
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering a Bacillus subtilis esterase for selective hydrolysis of D, L-menthyl acetate in an organic solvent-free system†
Jingjing Qiaoa,
Duxia Yanga,
Yingting Fenga,
Wan Weia,
Xun Liua,
Yinjun Zhang a,
Jianyong Zheng*a and
Xiangxian Ying*b
a,
Jianyong Zheng*a and
Xiangxian Ying*b
aKey Laboratory of Bioorganic Synthesis of Zhejiang Province, College of Biotechnology and Bioengineering, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: zjy821212@zjut.edu.cn
bHangzhou Hyproven Biopharm Co.,Ltd., Hangzhou 311107, China. E-mail: yingxx@zjut.edu.cn
First published on 3rd April 2023
Abstract
Esterase/lipase-catalyzed selective hydrolysis of D, L-menthyl esters has become one of the promising approaches for producing L-menthol, one of the most important flavoring chemicals with extensive uses. However, the activity and L-enantioselectivity of the biocatalyst are not sufficient for meeting the industrial requirements. Herein, a highly active para-nitrobenzyl esterase from Bacillus subtilis 168 (pnbA-BS) was cloned and then engineered to enhance its L-enantioselectivity. On the basis of the strategy tailoring the steric exclusion effect and structural flexibility of the region adjacent to the substrate, the substitution of Ala400 to Pro caused a remarkable improvement in the E value from 1.0 to 466.6. The variant A400P was purified and further confirmed with strict L-enantioselectivity in the selective hydrolysis of D, L-menthyl acetate, whereas the improved L-enantioselectivity caused decreased activity. To develop an efficient, easy-to-use, and green methodology, organic solvent was omitted and substrate constant feeding was integrated into the whole-cell catalyzed system. During the catalytic process, the selective hydrolysis of 1.0 M D, L-menthyl acetate in 14 h offered a conversion of 48.9%, e.e.p value of >99%, and space-time yield of 160.52 g (l d)−1.
1 Introduction
Esterases (E.C. 3.1.1.1) and lipases (E.C. 3.1.1.3) break ester bonds with frequently high activities and require no factors, representing a class of biocatalysts for the synthesis of optically pure chemicals.1–3 In the recent two decades, a plethora of studies on esterase/lipase-catalyzed chiral resolution of D, L-menthyl esters have made the approach promising for the biocatalytic synthesis of L-menthol.4–8 Menthol has eight possible isomers but the characteristic peppermint odour and the typical cooling/refreshing effect are only attributed to L-menthol.9,10 Therefore, the high optical purity of L-menthol (usually e.e.p > 98%) is critical for its commercial use and logically requires the use of esterases/lipases with excellent L-enantioselectivity. However, the L-enantioselectivity of known highly active esterases/lipases is usually not satisfying. The recombinant lipase from Candida rugosa exhibited strict L-enantioselectivity in the chiral resolution of D, L-menthyl benzoate (e.e.p > 99%), despite relatively low catalytic efficiency.4Activity, stability, and enantioselectivity of esterases/lipases are affected by various structural factors, i.e., steric exclusion, hydrogen bonding, hydrophobic interaction, and regional flexibility.11,12 Structure-guided protein engineering has proven to be a powerful tool for improving enzyme properties.13,14 In the case of Pseudomonas alcaligenes lipase, the residue Ala272 was identified as a hot spot for controlling diastereopreference.7 The substitution of Ala272 to Phe with a bulky side group increased the steric exclusion effect and decreased the flexibility of the region adjacent to the substrate, enhancing the diastereopreference in the resolution of racemic menthyl propionate. Similar to P. alcaligenes lipase, the engineering of an esterase from Bacillus aryabhattai was focused on the substrate binding pocket.15 The resulting variant L86Q/G284E increased the E value from 16.6 to 216.4 in the resolution of (R, S)-ethyl indoline-2-carboxylate. In recent years, rational surface engineering of esterases/lipases has emerged to improve organic solvent tolerance.12,16–18 The latest study on Bacillus subtilis lipase A demonstrated that the surface polar engineering strategy by substituting aromatic residue(s) to polar one(s) was powerful to generate beneficial variants tolerant to organic solvent.19
Protein engineering improves one of the catalytic properties of esterases/lipases; meanwhile, it might cause a negative impact on another catalytic property. It is not uncommon that the enantioselectivity improvement led to decreased activity.7,15 Overcoming the trade-off effect between enantioselectivity and activity remains challenging. Besides biocatalyst engineering, substrate/process engineering is also essential to apply biocatalysis for the efficient and sustainable production of organic compounds.20–23 For instance, water-miscible organic solvents could improve L-enantioselectivity and conversion in the hydrolysis of D, L-menthyl acetate.6 In the other case, the organic cosolvent for the solubilization of high-concentration substrate in an aqueous buffer can be omitted.24 In addition, the substrate-feeding strategy was effective in increasing the substrate loading when substrate inhibition occurred.25,26
Herein, a para-nitrobenzyl esterase from B. subtilis 168 (pnbA-BS) was cloned and over-expressed in E. coli BL21(DE3), which harbored high activity but low L-enantioselectivity in the resolution of D, L-menthyl acetate. Its enantioselectivity deficiency was overcome by tailoring its steric exclusion effect and structural flexibility. Finally, the resulting variant with improved L-enantioselectivity was accompanied by process engineering to develop an efficient, easy-to-use, and green process for the selective resolution of D, L-menthyl acetate to optically pure L-menthol (Scheme 1).
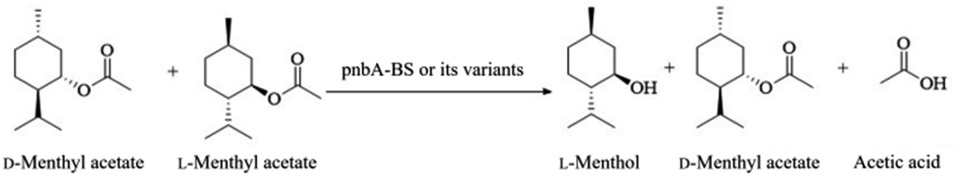 | ||
| Scheme 1 Esterase-catalyzed selective hydrolysis of D, L-menthyl acetate to L-menthol. | ||
2 Experimental section
2.1. Chemicals, enzymes, plasmids, and strain
The standards D, L-menthyl acetate, D-menthol, and L-menthol were purchased from Shanghai Macklin Biochemical Co., Ltd. Unless otherwise stated, all other reagents and chemicals were commercially available. Both DNA polymerase and the restriction enzyme Dpn I were supplied by Vazyme Biotech (Nanjing, China). The synthesis of oligonucleotide and the sequencing of PCR products were conducted by Tsingke Biological Technology (Beijing, China). The plasmid pET28a was used for esterase over-expression in the host strain E. coli BL21 (DE3).2.2. Cloning and overexpression of the esterase pnbA-BS
The gene encoding the esterase pnbA-BS (NCBI reference sequence: WP_100276345.1) was codon-optimized, and synthesized at Vazyme Biotech Co., Ltd (Nanjing, China) (Fig. S1†). The gene fragment was inserted into the sites Nco I and Xho I of the expression vector pET28a (Novagen, Shanghai), constructing the recombinant plasmid pET28a-pnbA-BS. The resulting recombinant plasmid was transformed into E. coli BL21(DE3) competent cells, forming the strain E. coli BL21(DE3)/pET28a-pnbA-BS. Unless otherwise stated, the recombinant E. coli strains were routinely grown in an LB medium containing 50 μg ml−1 kanamycin at 37 °C until the OD600 of 0.6. Strains were induced by adding 0.4 mM IPTG and cultured at 24 °C for 16 h. Cells were washed twice using 50 mM Tris–HCl buffer (pH 8.0) and then harvested by 8000g centrifugation at 4 °C for 10 min. Finally, lyophilized cells were obtained by freeze-drying and stored at −20 °C for further use.2.3. Site-directed/saturation mutagenesis
According to the whole plasmid mutagenesis protocol,27 site-directed/saturation mutagenesis of the esterase pnbA-BS was performed using the recombinant plasmid pET28a-pnbA-BS as a template. The PCR reaction system (50 μl) consisted of 1 μl forward primer (100 μM), 1 μl reverse primer (100 μM), 25 μl 2 × Phanta Buffer, 1 μl dNTP mixture (each 10 mM), 1 μl plasmid template, 1 μl DNA polymerase, and 21 μl ultrapure water. The PCR conditions: 95 °C for 5 min, and then 30 cycles (95 °C for 15 s, 55 °C for 15 s, 72 °C for 6 min), 72 °C extension for 10 min. The PCR product (2 μl) was digested with Dpn I at 37 °C for 2 h. After digestion, the resulting plasmids were transformed into E. coli BL21 (DE3) competent cells to generate the mutant strains.2.4. Homology modeling and molecular docking
The homology model of pnbA-BS was built based on the crystal structure of B. subtilis esterase 56C8 (PDB: 1C7J), both of which share 91% amino acid sequence identity. All molecular docking analyses of esterase and substrate were performed using Autodock Vina.28 Rigid receptor-flexible ligand docking was carried out with the standard parameters for interactive growing and subsequent scoring. Pymol was used to visualize the ligand docking results.292.5. Protein purification
The lyophilized cells were re-suspended in 100 mM PBS buffer (pH 7.0) at a concentration of 50 g l−1 and then sonicated for cell disruption. The lysate was collected by 8000g centrifugation at 4 °C for 10 min. The collected supernatant was filtered through a 0.45 μm membrane. The filtered samples were loaded onto a DEAE-Sepharose anion exchange column equilibrated with buffer A (50 mM Tris–HCl, pH 8.0). A linear gradient (0 to 0.5 M NaCl) was applied at a flow rate of 3 ml min−1, and esterase was eluted from the column at a concentration of 0.25 M NaCl. Fractions containing esterase activity were desalted and concentrated using the 10 kDa ultra-filtration membrane. The concentrated samples were loaded onto a Superdex 200 gel filtration column equilibrated with buffer A at a flow rate of 0.5 ml min−1. Esterase was eluted using buffer A and the collected esterase fractions were concentrated using the 10 kDa ultra-filtration membrane. The purity of the purified esterase samples was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as described previously.302.6. Activity assay and determination of kinetic parameters
The standard para-nitrophenyl butyrate was used as the substrate and its stock solution was 200 mM substrate dissolved in dimethyl sulfoxide. Unless stated otherwise, the 200 μl assay mixture contained 0.5 mM para-nitrophenyl butyrate, 3 μg purified esterase pnbA-BS or its variant A400P, and 100 mM PBS buffer (pH 9.0). The reaction was activated by adding the enzyme and conducted at 30 °C. The absorbance change at 405 nm was detected by the microplate reader. One unit of enzyme activity was defined as the amount of enzyme required for catalyzing the hydrolysis of 1 μmol of para-nitrophenyl butyrate per minute. All assays were conducted in triplicate. Kinetic parameters of the purified pnbA-BS and its variant A400P were determined within the substrate concentration range from 0 to 50 mM. According to Michaelis–Menton kinetics, the parameters Km and Vmax were calculated through the curve fitting using Origin Pro software (Version 8.5).2.7. Resolution of D, L-menthyl acetate, and GC analysis
The optimized mixture of whole-cell-catalyzed resolution of D, L-menthyl acetate (10 ml) included 0.2 g lyophilized cells expressing pnbA-BS or its variants, 500 mM substrate, and 100 mM PBS buffer (pH 7.0). The reaction was maintained at 30 °C and constant pH of 7.0 through the titration of 1 M NaOH. The reaction was activated by adding a lyophilized whole-cell catalyst and terminated by adding ethanol at a final concentration of 50% (v/v). The reactants were extracted by ethyl acetate, dried over anhydrous sodium sulfate, and subjected to GC analyses. All reactions were carried out in triplicate.The substrate and the products were determined using a GC (Agilent 6890N) equipped with an FID detector and chiral capillary CP7501 column (50 m × 250 μm × 0.25 μm). The flow rate of N2 as the carrier gas was set as 1.0 ml min−1. Both the injector and detector were kept at 225 °C. The injection volume was 1 μl. The column temperature program was listed as follows: initial temperature of 100 °C for 8 min, 4 °C min−1 ramp to 140 °C for 5 min, and 40 °C min−1 ramp to 180 °C for 1 min. The retention times of the standards are listed as follows: D-menthol, 20.615 min; L-menthol, 20.774 min; L-menthyl acetate, 21.097 min; D-menthyl acetate, 21.638 min (Fig. S2†).
Substrate enantiomer excess (e.e.s), product enantiomer excess (e.e.p), conversion rate (C), and enantiomer ratio (E) were calculated according to the following formulas.31
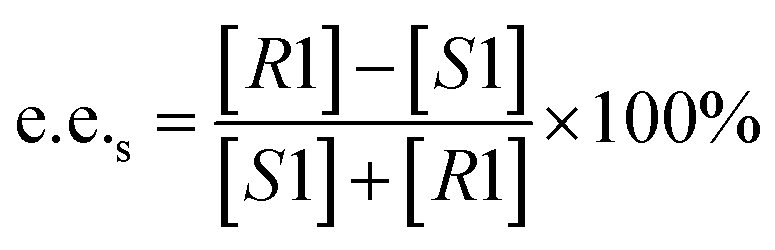
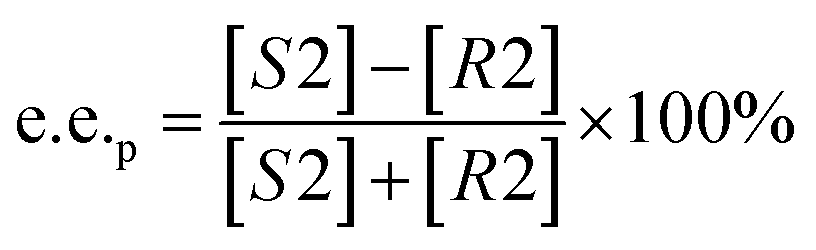
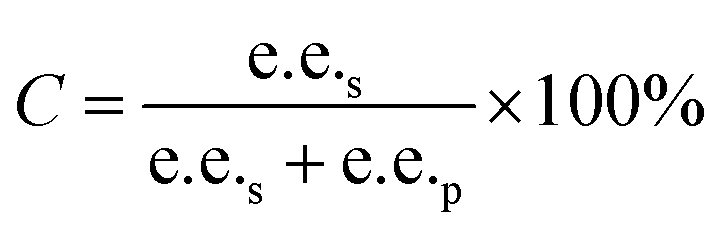
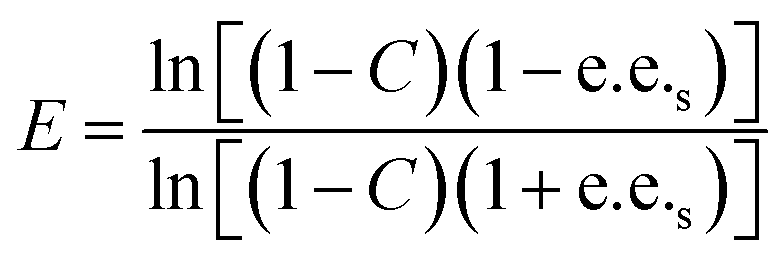
2.8. Preparation of L-menthol in the manner of substrate constant feeding
A 10 ml initial reaction mixture contained 250 mM D, L-menthyl acetate, 20 g l−1 lyophilized cells expressing the variant A400P and 100 mM PBS buffer (pH 7.0, 30 °C, and 600 rpm). Using a syringe pump, the rest of the required substrate was constantly fed within 10 h to the corresponding substrate loading (1 M, 1.1 M, 1.2 M, 1.3 M, 1.4 M, or 1.7 M). After that, the reaction was continued for 4–14 h for achieving higher conversions. At the end of each reaction, the reaction mixture was centrifuged to remove the cells and the organic phase was collected. The collected organic phase was further filtered using a 0.22 μm membrane for the organic system and then dried over anhydrous Na2SO4. The substrate and product in the organic phase were separated by column chromatography on silica gel using n-hexane/ethyl acetate (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as an elution solution. Finally, the product L-menthol was obtained by the removal of the solvent under reduced pressure and dried under a vacuum. The conversion and optical purity of L-menthol were monitored by chiral GC analysis. The product L-menthol was verified by the combination of GC-MS (Fig. S3†), 1H NMR, and 13C NMR analyses (Fig. S4†). 1H NMR (600 MHz, CDCl3) δ 3.42 (td, J = 10.5, 4.3 Hz, 1H), 2.25–2.14 (m, 1H), 2.01–1.83 (m, 1H), 1.75–1.64 (m, 1H), 1.64–1.59 (m, 1H), 1.58–1.34 (m, 2H), 1.18–1.07 (m, 1H), 1.03–0.95 (m, 2H), 0.93 (dd, J = 9.3, 6.8 Hz, 6H), 0.89–0.84 (m, 1H), 0.82 (d, J = 7.0 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 71.52 (s), 50.14 (s), 45.06 (s), 34.55 (s), 31.64 (s), 25.82 (s), 23.14 (s), 22.21 (s), 21.01 (s), 16.09 (s).
:1, v/v) as an elution solution. Finally, the product L-menthol was obtained by the removal of the solvent under reduced pressure and dried under a vacuum. The conversion and optical purity of L-menthol were monitored by chiral GC analysis. The product L-menthol was verified by the combination of GC-MS (Fig. S3†), 1H NMR, and 13C NMR analyses (Fig. S4†). 1H NMR (600 MHz, CDCl3) δ 3.42 (td, J = 10.5, 4.3 Hz, 1H), 2.25–2.14 (m, 1H), 2.01–1.83 (m, 1H), 1.75–1.64 (m, 1H), 1.64–1.59 (m, 1H), 1.58–1.34 (m, 2H), 1.18–1.07 (m, 1H), 1.03–0.95 (m, 2H), 0.93 (dd, J = 9.3, 6.8 Hz, 6H), 0.89–0.84 (m, 1H), 0.82 (d, J = 7.0 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 71.52 (s), 50.14 (s), 45.06 (s), 34.55 (s), 31.64 (s), 25.82 (s), 23.14 (s), 22.21 (s), 21.01 (s), 16.09 (s).
3 Results and discussion
3.1. Semi-rational design of the esterase pnbA-BS
The gene encoding the esterase pnbA-BS with a length of 489 aa was cloned and over-expressed in E. coli BL21(DE3) (Fig. S5†). When the lyophilized cells expressing the esterase pnbA-BS were used as whole-cell catalysts, the hydrolysis of 1 M D, L-menthyl acetate led to 48.4% conversion in 5 h. Unfortunately, the corresponding e.e.p value was only 0.4% (L). Recent examples indicated the effectiveness of improving enantioselectivity through structure-oriented engineering of substrate-binding pockets.15,32,33 Blast analysis predicted that the substrate binding pocket of the esterase pnbA-BS constituted 13 key residues (G105, G106, A107, E188, S189, A190, M193, T326, A330, L331, M358, A400, L403). Except for S189 as one of the catalytic triads, those residues were subjected to alanine scanning by site-directed mutagenesis. In the case of alanine residues, the substitution was either valine or glycine in order to slightly change the side chain size. The resulting variants were expressed in E. coli BL21(DE3) in a soluble form at a similar expression level (Fig. S5†). The variant G106A was not active (Table 1), indicating its pivotal role in activity maintenance. Compared with the wild-type pnbA-BS, the variants G105A and A190V showed an improvement in E values from 1.0 to 38.5 and 39.8, respectively, suggesting that adapting the side chain size of the selected residues could facilitate L-enantioselectivity improvement. However, tackling the role of those residues on L-enantioselectivity would lead to a large mutant library and further tedious screening. Thus, it is necessary to attempt a more efficient strategy for constructing a smaller and smarter mutant library.| Enzyme | e.e.p (%) | Conversion (%) | E |
|---|---|---|---|
| a The reaction mixture (10 ml) contained 100 mM D, L-menthyl acetate, 50 g l−1 wet cells expressing pnbA-BS or its variant, 10% (v/v) ethanol, and 100 mM PBS buffer solution (pH 7.0). The reaction was conducted in an orbital shaker (600 rpm, 30 °C) for 6 h, in the meantime, the pH was constantly maintained by the titration of 1 M NaOH. Data present mean values ± SD from three independent experiments.b The host strain was E. coli BL21(DE3) as the control without the expression of pnbA-BS or its variants. | |||
| pnbA-BS | 0.4 ± 0.5 | 90.5 ± 3.3 | 1.0 |
| G105A | 89.2 ± 1.3 | 37.6 ± 0.9 | 38.5 |
| G106A | No activity | ||
| A107G | 3.2 ± 0.9 | 96.9 ± 2.2 | 1.5 |
| A107V | 80.2 ± 1.2 | 35.6 ± 1.8 | 17.1 |
| E188A | 18.9 ± 0.3 | 66.6 ± 1.7 | 2.2 |
| A190G | 49.3 ± 1.9 | 33.2 ± 1.7 | 3.7 |
| A190V | 76.9 ± 2.1 | 58.5 ± 1.9 | 39.8 |
| M193A | 54.5 ± 1.6 | 54.3 ± 1.9 | 6.5 |
| T326A | 14.2 ± 0.6 | 88.5 ± 2.4 | 2.4 |
| A330G | 15.3 ± 0.4 | 88.7 ± 2.6 | 2.4 |
| A330V | 10.7 ± 1.7 | 79.9 ± 3.5 | 1.7 |
| L331A | 21.5 ± 1.3 | 62.7 ± 2.1 | 2.1 |
| M358A | 1.1 ± 0.6 | 92.0 ± 2.6 | 1.3 |
| A400G | 1.8 ± 0.7 | 98.0 ± 1.6 | 1.9 |
| A400V | 13.0 ± 1.6 | 82.8 ± 1.2 | 2.2 |
| L403A | 21.9 ± 0.8 | 64.6 ± 2.3 | 2.1 |
| Host strain b | 89.6 ± 1.4 | 4.5 ± 0.5 | 18.8 |
Virtual mutagenesis analysis is helpful to increase the accuracy of semi-rational design and reduce the screening workload of the variants. The three-dimensional modeling of pnbA-BS was built, in which the residues S189, E310, and H399 formed the catalytic triad.34 In the study on engineering a lipase from P. alcaligenes, its catalytic triad constituted of S133, D243, and H271 and the residue A272 was one of the hot spots determining the enantioselectivity.8 The substitution of A272 to a residue with a bulky side chain such as Phe increased the steric exclusion effect and decreased structural flexibility, achieving higher diastereoselectivity in the resolution of racemic menthyl propionate to produce L-menthol. Inspired by the work, careful investigation of the residue A400 of pnbA-BS adjacent to H399 was conducted through virtual saturation mutagenesis. The enzyme pnbA-BS was docked with D-menthyl acetate and L-menthyl acetate as ligands, respectively (Fig. S6†). Hydrogen bonds were formed between the hydroxyl group of S189 and the carbonyl group on L-menthyl acetate or D-menthyl acetate with a distance of 2.8 Å, demonstrating similar preference on either L-menthyl acetate or D-menthyl acetate as the substrate. The information was consistent with the low L-enantioselectivity of the esterase pnbA-BS in the hydrolysis of D, L-menthyl acetate. The virtual saturation mutagenesis analyses of the residue A400 were conducted to evaluate the effect of nineteen different substitutions. Among them, the substitution of Ala400 for Pro would be remarkable. On the one hand, the hydrogen bond between the hydroxyl group of S189 and the carbonyl group on L-menthyl acetate was changed from 2.8 Å to 3.1 Å, implying that the substitution of Ala400 to Pro would cause the activity decrease. On the other hand, this variant could be inactive on D-menthyl acetate since it seemed difficult to form the hydrogen bond between the hydroxyl group of S189 and the carbonyl group. Ideally, the variant A400P could solely function on L-menthyl acetate and thus possess strict L-enantioselectivity in the resolution of D, L-menthyl acetate.
The lack of precise crystal structure restricts the accuracy and precision of the docking analyses and it is necessary to verify the pivotal role of the residue Ala400 speculated from virtual saturation mutagenesis analysis. The real saturation mutagenesis of the residue A400 was performed and all the resulting 19 variants were expressed in E. coli BL21(DE3) (Fig. S7†). It was the variant A400P that showed the highest L-enantioselectivity with an E value of 466.6 and an e.e.p of 97.1% (L) (Table 2). The whole cells expressing the variant A400P did not fulfill the expected strict L-enantioselectivity, which should be attributed to the presence of isozyme(s) in the host strain E. coli BL21(DE3) (Table 1). Besides A400P, other seven variants A400P, A400K, A400Q, A400R, A400H, A400M, and A400L exhibited enhanced L-enantioselectivity (Table 2). The variants A400Y and A400W were not active, suggesting that the controlling of activity and enantioselectivity was subtle. Except for A400G, the variants showed decreased activity to some extent, suggesting the determining role of the residue A400 in enantioselectivity and activity.
| Enzyme | e.e.p (%) | Conversion (%) | E |
|---|---|---|---|
| a The reaction mixture (10 ml) contained 100 mM D, L-menthyl acetate, 50 g l−1 wet cells expressing pnbA-BS or its variant, 10% (v/v) ethanol, and 100 mM PBS buffer solution (pH 7.0). The reaction was conducted in an orbital shaker (600 rpm, 30 °C) for 6 h meanwhile constant pH was maintained by the titration of 1 M NaOH. Data present mean values ± SD from three independent experiments. | |||
| A400P | 97.1 ± 0.4 | 46.8 ± 1.0 | 466.6 |
| A400K | 94.8 ± 1.2 | 46.6 ± 0.9 | 56.8 |
| A400Q | 95.7 ± 1.6 | 11.8 ± 0.2 | 52.2 |
| A400R | 93.7 ± 2.8 | 25.7 ± 1.5 | 42.2 |
| A400H | 91.4 ± 0.6 | 46.0 ± 1.3 | 48.3 |
| A400M | 89.3 ± 2.3 | 7.9 ± 1.4 | 38.6 |
| A400L | 88.9 ± 0.9 | 47.7 ± 0.8 | 36.8 |
| A400E | 82.6 ± 1.6 | 45.9 ± 1.4 | 19.5 |
| A400T | 65.6 ± 1.2 | 58.7 ± 2.4 | 15.4 |
| A400I | 62.9 ± 1.3 | 59.6 ± 2.9 | 14.2 |
| A400N | 31.6 ± 0.6 | 74.3 ± 1.9 | 5.3 |
| A400F | 20.1 ± 0.8 | 78.8 ± 0.4 | 2.9 |
| A400D | 11.3 ± 1.0 | 86.6 ± 2.7 | 2.2 |
| A400V | 9.8 ± 0.6 | 88.7 ± 2.3 | 2.2 |
| A400C | 6.7 ± 0.4 | 89.1 ± 1.5 | 1.7 |
| A400S | 3.2 ± 0.3 | 88.8 ± 1.7 | 1.2 |
| A400G | 1.8 ± 0.7 | 98.0 ± 2.6 | 1.9 |
| pnbA-BS | 0.4 ± 0.5 | 90.5 ± 3.3 | 1.0 |
| A400Y | No activity | ||
| A400W | No activity | ||
3.2. Purification and characterization of the esterase pnbA-BS and its variant A400P
Since the host strain E. coli BL21(DE3) possessed the hydrolysis activity toward D, L-menthyl acetate, it is necessary to purify pnbA-BS and its variant A400P for accurate assessment of L-enantioselectivity improvement and activity decrease. Both pnbA-BS and its variant A400P were purified through a combination of anion exchange and subsequent gel-filtration chromatography (Fig. 1), and the kinetic parameters of the purified enzymes were determined. The kcat/Km values of pnbA-BS and its variant A400P were 44.26 mM−1 s−1 and 1.94 mM−1 s−1, respectively, verifying the speculation that increased L-enantioselectivity led to decreased activity (Table 3). When the purified pnbA-BS or its variant A400P was used as a biocatalyst in the hydrolysis of 500 mM L-menthyl acetate, the conversions were 97.3% and 65.9% in 24 h (Fig. S8†), respectively. Particularly, the purified A400P-catalyzed hydrolysis of D, L-menthyl acetate showed a sole product peak corresponding to L-menthol in the chiral GC chromatography (Fig. 2), confirming the notable improvement from extremely low L-enantioselectivity to strict L-enantioselectivity in the hydrolysis of D, L-menthyl acetate. Overcoming the trade-off effect between activity and enantioselectivity remains challenging. Thus, process engineering was subsequently attempted to further improve the catalytic efficiency.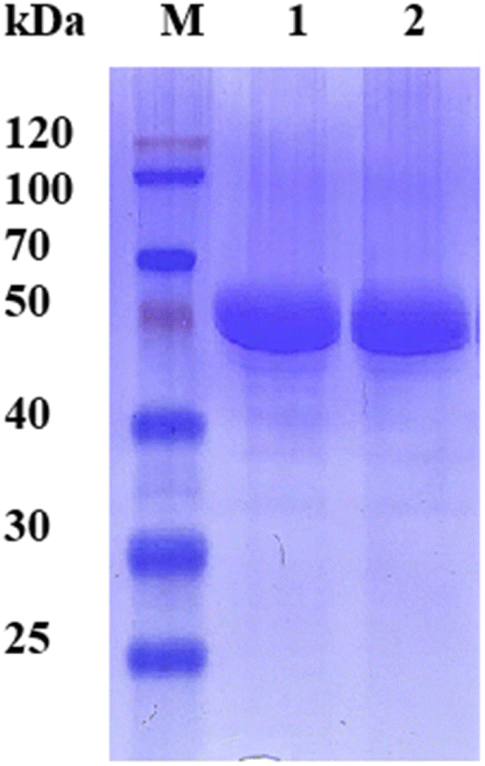 | ||
| Fig. 1 SDS-PAGE (12%) analyses of the purified esterase pnbA-BS and its variant A400P. Lane M, standard molecular mass proteins; lane 1, the esterase pnbA-BS; lane 2, the variant A400P. The proteins were visualized by staining with Coomassie brilliant blue R-250. | ||
| Parameter | pnbA-BS | pnbA-BS A400P |
|---|---|---|
| a The activity was determined using para-nitrobenzyl butyrate as a substrate in 100 mM PBS buffer (pH 9.0, 30 °C). In the assay mixture (200 μl), the assay was initiated by adding 3 μg of purified esterase pnbA-BS or its variant A400P. The substrate concentrations varied within the range from 0 to 50 mM. All activity assays were conducted in triplicate. | ||
| Km (mM) | 1.58 ± 0.13 | 0.94 ± 0.11 |
| Vmax (U mg−1) | 77.98 ± 0.35 | 2.03 ± 0.05 |
| kcat (s−1) | 69.93 ± 0.32 | 1.82 ± 0.04 |
| kcat/Km (mM−1 s−1) | 44.26 ± 0.20 | 1.94 ± 0.04 |
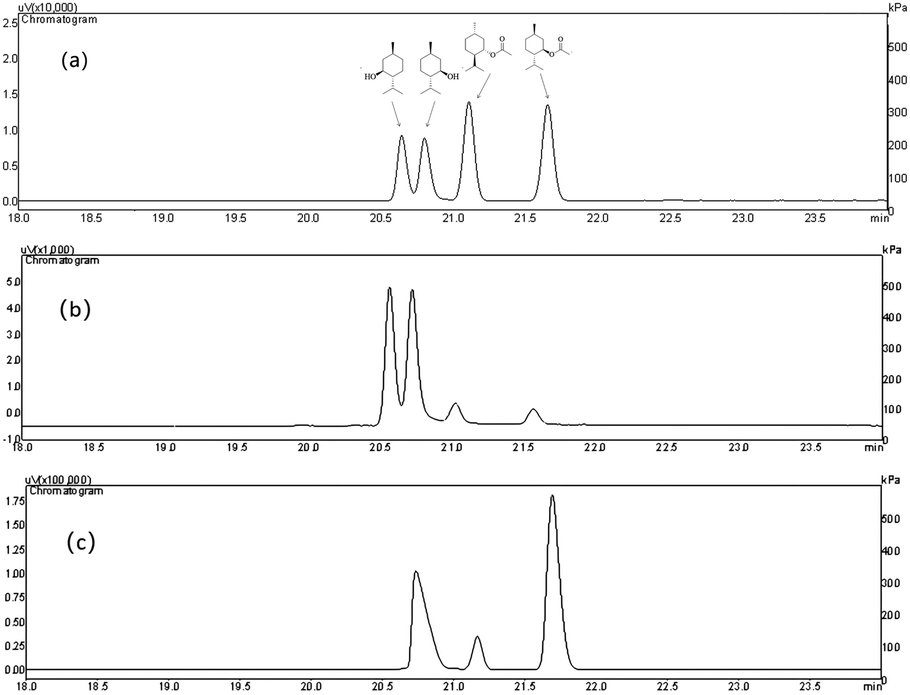 | ||
| Fig. 2 Comparison of L-enantioselectivity of pnbA-BS and its variant A400P through chiral GC chromatography. The reaction mixture (10 ml) contained 500 mM D, L-menthyl acetate, 35 mg purified pnbA-BS or its variant A400P, and 100 mM PBS buffer (pH 8.0). The reaction was conducted in an orbital shaker (600 rpm, 25 °C) for 24 h, and constant pH was maintained by the titration of 1 M NaOH. (a) The standards of D-menthol, L-menthol, L-menthyl acetate, and D-menthyl acetate (from left to right); (b) the reaction mixture catalyzed by the esterase pnbA-BS; (c) the reaction mixture catalyzed by the esterase pnbA-BS A400P. | ||
3.3. Whole-cell catalyzed hydrolysis of D, L-menthyl acetate to L-menthol
The L-enantioselectivity of the purified esterase pnbA-BS A400P was superior to that of the whole cells expressing the esterase pnbA-BS A400P. However, the catalytic efficiency of the whole-cell biocatalyst was greater than that of the purified enzyme. Whole-cell biocatalyst catalyzed the hydrolysis of 500 mM D, L-menthyl acetate leading to the conversion of 43.4% in 10 h, while it took the equivalent amount of the purified enzyme 24 h to reach the conversion of 42.2%. The cell envelope offered the protection of intracellular enzymes but also the permeability of hydrophobic substrates to keep the reaction going.35–37 Compared with isolated enzymes, the use of whole-cell catalysts could simplify the procedure of biocatalyst preparation and facilitate the biocatalyst reuse, offering a cost-effective advantage. More importantly, further catalytic experiments revealed that the e.e.p values maintained >99% (L) when the original substrate concentration was greater than 250 mM, suggesting that the negative effect of isozyme(s) in the host strain could be totally overcome. Thus, lyophilized whole cells expressing pnbA-BS A400P and 500 mM substrate were used for subsequent studies, unless stated otherwise.To achieve optimal catalytic efficiency, various factors, such as temperature, pH, co-solvent concentration, and substrate concentration, were investigated. The influence of the reaction temperature was determined over a range of 20–50 °C, and the highest conversion of D, L-menthyl acetate was observed at 30 °C (Fig. S9a†). When the temperature was greater than 30 °C, the conversion decreased as the temperature rose. To determine the optimal pH, the reaction was carried out at pH levels ranging from 5.5 to 8.5 at 30 °C. The highest conversion was detected at pH 7.0 (Fig. S9b†). The use of organic solvent could have a profound effect on the specific activity and enantioselectivity.5,6 The effect of various readily-available and cost-effective organic solvents on both activity and catalytic performance was tested. The tested organic solvents inhibited the specific activity, of which ethanol was the least harmful (Fig. 3a). When the ethanol concentration was set as 0%, 2%, 4%, 6% and 8% (v/v), the highest conversion was observed at the concentration of 6% (v/v). It was noted that high conversion could be achieved with time extension even though no co-solvent was supplemented (Fig. 3b). The organic solvent-free system could reduce the use of chemicals, simplify the product separation and enhance atom economy. Therefore, the organic solvent was omitted in the subsequent selective hydrolysis of D, L-menthyl acetate.
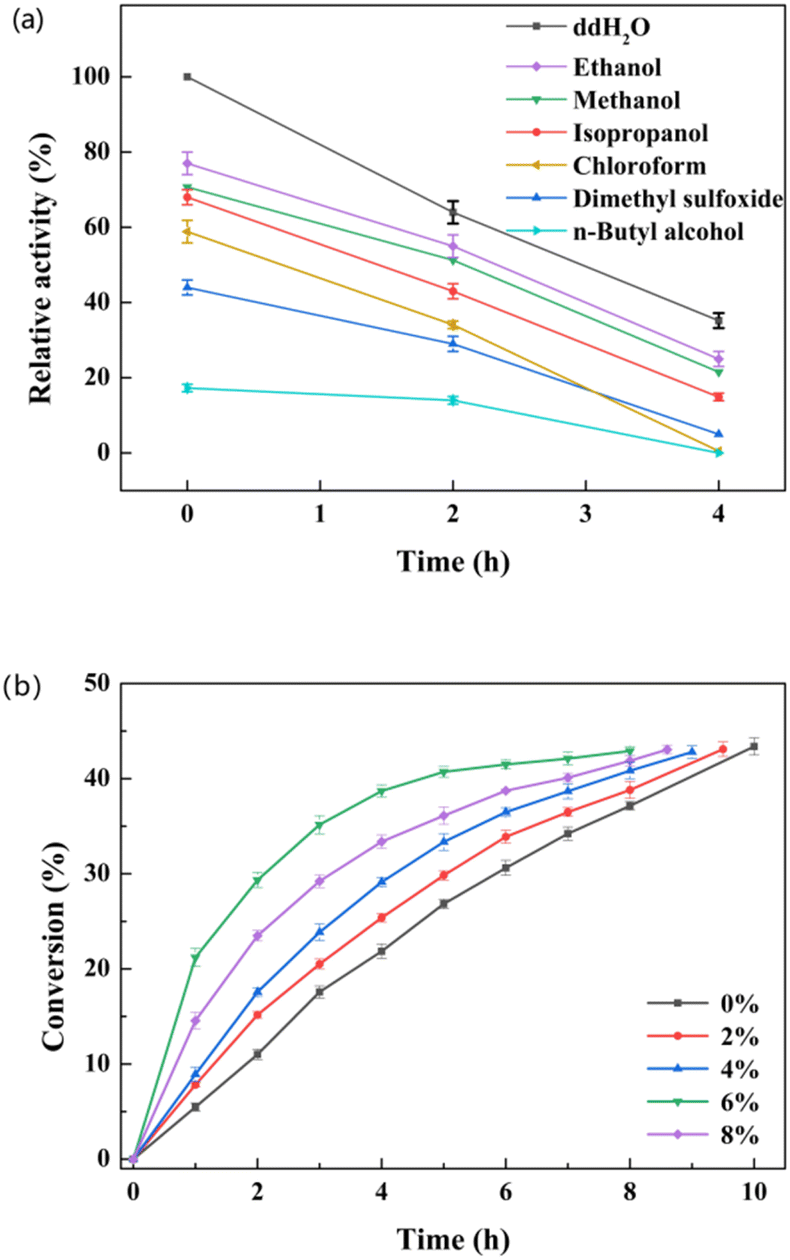 | ||
| Fig. 3 Effect of organic solvents on the activity (a) and catalytic performance (b) in the whole-cell catalyzed hydrolysis of D, L-menthyl acetate. (a) The activity assay mixture (200 μl) contained 0.5 mM para-nitrobenzyl butyrate as substrate, 3 μg purified esterase pnbA-BS A400P, and 100 mM PBS buffer (pH 9.0, 30 °C). (b) The reaction mixture (10 ml) contained 500 mM D, L-menthyl acetate, 0.2 g lyophilized cells expressing the esterase pnbA-BS A400P, and 100 mM PBS buffer solution (pH 7.0). The reaction was conducted in an orbital shaker (600 rpm, 30 °C) for 8 h meanwhile constant pH was maintained by the titration of 1 M NaOH. Data present mean values ± SD from three independent experiments. | ||
When the substrate concentration was increased in a stepwise manner, typical time courses under biocatalyst loading of 0.2 g lyophilized cells in a 10 ml reaction mixture are shown in Fig. 4. The times required to achieve >45% conversion for 100 mM, 250 mM, and 500 mM substrate were 4 h, 6 h, and 12 h, respectively. A further increase of the substrate concentration to 750 and 1000 mM resulted in decreased conversion within 14 h, and the decrease in catalytic efficiency might be attributed to the inhibition of the specific activity under the environment of high-concentration hydrophobic substrate.35
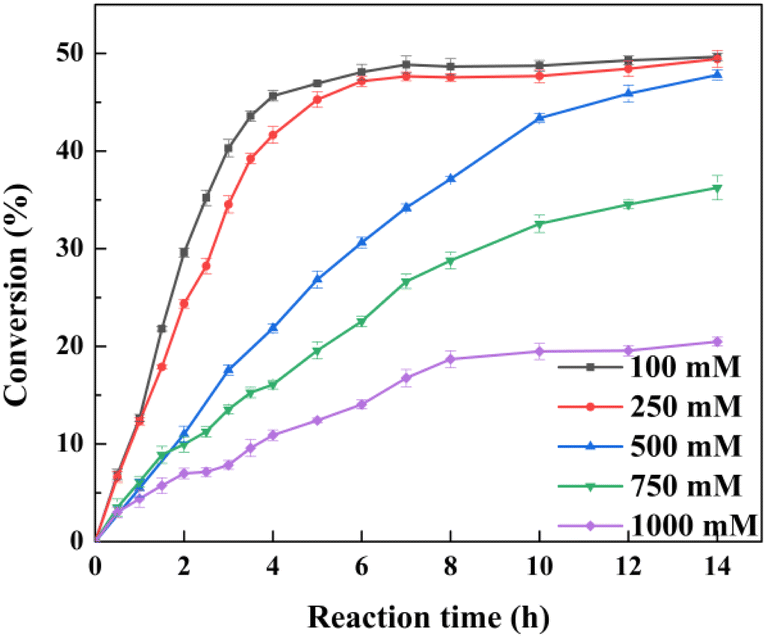 | ||
| Fig. 4 Whole-cell catalyzed selective hydrolysis of various concentrations of D, L-menthyl acetate. The reaction mixture (10 ml) contained 500 mM D, L-menthyl acetate, 0.2 g lyophilized cells expressing the esterase pnbA-BS A400P, and 100 mM PBS buffer solution (pH 7.0). The reaction was conducted in an orbital shaker (600 rpm, 30 °C) for 14 h meanwhile constant pH was maintained by the titration of 1 M NaOH. Data present mean values ± SD from three independent experiments. | ||
3.4. Enhancement of substrate loading and product accumulation through the substrate feeding strategy
It was expected that the substrate constant feeding would relieve the substrate inhibition on activity and benefit from the accumulation of higher concentration product.25 The reaction was conducted at pH 7.0 and 30 °C using 20 g l−1 of the lyophilized cells. The initial substrate concentration was 250 mM to abolish the negative effect of intracellular isozyme(s) and the rest of the substrate was constantly fed within 10 h to the corresponding substrate loading (1 M, 1.1 M, 1.2 M, 1.3 M, 1.4 M, or 1.7 M). After that, the reaction was extended for 4–14 h to achieve higher product accumulation. In the manner of substrate constant feeding, the substrate loading could reach up to 1700 mM with a conversion of 31.65% after 24 h reaction (Fig. 5). The highest product accumulation of 600.6 mM (93.8 g l−1) was observed when the substrate loading was 1300 mM. When 1000 mM substrate loading was applied, the conversion was 48.9% at 12 h, corresponding to the space-time yield of 160.52 g (l d)−1. Thus, substrate constant feeding was a simple and effective way to enhance product accumulation. When each reaction was terminated, water-immiscible substrate and product as the organic phase were simply recovered through centrifugation, requiring no frequently-applied solvent extraction. The collected substrate and product were separated through silica gel column chromatography. Finally, the resulting product was verified to be optically pure L-menthol (>99% e.e.p) through the combination of chiral GC, GC-MS, 1H NMR, and 13C NMR analyses.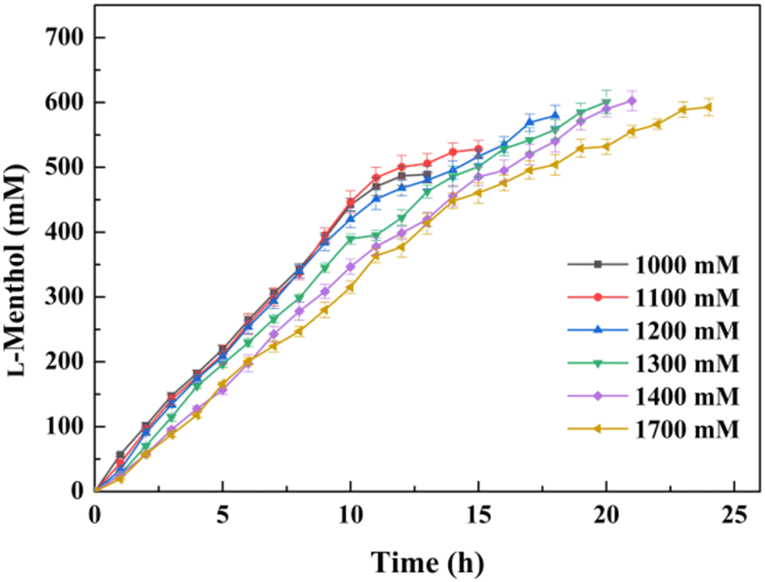 | ||
| Fig. 5 Selective hydrolysis of D, L-menthyl acetate through a substrate constant feeding strategy. The original reaction mixture (10 ml) contained 250 mM D, L-menthyl acetate, 0.2 g lyophilized cells expressing pnbA-BS A400P, and 100 mM PBS buffer (pH 7.0). The rest of the required substrate was constantly fed within 10 h. The reaction was conducted in an orbital shaker (600 rpm, 30 °C) for 24 h meanwhile constant pH was maintained by the titration of NaOH. Data present mean values ± SD from three independent experiments. | ||
4 Conclusions
The highly-active esterase pnba-BS was over-expressed in E. coli and engineered to improve its L-enantioselectivity in the selective hydrolysis of D, L-menthyl acetate to L-menthol. The variant A400P demonstrated strict L-enantioselectivity in the hydrolysis of D, L-menthyl acetate. In comparison with the esterase pnbA-BS, the substitution of Ala400 to Pro increased L-enantioselectivity but decreased the kcat/Km value, indicating the trade-off effect between activity and enantioselectivity. The negative effect of biacatalyst engineering on activity could be counter-weighed by the efforts of process engineering while maintaining strict L-enantioselectivity. The omitting of organic solvent from the reaction system could benefit the specific activity, simplify the product work-up, and reduce the use of chemicals. Moreover, the substrate constant feeding was applied to relieve the substrate inhibition. When the substrate loading was set as 1.0 M in the manner of substrate constant feeding, the e.e.p value was >99% and the space-time yield reached up to 160.52 g (l d)−1 after the 12 h reaction, representing an efficient, easy-to-use and green process for the L-menthol production.Author contributions
Jingjing Qiao: investigation, analysis, resources, data curation, visualization, writing–original draft. Duxia Yang: investigation, analysis, resources, visualization. Yingting Feng: investigation, analysis, validation. Wan Wei: analysis, validation. Xun Liu: analysis, validation. Yinjun Zhang: analysis, validation, writing–review and editing. Jianyong Zheng: conceptualization, analysis, resources, data curation, writing–review and editing, funding acquisition. Xiangxian Ying: conceptualization, analysis, resources, data curation, visualization, writing–original draft, review and editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Key Projects of Technological Innovation and Application Development in Chongqing City, China (No. cstc2021jscx-jbgs X0002) and The Natural Science Foundation of Zhejiang Province, China (No. LY18B020021).Notes and references
- C. A. Godoy, J. S. Pardo-Tamayo and O. Barbosa, Int. J. Mol. Sci., 2022, 23, 9933 CrossRef CAS PubMed.
- P. Fojan, P. H. Jonson, M. T. N. Petersen and S. B. Petersen, Biochimie, 2000, 82, 1033–1041 CrossRef CAS PubMed.
- N. Barzkar, M. Sohail, S. T. Jahromi, M. Gozari, S. Poormozaffar, R. Nahavandi and M. Hafezieh, Appl. Biochem. Biotechnol., 2021, 193, 1187–1214 CrossRef CAS PubMed.
- S. Vorlová, U. T. Bornscheuer, I. Gatfield, J.-M. Hilmer, H.-J. Bertram and R. D. Schmid, Adv. Synth. Catal., 2002, 344, 1152–1155 CrossRef.
- G.-W. Zheng, H.-L. Yu, J.-D. Zhang and J.-H. Xu, Adv. Synth. Catal., 2009, 351, 405–414 CrossRef CAS.
- G.-W. Zheng, J. Pan, H.-L. Yu, M.-T. Ngo-Thi, C.-X. Li and J.-H. Xu, J. Biotechnol., 2010, 150, 108–114 CrossRef CAS PubMed.
- H. Chen, J. Wu, L. Yang and G. Xu, Biochim. Biophys. Acta Protein Proteonomics, 2013, 1834, 2494–2501 CrossRef CAS PubMed.
- H. Chen, J. Wu, L. Yang and G. Xu, J. Mol. Catal. B: Enzym., 2014, 102, 81–87 CrossRef CAS.
- M. B. Ansorge-Schumacher and O. Thum, Chem. Soc. Rev., 2013, 42, 6475–6490 RSC.
- D. Brady, S. Reddy, B. Mboniswa, L. H. Steenkamp, A. L. Rousseau, C. J. Parkinson, J. Chaplin, R. K. Mitra, T. Moutlana, S. F. Marais and N. S. Gardiner, J. Mol. Catal. B: Enzym., 2012, 75, 1–10 CrossRef CAS.
- H. Chen, X. Meng, X. Xu, W. Liu and S. Li, Appl. Microbiol. Biotechnol., 2018, 102, 3487–3495 CrossRef CAS PubMed.
- K. Min, H. T. Kim, S. J. Park, S. Lee, Y. J. Jung, J.-S. Lee, Y. J. Yoo and J. C. Joo, Bioresour. Technol., 2021, 337, 125394 CrossRef CAS PubMed.
- M. Kataoka, T. Miyakawa, S. Shimizu and M. Tanokura, Appl. Microbiol. Biotechnol., 2016, 100, 5747–5757 CrossRef CAS PubMed.
- Z. Yu, H. Yu, H. Tang, Z. Wang, J. Wu, L. Yang and G. Xu, ChemCatChem, 2021, 13, 1–12 CrossRef.
- H. Zhang, Z. Cheng, L. Wei, X. Yu, Z. Wang and Y. Zhang, Bioorg. Chem., 2022, 120, 105602 CrossRef CAS PubMed.
- H. J. Park, J. C. Joo, K. Park, Y. H. Kim and Y. J. Yoo, J. Biotechnol., 2013, 163, 346–352 CrossRef CAS PubMed.
- V. Stepankova, S. Bidmanova, T. Koudelakova, Z. Prokop, R. Chaloupkova and J. Damborsky, ACS Catal., 2013, 3, 2823–2836 CrossRef CAS.
- H. Cui, L. Zhang, L. Eltoukhy, Q. Jiang, S. K. Korkunç, K.-E. Jaeger, U. Schwaneberg and M. D. Davari, ACS Catal., 2020, 10, 14847–14856 CrossRef CAS.
- M. D. Davari, H. Cui, M. Vedder, L. Zhang, K.-E. Jaeger and U. Schwaneberg, ChemSusChem, 2022, 15, e202102551 Search PubMed.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- K. V. K. Boodhoo, M. C. Flickinger, J. M. Woodley and E. A. C. Emanuelsson, Chem. Eng. Process., 2022, 172, 108793 CrossRef CAS.
- A. I. Benítez-Mateos, D. R. Padrosa and F. Paradisi, Nat. Chem., 2022, 14, 489–499 CrossRef PubMed.
- U. Hanefeld, F. Hollmann and C. E. Paul, Chem. Soc. Rev., 2022, 51, 594–627 RSC.
- I. Oroz-Guinea, C. Winkler, S. Glueck, K. Ditrich, M. Weingarten, M. Breuer, D. Schachtschabel and W. Kroutil, ChemCatChem, 2022, 14, e202101557 CrossRef CAS.
- M. Zhao, L. Gao, L. Zhang, Y. Bai, L. Chen, M. Yu, F. Cheng, J. Sun, Z. Wang and X. Ying, Biotechnol. Lett., 2017, 39, 741–1746 Search PubMed.
- A. Millán, N. Sala, M. Torres and R. Canela-Garayoa, Catalysts, 2021, 11, 216 CrossRef.
- X. Ying, S. Yu, M. Huang, R. Wei, S. Meng, F. Cheng, M. Yu, M. Ying, M. Zhao and Z. Wang, Molecules, 2019, 24, 1057 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- D. Seeliger and B. L. de Groot, J. Comput. Aided Mol. Des., 2010, 24, 417–422 CrossRef CAS PubMed.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS PubMed.
- J. Rakels, A. Straathof and J. Heijnen, Enzyme Microb. Technol., 1993, 15, 1051–1056 CrossRef CAS PubMed.
- W. Tang, L. Chen, J. Deng, Y. Kuang, C. Chen, B. Yin, H. Wang, J. Lin and D. Wei, Catal. Sci. Technol., 2020, 10, 7512–7522 RSC.
- B. Zhang, H. Du, Y. Zheng, J. Sun, Y. Shen, J. Lin and D. Wei, Microb. Biotechnol., 2022, 15, 1486–1498 CrossRef CAS PubMed.
- B. Spiller, A. Gershenson, F. H. Arnold and R. C. Stevens, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12305–12310 CrossRef CAS PubMed.
- R. Kratzer, J. M. Woodley and B. Nidetzky, Biotechnol. Adv., 2015, 33, 1641–1652 CrossRef CAS PubMed.
- Y. Qiao, C. Wang, Y. Zeng, T. Wang, J. Qiao, C. Lu, Z. Wang and X. Ying, Microb. Cell Fact., 2021, 20, 17 CrossRef CAS PubMed.
- Y. Jia, Q. Wang, J. Qiao, B. Feng, X. Zhou, L. Jin, Y. Feng, D. Yang, C. Lu and X. Ying, Catalysts, 2021, 11, 931 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00490b |
| This journal is © The Royal Society of Chemistry 2023 |