
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy on molecular mechanisms of CD4 dependency and independency of HIV-1 gp120†
Meng-Ting Liuab,
Jian-Xin Shene,
Xin-Wei Liab,
Li Yangd,
Yi Li d,
Peng Sang*abc and
Li-Quan Yang*abc
d,
Peng Sang*abc and
Li-Quan Yang*abc
aCollege of Agriculture and Biological Science, Dali University, Dali 671000, China. E-mail: pengsang@dali.edu.cn; ylqbioinfo@dali.cdu.cn
bKey Laboratory of Bioinformatics and Computational Biology of the Department of Education of Yunnan Province, Dali University, Dali 671000, China
cYunnan Key Laboratory of Screening and Research on Anti-pathogenic Plant Resources from West Yunnan, Dali University, Dali 671000, China
dCollege of Mathematics and Computer Science, Dali University, Dali 671000, China
eState Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, School of Life Sciences, Yunnan University, Kunming, China
First published on 21st February 2023
Abstract
Different HIV-1 strains have different antibody neutralization phenotypes (or CD4-dependencies). However, the molecular mechanisms underlying these differences remain to be elucidated. In this study, we constructed gp120 structural models from the CD4-dependent, neutralization-resistant JR-FL strain and the CD4-independent, neutralization-sensitive R2 strain and carried out several conventional molecular dynamics (MD) simulations and free energy landscape (FEL) constructions. Comparative analyses of the MD simulations and FELs indicated that R2 gp120 had higher global structural flexibility and greater conformational diversity than JR-FL gp120. This provides the preconditions for R2 gp120 to adopt a more open conformation than JR-FL gp120. Essential dynamics (ED) analysis showed that the collective motions of R2 gp120 tend towards an open state while those of JR-FL gp120 tend to retain a closed state. Based on conformational selection theory, R2 gp120's more readily sampled open state makes it more sensitive to neutralizing antibodies (or more CD4-independent) than JR-FL gp120, which may explain why the HIV-1 R2 and JR-FL strains show CD4-independent and -dependent phenotypes, respectively. Our study provides thermodynamic and kinetic insights into the CD4-dependent and -independent molecular mechanisms of HIV-1 gp120 and helps shed light on HIV-1 immune evasion.
1. Introduction
Human immunodeficiency virus type 1 (HIV-1) is the main pathogen that causes acquired immunodeficiency syndrome (AIDS). Enveloped virus surface glycoproteins play critical roles in the initial viral infection events, mediating virion attachment to cells and fusion of the viral and cellular membranes. The HIV-1 envelope (Env) spike is comprised of three gp120–gp41 glycoprotein heterodimers and plays a key role in viral entry and immune escape.1–4 The gp120 and gp41 subunits are generated by proteolytic cleavage of a precursor polypeptide, gp160.5,6 Gp120 directs target-cell recognition and viral tropism by interacting with the cell-surface receptor CD4 and one of several chemokine receptor family member coreceptors.7–9 The gp41 subunits then promote viral and cellular membrane fusion, a process that results in viral genome release into the host cell.10 In the Env, the three transmembrane gp41 molecules are obscured by the three extra-membrane gp120 molecules, while most of the gp120 structural region is exposed on the viral surface. After viral infection, most of an infected person's serum antibodies are against specific gp120 epitopes.10 Thus, gp120 is the most important Env immunogen.Previous studies have successively characterized the crystal structures of many HIV-1 outer membrane glycoprotein gp120 monomers and their complexes when combined with various antibodies. For example, in 1998, Kwong et al.11 and Wyatt et al.12 analyzed the complex crystal structure of the HIV-1 gp120 structural core combined with CD4 and antibody 17b. Subsequently, many more crystal structures of the gp120 core in complex with CD4 and antibody 17b or other antibodies have been identified.13–18 Before the Env trimer binds to the CD4 molecule, the trimer is in a “closed” state because the three gp120 molecules' V1/V2 ring regions are in close proximity to each other near the trimer's central axis. During the infection process, gp120 interacts with CD4 and undergoes large conformational changes.19 Then, the coreceptor (CCR5 or CXCR4) binding sites (V3 loops and bridge sheets) are exposed, while the V1/V2 regions move away from the central axis via rotational and translational movements. This process transforms the Env trimer from a “closed” state to an “open” state.
Structural studies have revealed the overall structure of the Env trimer in its CD4-bound state, including the arrangement of variable rings at the trimer vertices.20–22 Previous computational studies pointed out that in this extreme conformational state, gp120 has significant differences in dynamic properties, molecular motions, and conformational distributions.23 The closed state has a more stable, compact structure and higher global structural rigidity, especially the V1/V2 region and V3 loop, exhibiting significantly lower conformational flexibility. Closed state gp120 represents a stable “ground state” both structurally and energetically. Compared with the closed state, the gp120 open state has more conformational sub-states, larger conformational entropy, and lower thermal stability. Moreover, the open state gp120 V1/V2 domains and V3 loops exhibite flexible collective molecular motions, indicating that these domains' spatial orientation relative to the core determines the two ends' conformational differences.
Because HIV-1 Env glycoprotein gp120 is dependent on CD4 molecules for host cell infection, Guttman et al.24 suggested that HIV-1 gp120 can be divided into CD4-dependent and -independent gp120. Generally, the lower the Env CD4 dependence (i.e., HIV-1 with a lower tier), the closer its conformational state is to the open state. Thus, CD4-independent HIV-1 envelope trimers often adopt the open conformation. Conversely, the more CD4-dependent gp120 is (i.e., HIV-1 with a higher tier), the more it adopts a closed conformational state. In order to protect HIV-1 encoded proteins' conserved functional sites from host neutralizing antibodies, gp120 utilizes its sequence variation and structural flexibility to evade host immune recognition,25 resulting in different neutralizing phenotypes.26
Single molecule fluorescence resonance energy transfer (smFRET) analysis showed that the ligand-free Env trimer on the HIV-1 virus particles surface was dynamic and observed in low-, intermediate-, and high-FRET states.27 The low-FRET state is the predominant state and reflects Env's closed ground-state conformation before fusion, which was confirmed by ground-state stabilizing mutation studies. In the absence of receptor induction, ligand-free Env can spontaneously switch between different states, and the difference in this conformational transition capability is responsible for different neutralization sensitivity and resistance phenotypes among HIV-1 isolates. Guttman et al. recently used hydrogen deuterium exchange (HDX) to study different neutralizing antibodies' interactions with the HIV-1 Env trimer and the effects of its conformational changes.24 The results showed that CD4-dependent HIV-1 strains are more resistant to various antibodies and can more easily evade immune system surveillance. In contrast, CD4-independent HIV-1 is easily neutralized by antibodies and exhibits strong antibody neutralization sensitivity. The closed state Env trimer tends to bind antibodies with strong neutralizing power, while less neutralizing antibodies bind to the Env trimer only when it is in the open state. At the same time, antibody neutralization-sensitive viruses are more likely to adopt an open state than resistant viruses, so they are more likely to be bound by antibodies with weaker neutralization ability.
Previous studies showed that the CD4-bound open conformation of the ligand-free Env/gp120 from the neutralization-sensitive strain NL4-3 was larger than the CD4-bound open conformation from the anti-neutralization strain JR-FL, but the unbound closed state was still the dominant conformation for both ligand-free Env/gp120s.28 However, the specific mechanisms responsible for the phenotypic differences between these two HIV-1 strains remain unclear.29 Determining the differences in HIV-1's molecular dependence on CD4 is critical to understanding HIV-1's immune evasion mechanisms and to developing anti-HIV-1 drugs or vaccines.
In this study, we constructed CD4-dependent (neutralization-resistant) strain (JR-FL) and CD4-independent (neutralization-sensitive) strain (R2) gp120 structural models and subjected them to 10 conventional MD simulations and free energy landscape (FEL) constructions. Our results indicate that JR-FL gp120 is more structurally stable, conformationally inflexible, and less prone to spontaneous transition to the CD4-bound state compared to R2 gp120. This provides a plausible explanation for the phenotypic differences in neutralization resistance/neutralization sensitivity exhibited by viral strains JR-FL and R2.
2. Materials and methods
2.1. Sequence preparation
We randomly selected HIV-1 strains JR-FL and R2 from experimentally calibrated phenotypic lineages, of which JR-FL is a CD4-dependent infection strain. JR-FL was derived from primary, non-syncytium-inducing, macrophage tropic HIV-1 strains that were isolated from brain tissue of San Francisco area patients who were infected with clade B viruses. It has strong neutralization resistance, and the virus grade is tier3.26,30 The R2 strain is a typical CD4-independent, neutralization-sensitive infection strain.26,31 Both the JR-FL and R2 strain gp120 sequences belong to the same clade (clade B), and phylogenetic study results indicated that JR-FL is evolutionarily close to the R2 strain.31 We obtained the gp160 amino acid sequences from the Swiss-Prot protein sequence database32 (https://www.uniprot.org) with the accession numbers Q75760 and Q9WPZ4, respectively. Therefore, using JR-FL and R2 viral strains may reduce background noise and increase study result reliability. For the two gp160 sequences, the segments corresponding to the signal peptide and gp41, and some N-terminal residues of gp120 were removed. For the JR-FL gp160 sequence, we deleted the signal peptide (amino acid residues 1–29), a gp41 fragment (amino acid residues 521–711), and some of the gp120 N-terminal residues. For the R2 gp160 sequence, we manually removed the corresponding fragments of the signal peptide (amino acid residues 1–30) and gp41 sequence (amino acid residues 539–720), as well as some of the C-terminal sequence fragments. The finally obtained gp120 sequences were at positions 30–492 and 31–509, respectively. We used the software package MODELLER V9.15 for homology modeling.33 The selected template had a high degree of sequence identity with the target sequence. Taking the molecular probability density function minimum value as the standard, the structure with the best stereochemical quality was selected as the final model from the 20 constructed candidate models.For clarity in subsequent analysis, the numbering of two gp120s needs to be consistent with the template sequence, and we numbered conventional secondary structure elements according to the HXBc2 crystal structure conventions (PDB: 3J70 and 1G9M).13,34 We performed structure-based multiple sequence alignments between JR-FL gp120 and R2 gp120 using R2 gp120 as the template to determine the structurally equivalent residue positions (Fig. 1). The alignment results were obtained using ESPript (https://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi).
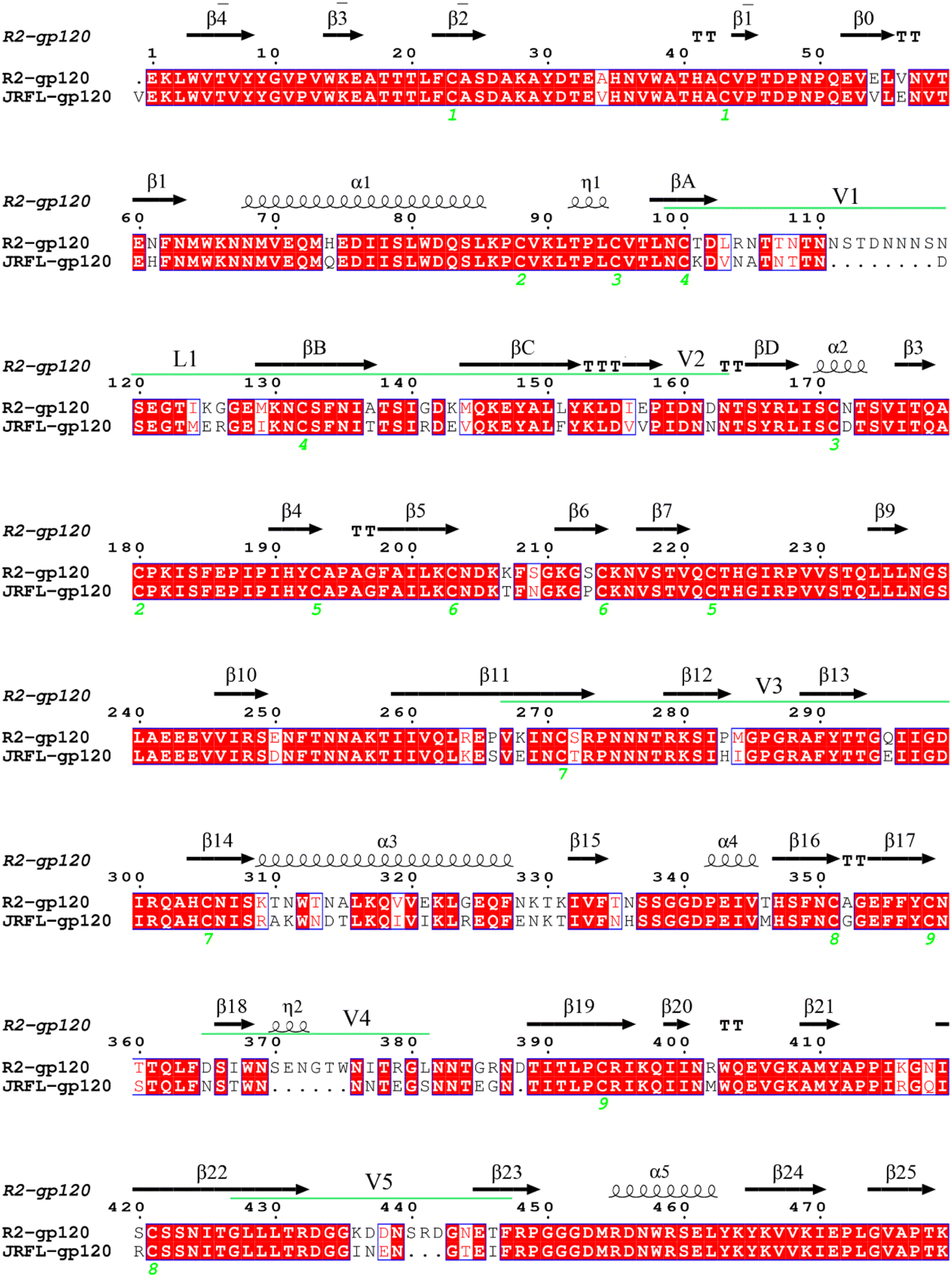 | ||
| Fig. 1 Target and template sequence alignment. R2 gp120 was used as the template sequence with known crystal structure. Residues are numbered according to the template sequence. Conserved residues are shaded in light red. Regular secondary structural elements are numbered according to the HXBc2 crystal structures (PDB IDs:1G9M and 3J70) with black arrows and black spirals representing β-strands and α-helices (or 3/10 helices), respectively. The variable regions (V1/V2, V3, V4, and V5) are indicated by green line segments above the alignment. The four β-strands in the V1/V2 region, designated A to D, are labelled βA to βD, respectively. The V1 and V2 loops are located between βA and βB and between βC and βD, respectively, and the loop connecting βB and βC is labelled L1. | ||
2.2. MD simulations
The selected CD4-dependent (neutralization-resistant) and -independent (neutralization-sensitive) gp120 structural models were individually used as starting structures for the MD simulations performed using the software GROMACS 2020.6 with the AMBER99SB-LIDN force field.35,36 Each starting structure was described with the TIP3P water model37 in a dodecahedron box with a protein-wall minimum distance of 0.8 nm. Na+ (126 and 130 for the JR-FL gp120 and R2 gp120 systems, respectively) and Cl− (122 and 132 for the two systems, respectively) ions were also introduced to achieve an electroneutral system at a salt concentration of 150 mM. In order to eliminate unreasonable contact between atoms and stereochemical conflicts, the simulation systems were subjected first to energy minimizations using the steepest descent algorithm (the number of steps of steepest descent and conjugate gradient was 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), and then to the conjugate gradient energy minimization until the system's potential energy no longer changed significantly (the maximum force ≤ 10 kJ mol−1 nm−1). To effectively soak the solute in the solvent, 200 ps position-restrained simulations were carried out in which the protein's heavy atoms were restrained by decreasing harmonic potential force.38 In the position-restrained molecular dynamics simulations, the harmonic position binding constants (Kposres 1/4 1000, 100, 10, and 0 kJ mol−1 nm−2) on the protein's heavy atoms were reduced, and the heavy atoms were subjected to a suppressive force of 1000 kJ mol−1 nm−2 to limit their fluctuations. The water molecules were not subjected to the suppressive force and were free to move. After the position-restrained simulations were completed, the system was subjected to MD production simulations for 100 ns.
000), and then to the conjugate gradient energy minimization until the system's potential energy no longer changed significantly (the maximum force ≤ 10 kJ mol−1 nm−1). To effectively soak the solute in the solvent, 200 ps position-restrained simulations were carried out in which the protein's heavy atoms were restrained by decreasing harmonic potential force.38 In the position-restrained molecular dynamics simulations, the harmonic position binding constants (Kposres 1/4 1000, 100, 10, and 0 kJ mol−1 nm−2) on the protein's heavy atoms were reduced, and the heavy atoms were subjected to a suppressive force of 1000 kJ mol−1 nm−2 to limit their fluctuations. The water molecules were not subjected to the suppressive force and were free to move. After the position-restrained simulations were completed, the system was subjected to MD production simulations for 100 ns.
The protocol and parameters for the production dynamics simulation were as follows: integration time step was 2 fs with the LINCS algorithm39 constraining all bond lengths; long-range electrostatic interactions were handled by the Particle Mesh Ewald (PME) method40 with a cut-off of 1.0 nm (for electrostatic and Lennard–Jones interactions); a twin-range cut-off was used for van der Waals (VDW) interaction calculations with short- and long-range cut-off distances set to 1.0 and 1.4 nm, respectively; solute and solvent temperatures were separately coupled to a 300 K heat bath with a coupling constant τt of 0.1 ps;15 and pressure was maintained at 1 atm using a Parrinello–Rahman barostat40,41 with a coupling constant τp of 0.5 ps. To improve the conformational sampling efficiency, we performed 10 conventional MD simulation strategies for each system. For each system, we performed ten 100 ns production MD simulations, each starting at a different initial atomic velocity assigned by the Maxwell distribution at 300 K.
2.3. Dynamic property analysis
The root mean square deviation (RMSD) and Cα root mean square fluctuation (RMSF) were calculated using the GROMACS tools “gmx rmsd” and “gmx rmsf”, respectively.36 The RMSD value can be used to measure the degree of deviation or conformational difference of a protein structure relative to the reference structure. For simulated trajectories, the RMSD value fluctuation can reflect the degree of protein structure stability during the simulation. For molecular dynamics simulation trajectories, the amino acids' mobility or flexibility during the simulation can be assessed by calculating the RMSF values of Cα or backbone atoms in amino acid residues.2.4. Essential dynamics analysis
Mathematically, principal component analysis (PCA) is an effective method for data dimensionality reduction analysis and is also called the essential dynamics (ED) method when applied to molecular dynamics simulation trajectory analyses.42 Gp120 collective motion was determined by PCA on a covariance matrix constructed from the Cα atomic fluctuations in the MD trajectory. A set of eigenvectors/principal components (PCs) and corresponding eigenvalues were obtained, representing the collective motion patterns and amplitudes of the protein structure's atomic fluctuations. Porcupine plots were produced along the protein motion of the first few eigenvectors, which were obtained using the “modevectors.py” script in PyMol with the two extreme eigenvector projection values as input. In the porcupine plots, the length and direction of the burr stretched on the Cα atom represented the atom's fluctuation amplitude and motion direction, respectively, and the protein's motion pattern along the eigenvectors could be clearly observed through the porcupine plots.2.5. Free energy landscape
A protein–solvent system's free energy landscape (FEL) can describe the correspondence between a protein's conformational state and its free energy. Therefore, the FEL can be used to characterize not only the protein's thermodynamic properties (i.e., the probability distribution of different conformational states), but also its energetic properties (i.e., conformational transitions between different states).43,44 In this study, the first and second eigenvectors were used as reaction coordinates to reconstruct the JR-FL gp120 and R2 gp120 free energy spectra. FELs with eigenvectors 1 and 2 as the reaction coordinates were constructed using the probability density function as follows:where kB is the Boltzmann's constant, T is the temperature, Ni is the population of bin i, Nmax is the population of the most populated bin, and F(s) is the free energy of state i.
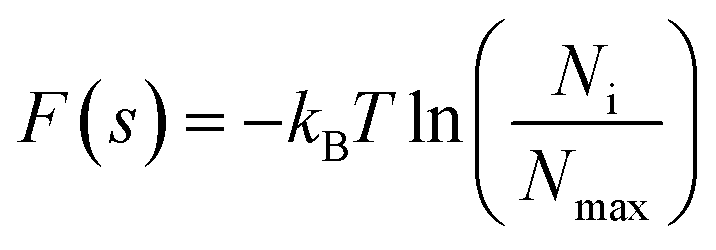
3. Results
3.1. Structural model description
The constructed JR-FL gp120 and R2 gp120 structural models are shown in Fig. 2. The two gp120 variants have 81% sequence identity, indicating that they are highly similar. The JR-FL and R2 gp120 structural models contain 463 and 479 amino acid residues, respectively. Overall, both structures can be divided into three parts: the inner domain, the outer domain, and a relatively small domain that lies beneath the juxtaposed inner and outer domains.45 Both gp120s include 5 major α-helices and 33 β-sheets. Their inner domains mainly consist of the N- and C-termini, a near-terminal seven-stranded β-sandwich (composed of β3, β0, β1, β5, β6, β7, and β25), and two α-helices (α1 and α5). Their outer domains are primarily composed of two end-to-end stacked β-barrels, one α-helix (α2), and two loop excursions (V4 and V5). The bridge layer domain comprises the V1/V2 region extending from the inner domain, the V3 loop extending from the outer domain, and the two bridging sheet elements.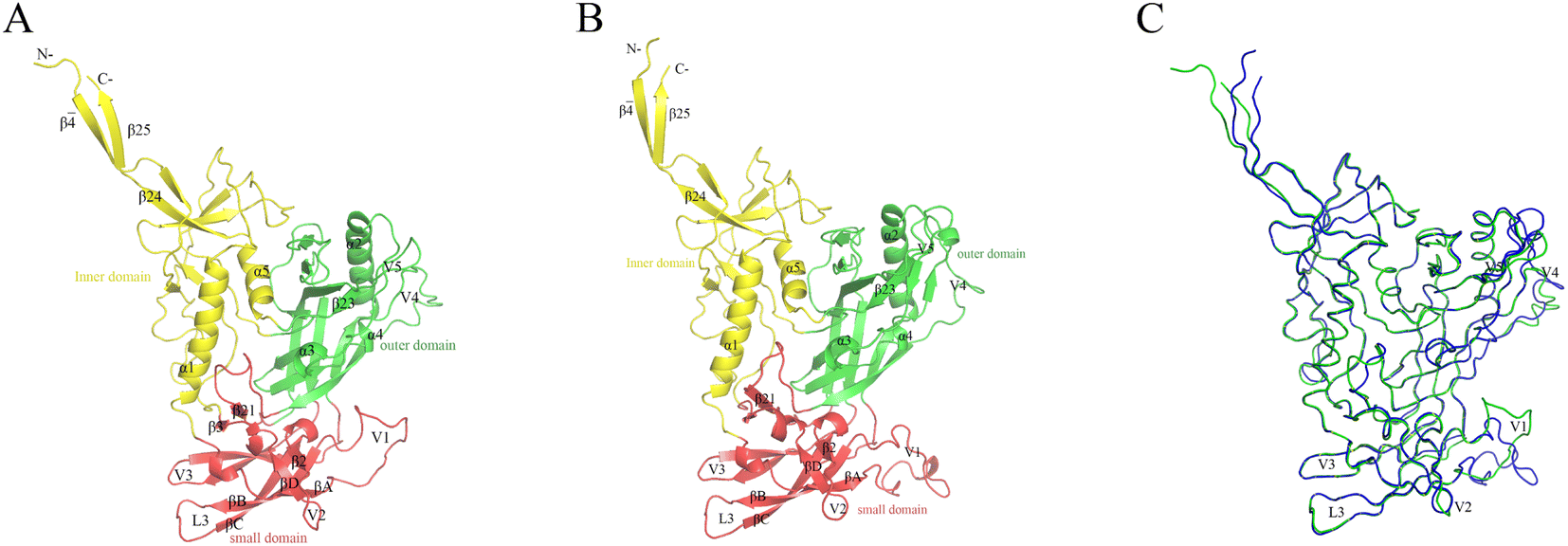 | ||
| Fig. 2 Structural models of the two near full-length gp120s in the unliganded state. These models served as the simulation system starting structures. (A) JR-FL gp120 model ribbon representation. (B) R2 gp120 ribbon representation. (C) Backbone superposition of JR-FL gp120 (green) and R2 gp120 (blue) models. | ||
3.2. Conformation stability and sampling convergence
To evaluate gp120s′ structural stability and deviation during simulations, we calculated the time evolution of the gp120 backbone RMSD values relative to their respective starting structural models (Fig. 3). It took 5 ns for each system to reach a relatively stable RMSD value. Starting from 5 ns until the end of the simulation, R2 gp120 exhibited higher RMSD values than JR-FL gp120, with most JR-FL gp120 trajectories having smaller fluctuations around 0.4 nm (Fig. 3A). The R2 gp120 replicas had wider RMSD ranges (about 0.3–0.65 nm) and their fluctuations were larger. We found that R2 gp120 experienced greater structural deviations and conformational changes during MD simulations, indicating that R2 gp120's structural stability was lower. In contrast, the JR-FL gp120 structure was more likely to stabilize. Indeed, JR-FL gp120 is usually considered to be in an anti-neutralization conformation after long-term immune selection.38,46 The lower a protein's structural stability, the higher its ability for conformational change. Thus, the less stable R2 gp120 may have a greater ability to disconnect from the unliganded state. After reaching a relatively stable RMSD equilibrium, both JR-FL and R2 gp120s fluctuated around mean RMSD values of 0.4 and 0.5 nm, respectively, in similar structural domains. In addition to subtle differences in RMSD temporal variation, the RMSD values of the two different gp120s were distributed over an approximately 0.3–0.65 nm range, indicating that gp120 can cover sufficient structural deviations to maintain essential viral infection functions while exhibiting different neutralizing phenotypes. This means that RMSD positively correlated with the degree of neutralization sensitivity, suggesting that gp120's neutralization-sensitive phenotype has a highly structural bias. The hypothesis that structural stability is negatively correlated with conformational changes is further supported by a study by Y. D. Kwon, M. Pancera et al.47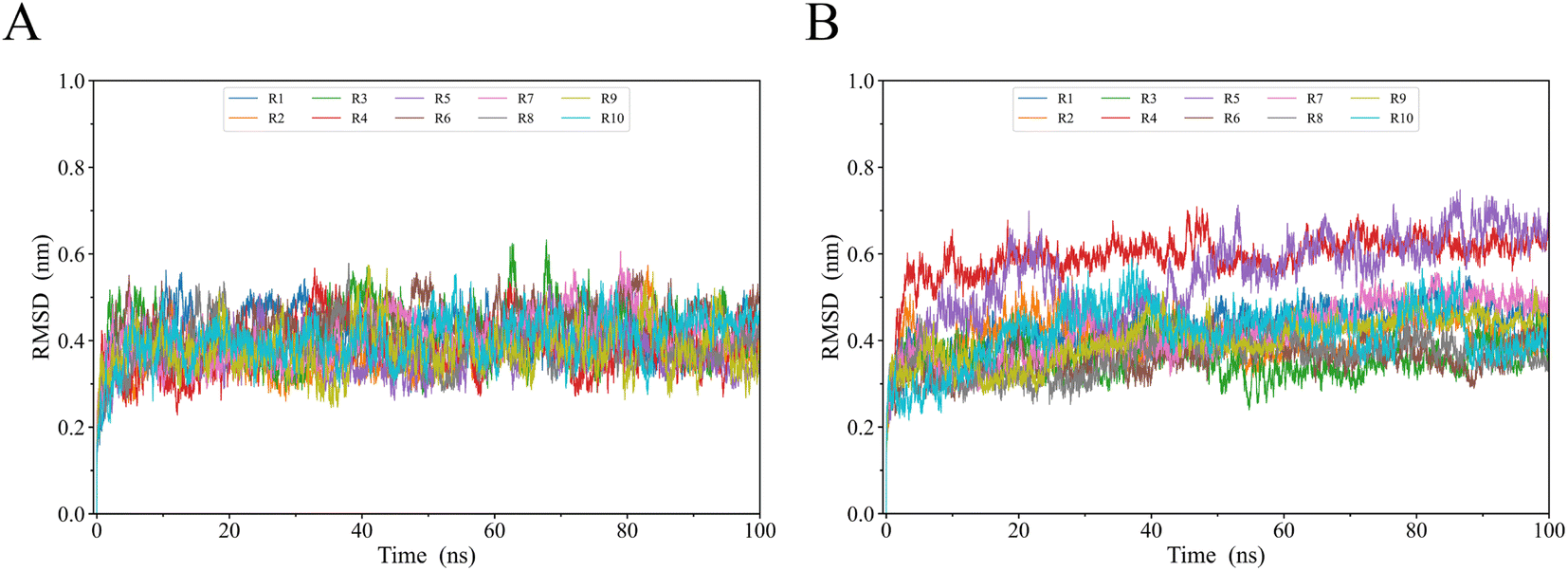 | ||
| Fig. 3 Temporal evolution of gp120 backbone RMSD values for (A) JR-FL gp120 and (B) R2 gp120 relative to their respective starting structures calculated from 8 MD simulation replicas (R1 to R10). | ||
To ensure that gp120's intrinsic properties are reflected for each simulated system, the balanced portions of each simulation (5–100 ns) were concatenated together to obtain a single 950 ns connected trace. The subsequent analysis was based on the two connections' equilibrium trajectories to ensure that the calculated parameters accurately reflected gp120's intrinsic properties.
3.3. Conformational flexibility comparison
The Cα atom RMSF value is commonly used as a protein's structural flexibility index and was calculated from the single linkage equilibrium trajectory of the two gp120s (Fig. 4). The average Cα atom RMSF value for JR-FL gp120 was 0.26 nm (dashed line in Fig. 4A), while the average Cα atom RMSF value of R2 gp120 was 0.31 nm, indicating that the CD4-independent R2 gp120 had higher global conformational flexibility compared to the CD4-dependent JR-FL gp120. The two gp120s had similar RMSF characteristics, with regular secondary structure regions N-termini, C-termini, and V1, V2, V3, V4, and V5 loops showing higher RMSF values than average ones. The ectodomain secondary structure regions had lower mean RMSF values.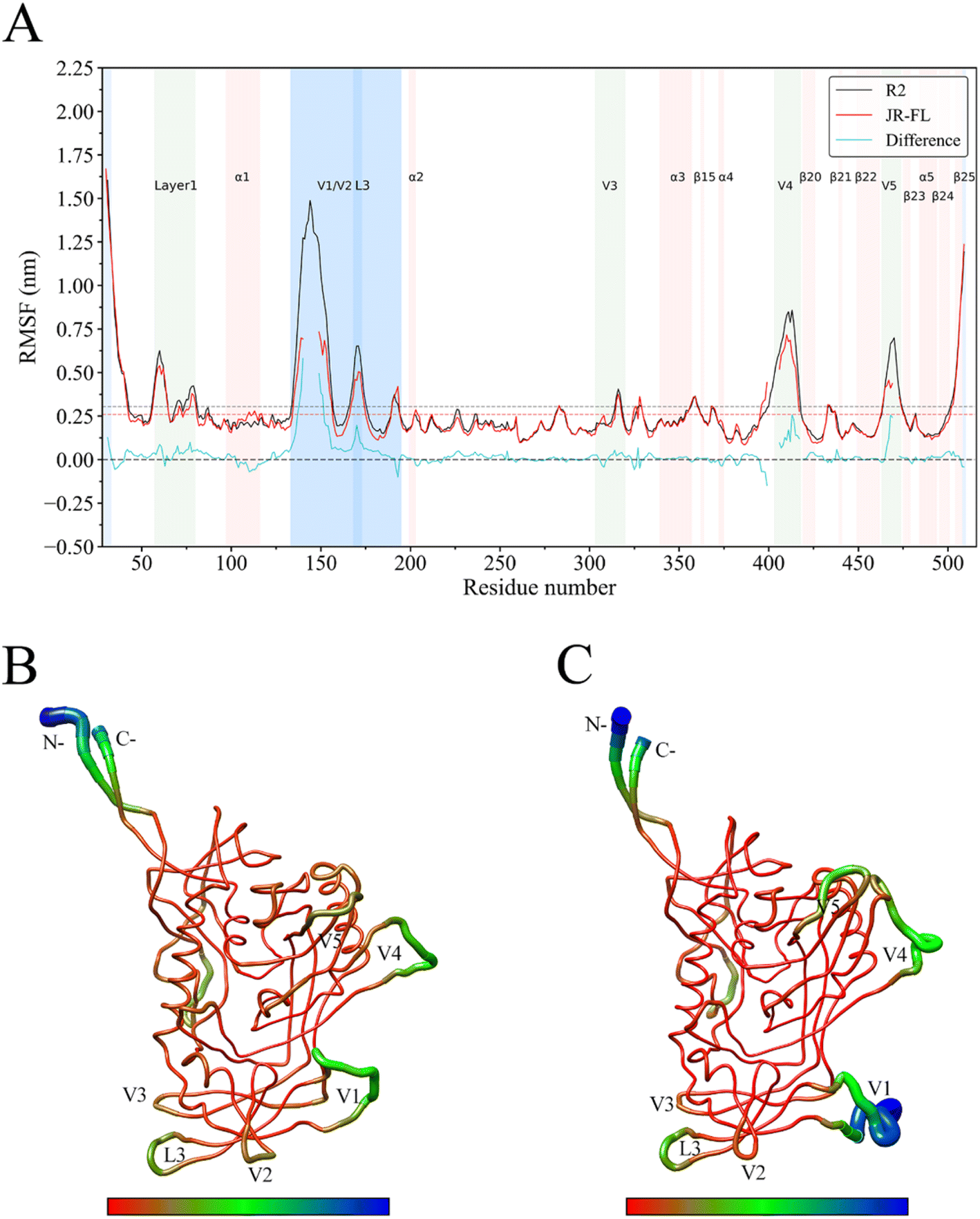 | ||
| Fig. 4 JR-FL gp120 and R2 gp120 structural flexibility comparison. (A) Per-residue average backbone RMSF profiles calculated from R2 gp120 (black line) and JR-FL gp120 (red line) MD trajectories. (B) JR-FL gp120 and (C) R2 gp120 3D backbone representation structures mapped with per-residue average backbone RMSF values. The backbone color ranges from red to blue, corresponding to line thickness and denote backbone RMSF values varying from lowest to highest. (B and C) were generated using UCSF Chimera.52 | ||
We observed that the R2 gp120 structural region RMSF was higher than that of JR-FL gp120, and we performed flexibility difference quantitative analysis by subtracting the JR-FL gp120 RMSF value from the R2 gp120 RMSF value at the structurally equivalent residue position (shown as cyan curves in Fig. 4A). The results showed that R2 gp120 had a higher conformational flexibility than JR-FL gp120 in the inner and outer regions or bridge-sheet domains (RMSF difference > 0). JR-FL gp120 showed higher flexibility (RMSF difference < 0) in only a few regions. These results indicated that CD4-independent (neutralization-sensitive) R2 gp120 had higher global conformational flexibility than CD4-dependent (neutralization-resistant) JR-FL gp120, consistent with the findings described by Guttman M., Kahn M., et al.48 This demonstrates that MD simulations can provide reliable kinetic information about proteins and that full-length ligand-free gp120 variants from different isolates have similar local structural conformational flexibility.
Larger RMSF differences were observed between residues 138–153, 169–174, 406–415, and 467–471. The marked flexibility that we observed in these regions in R2 gp120 may make disrupting subunit interactions easier, increasing the possibility that R2 gp120 opens the trimeric crown when unliganded. Compared with JR-FL gp120, R2 gp120's V4/V5 loop region has significantly higher flexibility. The greater V4/V5 mobility in R2 gp120 contributes more to β20–β21 and α5 fluctuations (V4/V5 are rigidly connected to the β20 sheet and the α5 helical layer, respectively). To more clearly show the differences in flexibility between the two gp120 variants, we mapped the CD4-dependent and -independent gp120 RMSF values to their respective three-dimensional backbone structures and represented flexibility differences by backbone color and thickness (Fig. 4B and C). Fig. 4C shows that the V1/V2 loop region had greater conformational flexibility in R2 gp120 than in JR-FL gp120.
The current analyses provide insights into gp120 unliganded trimer structural dynamics. Both the cryo-EM trimer structure in complex with antibody PGV04 and the crystal structure in complex with antibody PGT122 were obtained using the same BG505 SOSIP.664 construct studied here.49,50 These structures reveal how the V1/V2 and V3 variable regions form intimate interactions at the trimer crown. We found that several V1/V2 regions indeed have a high degree of HDX protection, which is consistent with the extensive β-sheet secondary structure present in the “Greek key” motif seen in the PG9-bound scaffolded V1/V2 structure51 and in the SOSIP.664 trimer structure. CD4-induced conformational changes in gp120 can be transmitted to HR1 via α1 and layer1, which in turn promotes gp41 precursors formation for cell membrane fusion.45 Thus, the high conformational flexibility of layer1 in R2 gp120 may promote gp41 fusion mechanisms more so than JR-FL gp120. The R2-Env initiation network is expected to open the trimeric corona more easily, exposing coreceptor binding sites and facilitating the gp41 fusion mechanism.
3.4. Essential dynamics analysis
Essential dynamics analysis can extract simulation trajectories of a protein molecule's most dominant motion patterns from the Cartesian coordinate system. These trajectories are often closely related to protein functions. Fig. 5 shows the R2 gp120 and JR-FL gp120 eigenvalues obtained from the essential dynamics analyses as a function of the eigenvector ordinates and the cumulative contribution of the eigenvectors to the total mean square fluctuations (TMSFs). We diagonalized the covariance matrices of Cα atomic position fluctuations in JR-FL gp120 and R2 gp120 equilibrium trajectories and obtained TMSF values of 123.14 and 141.04 nm2, respectively. The Cα atomic fluctuation of CD4-independent R2 strain was significantly larger than that of the CD4-dependent JR-FL strain. This indicates that R2 gp120 has stronger structural fluctuations, consistent with the previous RMSD and RMSF results.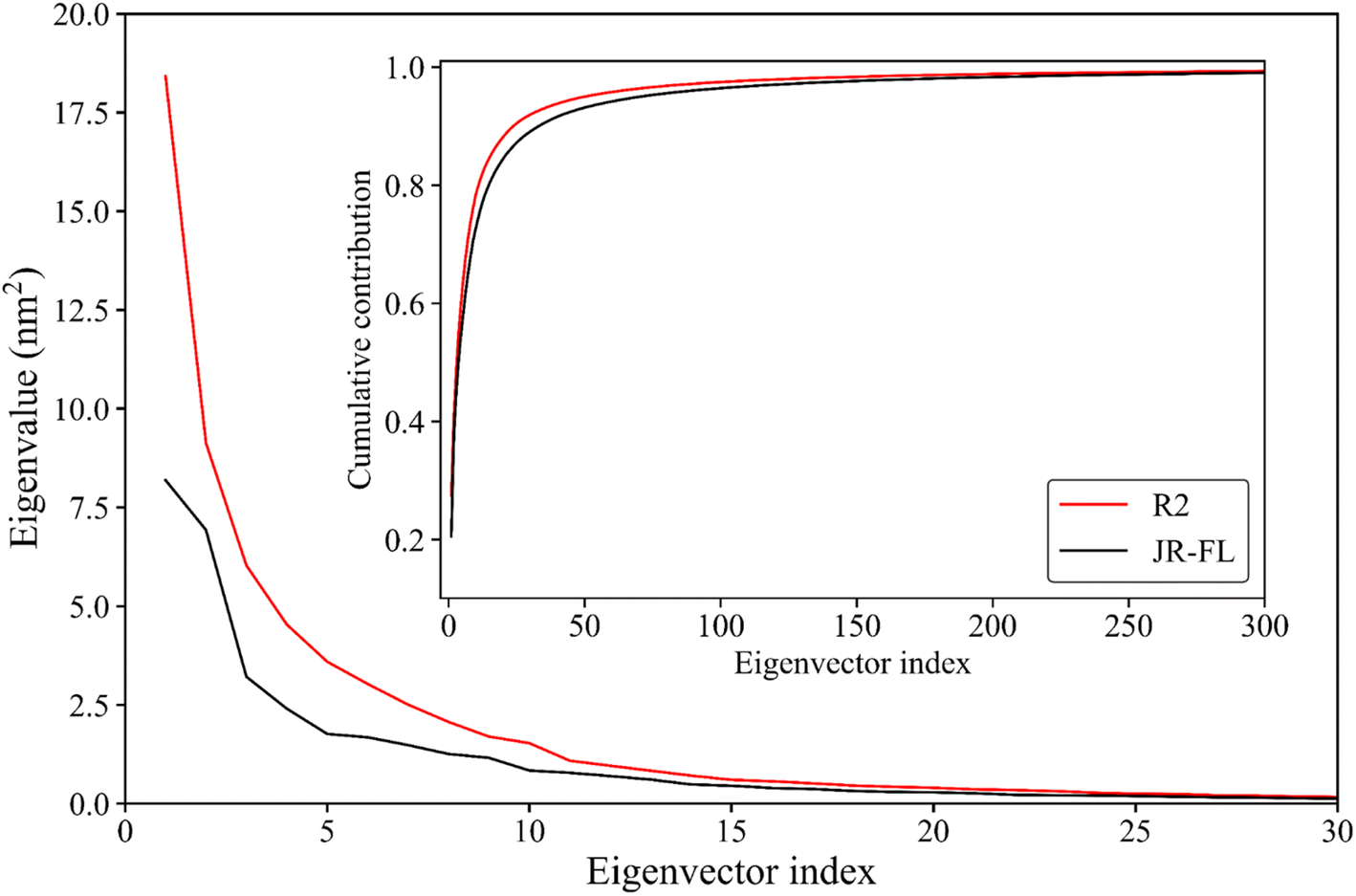 | ||
| Fig. 5 Eigenvalues of the first 30 eigenvectors (main plot) and cumulative contribution of all eigenvectors to the total mean square fluctuations (inset plot) for JR-FL gp120 (black line) and R2 gp120 (red line). | ||
The first R2 gp120 and JR-FL gp120 eigenvectors had the largest eigenvalue. With increasing eigenvector number, the corresponding eigenvalue decreased. From the fifth eigenvector, the eigenvalue decreased gently (Fig. 5). The top 15 R2 gp120 eigenvector eigenvalues were significantly higher than the corresponding JR-FL gp120 eigenvalues. The first five were especially high, indicating that R2 gp120 motion along the first five eigenvectors had significantly larger atomic fluctuation amplitudes compared to JR-FL gp120 or more severe large-scale coordinated motion. The inset of Fig. 5 shows the cumulative contribution rate of all eigenvectors to TMSF. The cumulative contribution rates of R2 gp120's first two and top five eigenvectors to TMSF were 41.09% and 62.21%, respectively. The cumulative contribution rates of JR-FL gp120's corresponding eigenvectors were 38.00% and 57.54%, respectively. The number of gp120 atoms used for the essential dynamics analysis was 479, leading to 1437 dimensions in gp120 conformational space (i.e., 3 × 479 eigenvectors). In this high-dimensional conformational space, the first ten eigenvectors (0.69% of the total number of vectors) contributed more than 75% to the TMSF (overall conformational fluctuations), indicating that our essential dynamics analysis successfully extracted the most dominant gp120 motion patterns. Compared to JR-FL gp120, R2 gp120 required a smaller number of feature vectors to achieve the same level of cumulative contribution.
3.5. Largest-amplitude collective motions
Fig. 6 shows the two gp120 variants' motion patterns along the first two eigenvectors in a porcupine diagram. The direction and length ratios of the cone drawn on the Cα atom indicate the motion direction and Cα fluctuation amplitude, respectively. Both gp120 variants' maximum vibrational fluctuations were mainly in the N-terminus, C-terminus, and V1/V2 ring regions, and the structural core and internal domain positions were rarely involved in the collective motion (Fig. 6A–D). As shown in Fig. 6A and B, the first two eigenvectors of the JR-FL gp120 describe a common rotation of the inner and outer domains (which constitute the gp120 core) in an anticlockwise direction around an axis parallel to both domains; the V3 loop and V1/V2 rotate in the same direction as that of the core. In contrast, the first two eigenvectors of the R2 gp120 (Fig. 6C and D) describes an approximately anticlockwise rotation of the gp120 core, but the V1/V2 moves in the down-right direction, exhibiting a trend to deviate from the core. We observed that R2 gp120 displacements at the same structural positions were larger, and a closer look revealed that the R2 gp120 loop regions and internal structure domains spanned more substructures involved in collective motion than JR-FL gp120 and were displaced to a greater extent. Additionally, the R2 gp120 V1/V2 and V3 loop downward movements were in the opposite direction to those observed in JR-FL gp120. The turbines rotating in two different directions cause core structure torsion, and the V1/V2 and V3 loop downshifts may facilitate the release of the V3 loop and the conformational transition from unliganded state to liganded state.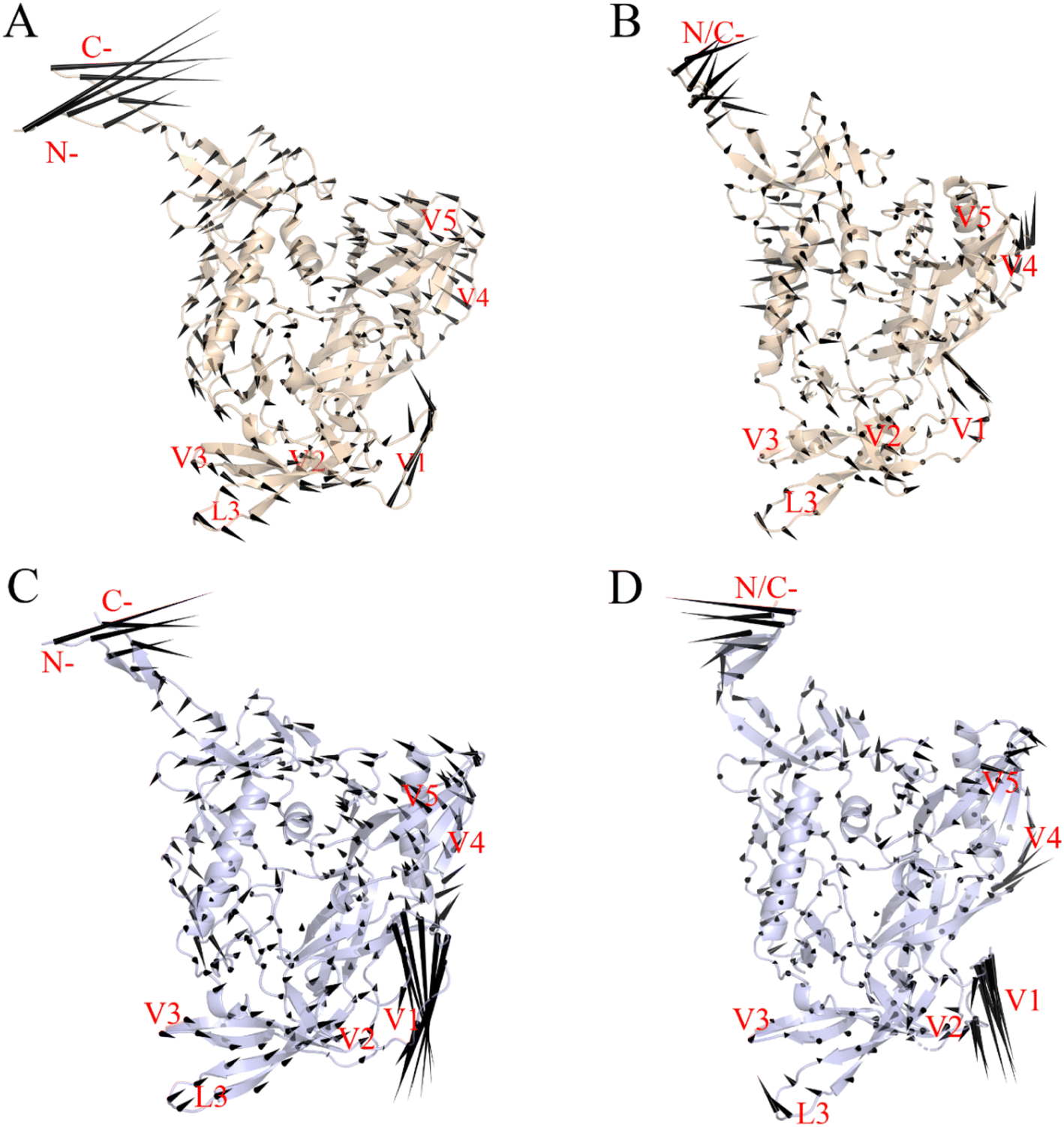 | ||
| Fig. 6 Porcupine plots showing the two gp120 structural models' largest-amplitude collective motions along the first two eigenvectors of gp120 in the JR-FL gp120 (A and B) and R2 gp120 (C and D) forms. The direction and length of the cones drawn on the Cα atoms represent the atom's fluctuation direction and amplitude, respectively. | ||
Langley et al.53 showed that β20–β21 hairpin rearrangement is a key initial step in triggering gp120's conformational shift from an unbound state to a CD4-bound state, which is a prerequisite for further V1/V2 repositioning and mature bridge formation. The gp120 V3 loop in the CD4-bound state is fully exposed and participates in mature coreceptor CD4i epitope formation,51,54 and the V1/V2 region motion along eigenvector 1 may help break its V3 loop binding. We conclude that the differences in movement direction, fluctuation magnitude, and spanning range of relevant structures involved in R2 gp120 versus JR-FL gp120 collective movements may result in different gp120 conformational presentation, i.e., CD4-dependent (neutralization-resistant) JR-FL gp120 has a greater ability to remain ligand-free, while CD4-independent (neutralization-sensitive) R2 gp120 is more likely to transit to the CD4-bound state.
3.6. Free energy landscape
We reconstructed JR-FL gp120 and R2 gp120 free energy landscapes (FELs) using the first and second eigenvector projections as reaction coordinates (Fig. 7A and B). Fig. 7C and D depict the representative structure of local energy minimum structure on the energy surface. We found that the JR-FL gp120 FEL spanned −6.2 to 8.5 nm and −8.0 to 8.9 nm on PC1 and PC2, respectively, and was elliptical, regular, and continuous. The R2 gp120 FEL spanned −10.8 to 7.6 nm and −8.9 to 8.0 nm on PC1 and PC2, respectively, and was irregular and divergent. Compared to the JR-FL gp120 FEL, the R2 gp120 FEL occupied a larger conformational subspace area composed of the first two eigenvectors. These results suggest that R2 gp120 has greater conformational entropy and more complex dynamic behavior than JR-FL gp120. In addition, the R2 gp120 FEL contains five local free energy minimum regions (free energy values below −14 kJ mol−1), while JR-FL gp120 FEL contains two. This indicates that R2 gp120 has a higher conformational diversity than JR-FL gp120. Greater conformational entropy implies higher conformational freedom or structural variability, which would allow R2 gp120 to sample more conformational states or sub-states; however, increased conformational freedom also leads to decreased R2 gp120 in the thermal and structural stability.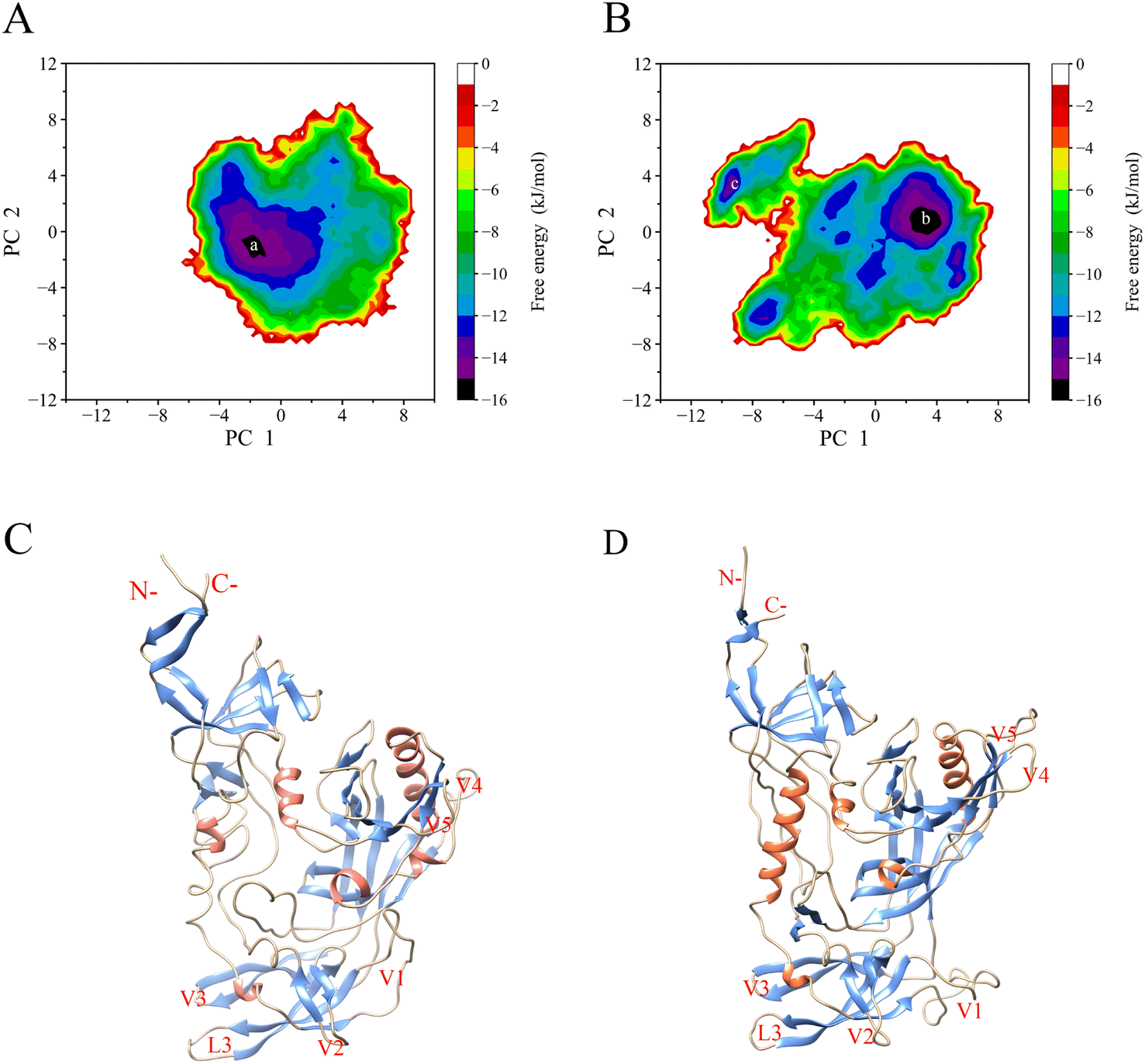 | ||
| Fig. 7 Constructed free energy landscapes (FELs) of the two forms of gp120 using projections of eigenvectors 1 and 2 as the reaction coordinates, and the representative structures of local energy minimum structure on the energy surface. (A) FEL of the JR-FL gp120. (B) FEL of the R2 gp120. (C) The representative structure of the minimum local energy structure on the JR-FL gp120 energy surface. (D) The representative structure of the minimum local energy structure on the R2 gp120 energy surface. The color bar denotes the relative free energy level in kJ mol−1. The basins/minima with free energy level lower than −15 kJ mol−1 are labelled with “a” in (A) and “b” to “c” in (B). | ||
Higher protein conformational flexibility often leads to lower thermal stability or higher thermal sensitivity. According to the FEL theory of protein–solvent systems, high conformational flexibility results in proteins having more conformational states that coexist in equilibrium as an ensemble of conformations in the free energy well at the bottom of the funnel-shaped free energy landscape harmony. The stronger a protein's conformational flexibility, the more free energy wells exist at the FEL bottom and lower free energy barrier between wells is, resulting in easier conformational transitions between different states or sub-states.7,9 In conclusion, CD4-independent R2 gp120 exhibited higher global and local conformational flexibility than CD4-dependent JR-FL gp120. This suggests that R2 gp120 has more enthalpic or entropic factors that contribute positively to its structural stability, which is consistent with previous structural and geometric property analyses of the two gp120 types.38 The higher the minimum value at the bottom of the FEL, the higher the probability that the protein can bind to the ligand.8 Highly flexible proteins interact more readily with a wide range of structurally different ligands and modulate the thermodynamics and dynamics of protein ligand recognition/binding; therefore, they are more susceptible to ligand activation.9 In summary, we found that the CD4-independent (neutralization-sensitive) R2 gp120 had greater conformational entropy, lower thermal stability, and more complex structural diversity with improved ability to converge to the CD4-bound state.
4. Discussion
The Env protein gp120 plays an important role in HIV-1 viral invasion and immune escape, and both theoretical and experimental results suggest that gp120's interaction with antibodies largely determines HIV-1's neutralizing phenotype.6,55 In addition, the Env trimer's transition from closed to open state and gp41's membrane penetration process during HIV-1 infection are directly related to gp120's conformational changes. Therefore, understanding gp120's structural dynamics and energetic behavior is important for gaining insight into HIV-1's immune escape and infection mechanisms. Previous studies have shown that many shorter trajectories are performed versus a single (or few) longer trajectory. A single trajectory is likely to become “bound’ to a single local basin of attraction and repeatedly explore a small region of conformational space.56 In the current study, we performed several MD simulations of gp120 in the CD4 ligand-free state to probe the effects of CD4 binding on gp120's dynamics and thermodynamics.Our study showed that CD4-dependent (neutralization-resistant) JR-FL and CD4-independent (neutralization-sensitive) R2 gp120 variants have similar core structures, with the main differences being in the variable loop region orientation and the bridge sheet layer. The structural and geometric property comparisons based on series balanced trajectories revealed that R2 gp120 had a greater overall conformational flexibility than JR-FL gp120. At the same time, we also performed a single longer trajectory (1 μs) of the two gp120s, and the RMSD indicated a trend close to the multiple repetitions of the conventional MD simulations (Fig. S8†). These results are consistent with the hydrogen/deuterium exchange (HDX) profiles of full-length monomeric gp120 from the SF162 isolate, indicating that on one hand, MD simulations can provide reliable information about protein dynamics, and on the other hand, full-length ligand-free gp120 variants from different isolates have similar local conformational flexibility and rigidity distributions.13 The essential dynamics analyses (Fig. 5 and S10†) showed CD4-dependent (neutralization-resistant) and CD4-independent (neutralization-sensitive) gp120 molecular movement patterns from which the resulting functional effects can be inferred. Both gp120 variants' main molecular movement patterns can be described as global or local regional structures rotating around a central axis, forming a counterclockwise or clockwise rotating turbine with small reside fluctuations in structures close to the core region (turbine center) and large residue fluctuations in the peripheral ring region. Differences in rotation direction can lead to twisting, separation, or proximity of related structural regions, and the largest-amplitude collective motion patterns are often closely related to the gp120's function. The differences between JR-FL and R2 gp120s in motion direction, fluctuation amplitude, and span of substructures involving collective motion may result in different gp120 conformations, i.e., JR-FL gp120's greater ability to remain unbound and R2 gp120's greater likelihood of transitioning to the CD4-bound state. In this state, the fully exposed V3 loop and the mature bridging plate allow for high affinity interactions with the co-receptor and efficient recognition by various CD4i or CD4bs neutralizing monoclonal antibodies.57
Our MD simulations also identified high flexibility regions, which were observed in both gp120 forms, i.e., the V1/V2 regions, the V3, V4, and V5 loops, and the N- and C-termini (Fig. 4A). For both gp120 neutralizing phenotypes, the V1/V2 region exhibited the highest flexibility in the MD simulations. In addition, the V1/V2 region exhibited the largest fluctuation amplitude and complex movement patterns along the first eigenvector (Fig. 6). Furthermore, the FEL analyses (Fig. 7A–D and S11A–D†) showed that CD4-independent gp120 had larger conformational entropy, more unstable structure, and higher conformational diversity than CD4-dependent gp120. The V1/V2 loop of the representative structure of the minimum local energy structure on the JR-FL gp120 energy surface tends to be closed (Fig. 7C). In contrast, the V1/V2 loop of the R2 gp120 tends to be open (Fig. 7D). This is consistent with the results obtained from the previous structural property analyses, conformational flexibility analyses, and essential dynamics analyses. Therefore, the CD4-independent neutralization-sensitive HIV-1 Env trimer gp120 could adopt a CD4-bound state with a certain probability and directly interact with the CCR or CXCR4 coreceptor through a conformational selection mechanism without CD4 induction. On the other hand, the CD4-dependent neutralization-resistant gp120 had high conformational rigidity due to its weak conformational flexibility, and it needs to bind to the CD4 receptor before it can further bind the co-receptor and complete cell infection.
Various approaches had previously indicated that the footprint of a coreceptor on gp120 would include the V3 loop and the bridging sheet,58,59 and that gp120 would contact the coreceptor by the N-terminus and ECL2 of CCR5. We also selected another typical CD4-dependent strain, H061.14 gp120,26 and performed similar multiple conventional MD simulations, essential dynamic analysis, and FEL reconstructions (Fig. S1–S7†). MD trajectory comparative analyses showed that R2 gp120 exhibited greater structural fluctuations/deviations and higher global conformational flexibility compared to H061.14 gp120. Close comparison of local conformational flexibility showed that some of the structural regions involving direct interactions with gp41 and adjacent gp120 subunits in the closed trimeric Env context exhibited significantly higher flexibility in R2 gp120 than in H061.14 gp120. Thus, the probability for R2 Env to open the trimer crown and prime gp41 fusogenic properties without induction by CD4 was increased. The collective motions derived from essential dynamic analysis revealed that R2 gp120 was prone to spontaneous transition to the neutralization-sensitive CD4-bound state while H061.14 gp120 tended to maintain a neutralization-resistant unliganded state. The bindings of neutralizing antibodies to these two types of gp120 is also different. The experiment of Guttman et al.24 confirmed that the binding of 17b to KNH1144 SOSIP.664 trimers occurs very slowly in the absence of CD4 but rapidly in the presence of CD4, which is consistent with our conjecture of H061.14. The BG505 SOSIP.664 trimer was used to ascertain the effects of trimerization on CD4i MAb binding, while the R2 strain can infect CD4-negative cell,60 constitutively exposes several CD4i epitopes,61 and therefore could theoretically bind CD4i MAbs in the absence of CD4.30 Finally, FEL comparisons revealed that R2 gp120 had larger conformational entropy, richer conformational diversity, and lower thermostability than H061.14 gp120. The lower structural stability and higher conformational flexibility of unliganded R2 gp120 when compared to the unliganded form of H061.14 gp120, as well as its higher propensity to shift to the CD4-bound state gives R2-Env an increased ability to sample the open state in the absence of CD4. Where co-receptor binding sites and conserved antibody neutralizing epitopes have been formed and exposed, thus allowing efficient recognition/binding of the relevant antibody and co-receptor via conformational selection. Both smFRET and HDX data support the viewpoint.24,28,62 This may explain why R2 gp120 was more structurally unstable and conformationally flexible and had a higher propensity to transition to the CD4-bound state.
The V1/V2 loop region localization or orientation had an important influence on the overall gp120 dynamic behavior, and most of the previously conducted gp120-related studies have been based on its core structure. The structural model in this study contained the V1/V2 loop region, using the gp120 structure from the Env protein trimer structure as a template. A gp120 structural model in the nearly full-length unliganded state was constructed, and the differences in geometric properties, conformational flexibility, molecular motions, and FELs between the CD4-independent (neutralization-sensitive) and CD4-dependent (neutralization-resistant) gp120 types were investigated by multiple molecular dynamics simulation iterations. These results are important for an in-depth understanding of how gp120's structural dynamics, function, and HIV-1 infection mechanism are related.
5. Conclusion
In this study, we constructed two gp120 structural models in the CD4 unliganded state from two different HIV-1 isolates (with CD4-dependent versus CD4-independent phenotypes) for MD simulations. Since multiple short time MD simulation repetitions can yield higher sampling efficiency in conformational space than single long time dynamics simulations, we performed ten 100 ns molecular dynamics simulations for each of the two structural models, and the concatenated equilibrium simulation trajectory length used for the analysis reached 0.95 μs. The comparative analyses of the dynamic and thermodynamic differences between the two different gp120 variants, combined with comparative analyses of RMSD, RMSF, ED, and MD equilibrium trajectory free energy led to the following conclusions: (i) CD4-independent (neutralization-sensitive) R2 gp120 had larger conformational flexibility, lower structural stability, and a greater number of conformational states or sub-states than typical CD4-dependent (neutralization-resistant) JR-FL gp120; (ii) comparing the molecular motions, we observed that R2 gp120 had more substructures exhibiting larger collective movement amplitudes, and different substructures' motion direction and amplitude led to conformational differences; (iii) JR-FL gp120 is more likely to maintain a neutralization-resistant unliganded state than R2 gp120, while R2 gp120 is more likely to maintain a neutralization-sensitive CD4-bound state; (iv) both gp120 variants exhibited significantly greater conformational flexibility in the V1/V2 and V3 loop regions, and V1/V2 loop reorientation ability is crucial for gp120 to transit from the liganded to unliganded state.Our MD simulations and constructed FELs revealed differences in the dynamics and energies of CD4-independent (neutralization-sensitive) and CD4-dependent (neutralization-resistant) gp120. These findings shed light on the molecular basis of the R2 gp120 and JR-FL gp120 HIV-1 isolate phenotypic differences. Based on these results, we speculate that modifying neutralization-resistant, CD4-dependent Env trimers to reduce CD4-induced opening could be a good strategy for treatment of AIDS. In conclusion, our results provide a detailed dynamic and thermodynamic interpretation of gp120, which further helps our understanding of HIV-1 infiltration and fusion mechanism and potentially guides anti-HIV-1 vaccine and drug development.
Author contributions
M.-T. L., P. S. and Y.-Q. C. performed the MD simulation, analyzed the data, and participated in designing the study and drafting the manuscript. M.-T. L., J.-X. S., L. Y. and X.-W. L. performed the BFEs calculation. Y. L. revised the manuscript. M.-T. L., P. S. and L.-Q. Y. designed the study, coordinated the work, and wrote and revised the manuscript. All authors contributed to and approved the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest for this work.Acknowledgements
This study was funded by grants (31960198, 31860243 and 62003068) from the National Natural Sciences Foundation of China, the Yunnan Applied Basic Research Projects (2019FB021). We thank High Performance Computer Center of Dali University for computational support.References
- M. Kowalski, J. Potz, L. Basiripour, T. Dorfman, W. C. Goh, E. Terwilliger, A. Dayton, C. Rosen, W. Haseltine and J. Sodroski, Science, 1987, 237, 1351–1355 CrossRef CAS PubMed.
- M. Lu, S. C. Blacklow and P. S. Kim, Nat. Struct. Biol., 1995, 2, 1075–1082 CrossRef CAS PubMed.
- X. Wei, S. K. Ghosh, M. E. Taylor, V. A. Johnson, E. A. Emini, P. Deutsch, J. D. Lifson, S. Bonhoeffer, M. A. Nowak and B. H. Hahn, Nature, 1995, 373, 117–122 CrossRef CAS PubMed.
- R. Wyatt and J. Sodroski, Science, 1998, 280, 1884–1888 CrossRef CAS PubMed.
- M. Pancera, T. Zhou, A. Druz, I. S. Georgiev, C. Soto, J. Gorman, J. Huang, P. Acharya, G.-Y. Chuang and G. Ofek, Nature, 2014, 514, 455–461 CrossRef CAS PubMed.
- Y. Li, L. Deng, S.-M. Ai, P. Sang, J. Yang, Y.-L. Xia, Z.-B. Zhang, Y.-X. Fu and S.-Q. Liu, RSC Adv., 2018, 8, 14355–14368 RSC.
- S.-Q. Liu, S.-X. Liu and Y.-X. Fu, J. Mol. Model., 2008, 14, 857–870 CrossRef CAS PubMed.
- H. Li, Y. Xie, C. Liu and S. Liu, Sci. China: Life Sci., 2014, 57, 287–302 CrossRef CAS PubMed.
- L.-Q. Yang, P. Sang, R.-P. Zhang and S.-Q. Liu, RSC Adv., 2017, 7, 42094–42104 RSC.
- D. C. Chan, D. Fass, J. M. Berger and P. S. Kim, Cell, 1997, 89, 263–273 CrossRef CAS PubMed.
- P. D. Kwong, R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski and W. A. Hendrickson, Nature, 1998, 393, 648–659 CrossRef CAS PubMed.
- R. Wyatt, P. D. Kwong, E. Desjardins, R. W. Sweet, J. Robinson, W. A. Hendrickson and J. G. Sodroski, Nature, 1998, 393, 705–711 CrossRef CAS PubMed.
- P. D. Kwong, R. Wyatt, S. Majeed, J. Robinson, R. W. Sweet, J. Sodroski and W. A. Hendrickson, Structure, 2000, 8, 1329–1339 CrossRef CAS PubMed.
- C.-c. Huang, M. Tang, M.-Y. Zhang, S. Majeed, E. Montabana, R. L. Stanfield, D. S. Dimitrov, B. Korber, J. Sodroski and I. A. Wilson, Science, 2005, 310, 1025–1028 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- T. Zhou, L. Xu, B. Dey, A. J. Hessell, D. Van Ryk, S.-H. Xiang, X. Yang, M.-Y. Zhang, M. B. Zwick and J. Arthos, Nature, 2007, 445, 732–737 CrossRef CAS PubMed.
- L. Chen, Y. Do Kwon, T. Zhou, X. Wu, S. O'Dell, L. Cavacini, A. J. Hessell, M. Pancera, M. Tang and L. Xu, Science, 2009, 326, 1123–1127 CrossRef CAS PubMed.
- L. Kong, J. H. Lee, K. J. Doores, C. D. Murin, J.-P. Julien, R. McBride, Y. Liu, A. Marozsan, A. Cupo and P.-J. Klasse, Nat. Struct. Mol. Biol., 2013, 20, 796–803 CrossRef CAS PubMed.
- A. L. Cunningham, H. Donaghy, A. N. Harman, M. Kim and S. G. Turville, Curr. Opin. Microbiol., 2010, 13, 524–529 CrossRef CAS PubMed.
- J. Liu, A. Bartesaghi, M. J. Borgnia, G. Sapiro and S. Subramaniam, Nature, 2008, 455, 109–113 CrossRef CAS PubMed.
- J.-P. Julien, A. Cupo, D. Sok, R. L. Stanfield, D. Lyumkis, M. C. Deller, P.-J. Klasse, D. R. Burton, R. W. Sanders and J. P. Moore, Science, 2013, 342, 1477–1483 CrossRef CAS PubMed.
- D. Lyumkis, J.-P. Julien, N. De Val, A. Cupo, C. S. Potter, P.-J. Klasse, D. R. Burton, R. W. Sanders, J. P. Moore and B. Carragher, Science, 2013, 342, 1484–1490 CrossRef CAS PubMed.
- Y. Li, L. Deng, J. Liang, G.-H. Dong, Y.-L. Xia, Y.-X. Fu and S.-Q. Liu, Phys. Chem. Chem. Phys., 2020, 22, 5548–5560 RSC.
- M. Guttman, A. Cupo, J.-P. Julien, R. W. Sanders, I. A. Wilson, J. P. Moore and K. K. Lee, Nat. Commun., 2015, 6, 1–11 Search PubMed.
- J. B. Munro and W. Mothes, J. Virol., 2015, 89, 5752–5755 CrossRef CAS PubMed.
- M. S. Seaman, H. Janes, N. Hawkins, L. E. Grandpre, C. Devoy, A. Giri, R. T. Coffey, L. Harris, B. Wood and M. G. Daniels, J. Virol., 2010, 84, 1439–1452 CrossRef CAS PubMed.
- W. Mothes, J. Gorman, J. Munro, X. Ma, P. Kwong and S. Blanchard, JAIDS, J. Acquired Immune Defic. Syndr., 2014, 67, 55 CrossRef.
- J. B. Munro, J. Gorman, X. Ma, Z. Zhou, J. Arthos, D. R. Burton, W. C. Koff, J. R. Courter, A. B. Smith III and P. D. Kwong, Science, 2014, 346, 759–763 CrossRef CAS PubMed.
- K. S. Appelberg, M. A. Wallet, J. P. Taylor, M. N. Cash, J. W. Sleasman and M. M. Goodenow, AIDS Res. Hum. Retroviruses, 2017, 33, 690–702 CrossRef CAS PubMed.
- G. Kaplan, A. Roitburd-Berman, G. K. Lewis and J. M. Gershoni, J. Virol., 2016, 90, 4481–4493 CrossRef CAS PubMed.
- G. V. Quinnan, P. F. Zhang, D. W. Fu, M. Dong and H. J. Alter, AIDS Res. Hum. Retroviruses, 1999, 15, 561–570 CrossRef CAS PubMed.
- A. Bairoch, R. Apweiler, C. H. Wu, W. C. Barker, B. Boeckmann, S. Ferro, E. Gasteiger, H. Huang, R. Lopez and M. Magrane, Nucleic Acids Res., 2005, 33, D154–D159 CrossRef CAS PubMed.
- B. Webb and A. Sali, Curr. Protoc. Bioinf., 2016, 54, 5.6.1–5.6.37 Search PubMed.
- M. Pancera, S. Majeed, Y.-E. A. Ban, L. Chen, C.-c. Huang, L. Kong, Y. D. Kwon, J. Stuckey, T. Zhou and J. E. Robinson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1166–1171 CrossRef CAS PubMed.
- A. E. Aliev, M. Kulke, H. S. Khaneja, V. Chudasama, T. D. Sheppard and R. M. Lanigan, Proteins: Struct., Funct., Bioinf., 2014, 82, 195–215 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- Y. Li, X.-L. Zhang, X. Yuan, J.-C. Hou, P. Sang and L.-Q. Yang, RSC Adv., 2020, 10, 30499–30507 RSC.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- S. Nosé and M. Klein, Mol. Phys., 1983, 50, 1055–1076 CrossRef.
- A. Amadei, A. B. Linssen and H. J. Berendsen, Proteins: Struct., Funct., Bioinf., 1993, 17, 412–425 CrossRef CAS PubMed.
- J. D. Bryngelson, J. N. Onuchic, N. D. Socci and P. G. Wolynes, Proteins: Struct., Funct., Bioinf., 1995, 21, 167–195 CrossRef CAS PubMed.
- K. Henzler-Wildman and D. Kern, Nature, 2007, 450, 964–972 CrossRef CAS PubMed.
- A. Šali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- Y. Do Kwon, M. Pancera, P. Acharya, I. S. Georgiev, E. T. Crooks, J. Gorman, M. G. Joyce, M. Guttman, X. Ma and S. Narpala, Nat. Struct. Mol. Biol., 2015, 22, 522–531 CrossRef CAS PubMed.
- M. Guttman, M. Kahn, N. K. Garcia, S.-L. Hu and K. K. Lee, J. Virol., 2012, 86, 8750–8764 CrossRef CAS PubMed.
- J.-P. Julien, J. H. Lee, A. Cupo, C. D. Murin, R. Derking, S. Hoffenberg, M. J. Caulfield, C. R. King, A. J. Marozsan and P. J. Klasse, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4351–4356 CrossRef CAS PubMed.
- M. Guttman, N. K. Garcia, A. Cupo, T. Matsui, J.-P. Julien, R. W. Sanders, I. A. Wilson, J. P. Moore and K. K. Lee, Structure, 2014, 22, 974–984 CrossRef CAS PubMed.
- H. Choe, M. Farzan, Y. Sun, N. Sullivan, B. Rollins, P. D. Ponath, L. Wu, C. R. Mackay, G. LaRosa and W. Newman, Cell, 1996, 85, 1135–1148 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- D. R. Langley, S. Roy Kimura, P. Sivaprakasam, N. Zhou, I. Dicker, B. McAuliffe, T. Wang, J. F. Kadow, N. A. Meanwell and M. Krystal, Proteins: Struct., Funct., Bioinf., 2015, 83, 331–350 CrossRef CAS PubMed.
- A. Andrianov and V. Veresov, Biochemistry, 2006, 71, 906–914 CAS.
- D. C. Chan and P. S. Kim, Cell, 1998, 93, 681–684 CrossRef CAS PubMed.
- L. S. Caves, J. D. Evanseck and M. Karplus, Protein Sci., 1998, 7, 649–666 CrossRef CAS PubMed.
- S.-H. Xiang, P. D. Kwong, R. Gupta, C. D. Rizzuto, D. J. Casper, R. Wyatt, L. Wang, W. A. Hendrickson, M. L. Doyle and J. Sodroski, J. Virol., 2002, 76, 9888–9899 CrossRef CAS PubMed.
- C. D. Rizzuto, R. Wyatt, N. Hernández-Ramos, Y. Sun, P. D. Kwong, W. A. Hendrickson and J. Sodroski, Science, 1998, 280, 1949–1953 CrossRef CAS PubMed.
- C.-c. Huang, S. N. Lam, P. Acharya, M. Tang, S.-H. Xiang, S. S.-u. Hussan, R. L. Stanfield, J. Robinson, J. Sodroski and I. A. Wilson, Science, 2007, 317, 1930–1934 CrossRef CAS PubMed.
- P. F. Zhang, P. Bouma, E. J. Park, J. B. Margolick, J. E. Robinson, S. Zolla-Pazner, M. N. Flora and G. V. Quinnan Jr, J. Virol., 2002, 76, 644–655 CrossRef CAS PubMed.
- K. R. Young, B. E. Teal, Y. Brooks, T. D. Green, J. F. Bower and T. M. Ross, AIDS Res. Hum. Retroviruses, 2004, 20, 1259–1268 CrossRef CAS PubMed.
- X. Du, Y. Li, Y.-L. Xia, S.-M. Ai, J. Liang, P. Sang, X.-L. Ji and S.-Q. Liu, Int. J. Mol. Sci., 2016, 17, 144 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00433c |
| This journal is © The Royal Society of Chemistry 2023 |