
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmploying non-canonical amino acids towards the immobilization of a hyperthermophilic enzyme to increase protein stability†
Hannah J. Switzer‡
,
Christina A. Howard‡,
John F. Halonski,
Emily M. Peairs,
Nolan Smith,
Maddy P. Zamecnik,
Sanjana Verma and
Douglas D. Young *
*
Department of Chemistry, William & Mary, Williamsburg, VA, USA. E-mail: dyoung01@wm.edu
First published on 14th March 2023
Abstract
A carboxylesterase derived from Sulfolobus solfataricus P1 was immobilized onto an epoxy-activated Sepharose resin via non-canonical amino acids. The immobilized enzyme exhibited heightened performance in organic solvents, recyclability, and stability at room temperature for over two years. The incorporation of a non-canonical amino acid afforded a high degree of control over the bioorthogonal immobilization reaction. These results indicate that the specificity conferred by genetic code expansion produces advantages in protein immobilization and broadens the utility of such proteins to non-biological settings.
Introduction
Proteins are versatile biomacromolecules that have diverse functions within the cell and are also widely utilized in industrial settings. However, the utility of proteins is limited by their sensitivity to non-physiological environments outside of biological systems. Proteins are highly sensitive to pH, salt concentration, temperature, and organic solvents when in solution.1–6 One method of conferring a higher degree of stability is via protein immobilization onto a solid-support.4,7–11 This affords a level of stabilization by lessening protein unfolding and aggregation. Thus, protein immobilization offers researchers a cost-effective avenue for generating proteins with increased stability, functionality, and recyclability. Methods of protein immobilization include adsorption, entrapment, and covalent coupling, each possessing unique advantages and disadvantages.11–15 However, the stability provided by covalent immobilization can provide improved recyclability, particularly when immobilized proteins are exposed to non-physiological conditions.16–18 One drawback of this method is the inconsistency of immobilization when utilizing only the 20 canonical amino acids. Most proteins contain multiple reactive residues, leading to a lack of control over the immobilization site and orientation (Fig. 1).12 The resulting random positioning of proteins on the solid support can lead to decreased functionality due to blockage of the active site. One remedy to this pitfall is the employment of non-canonical amino acids (ncAAs), as they can be engineered to possess a unique functional handle that facilitates a higher degree of control over the immobilization reaction.18–22 Moreover, the site of ncAA incorporation can be genetically controlled to minimize any perturbation to protein function.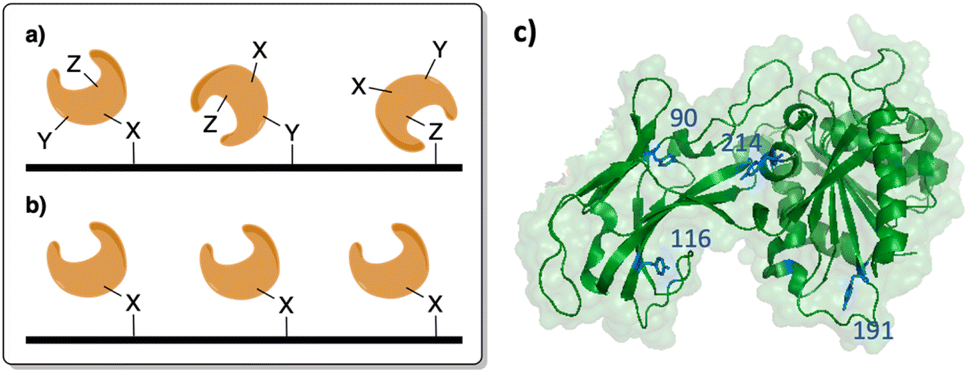 | ||
| Fig. 1 (a) Protein immobilization utilizing the 20 canonical amino acids results in random orientation, possibly blocking the active site of a protein. (b) Protein immobilization using ncAAs allows for increased control over surface attachment. (c) Crystal structure of SSo P1 EST analog with key tyrosine residues highlighted (blue; PDB 3DOH). | ||
Genetic code expansion technologies to introduce ncAAs have been well established and employed towards numerous applications including protein immobilization.23–25 Consequently, a number of ncAAs with unique chemical functionality have been genetically encoded.26–28 Additionally, numerous bioorthogonal reactions have been developed that can be applied to the site-specific immobilization of proteins.29–35 Ideally, this approach will result in a more homogenous orientation and allow higher retention of enzymatic activity. Previous research involving the ncAA-based immobilization of GFP demonstrated the feasibility of this approach; however, this research aims to expand this methodology towards the immobilization of enzymes, and their utilization in non-biological conditions.18
Hyperthermophilic enzymes play a key role in industrial processes.36–38 We selected the hyperthermophilic enzyme Sulfolobus solfataricus carboxylesterase P1 (SSo EST1) as a model enzyme due to its high stability and existence of a simple enzymatic activity assay.39,40 SSo EST1 is a hyperthermophilic enzyme involved in hydrolysis of ester into an alcohol and a carboxylic acid. This enzymatic activity can be easily detected with standard spectrophotometric assays, allowing for monitoring of any alterations in enzyme function. This assay can also be employed to monitor enzyme stability across different time points and investigate the ability of the enzyme to be recycled through numerous assays. Additionally, the robustness of this protein allows us to probe the extremes of immobilized enzymatic stability at high temperatures, in organic solvents, and under microwave irradiation. Specifically, our research explores the covalent immobilization of SSo EST1 onto epoxy-activated Sepharose resin using ncAAs. Ultimately this work aims to develop a generalizable methodology for the improved immobilization of enzymes that will facilitate their widespread utilization in non-biological applications.
Results and discussion
First, we sought to select appropriate residues of SSo EST1 for immobilization. Analysis of the crystal structure identified four surface-exposed tyrosine residues that were candidates for mutation to genetically encode a ncAA: Sso EST1 Y90TAG Y116TAG, Y191TAG, and Y214TAG (Fig. 1b). The tyrosine residues were selected in order to make minimal perturbations to the overall protein structure as the ncAAs are tyrosine derivatives. Several sites were selected as ncAA incorporation site has varying effects both on expression and function of the protein, requiring screening of multiple locations. Mutants were then generated using a Quik Change PCR Mutagenesis Kit (Qiagen) and successful insertion of the TAG codon was confirmed by sequencing (Genewiz). Wild-type SSo EST1 and the four mutant proteins were expressed in E. coli to ascertain if the alteration of the natural tyrosine with a ncAA affected enzymatic activity. As a result of the SSo EST1 protein being encoded on a pQE-30 plasmid that employs a T5 RNA promoter, protein expressions in BL-21(DE3) E. coli required the addition of a pREP4 plasmid to introduce more lac repressor and allow the protein induction to become inducible. Consequently, BL-21(DE3) E. coli were co-transformed with a plasmid containing the SSo EST1 (pQE-30; Amp), a plasmid containing the translational machinery for ncAA incorporation (pEVOL-pPrF; Chlor), and the pREP4 plasmid (Kan) and grown on the triple antibiotic agar. Colonies were selected and used to induce expression cultures of the SSo EST1 protein containing p-propargyloxyphenylalanine (pPrF) at the site of the TAG mutation (Fig. 2a). Expressions afforded highest protein yields after 16 hours at 37 °C, followed by purification of SSo EST1 via heat denaturation as described in the literature.39 Protein yield and purity were assessed by SDS-PAGE, indicating that some mutation sites afforded lower levels of expression, potentially due to destabilizing effects of ncAA incorporation at that site (Fig. 2c). Incorporation of pPrF at sites 90 and 116 resulted in similar or slightly lower protein yields than wild-type expressions; however, incorporation at residues 191 and 214 resulted in significantly decreased yields. These results indicate the importance of residue selection for ncAA incorporation into proteins and demonstrate the need to produce multiple mutants to ensure genetic code expansion. Incorporation of the ncAA was also confirmed by sequential bioconjugation with an alkynyl fluorophore under previously described Glaser–Hay conditions.33,41,42 Conjugates were analyzed by SDS-PAGE to confirm the generation of fluorescent SSo EST1, indicating the presence of an alkynyl ncAA needed for successful reaction (see ESI†). | ||
| Fig. 2 Experimental approach to SSo-EST genetic code expansion and immobilization. (a) Incorporation of pPrF in response to a TAG codon facilitates site-specific incorporation of an immobilization handle. (b) Glaser–Hay bioconjugation to utilize the covalent diyne to immobilize enzymes. (c) SDS-PAGE of SSo-EST mutant expressions. Amber suppression was observed for all mutants, but with variable levels of success depending on mutation site. (d) SSo-EST1 enzymatic assay to assess the activity of each mutant. Assays were conducted in triplicate and normalized to protein concentrations. | ||
Other than expression, the other potential issue in ncAA incorporation is potentially reducing protein functionality. To characterize each mutant, the SSo EST1 proteins were subjected to a colorimetric assay based on their ability to hydrolyze esters. Prior work with SSo EST1 has demonstrated that p-nitrophenyl hexanoate (pNp-Hex) is a substrate for the enzyme, allowing for monitoring of the ester hydrolysis at 405 nm via the production of the yellow nitrophenolate anion.39 Proteins were incubated in the presence of pNp-Hex for one hour at 80 °C followed by reaction quenching with saturated sodium bicarbonate and analysis on a spectrophotometer (Fig. 2d). Contol samples without protein were used to correct for non-enzymatic hydrolysis. When corrected for protein concentration, differences were observed in the various mutation sites. The SSo EST1-116 mutant maintained comparable activity to the wild-type protein suggesting minimal perturbations caused by pPrF, whereas the SSo EST1-214 mutant shows significantly reduced activity. This may be due to its proximity to the active site or due to other structural alterations that may arise from its incorporation.
With functional SSo EST1-pPrF proteins in hand, the next step involved their immobilization on a solid-support to determine if covalent immobilization via a ncAA enhances protein stability in non-biological environments (Fig. 1b). While previous research has indicated that this immobilization promotes enzyme stability, it has not been extensively studied in the context of site-specific immobilization mediated by ncAAs. Earlier work in our laboratory used this approach with a 1,3-dipolar cycloaddition to immobilize GFP as a proof-of-concept. Initial findings indicated that fluorescence was maintained in non-aqueous solvents; however, GFP is not enzymatic so catalytic potential could not be assessed. Given the versatility of pPrF for numerous bioorthogonal reactions, it is the ideal candidate for immobilization investigations. Given the previous experience in our laboratory with the development and optimization of bioorthogonal Glaser–Hay alkynyl couplings, these optimized conditions were initially identified as the starting point for enzymatic immobilizations. To generate an alkynyl coupling partner on the solid-support, epoxy-activated Sepharose 6B resin was derivatized with alkynyl alcohols of various lengths: propargyl alcohol, 5-hexyn-1-ol, and 10-undecyn-1-ol. SSo EST-191 was then immobilized to the resin using a Glaser–Hay coupling. As a control, SSo EST1-WT was also subjected to Glaser–Hay conditions to examine non-specific association of SSo EST1 and resin. Previous work with GFP and the resins indicated that resin loading with protein using this approach resulted in reproducible resin loadings based on protein concentration. Loading of the resin was quantified using the intrinsic fluorescence of GFP. We assumed that SSo EST1 was loaded at a similar rate due to the solvent-exposed nature of the ncAAs in both GFP and SSo EST1. The resins were then assayed using the previously described pNp-Hex substrate and supernatant was monitored at 405 nm. While all three resins displayed enzymatic activity relative to the negative controls, the highest enzymatic activity was observed with propargyl alcohol. This is in agreement with previous GFP immobilization results, and thus propargyl alcohol derivatized Sepharose 6B resin was employed in all subsequent immobilizations (see ESI†). To determine if protein immobilization affected enzymatic activity, each mutant was immobilized via Glaser–Hay coupling to propargyl alcohol derivatized Sepharose resin. The individual resins harboring each SSo EST1-pPrF mutant were then assayed as previously described (Fig. 3a). Gratifyingly, all 4 resins exhibited enzymatic activity relative to a control where the wild-type protein was subjected to a Glaser–Hay reaction with the resin. All resin absorbances were normalized to resin alone that had never been exposed to protein. Despite the differences in solution phase activities, the resins exhibited comparable catalysis between all mutants over the duration of the assay. This is likely due to the decreased concentration of proteins on the resins, resulting in saturation of the protein with relatively small amounts of substrate. Thus, the catalysis became diffusion limited. All assays were performed in triplicate on unique resin samples to demonstrate reproducibility. Resin coupling reactions were also performed with GFP-151-pPrF to ensure that Glaser–Hay conditions facilitated protein immobilization by observing protein fluorescence on the resin (see ESI†).
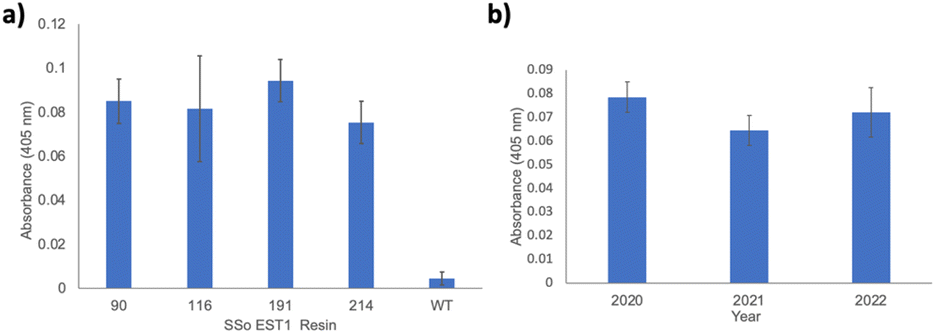 | ||
| Fig. 3 Immobilized SSo-EST1 assays. (a) Enzymatic assay of immobilized SSo-EST1 mutants. All assays were performed in triplicate on different resins. (b) Recyclability of the SSo EST1-116pPrF resin over a three-year period stored at room temperature. | ||
With immobilized and catalytically active SSo EST1 prepared, we next examined the advantages of the solid-support. One major advantage conferred to immobilized enzymes is a degree of recyclability. Consequently, the SSo EST1-116-pPrF resin was subjected to multiple assays over the course of three years and its activity was monitored. Moreover, due to the pandemic, this resin was accidently left on the benchtop for 6 months at room temperature from February 2020 to July 2020. Normally, the resins are stored in PBS buffer at 4 °C when not being actively assayed. Interestingly, despite these factors very little activity was lost. A sample of wild type protein that was also on the benchtop for 6 months demonstrated a 80% reduction in activity relative to freshly expressed enzyme. Additionally, this resin was subjected to assay conditions 18 times over the three years with little loss in activity (Fig. 3b). These results demonstrate both the stability and recyclability of the immobilized SSo EST1 over multiple years, as resins were simply washed with buffers and stored between each assay.
In an attempt to leverage the increased stability of the immobilized protein, we next became interested in determining its activity in non-aqueous solvents. This has distinct applications to expanding the utilization of enzymes to industrial applications. Previous work demonstrated increased GFP stability in a range of organic solvents, and ideally this stability should extend to SSo EST1. Moreover, several other studies have demonstrated solution-based enzymatic activity of thermophilic enzymes in organic solvents; however, the studies were limited to low percentages of organic solvents in aqueous solutions. To perform an adequate comparison, the wild-type enzyme was first examined in solution to assess activity. Due to experimental conditions and miscibility considerations, we limited this initial study to THF. The enzyme was assayed in 0%, 10%, 25% and 50% THF under identical assay conditions (60 min, 60 °C) and demonstrated a gradually decreased activity with increasing THF (Fig. 4a). As a consequence of the protein being already in an aqueous solution, trials were limited to 50% THF. Interestingly, when immobilized SSo EST1-191-pPrF was subjected to 0%, 50% and 100% THF, activity of the enzyme was shown to increase (Fig. 4b). Due to background hydrolysis of the substrate in THF, all data points were normalized to controls using mixtures containing the pNp-Hex without resin or Sso EST1. We initially hypothesized that the surprising increase in hydrolysis was due to non-specific acid–base catalysis occurring with amino acid residues on the protein. To ensure that the activity was SSo EST1 specific, the assay was also performed in the different solvents with a GFP immobilized resin. No additional substrate hydrolysis was observed beyond background, indicating that the observed activity is due specifically to the SSo EST1 enzyme. It is possible that the more organic environment could assist with the catalysis of this specific reaction due to the altered polarity of the solvent. Additionally, the high concentrations of organics could cause perturbation of non-covalent interactions within the hyperthermophilic protein that increase its ability to bind and hydrolyze the substrate.
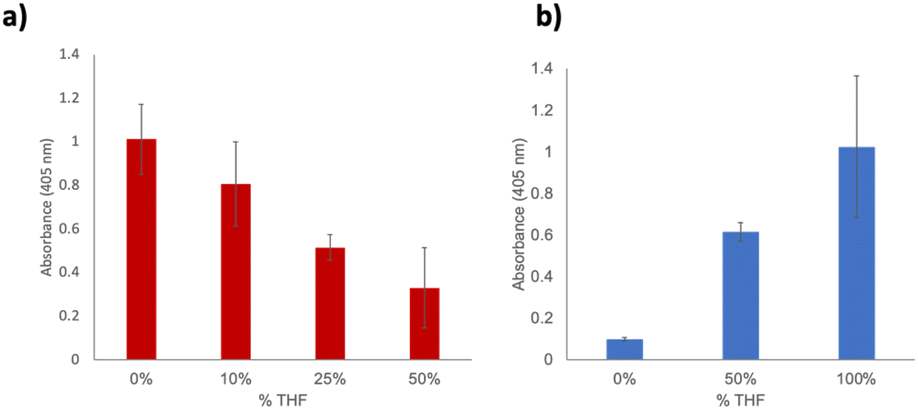 | ||
| Fig. 4 Assays of SSo EST1 in organic solvents. (a) Behavior of wild type SSo EST1 in solutions of varying THF concentrations, as THF percentage increases, enzymatic activity drops. All assays were conducted in triplicate. (b) Behavior of SSo EST-191pPrF mutant immobilized on a Sepharose resin. In this case with increased THF concentrations the immobilized enzyme performs more efficiently leading to higher activities. All assays were conducted in triplicate. | ||
Finally, given the ability of immobilized SSo EST1 to retain catalytic properties in 100% THF, we investigated other conditions where immobilization conferred increased stability of SSo EST1. Previous research has employed this enzyme in the investigation of enzymatic microwave catalysis.43 This enzyme was able to catalyze ester hydrolysis under microwave conditions at very low temperatures (−20 °C). This study was limited to aqueous solutions which were not microwave transparent, and thus required the low temperatures to prevent rapid heating of the solution. This necessitated specialized microwave reactors and made it difficult to deconvolute thermal versus microwave effects on the reaction. With the immobilized variant of the enzyme, it becomes feasible to conduct the microwave irradiation in pure THF, a microwave-transparent solvent. This allows for microwave input without solvent heating and can be used to accelerate reactions. Using a conventional CEM Discover microwave in power mode (fixing the microwave input power and allowing temperature to fluctuate), various conditions were examined, and some activity was detected even at low microwave inputs. Due to the microwave transparency of THF, reaction temperatures were limited to 53 °C. Highest enzymatic activity was observed at the maximum microwave power setting of 300 W for 30 min (Fig. 5). While continuing investigations into microwave irradiation are ongoing, these proof-of-concept experiments demonstrate the ability to conduct enzymatic catalysis under microwave conditions in organic solvents. Control reactions were performed in the absence of resin, and in the presence of immobilized GFP to ensure that the catalytic effects were due to SSo EST1.
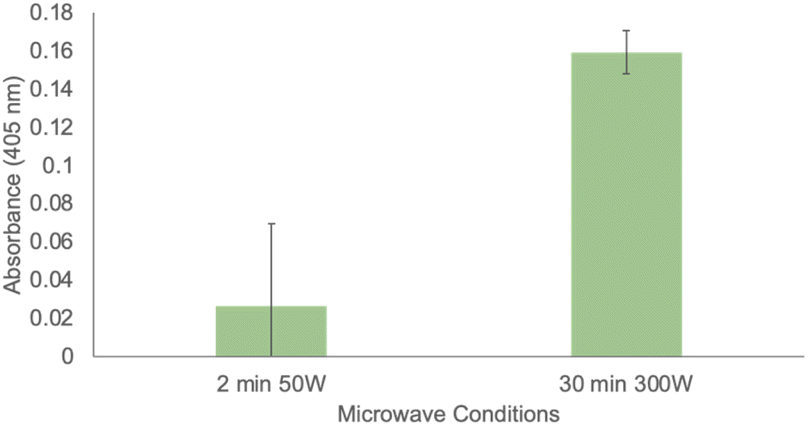 | ||
| Fig. 5 Enzymatic activity of immobilized SSo EST protein under microwave conditions. All assays were conducted in triplicate. | ||
Experimental
Expression and purification of SSo EST1
A pQE-30 plasmid harboring a variable SSo EST1 TAG mutant (0.33 μL) was co-transformed with a M15 pREP4 plasmid (0.33 μL) and a pEVOL-pPrF plasmid (0.33 μL) into BL21 (DE3) cells using an Eppendorf eporator electroporator. The cells were then plated and grown on LB agar in the presence of chloramphenicol (0.034 mg mL−1), ampicillin (0.05 mg mL−1) and kanamycin (0.01 mg mL−1) at 37 °C overnight. One colony was then used to inoculate LB media (4 mL) containing ampicillin, chloramphenicol and kanamycin. The culture was incubated at 37 °C overnight and used to inoculate an expression culture (10 mL LB media, 0.05 mg mL−1 Amp, 0.034 mg mL−1 Chlor, 0.01 mg mL−1 Kan) at an OD600 0.1. The cultures were incubated at 37 °C to an OD600 between 0.6 and 0.8 at 600 nm, and protein expression was induced by addition of pPrF (100 mL, 100 mM) and 20% arabinose (10 mL) and 0.8 mM isopropyl β-D-1-thiogalactopyranoside (IPTG; 10 mL). The cultures were allowed to shake at 30 °C for 16–20 h then centrifuged at 5000 rpm for 10 minutes and stored at −80 °C for 3 hours. The cell pellet was re-suspended using 500 μL of Bugbuster (Novagen) containing lysozyme and 200 μL Lysis buffer. The mixture was incubated at 37 °C for 20 minutes. The solution was transferred to an Eppendorf tube and centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 minutes. The supernatant was then incubated at 80 °C for 10 minutes and centrifuged at 15000 rpm for 10 minutes. Purified SSo EST1 was analyzed by SDS-PAGE (10%) and employed without further purification. Protein concentrations were determined by UV-vis absorbance at 280 nm and via a BCA assay (VWR).
000 rpm for 10 minutes. The supernatant was then incubated at 80 °C for 10 minutes and centrifuged at 15000 rpm for 10 minutes. Purified SSo EST1 was analyzed by SDS-PAGE (10%) and employed without further purification. Protein concentrations were determined by UV-vis absorbance at 280 nm and via a BCA assay (VWR).
Aqueous esterase activity assay
The reaction mixture was prepared in triplicate by adding 2 μL of SSo EST1 TAG variant protein to a mixture of 76 μL of 100 mM Acetate buffer, 4 μL of 4-nitrophenol-hexanoate (0.14 g mL−1), and 18 μL of 1× PBS (pH 7.2). A control mixture was also made in triplicate, which did not contain protein. The control and reaction mixtures were incubated at 80 °C for 60 min. After incubation, 100 μL of saturated sodium bicarbonate was added to each Eppendorf tube. The absorbance of each assay was measured by UV-vis spectroscopy on either a Thermo Scientific Nanodrop 2000 spectrophotometer or a BioTek Synergy HT microplate reader equipped with a BioTek take3 plate.General Glaser–Hay immobilization conditions
CuI (500 mM in H2O, 5 μL) and tetramethylethylenediamine (500 mM, TMEDA, 5 μL) were added to a sterile 1.5 mL Eppendorf tube and allowed to equilibrate for 10 minutes. Next, 30 mg derivatized Sepharose 6B resin (GE Healthcare) was added and allowed to equilibrate for an additional 10 minutes followed by the addition of 20 μL SSo EST1/pPrF or GFP/pPrF. The reactions were then diluted in phosphate buffered saline solution at pH 7.4 (PBS, 10 μL). A control reaction was prepared using 20 μL SSo EST1 WT in place of SSo EST/pPrF. A second control reaction was prepared using 20 μL SSo EST1/pAzF, 30 mg derivatized Sepharose 6B resin (GE Healthcare), and 10 μL of PBS buffer in place of the catalyst system. The reaction was shaken at 200 rpm for 6 hours at 37 °C. The reactions were then transferred to an empty spin column and washed with 1× PBS buffer (10 × 200 μL) and centrifuged at 3600 rpm for 1 minute. Resins were then diluted in PBS buffer (200 μL) and analyzed for protein activity via the previously described assay. Immobilized protein assays were performed previously described but replacing the 30 μL SSo EST1 protein with 30 mg SSo EST1 resin.Conclusions
This research demonstrates the application of genetic code expansion toward protein immobilization. The site-specific nature of ncAA incorporation allows for engineering of a unique handle into proteins that facilitates highly controlled immobilization reactions. Moreover, this covalent linkage has been demonstrated to dramatically increase the recyclability and stability of the immobilized protein. The hyperthermophilic SSo EST1 protein not only retained its activity over the course of 18 assays across 3 years, but also remained stable when stored at room temperature for 6 months. Additionally, immobilization facilitated the transition to enzymatic catalysis in organic solvents and under microwave conditions. The ability to utilize a biological macromolecule under these abiotic conditions has far-reaching applications to exploit enzymes in numerous settings that were previously precluded due to aqueous requirements. Moreover, much of this work is transferable to other enzymes that catalyze more industrially useful transformations. Future work is ongoing to further examine the increased stability of the enzyme, investigate the mechanisms associated with non-aqueous catalysis, and probe the advantages of the site-specific immobilization with respect to protein orientation.Conflicts of interest
There are no conflicts to declare.Notes and references
- J. L. Adrio and A. L. Demain, Microbial enzymes: tools for biotechnological processes, Biomolecules, 2014, 4(1), 117–139 CrossRef PubMed.
- A. R. Ismail, H. Kashtoh and K. H. Baek, Temperature-resistant and solvent-tolerant lipases as industrial biocatalysts: Biotechnological approaches and applications, Int. J. Biol. Macromol., 2021, 187, 127–142 CrossRef CAS PubMed.
- J. L. Porter, R. A. Rusli and D. L. Ollis, Directed Evolution of Enzymes for Industrial Biocatalysis, Chembiochem, 2016, 17(3), 197–203 CrossRef CAS PubMed.
- S. Datta, R. L. Christena and Y. R. Rajaram, Enzyme immobilization: an overview on techniques and support materials, Biotech, 2013, 3(1), 1–9 Search PubMed.
- M. Padariya, M. Baginski, M. Babak and U. Kalathiya, Organic solvents aggregating and shaping structural folding of protein, a case study of the protease enzyme, Biophys. Chem., 2022, 291, 106909 CrossRef CAS PubMed.
- H. Cui, L. Eltoukhy and L. Zhang, Less Unfavorable Salt Bridges on the Enzyme Surface Result in More Organic Cosolvent Resistance, Angew. Chem., Int. Ed. Engl., 2021, 60(20), 11448–11456 CrossRef CAS PubMed.
- J. Lee, J. H. Ko, E. W. Lin, P. Wallace, F. Ruch and H. D. Maynard, Trehalose hydrogels for stabilization of enzymes to heat, Polym. Chem., 2015, 6(18), 3443–3448 RSC.
- M. Meldal and S. Schoffelen, Recent advances in covalent, site-specific protein immobilization, F1000Research, 2016, 5, 2303 Search PubMed.
- M. Rashidian, J. M. Song, R. E. Pricer and M. D. Distefano, Chemoenzymatic reversible immobilization and labeling of proteins without prior purification, J. Am. Chem. Soc., 2012, 134(20), 8455–8467 CrossRef CAS PubMed.
- C. Mateo, J. Palomo, G. Fernandez-Lorente, J. Guisan and R. Fernandez-Lafuente, Improvement of enzyme activity, stability and selectivity via immobilization techniques, Enzyme Microb. Technol., 2007, 40(6), 1451–1463 CrossRef CAS.
- D. Brady and J. Jordaan, Advances in enzyme immobilisation, Biotechnol. Lett., 2009, 31(11), 1639–1650 CrossRef CAS PubMed.
- M. Wilchek and T. Miron, Oriented versus random protein immobilization, J. Biochem. Biophys. Methods, 2003, 55(1), 67–70 CrossRef CAS.
- F. Rusmini, Z. Zhong and J. Feijen, Protein immobilization strategies for protein biochips, Biomacromolecules, 2007, 8(6), 1775–1789 CrossRef CAS.
- T. Elgren, O. Zadvorny and E. Brecht, Immobilization of active hydrogenases by encapsulation in polymeric porous gels, Nano Lett., 2005, 5(10), 2085–2087 CrossRef CAS.
- N. Nawani, R. Singh and J. Kaur, Immobilization and stability studies of a lipase from thermophilic Bacillus sp: The effect of process parameters on immobilization of enzyme, Electron. J. Biotechnol., 2006, 9(5) DOI:10.4067/S0717-34582006000500011.
- K. Polizzi, A. Bommarius, J. Broering and J. Chaparro-Riggers, Stability of biocatalysts. Review, Curr. Opin. Chem. Biol., 2007, 11(2), 220–225 CrossRef CAS PubMed.
- M. T. Smith, J. C. Wu, C. T. Varner and B. C. Bundy, Enhanced protein stability through minimally invasive, direct, covalent, and site-specific immobilization, Biotechnol. Prog., 2013, 29(1), 247–254 CrossRef CAS PubMed.
- B. K. Raliski, C. A. Howard and D. D. Young, Site-specific protein immobilization using unnatural amino acids, Bioconjug. Chem., 2014, 25(11), 1916–1920 CrossRef CAS PubMed.
- S. I. Lim, Y. Mizuta, A. Takasu, Y. H. Kim and I. Kwon, Site-specific bioconjugation of a murine dihydrofolate reductase enzyme by copper(I)-catalyzed azide-alkyne cycloaddition with retained activity, PLoS One, 2014, 9(6), e98403 CrossRef PubMed.
- M. H. Seo, J. Han, Z. Jin, D. W. Lee, H. S. Park and H. S. Kim, Controlled and oriented immobilization of protein by site-specific incorporation of unnatural amino acid, Anal. Chem., 2011, 83(8), 2841–2845 CrossRef CAS PubMed.
- K. Hernandez and R. Fernandez-Lafuente, Control of protein immobilization: coupling immobilization and site-directed mutagenesis to improve biocatalyst or biosensor performance, Enzyme Microb. Technol., 2011, 48(2), 107–122 CrossRef CAS PubMed.
- A. Ikeda-Boku, K. Kondo and S. Ohno, Protein fishing using magnetic nanobeads containing calmodulin site-specifically immobilized via an azido group, Biochem. J., 2013, 154(2), 159–165 CrossRef CAS.
- D. D. Young and P. G. Schultz, Playing with the Molecules of Life, ACS Chem. Biol., 2018, 13(4), 854–870 CrossRef CAS.
- T. S. Young and P. G. Schultz, Beyond the Canonical 20 Amino Acids: Expanding the Genetic Lexicon, J. Biol. Chem., 2010, 285(15), 11039–11044 CrossRef CAS.
- C. Liu, P. Schultz, R. Kornberg, C. Raetz, J. Rothman and J. Thorner, Adding New Chemistries to the Genetic Code. Review|Book Chapter, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS PubMed.
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Expanding the genetic code of E. coli, Science, 2001, 292(5516), 498–500 CrossRef CAS PubMed.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli, J. Am. Chem. Soc., 2002, 124(31), 9026–9027 CrossRef CAS PubMed.
- A. Deiters and P. G. Schultz, In vivo incorporation of an alkyne into proteins in Escherichia coli, Bioorg. Med. Chem. Lett., 2005, 15(5), 1521–1524 CrossRef CAS.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition, J. Am. Chem. Soc., 2003, 125(11), 3192–3193 CrossRef CAS.
- E. Sletten and C. Bertozzi, Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality, Angew. Chem. Int. Ed., 2009, 48(38), 6974–6998 CrossRef CAS.
- J. Kalia and R. Raines, Advances in Bioconjugation, Curr. Org. Chem., 2010, 14(2), 138–147 CrossRef CAS PubMed.
- K. Nwe and M. W. Brechbiel, Growing applications of "click chemistry" for bioconjugation in contemporary biomedical research, Cancer Biother. Radiopharm., 2009, 24(3), 289–302 CrossRef CAS PubMed.
- J. S. Lampkowski, J. K. Villa, T. S. Young and D. D. Young, Development and Optimization of Glaser-Hay Bioconjugations, Angew. Chem., Int. Ed. Engl., 2015, 54, 9343–9346 CrossRef CAS.
- J. Maza, Z. Nimmo and D. D. Young, Expanding the scope of alkyne-mediated bioconjugations utilizing unnatural amino acids, Chem. Commun., 2016, 52(1), 88–91 RSC.
- C. Travis, G. Gaunt, E. King and D. D. Young, Genetic encoding of a bioconjugation handle for [2+2+2] cycloaddition reactions, Chembiochem, 2020, 21, 310–314 CrossRef CAS PubMed.
- D. Cowan, Thermophilic proteins: Stability and function in aqueous and organic solvents, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 1997, 118(3), 429–438 CrossRef CAS PubMed.
- H. Radianingtyas and P. Wright, Alcohol dehydrogenases from thermophilic and hyperthermophilic archaea and bacteria, FEMS Microbiol. Rev., 2003, 27(5), 593–616 CrossRef CAS.
- G. D. Haki and S. K. Rakshit, Developments in industrially important thermostable enzymes: a review, Bioresour. Technol., 2003, 89(1), 17–34 CrossRef CAS PubMed.
- A. Sehgal and R. Kelly, Enantiomeric resolution of 2-aryl propionic esters with hyperthermophilic and mesophilic esterases: Contrasting thermodynamic mechanisms, J. Am. Chem. Soc., 2002, 124(28), 8190–8191 CrossRef CAS PubMed.
- D. Comfort, S. Chhabra and S. Conners, Strategic biocatalysis with hyperthermophilic enzymes, Green Chem., 2004, 6(9), 459–465 RSC.
- C. R. Travis, L. E. Mazur, E. M. Peairs, G. H. Gaunt and D. D. Young, Mechanistic investigation and further optimization of the aqueous Glaser-Hay bioconjugation, Org. Biomol. Chem., 2019, 17(13), 3396–3402 RSC.
- J. C. Maza, Z. M. Nimmo and D. D. Young, Expanding the scope of alkyne-mediated bioconjugations utilizing unnatural amino acids, Chem. Commun., 2016, 52(1), 88–91 RSC.
- D. D. Young, J. Nichols, R. M. Kelly and A. Deiters, Microwave activation of enzymatic catalysis, J. Am. Chem. Soc., 2008, 130(31), 10048 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00392b |
| ‡ Authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2023 |