whilst the last weight loss appears at ∼50 °C lower temperature (between 375 °C and 440 °C) compared to the untreated wood. The changes in thermal behaviour were most likely due to the alteration of wood material induced by the acidic medium during the treatment. Furthermore, the residual mass of the material after the burn-off was 7%, 11% and 24% higher for the DCPD(1)-, DCPD(2) and DCPD(3)-wood samples compared to that of the untreated wood indicating that treatment had a significant effect on the mass change. Moreover, after releasing of absorbed water at ∼67 °C another dehydration occurred with enthalpy values 15.2 mJ mg−1 (peak I, 185 °C) for DCPD(1)-wood, 22.2 mJ mg−1 (peak I, 186 °C) for DCPD(2)-wood, and 61.4 mJ mg−1 (peak I, 187 °C) for DCPD(3)-wood. The untreated wood exhibited no such dehydration at this temperature (Fig. 8). The huge exothermic reactions on DSC curves (double peak starting at approx. 250 °C) also remain coupled with main mass decrease on TG curves. High amount of heat is released during this burning process, i.e. 5.13 J mg−1 (peak II + peak III) for DCPD(1)-wood, 5.26 J mg−1 for DCPD(2)-wood, 4.13 J mg−1 for DCPD(3)-wood, and 4.96 J mg−1 (peak IIW + peak IIIW) for the untreated wood.
To comprehend the thermal degradation and gain additional information on the material dehydration, mineral phase transition as well as volatile products released during the degradation and final residues the TG/DSC/DTG and TG/DTA coupled with MS analyses were performed for the brushite-mineralised wood samples under pyrolysis conditions. The TG/DTG and DSC curves of the DCPD(1)-wood composite (ESI Fig. S5†), show that thermal decomposition under pyrolysis conditions progresses in four main steps. The first weight loss was observed at temperatures up to 100 °C, and a second weight loss occurred upon further heating the samples up to 200 °C (a small endothermic event with a peak max. at 188 °C). These were attributed to the removal of water and to the dehydration of mineral present within wood matrix. The third weight loss of ∼55% for DCPD(1)-wood sample takes place up to 380 °C and can mainly be assigned to the decomposition of large complex carbohydrate molecules, starting with hemicelluloses around 200 °C, and the removal of CO and CO2 as the major components as well as H2, CH4, C2H4 and H2O as the minor components.7,67 The cellulose pyrolysis occurs at higher temperatures (315–400 °C), while lignin, as mentioned previously, decompose slowly over a wider temperature range (150–900 °C).67,68 In addition, the volatile products like NH3, H2O and CO2 can evolve due to the thermal decomposition of initial inorganic salts.69,70 Heating to 855 °C led to 18% decrease in weight and this was attributed to the further degradation of organic components of wood and the formation of char (a solid residue left after the devolatilization is complete). The phase transformation within the ceramic also proceeds in this temperature region and such events can be seen with small changes in the DCS curve (around 550 °C and 700 °C). The loss of lattice water associated with formed mineral also appears as CaHPO4 upon pyrolysis at higher temperatures transforms into pyrophosphate (Ca2P2O7) and H2O.71 The remaining solid residue, consisting of the carbon-rich and calcium phosphate-based materials, was 17%. A similar thermal behaviour was observed for the brushite-wood composites prepared from solutions of higher concentration (TG/DTG and DSC curves are shown in ESI Fig S6 and S7†) and showed a correlation between initial concentration of the solution and the mineral content, i.e. the remaining solid residue after the burn-off at 855 °C was ∼21% and 28% for DCPD(2)-wood and DCPD(3)-wood samples, respectively (inset in ESI Fig. S5†). In addition, the thermal behaviour of the surface layer of the mineralised wood blocks was also studied. The TG/DTG and DSC curves exhibited similar features and an average residue mass of the DCPD(1)-, DCPD(2)- and DCPD(3)-wood after the burn-off at 855 °C was 24.13% ± 3.15 (data not presented). This result indicates a complete saturation of the Scots pine sapwood blocks with the initial solutions during the cyclic-treatment process and relevantly homogeneous formation of the solid precipitate within the entire wood matrix.
Evolution of volatile products during the thermal decomposition of the brushite-mineralised wood was further evaluated by TG/DTA coupled with MS. Fig. 9 shows the MS curves recorded of the untreated and modified wood, and the corresponding TG/DTA curves are presented in ESI Fig. S8.† The MS ion curves for N2 (m/z = 14), H2O (m/z = 17–18), O2 (m/z = 32), C2O (m/z = 39–40), and CO2 (m/z = 44) show peaks that correspond to the individual TGA steps. It can be observed that for the unmodified Scots pine wood sample the major evolution of H2O, O2 and CO2 gases occurs between 250 and 390 °C, and this, as mentioned earlier, can be attributed to the degradation of wood constituents. The MS curves for the brushite-mineralised wood samples exhibited different behaviour. The first significant peak in the MS curves was observed at ∼190 °C, and it can be assigned to the removal of ammonia and water (NH3+/H2O, m/z = 17–18) due to the degradation of ammonia-containing compounds, bound water to the wood matrix as well as to the loss of the crystal water from the brushite molecules. The second peak was observed in the region of 250–380 °C (with maximum at around 350 °C). This could be attributed to the decomposition of the actual wood material. Gas evolution events are consistent with the events observed in the TGA/DTA curves (ESI Fig. S8†). The third broad peak in the MS curve of NH3+/H2O was detected when heating the materials up to 500 °C and was assigned to the further degradation of the inorganic additives present within the wood matrix. One can also observe that the ratio of peak I (at 190 °C) and peak II (∼350–355 °C) (Fig. 9) is significantly changing, i.e. the intensity of peak I is increasing with an increase in concentration of inorganic additives. This suggests that initial inorganic chemicals used to treat wood and subsequently formed mineral within wood matrix might be suitable additives aiming to alter thermal degradation behaviour of wood material. The other ionic/molecular species detected in the MS spectra of brushite-mineralised wood exhibited similar evolution patterns. Additionally, the small peaks in the MS curves corresponding to the chemical species of higher molecular weight (m/z = 55, and m/z = 57) at 355 °C were also observed; these could be due to the organic phase products released during the wood degradation. The residue mass values of the mineralised wood samples studied under different experimental conditions (i.e. heating rate and atmosphere over the sample) were similar (Fig. 8; and S5 (inset)†). These results indicate that the mineralization of wood when an insoluble material is formed within wood matrix via wet-chemistry route might be a worthwhile strategy seeking both low-environmental impact and enhanced-performance wood materials with tailored properties.
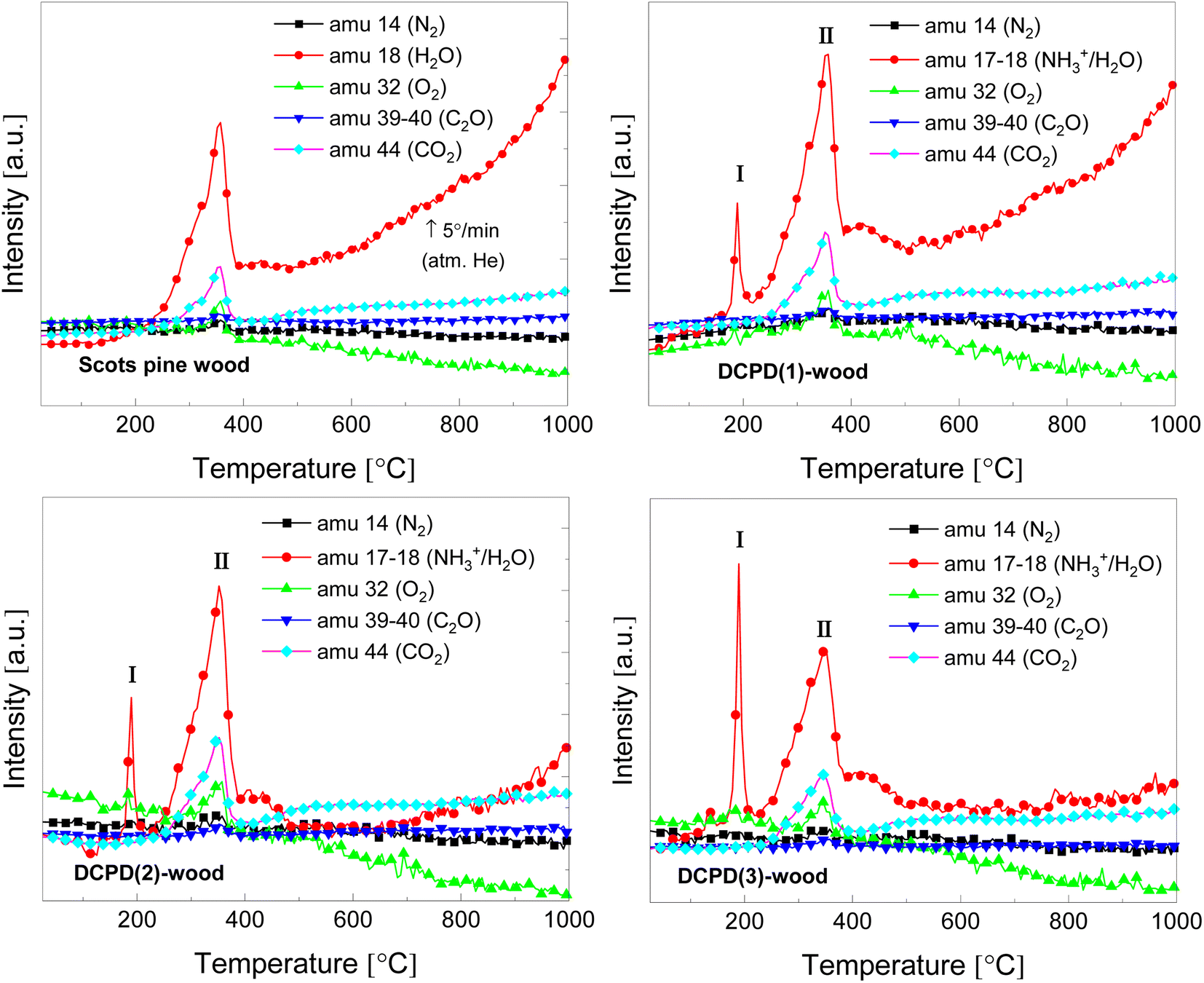 | ||
| Fig. 9 MS curves of the untreated and the brushite-mineralised Scots pine sapwood samples (amu – the atomic mass unit). | ||
3.5. Mechanical properties
The impact of brushite on the mechanical properties of wood was evaluated by determining compressive and bending strengths. Compressive strength properties of the brushite-mineralised wood were investigated in both perpendicular and parallel to the fibre directions (Fig. 10(a and b)). The density of wood increased from 499 to 534, 580 and 634 kg m−3 for DCPD(1)-wood, DCPD(2)-wood and DCPD(3)-wood specimens, respectively. For compressive strength in the perpendicular to the fibre direction, the strength increased by 20.4%, from 4.9 MPa for untreated wood to 5.9 MPa for DCPD(1)-wood and DCPD(2)-wood specimens. For DCPD(3)-wood specimens the compressive strength was 4.6 MPa. For compression in the parallel to the fibre direction, the strength decreased by 54.0% from 42.1 MPa to 27.3 MPa for DCPD(1)-wood specimens, but slightly increased to 43.7 MPa (by 3.8%) and 44.5 MPa (by 5.7%) for DCPD(2)-wood and DCPD(3)-wood specimens, respectively. The compressive strength test showed that mineralization with brushite did not induce a significant increase or decrease in strength.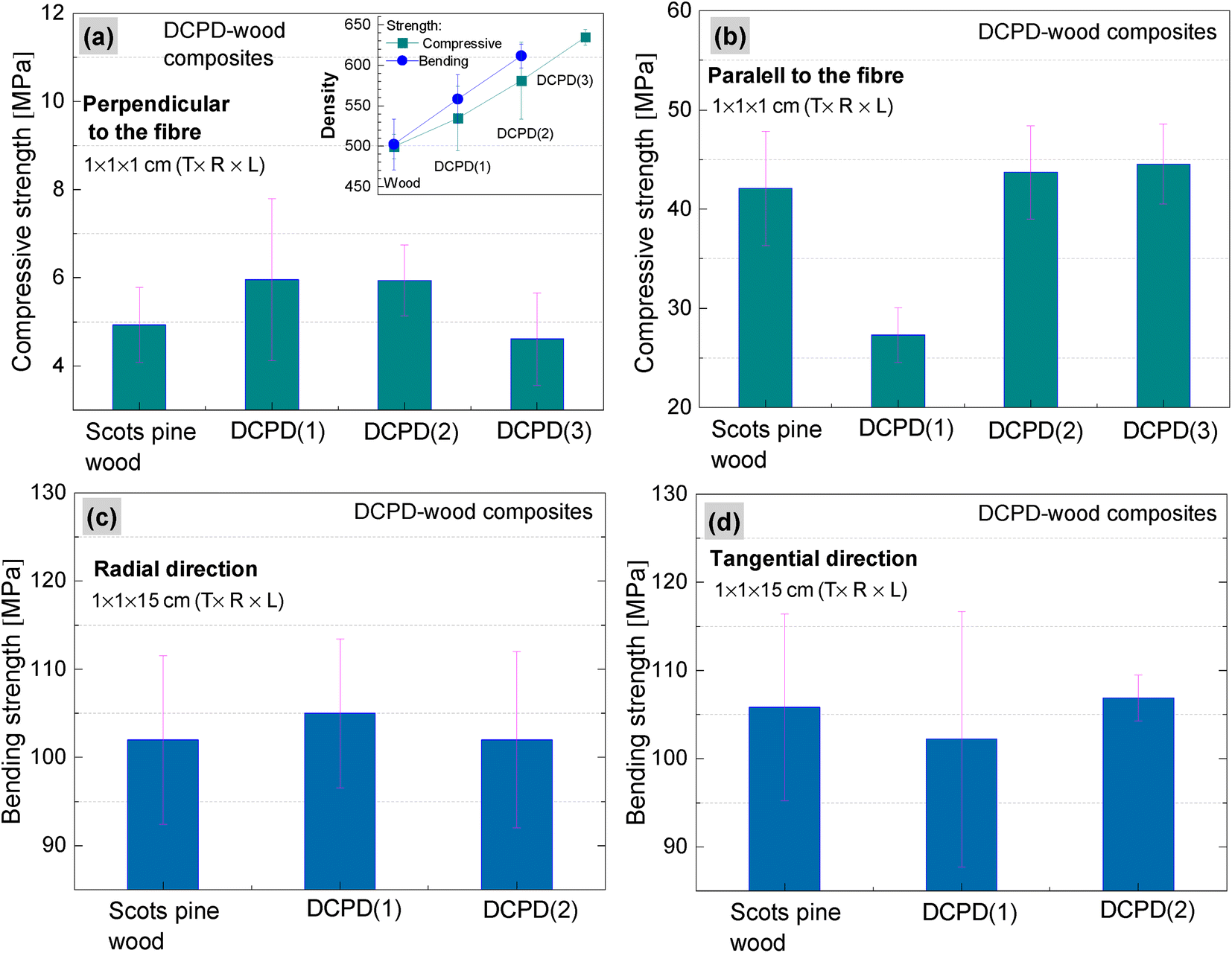 | ||
| Fig. 10 Mechanical properties: (a and b) compressive strength and (c and d) bending strength of the untreated and the brushite-mineralised Scots pine sapwood samples (inset: the average density values of the samples). | ||
Regarding the bending strength (Fig. 10(c and d)), very similar values were obtained for the mineralised wood as compared to the untreated wood. The bending strength in the radial direction was 102 MPa for the untreated wood, and 105 MPa and 102 MPa for DCPD(1)-wood and DCPD(2)-wood specimens, respectively. The bending strength in tangential direction was 105 MPa for the untreated wood and 102 MPa and 107 MPa for the DCPD(1)-wood and DCPD(2)-wood specimens, respectively. Guo et al. reported struvite-mineralised wood where process included a vacuum impregnation of the KH2PO4 and MgSO4 salt solution into the cavities of Norway Spruce (Picea abies) wood specimens followed by ammonium fumigation, which ensured mineral struvite precipitation.17 The reported studies of mechanical properties showed that mineralization had a minor effect on mechanical properties, i.e. compressive strength correlated with the loading direction, while the three-point bending test revealed a decrease in the strength.17 Such changes in values of compressive and bending strengths for the brushite-mineralised wood might be attributed to the mechanical properties of the actual ceramic material.71–75 CaP ceramics are brittle materials, and the compressive strength ranges significantly, depending on their composition, crystallinity, particle and crystal sizes and/or porosity, and these are strongly influenced by the material processing conditions.71,76–78 On the other hand, wood is anisotropic material with complex structure. The inherent microstructure of wood, density as well as moisture content are highly significant factors in achieving homogeneous saturation of matrix with coprecipitated ceramic material. Thus, structure-sensitive properties of ceramics as well as synergistic effects of materials with conflicting properties, i.e. ceramics and wood, will impact the mechanical behaviour of ceramic-reinforced wood material.
Furthermore, the formation of precipitate from the solutions of higher concentrations proceeds instantly and leads to the formation of higher mineral amount on the wood-block surface during the treatment process (cycle-II impregnation) compared to those blocks mineralised from solutions of lower concentrations. As a result, fewer reactive species might diffuse into the wood block. As mentioned above, the SEM analysis showed that the marginal change in filling of wood cell lumina was observed within the internal layers cut from the middle of the wood blocks produced from solutions of different concentrations. This, subsequently, induce a minor changes on mechanical properties for brushite-mineralised wood produced from the 1.0 M Ca(CH3COO)2/0.6 M NH4H2PO4, and 1.5 M Ca(CH3COO)2/0.9 M NH4H2PO4 systems compared to the wood mineralised from the 0.5 M Ca(CH3COO)2/0.3 M NH4H2PO4 system. Therefore, possibilities to increase the degree of saturation of the entire wood block with homogeneous precipitation of the mineral and how such a process should be up-scaled for large-scale fabrication, are needed to be further studied.
Overall results imply that proposed wet-chemistry processing route to CaP-based-wood composites opens possibilities for the upscaling, due to its simplicity, low-cost and established industrial wood treatment techniques that are available. This further suggests that if extremely benign and technologically feasible methods, when stable inorganic materials are crystalized within wood cell-wall cavities can be developed, the pristine structure and durability of final-wood product can be retained with relatively good confidence. This is especially attractive for the construction sector.
4. Conclusions
Scots pine sapwood was successfully mineralised with CaHPO4·2H2O (brushite) via wet-chemistry processing route from aqueous solutions of inorganic salts. It was revealed from in situ XRD studies conducted to monitor the formation of brushite phase that the active crystallization of the mineral in the 0.5 M Ca(CH3COO)2 and 0.3 M NH4H2PO4 system proceeds between 100 and 120 min since the beginning of reaction. The SEM/EDS analysis showed cell wall saturation with precipitate and the formation of plate-like laminal crystals within wood cell lumina before and after the dissolution. The results suggest that initial ion concentrations and medium pH were the defining factors for the formation of the plate-like brushite crystals within wood matrix. The FTIR analysis performed on the mineralised wood before and after the dissolution tests further confirmed an adequate stability of the mineral intercalated within wood matrix. Thermogravimetric analysis and mass spectroscopy revealed significant increase in the residue mass after the burn-off under pyrolysis conditions, while the identification of evolved gaseous species showed relatively enhanced water release from the mineralised wood thereby implying the material fire retardancy properties. Mechanical property measurements showed similar strength for brushite-wood produced from solutions of different concentrations as compared to those of the untreated wood. The results indicate that proposed simple and viable wet-chemistry processing route has a potential in producing environmentally friendly CaP-mineralised wood composites that could be used in harsher environments like elevated moisture or as fire-retardant materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Edita Garskaite acknowledge the Swedish Research Council FORMAS (project “Utilization of solid inorganic waste from the aquaculture industry as wood reinforcement material for flame retardancy” (grant no. 2018-01198)) and Brandforsk foundation (project no. 2021-13) for the financial support of this work. The research done in the University of Aveiro was supported by the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020, UIDP/50011/2020 & LA/P/0006/2020, financed by national funds through the FCT/MEC (PIDDAC).References
- M. H. Ramage, H. Burridge, M. Busse-Wicher, G. Fereday, T. Reynolds, D. U. Shah, G. Wu, L. Yu, P. Fleming, D. Densley-Tingley, J. Allwood, P. Dupree, P. F. Linden and O. Scherman, Renewable Sustainable Energy Rev., 2017, 68, 333–359 CrossRef.
- C. Howard, C. C. Dymond, V. C. Griess, D. Tolkien-Spurr and G. C. van Kooten, Carbon Balance Manage., 2021, 16, 9 CrossRef PubMed.
- L. A. Berglund and I. Burgert, Adv. Mater., 2018, 30, 1704285 CrossRef PubMed.
- P. Bonfield, MRS Bull., 2008, 33, 454–456 CrossRef.
- V. A. Dakwale, R. V. Ralegaonkar and S. Mandavgane, Sustain. Cities Soc., 2011, 1, 211–218 CrossRef.
- I. Burgert, E. Cabane, C. Zollfrank and L. Berglund, Int. Mater. Rev., 2015, 60, 431–450 CrossRef CAS.
- E. Garskaite, O. Karlsson, Z. Stankeviciute, A. Kareiva, D. Jones and D. Sandberg, RSC Adv., 2019, 9, 27973–27986 RSC.
- C.-M. Popescu and A. Pfriem, Fire Mater., 2020, 44, 100–111 CrossRef CAS.
- E. Sjöström, in Wood Chemistry, ed. E. Sjöström, Academic Press, San Diego, 2nd edn, 1993, pp. 1–20, DOI:10.1016/B978-0-08-092589-9.50005-X.
- E. Toumpanaki, D. U. Shah and S. J. Eichhorn, Adv. Mater., 2020, 2001613 Search PubMed.
- M. F. Ashby and D. R. H. Jones, in Engineering Materials 2, ed. M. F. Ashby and D. R. H. Jones, Butterworth-Heinemann, Boston, 4th edn, 2013, pp. 493–508, DOI:10.1016/B978-0-08-096668-7.00029-2.
- B. Mahltig, C. Swaboda, A. Roessler and H. Böttcher, J. Mater. Chem., 2008, 18, 3180–3192 RSC.
- C. Namyslo Jan, E. Kaufmann Dieter, C. Mai and H. Militz, Holzforschung, 2015, 69, 595 CrossRef.
- V. Merk, M. Chanana, T. Keplinger, S. Gaan and I. Burgert, Green Chem., 2015, 17, 1423–1428 RSC.
- V. Merk, J. K. Berg, C. Krywka and I. Burgert, Cryst. Growth Des., 2017, 17, 677–684 CrossRef CAS.
- H. Guo, M. Luković, M. Mendoza, C. M. Schlepütz, M. Griffa, B. Xu, S. Gaan, H. Herrmann and I. Burgert, ACS Appl. Mater. Interfaces, 2019, 11, 5427–5434 CrossRef CAS PubMed.
- H. Guo, M. Özparpucu, E. Windeisen-Holzhauser, C. M. Schlepütz, E. Quadranti, S. Gaan, C. Dreimol and I. Burgert, ACS Sustainable Chem. Eng., 2020, 8, 10402–10412 CrossRef CAS.
- N. Eliaz and N. Metoki, Materials, 2017, 10(4), 334 CrossRef PubMed.
- E. Boanini, F. Silingardi, M. Gazzano and A. Bigi, Cryst. Growth Des., 2021, 21, 1689–1697 CrossRef CAS.
- K. Ishikawa, in Comprehensive Biomaterials II, ed. P. Ducheyne, Elsevier, Oxford, 2017, pp. 368–391, DOI:10.1016/B978-0-12-803581-8.10170-5.
- R. Z. LeGeros, S. Lin, R. Rohanizadeh, D. Mijares and J. P. LeGeros, J. Mater. Sci.: Mater. Med., 2003, 14, 201–209 CrossRef CAS PubMed.
- S. E. Lobo and T. L. Arinzeh, Materials, 2010, 3, 815–826 CrossRef CAS.
- L. Sinusaite, I. Grigoraviciute-Puroniene, A. Popov, K. Ishikawa, A. Kareiva and A. Zarkov, Ceram. Int., 2019, 45, 12423–12428 CrossRef CAS.
- M. Mathew and S. Takagi, J. Res. Natl. Inst. Stand. Technol., 2001, 106, 1035–1044 CrossRef CAS PubMed.
- E. L. Prien and E. L. Prien, Jr, Am. J. Med., 1968, 45, 654–672 CrossRef CAS PubMed.
- M. A. Miller, M. R. Kendall, M. K. Jain, P. R. Larson, A. S. Madden and A. C. Tas, Advances in Bioceramics and Porous Ceramics VI, 2013, ch. 7, pp. 77–91, DOI:10.1002/9781118807811.
- A. Hirsch, I. Azuri, L. Addadi, S. Weiner, K. Yang, S. Curtarolo and L. Kronik, Chem. Mater., 2014, 26, 2934–2942 CrossRef CAS.
- F. Tamimi, Z. Sheikh and J. Barralet, Acta Biomater., 2012, 8, 474–487 CrossRef CAS PubMed.
- G. Cama, B. Gharibi, M. S. Sait, J. C. Knowles, A. Lagazzo, S. Romeed, L. Di Silvio and S. Deb, J. Mater. Chem. B, 2013, 1, 958–969 RSC.
- M. S. A. Johnsson and G. H. Nancollas, Crit. Rev. Oral Biol. Med., 1992, 3, 61–82 CrossRef CAS PubMed.
- S. Mandel and A. C. Tas, Mater. Sci. Eng., C, 2010, 30, 245–254 CrossRef CAS PubMed.
- M. A. Miller, M. R. Kendall, M. K. Jain, P. R. Larson, A. S. Madden and A. C. Tas, J. Am. Ceram. Soc., 2012, 95, 2178–2188 CrossRef CAS.
- A. C. Tas, J. Am. Ceram. Soc., 2016, 99, 1200–1206 CrossRef CAS.
- J. D. Termine, R. A. Peckauskas and A. S. Posner, Arch. Biochem. Biophys., 1970, 140, 318–325 CrossRef CAS PubMed.
- O. Borkiewicz, J. Rakovan and C. L. Cahill, Am. Mineral., 2010, 95, 1224–1236 CrossRef CAS.
- M. Okada, E. S. Hara, D. Kobayashi, S. Kai, K. Ogura, M. Tanaka and T. Matsumoto, ACS Appl. Bio Mater., 2019, 2, 981–986 CrossRef CAS PubMed.
- A. J. Hoeher, S. T. Mergelsberg, O. J. Borkiewicz and F. M. Michel, Cryst. Growth Des., 2021, 21, 3736–3745 CrossRef CAS.
- M. Bohner, B. L. G. Santoni and N. Döbelin, Acta Biomater., 2020, 113, 23–41 CrossRef CAS PubMed.
- I. Grigoraviciute-Puroniene, Y. Tanaka, V. Vegelyte, Y. Nishimoto, K. Ishikawa and A. Kareiva, Ceram. Int., 2019, 45, 15620–15623 CrossRef.
- I. Grigoraviciute-Puroniene, A. Zarkov, K. Tsuru, K. Ishikawa and A. Kareiva, J. Sol-Gel Sci. Technol., 2019, 91, 63–71 CrossRef CAS.
- T. Toshima, R. Hamai, M. Tafu, Y. Takemura, S. Fujita, T. Chohji, S. Tanda, S. Li and G. W. Qin, J. Asian Ceram. Soc., 2014, 2, 52–56 CrossRef.
- O. Mekmene, S. Quillard, T. Rouillon, J.-M. Bouler, M. Piot and F. Gaucheron, Dairy Sci. Technol., 2009, 89, 301–316 CrossRef CAS.
- M. S. Johnsson and G. H. Nancollas, Crit. Rev. Oral Biol. Med., 1992, 3, 61–82 CrossRef CAS PubMed.
- A. K. Lynn and W. Bonfield, Acc. Chem. Res., 2005, 38, 202–207 CrossRef CAS PubMed.
- I. T. Hafez, C. A. Paraskeva, A. Toliza, P. G. Klepetsanis, P. G. Koutsoukos, Ø. Gustavsen, T. Østvold and A. C. Payatakes, Cryst. Growth Des., 2006, 6, 675–683 CrossRef CAS.
- A. Geffert, J. Geffertova and M. Dudiak, Forests, 2019, 10, 852 CrossRef.
- Y. Yamakoshi, in Reference Module in Biomedical Sciences, Elsevier, 2014, DOI:10.1016/B978-0-12-801238-3.00037-4.
- R. Tang, C. A. Orme and G. H. Nancollas, J. Phys. Chem. B, 2003, 107, 10653–10657 CrossRef CAS.
- P.-T. Cheng and K. P. H. Pritzker, Calcif. Tissue Int., 1983, 35, 596–601 CrossRef CAS PubMed.
- G. Montes-Hernandez and F. Renard, J. Phys. Chem. C, 2020, 124, 15302–15311 CrossRef CAS.
- J. W. Murray and R. V. Dietrich, Am. Mineral., 1956, 41, 616–626 CAS.
- T. Funda, I. Fundova, A. Gorzsás, A. Fries and H. X. Wu, Wood Sci. Technol., 2020, 54, 289–311 CrossRef CAS.
- E. Garskaite, S. L. Stoll, F. Forsberg, H. Lycksam, Z. Stankeviciute, A. Kareiva, A. Quintana, C. J. Jensen, K. Liu and D. Sandberg, ACS Omega, 2021, 6(33), 21719 CrossRef CAS PubMed.
- T. K. Ronson and A. J. McQuillan, Langmuir, 2002, 18, 5019–5022 CrossRef CAS.
- C. Drouet, BioMed Res. Int., 2013, 2013, 490946 Search PubMed.
- M. Trpkovska, B. Šoptrajanov and P. Malkov, J. Mol. Struct., 1999, 480–481, 661–666 CrossRef CAS.
- M. M. Mirković, T. D. L. Pašti, A. M. Došen, M. Ž. Čebela, A. A. Rosić, B. Z. Matović and B. M. Babić, RSC Adv., 2016, 6, 12219–12225 RSC.
- T. Seki, K.-Y. Chiang, C.-C. Yu, X. Yu, M. Okuno, J. Hunger, Y. Nagata and M. Bonn, J. Phys. Chem. Lett., 2020, 11, 8459–8469 CrossRef CAS PubMed.
- C.-C. Yu, K.-Y. Chiang, M. Okuno, T. Seki, T. Ohto, X. Yu, V. Korepanov, H.-O. Hamaguchi, M. Bonn, J. Hunger and Y. Nagata, Nat. Commun., 2020, 11, 5977 CrossRef CAS PubMed.
- A. T. Pemberton, D. B. Magers and D. A. King, J. Chem. Educ., 2019, 96, 132–136 CrossRef CAS.
- F. P. Rotzinger, J. M. Kesselman-Truttmann, S. J. Hug, V. Shklover and M. Grätzel, J. Phys. Chem. B, 2004, 108, 5004–5017 CrossRef CAS.
- S. V. Dorozhkin, Biomater. Sci., 2021, 9, 7748–7798 RSC.
- J. V. Rau, M. Fosca, V. S. Komlev, I. V. Fadeeva, V. Rossi Albertini and S. M. Barinov, Cryst. Growth Des., 2010, 10, 3824–3834 CrossRef CAS.
- S. Gebke, K. Thümmler, R. Sonnier, S. Tech, A. Wagenführ and S. Fischer, Molecules, 2020, 25, 335 CrossRef CAS PubMed.
- A. Dosen and R. F. Giese, Am. Mineral., 2011, 96, 368–373 CrossRef CAS.
- R. L. Frost and S. J. Palmer, Thermochim. Acta, 2011, 521, 14–17 CrossRef CAS.
- X. Zhou, W. Li, R. Mabon and L. J. Broadbelt, Energy Technol., 2017, 5, 52–79 CrossRef CAS.
- H. Yang, R. Yan, H. Chen, D. H. Lee and C. Zheng, Fuel, 2007, 86, 1781–1788 CrossRef CAS.
- R. S. Viswanath and P. J. Miller, Solid State Commun., 1979, 32, 703–706 CrossRef CAS.
- N. Gao, A. Li, C. Quan, L. Du and Y. Duan, J. Anal. Appl. Pyrolysis, 2013, 100, 26–32 CrossRef CAS.
- A. Bolarinwa, U. Gbureck, P. Purnell, M. Bold and L. Grover, Adv. Appl. Ceram., 2009, 109(5), 291 CrossRef.
- D. Bigoni, R. Cavuoto, D. Misseroni, M. Paggi, A. Ruffini, S. Sprio and A. Tampieri, Mater. Today Bio, 2020, 5, 100032 CrossRef CAS PubMed.
- E. Charrière, S. Terrazzoni, C. Pittet, P. Mordasini, M. Dutoit, J. Lemaître and P. Zysset, Biomaterials, 2001, 22, 2937–2945 CrossRef PubMed.
- E. Konofalska, P. Kozakiewicz, W. Buraczyk, H. Szeligowski and H. Lachowicz, Forests, 2021, 12, 619 CrossRef.
- F. Barthelat, Z. Yin and M. J. Buehler, Nat. Rev. Mater., 2016, 1, 16007 CrossRef CAS.
- J. Engstrand, C. Persson and H. Engqvist, J. Mech. Behav. Biomed. Mater., 2014, 29, 81–90 CrossRef CAS PubMed.
- M. P. Hofmann, A. R. Mohammed, Y. Perrie, U. Gbureck and J. E. Barralet, Acta Biomater., 2009, 5, 43–49 CrossRef CAS PubMed.
- H. Moussa, W. Jiang, A. Alsheghri, A. Mansour, A. E. Hadad, H. Pan, R. Tang, J. Song, J. Vargas, M. D. McKee and F. Tamimi, Acta Biomater., 2020, 106, 351–359 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00305a |
| This journal is © The Royal Society of Chemistry 2023 |