
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards hard-magnetic behavior of CoFe2O4 nanoparticles: a detailed study of crystalline and electronic structures, and magnetic properties
D. H. Manh *a,
T. D. Thanha,
T. L. Phanbc and
D. S. Yangd
*a,
T. D. Thanha,
T. L. Phanbc and
D. S. Yangd
aInstitute of Materials Science, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Hanoi, Viet Nam. E-mail: manhdh.ims@gmail.com
bDepartment of Physics, Hankuk University of Foreign Studies, Yongin 449-791, South Korea
cFaculty of Engineering Physics and Nanotechnology, VNU-University of Engineering and Technology, 144 Xuan Thuy, Cau Giay, Ha Noi, Viet Nam
dDepartment of Science Education, Chungbuk National University, Cheongju 360-763, South Korea
First published on 13th March 2023
Abstract
We have used the coprecipitation and mechanical-milling methods to fabricate CoFe2O4 nanoparticles with an average crystallite size (d) varying from 81 to ∼12 nm when changing the milling time (tm) up to 180 min. X-ray diffraction and Raman-scattering studies have proved the samples crystalizing in the spinel structure. Both the lattice constant and residual strain tend to increase when tm(d) increases (decreases). The analysis of magnetization data has revealed a change in the coercivity (Hc) towards the hard-magnetic properties. Specifically, the maximum Hc is about 2.2 kOe when tm = 10 min corresponding to d ≈ 29 nm; beyond this tm(d) value, Hc gradually decreases. Meanwhile, the increase of tm always reduces the saturation magnetization (Ms) from ∼69 emu g−1 for tm = 0 to 35 emu g−1 for tm = 180 min. The results collected as analyzing X-ray absorption data have indicated a mixed valence state of Fe2+,3+ and Co2+ ions. We think that the migration and redistribution of these cations between the tetrahedral and octahedral sites together with lattice distortions and defects induced by the milling process have impacted the magnetic properties of the CoFe2O4 nanoparticles.
1 Introduction
Spinel-type ferrite oxides are important materials used widely in many technological applications, including permanent magnets, high-density data storage, rechargeable lithium-ion batteries, microwave absorbers, electromagnetic generators, aqueous supercapacitors, and biomedical labeling.1–5 Their chemical formula is generalized as AB2O4, where A and B are usually divalent and trivalent cations, respectively. They crystallize in a cubic lattice, and belong to the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. Their unit cell consists of eight formula units, with two types of interstitials: (i) tetrahedral interstitials are surrounded by four oxygen atoms (called A sites), and (ii) octahedral ones are surrounded by six oxygen atoms (B sites). A and B metal cations can reside in eight tetrahedral sites and 16 octahedral sites, respectively. We would obtain a normal or inverse spinel type depending on the residence ratio of A and B in these sites. The structure is called a normal spinel if the A cations completely occupy the tetrahedral sites and the B cations completely occupy the octahedral sites. In contrast, if the tetrahedral sites are occupied half by the A cations, and the octahedral sites are occupied by all the B cations and half of the A cations, the structure is called an inverse spinel.1,3 However, when modifying synthesis conditions and the crystal size, a random distribution of A and B at the tetrahedral and octahedral sites would happen, leading to the so-called mixed spinel type. Its chemical formula is thus defined as (Aδ2+B1−δ3+)[A1−δ2+B1+δ3+]O42−, where δ is the degree of inversion, and the round and square brackets are indicative of the A (tetrahedral) and B (tetrahedral) sites, respectively. This formula can be generalized for all spinel types: a normal spinel with δ = 1, an inverse spinel with δ = 0, and a mixed spinel with 0 < δ < 1.6 Apart from the changes related to the particle size and the valence of the cations (because A and B ions can have different oxidation states), the change in value of δ (the cation distribution) also influences strongly the optical, electrical and magnetic properties of spinel ferrites.
m space group. Their unit cell consists of eight formula units, with two types of interstitials: (i) tetrahedral interstitials are surrounded by four oxygen atoms (called A sites), and (ii) octahedral ones are surrounded by six oxygen atoms (B sites). A and B metal cations can reside in eight tetrahedral sites and 16 octahedral sites, respectively. We would obtain a normal or inverse spinel type depending on the residence ratio of A and B in these sites. The structure is called a normal spinel if the A cations completely occupy the tetrahedral sites and the B cations completely occupy the octahedral sites. In contrast, if the tetrahedral sites are occupied half by the A cations, and the octahedral sites are occupied by all the B cations and half of the A cations, the structure is called an inverse spinel.1,3 However, when modifying synthesis conditions and the crystal size, a random distribution of A and B at the tetrahedral and octahedral sites would happen, leading to the so-called mixed spinel type. Its chemical formula is thus defined as (Aδ2+B1−δ3+)[A1−δ2+B1+δ3+]O42−, where δ is the degree of inversion, and the round and square brackets are indicative of the A (tetrahedral) and B (tetrahedral) sites, respectively. This formula can be generalized for all spinel types: a normal spinel with δ = 1, an inverse spinel with δ = 0, and a mixed spinel with 0 < δ < 1.6 Apart from the changes related to the particle size and the valence of the cations (because A and B ions can have different oxidation states), the change in value of δ (the cation distribution) also influences strongly the optical, electrical and magnetic properties of spinel ferrites.
So far, many intriguing electrical and magnetic properties in spinel ferrites have been discovered, such as (A, B)-sublattice-related exchange interactions, orbital-/charge-ordering, metal-insulator transition, magnetic frustration, spin-lattice coupling, and Verwey transition,3,7 which could be controlled by the substitutions at that A and B sites. These properties together with technological application potentials have attracted intensive interest of the solid-state-physics community, specially nanostructured spinel ferrites that have more unique characters different from bulk counterparts.1,8–11 It has been believed that a surface-area-to-volume ratio of nanostructures is large, and additionally provides the structural degrees of freedom. Among nanostructured spinel ferrites, cobalt spinel ferrite (CoFe2O4) has attracted much more research interest since it has outstanding magnetic, magneto-elastic/optical, magnetophototronic, and photo-magnetic properties.12–14 Most markedly, it is classified as a semi-hard material with a large magneto-crystalline anisotropy and saturation magnetization that can used to fabricate rare-earth-free permanent magnets.15–17 Similar to NiFe2O4, CoFe2O4 is well known as an inverse spinel (notably, ZnFe2O4 is a normal spinel while (Mg,Zn)Fe2O4 is a mixed spinel),18 in which the Co2+ cations occupy the B sites while the Fe3+ cations occupy both the A and B sites, meaning (Fe3+)[Co2+Fe3+]O4.19,20 Having modified fabrication conditions and the crystal size, the proportion of Co2+ and Fe3+ in the A and B sites would be changed, consequently the degree of inversion δ. Together with δ, surficial defects and lattice distortions also strongly influence the magnetic properties of CoFe2O4, which are characterized by the magnitudes of the coercivity (Hc), saturation magnetization (Ms), and remanent magnetization (Mr).
It has been used various techniques to synthesize nanostructured CoFe2O4 materials, such as magnetron sputtering,21 hydrothermal method,11,22,23 sol–gel method,23 and combustion, coprecipitation and precipitation methods.24 Depending on synthesis methods and conditions, the values of Hc, Mr, and Ms would be different. Among these parameters, Hc is considered as a key parameter characteristic of the magnetic hardness of cobalt ferrite, which is ascribed to the occupation of Co2+ because it is ascribed to be a highly anisotropic ion.25,26 In practice, it can be tuned Hc via fabricating core/shell nanomaterials,27 nanocomposites,17,28 and nanostructures (nanoparticles, nanowires, nanofibers, nanoribbons, and thin films).21,23,24,29,30 It is clear that an enhancement in Hc can be realized upon changing the crystal shape and particle size, and/or creating a high level of residual lattice strain and distortions inside particles. Because of these features, some research groups prepared CoFe2O4 by using hydrothermal, coprecipitation and sol–gel methods, and then carried out high-energy ball milling to produce CoFe2O4 nanoparticles (NPs) with different sizes and different levels of lattice strain and distortions upon controlling the milling time (tm), consequently high magnetic performance.13,16,31–33 In those studies, the researchers employed X-ray/electron diffraction and/or Mössbauer spectroscopy to figure out the relations between the particle size, microstructures and cation distribution, and the magnetic properties, without directly using sensitive tools in order to identify Co and Fe oxidation states. To get more knowledge about CoFe2O4 NPs prepared by the mechanical ball milling method, we shall use X-ray diffraction accompanied with Rietveld refinement, Raman spectroscopy, and X-ray absorption spectroscopy to analyze the structural characterization, and electronic structure versus the milling process (tm), and then evaluate carefully their impacts on the magnetic properties.
2 Experimental details
First, a large amount of CoFe2O4 in powder was synthesized by a conventional coprecipitation method. Initial chemicals including CoCl2·6H2O and FeCl2·4H2O (98%) ordered from Sigma-Aldrich were used as precursors. To produce Co2+ and Fe2+ containing solutions, we dissolved 4 mmol CoCl2·6H2O and 8 mmol FeCl2·4H2O in 70 ml and 30 mL distilled water, respectively. After that, the Fe2+ solution was dropped into the Co2+ solution. The pH of the mixture was adjusted by controlling NH4OH amount. The mixture was firstly stirred at room temperature for 30 min, and then transferred to Teflon-line autoclaves. The autoclaves were tightly sealed, heated to 180 °C and kept at this temperature for 24 h. After completing chemical reactions, precipitation was collected upon centrifuging at 8000 rpm, and washed several times to remove excess precursors. The collected powder sample was treated at 200 °C for 3 h to evaporate water. After dried, it was divided into small parts of ∼0.3 g. These parts were in turn taken to mill in normal atmosphere for different milling times (tm) ranging from 1 to 180 min (hereafter, the as-prepared sample is labelled as tm = 0), see Table 1 for more details. The milling process was carried out in air using a SPEX SamplePrep Mixer/Mill, and the set of stainless-steel grinding media (vial and balls).| tm (min) | d (nm) | ε | B × 10−6 (Oe2) | Ha (kOe) | K1 × 10−6 (erg g−1) |
|---|---|---|---|---|---|
| 0 | 80.9 | 0.0017 | 2.52 | 6.15 | 0.210 |
| 1 | 80.2 | 0.0015 | 2.51 | 6.13 | 0.198 |
| 2 | 50.6 | 0.0027 | 2.62 | 6.26 | 0.200 |
| 4 | 55.7 | 0.0025 | 2.47 | 6.08 | 0.197 |
| 8 | 40.4 | 0.0034 | 2.49 | 6.12 | 0.192 |
| 10 | 29.3 | 0.0047 | 2.95 | 6.65 | 0.200 |
| 20 | 17.9 | 0.0075 | 3.69 | 7.44 | 0.201 |
| 40 | 12.8 | 0.0107 | 3.39 | 7.13 | 0.170 |
| 60 | 12.3 | 0.0111 | 2.99 | 6.70 | 0.139 |
| 120 | 11.6 | 0.0118 | 2.90 | 6.59 | 0.130 |
| 180 | 12.4 | 0.0111 | 2.68 | 6.34 | 0.111 |
After milled, the final products were checked the particle morphology by a field emission scanning electron microscope (SEM, JSM-5410LV) that was linked with X-ray energy dispersive spectroscopy (EDS). Their crystal structure was characterized by a Bruker X-ray diffractometer equipped with the copper Kα radiation. The Rietveld refinement method was also used to analyze X-ray diffraction (XRD) patterns, allowing us to evaluate the structural parameters, crystallite size, and lattice strain. In addition to XRD, an XploRA PLUS Raman spectrometer (Horiba) was used to judge structure phases, in which a laser wavelength of 785 nm was used as an excitation source. To identify the oxidation state of the cations in CoFe2O4, we utilized X-ray absorption spectroscopy (XAS). XAS measurements on the powder samples were carried out under the transmission configuration. The magnetic properties were investigated through magnetic-field-dependent magnetization, M(H), measurements, which were performed on a conventional vibrating sample magnetometer.
3 Results and discussion
We have selected typical samples to check their particle size and surface morphology by means of an electron microscope. Fig. 1(a)–(f) show representative micrographs of the samples with tm = 0, 4, 10, 20, 60, and 180 min, which were recorded with the same scale bar. One can see that particles in the samples have irregular shapes and random orientation. While the particle size of the as-prepared sample (tm = 0) is about 200–400 nm, see Fig. 1(a), that of the other samples is much smaller. It is hard to exactly evaluate the size distribution if basing on the micrographs, particularly for the samples with tm > 10 min, see Fig. 1(d)–(f). For the milled samples, their NPs aggregate to form large clusters. This phenomenon is popular for nanomaterials,15,34 and assigned to attractive magnetic and/or electrostatic forces existing among NPs.35,36 | ||
| Fig. 1 Micrographs of typical NP samples with tm = 0 (a), 4 (b), 10 (c), 20 (d), 60 (e) and 180 (f) min, which were recorded as fixing scale bars of 1 μm. | ||
Together with the inspection of the particle morphology, the presence of elements in the samples has also been checked. Fig. 2 shows typical EDS spectra of the samples with tm = 0, 60 and 180 min, which were recorded within an area of ∼0.02 × 0.02 mm2, see the inset of Fig. 2. Apart from a weak peak of carbon (C) at ∼0.25 keV, some strong peaks at energies E = 0.4–8.1 keV are from Co, Fe and O of cobalt ferrites. For the tm = 0 sample, weight (atomic) percentage values of Co, Fe and O averaged from different measurement positions are about 20.3 (9.8), 40.4 (20.6), and 39.3 (69.6)%, respectively. For the samples with tm = 60 and 180 min, the concentration of these elements changed within few percentages. This could be due to oxygen absorbency (from air) at the surface, and grinding-media-related contaminations that happen as decreasing the NP size.
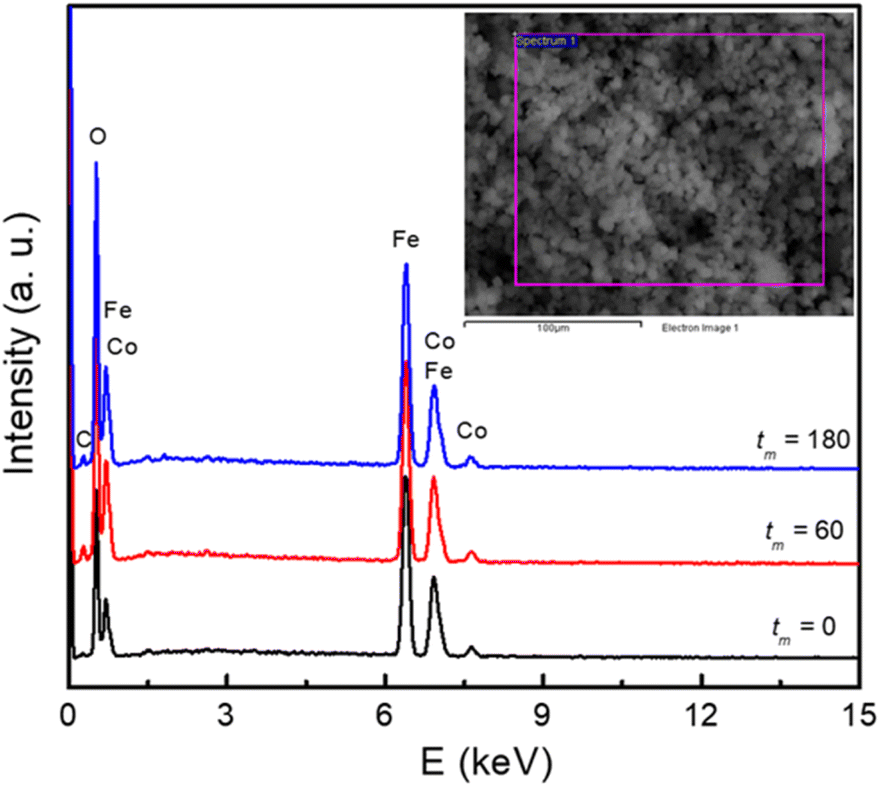 | ||
| Fig. 2 EDS spectra of typical samples with tm = 0, 60, and 180 min. Inset: a selected area for recording EDS data. | ||
The crystal structure has also been characterized by using an X-ray diffractometer. Diffraction patterns in the angle range of 2θ = 15–80° are shown in Fig. 3. The analysis has revealed that the XRD pattern of the as-prepared CoFe2O4 sample is fully indexed to the spinel cubic structure with the Fdm symmetry and lattice constants a = b = c ≈ 8.4 Å, consistent with previous studies.15,16,22,23,25 Furthermore, the Rietveld refinement using a structural model reported for the cubic-spinel phase37 provided a satisfactory fit to the experimental data (see Fig. 3), confirming a single phase of the tm = 0 sample. As increasing tm, the feature of diffraction patterns remains unchanged, except the peak broadening due to a decrease in the crystallite size as discussed below. This proves the stability of the cubic spinel phase of CoFe2O4 NPs versus the mechanical milling.
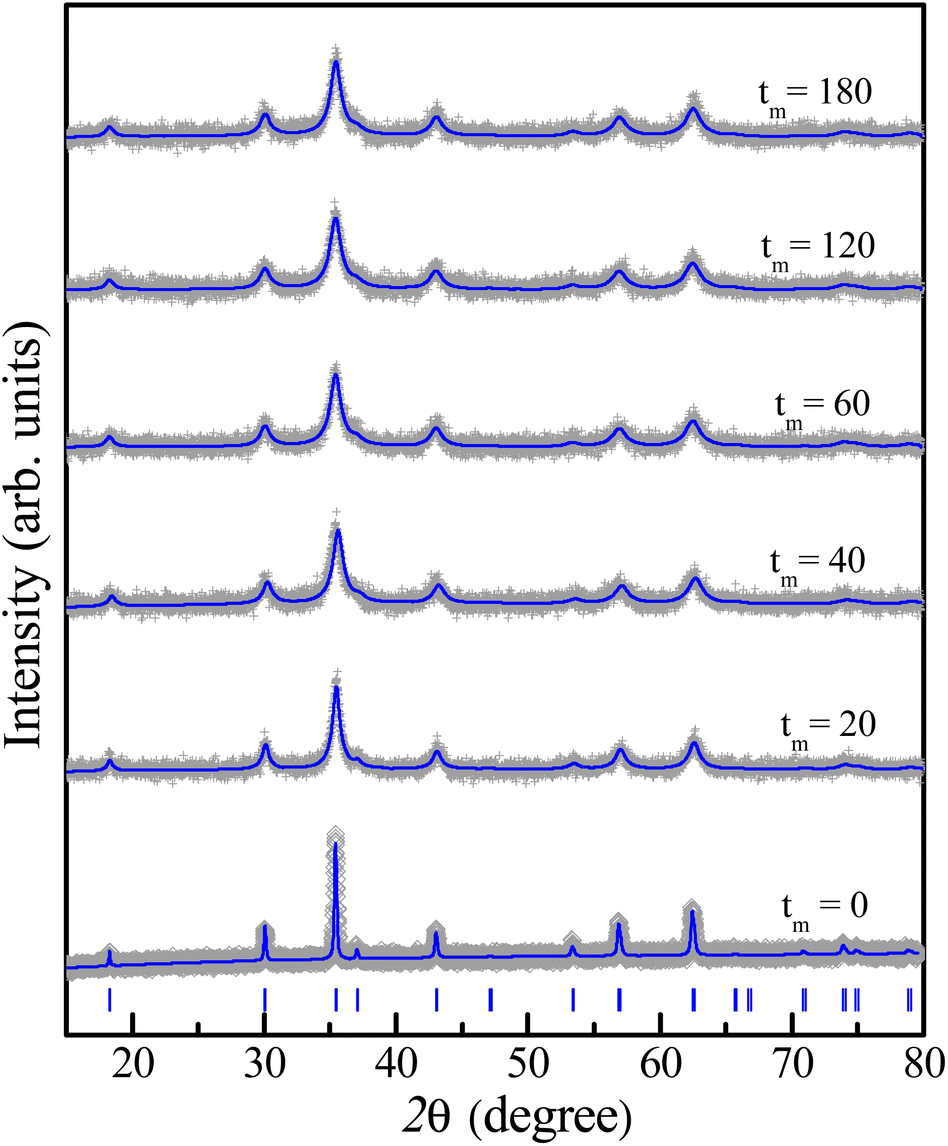 | ||
| Fig. 3 Rietveld refinement of XRD patterns of some representative samples with different milling times tm, in which blue vertical bars show standard diffraction lines of the spinel structure. | ||
Based on 2θ-position dependences and the peak broadening value (β) after subtracting instrumental broadening contributions, the crystallite size (d) and lattice strain (micro-deformation, ε) as a function of tm were estimated upon the Williamson–Hall (WH) theory generalized by the equation of β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ = (kλ/d) + 2εsinθ, where k = 0.9 is the shape factor.38 The obtained d and ε data are listed in Table 1 for reference. We have found that d and ε data can be described by tm-dependent exponential functions of d(tm) = 67.74 × exp(−0.129 × tm) + 12.36 and ε(tm) = −0.011 × exp(0.045 × tm) + 0.012, respectively, see Fig. 4. A rapid decrease (increase) of d(ε) takes place when tm changes from 1 to 40 min. As tm > 40 min, d and ε are less changed and approach to the critical values of about 12 nm and 0.011, respectively. Change tendencies of d and ε with respect to tm are also analogous to those recorded for other CoFe2O4 NPs fabricated by mechanical milling.13,31,33 Depending on the size of initial CoFe2O4 powders, the mass ratio of balls to powders, and tm value, it could be decreased d to a critical value of dc ≈ 9 nm.31,33
cosθ = (kλ/d) + 2εsinθ, where k = 0.9 is the shape factor.38 The obtained d and ε data are listed in Table 1 for reference. We have found that d and ε data can be described by tm-dependent exponential functions of d(tm) = 67.74 × exp(−0.129 × tm) + 12.36 and ε(tm) = −0.011 × exp(0.045 × tm) + 0.012, respectively, see Fig. 4. A rapid decrease (increase) of d(ε) takes place when tm changes from 1 to 40 min. As tm > 40 min, d and ε are less changed and approach to the critical values of about 12 nm and 0.011, respectively. Change tendencies of d and ε with respect to tm are also analogous to those recorded for other CoFe2O4 NPs fabricated by mechanical milling.13,31,33 Depending on the size of initial CoFe2O4 powders, the mass ratio of balls to powders, and tm value, it could be decreased d to a critical value of dc ≈ 9 nm.31,33
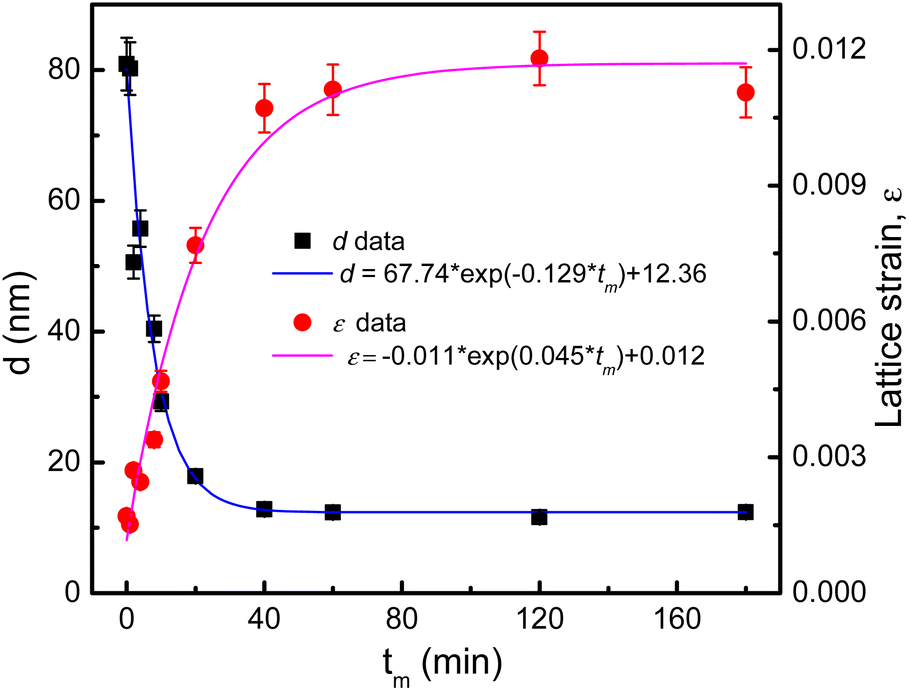 | ||
| Fig. 4 Variations of d and ε data according to exponential functions of tm. | ||
Another interesting result obtained from the XRD refinement is an anomalous evolution of the lattice parameter (a) as a function of tm(d) as shown Fig. 5(a) and its inset. In the interval of tm = 1–10 min (d reduces from ∼80 to ∼29 nm), a slightly increases with increasing tm. Beyond tm = 10 min (d ≈ 29 nm), it sharply increases with further increasing tm (i.e., decreasing d). A similar expansion of the lattice parameters versus decreasing the particle size has also been reported on CeO2−x and BaTiO3 NPs, which was attributed to a decreased electrostatic force caused by valence reduction of Ce and an increased ionicity of Ti, respectively.39 On the other hand, we have also observed an significant change in the intensity ratio of I(220)/I(222) with respect to tm(d), where I(220) and I(222) are the integrated intensities of (220) and (222) peaks, respectively, see Fig. 5(b) and its inset. This ratio reflects the cation distribution at the A and B sites in cobalt ferrite.40 Concretely, it displays a maximum at tm = 10 min (d ≈ 29 nm), and beyond this tm(d) value, it gradually decreases. We believe that the cation distribution is a main factor influencing the lattice properties of the milled CoFe2O4 NPs.
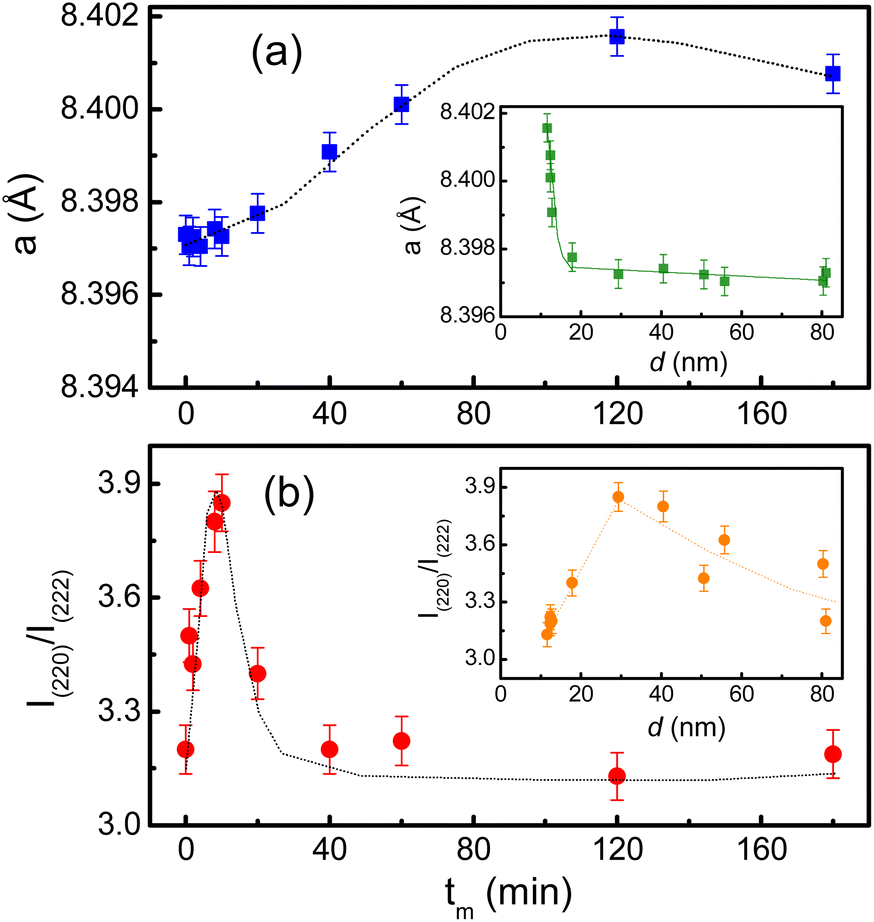 | ||
| Fig. 5 Variations of (a) the lattice constant, and (b) I(220)/I(222) as a function of tm. The insets show the variation of these parameters with respect to the crystallite size d. | ||
We have also used Raman scattering (RS) spectroscopy to consider how characteristic phonon modes of CoFe2O4 NPs changes with respect to tm. This is known as a sensitive and non-destructive tool accompanied with XRD to assess the crystalline structure. According to the group theory's prediction, near the center of Brillouin zone (q = 0, the conservation of momentum vector), cobalt ferrite CoFe2O4 with the Fdm spinel structure (Oh7 symmetry) has five Raman active modes, Γ = A1g + Eg + 3T2g, associated with the motion of O and A-/B-site atoms.41 Energy order of the modes is addressed as follows: A1g(1) > A1g(2) > T2g(1) > T2g(2) > Eg > T2g(3).42 In the current work, we have observed all of these modes in RS spectra of CoFe2O4 NPs, see Fig. 6 that plots RS data of some representative samples, normalized to the T2g(2)-mode intensity. For the tm = 0 sample, its spectrum consists of the modes peaked at about 193, 303, 475, 566, 617 and 686 cm−1 associated to T2g(3), Eg, T2g(2), T2g(1), A1g(2) and A1g(1) symmetries, respectively, as labelled in Fig. 6. Basically, 617 cm−1 (A1g(2)) and 686 cm−1 (A1g(1)) are associated with symmetric stretching of O atoms versus Co and/or Fe atoms in the tetrahedral (AO4) sites.41,43 It should be noticed that as studying Fe3O4, at frequencies above 600 cm−1, it is only observed one mode due to A1g.44,45 The A1g-band splitting into two sub-bands A1g(1) and A1g(2) in CoFe2O4 hints that 686 cm−1 (A1g(1)) and 617 cm−1 (A1g(2)) are related to FeO4 and CoO4 tetrahedra, respectively.41,46 One can thus rely upon their intensity ratio (A1g(1)/A1g(2)) to assess the cation distribution at the A and B sites. Meanwhile, the other modes occupying at lower frequencies, Eg and T2g(3), are attributed to the symmetric and anti-symmetric bending of O atoms in the Fe-/Co–O bonds at the octahedral (BO6) sites.41,43,46
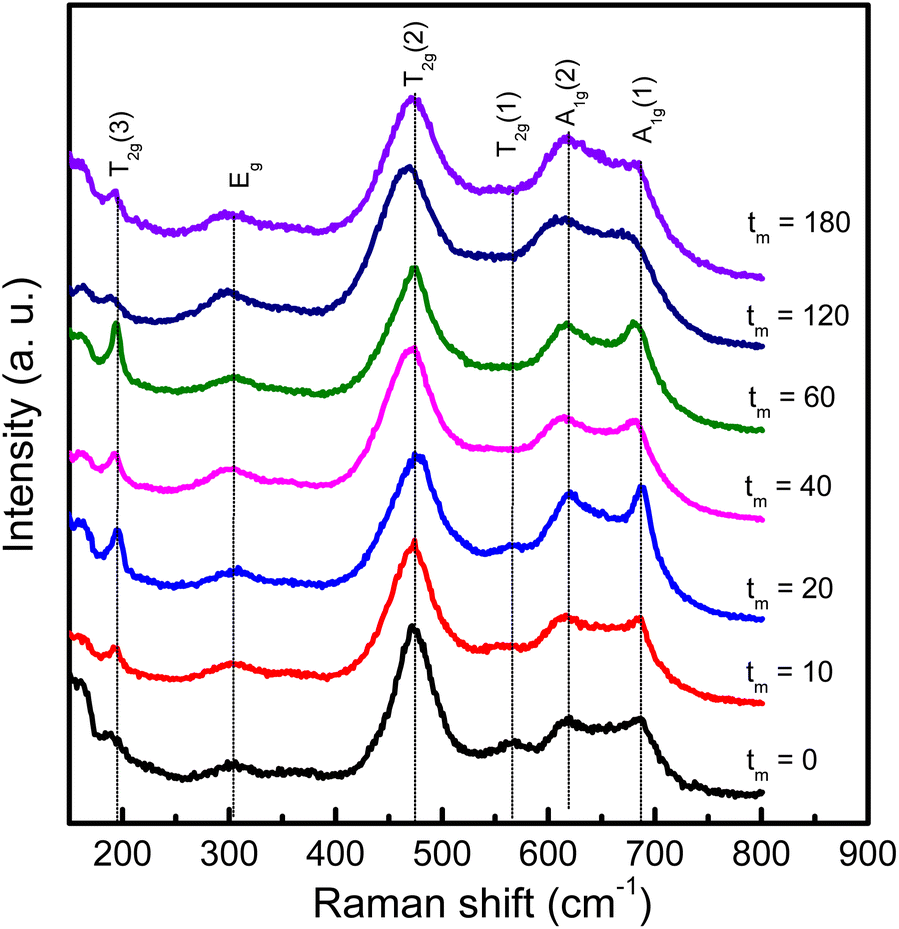 | ||
| Fig. 6 RS spectra of representative CoFe2O4 NPs with different tm values excited by a laser wavelength of 785 nm, which were normalized to the intensity of the T2g(2) mode at ∼475 cm−1. | ||
As considering the RS spectra of CoFe2O4 NPs versus various the milling process, the vibration mode at ∼566 cm−1 (T2g(1)) becomes invisible as tm = 40 min. Meanwhile, the other modes still persist, and their peak position tends to shift (about 4–9 cm−1, depending on mode types) towards lower frequencies and their spectral linewidth becomes gradually broadened when tm increases, particularly the samples with tm > 20 min, see Fig. 6. Basically, the redshift of the modes can be qualitatively explained via the expansion of the lattice parameter a versus increasing tm, as shown above. An increase of a reflects an increase of Fe-/Co–O bond distances, which reduces the coupling constant k. Approximately, phonon vibration frequency can be expressed as 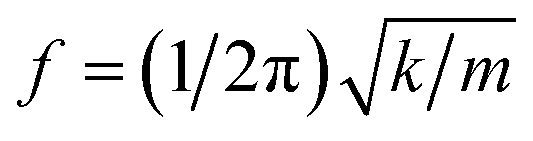 , where m is mass of an atom, and the decreased k causes the redshift of the modes. Alternatively, when tm increases, high pressure and local heating during the milling generate lattice disorders/defects, increasing grain boundaries (due to the reduced d). Broken long-range order of CoFe2O4 lattice host allows the q ≠ 0 scattering contributions.41 Concurrently, some modes with weak intensity (such as T2g(1) in our work) would be extinguished, while the linewidth and peak position of persisting modes broadens and shift, respectively.
, where m is mass of an atom, and the decreased k causes the redshift of the modes. Alternatively, when tm increases, high pressure and local heating during the milling generate lattice disorders/defects, increasing grain boundaries (due to the reduced d). Broken long-range order of CoFe2O4 lattice host allows the q ≠ 0 scattering contributions.41 Concurrently, some modes with weak intensity (such as T2g(1) in our work) would be extinguished, while the linewidth and peak position of persisting modes broadens and shift, respectively.
It is worth noticing that the intensity ratio between two modes of 617 cm−1 (A1g(2) associated with Co at the tetrahedral site) and 475 cm−1 (T2g(2), associated with Co at the octahedral site14) also varies versus tm, similar to the comparison of the intensity ratio between 617 cm−1 (A1g(2)) and 686 cm−1 (A1g(1)) mentioned above. These features reflect the migration of Co and Fe ions between the tetrahedral (A) and octahedral (B) sites as varying tm. As studying nanostructured cobalt ferrites, it has been suggested that Co2+ and Fe3+ can occupy both A and B sites (i.e., 0 < δ < 1). For spinel-structured materials, however, divalent and trivalent elements usually occupy the A and B sites, respectively.1,3 The valence identification of Co and Fe in our fabricated cobalt ferrites is thus necessary to clarify the lattice-constant change as well as magnetic behaviors with respect to tm.
Concerning the valence identification, we have utilized XAS and recorded the Fe and Co K-edge spectra of typical samples, and reference oxides including Fe2O3, Fe3O4 and CoO. Before measurements, Fe (E0 = 7112 eV) and Co (E0 = 7709 eV) foils were used to calibrate the Fe and Co K-edge energies, respectively. Fig. 7(a) and (b) show the Fe K-edge XAS and first-order derivative spectra of the samples with tm = 0, 60 and 180 min. In the pre-edge region with E = 7110–7118 eV, as graphed the inset of Fig. 7(a), a broad hump peaked at ∼7114 eV, corresponding to an inflection point in Fig. 7(b), is observed. Basically, the pre-edge peak is associated to the electric quadrupole–forbidden transitions from the O 1s level to 3d transition metals (Fe and Co in the present case). Following the pre-edge region, there is a rapid increase in the XAS intensity of the absorption K-edge. From Fig. 7(b), one can see that the absorption edge of the CoFe2O4 NPs at ∼7122 eV is less independent of tm, and locates between the absorption edges of Fe3O4 (7121 eV) and Fe2O3 (∼7123 eV). This reflects that both Fe2+ and Fe3+ ions coexist in our CoFe2O4 NPs.
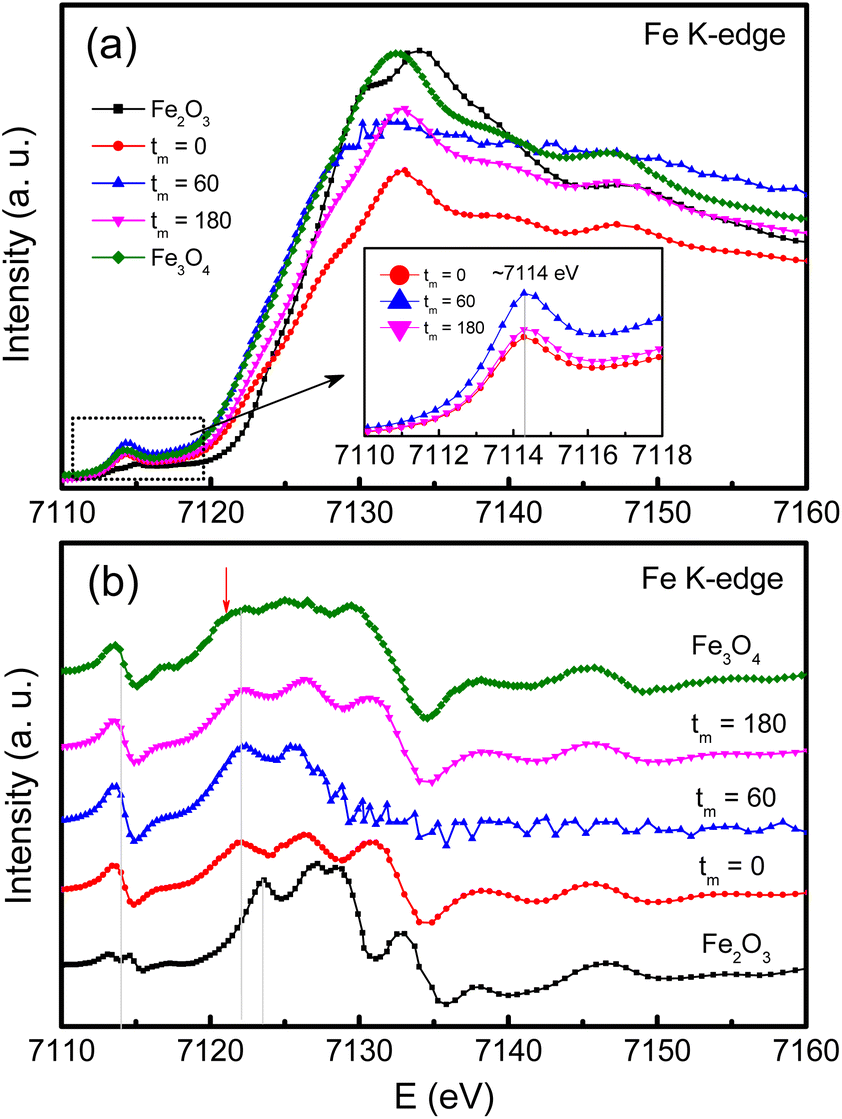 | ||
| Fig. 7 (a) Fe K-edge XAS data, and (b) their derivative data of typical CoFe2O4 NPs, tm = 0, 60 and 180 min, compared with those of Fe2O3 and Fe3O4 references. The inset of (a) plots an enlarged view of the re-edge region. | ||
For the case of Co, its K-edge XAS and first-order derivative spectra of the samples with tm = 0, 10, 60 and 180 min, and CoO are shown in Fig. 8(a)and (b). Evidently, the absorption edges of milled CoFe2O4 NPs locating at ∼7717 eV are almost unchanged versus tm, and well match the absorption edge of CoO, see Fig. 8(b). These results suggest that only Co2+ is present in our samples. Notably, in the pre-edge region, the Co K-edge XAS spectrum of the tm = 0 sample has two weak-intensity humps peaked at ∼7709.3 (named P1) and 7712.8 eV (P2), see the inset of Fig. 8(a). With increasing tm, P1 still persists while P2 becomes invisible as tm > 60 min. Because these pre-edge humps are associated with the occupancy of Co2+ in the octahedral and tetrahedral crystal fields,47 the intensity change of P1 and P2 proves the redistribution of Co2+ ions in these fields, and milling-induced lattice distortions. Normally, the pre-edge peak due to the octahedral site is broad and more diffused than that due to the tetrahedral site,37 it is reasonable to assign that P1 and P2 are associated with Co2+ at the A and B sites, respectively. The intensity decrease of P2 with increasing tm indicates the migration of Co2+ from the B site to the A site. Additionally, if comparing the pre-edge peaks in the Fe and Co K-edge XAS spectra of all the samples, it comes to our attention that the pre-edge peak at the Fe K-edge is stronger than that at the Co K-edge. It means that at the A site the fraction of Fe2+,3+ ions is larger than that of Co2+ ions.48 In other words, the XAS analyses have demonstrated the mixed valence state of Fe2+,3+ and Co2+ ions in all CoFe2O4 NPs, and they are present in both the A and B sites, resulting in the mixed spinel type of (Coδ2+Fe1−δ2+,3+)[Co1−δ2+Fe1+δ2+,3+]O42−. We think that the migration and redistribution of these ions at the A and B sites together with milling-induced lattice disorders and defects would strongly influence the magnetic behaviors of milled CoFe2O4 NPs. As studying cobalt ferrites, Singh et al.37 found a coexistence of Fe2+,3+ and Co2+,3+ ions, but Londo et al.49 found Co2+,3+ and Fe3+ ions. Meanwhile, some works only found Co2+ and Fe3+ ions in cobalt ferrites.41,43,46 Reviewing the previous works on mechanically-milled CoFe2O4 NPs,13,16,32,33 one can see that the analyses of electronic structure based on sensitive tools such as X-ray photoelectron spectroscopy (XPS) and XAS were less carried out. Cedeño-Mattei et al.31 studied Mössbauer spectra and suggested the presence of Co2+ and Fe3+ ions only. Such contradictory results are generated due to the differences in sample fabrication methods and processing conditions, which influence the magnetic properties of cobalt ferrites.
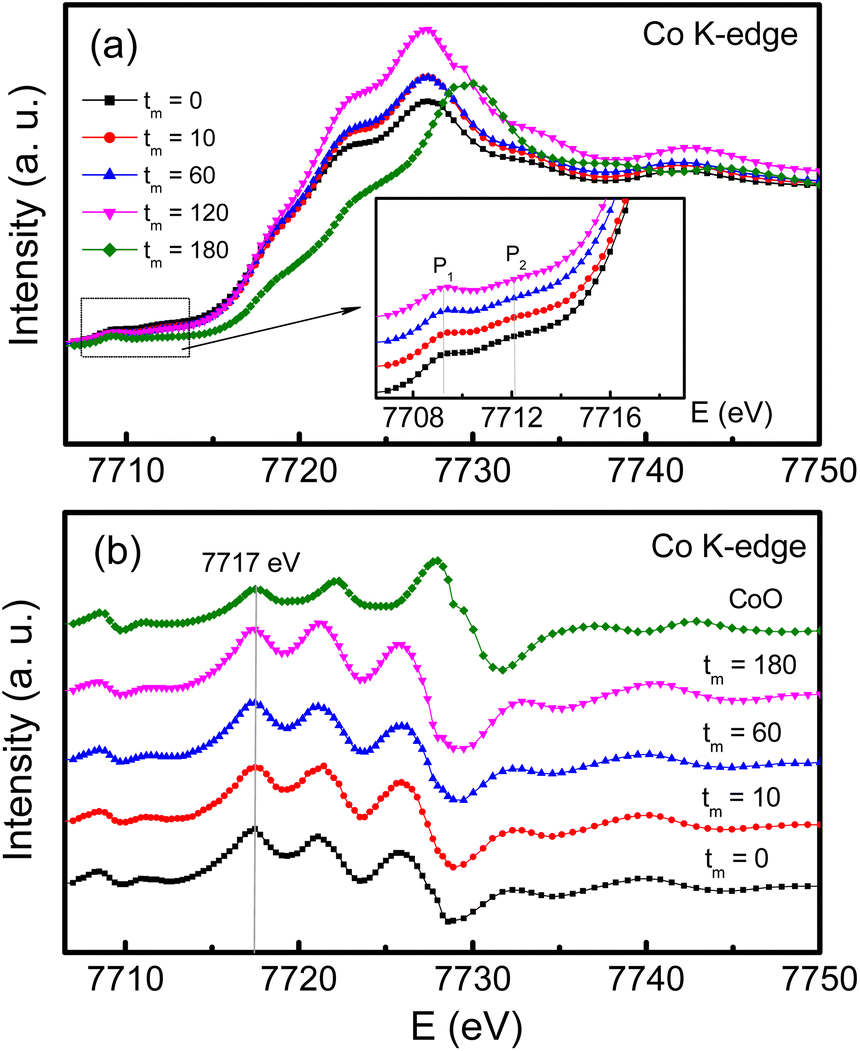 | ||
| Fig. 8 (a) Co K-edge XAS data, and (b) their derivative data of CoFe2O4 NPs with tm = 0, 10, 60 and 180 min compared with those of CoO. The inset of (a) is an enlarged view of the pre-edge region. | ||
To study the magnetic behaviors, we have measured the field-dependent magnetization, M(H). The M(H) data of representative samples graphed in Fig. 9 reveal all of them exhibiting the hysteresis character. Hysteresis loops have been recorded in the field range |H| = 0–7.5 kOe. Their shape is almost unchanged versus tm, excepting the characteristic parameters such as Ms, Mr, and Hc. For Mr and Hc, their values were directly obtained from the intersection points between a M(H) curve and the vertical (M) and horizontal (H) axes, respectively, as illustrated in Fig. 9. Determination of Ms can be based on the law of approach to saturation (LAS), in which H-dependent M obeys the following expression:50
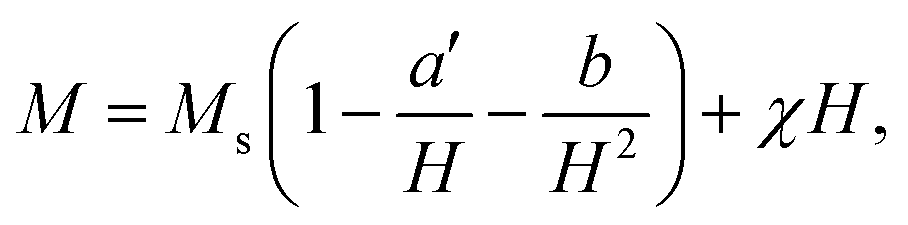 | (1) |
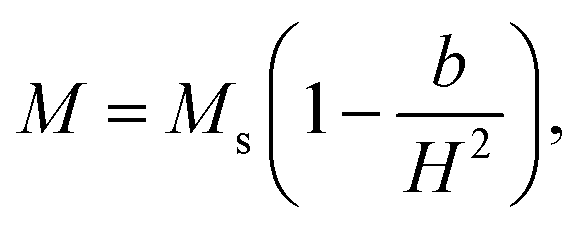 | (2) |
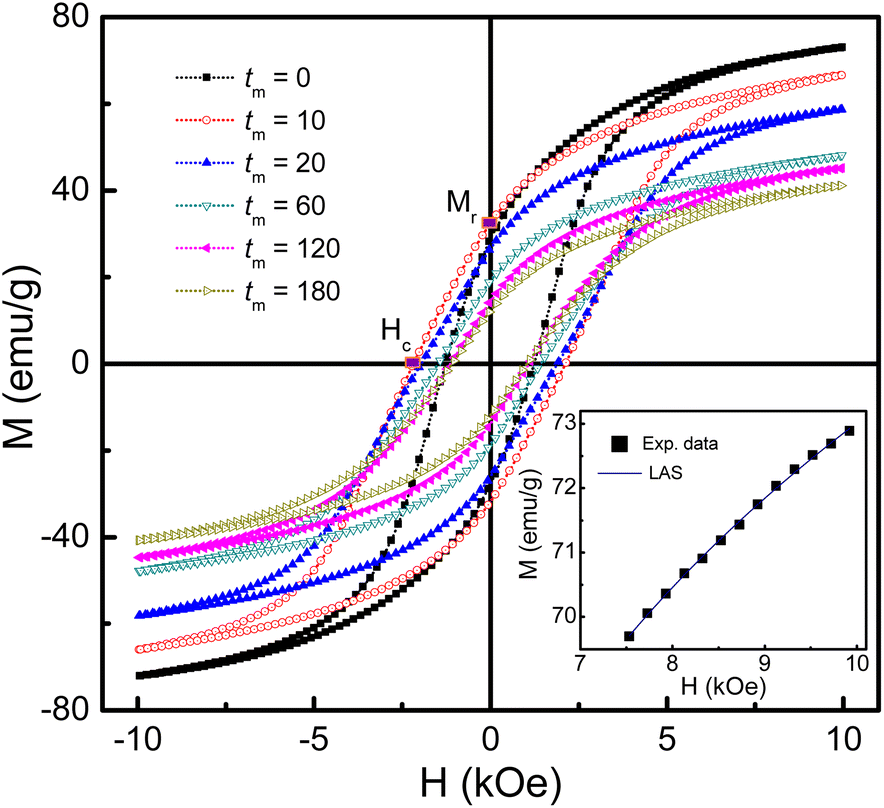 | ||
| Fig. 9 M(H) hysteresis loops of some representative samples of mechanically-milled CoFe2O4 NPs. Inset: M(H) data of the as-prepared sample (tm = 0) at fields H > 7.5 kOe fitted to the LAS model. | ||
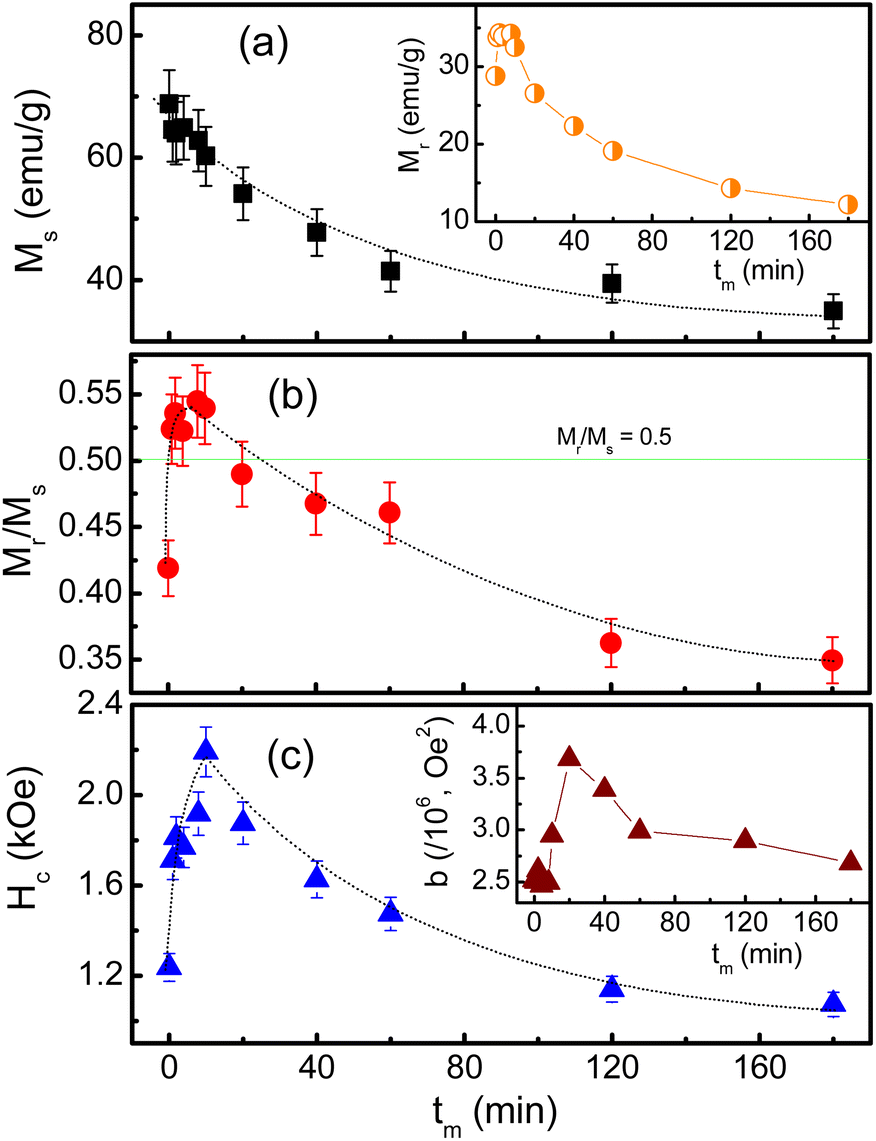 | ||
| Fig. 10 Variations of (a) Ms, (b) Mr/Ms and (c) Hc as a function of tm. The insets in (a) and (c) plot tm-dependent Mr and b data, respectively. Dotted lines are to guide the eyes only. | ||
Notably, since the crystallite size d gradually decreases as increasing tm, tm-dependent interparticle interactions can also influence Ms. It can be could be assessed interparticle interactions based on the Mr/Ms ratio.51 Mr determined from Fig. 9 reveals its maximum value being about ∼34 emu g−1 as tm = 4–8 min, and below and above these tm values, Mr decreases, see in the inset of Fig. 10(a). From the Mr data, we calculated tm-dependent Mr/Ms values. As shown in Fig. 10(b), the variation tendency of Mr/Ms is similar to that of Mr. As milling with tm = 1–10 min, Mr/Ms > 0.5 reflects the exchange coupling between neighboring grains/NPs.52 Beyond these tm values, the magnetostatic interaction between grains plays a dominant role because Mr/Ms < 0.5. Surprisingly, the as-prepared sample (tm = 0) also belongs to the magnetostatic interaction (Mr/Ms < 0.5). This could be due to the magnetostatic interaction additionally dependent on other factors, such as the surface state, isotropy, and aggregation of NPs, apart from the grain size, since SEM images of the samples shown in Fig. 1(a)–(f) have different features. It is necessary to say that Mr/Ms = 0.5 corresponds to randomly oriented non-interacting grains (like single domains).51,53
Such changes in the exchange and magnetostatic interactions versus tm would also influence Hc. This can be clearly seen in Fig. 10(c), where variation tendencies of Hc and Mr/Ms are almost the same. For the tm = 0 sample, Hc is about 1.24 kOe. It rapidly increases to ∼2.19 kOe as tm = 10 min (d ≈ 29 nm), corresponding to the dominancy of exchange interactions between grains/NPs. As milling with tm longer than 10 min, the transformation of exchange–magnetostatic interactions reduces gradually Hc to ∼1.07 kOe (for tm = 180 min).
These changes of Hc (as well as Mr and Mr/Ms) are in good agreement with those of the lattice constant a and the ratio I(220)/I(222) with respect to tm, see Fig. 5(a) and (b) and their inset. It means that Hc of milled CoFe2O4 NPs is also associated with the distribution of Co2+ in the A and B site. As comparing with previous works on mechanically-milled CoFe2O4 NPs, we have found that the variations of Hc(tm) in the current work are in good accordance to those reported by Liu and co-workers.13 However, the differences in Hc(tm) values reported in literature13,32,33 are mainly due to initial powders, which may have different sizes, particle morphology, and concentrations of Fe and Co ions at the A and B sites (in which Fe and Co can have oxidation numbers 2+ and 3+, even 4+). These factors are sensitive to sample synthesis methods and processing conditions. In fact, Hc is dependent on not only microstructures, lattice strain/defects,13,32,54 Co2+ distribution and interparticle interactions,51 but also magnetocrystalline anisotropy characterized by the parameter b. An analogous variation between b and Hc shown in Fig. 10(c) and the inset of Fig. 10(c) proves this statement. It has also been suggested b having the correlation with the anisotropy field (Ha) and anisotropy constant (K1) as follows:
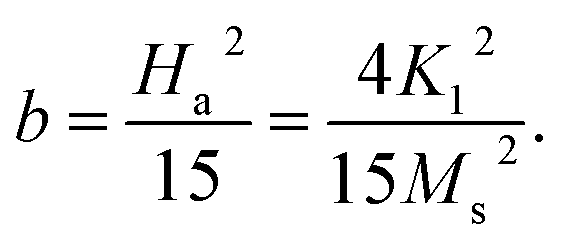 | (3) |
With the obtained b and Ms data, we could calculate Ha and K1 as using eqn (3). The results shown in Table 1 indicate the variation tendency of Ha similar to that of Hc and b, meaning that Ha reaches to the maximum value at tm = 10 min (d ≈ 29 nm). Meanwhile, because K1 is a function of b and Ms, we have found that a less change of K1 as tm = 0–20 min becomes gradually decreased as tm > 20 min, which is fairly the same as the variation tendency of Ms. Evidently, the data of the magnetic parameters reveal an anomalous variation around the milling time tm = 10 min (d ≈ 29 nm), where there is the transformation of exchange-to-magnetostatic interactions, and more Co2+ migrates to the A site. As tm > 10 min, an increase of the surface-to-volume ratio and surface defects enhances the magnetic dead layer, and causes short-range magnetic order, leading to the reduction of the magnetic parameters. With the above results, it is necessary to systematically study other nanostructured cobalt ferrites with different sizes using both XPS and XAS (or Mössbauer) techniques in order to identify the oxidation state and concentration of Fe and Co occupying the A and B sites. Based on these data, one can figure out the origin of the hard-magnetic properties of cobalt ferrites.
4 Conclusion
We based on a coprecipitation method to prepare CoFe2O4 NPs with d ≈ 81 nm. These powders were then mechanically milled in air for tm = 1–180 min to decrease d down to ∼12 nm. Both X-ray and RS studies indicated all the fabricated samples having the cubic-spinel structure. XAS analyses proved the coexistence of Fe2+,3+ and Co2+ ions in the samples. They occupy both the A and B sites, resulting in the formation of (Coδ2+Fe1−δ2+,3+)[Co1−δ2+Fe1+δ2+,3+]O42− mixed spinel compounds. The features of some characteristic XRD peaks and Raman modes, and the pre-edge XAS spectra revealed the migration and redistribution of the cations between the A and B sites, which would influence the lattice constant a of milled CoFe2O4 NPs. Particularly, as changing tm from 1 to 10 min, we found Hc of CoFe2O4 NPs shifts towards the hard-magnetic behavior, which is related to the exchange coupling between neighboring grains, due to Mr/Ms > 0.5. Meanwhile, the decrease of Hc as tm > 10 min is related to the magnetostatic interaction between grains, due to Mr/Ms < 0.5. For Ms, its gradual decrease with respect to tm is mainly due to enhancements of surface defects and the thickness of magnetic-dead layers, which cause short-range magnetic order, consequently the reduction of Ms.Author contributions
D. H. Manh, and T. D. Thanh: Conceptualization, Methodology and Writing; T. L. Phan, and D. S. Yang: Investigation and Reviewing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Excellence Research Team Development Program at the Vietnam Academy of Science and Technology NCXS01.04/22-24.References
- T. Tatarchuk, M. Bououdina, J. Judith Vijaya and L. John Kennedy, Spinel Ferrite Nanoparticles: Synthesis, Crystal Structure, Properties, and Perspective Applications, 2017, pp. 305–325, DOI:10.1007/978-3-319-56422-7_22.
- Y. Oh, M. Sahu, S. Hajra, A. M. Padhan, S. Panda and H. J. Kim, Spinel Ferrites (CoFe2O4): Synthesis, Magnetic Properties, and Electromagnetic Generator for Vibration Energy Harvesting, J. Electron. Mater., 2022, 51, 1933–1939, DOI:10.1007/s11664-022-09551-5.
- V. Tsurkan, H.-A. Krug von Nidda, J. Deisenhofer, P. Lunkenheimer and A. Loidl, On the complexity of spinels: Magnetic, electronic, and polar ground states, Phys. Rep., 2021, 926, 1–86, DOI:10.1016/j.physrep.2021.04.002.
- K. Malaie and M. R. Ganjali, Spinel nano-ferrites for aqueous supercapacitors; linking abundant resources and low-cost processes for sustainable energy storage, J. Energy Storage, 2021, 33, 102097, DOI:10.1016/j.est.2020.102097.
- (a) T. N. Pham, T. Q. Huy and A. T. Le, Spinel ferrite (AFe2O4)-based heterostructured designs for lithium-ion battery, environmental monitoring, and biomedical applications, RSC Adv., 2020, 10, 31622–31661, 10.1039/D0RA05133K; (b) X. Meng, L. He, Y. Liu, Y. Yu and W. Yang, Carbon-coated defect-rich MnFe2O4/MnO heterojunction for high-performance microwave absorption, Cacbon, 2022, 194, 207–219, DOI:10.1016/j.carbon.2022.03.075.
- R. K. Kotnala and J. Shah, Chapter 4 - Ferrite Materials: Nano to Spintronics Regime, Handbook of Magnetic Materials, 2015, vol. 23, pp. 291–379, DOI:10.1016/B978-0-444-63528-0.00004-8.
- (a) M. Matsuda, H. Ueda, A. Kikkawa, Y. Tanaka, K. Katsumata, Y. Narumi, T. Inami, Y. Ueda and S. H. Lee, Spin–lattice instability to a fractional magnetization state in the spinel HgCr2O4, Nat. Phys., 2007, 3, 397–400, DOI:10.1038/nphys586; (b) M. A. Ali, M. M. Uddin, M. N. I. Khan, F. U. Z. Chowdhury and S. M. Haque, Structural, morphological and electrical properties of Sn-substituted Ni-Zn ferrites synthesized by double sintering technique, J. Magn. Magn. Mater., 2017, 424, 148–154, DOI:10.1016/j.jmmm.2016.10.027.
- K. R. Sanchez-Lievanos, J. L. Stair and K. E. Knowles, Cation Distribution in Spinel Ferrite Nanocrystals: Characterization, Impact on their Physical Properties, and Opportunities for Synthetic Control, Inorg. Chem., 2021, 60, 4291–4305, DOI:10.1021/acs.inorgchem.1c00040.
- F. Xu, X. Li, G. Gan, S. Fan, Z. Yin, L. Wang, M. Qin, M. O. Tadé and S. Liu, Manganese-Based Spinel Core–Shell Nanostructures for Efficient Electrocatalysis of 1,2-Dichloroethane, ACS Appl. Nano Mater., 2020, 3, 10778–10786, DOI:10.1021/acsanm.0c02099.
- N. Garg, Menaka, K. V. Ramanujachary, S. E. Lofland and A. K. Ganguli, Nanostructured dimagnesium manganese oxide (Spinel): Control of size, shape and their magnetic and electro catalytic properties, J. Solid State Chem., 2013, 197, 392–397, DOI:10.1016/j.jssc.2012.08.063.
- S. C. Goh, C. H. Chia, S. Zakaria, M. Yusoff, C. Y. Haw, S. Ahmadi, N. M. Huang and H. N. Lim, Hydrothermal preparation of high saturation magnetization and coercivity cobalt ferrite nanocrystals without subsequent calcination, Mater. Chem. Phys., 2010, 120, 31–35, DOI:10.1016/j.matchemphys.2009.10.016.
- C. Zhao, A. Gao, Y. Yang, C. Tu, A. Bhutani, K. A. Walsh, S. Gong and D. P. Shoemaker, High-quality CoFe2O4 thin films with large coercivity grown via a wet chemical route, AIP Adv., 2019, 9, 035126, DOI:10.1063/1.5085232.
- B. H. Liu, J. Ding, Z. L. Dong, C. B. Boothroyd, J. H. Yin and J. B. Yi, Microstructural evolution and its influence on the magnetic properties of CoFe2O4 powders during mechanical milling, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 184427, DOI:10.1103/PhysRevB.74.184427.
- S. Singh and N. Khare, Low field magneto-tunable photocurrent in CoFe2O4 nanostructure films for enhanced photoelectrochemical properties, Sci. Rep., 2018, 8, 6522, DOI:10.1038/s41598-018-24947-2.
- N. Hosni, K. Zehani, T. Bartoli, L. Bessais and H. Maghraoui-Meherzi, Semi-hard magnetic properties of nanoparticles of cobalt ferrite synthesized by the co-precipitation process, J. Alloys Compd., 2017, 694, 1295–1301, DOI:10.1016/j.jallcom.2016.09.252.
- F. J. Pedrosa, J. Rial, K. M. Golasinski, M. N. Guzik, A. Quesada, J. F. Fernández, S. Deledda, J. Camarero and A. Bollero, Towards high performance CoFe2O4 isotropic nanocrystalline powder for permanent magnet applications, Appl. Phys. Lett., 2016, 109, 223105, DOI:10.1063/1.4969064.
- P. Maltoni, T. Sarkar, G. Barucca, G. Varvaro, F. Locardi, D. Peddis and R. Mathieu, Tuning the Magnetic Properties of Hard–Soft SrFe12O19/CoFe2O4 Nanostructures via Composition/Interphase Coupling, J. Phys. Chem. C, 2021, 125, 5927–5936, DOI:10.1021/acs.jpcc.1c00355.
- (a) Z. Ž. Lazarević, Č. Jovalekić, A. Milutinović, D. Sekulić, V. N. Ivanovski, A. Rečnik, B. Cekić and N. Ž. Romčević, Nanodimensional spinel NiFe2O4 and ZnFe2O4 ferrites prepared by soft mechanochemical synthesis, J. Appl. Phys., 2013, 113, 187221, DOI:10.1063/1.4801962; (b) S. Iraqui, S. S. Kashyap and M. H. Rashid, NiFe2O4 nanoparticles: an efficient and reusable catalyst for the selective oxidation of benzyl alcohol to benzaldehyde under mild conditions, Nanoscale Adv., 2020, 2, 5790–5802, 10.1039/D0NA00591F; (c) C. Yao, Q. Zeng, G. F. Goya, T. Torres, J. Liu, H. Wu, M. Ge, Y. Zeng, Y. Wang and J. Z. Jiang, ZnFe2O4 Nanocrystals: Synthesis and Magnetic Properties, J. Phys. Chem. C, 2007, 111, 12274–12278, DOI:10.1021/jp0732763; (d) M. A. Ali, M. N. I. Khan and F. Chowdhury, et al., Yttrium-substituted Mg–Zn ferrites: correlation of physical properties with Yttrium content, J. Mater. Sci.: Mater. Electron., 2019, 30, 13258–13270, DOI:10.1007/s10854-019-01689-z.
- J. L. Ortiz-Quiñonez, U. Pal and M. S. Villanueva, Structural, Magnetic, and Catalytic Evaluation of Spinel Co, Ni, and Co–Ni Ferrite Nanoparticles Fabricated by Low-Temperature Solution Combustion Process, ACS Omega, 2018, 3, 14986–15001, DOI:10.1021/acsomega.8b02229.
- Y. H. Hou, Y. J. Zhao, Z. W. Liu, H. Y. Yu, X. C. Zhong, W. Q. Qiu, D. C. Zeng and L. S. Wen, Structural, electronic and magnetic properties of partially inverse spinel CoFe2O4: a first-principles study, J. Phys. D: Appl. Phys., 2010, 43, 445003, DOI:10.1088/0022-3727/43/44/445003.
- S. E. Shirsath, D. Wang, J. Zhang, A. Morisako, S. Li and X. Liu, Single-Crystal-like Textured Growth of CoFe2O4 Thin Film on an Amorphous Substrate: A Self-Bilayer Approach, ACS Appl. Electron. Mater., 2020, 2, 3650–3657, DOI:10.1021/acsaelm.0c00716.
- S. Moosavi, S. Zakaria, C. H. Chia, S. Gan, N. A. Azahari and H. Kaco, Hydrothermal synthesis, magnetic properties and characterization of CoFe2O4 nanocrystals, Ceram. Int., 2017, 43, 7889–7894, DOI:10.1016/j.ceramint.2017.03.110.
- A. Hajalilou, S. A. Mazlan, M. Abbasi and H. Lavvafi, Fabrication of spherical CoFe2O4 nanoparticles via sol–gel and hydrothermal methods and investigation of their magnetorheological characteristics, RSC Adv., 2016, 6, 89510–89522, 10.1039/C6RA13493A.
- M. Houshiar, F. Zebhi, Z. J. Razi, A. Alidoust and Z. Askari, Synthesis of cobalt ferrite (CoFe2O4) nanoparticles using combustion, coprecipitation, and precipitation methods: A comparison study of size, structural, and magnetic properties, J. Magn. Magn. Mater., 2014, 371, 43–48, DOI:10.1016/j.jmmm.2014.06.059.
- Z. Zi, Y. Sun, X. Zhu, Z. Yang, J. Dai and W. Song, Synthesis and magnetic properties of CoFe2O4 ferrite nanoparticles, J. Magn. Magn. Mater., 2009, 321, 1251–1255, DOI:10.1016/j.jmmm.2008.11.004.
- H. Latiff, M. Kishimoto, J. Inoue, E. Kita, H. Yanagihara and T. Devillers, Coercivity analysis of cubic and tetragonal (Cu,Co) ferrite particles within the global model, J. Magn. Magn. Mater., 2019, 489, 165380, DOI:10.1088/1361-6463/abe5e1.
- E. Lima, E. L. Winkler, D. Tobia, H. E. Troiani, R. D. Zysler, E. Agostinelli and D. Fiorani, Bimagnetic CoO Core/CoFe2O4 Shell Nanoparticles: Synthesis and Magnetic Properties, Chem. Mater., 2012, 24, 512–516, DOI:10.1021/cm2028959.
- L. Pan, D. Cao, P. Jing, J. Wang and Q. Liu, A novel method to fabricate CoFe2O4/SrFe12O19 composite ferrite nanofibers with enhanced exchange coupling effect, Nanoscale Res. Lett., 2015, 10, 131, DOI:10.1186/s11671-015-0829-z.
- Z. Wang, X. Liu, M. Lv, P. Chai, Y. Liu, X. Zhou and J. Meng, Preparation of One-Dimensional CoFe2O4 Nanostructures and Their Magnetic Properties, J. Phys. Chem. C, 2008, 112, 15171–15175, DOI:10.1021/jp802614v.
- Y. C. Wang, J. Ding, J. B. Yi, B. H. Liu, T. Yu and Z. X. Shen, High-coercivity Co-ferrite thin films on (100)-SiO2(100)-SiO2 substrate, Appl. Phys. Lett., 2004, 84, 2596–2598, DOI:10.1063/1.1695438.
- Y. Cedeño-Mattei, O. Perales-Pérez and O. N. C. Uwakweh, Show more Effect of high-energy ball milling time on structural and magnetic properties of nanocrystalline cobalt ferrite powders, J. Magn. Magn. Mater., 2013, 341, 17–24, DOI:10.1016/j.jmmm.2013.04.015.
- A. S. Ponce, E. F. Chagas, R. J. Prado, C. H. M. Fernandes, A. J. Terezo and E. Baggio-Saitovitch, High coercivity induced by mechanical milling in cobalt ferrite powder, J. Magn. Magn. Mater., 2013, 344, 182–187, DOI:10.1016/j.jmmm.2013.05.056.
- F. J. Pedrosa, J. Rial, K. M. Golasinski, M. Rodríguez-Osorio, G. Salas, D. Granados, J. Camarero and A. Bollero, Tunable nanocrystalline CoFe2O4 isotropic powders obtained by co-precipitation and ultrafast ball milling for permanent magnet applications, RSC Adv., 2016, 6, 87282–87287, 10.1039/C6RA15698C.
- B. J. Rani, M. Ravina, B. Saravanakumar, G. Ravi, V. Ganesh, S. Ravichandran and R. Yuvakkumar, Ferrimagnetism in cobalt ferrite (CoFe2O4) nanoparticles, Nano-Struct. Nano-Objects, 2018, 14, 84–91, DOI:10.1016/j.nanoso.2018.01.012.
- S. Li, T. Zhang, R. Tang, H. Qiu, C. Wang and Z. Zhou, Solvothermal synthesis and characterization of monodisperse superparamagnetic iron oxide nanoparticles, J. Magn. Magn. Mater., 2015, 379, 226–231, DOI:10.1016/j.jmmm.2014.12.054.
- S. C. Endres, L. C. Ciacchi and L. Mädler, A review of contact force models between nanoparticles in agglomerates, aggregates, and films, J. Aerosol Sci., 2021, 153, 105719, DOI:10.1016/j.jaerosci.2020.105719.
- M. N. Singh, A. K. Sinha and H. Ghosh, Determination of transition metal ion distribution in cubic spinel Co1.5Fe1.5O4 using anomalous x-ray diffraction, AIP Adv., 2015, 5, 087115, DOI:10.1063/1.4928425.
- G. K. Williamson and W. H. Hall, X-ray line broadening from filed aluminium and wolfram, Acta Metall., 1953, 1, 22–31, DOI:10.1016/0001-6160(53)90006-6.
- S. Tsunekawa, K. Ishikawa, Z.-Q. Li, Y. Kawazoe and A. Kasuya, Origin of Anomalous Lattice Expansion in Oxide Nanoparticles, Phys. Rev. Lett., 2000, 85, 3440–3443, DOI:10.1103/PhysRevLett.89.249905.
- V. Šepelák, D. Baabe, D. Mienert, D. Schultze, F. Krumeich, F. J. Litterst, K. D. Becker, V. Šepelák, D. Baabe, D. Mienert, D. Schultze, F. Krumeich, F. J. Litterst and K. D. Becker, J. Magn. Magn. Mater., 2003, 257, 377–386, DOI:10.1016/S0304-8853(02)01279-9.
- P. Chandramohan, M. P. Srinivasan, S. Velmurugan, S. V. Narasimhan, P. Chandramohan, M. P. Srinivasan, S. Velmurugan and S. V. Narasimhan, J. Solid State Chem., 2011, 184, 89–96, DOI:10.1016/j.jssc.2010.10.019.
- Pressure-Induced Phase Transitions in AB2X4 Chalcogenide Compounds, ed. F. J. Manjon, I. Tiginyanu and V. Ursaki, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, vol. 189 Search PubMed.
- S. M. Ansari, B. B. Sinha, D. Phase, D. Sen, P. U. Sastry, Y. D. Kolekar and C. V. Ramana, Particle Size, Morphology, and Chemical Composition Controlled CoFe2O4 Nanoparticles with Tunable Magnetic Properties via Oleic Acid Based Solvothermal Synthesis for Application in Electronic Devices, ACS Appl. Nano Mater., 2019, 2, 1828–1843, DOI:10.1021/acsanm.8b02009.
- D. M. Phase, S. Tiwari, R. Prakash, A. Dubey, V. G. Sathe, R. J. Choudhary, D. M. Phase, S. Tiwari, R. Prakash, A. Dubey, V. G. Sathe and R. J. Choudhary, J. Appl. Phys., 2006, 100, 123703, DOI:10.1063/1.2403849.
- M. Baghaie Yazdi, K.-Y. Choi, D. Wulferding, P. Lemmens and L. Alff, Raman study of the Verwey transition in magnetite thin films, New J. Phys., 2013, 15, 103032, DOI:10.1088/1367-2630/15/10/103032.
- S. M. Ansari, K. C. Ghosh, R. S. Devan, D. Sen, P. U. Sastry, Y. D. Kolekar and C. V. Ramana, Eco-Friendly Synthesis, Crystal Chemistry, and Magnetic Properties of Manganese-Substituted CoFe2O4 Nanoparticles, ACS Omega, 2020, 5, 19315–19330, DOI:10.1021/acsomega.9b02492.
- S. Ammar, A. Helfen, N. Jouini, F. Fiévet, I. Rosenman, F. Villain, P. Molinié and M. Danot, Magnetic properties of ultrafine cobalt ferrite particles synthesized by hydrolysis in a polyol medium, J. Mater. Chem., 2001, 11, 186–192, 10.1039/b003193n.
- D. Carta, A. Corrias, A. Falqui, R. Brescia, E. Fantechi, F. Pineider and C. Sangregorio, EDS, HRTEM/STEM, and X-ray absorption spectroscopy studies of co-substituted maghemite nanoparticles, J. Phys. Chem. C, 2013, 117, 9496–9506, DOI:10.1021/jp401706c.
- S. Londo, S. Biswas, J. Husek, I. V. Pinchuk, M. J. Newburger, A. Boyadzhiev, A. H. Trout, D. W. McComb, R. Kawakami and L. R. Baker, Ultrafast Spin Crossover in a Room-Temperature Ferrimagnet: Element-Specific Spin Dynamics in Photoexcited Cobalt Ferrite, J. Phys. Chem. C, 2020, 124, 11368–11375, DOI:10.1021/acs.jpcc.0c03736.
- J. Lee, E. J. Lee, T.-Y. Hwang, J. Kim and Y.-H. Choa, Anisotropic characteristics and improved magnetic performance of Ca–La–Co-substituted strontium hexaferrite nanomagnets, Sci. Rep., 2020, 10, 15929, DOI:10.1038/s41598-020-72608-0.
- S. H. C. Sushin Chikazumi, Physics of Magnetism, Krieger Pub Co, 1978 Search PubMed.
- J. N. Dahal, D. Neupane and S. R. Mishra, Exchange-Coupling Behavior in SrFe12O19/La0.7Sr0.3MnO3 Nanocomposites, Ceramics, 2019, 2, 100–111, DOI:10.3390/ceramics2010010.
- E. C. Stone and E. P. Wohlfarth, A mechanism of magnetic hysteresis in heterogeneous alloys, Philos. Trans. R. Soc. London, Ser. A, 1948, 240, 599–642, DOI:10.1098/rsta.1948.0007.
- S. Singh, S. Munjal and N. Khare, Strain/defect induced enhanced coercivity in single domain CoFe2O4 nanoparticles, J. Magn. Magn. Mater., 2015, 386, 69–73, DOI:10.1016/j.jmmm.2015.03.057.
| This journal is © The Royal Society of Chemistry 2023 |