
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiving ring-opening polymerization of β-butyrolactone initiated by mononuclear zirconium compounds containing sterically hindered N,O-chelate and anionic dimethylamide ligands†
Jianwei Jiang,
Jihyun Choi and
Sungho Yoon *
*
Department of Chemistry, Chung-Ang University, Seoul 06974, Republic of Korea. E-mail: sunghoyoon@cau.ac.kr
First published on 3rd April 2023
Abstract
The ring-opening polymerization of β-lactones into polyhydroxyalkanoates (PHA), biodegradable polymers with high molecular weight and narrow polydispersity, is of significant interest. The mononuclear zirconium compound containing sterically hindered N,O-chelate and anionic dimethylamide ligands was used as an initiator for the polymerization of β-butyrolactone (BBL), resulting in polyhydroxylbutyrate (PHB) with a number-average molecular weight of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. Kinetic studies demonstrate a first-order dependence on β-butyrolactone (BBL) concentration at room temperature, accompanied by narrow molecular weight distributions (ca. 1.03–1.07), indicating a well-controlled living polymerization.
000 g mol−1. Kinetic studies demonstrate a first-order dependence on β-butyrolactone (BBL) concentration at room temperature, accompanied by narrow molecular weight distributions (ca. 1.03–1.07), indicating a well-controlled living polymerization.
Introduction
Poly (3-hydroxybutyrate) (PHB) is the most widely studied polyhydroxyalkanoate (PHA), which is a biodegradable polymer with potential applications in tissue engineering and regenerative medicine, food packaging, and drug delivery systems. The preparation methods of PHB include the biological route,1,2 direct copolymerization of propylene oxide/CO,3,4 and ring-opening polymerization (ROP) of β-butyrolactone (BBL).5–8 Particularly, metal-compound-initiated ROP of BBL is of interest due to its high molecular weight, narrow polydispersity, and controlled tacticity of the resulting PHB.4–36Main-group metals such as Sn,9–11 Al,12–14 and In,15–17 as well as transition metals like Zn,18–21 Cr,3,4,22,23 Ti,24–26 and Zr,26–30 and rare earth metal compounds have all been used in BBL polymerization.31–33 Zr compounds, which can have coordination numbers of 6, 7, or 8, have been of particular interest due to their low toxicity,34–36 colorless character of the initiator, and high activity. The tenfold lower toxicity of Zr compounds compared to analogous Sn compounds has resulted in their approval by the Food and Drug Administration (FDA).34 The bis(imino)phenoxide Zr compound (Chart 1A) displayed a high level of efficiency for BBL ROP, and nearly all BBL (200 equiv.) was consumed within 10 minutes under neat conditions.27 The (R,R)-(−)-N,N′-bis(3,5-di-t-butylsalicydidene)-1,2-cyclohexanediamine (salen) Zr compound (Chart 1B) was able to catalyze ROP of BBL under neat conditions, yielding PHB with a low polydispersity index (PDI, 1.01).28 The syndio-enriched structure of stereoregular PHB was obtained from racemic BBL polymerization in toluene using amine tris(phenolate) Zr compound (Chart 1C).29 Thioetherphenolate Zr compound (Chart 1D) was found to be effective in the co-polymerization of BBL with lactide.30 Despite their efficiency, the tedious preparation and purification of Zr compounds is a major drawback. Furthermore, since most of these Zr compounds are aryloxide-based complexes, the resulting PHB is colored, which limits its industrial applications.27,28,30 A straightforward and economical synthetic method for producing a colorless Zr compound could be highly beneficial for its potential use in PHB production.
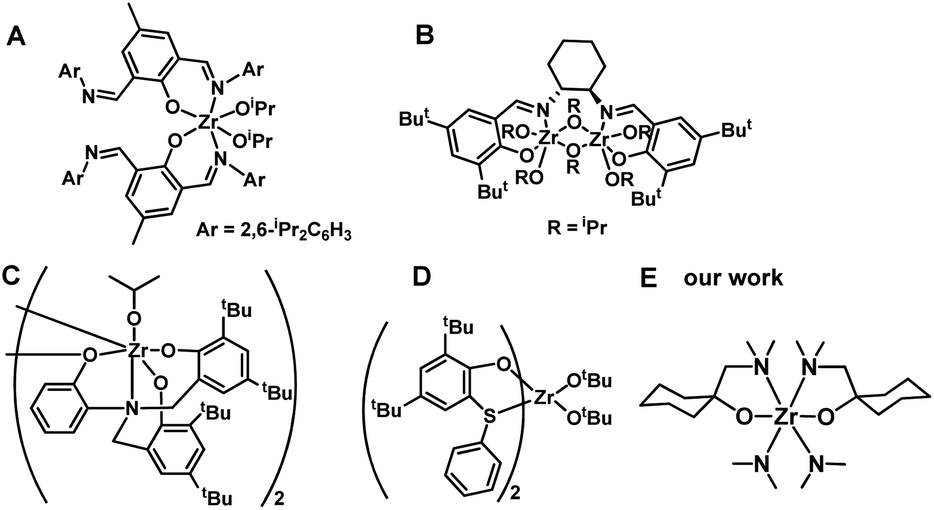 | ||
| Chart 1 (A–E) Zr-compounds for BBL polymerization. | ||
The coordination–insertion mechanism (Scheme 1) is typically used in metal-compound-initiated lactone polymerization, as documented.5–9,33 The process begins with the coordination of lactone to the unsaturated metal center, followed by a nucleophilic attack of the active group (X) on the carbonyl carbon of the lactone, resulting in the insertion of the lactone into the M–X bond (X = alkoxide, amide, etc.). The migration of metal to the ethereal oxygen, followed by a rearrangement of the four-membered cyclic intermediate, results in the cleavage of the acyl–oxygen bond and the formation of metal alkoxide species. This is an accurate catalytic site for lactone polymerization. If a structurally well-defined compound meets three requirements, including (1) a coordination unsaturated metal center; (2) one or more nucleophilic attachments to the metal center; and (3) the ability of ancillary ligand to stabilize the whole metal compound, then it may initiate BBL polymerization in a manner that is controlled by the proposed mechanism.
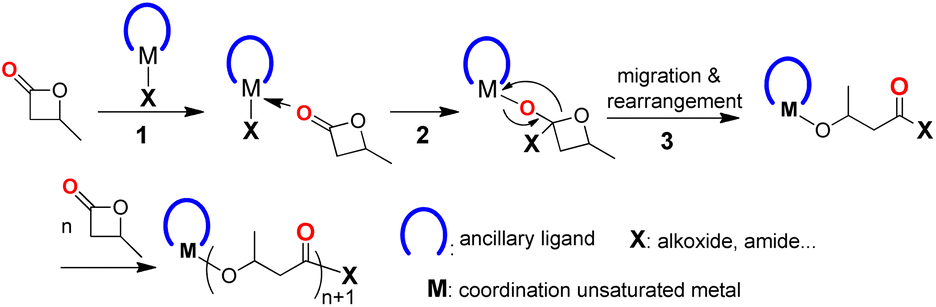 | ||
| Scheme 1 BBL polymerization by coordination–insertion mechanism. | ||
To validate our hypothesis, we chose a structurally well-defined and colorless Zr compound, dimethylamide Zr(IV) with N,O-chelate ligand, to initiate BBL polymerization (Chart 1E). We hypothesized that the Zr center in the compound (bis(dimethylamido)bis[1-((dimethylamino)methyl)cyclohexanolate]zirconium) could act as an electrophile due to its unsaturated coordination, while the anionic –NMe2 group could serve as a nucleophile. The two N,O-chelate ligands, which have a sterically hindered moiety, can stabilize the Zr center, thus allowing this mononuclear Zr compound to act as an effective initiator for BBL ROP via a typical coordination polymerization mechanism, resulting in the formation of colorless PHB.
Experimental sections
Materials and methods
BBL was purchased from Sigma-Aldrich, and purified by drying using CaH2 (4 wt%) at 35 °C for 3 days followed by distillation (40–50 °C, 30 mbar). Zr initiator was synthesized according to our previously reported procedure.37 1H and 13C NMR spectra were recorded in deuterated chloroform using a Bruker instrument (600 or 300 MHz). The molecular weight and polydispersity index (Mw/Mn, PDI) of products were determined by Gel Permeation Chromatography (GPC) using a Waters 2410 refractive index detector (Milford, MA, USA) with a Waters 515 HPLC pump. The GPC column was eluted with THF at a flow rate of 1.0 mL min−1. GPC curves were calibrated using a polystyrene standard with molecular weights ranging from 162 to 6570000 g mol−1.
BBL polymerization procedure
In a glove box, Zr initiator (10.0 mg), BBL (350 mg), and toluene-d8 (1.546 g) were added into a vial of 4 mL, and the reaction was run under room temperature at a stirring rate of 600 rpm. 1H NMR sample was taken from the solution at different polymerization time. The conversion was calculated based on the integration area of methine for PHB and methyl for BBL.GPC sample was prepared as follows
After measuring 1H NMR of these samples, they were quenched by adding two drops of acetic acid. The resulting solution was filtered over Celite, and the filtrate was vacuumed under 40 °C for several hours. The residue compound was diluted with THF (approximately 1.0 mg mL−1), which was used for GPC measurement after the filtration using PTFE filter (0.2 μm).MALDI-TOF characterization of the oligomer PHB
Mass spectra were recorded on a Bruker Autoflex MALDI-ToF (time-of-flight mass (ToF) spectrometer equipped with MALDI ion source). The oligomer PHB, dithranol matrix and sodium trifluoroacetate (cation source) were dissolved in THF at 10 mg mL−1, respectively. The solutions were mixed in a 2:2:1 volume ratio. 2 μL of the resulting mixture was spotted on the sample plate and submitted for MALDI-TOF mass analysis.
Results and discussion
According to our recently developed method, the Zr(IV) compound, bis(dimethylamido)bis[1-((dimethylamino)methyl)cyclohexanolate]zirconium, was synthesized by inserting epoxide into tetrakis(dimethylamido)zirconium at room temperature (Scheme 2).37 X-ray diffraction of a single crystal revealed that the Zr compound was a mononuclear sphere with six coordination sites, composed of two –NMe2 and two N,O-chelate ligands. The incorporation of N,O-chelate ligand significantly augmented the thermal stability of the Zr compound, thus making it possible to successfully deposit the compound on ZrO2 thin film through atomic layer deposition. This method is highly simple, cost-effective, eco-friendly.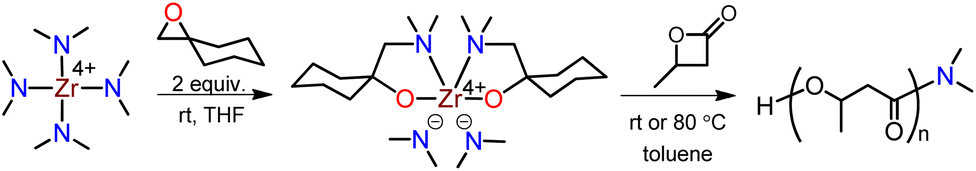 | ||
| Scheme 2 Schematic representation for the synthesis of the Zr-compound and PHB. | ||
The initial evaluation of the effect of the Zr compound on PHB synthesis was conducted at room temperature, with a molar ratio of 200:1 (BBL:initiator) and a BBL concentration of approximately 2.5 M, in toluene-d8, for a period of one day (Fig. S1†). The integration area of the PHB (methine peak, 5.3 ppm) and BBL peaks (methyl peak, 1.6 ppm) in Fig. 1 demonstrated that 28% of BBL was converted to PHB.
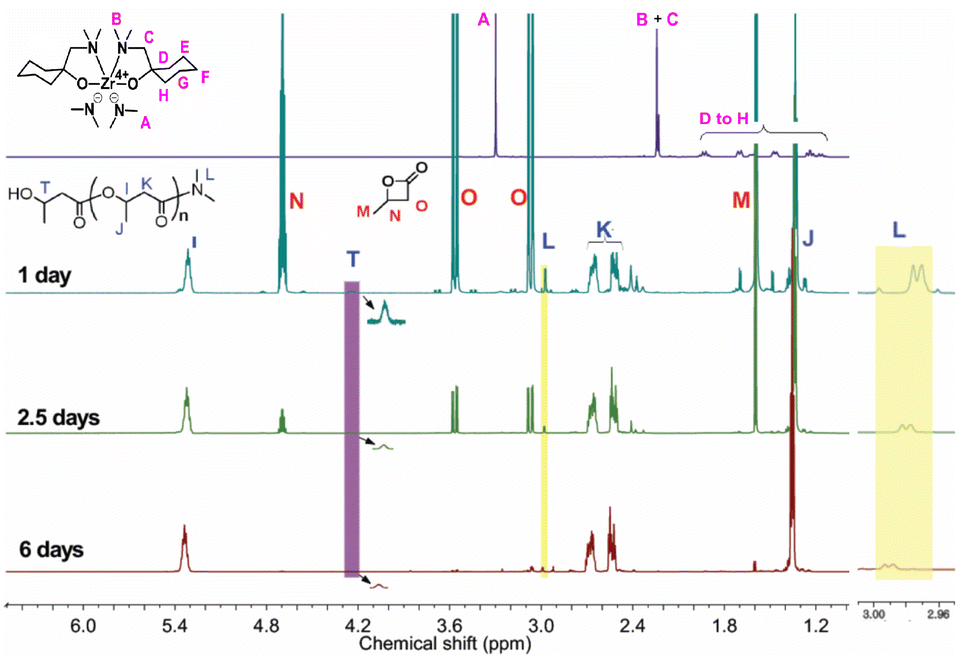 | ||
| Fig. 1 1H NMR spectra (CDCl3, 600 MHz) measured at various reaction times. | ||
Fig. S2† of the 13C NMR spectrum showed that PHB had an atactic configuration, with the carbonyl peak displaying equal intensities of meso and racemic diad signals. As the reaction progressed, the usage of BBL increased, leading to a conversion rate of 98% within 6 days. Fig. 2a showed a linear relationship between ln([BBL]0/[BBL]t) and the reaction time, with an apparent propagation rate constant (kapp) of 0.03 ± 1.0 × 10−3 h−1. Additionally, Fig. 2b revealed that the number-average molecular weights (Mn) of PHB increased linearly with BBL conversion, as determined by GPC. The PHB samples all had a narrow PDI (1.03–1.07), with monomodal and symmetrical GPC curves (Fig. 2c), which suggests that the Zr center promoted negligible elimination and transesterification reactions. These results clearly show that the BBL polymerization initiated by the Zr compound at room temperature has an excellent living feature.
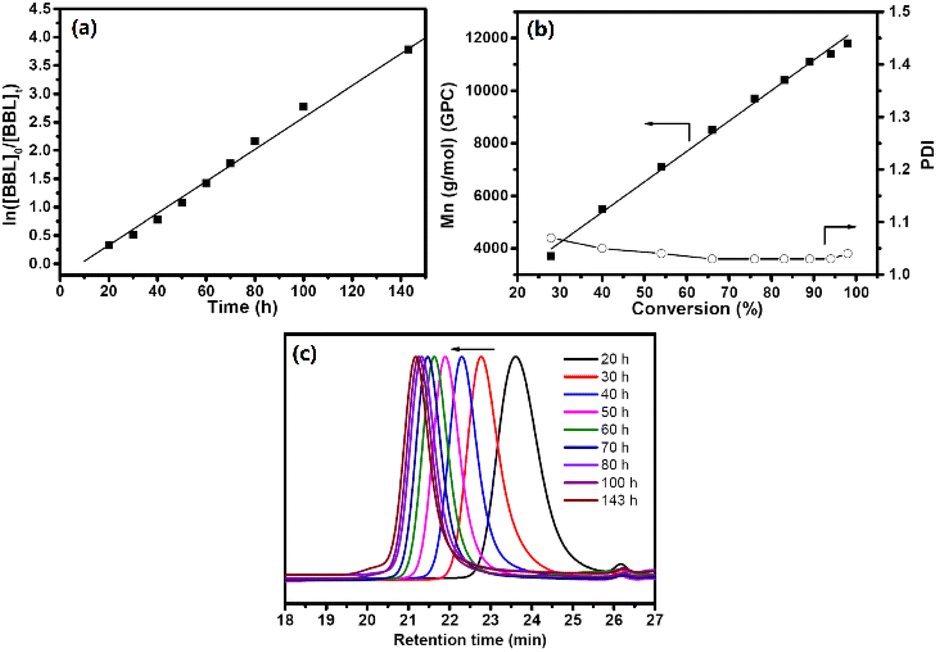 | ||
| Fig. 2 (a) Plot of ln([BBL]0/[BBL]t) versus time. (b) Plot of PHB Mn and PDI as a function of conversion. (c) GPC curves obtained from kinetics experiments. Reaction condition: BBL/initiator = 200, [BBL] = 2.5 M, room temperature, toluene-d8. | ||
In order to investigate the active site of the initiator on the BBL polymerization, a 1H NMR study was conducted in detail. The peaks of –NMe2 (A) and Me2N–CH2– (B and C) on the initiator bis(dimethylamido)bis[1-((dimethylamino)methyl)cyclohexanolate]zirconium, were observed at 3.30 ppm and 2.23–2.24, respectively (Fig. 1). The position of peaks A, B and C changed after polymerization, suggesting that the initiator was involved in the BBL polymerization and thus altering the structure of the initiator. The doublet peak at 2.98 ppm, which is very close to the CH3– peak in DMF (HCONMe2, doublet peak at 2.96 and 2.88 ppm), is likely due to –CONMe2 amide formation. Furthermore, the decrease in the ratio of peak intensity between 2.98 ppm and 5.3 ppm with an increase in PHB length further supports the amide formation. After 6 days of running, the product was hydrolyzed with dilute HCl in methanol, resulting in the isolation of a white viscous polymer. The 1H NMR spectrum of this compound is shown in Fig. S3.† It was interesting to note that peaks corresponding to PHB were observed, along with a peak of very weak intensity at 2.98 ppm, indicating the formation of an amide group. The absence of chelate ligand peaks in the purified PHB implied that the sterically hindered chelate ligand did not act as the nucleophile for BBL polymerization.
By treating our Zr compound with isopropanol (Fig. S4†), we synthesized another Zr compound, isopropoxide Zr with N,O-chelate ligand, and tested its activity for BBL polymerization, in the absence of Zr–NMe2 group. The 1H NMR spectrum showed that the amide peak of PHB at around 2.98 ppm had disappeared (Fig. S5†), and the newly observed peaks were attributed to the –(CO)–OiPr group. Interestingly, if TDMAZ was used as initiator for BBL polymerization at room temperature for 24 h, no PHB was detected in the resulting mixture (Fig. S6†). From these results, it can be inferred that the anionic –NMe2 group on the Zr compound acted as the nucleophilic group, which was responsible for the cleavage of the acyl–oxygen bond in BBL and thus initiated BBL polymerization.
From the 1H NMR spectra (Fig. 1), a broad peak at 4.2 ppm was observed on PHB, which was attributed to the methine of the end group HOCH(CH3)–CH2–CO–;30 we hypothesized that the Zr-alkoxide species breaks the BBL ring at the acyl–oxygen bond, thus enabling PHB growth, and this was confirmed by using it as the precise catalytic site for BBL polymerization.
MALDI-TOF measurement was performed to confirm the structure of the resulting PHB. The sample was synthesized from ROP of BBL at room temperature for 8 h, with a molar ratio of 200/1 (BBL/initiator). A nearly monomodal distribution of Na+ adducts was detected in the range of 400–1200 Da with a regular mass-to-mass peak increment of 86 Da (Fig. S7†). These peaks were consistent with the linear oligomer, which was end capped with –OH and –CONMe2 amide groups.
Table 1 entries 1–3 demonstrate that when the reaction was conducted at 60 °C for 16 hours in toluene-d8 and C6D6, the BBL conversion was 99% and 97%, respectively. However, the conversion rate in THF under the same reaction conditions was only 43%. This prompted further investigation into the activity of the Zr initiator. The classic coordination–insertion mechanism states that weak coordinating solvents, such as toluene or benzene, usually have higher activity than the more strongly coordinating solvent, such as THF, and this result is in agreement with that.
| Entry | BBL/initiator | Solvent | Temp. (°C) | Time (h) | Conversion (%) | Mn (g mol−1) | PDI |
|---|---|---|---|---|---|---|---|
| 1 | 50 | Toluene-d8 | 60 | 16 | >99 | — | — |
| 2 | 50 | C6D6 | 60 | 16 | 97 | — | — |
| 3 | 50 | THF-d8 | 60 | 16 | 43 | — | — |
| 4 | 300 | Toluene-d8 | rt | 24 | 21 | 4800 | 1.04 |
| 5 | 300 | Toluene-d8 | 40 | 24 | 43 | 8500 | 1.03 |
| 6 | 300 | Toluene-d8 | 80 | 24 | 62 | 11400 |
1.03 |
| 7 | 200 | Toluene-d8 | 80 | 0.5 | 20 | 3100 | 1.04 |
| 8 | 200 | Toluene-d8 | 80 | 2 | 56 | 7100 | 1.04 |
| 9 | 200 | Toluene-d8 | 80 | 6 | 81 | 10400 |
1.04 |
| 10 | 200 | Toluene-d8 | 80 | 24 | 85 | 11000 |
1.04 |
At room temperature, the BBL conversion was only 21% after one day, however, when the temperature was increased to 40 °C and 80 °C, the conversion increased two and three times respectively, to 43% and 62% (Table 1, entries 4–6). Kinetic experiments were conducted using the BBL/initiator = 200 in toluene-d8 to investigate the correlation between BBL conversion and reaction time at 80 °C (Table 1, entries 7–10). After 0.5 h, the BBL conversion was 12%, and it quickly rose to 64% within 2 h. The reason for the accelerating process may be the greater nucleophilicity of the newly generated alkoxide ligand versus the amide ligand, which exist in the initiating state. As the reaction time was extended further (>2 h), the BBL gradually converted to PHB, with the conversion being 83% in 6 h and 85% in 24 h. The results suggest that no significant improvement of BBL conversion was observed at extended reaction times, which may be due to side reactions occurring.
To investigate the potential side reactions, an oligomerization reaction was conducted at 80 °C using BBL/initiator = 20. The 1H NMR spectrum (Fig. S8†) showed clear trans-crotonate peaks of the product, indicating β-proton elimination. This elimination reaction also led to the formation of Zr–OH species, which were then converted to H2O and Zr–O–Zr species through a condensation process due to its thermodynamic instability.38 Our findings suggest that β-proton elimination was the predominant mechanism in Zr-compound-initiated BBL polymerization at elevated temperature for PHB synthesis, due to the detrimental effect of the generated H2O on the Zr-initiator and the insufficient strength of the Zr–O–Zr species to initiate the polymerization.
Conclusions
A structurally well-defined Zr compound was proposed to initiate BBL polymerization for obtaining PHB, which was readily synthesized from the epoxide insertion into TDMAZ using our recently developed method. The compound showed living-polymerization of BBL at room temperature with a narrow PDI (1.03–1.07). PHB end-group study and solvent dependence of initiator activity revealed that BBL polymerization proceeded by coordination–insertion mechanism. We discovered that the decrease in Zr-initiator activity at higher temperatures was likely caused by β-proton elimination. Compared to other initiators (Zn, Sn, Cr, ln, and rare earth metals),19 Zr compound has a comparatively slower reaction rate for BBL ROP. We are currently working on improving the reaction rate using other colorless metal compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the C1 Gas Refinery Program (2018M3D3A1A01018006) and carbon to X Project (2020M3H7A1098259) through the National Research Foundation of Korea funded by the Ministry of Science, ICT, and Future Planning, Republic of Korea.References
- H. M. Müller and D. Seebach, Angew. Chem., Int. Ed., 1993, 32, 477–502 CrossRef.
- S. Mecking, Angew. Chem., Int. Ed., 2004, 43, 1078–1085 CrossRef CAS PubMed.
- J. Jiang, S. Rajendiran, L. Piao and S. Yoon, Top. Catal., 2017, 60, 750–754 CrossRef CAS.
- J. C. Yang, J. Yang, W. B. Li, X. B. Lu and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202116208 CAS.
- J. F. Carpentier, Macromol. Rapid Commun., 2010, 31, 1696–1705 CrossRef CAS PubMed.
- C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- R. Reichardt and B. Rieger, Adv. Polym. Sci., 2012, 245, 49–90 CrossRef CAS.
- H. Li, R. M. Shakaroun, S. M. Guillaume and J.-F. Carpentier, Eur. J. Chem., 2020, 26, 128–138 CrossRef CAS PubMed.
- J. E. Kemnitzer, S. P. McCarthy and R. A. Gross, Macromolecules, 1993, 26, 1221–1229 CrossRef CAS.
- M. Möller, R. Kånge and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2067–2074 CrossRef.
- Z. Wei, L. Liu and M. Qi, React. Funct. Polym., 2006, 66, 1411–1419 CrossRef CAS.
- N. Nomura, T. Aoyama, R. Ishii and T. Kondo, Macromolecules, 2005, 38, 5363–5366 CrossRef CAS.
- E. D. Cross, L. E. N. Allan, A. Decken and M. P. Shaver, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1137–1146 CrossRef CAS.
- F. M. García-Valle, V. Tabernero, T. Cuenca, M. E. G. Mosquera, J. Cano and S. Milione, Organometallics, 2018, 37, 837–840 CrossRef.
- C. Xu, I. Yu and P. Mehrkhodavandi, Chem. Commun., 2012, 48, 6806–6808 RSC.
- S. M. Quan and P. L. Diaconescu, Chem. Commun., 2015, 51, 9643–9646 RSC.
- J. Bruckmoser, D. Henschel, S. Vagin and B. Rieger, Catal. Sci. Technol., 2022, 12, 3295–3302 RSC.
- L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239–15248 CrossRef CAS PubMed.
- T. Ebrahimi, D. C. Aluthge, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2016, 49, 8812–8824 CrossRef CAS.
- M. Shaik, J. Peterson and G. Du, Macromolecules, 2019, 52, 157–166 CrossRef CAS.
- W. Gruszka, L. C. Walker, M. P. Shaver and J. A. Garden, Macromolecules, 2020, 53, 4294–4302 CrossRef CAS.
- M. Zintl, F. Molnar, T. Urban, V. Bernhart, P. Preishuber-Pflügl and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 3458–3460 CrossRef CAS PubMed.
- S. Vagin, M. Winnacker, A. Kronast, P. T. Altenbuchner, P. Deglmann, C. Sinkel, R. Loos and B. Rieger, ChemCatChem, 2015, 7, 3963–3971 CrossRef CAS.
- D. Dakshinamoorthy and F. Peruch, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5176–5185 CrossRef CAS.
- H. J. Altmann, M. R. Machat, A. Wolf, C. Gürtler, D. Wang and M. R. Buchmeiser, J. Polym. Sci., 2021, 59, 274–281 CrossRef CAS.
- T. K. Saha, M. Mandal, D. Chakraborty and V. Ramkumar, New J. Chem., 2013, 37, 949–960 RSC.
- T. K. Saha, B. Rajashekhar, R. R. Gowda, V. Ramkumar and D. Chakraborty, Dalton Trans., 2010, 39, 5091–5093 RSC.
- T. K. Saha, V. Ramkumar and D. Chakraborty, Inorg. Chem., 2011, 50, 2720–2722 CrossRef CAS PubMed.
- B. J. Jeffery, E. L. Whitelaw, D. Garcia-Vivo, J. A. Stewart, M. F. Mahon, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 12328–12330 RSC.
- E. Luciano, A. Buonerba, A. Grassi, S. Milione and C. Capacchione, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3132–3139 CrossRef CAS.
- A. Amgoune, C. M. Thomas, S. Ilinca, T. Roisnel and J.-F. Carpentier, Angew. Chem., Int. Ed., 2006, 45, 2782–2784 CrossRef CAS PubMed.
- Z. Zhuo, C. Zhang, Y. Luo, Y. Wang, Y. Yao, D. Yuan and D. Cui, Chem. Commun., 2018, 54, 11998–12001 RSC.
- D. M. Lyubov, A. O. Tolpygin and A. A. Trifonov, Coord. Chem. Rev., 2019, 392, 83–145 CrossRef CAS.
- M. Bero, P. Dobrzyński and J. Kasperczyk, Polym. Bull., 1999, 42, 131–139 CrossRef CAS.
- K. T. Kalinin, N. G. Sedush, P. V. Dmitryakov and S. N. Chvalun, ChemistryOpen, 2020, 9, 1027–1032 CrossRef CAS PubMed.
- S. Impemba, G. Roviello, S. Milione and C. Capacchione, Inorg. Chem., 2021, 60, 7561–7572 CrossRef CAS PubMed.
- J. Jiang, S. Choi, J. Oh, J. Choi, H. J. Sun and S. Yoon, Dalton Trans., 2022, 51, 5315–5321 RSC.
- B. Sharma, T. M. Callaway, A. C. Lamb, C. A. Steren, S.-J. Chen and Z. L. Xue, Inorg. Chem., 2013, 52, 11409–11421 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00338h |
| This journal is © The Royal Society of Chemistry 2023 |