
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical study on the mechanism of water oxidation catalyzed by a mononuclear copper complex: important roles of a redox non-innocent ligand and HPO42− anion†
Ying-Ying Li *a,
Xiao-Yan Wanga,
Hui-Ji Lia,
Jia-Yi Chenb,
Yao-Hua Koua,
Xiao Lia and
Yaping Wanga
*a,
Xiao-Yan Wanga,
Hui-Ji Lia,
Jia-Yi Chenb,
Yao-Hua Koua,
Xiao Lia and
Yaping Wanga
aSchool of Chemistry and Chemical Engineering, Zhengzhou Normal University, Zhengzhou 450044, China. E-mail: liyingying@zznu.edu.cn
bKey Laboratory of Material Chemistry for Energy Conversion and Storage, Ministry of Education, Hubei Key Laboratory of Bioinorganic Chemistry and Materia Medica, Hubei Key Laboratory of Materials Chemistry and Service Failure, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, P. R. China
First published on 14th March 2023
Abstract
The water oxidation reaction is the bottleneck problem of the artificial photosynthetic system. In this work, the mechanism of water oxidation catalyzed by a mononuclear copper complex in alkaline conditions was studied by density functional calculations. Firstly, a water molecule coordinating with the copper center of the complex (CuII, 1) generates CuII–H2O (2). 2 undergoes two proton-coupled electron transfer processes to produce intermediate (4). The oxidation process occurs mainly on the ligand moiety, and 4 (˙L–CuII–O˙) can be described as a CuII center interacting with a ligand radical antiferromagnetically and an oxyl radical ferromagnetically. 4 is the active species that can trigger O–O bond formation via the water nucleophilic attack mechanism. This process occurs in a step-wise manner. The attacking water transfers one of the protons to the HPO42− coupled with an electron transfer to the ligand radical, which generates a transient OH˙ interacting with the oxyl radical and H2PO4−. Then the O–O bond is formed through the direct coupling of the oxo radical and the OH radical. The triplet di-oxygen could be released after two oxidation processes. According to the Gibbs free energy diagram, the O–O bond formation was suggested to be the rate-limiting step with a calculated total barrier of 19.5 kcal mol−1. More importantly, the copper complex catalyzing water oxidation with the help of a redox non-innocent ligand and HPO42− was emphasized.
Introduction
Water splitting driven by sunlight stands out as one of the promising tactics for solving the energy crisis. Water splitting (eqn (1)) consists of a water oxidation reaction (eqn (2)) and a proton reduction reaction (eqn (3)). The water oxidation lies at the heart of the reaction since it is both thermodynamically and kinetically demanding. Developing highly efficient, stable, and economical water oxidation catalysts (WOCs) remains challenging. Heterogeneous WOCs have advantages in industrial applications, while homogeneous WOCs are beneficial for mechanism studies, such as possibly detectable intermediates and controllable catalytic properties.1–3 To date, WOCs based on noble metals, such as Ru and Ir, show relatively high activities and stabilities, while their widespread applications on the scale of industrialization are limited.4–10 Due to the low cost and high earth abundance, copper-based complexes are good candidates for catalyzing water oxidation. Great progress has been made during the last few decades since the Mayer group reported the first molecular Cu-based catalyst.11–19 Computational calculations are beneficial for detailed mechanistic investigations, which can provide important insights for the design of water oxidation catalysts.20–34| 2H2O → 2H2 + O2 | (1) |
| 2H2O → O2 + 4H+ + 4e− E0 = 1.23 V vs. SHE at pH = 0 | (2) |
| 4H+ + 4e− → 2H2 E0 = 0 V vs. SHE at pH = 0 | (3) |
Among transition metal-based WOCs, Cu catalysts are particularly interesting in terms of the O–O bond formation process. Conventionally, H2O or OH− nucleophilic attack (WNA, Fig. 1a) on the formal CuIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O through a single site mechanism is proposed for most of Cu–WOCs.32,35,36 Koepke and co-workers revealed that the O–O bond could form via the redox isomerization mechanism for the bis(μ-oxo) linkage di-copper catalyst (Fig. 1b)37. The intramolecular coupling between the bridging oxo group and the terminal OH moiety was the most favorable for the dinuclear copper complex (Fig. 1c)38. Mao and co-workers reported that the copper-bound terminal oxyl radical ligand and the adjacent oxyanion on the bipyridine group trigger the O–O bond formation for the mononuclear copper WOC with bipyridine ligands (Fig. 1d)39. In addition, the single electron transfer-water nucleophilic attack (SET-WNA) mechanism has been suggested to be more favorable for a series of mono-copper catalysts and the tricopper-containing polyoxometalate (Fig. 1e)24,40–42. The O–O bond formation prefers the unusual coupling of the bridging oxyl radical and the CO3−˙ for the bioinspired trinuclear copper catalyst (Fig. 1f)36. Recently, the OH− nucleophilic attack onto the ˙+L–CuII–OH− was calculated to be the most favorable scenario for the mononuclear copper catalyst (Fig. 1g)23. Depending on the specific catalytic environment, the specified O–O bond formation process is normally decisive for catalytic efficiency.
O through a single site mechanism is proposed for most of Cu–WOCs.32,35,36 Koepke and co-workers revealed that the O–O bond could form via the redox isomerization mechanism for the bis(μ-oxo) linkage di-copper catalyst (Fig. 1b)37. The intramolecular coupling between the bridging oxo group and the terminal OH moiety was the most favorable for the dinuclear copper complex (Fig. 1c)38. Mao and co-workers reported that the copper-bound terminal oxyl radical ligand and the adjacent oxyanion on the bipyridine group trigger the O–O bond formation for the mononuclear copper WOC with bipyridine ligands (Fig. 1d)39. In addition, the single electron transfer-water nucleophilic attack (SET-WNA) mechanism has been suggested to be more favorable for a series of mono-copper catalysts and the tricopper-containing polyoxometalate (Fig. 1e)24,40–42. The O–O bond formation prefers the unusual coupling of the bridging oxyl radical and the CO3−˙ for the bioinspired trinuclear copper catalyst (Fig. 1f)36. Recently, the OH− nucleophilic attack onto the ˙+L–CuII–OH− was calculated to be the most favorable scenario for the mononuclear copper catalyst (Fig. 1g)23. Depending on the specific catalytic environment, the specified O–O bond formation process is normally decisive for catalytic efficiency.
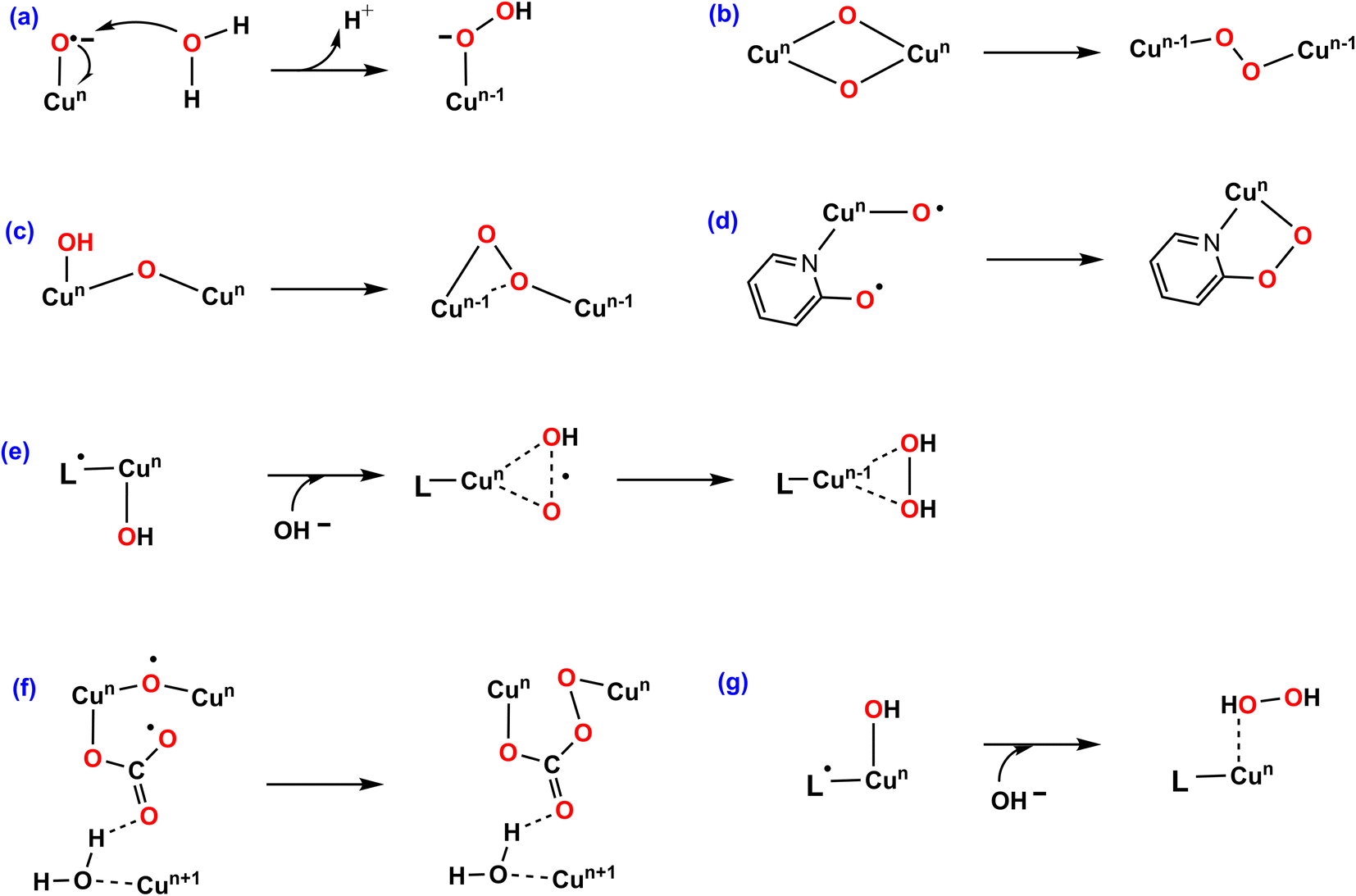 | ||
| Fig. 1 Proposed O–O bond formation mechanisms. (a) Water nucleophilic attack (WNA). (b) Redox isomerization. (c) Intramolecular coupling. (d) Coupling of the oxyl radical ligand and the adjacent oxyanion. (e) Single electron transfer-water nucleophilic attack (SET-WNA). (f) Coupling of the bridging oxyl radical and the CO3− radical. (g) OH− nucleophilic attack. | ||
Recently, Zhang and coworkers reported a copper complex with a pentadentate amine-pyridine ligand (Scheme 1) that can catalyze water oxidation in pH 11.0 phosphate buffer solution, and a kcat of 0.81 s−1 was reported.43 The experiment study showed an overpotential of 831 mV. The stability of the catalyst was examined by a controlled potential electrolysis experiment, which suggested the copper complex remained stable over 3 hours.43 In this work, the catalytic reaction mechanism was fully calculated via density functional calculations. The redox non-innocent role of the ligand was suggested. The most important O–O bond formation step was investigated. The possible WNA mechanism was studied and a step-wise mechanism was proposed. The unique role of HPO42− in the working solution was confirmed.
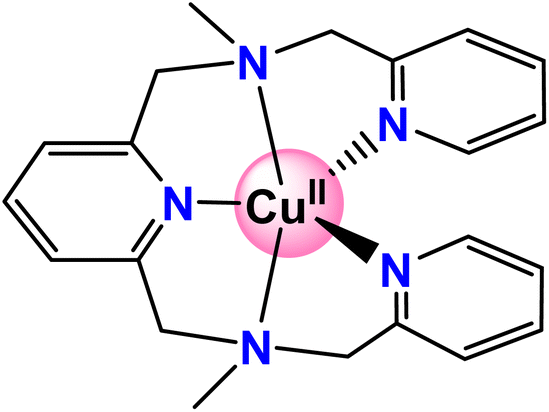 | ||
| Scheme 1 Schematic representation of the copper-WOC. | ||
Computational details
All calculations were performed using Gaussian 16 program.44 The B3LYP45 functional coupled with the Grimme D3 (ref. 46) dispersion correction were used for the geometries optimization in conjunction with the SMD continuum solvation model.47 All atoms were described by def2-SVP basis set.48 Based on the geometries optimization, analytical frequency calculations at the same level of theory were performed to confirm the nature of various species. The absence of the imaginary frequency was used to verify that the structure is at the minimum. While only one imaginary frequency is presented for the transition state. The final energy was obtained via a single-point calculation with a larger basis set of def2-TZVP.48The eqn (4) is used to calculate the redox potential for a reduction reaction.
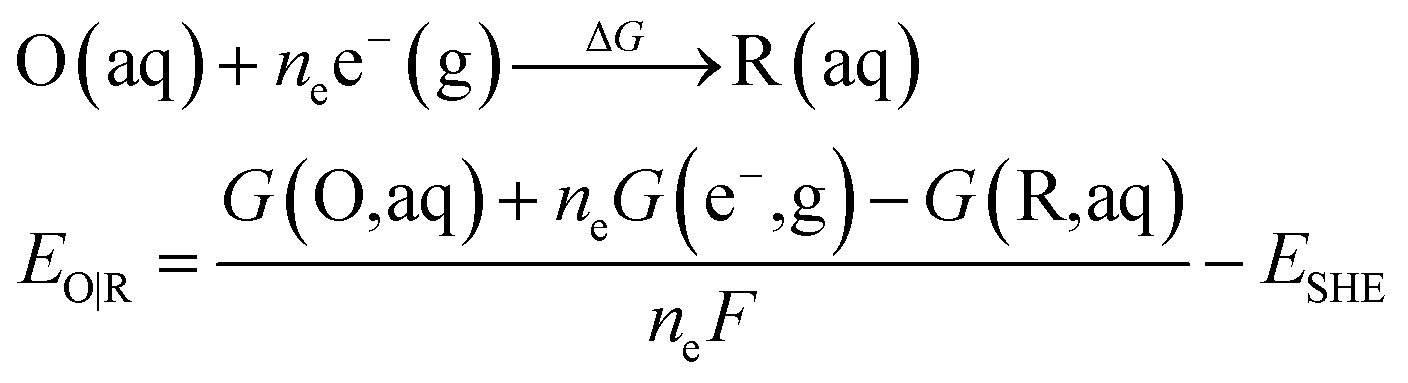 | (4) |
where EO|R is the redox potential, ΔG is the Gibbs free energy change of the reduction reaction, G(X, aq) is the free energy of the X with the SMD solvation model in aqueous solution. ne is the number of the transferred electrons in the reduction reaction, F is the Faraday constant.33 The absolute standard hydrogen potential of 4.281 V (ESHE) corresponding to an electron affinity of 98.7 kcal mol−1 from the experiment was taken as a reference for the calculation of redox potentials.49
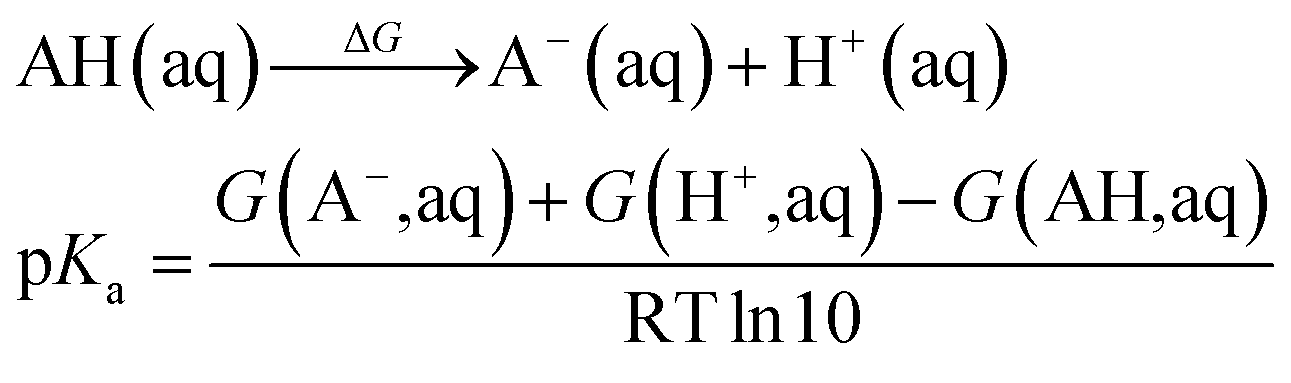 | (5) |
The pKa values were calculated in order to determine the protonation state of all possible intermediates, where eqn (5) was used. The experimental solvation energy of −264.0 kcal mol−1 of the H+ was employed.50 So the value of −270.3 kcal mol−1 was used for the G(H+, aq) when the statistical gas-phase free energy of formation of H+ of −6.3 kcal mol−1 was added. For the calculations of solvation free energy for all species in aqueous solution as defined as the free energy of transfer of the solute from the gas phase (1 atm, 24.5 L mol−1) to the aqueous phase (1 M) at 298 K, a concentration correction of 1.9 kcal mol−1 (derived from RTln(24.5)) at 298.15 K was added. In the case of water, a correction value of 4.3 kcal mol−1 since it has a standard state of 55.56 mol L−1.
Results and discussion
The present work started from the geometry optimization of the copper-based water oxidation catalyst (labeled as 1, Fig. 2). 1 has a total charge of +2. In 1, nitrogen atoms from three pyridines and two amide groups coordinate with the CuII center. 1 has a ground state of doublet state, where the spin density on the copper center is 0.58, and the electron is partially delocalized to the ligand. Before initiating the water oxidation process, one water molecule has to bind with the CuII, which generates 2 (CuII–OH2, Fig. 2). This process is endergonic by 3.7 kcal mol−1. In 2, the distance between the copper center and the water oxygen is 2.04 Å. The spin density on the Cu center is 0.62. The coordination of water leads to the dissociation of two of the N atoms, which suggests that the copper complex has to go through a conformational change before initiate the water oxidation. 2 is the starting point of the catalytic cycle (vide infra).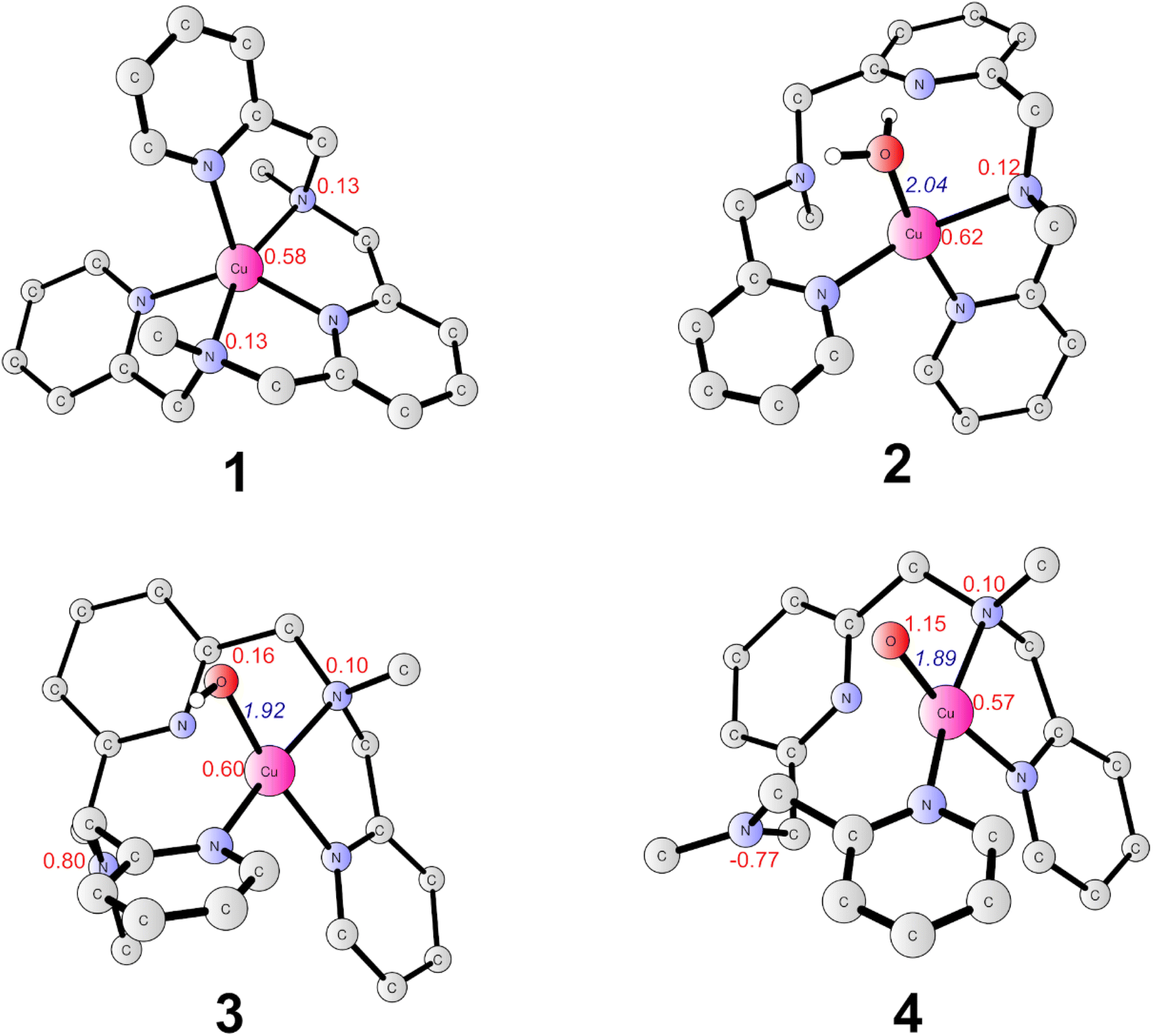 | ||
| Fig. 2 Optimized structures of 1(CuII, doublet), 2 (CuII, doublet), 3 (CuIII, triplet) and 4 (CuIV, doublet). Selected distances are shown in Angstrom in blue italic. Spin densities on selected atoms are presented in red. For clarity, non-important hydrogen atoms are not shown. | ||
The oxidation of 2 is a proton-coupled electron transfer (PCET) process, during which one of the protons of the water ligand is released. This is consistent with the experimental phenomenon that the first oxidation of the catalyst in phosphate buffer is pH-dependent, with a slope of 59.8 mV per pH unit.43 This oxidation process leads to the generation of the triplet 3 (˙L–CuII–OH, Fig. 2). The singlet lies at +3.6 kcal mol−1 above the ground triplet state. In 3, the Cu–O bond has a distance of 1.92 Å. The spin densities on the Cu center and ligand were calculated to be 0.60 and 0.90, respectively. This suggests that the first oxidation mainly occurs on the ligand moiety. The electronic structure of 3 can be interpreted as a divalent copper (SCu = 1/2) center interacting ferromagnetically with a ligand radical (SLigand = 1/2). The redox potential of the PCET process was calculated to be 1.03 V. Taking the applied potential of 1.41 V as a reference. This step is exergonic by 8.8 kcal mol−1 (Fig. 3). The pKa of 3pt (˙L–CuII–H2O, Fig. S1†) was calculated to be 11.2, which is close to the working pH of 11. Therefore, it is difficult to determine whether 3 or 3pt dominates in the working conditions. The one-electron oxidation process of 2 is also possible. The redox potential of an electron releasing process is 1.02 V, which would compete with the PCET process.
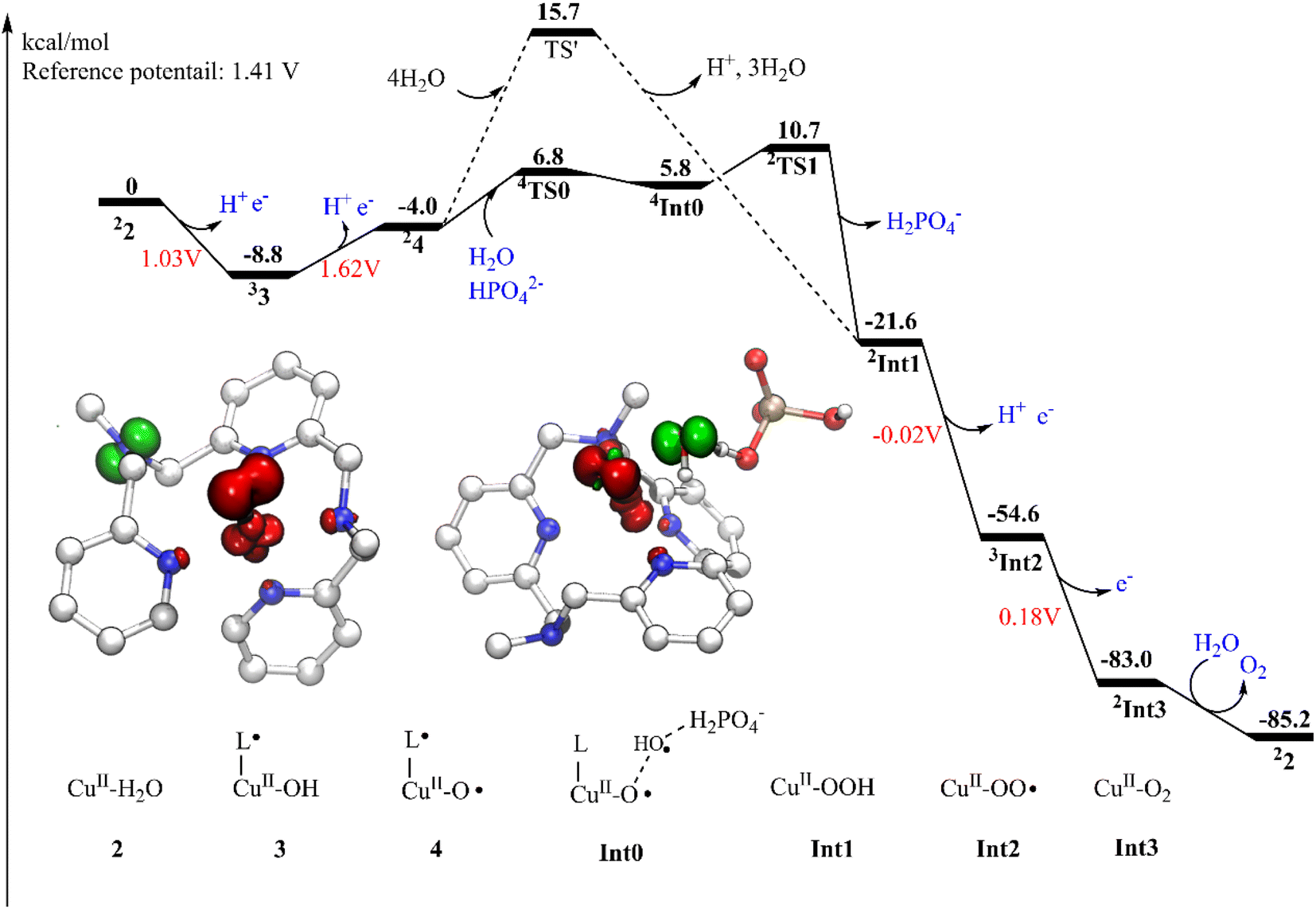 | ||
| Fig. 3 Gibbs energy diagram of water oxidation catalyzed by the copper-based catalyst. Inset: the spin density populations of 4 (left) and TS1 (right) are shown. The left superscripts indicate the multiplicity of the ground state of specified species. | ||
Next, a subsequent PCET of 3 leads to the formation of 4 (˙L–CuIII–O˙, Fig. 2) with an associated redox potential of 1.62 V. The protonation state of 4 (4pt, ˙L–CuIII–OH, Fig. S1†) has a pKa of 10.6. 4 is a doublet, and the quartet is only 0.39 kcal mol−1 higher than the ground state. In 4, the spin densities on Cu, ligand, and O are 0.57, −0.67, and 1.15, respectively (Fig. 2 and 3). According to the spin population, 4 can be described as a CuII (SCu = 1/2) center coupling to a ligand radical (SLigand = −1/2) antiferromagnetically and an oxyl radical (SO = 1/2) ferromagnetically. The distance of Cu–O in 4 is 1.89 Å (Fig. 2). This oxidation process is endergonic by 4.8 kcal mol−1 with an applied potential of 1.41 V (Fig. 3). The oxidation processes from 2 to 4 occur mainly on the ligand moiety, which emphasizes the redox non-innocent role of the ligand.
The formal CuIV complex (4) was proposed to be the active species to trigger the O–O bond formation, as suggested by previous studies.23,26,42,51 The water nucleophilic attack (WNA) mechanism was investigated. The calculation results suggest a step-wise mechanism when phosphate ion behaves as a proton acceptor. Firstly, the proton of attacking water transfers to the HPO42− ion coupled with an electron transfer via TS0 (Fig. 4). TS0 prefers to be a quartet. The spin density on Cu, O1, and O2 are 0.59, 1.60, and 0.43, respectively. During this PCET process, a β electron of attacking water transfers to the ligand radical. In TS0, the O2–H1 and O3–H1 distances were calculated to be 1.14 Å and 1.29 Å, respectively. The O1–O2 and Cu–O1 have distances of 2.14 Å and 1.95 Å, respectively. The only imaginary frequency of 315.42i cm−1 corresponding to the proton transfer to the HPO42− confirms that it is a true transition state. The doublet TS0 is 1.0 kcal mol−1 higher than the quartet state. This PECT process leads to the quartet Int0 (Fig. 4). Int0 is 1.0 kcal mol−1 below the TS0. In Int0, the spin densities on Cu, O1 and O2 are 0.58, 1.48 and 0.62, respectively, which suggests a transient OH˙ interacting with the L–CuII–O˙ and the generated H2PO4−. The O2–H1 and O3–H1 distances are 1.57 Å and 1.02 Å, respectively. The doublet Int0 is 2.0 kcal mol−1 higher. However, the electron evolution during this process is slightly different for the doublet state. The β electron of the attacking water may transfer firstly to the copper center while the α electron from the Cu transfers to the ligand (Fig. S2†) TS0 lies at +10.8 kcal mol−1 above the 4. (Fig. 3).
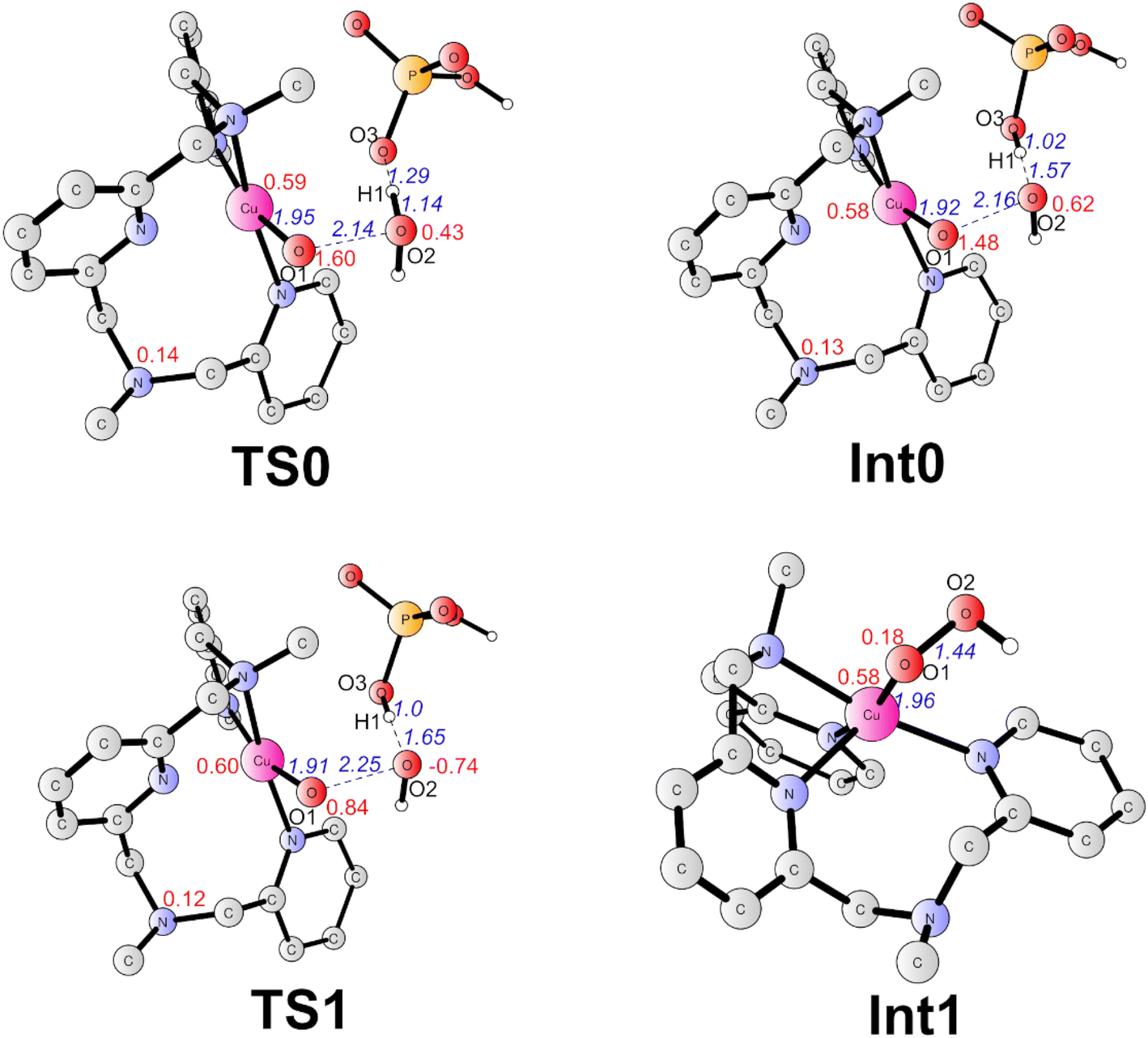 | ||
| Fig. 4 Optimized structures of TS0 (quartet), Int0 (quartet), TS1 (doublet), and Int1 (doublet). Selected distances are shown in Angstrom in blue italic. Spin densities on selected atoms are presented in red. For clarity, non-important hydrogen atoms are not shown. The imaginary frequencies of TS0 and TS1 are 315.42i cm−1 and 332.76i cm−1, respectively. | ||
From Int0, the direct coupling of the oxo radical and the OH radical at doublet via TS1 generates the hydro-peroxide intermediate (Int1, Fig. 4). The opposite spins (0.84 on O1 and -0.74 on O2 in TS1) are required for the O–O bond formation. A spin crossing from the quartet to the doublet is necessary for this process. In TS1, the nascent O–O distance is 2.25 Å. The spin density on the copper center is 0.60. The optimized Cu–O distance is 1.91 Å. TS1 lies at +4.9 kcal mol−1 above the Int0. For the step-wise O–O bond formation process, the proton transfer requires a higher energy barrier than the hydroxide coupling with the Cu-oxo, which is similar with the reported mononuclear copper WOC.52,53 Taking the energy penalty of the formation of 4, the whole O–O bond formation process has a total energy barrier of 19.5 kcal mol−1. Downhill from the TS1, a hydro-peroxide intermediate (Int1, Fig. 4) was generated. Int1 is a doublet, in which the spin density on Cu and O1 are 0.58 and 0.18, respectively. Int1 is 32.3 kcal mol−1 lower than the TS1. In Int1, the O1–O2 and Cu–O1 distances are 1.44 and 1.96 Å, respectively.
The alternative situation for the O–O bond formation via the WNA mechanism while the water molecule as a proton acceptor was also investigated. The transition state (TS', Fig. 5) for this pathway prefers to be a doublet. At TS', the O1–O2′ has a distance of 1.97 Å. The leaving proton is at a distance of 1.02 Å from the O2′, while it is 1.56 Å far away from the proton acceptor oxygen (O3′). TS' was confirmed to be a true transition state with only an imaginary frequency of 755.42i cm−1 that corresponds to the proton transfer to the second water and the O–O bond formation concertedly. TS' lies at +5.0 kcal mol−1 above the TS1 (Fig. 3). Therefore, this pathway is less favorable in the working conditions, which emphasizes that the buffer anion HPO42− ion plays a very important role for the water oxidation. The pronounced role of HPO42− ion is consistent with the experimental study that the kcat value is linear with the concentration of HPO42−.43 The deuterium kinetic isotope effect (kcatH2O/kcatD2O) for the WNA process assisted by was HPO42− calculated. And the calculation gives a KIE value of 4.3, which is consistent with the value of 2.1 from the KIE experiment.43 This also supports the proposed mechanism. For the O–O bond formation process, the present calculations suggest a step-wise WNA scenario where the H atom released from the attacking water firstly, then the O–O bond was formed via the direct coupling of the OH radical and Cu–O˙. This is different with a pathway in Fig. 1 where water nucleophilic attack occurs in a concerted way. HPO42− assisted the O–O bond formation via WNA mechanism was also suggested by reported Ni, Co and Ru-based WOCs both by experimental and computational research, where the role of buffer hydrogen phosphate anion was proven to accept the proton during the O–O bond formation process via WNA mechanism.27,54–56 Experimental studies like 18O isotope labeling and radical indicator experiment or Ampliflu red titrant experiment for OH˙ experiment for testing OH˙ are useful to verify our proposed mechanism.
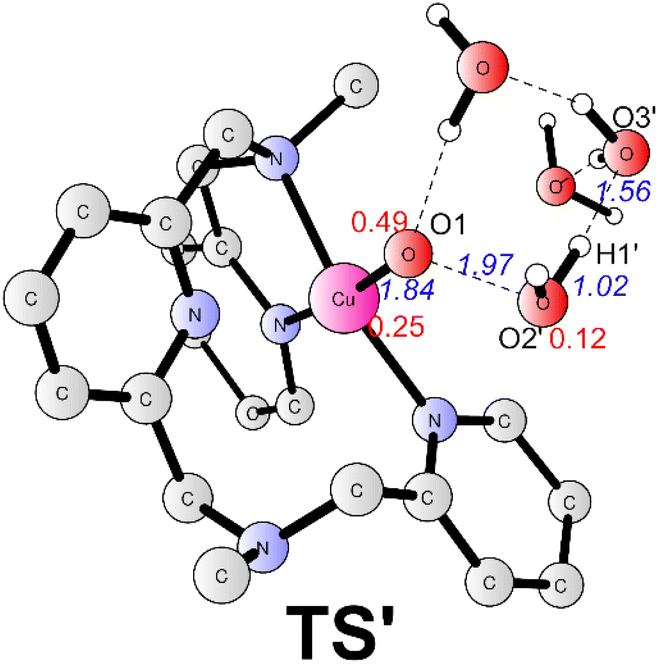 | ||
| Fig. 5 Optimized structures of TS' (doublet). Selected distances are shown in Angstrom in blue italic. Spin densities on selected atoms are presented in red. For clarity, non-important hydrogen atoms are not shown. The imaginary frequency of TS’ is 755.42i cm−1. | ||
Subsequently, a PCET of Int1 affords to the triplet Int2 (Fig. 6). The singlet is 2.1 kcal mol−1 higher than the ground state. In Int2, the super-oxide group coordinates with the CuII center, which was suggested by the spin densities on the two oxygen atoms (0.67 on O1 and 0.70 on O2). The O1–O2 and Cu–O1 distances are 1.28 and 2.05 Å, respectively. This step has a redox potential of −0.02 V. Finally, another one-electron oxidation process with a redox potential of 0.18 V prompts the generation of the triplet dioxygen that is loosely connected to the copper center (Int3, Fig. 6). In Int3, the spin densities on the two oxygen atoms are 0.99 and 1.00, respectively. The Cu–O1 distance is 2.93 Å. The triplet dioxygen can easily release from the Int3 and then the second water coordination with the copper center regenerate 2. 2 can reenter the next catalytic cycle to catalyze water oxidation reaction.
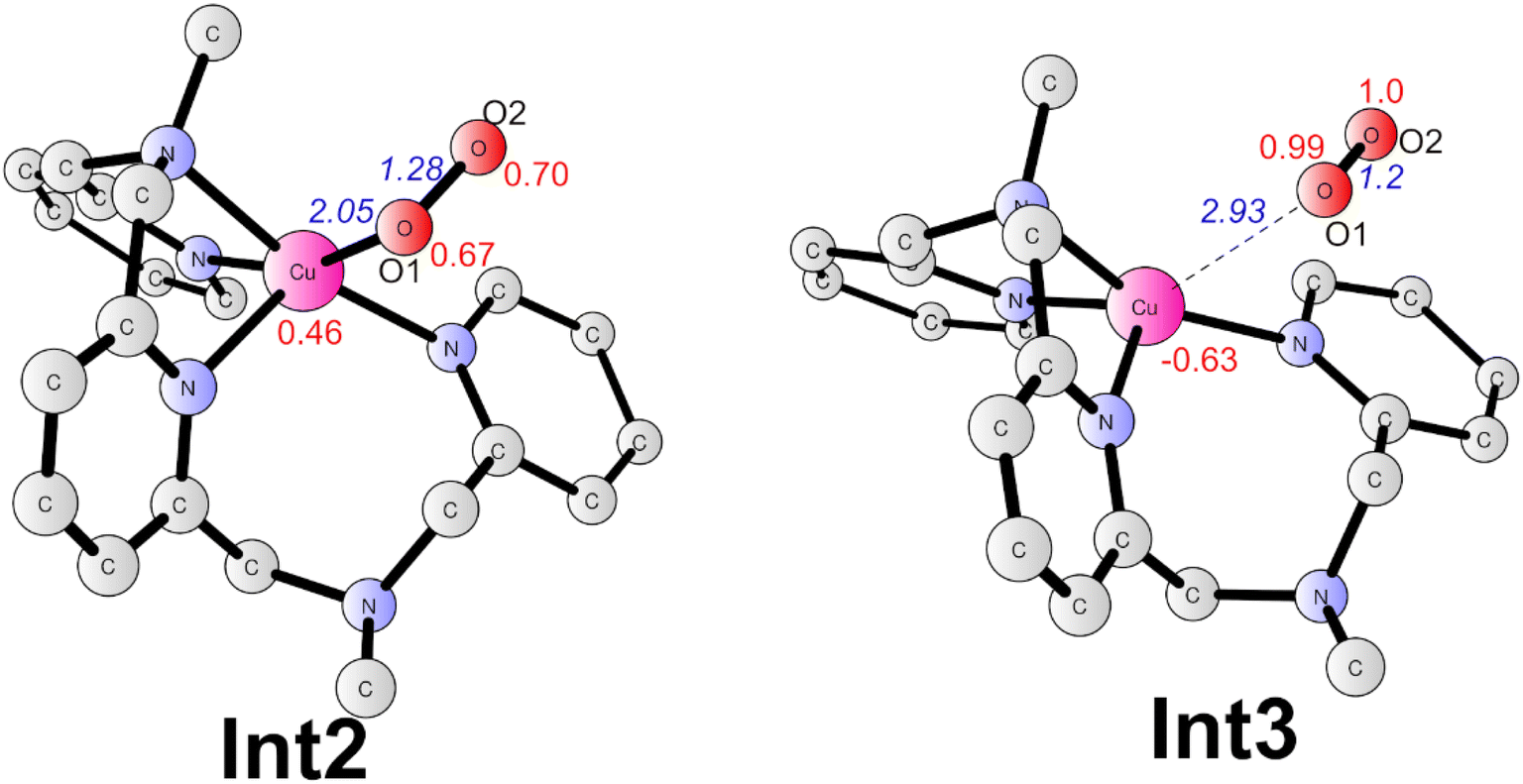 | ||
| Fig. 6 Optimized structures of Int2 (triplet) and Int3 (doublet). Selected distances are shown in Angstrom in blue italic. Spin densities on selected atoms are presented in red. For clarity, non-important hydrogen atoms are not shown. | ||
Single-point calculations with B3LYP*-D3 (15% Hartree–Fock exchange),57 PBE0-D3,58 and M06-D3,59 were also carried out to illustrate how sensitive the calculated redox potentials and barriers are in terms of the choice of the different density functionals. The results are shown in Table 1. The Gibbs energy diagrams for catalytic cycle are shown Fig. S3–S5.† For all functionals, calculated oxidation potentials for 2 (CuII–OH2) to 3 (˙L–CuII–OH) and 3 to 4 (˙L–CuIII–O˙) are around 1.0 V and 1.6 V, respectively. Importantly, the HPO42− assisted WNA mechanism is energy favorable, which suggest the important role of buffer HPO42− ion. While for the B3LYP*-D3 functional, these two pathways have quantitatively similar barriers.
Conclusion
In the present work, density functional calculations were employed to investigate the mechanism of water oxidation catalyzed by a mononuclear copper water oxidation catalyst. As shown in Fig. 7, the catalytic cycle starts after the coordination of a water molecule with the copper center, which leads to the generation of 2 (CuII–OH2). After two PCET processes, the active species (L˙-CuII–O˙, 4) was generated. The first oxidation process mainly occurs on the ligand, while the second electron is released from the OH moiety. So the redox non-innocent role of the ligand is important for the catalytic process. 4 triggers the O–O bond formation via the water nucleophilic attack mechanism, during which the HPO42− works as the proton acceptor. The WNA first undergoes a PCET process, during which one of the protons of attacking water transfers to the phosphate anion coupled with an electron transfer. Then, the O–O bond forms via the direct coupling between the oxo radical and the generated OH radical. This phosphate-assisted WNA mechanism has a total barrier of 19.5 kcal mol−1. The alternative pathway of solvent water accepting the H+ to afford the O–O bond formation was calculated to be energy unfavorable. According to DFT calculations, the strong ability of HPO42− to abstract a proton from the attacking water to prompt the O–O bond formation was emphasized. The WNA process leads to the hydro-peroxide intermediate (Int1), from which the triplet di-oxygen can be released after two oxidation processes.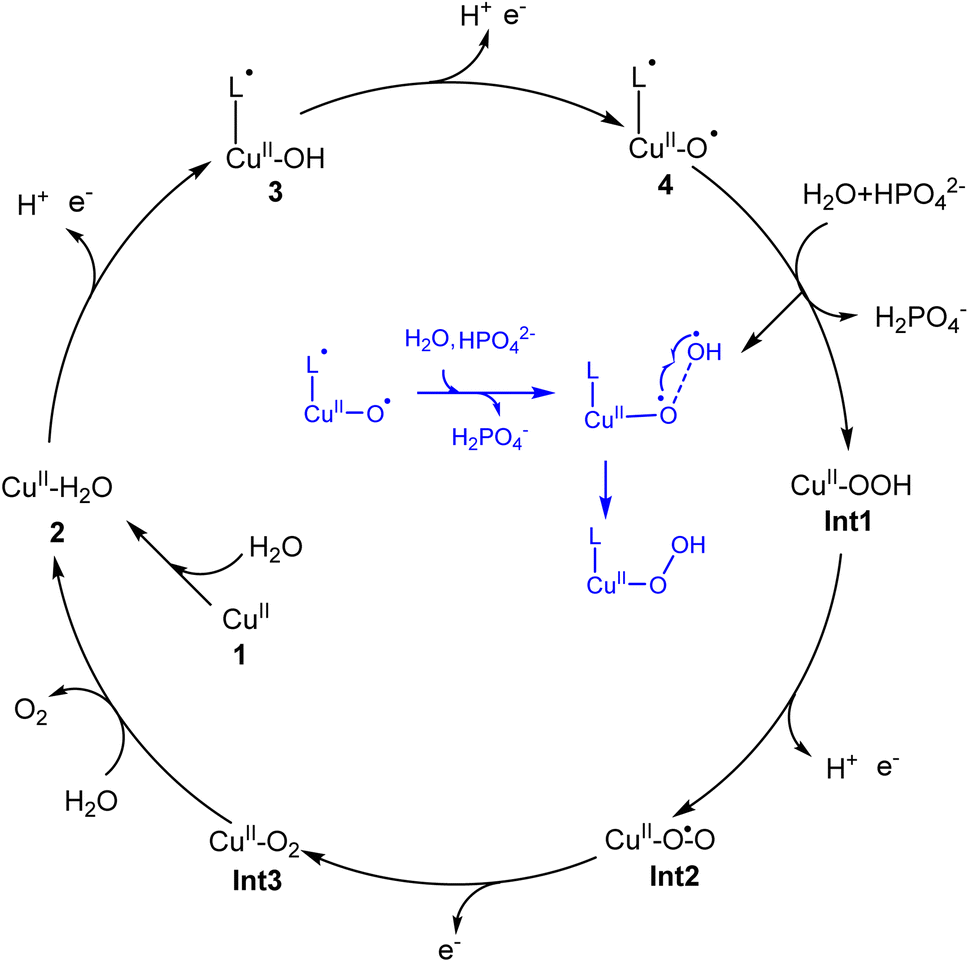 | ||
| Fig. 7 Proposed catalytic cycle for water oxidation catalyzed by the copper complex. Inset: The O–O bond formation process is described. | ||
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Key Scientific Research Projects of Colleges and Universities in Henan Province (No. 23A150041 and No. 21B150021) and College Students' innovation and entrepreneurship training program of Zhengzhou local university (ZDC2022003) and Zhengzhou Normal University (DCY2021046). Henan Provincial Scientific and Technological Projects (No. 222102320190). Authors thank Prof. Rong-Zhen Liao for valuable discussions.References
- L. H. Zhang, S. Mathew, J. Hessels, J. N. H. Reek and F. Yu, ChemSusChem, 2021, 14, 234–250 CrossRef CAS PubMed.
- J. Li, C. A. Triana, W. Wan, D. P. Adiyeri Saseendran, Y. Zhao, S. E. Balaghi, S. Heidari and G. R. Patzke, Chem. Soc. Rev., 2021, 50, 2444–2485 RSC.
- R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Suriñach and A. Llobet, Nat. Rev. Chem., 2019, 3, 331–341 CrossRef CAS.
- N. Noll, A.-M. Krause, F. Beuerle and F. Würthner, Nat. Catal., 2022, 5, 867–877 CrossRef CAS.
- J. Yang, L. Wang, S. Zhan, H. Zou, H. Chen, M. S. G. Ahlquist, L. Duan and L. Sun, Nat. Commun., 2021, 12, 373 CrossRef CAS PubMed.
- N. Vereshchuk, R. Matheu, J. Benet-Buchholz, M. Pipelier, J. Lebreton, D. Dubreuil, A. Tessier, C. Gimbert-Surinach, M. Z. Ertem and A. Llobet, J. Am. Chem. Soc., 2020, 142, 5068–5077 CrossRef CAS PubMed.
- D. W. Shaffer, Y. Xie, D. J. Szalda and J. J. Concepcion, J. Am. Chem. Soc., 2017, 139, 15347–15355 CrossRef CAS PubMed.
- L. Tong and R. P. Thummel, Chem. Sci., 2016, 7, 6591–6603 RSC.
- A. Kundu, S. K. Barman and S. Mandal, Inorg. Chem., 2022, 61, 1426–1437 CrossRef CAS.
- A. Bucci, G. Menendez Rodriguez, G. Bellachioma, C. Zuccaccia, A. Poater, L. Cavallo and A. Macchioni, ACS Catal., 2016, 6, 4559–4563 CrossRef CAS.
- G. Ruan, N. Fridman and G. Maayan, Chem. – Eur. J., 2022, 66, 1–6 Search PubMed.
- G. Ruan, P. Ghosh, N. Fridman and G. Maayan, J. Am. Chem. Soc., 2021, 143, 10614–10623 CrossRef CAS PubMed.
- Y. Liu, Y. Han, Z. Zhang, W. Zhang, W. Lai, Y. Wang and R. Cao, Chem. Sci., 2019, 10, 2613–2622 RSC.
- X. Jiang, J. Li, B. Yang, X. Z. Wei, B. W. Dong, Y. Kao, M. Y. Huang, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed. Engl., 2018, 57, 7850–7854 CrossRef CAS PubMed.
- K. J. Fisher, K. L. Materna, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2017, 7, 3384–3387 CrossRef CAS.
- T. Zhang, C. Wang, S. Liu, J.-L. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136, 273–281 CrossRef CAS PubMed.
- M. K. Coggins, M. T. Zhang, Z. Chen, N. Song and T. J. Meyer, Angew. Chem., Int. Ed., 2014, 53, 12226–12230 CrossRef CAS PubMed.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS.
- F. Yu, F. Li, J. Hu, L. Bai, Y. Zhu and L. Sun, Chem. Commun., 2016, 52, 10377–10380 RSC.
- P. E. M. Siegbahn, J. Phys. Chem. A, 2020, 124, 5849–5855 CrossRef CAS.
- Y. Y. Li, E. J. Meijer and R. Z. Liao, ChemCatChem, 2021, 13, 4251–4259 CrossRef CAS.
- M. Schilling and S. Luber, Front. Chem., 2018, 6, 1–21 CrossRef.
- P. Wu, S. Yan, W. Fang and B. Wang, ChemSusChem, 2022, e202102508 CAS.
- I. Funes-Ardoiz, P. Garrido-Barros, A. Llobet and F. Maseras, ACS Catal., 2017, 7, 1712–1719 CrossRef CAS.
- Y. Kurashige, M. Saitow, J. Chalupsky and T. Yanai, Phys. Chem. Chem. Phys., 2014, 16, 11988–11999 RSC.
- R.-Z. Liao and P. E. M. Siegbahn, ChemSusChem, 2017, 10, 4236–4263 CrossRef CAS PubMed.
- Y.-Y. Li and R.-Z. Liao, Chin. Chem. Lett., 2022, 33, 358–361 CrossRef CAS.
- S. Zhan, J. A. De Gracia Trivino and M. S. G. Ahlquist, J. Am. Chem. Soc., 2019, 141, 10247–10252 CrossRef CAS PubMed.
- S. Hu, P. Xu, R. X. Xu and X. Zheng, Inorg. Chem., 2021, 60, 7297–7305 CrossRef CAS PubMed.
- M. Sutradhar, A. J. L. Pombeiro and J. A. L. da Silva, Coord. Chem. Rev., 2021, 439, 213911 CrossRef CAS.
- P. Xu, S. Hu, H. D. Zhang and X. Zheng, Phys. Chem. Chem. Phys., 2018, 20, 14919–14926 RSC.
- J. M. de Ruiter and F. Buda, Phys. Chem. Chem. Phys., 2017, 19, 4208–4215 RSC.
- A. V. Marenich, J. Ho, M. L. Coote, C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2014, 16, 15068–15106 RSC.
- J. A. de Gracia Triviño and M. S. G. Ahlquist, ACS Catal., 2023, 13, 1270–1279 CrossRef.
- F. Chen, N. Wang, H. Lei, D. Guo, H. Liu, Z. Zhang, W. Zhang, W. Lai and R. Cao, Inorg. Chem., 2017, 56, 13368–13375 CrossRef CAS PubMed.
- Q. F. Chen, Z. Y. Cheng, R. Z. Liao and M. T. Zhang, J. Am. Chem. Soc., 2021, 143, 19761–19768 CrossRef CAS PubMed.
- S. J. Koepke, K. M. Light, P. E. VanNatta, K. M. Wiley and M. T. Kieber-Emmons, J. Am. Chem. Soc., 2017, 139, 8586–8600 CrossRef CAS PubMed.
- X. J. Su, M. Gao, L. Jiao, R. Z. Liao, P. E. Siegbahn, J. P. Cheng and M. T. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4909–4914 CrossRef CAS PubMed.
- Q.-Y. Mao, Y.-J. Pang, X.-C. Li, G.-J. Chen and H.-W. Tan, ACS Catal., 2019, 9, 8798–8809 CrossRef CAS.
- X.-F. Su, W. Guan, L.-K. Yan and Z.-M. Su, J. Catal., 2020, 381, 402–407 CrossRef CAS.
- P. Garrido-Barros, I. Funes-Ardoiz, S. Drouet, J. Benet-Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758–6761 CrossRef CAS PubMed.
- M. Gil-Sepulcre, P. Garrido-Barros, O. Jan, I. Funes-Ardoiz, R. Bofill, X. Sala, B.-B. Jordi and A. Llobet, Angew. Chem., Int. Ed., 2021, 60, 18639–18644 CrossRef CAS PubMed.
- Z. Xu, Z. Zheng, Q. Chen, J. Wang, K. Yu, X. Xia, J. Shen and Q. Zhang, Dalton Trans., 2021, 50, 10888–10895 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2009, 5, 2447–2464 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. A. Isse and A. Gennaro, J. Phys. Chem. B, 2010, 114, 7894–7899 CrossRef CAS PubMed.
- D. M. Camaioni and C. A. Schwerdtfeger, J. Phys. Chem. A, 2005, 109, 10795–10797 CrossRef CAS PubMed.
- N. Wang, H. Zheng, W. Zhang and R. Cao, Chin. J. Catal., 2018, 39, 228–244 CrossRef CAS.
- K. R. Gorantla and B. S. Mallik, J. Phys. Chem. A, 2021, 125, 6461–6473 CrossRef CAS PubMed.
- J. Shen, M. Wang, P. Zhang, J. Jianga and L. Sun, Chem. Commun., 2017, 53, 4374–4377 RSC.
- J. Shi, Y. H. Guo, F. Xie, Q. F. Chen and M. T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 4000–4008 CrossRef CAS PubMed.
- L. Wang, L. Duan, R. B. Ambre, Q. Daniel, H. Chen, J. Sun, B. Das, A. Thapper, J. Uhlig, P. Diner and L. Sun, J. Catal., 2016, 335, 72–78 CrossRef CAS.
- H. Pan, L. Duan and R. Z. Liao, ChemCatChem, 2019, 12, 219–226 CrossRef.
- M. Reiher, O. Salomon and B. A. Hess, Theor. Chem. Acc., 2001, 107, 7894–7899 Search PubMed.
- C. Adam and V. Barone, J. Chem. Phys., 1999, 130, 6158–6170 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00648d |
| This journal is © The Royal Society of Chemistry 2023 |