
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalysed one-pot three component synthesis of 3,3′-disubstituted oxindoles featuring an all-carbon quaternary center and spiro[2H-pyran-3,4′-indoline]†
Chandrakant B. Nichinde‡
 ab,
Baliram R. Patil‡ab,
Suryakant S. Chaudhariab,
Bhupendra P. Malibc,
Rajesh G. Gonnadebc and
Anil K. Kinage*ab
ab,
Baliram R. Patil‡ab,
Suryakant S. Chaudhariab,
Bhupendra P. Malibc,
Rajesh G. Gonnadebc and
Anil K. Kinage*ab
aChemical Engineering and Process Development Division, CSIR-National Chemical Laboratory, Pune, India-410 008. E-mail: ak.kinage@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201 002, India
cPhysical and Material Chemistry Division, CSIR-National Chemical Laboratory, Pune, India-410 008
First published on 28th April 2023
Abstract
A simple and efficient methodology for the one-pot synthesis of 3,3′-disubstituted oxindoles featuring an all-carbon quaternary center has been demonstrated through L-proline catalysed three-component reaction based on sequential Knoevenagel condensation/Michael addition and also one-pot synthesis of spiro[2H-pyran-3,4′-indoline] through consecutive Knoevenagel condensation/Michael addition/reduction/cyclization reactions from readily available isatin derivatives, malononitrile, and ketones. The present methodology presents several advantages, including simple reaction set-up, short reaction times, and easy to work-up. Also, this strategy offers broad substrate scope with excellent yields and high atom economy, under mild reaction conditions.
Introduction
3,3′-Disubstituted oxindoles and spiro[2H-pyran-3,4′-indoline] framework featuring an all-carbon quaternary center at C-3 position have been frequently identified as core structurally important heteroaromatic motifs for the variability of drug molecules, biologically active natural products, synthetic pharmaceuticals and have been widely utilized for alkaloid synthesis.1 These are powerful synthetic intermediates for the synthesis of complex biologically active molecules.2 The oxindole framework bearing an all-carbon quaternary center at C-3 and spiro[2H-pyran-3,4′-indoline] is found in a specific class of medicinally favourable molecules.3,4 Furthermore, several oxindole and spirooxindole equivalents show an extensive range of biological activities as shown in Fig. 1 (antidepressant effect,5 anti-Alzheimer,6 5-HT7 receptor antagonists,7 anticancer,8 antibacterial,9 p38a inhibitor,10 treatment of hemorrhagic fever with renal syndrome,11 etc.). Because of its many advantages chemists are inspired to develop synthetic methods towards 3,3-disubstituted oxindoles and spirooxindoles frameworks.12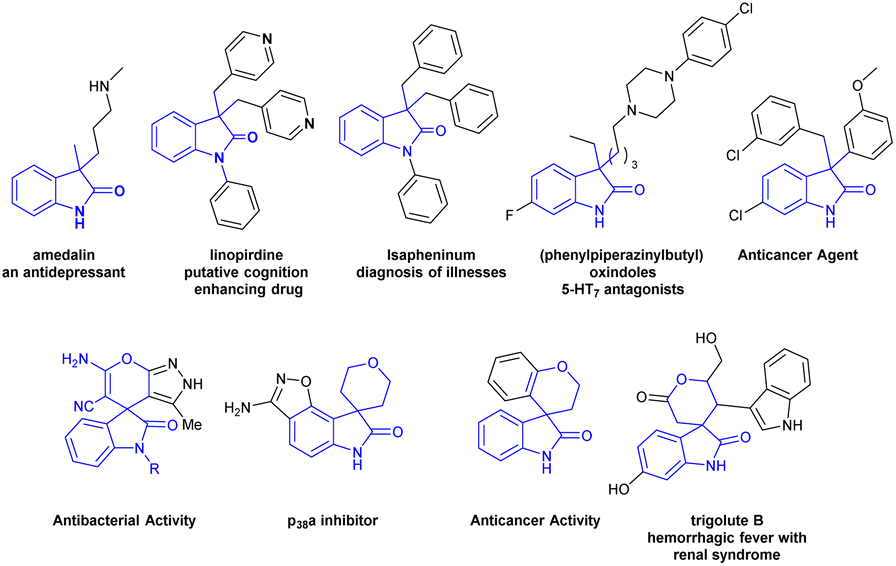 | ||
| Fig. 1 3,3′-Disubstituted oxindoles featuring an all-carbon quaternary center and spirooxindole fragments present in natural products and pharmaceuticals. | ||
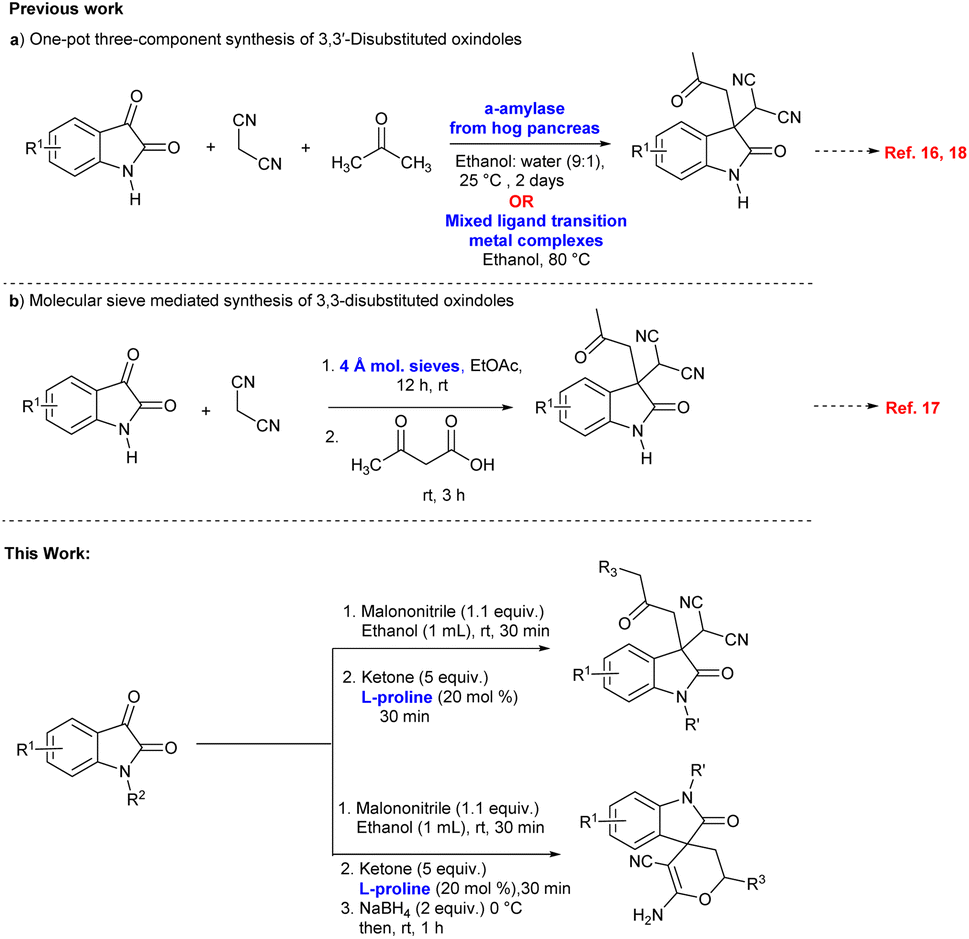
The multicomponent reactions (MCRs) are favourable methodology for building structurally different molecules and offer important advantages for finalized conventional linear-type synthesis due to its convergent, flexible and atom-efficient nature.13 In the past few years there has been prodigious development in multi-component reactions, and attempts continue to be made to develop new MRCs.14 L-Proline is one of the most important, well-known, inexpensive and radially available organocatalyst, demonstrating efficacy in various C–C and C–heteroatom asymmetric bond-forming reactions.15
In recent years one-pot multicomponent synthetic protocols have been reported for the construction of 3,3′-disubstituted oxindoles with an all-carbon quaternary center using a three-component Knoevenagel/Michael addition reaction of isatin, malononitrile and ketoacids or ketones (Scheme 1).16–18 Furthermore, few synthetic methods have been established for the enantioselective synthesis of 3,3′-disubstituted oxindoles via the Michael reaction of ketones to isatylidene malononitrile.19–21 Also, spiro[2H-pyran-3,4′-indoline] oxindoles as a type of heterocyclic spirooxindoles have attracted considerable attention and few synthetic protocols have been reported for the construction of the compounds via, Michael addition/cascade reduction/cyclization.22
 | ||
| Scheme 1 One pot synthetic approach towards disubstituted oxindoles featuring an all-carbon quaternary center. | ||
To the best of our knowledge, one-pot three-component L-proline catalysed sequential Knoevenagel condensation/Michael addition reaction via enamine activation has been not investigated so far for the synthesis of 3,3′-disubstituted oxindoles featuring an all-carbon quaternary center. Moreover, this strategy has been significantly extended to the synthesis of spiro[2H-pyran-3,4′-indoline] in one-pot by sequential Knoevenagel condensation/Michael addition/reduction/cyclization reaction.
Results and discussion
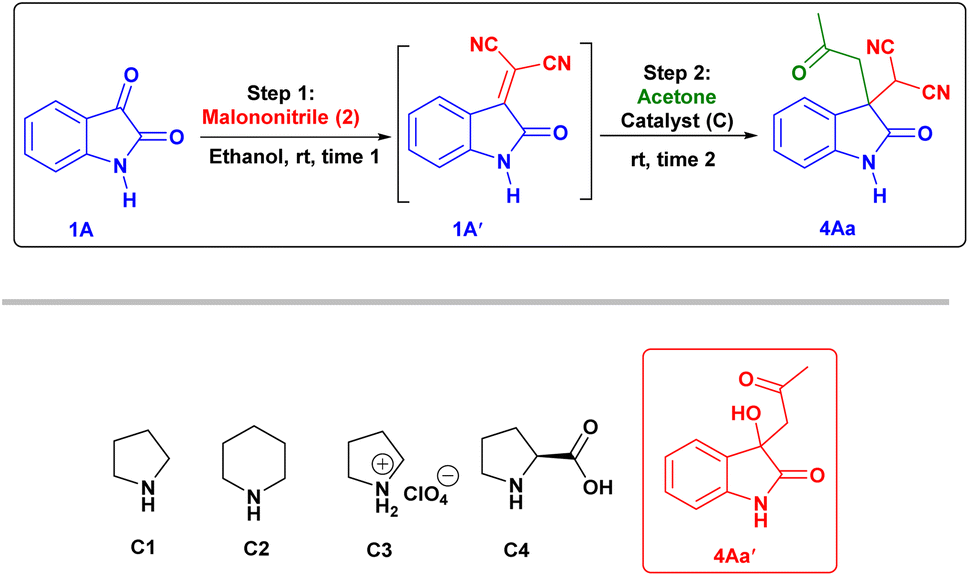
First, we studied the effect of different solvents on the reaction of isatin and malononitrile for in situ Knoevenagel condensation (see ESI Table 1†) to give isatylidene malononitrile (1A′). Then we envisioned a suitable catalyst for the formation of enamine (INT-C), which can attack further on reactive isatylidene malononitrile (1A′) formed in step-1 to give the expected product through Michael addition.To explore the one-pot synthesis, isatin, malononitrile, acetone and catalyst were stirred at room temperature (RT) in a reaction vessel for 1 h. It was observed that along with desired product undesirable aldol adduct product 4Aa′ is also formed. Therefore, to avoid the formation of undesired product we first added isatin (1A) and malononitrile (2) as an active methylene group for Knoevenagel condensation to give isatylidene malononitrile (1A′) (Michael acceptor) and after complete formation (ESI†) of isatylidene malononitrile (1A′), we added acetone in presence of catalyst in a same reaction vessel. In this way, we obtained complete conversion to the desired product. Based on enamine forming capacity various catalysts were further tested. The rate of reaction for step-2 (Michael addition) was slow for pyrrolidine, piperidine, pyrrolidine with chloroacetic acid additive, and pyrrolidinium salts as a catalyst. Where we observed a trace amount of 4Aa on thin layer chromatography (TLC) (Table 1, entry 2–5 and 9). However, reaction worked efficiently when we Used L-proline as a catalyst at RT, reaction completed within 1 h and product 4Aa with 97% yield. We further carried out all the reactions using L-proline catalyst in different solvents (see Table 4, entries 2–8) and we found that ethanol is an ideal solvent for one-pot reaction (Table 1, entry 6). Further, we studied the effect of catalyst concentration by reducing the L-proline concentration from 20 to 10 or 5 mol%, we observed that slight decrease in the product yield, and the reaction took a longer time (Table 1, entries 7 and 8). With an optimized reaction condition in hand, we further used this methodology for different isatin (1) and ketone derivatives (3). Gratifyingly, we observed high to excellent yield in all reactions to give the 3,3-disubstituted oxindoles (up to 98% isolated yield). Further, we studied the isatin with different electron-donating groups viz. the 5-Me, 5-OMe which showed excellent yield (4Ab, 4Ac).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent (1 mL) | Time (1) h | Time (2) h | Yieldsc 4Aa (%) |
a Unless otherwise noted, a mixture of 1A (0.5 mmol, 1 equiv.), 2 (0.55 mmol, 1.1 equiv.) in solvent (1 mL), stirred at rt for respective time 1, Then added 3 (2.5 mmol) and catalyst (mol%) in same pot and stirred for time 2.b 1A′ remain in reaction mixture confirmed by 1H and 13C NMR.c Isolated yield.d Checked HPLC for product (column: chiral pack AD-H. Solvent system: 30 IPA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 n-hexane).e Added additive chloroacetic acid (20 mol%).f 4 equiv. of acetone was used.g Observed trace amount of 4Aa on TLC. :70 n-hexane).e Added additive chloroacetic acid (20 mol%).f 4 equiv. of acetone was used.g Observed trace amount of 4Aa on TLC. |
|||||
| 1b | — | EtOH | 0.5 | 72 | N.R.g |
| 2b | C1 (20) | EtOH | 0.5 | 48 | <5g |
| 3b | C2 (20) | EtOH | 0.5 | 48 | <5g |
| 4b,e | C1 (20) | EtOH | 0.5 | 48 | <5g |
| 5b | C3 (20) | EtOH | 0.5 | 48 | <5g |
| 6d | C4 (20) | EtOH | 0.5 | 0.5 | 97 |
| 7d | C4 (5) | EtOH | 0.5 | 5 | 81 |
| 8d | C4 (10) | EtOH | 0.5 | 3 | 91 |
| 9d,f | C4 (20) | EtOH | 0.5 | 2 | 86 |
| 10d | C4 (20) | — | 2 | 24 | 37 |
Isatin with halogen substitute at different positions (5-F, 5-Cl, 5-Br, 5-I, 7-F and 7-Cl) was studied and showed excellent yield (up to 97% 4Ad–4Ai) (Fig. 2). Multi-halogen substituted isatin like 4,7-dichloro substituted isatin also gave an excellent yield 94% (4Aj) (Table 2).
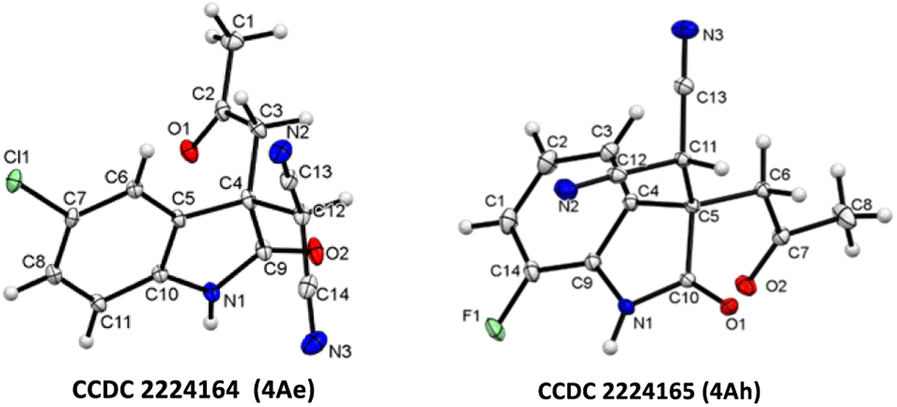 | ||
| Fig. 2 X-ray crystallographic analysis of 4Ae and 4Ah.26 | ||
| a Unless otherwise noted, a mixture of 1A (0.5 mmol, 1 equiv.), 2 (0.55 mmol, 1.1 equiv.) in ethanol (1 mL) was stirred at rt for 30 minutes, followed by addition of 3 (2.5 mmol, 5.0 equiv.) and L-proline (0.1 mmol. 20 mol%), stirred continuously for next 30 min; isolated yields.b Reaction required more time for total conversion from optimize time are mentioned.c 1A′ remain in reaction mixture confirmed by 1H and 13C NMR. |
|---|
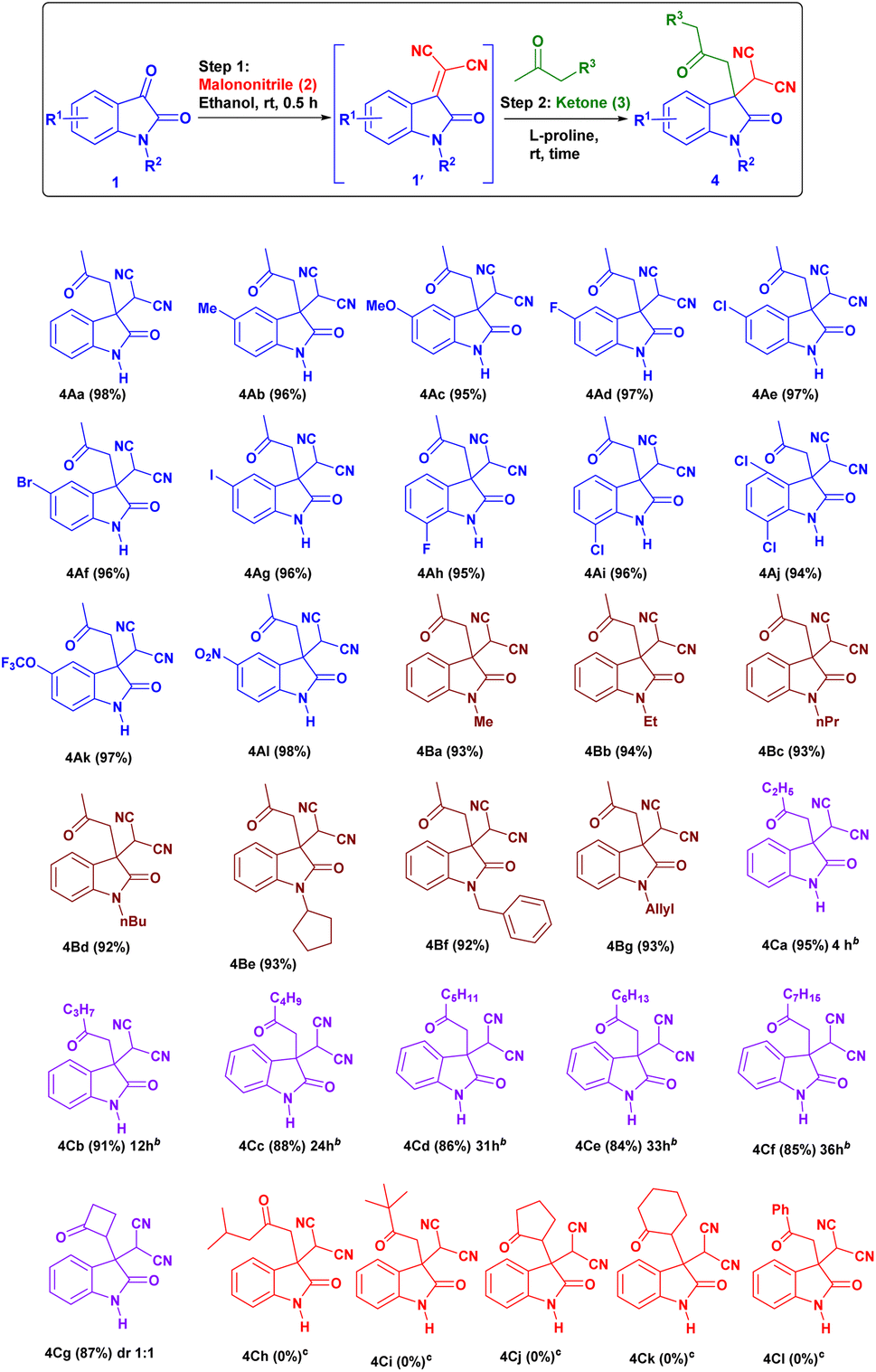 |
Isatin with electron withdrawing group 5-OCF3, 5-NO2 gave excellent yields up to 98% (4Ak, 4Al).
The N-substituted isatin23 like N-methyl, N-ethyl, N-propyl, N-butyl, N-cyclopentyl, N-allyl, N-benzyl were also evaluated in which all gave an excellent yield of product (92–94%, 4Ba–4Bg). Then we focus our courtesy on one-pot reaction using different ketones (3). We obtained desired product in high to excellent yield. However, with an elongated carbon chain reaction yield decrease (up to 84%) and the rate of reaction becomes slow (4Ca–4Cf).24 Unfortunately, in the case of sterically hindered ketones like isobutyl methyl ketone (4Ch) and tert-butyl methyl ketone (4Ci) have not given the expected product at optimized reaction condition. As well as in case of cyclopentanone (4Cj), cyclohexanone (4Ck) and acetophenone (4Cl) we observed only isatylidene malononitrile (1A′) (step 1) instead of the expected desired product. Surprisingly, by use of cyclobutanone, we got the expected desired product (4Cg) in good yield (87%) with approximately 1:1 diastereomeric ratio calculated from proton NMR.
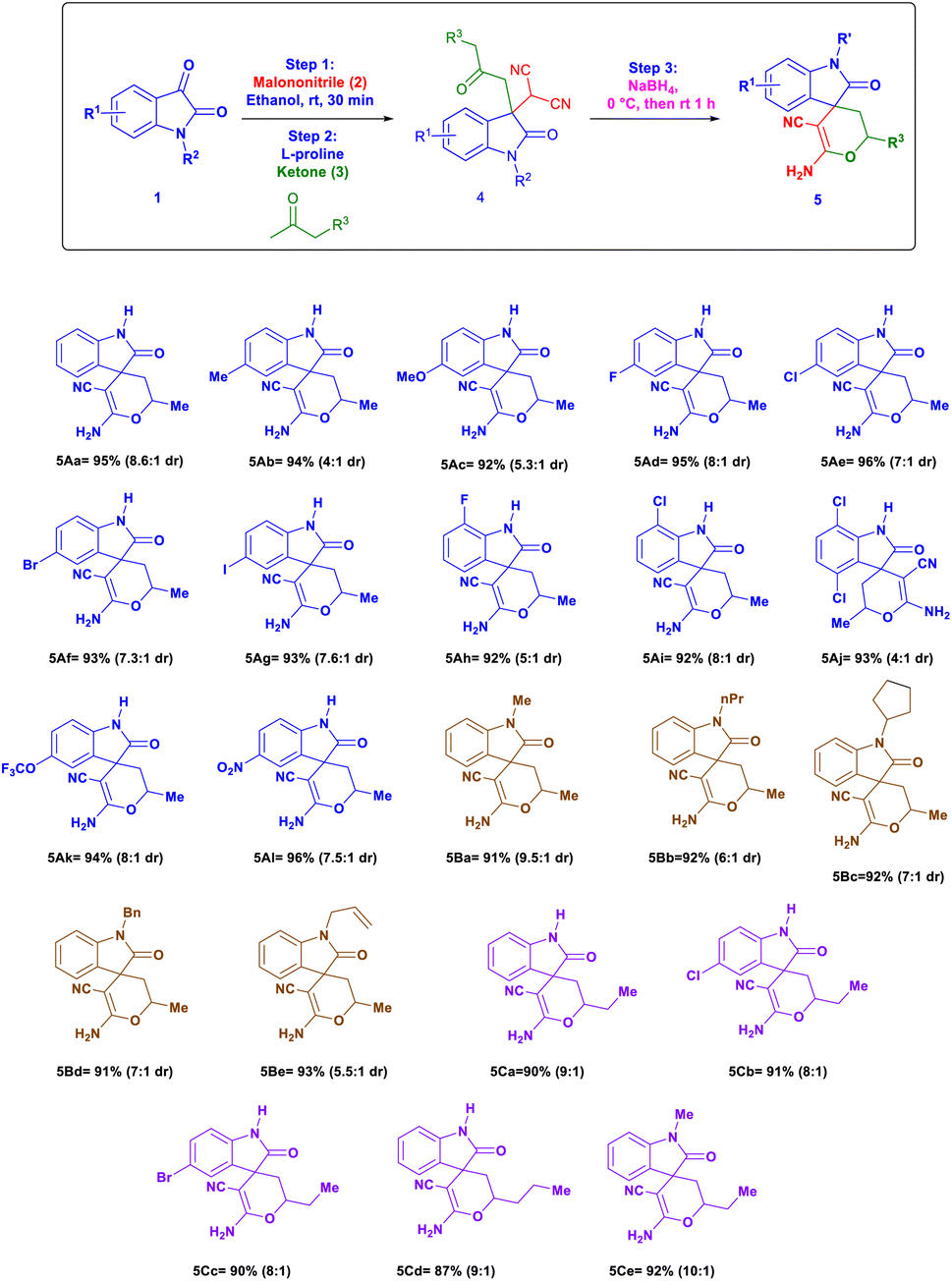
After developing the above effective one-pot reaction for the synthesis of 3,3-disubstituted oxindoles, our next goal is to extend this methodology for cyclization to the synthesis of spiro[2H-pyran-3,4′-indoline] by following a strategy of reduction/cyclization. The reduction of 3,3-disubstituted oxindoles (4) with sodium borohydride in the same reaction pot gave a cyclization product with high to excellent yield. This reaction was carried out in one pot by using isatin 1, malononitrile 2, and acetone to obtain 3,3-disubstituted oxindoles in the same pot sodium borohydride was added we obtained cyclization product 5Aa with 95% yield (combine yield of two diastereomers), 8.6:1 dr (Table 3).25 Isatin with different electron-donating groups like 5-Me (5Ab, 94% yield, 4:1 dr), 5-OMe (5Ac, 92% yield, 5.3:1 dr) and halogen-substituted isatin at a different position like 5-F, 5-Cl, 5-Br, 5-I, 7-F, 7-Cl and 4,7-dichloro substituted substrates shows excellent yields up to 96% and dr from 7:1 to 8:1 (5Ad to 5Aj). Optimized procedure equally worked smoothly with isatin containing electron withdrawing groups like 5-OCF3 (5Ak, 94% yield, 8:1 dr) and 5-NO2 (5Al, 96% yield, 7.5:1 dr). The reaction with different N-substituted isatin like N-methyl, N-propyl, N-cyclopentyl, N-benzyl and N-allyl also gave yield up to 93% and dr ratio up to 9.5:1 (5Ba–5Be). Then we check the applicability of reaction to different substituted isatin derivatives and elongated linear ketones like butanone or pentanone which also showed a good yield up to 93% with 9:1 dr (5Ca–5Cd). We also tried N-methyl isatin with butanone we observed that reaction work equally like other substrates with 92% yield, 10:1 dr (5Ce) (Fig. 3).
| a Unless otherwise noted, a mixture of 1A (0.5 mmol, 1 equiv.), 2 (0.55 mmol, 1.1 equiv.) in ethanol (1 mL) was stirred at rt for 30 minutes, followed by addition of 3 (2.5 mmol, 5.0 equiv.) and L-proline (0.1 mmol. 20 mol%), stirred continuously till get formation of 4, then added NaBH4 (1 mmol) at 0 °C stirred for 10 min and then stirred reaction mixture at rt for 1–2 h. dr mentioned above are calculated by 1H NMR of crude product. Yields shown above are the combine yield of two diastereomers. We separate two diastereomer by silica gel column chromatography and in ESI characterisations provided are for major diastereomer only. |
|---|
 |
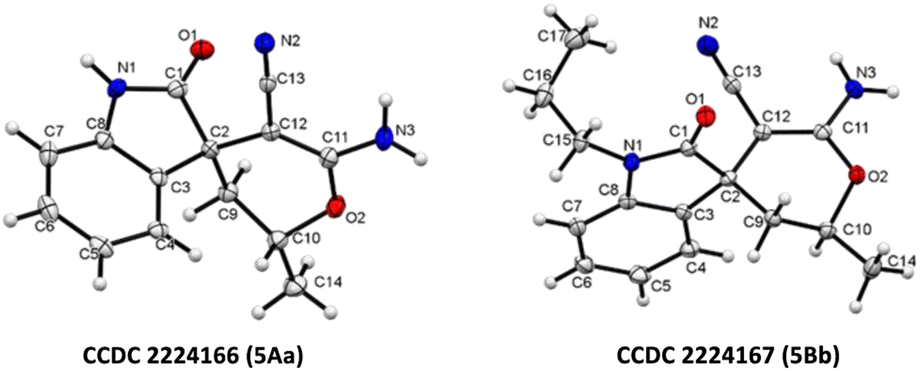 | ||
| Fig. 3 X-ray crystallographic analysis of 5Aa and 5Bb.27 | ||
Further, we tried the feasibility of this one-pot reaction for asymmetric synthesis of 3,3′-disubstituted oxindoles. For this we have chosen ideal substrate isatin (1), malononitrile (2) and acetone in presence of different solvents, but we observed very poor enantioselectivity in all solvents (Table 4, entries 2–9). Then we tried few bulky versions of L-proline catalyst, however, the product (4Aa) obtained with very poor enantioselectivity (<5%) (Table 4, entries 11–13). This outcome showed that it is difficult to obtain the enantioselective product at the current optimized reaction condition by using a bulkier group containing L-proline as a catalyst.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (20 mol%) | Solvent | Time 1, h | Time 2 | Yieldb, % | eec, % |
| a Unless otherwise noted, a mixture of 1A (0.5 mmol, 1 equiv.), 2 (0.55 mmol, 1.1 equiv.) in ethanol (1 mL) was stirred at rt for 30 minutes, followed by addition of acetone (2.5 mmol, 5.0 equiv.) and chiral organocatalyst (0.1 mmol. 20 mol%), stirred continuously till get total conversion.b Isolated yields.c Determinated by chiral HPLC analysis (column: chiral pack AD-H; solvent system – 30 IPA:70 n-hexane).d At −20 °C. |
||||||
| 1 | Cat-I | EtOH | 0.5 | 0.5 h | 97 | 3 |
| 2 | Cat-I | DMF | 6 | 5 | 67 | 4 |
| 3 | Cat-I | CH3CN | 14 | 24 | 54 | 4 |
| 4 | Cat-I | DCM | 12 | 24 | 40 | 3 |
| 5 | Cat-I | MeOH | 0.5 | 2 | 92 | 3 |
| 6 | Cat-I | Et2O | 24 | 48 | N.R. | — |
| 7 | Cat-I | DMSO | 6 | 4 | 87 | 4 |
| 8 | Cat-I | EtOAc | 13 | 5 | 90 | 3 |
| 9d | Cat-I | EtOH | 0.5 | 14 h | 96 | 4 |
| 11 | Cat-II | EtOH | 0.5 | 5 days | 95 | 4 |
| 11 | Cat-III | EtOH | 0.5 | 6 days | 93 | 4 |
| 12 | Cat-IV | EtOH | 0.5 | 7 days | 92 | 2 |
| 13 | Cat-V | EtOH | 0.5 | 4 days | 94 | 3 |
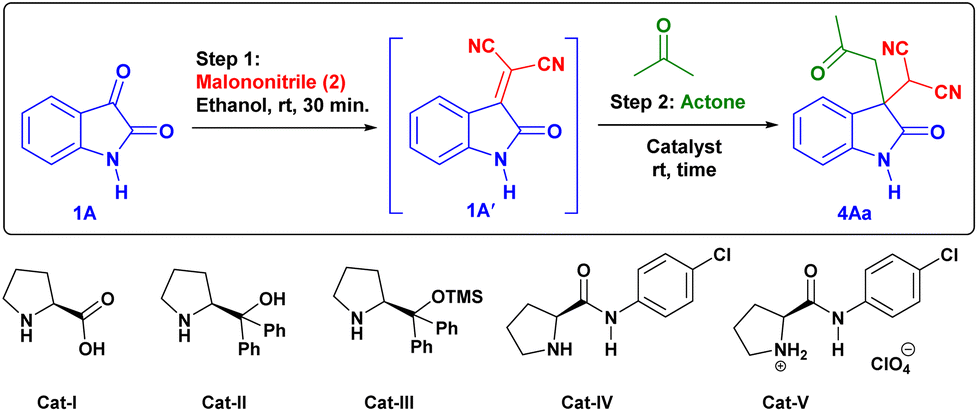
With the help of previously reported mechanisms,28 we proposed a plausible mechanism based on enamine catalysis (Scheme 2), as an enamine (INT-C) via, carbinoalamine (INT-A), iminium ion (INT-B) intermediates. Enamine intermediate (INT-C) attack to isatylidene malononitrile (1A′) which is already formed in step 1 and leads to a nine-membered ring transition state (T. S.). Further this transition state converts to iminium ion (INT-D) with construction of new C–C bond and intramolecular proton transfer. Which on hydrolysis gives required product (4) and the catalyst is regenerated for the next catalytic cycle via INT-E. Although L-proline's α-chirality is ineffective to introduced enantioselectivity in the final product, but it indicates that the carboxylate functionality is essential for catalysis. As pyrrolidine (C1) catalyst is unable to complete reaction. Therefore, L-proline not only acts as an enamine catalyst but also at the same time act as a Brønsted acid co-catalyst. In the same way, we proposed a plausible mechanism for spiro[2-H-pyran-3,4′-indoline] (Scheme 3). As we previously discussed a mechanism for 3,3′-disubstituted oxindoles featuring an all-carbon quaternary center (Scheme 2). After the addition of sodium borohydride ketone group present in 4 will reduce to alcohol and converts to INT-I. Rapid intramolecular attack of an alcoholic group on nitrile group form INT-II, which converts to final product 5 after tautomerization.
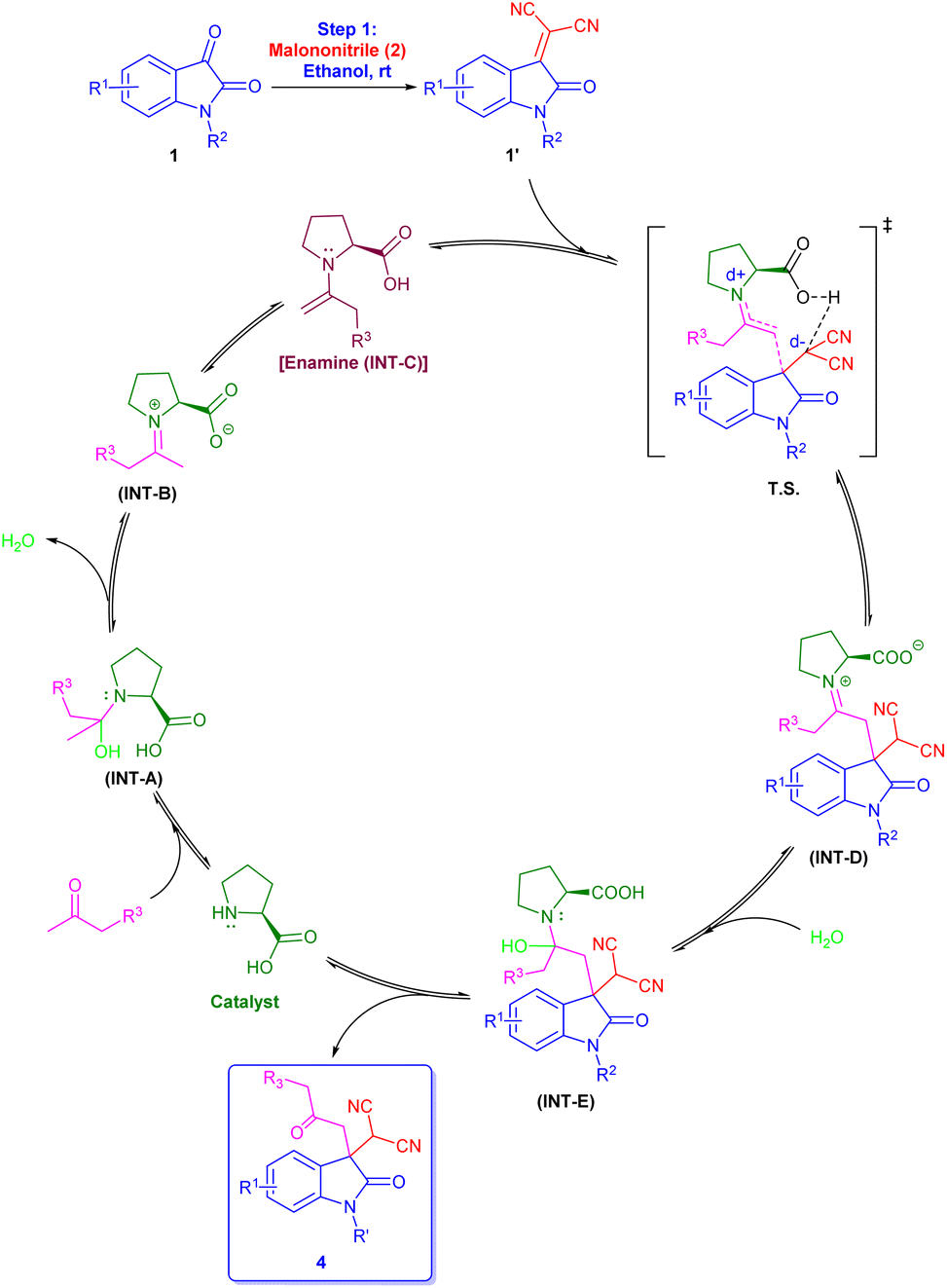 | ||
| Scheme 2 Plausible reaction mechanism for L-proline catalysed 3,3′-disubstituted oxindoles featuring an all-carbon quaternary center. | ||
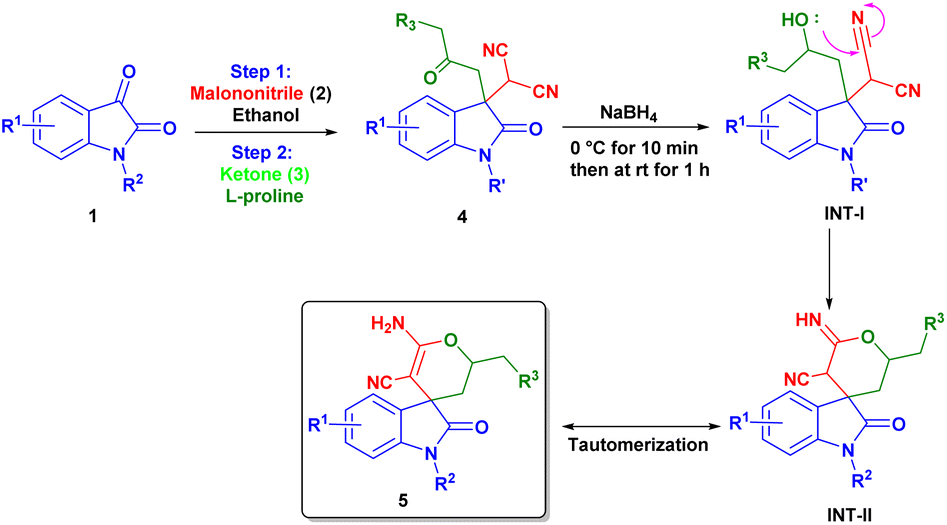 | ||
| Scheme 3 Plausible reaction mechanism for spiro[2-H-pyran-3,4′-indoline]. | ||
Conclusions
In conclusion, we effectively show a one-pot, L-proline catalyzed consecutive Knoevenagel condensation/Michael addition reaction and successive Knoevenagel condensation/Michael addition/reduction/cyclization reaction of ketones with isatylidene malononitrile generated in situ from commercially available isatins and malononitrile. The reported synthetic procedures were highly efficient, sustainable, and broad in substrate scope. Significantly, we had shown efficient one-pot methodology for the synthesis of 3,3′-disubstituted oxindoles with all-carbon quaternary center and spiro[2H-pyran-3,4′-indoline] at very mild conditions using organocatalyst. Further extension of asymmetric work of this methodology is going on in our laboratory.Author contributions
C. B. N. and B. R. P. executed the experiment's session. B. P. M. performed the X-ray single crystal diffraction analysis in consultation with R. G.; S. S. C. finalized the data. A. K. K. directed the project, and C. B. N. wrote the manuscript with the help of A. K. K. and feedback from other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. B. N. thanks CSIR-New Delhi, India, for the fellowship. B. R. P. and S. S. C. thank UGC for a fellowship. The support from central NMR, HRMS facility, and NCL is greatly acknowledged.Notes and references
- (a) J. J. Badillo, N. V. Hanhan and A. K. Franz, Curr. Opin. Drug Discovery Dev., 2020, 13, 758 Search PubMed; (b) C. Marti and E. M. Carrerira, Eur. J. Org. Chem., 2003, 2209 CrossRef CAS; (c) S. Hibino and T. Choshi, Nat. Prod. Rep., 2002, 18, 66 RSC; (d) H. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 36 CrossRef CAS PubMed; (e) M. Pettersson, D. Knueppel and S. F. Martin, Org. Lett., 2007, 9, 4623 CrossRef CAS PubMed; (f) D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999 RSC; (g) N. Ye, H. Chen, E. A. Wold, P. Y. Shi and J. Zhou, ACS Infect. Dis., 2016, 2, 382 CrossRef CAS PubMed; (h) C. V Galliford and K. A. Sheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef PubMed; (i) S. Wang, Y. jiang, S. Wu, G. Dong, Z. Miao, W. Zhang and C. Sheng, Org. Lett., 2016, 18, 1028 CrossRef CAS PubMed; (j) D. Cheng, Y. Ishihara, B. Tan and C. F. Barbas lll, ACS Catal., 2014, 4, 743 CrossRef CAS; (k) M. M. Santos, Tetrahedron, 2014, 70, 9735 CrossRef CAS.
- A. Fensonme, W. R. Admas, T. J. Berrodin, J. Cohen, C. Huselton, A. lllenaberger, J. C. Kern, V. A. Hudak, M. A. Marella, E. G. Melenski, C. C. McComs, C. A. Mugford, O. D. Slayden, M. Yudt, Z. Zhang, P. Zhang, Y. Zhu, R. C. Winnekar and J. E. Wrobel, J. Med. Chem., 2008, 51, 1861 CrossRef PubMed.
- S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20 CrossRef CAS.
- A. Fensome, W. R. Adams, T. J. Berrodin, J. Cohen, C. Huselton, A. lllenbrger, J. C. Kern, V. A. Hudak, M. A. Marella, E. G. Melenski, C. C. McComas, C. A. Mugfold, O. D. Slayden, M. Yudt, Z. Zhang, P. Z hang, Y. Zhu, R. C. Winneker and J. E. Wrobel, J. Med. Chem., 2008, 51, 1861 CrossRef CAS PubMed.
- (a) A. Canas-Rodriguez and P. R. Leeming, J. Med. Chem., 1972, 15, 762 CrossRef CAS PubMed; (b) M. K. Uddin, S. G. Reignier, T. Coulter, C. Montalbetti, C. Granas, S. Butcher, C. K. Jensen and J. Feilding, Bioorg. Med. Chem. Lett., 2007, 17, 2854 CrossRef CAS PubMed.
- S. W. Tam and R. Zaczek, Adv. Exp. Med. Biol., 1995, 363, 47 CrossRef CAS PubMed.
- (a) B. Volk, J. Barkoczy, E. Hegedus, S. Udvari, I. Gacsalyi, T. Meezei, K. Pallagi, H. Kompagne, G. Levay, A. Egyed, L. G. Harsing, M. Spedding and G. Siming, J. Med. Chem., 2008, 51, 25522 CrossRef PubMed; (b) B. Volk, I. Gacasalyi, K. Pallagi, L. Posszvacz, I. Gynos and E. Szabo, J. Med. Chem., 2011, 54, 6657 CrossRef CAS PubMed.
- B. Yu, D. Q. Yu and H. M. Liu, Eur. J. Med. Chem., 2015, 97, 673 CrossRef CAS PubMed.
- G. G. Ladani and M. P. Patel, Heterocycl. Lett., 2016, 6, 393 Search PubMed.
- P. Eastwood, J. González, E. Gómez, F. Caturla, N. Aguilar, M. Mir, J. Aiguadé, V. Matassa and C. Balagué, Med. Chem. Lett., 2011, 21, 6253 CrossRef CAS PubMed.
- (a) J. Z. Huang, C. L. Zhang, Y. F. Zhu, L. L. Li, D. F. Chen, Z. Y. Han and L. Z. Gong, Chem.–Eur. J., 2015, 21, 8389 CrossRef CAS PubMed; (b) B. N. Reddy and C. V. Ramana, Tetrahedron, 2017, 73, 888 CrossRef CAS.
- (a) A. Millemaggi and R. J. K. Taylor, Eur. J. Org. Chem., 2010, 2010, 4527 CrossRef; (b) F. Zhou, Y. L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CrossRef CAS; (c) T. Zhang, L. Cheng, S. Hameed, L. Liu, D. Wang and Y. J. Chen, Chem. Commun., 2011, 47, 6644 RSC; (d) G. Bencivenni, L. Y. Wu, A. Mazzanti, B. Giannichi, F. Pesciaioli, M. P. Song, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7200 CrossRef CAS PubMed; (e) L. L. Wang, L. Peng, J. F. Bai, L. N. Jia, X. Y. Luo, Q. C. Huang, X. Y. Xu and L. X. Wang, Chem. Commun., 2011, 47, 5593 RSC; (f) X. Jiang, Y. Sun, J. Yao, Y. Cao, M. Kai, N. He, X. Zhang, Y. Wang and R. Wang, Adv. Synth. Catal., 2012, 354, 917 CrossRef CAS; (g) S. J. Chai, Y. F. Lai, J. C. Xu, H. Zheng, Q. Zhu and P. F. Zhang, Adv. Synth. Catal., 2011, 353, 371 CrossRef CAS; (h) W. B. Chen, Z. J. Wu, Q. L. Pei, L. F. Cun, X. M. Zhang and W. C. Yuan, Org. Lett., 2010, 12, 3132 CrossRef CAS PubMed; (i) Y. B. Lan, H. Zhao, Z. M. Liu, G. G. Liu, J. C. Tao and X. W. Wang, Org. Lett., 2011, 13, 4866 CrossRef CAS PubMed; (j) D. B. Ramachary, C. Venkaiah and R. Madhavachary, Org. Lett., 2013, 15, 3042 CrossRef CAS PubMed; (k) Z. An, Y. Guo, L. Zhao, Z. Li and J. He, ACS Catal., 2014, 4, 2566 CrossRef CAS; (l) J. Kaur, A. Kumari and S. S. Chimni, Tetrahedron, 2017, 73, 802 CrossRef CAS; (m) N. Gupta, T. Roy, D. Ghosh, S. H. Abdi, R. I. Kureshy, H. K. Noor-ul and H. C. Bajaj, RSC Adv., 2015, 5, 17843–17850 RSC.
- (a) B. B. Touré and D. G. Hall, Chem. Rev., 2009, 109, 4439 CrossRef PubMed; (b) B. Eftekhari-Sis, M. Zirak and A. Akbari, Chem. Rev., 2013, 113, 2958 CrossRef CAS PubMed.
- (a) P. Brandaõ, C. S. Marques, E. P. Carreiro, M. Pineiro and A. Burke, Chem. Rec., 2021, 21, 924 CrossRef PubMed; (b) B. Yu, D. Q. Yu and H. M. Liu, Eur. J. Med. Chem., 2015, 97, 673 CrossRef CAS PubMed; (c) M. Zhang, W. Yang, K. Li, K. Sun, J. Ding, L. Yang and C. Zhu, Synthesis, 2019, 51, 3847 CrossRef CAS; (d) D. Charvin, R. Medori, R. A. Hauser and O. Rascol, Nat. Rev. Drug Discovery, 2018, 17, 804 CrossRef CAS PubMed.
- (a) B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS; (b) K. Sakthivel, W. Notz, T. Bui and C. F. Barbas III, J. Am. Chem. Soc., 2001, 123, 5260 CrossRef CAS PubMed; (c) W. Notz and B. List, J. Am. Chem. Soc., 2000, 122, 7386 CrossRef CAS; (d) A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 6798 CrossRef CAS PubMed; (e) A. B. Northrup, I. K. Mangion, F. Hettche and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2004, 43, 2152 CrossRef CAS PubMed; (f) C. Pidathala, L. Hoang, N. Vignola and B. List, Angew. Chem., Int. Ed., 2003, 42, 2785 CrossRef CAS PubMed.
- T. He, Q. Q. Zeng, D. C. Yang, Y. H. He and Z. Guan, RSC Adv., 2015, 5, 37843 RSC.
- Y. L. Guo, Y. H. Li, H. H. Chang, T. S. Kuo and J. H. Han, RSC Adv., 2016, 6, 74683 RSC.
- S. Hegade, G. Gaikwad, Y. Jadhav, A. Pore and A. Mulik, Monatsh. Chem., 2022, 153, 95 CrossRef CAS.
- L. Liu, D. Wu, X. Li, S. Wang, H. Li, J. Li and W. Wang, Chem. Commun., 2012, 48, 1692 RSC.
- H. Zhao, Y. B. Lan, Z. M. Liu, Y. Wang, X. W. Wang and J. C. Tao, Eur. J. Org. Chem., 2012, 2012, 1935 CrossRef CAS.
- A. Kumar and S. S. Chimni, Beilstein J. Org. Chem., 2014, 10, 929 CrossRef PubMed.
- (a) X. Jiang, Y. Sun, J. Yao, Y. Cao, M. Kai, N. He, X. Zhang, Y. Wang and R. Wang, Adv. Synth. Catal., 2012, 354, 917 CrossRef CAS; (b) S. J. Chai, Y. F. Lai, J. C. Xu, H. Zheng, Q. Zhu and P. F. Zhang, Adv. Synth. Catal., 2011, 353, 371 CrossRef CAS; (c) W. B. Chen, Z. J. Wu, Q. L. Pei, L. F. Cun, X. M. Zhang and W. C. Yuan, Org. Lett., 2010, 12, 3132 CrossRef CAS PubMed; (d) Y. Li, H. Chen, C. Shi, D. Shi and S. Ji, J. Comb. Chem., 2010, 12, 231 CrossRef CAS PubMed.
- Synthesis of N-protected isatin: to a round bottom flask was added isatin (10 mmol, 1 equiv.), DMF (10 ml), and K2CO3 (12 mmol, 1.2 equiv.) and the solution was stirred at r.t for 30 minutes. The solution turns rapidly dark purple. Methyl iodide, alkyl bromide or benzyl bromide, (11 mmol, 1.1 equiv.) was added in one portion. The reaction mixture rapidly change colour. After 12 h, water (20 ml) was added, then the suspension was filtered, and purified by flash column chromatography on silica gel to afford the product.
- A reaction conditions for synthesis of 3,3′-disubstituted oxindoles: mixture of 1 (0.5 mmol, 1 equiv.), 2 (0.55 mmol, 1.1 equiv.) in ethanol (1 ml) was stirred at room temperature for 30 minutes, followed by addition of ketone (2.5 mmol, 5.0 equiv.) and L-proline (0.1 mmol, 20 mol%), continued stirring at rt till to get total conversion of 1′, the product observed by TLC.
- A reaction conditions for synthesis of spiro[2-H-pyran-3,4′-indoline]: mixture of 1 (0.5 mmol, 1 equiv.), 2 (0.55 mmol, 1.1 equiv.) in ethanol (1 ml) was stirred at room temperature for 30 minutes, followed by addition of ketone (2.5 mmol, 5.0 equiv.) and L-proline (0.1 mmol, 20 mol%), continued stirring at rt till to get total conversion of 1′, the product observed by TLC, then added NaBH4 (1 mmol, 2 equiv.) at 0 °C stirred mixture for 15 min then stirred at rt for next 1 hr. dr mentioned are calculated by 1H NMR of crude product. Yields shown above are the combine yield of two diastereomers. We separate two diastereomer by silica gel column chromatography and in ESI† characterisations provided are for major diastereomer only.
- CCDC 2224164 (4Ae) and CCDC 2224165 (4Ah) contains the supplementary crystallographic data for the paper.†.
- CCDC 2224166 (5Aa) and CCDC 2224167 (5Bb) contains the supplementary crystallographic data for this paper.†.
- B. List, Acc. Chem. Res., 2004, 37, 548 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2224164–2224167. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra00510k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |