
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent analytical methodologies for the determination of anti-covid-19 drug therapies in various matrices: a critical review
Hadeel A. Khalil,
Nermeen A. Hassanein and
Amira F. El-Yazbi *
*
Department of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, University of Alexandria, P.O.Box: 21521, El-Messalah, Alexandria 21521, Egypt. E-mail: amira.elyazbi@alexu.edu.eg
First published on 28th April 2023
Abstract
Since the discovery of the first case infected with severe acute respiratory syndrome coronavirus-2 (SARS CoV-2) in Wuhan, China in December 2019, it has turned into a global pandemic. According to the World Health Organization (WHO) statistics, about 603.7 million confirmed coronavirus cases and 6.4 million deaths have been reported. Remdesivir (RMD) was the first U.S. Food and Drug Administration (FDA) approved antiviral drug for the treatment of coronavirus in pediatrics and adults with different disease severities, ranging from mild to severe, in both hospitalized and non-hospitalized patients. Various drug regimens are used in Covid-19 treatment, all of which rely on the use of antiviral agents including ritonavir (RTN)/nirmatrelvir (NTV) combination, molnupiravir (MLP) and favipiravir (FVP). Optimizing analytical methods for the selective and sensitive quantification of the above-mentioned drugs in pharmaceutical dosage forms and biological matrices is a must in the current pandemic. Several analytical techniques were reported for estimation of antivirals used in Covid-19 therapy. Chromatographic methods include Thin Layer Chromatography (TLC) densitometry, High Performance Thin Layer Chromatography (HPTLC), Reversed Phase-High Performance Liquid Chromatography (RP-HPLC), High Performance Liquid Chromatography Tandem Mass Spectrometry (HPLC-MS/MS) or Ultraviolet detectors (HPLC-UV), Ultra High-Performance Liquid Chromatography (UHPLC-MS/MS) or (UPLC-UV) and Micellar Liquid Chromatography (MLC). In addition to other spectroscopic methods including Paper Spray Mass Spectrometry (PS-MS), UV-Visible Spectrophotometry, and Spectrofluorimetry. Herein, we will focus on the clarification of trendy, simple, rapid, accurate, precise, sensitive, selective, and eco-friendly analytical methods used for the analysis of anti-Covid-19 drugs in dosage forms as well as biological matrices.
1. Introduction
In December 2019, China was invaded by the highly contagious severe acute respiratory syndrome coronavirus-2 (SARS CoV-2), declaring the beginning of one of the most vicious pandemics that has infested the whole world, Covid-19. It only took less than 100 days for Covid-19 to be defined as a global pandemic by the world health organization (WHO).1 Up to 7 August 2022, WHO reported about 603.7 million confirmed Covid-19 cases including about 6.4 million deaths.2SARS CoV-2 is an enveloped single-stranded RNA virus that enters the cell via the binding of its glycosylated S proteins and the host human receptors called angiotensin-converting enzyme 2 (ACE-2).3,4 ACE-2 receptors are vastly distributed in the body with varying intensities.5 Various disease severities were observed as asymptomatic ranging from mild (involving high temperature, headache, dry cough, and sore throat),6 moderate to severe involving pneumonia and multi-organ damage.7 Even after the acute phase of the disease had been cleared, many cases had been diagnosed with long Covid-19 syndrome which persists for several weeks or months leading to complications and disabilities.8
Unfortunately, despite the continuous efforts invested in the development of several anti-Covid 19 vaccines, their effectiveness is counteracted by the tendency of SARS-CoV-2 to change its amino acids leading to mutations.9 Scientists focused on repurposing already approved drugs for the treatment of other viral infections, since developing a novel, safe and effective drug is a tedious and time-consuming process. Several drugs have been used for Covid-19 since its spread, however, few are currently used, most of which belong to antivirals, namely, remdesivir (RMD), favipiravir (FVP), molnupiravir (MLP), ritonavir (RTN), and nirmatrelvir (NTV). Studies conducted on the proposed drugs since the beginning of the outbreak up till now are demonstrated in (Fig. 1).
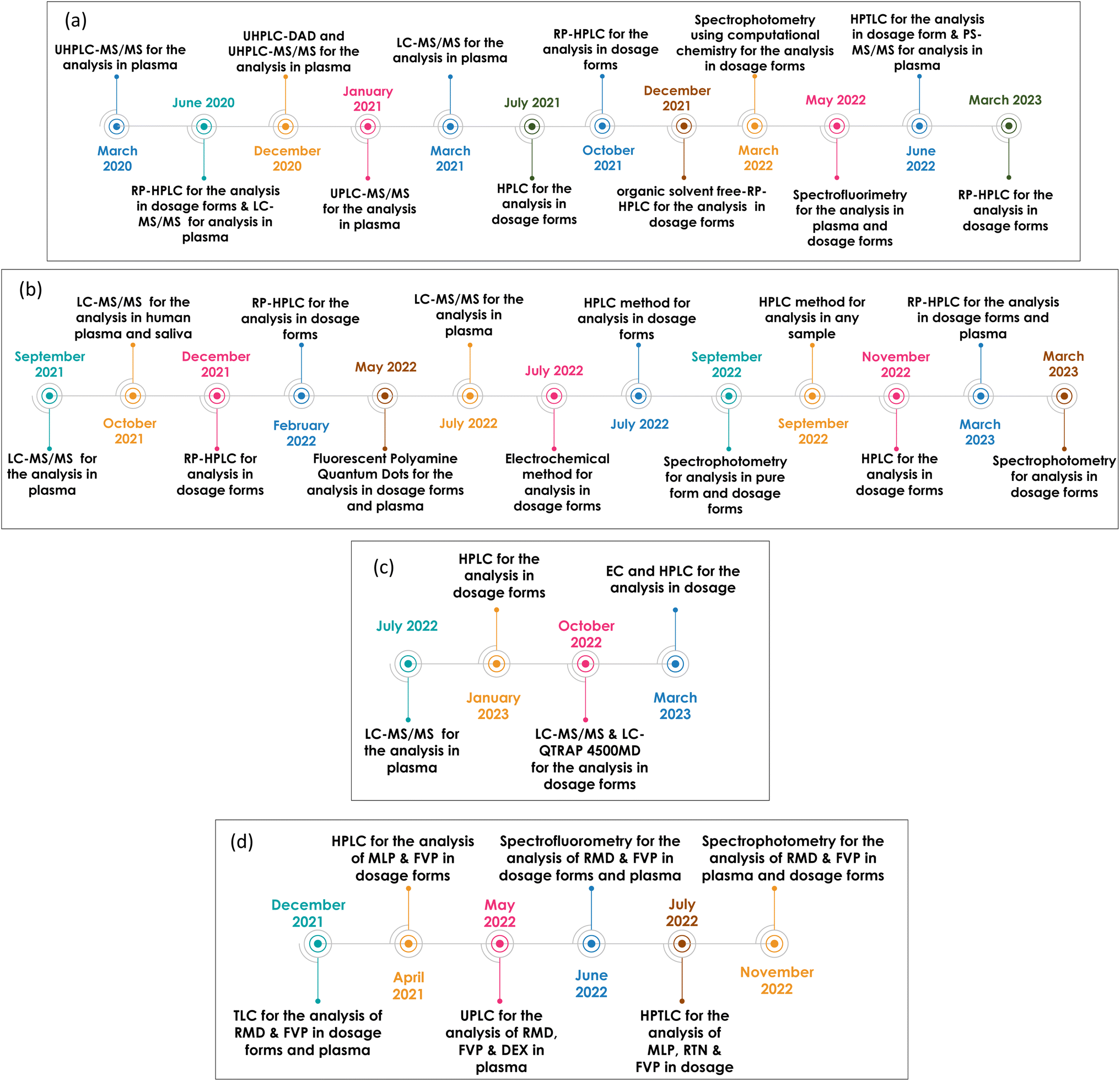 | ||
| Fig. 1 Recent studies conducted on (a) RMD, (b) MLP, (c) NTV & RTN and (d) various antiviral drug combinations, since the beginning of the outbreak up till now. | ||
Remdesivir (RMD), 2-ethylbutyl (2S)-2-[[[(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxyoxolan-2-yl]methoxyphenoxyphosphoryl]amino]propanoate] (Fig. 2) is a broad-spectrum adenosine analog prodrug.10 It is activated intracellularly into the pharmacologically active metabolite (GS-443902). RMD is an RNA-dependent RNA polymerase inhibitor (RdRpI). RMD's binding to the viral RNA leads to the permanent inhibition of viral genome replication.11 The mechanism of its action is demonstrated in Fig. 3. It is the first antiviral drug to be approved by the Food and Drug Administration (FDA) for the treatment of confirmed Covid-19 cases after the positivity of two successive tests. It was used in the treatment of Covid-19 cases ranging from 28 days-old pediatric patients (≥3 Kg) to adults in all phases of the disease.12
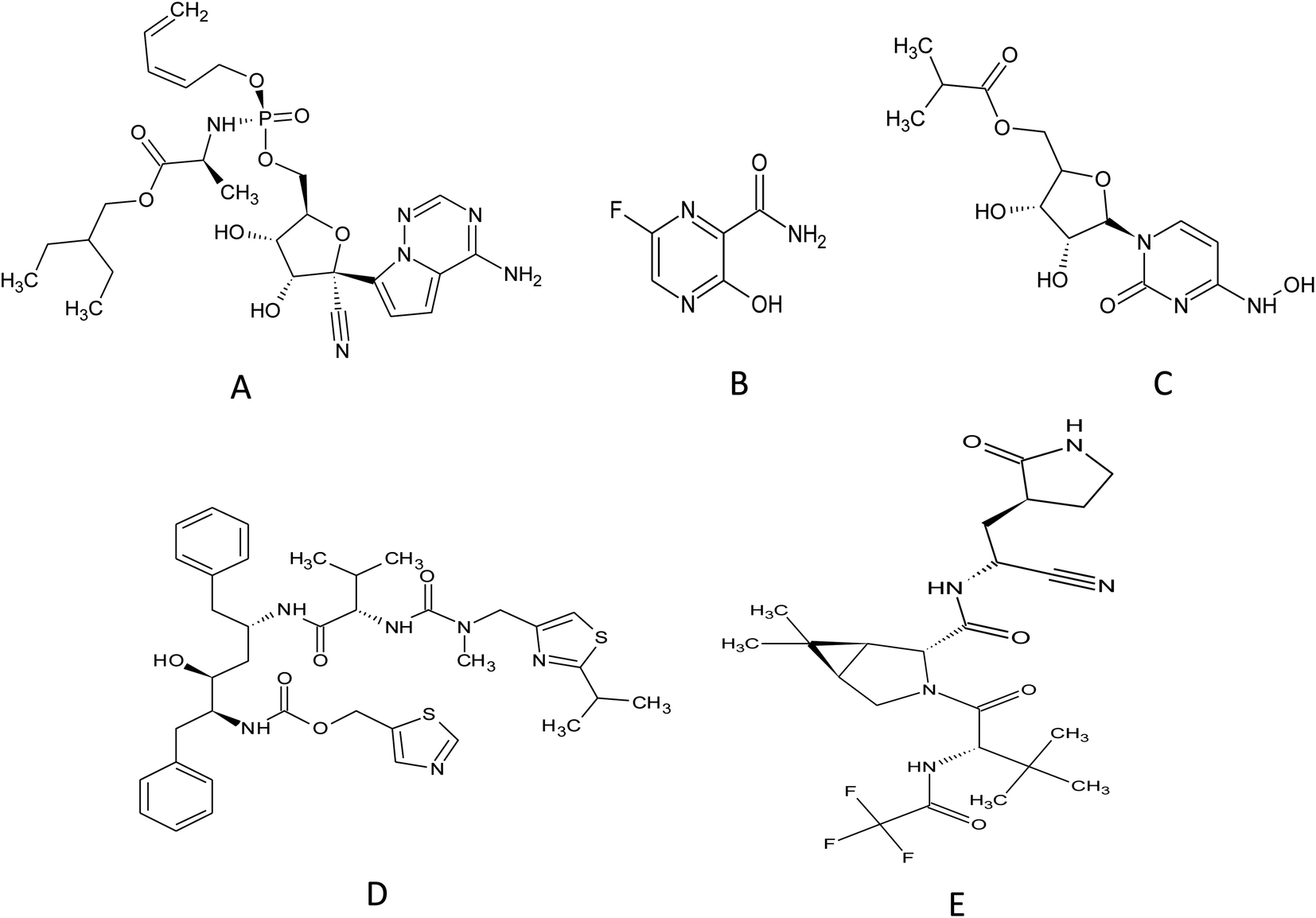 | ||
| Fig. 2 Chemical structure of (A) remdesivir, (B) favipiravir, (C) molnupiravir, (D) ritonavir and (E) nirmatrelvir. | ||
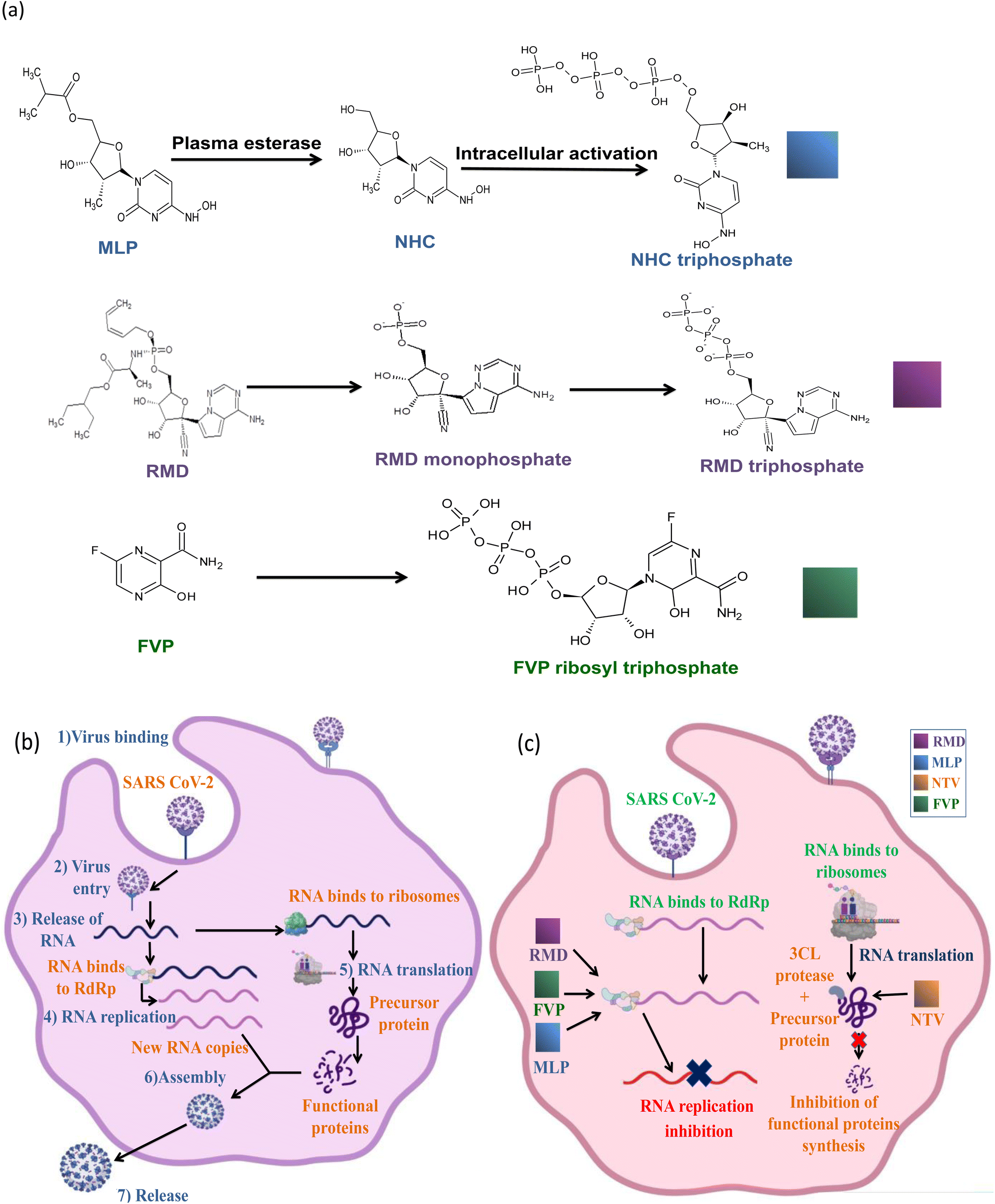 | ||
| Fig. 3 Inhibition mechanisms of SARS-COV-2 by the selected drugs, (a) the mechanism of intracellular activation of MLP, RMD and FVP, (b) schematic diagram demonstrating the binding, viral cell entry, replication and spreading of SARS-COV-2 in the host cell and (c) schematic diagram presenting the effect of each drug to stop SARS-COV-2 replication. | ||
Originally, Favipiravir (FVP), 5-fluoro-2-oxo-1H-pyrazine-3-carboxamide (Fig. 2) was developed for the treatment of the resistant cases of influenza by Toyama Chemical Co., Ltd, London in Japan. Favipiravir (FVP) is a modified pyrazine analog prodrug which is currently under investigation for the treatment of Covid-19. After entering the cell, it is activated by ribosylation and phosphorylation into FVP-ribose triphosphate (FVP-RTP). The activated form acts as an RNA-dependent RNA polymerase inhibitor (RdRpI)13 and leads to the permanent inhibition of viral genome replication (Fig. 3).
Owing to the absence of sufficient safety data, FDA has categorized molnupiravir (MLP), ((2R,3S,4R,5R)-3,4-dihydroxy-5-(4-(hydroxyamino)-2-oxopyrimidin-1(2H)-yl) tetrahydrofuran-2-yl) methyl isobutyrate (Fig. 2), as an Emergency Use Aauthorization (EUA) medication during the current pandemic. MLP is the first orally administrated antiviral drug to be used in Covid-19 treatment. MLP is a pyrimidine ribonucleoside analog which is a prodrug of N-hydroxy cytidine (NHC). It is used in the early stages of the disease to reduce the risk of hospitalization.14 By intracellular phosphorylation, NHC is activated into NHC triphosphate. NHC triphosphate displaces guanosine or adenosine through binding to viral RNA polymerase; leading to viral RNA mutagenesis disrupting the replication ability of SARS-CoV-2 (ref. 15) as demonstrated in (Fig. 3).
Similarly, a binary combination of Ritonavir (RTN), 1,3-thiazol-5-ylmethyl N-[(2S,3S,5S)-3-hydroxy-5-[[(2S)-3-methyl-2-[[methyl-[(2-propan-2-yl-1,3-thiazol-4-yl) methyl] carbamoyl] amino] butanoyl] amino-1,6-diphenylhexan-2-yl] carbamate and Nirmatrelvir (NTV), (1R,2S,5S)-N-[(1S)-1-cyano-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl]-3-[(2S)-3,3-dimethyl-2-[(2,2,2 trifluoroacetyl)amino]butanoyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide (Fig. 2) is employed as an Emergency Use Authorization (EUA). This combination is used to resolve mild to moderate Covid-19 cases within 5 days from symptoms appearance. It is available commercially as oral tablets consisting of 100 mg RTN and 150 mg NTV. NTV is a viral protease (3CLpro) inhibitor that prevents precursor protein cleavage; those proteins are used for new infectious particle synthesis,16 thus, ceases infection (Fig. 3). On the other hand, RTN is used as an inhibitor of CYP3A to help prolong the half-life of NTV and maintain its plasma levels, hence, NTV is considered as the anti-Covid-19 active component in such combination.17
Optimizing analytical methods capable of selectively and sensitively quantifying drugs used in the treatment of Covid-19 in pharmaceutical dosage forms and plasma samples is an essential step for the accurate determination of such drugs in various matrixes during this existing pandemic. In this review, we will focus on recently reported analytical methods employed in the assay of various medications that are currently approved for the treatment of Covid-19.
Nowadays, several concerns are being raised concerning the health hazards posed by the application of analytical procedures. Green analytical chemistry (GAC) protocols were developed as an alternative solution that minimizes the ecological burden without affecting the efficiency of those procedures. Various GAC metrics have been implemented to evaluate the greenness of analytical procedures, namely; National Environmental Method Index (NEMI), General Analytical Procedure Index (GAPI), Analytical Eco scale, and Analytical Greenness (AGREE).
The National Environmental Methods Index (NEMI) was first reported, and the results were represented in a simple pictogram, divided into four parts. Each part reflects a different rubric (generation of waste, reagents that are persistent, bio accumulative, or toxic, whether reagents are hazardous, whether the conditions are corrosive).18 These rubrics are evaluated in a binary way: if a value of a rubric is met, the designated part of the pictogram is colored in green; otherwise, it is kept uncolored.
The General Analytical Procedure Index (GAPI) consists of a five-pentagram figure that assesses the greenness of the procedure according to 15 parameters by displaying (red, yellow and green) representing high, medium to low impact, respectively.19 GAPI evaluates the analytical procedure thoroughly by assessing the sample preparation, reagents and solvents, instrumentation used with a circle in the middle that indicates if the method can be utilized for qualification or quantification purposes.
Another broad-spectrum tool is the Analytical Greenness (AGREE) which evaluates the analytical procedure according to the twelve principles of green analytical chemistry (GAC). AGREE can be simply estimated through an open access calculator.20 The results are displayed as a circular pictogram showing the score (zero to one) at the center and is divided into twelve sectors. The degree of agreement with the GAC principles is illustrated by giving a certain color to each one of the sectors.
The Analytical eco-scale is another metric system that depends on the quantity, possible safety and/or health hazards of the chemical reagents and the energy involved during application of the procedure under investigation.21–23 Analytical eco-scale is calculated by subtracting the total penalty points from 100.
2. Methods
2.1 Remdesivir (RMD)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :distilled water (55:45 v/v) at pH 4. A diode array detector DAD was set at 240 nm to detect RMD and a fluorescence detector at λex/λem 245/390 nm was used to detect the degradation products. The reached linearity ranges were as follows 0.1–15 μg mL−1 and 0.05–15 μg mL−1 for RMD and the degradation products, respectively. Additionally, the relatively low limit of detections (LOD) of 0.03 and 0.015 μg mL−1 for RMD and the degradation products, respectively confirms the high sensitivity of this method.24
:distilled water (55:45 v/v) at pH 4. A diode array detector DAD was set at 240 nm to detect RMD and a fluorescence detector at λex/λem 245/390 nm was used to detect the degradation products. The reached linearity ranges were as follows 0.1–15 μg mL−1 and 0.05–15 μg mL−1 for RMD and the degradation products, respectively. Additionally, the relatively low limit of detections (LOD) of 0.03 and 0.015 μg mL−1 for RMD and the degradation products, respectively confirms the high sensitivity of this method.24Akbel et al. have compared between two commonly used analytical techniques for the quantification of RMD. The first was a RP-HPLC method using a mobile phase composed of phosphate buffer and acetonitrile adjusted to pH 7. Detection of RMD was performed at 247 nm using UV-visible detector. On the other hand, the second technique employed UV-spectrophotometry at λmax 247 nm. RMD was dissolved in no solvents but deionized water. Good linearity was observed over the range of 10–60 μg mL−1, trueness was adequate as the mean recoveries were close to 100% and the correlation coefficients in both techniques were above 0.99. However, the superiority of the spectrophotometric method was displayed in the lower LOD obtained (2.4 μg mL−1) versus (3 μg mL−1) by the HPLC method and in its greenness as it involves the use of an eco-friendly diluent (water) and involves a higher through put of samples. On the opposite, the HPLC method consumes highly hazardous organic solvents such as methanol and acetonitrile in large volumes posing a greater environmental concerns.25
Sayed et al. have resolved the greenness issue by developing an organic solvent free RP-HPLC method for the analysis of RMD in the presence of forced degradation products. This time, the mobile phase composition was different as it was a mixed micellar mobile phase composed of Brij-35 (non-ionic polyoxyethylene surfactant), SLS (sodium lauryl sulfate is an anionic surface-active agent), and disodium hydrogen phosphate, which was then dissolved in water, and adjusted to pH 6 using phosphoric acid. Brij-35 and SLS were used to enhance the elution and maintain its efficiency. Samples injected on RP-C18 column, peaks were detected at 244 nm. RMD was exposed to different harsh conditions such as alkaline hydrolysis, acidic hydrolysis, oxidative degradation, and photolytic degradation. For alkaline hydrolysis, complete degradation was achieved at 25 °C for 3 h or at 45 °C for 1 h. As for acidic hydrolysis, degradation was carried out using 0.1 N HCl for 1 h at 40 or 50 °C, for oxidative degradation 4.5% H2O2 was used at 40 and 50 °C for 1 h and for photolytic degradation, the sample was exposed to UV-lamp at 365 nm for 1 day and no significant changes were observed. All the impurities resulting from the degradation showed no interference with the peak of RMD which confirmed the specificity of the method. The linearity was observed over the range of 5–100 μg mL−1, LOD was 0.5 μg mL−1, and LOQ was 2 μg mL−1. This HPLC method was found to be not only accurate and precise but has also succeeded in establishing a green eco-friendly system for the assay of RMD as displayed by the GAPI and AGREE systems (score of 0.77).26 It is worth mentioning that the disposal of surfactants in great quantities can be harmful to the environment, yet, considering the fact that a conventional LC system can generate around 0.5 L of organic waste daily, replacing large volumes of hazardous organic solvents with minimal volumes of surfactants may be the best available solution. Mixed-micellar mobile phases have demonstrated adequate performance at replacing hazardous organic solvents as it improves the elution power in RP stationary phases without the loss of their separation efficiency, involves much lower cost, toxicity and environmental harm and hence the use of minimal volumes of surfactants represents a beneficial compromise in comparison with other hazardous organic solvents.
A spectrophotometric technique employing computational chemistry for the quantification of RMD was developed by Ramzy et al. Computational chemistry is applied for predicting the results before performing the practical experiment, this reduces the required time and expenses by cutting down the number of possible trials. Theoretical studies were used to find the optimum acid dye that produces the highest calculated interaction energy with RMD using Gaussian 03 software with the density functional theory (DFT). The binding energy resulted from the interaction between RMD and numerous acid dyes were calculated with the following equation:
| ΔE = EA–B − EA − nEB |
Bromophenol blue (BPB) was the acid dye that fulfilled the required criteria. The yellow ion-pair complex formed by the interaction between RMD and BPB was quantified by measuring the absorbance using a UV-Visible Spectrophotometer at λ = 418 nm. Beer's law applied to concentrations from 2–12 μg mL−1. This technique represented great accuracy and precision illustrated in percentage recovery (99.0%) and low values of percentage RSD (0.752%).27
A simple and green HPTLC method for quantification of RMD and studying the stability profile in both bulk form and dosage forms was proposed by El-Kafrawy et al. Both powder and dosage form were dissolved in 50% aqueous ethanol. Degradation products were prepared by exposing the drug to different stress conditions such as acidic, basic, neutral, photolytic, and oxidative degradation. 5 μL of each sample and degradant were spotted on pre-activated TLC silica gel aluminum plates 60 F254 and the elution was completed using ethyl acetate and ethanol (96:4 v/v) then the plates were detected using a densitometer at 245 nm. The method has showed good linearity (6–100 μg mL−1) with low % RSD at this range, sensitivity (LOD was 1.67 μg mL−1 and LOQ was 5.55 μg mL−1), selectivity (no overlapping with the excipient or the degradation products), cost-effectiveness and greenness according to GAPI and AGREE metric systems (score of 0.77).28
Rawat et al. have developed a simple, rapid, selective and sensitive RP-HPLC method for estimation of RMD in intravenous marketed dosage forms. They used methanol for dissolving and diluting solutions, 10 μL of the solution were injected on Zorbax Eclipse plus C18 column (4.6 mm × 150 mm, 5.0 μm), it was isocratically eluted using acetonitrile and 0.1% formic acid in water (45:55, v/v) with a flow rate of 0.7 mL min−1. DAD was used at 245 nm for detection. The method was validated according to the ICH guidelines, linearity range was 2–100 μg mL−1, LOD was 0.57 μg mL−1, LOQ was 1.73 μg mL−1 and % RSD was 0.571, 0.579 for interday and intraday, respectively.29
Larabi et al. developed and validated a LC-MS/MS method for the quantification of RMD and GS-441524 in plasma. Plasma samples were obtained from Covid-19 treated patients after the administration of the loading dose. Sodium fluoride was added to increase the stability of RMD and GS-441524 in plasma. This was followed by the addition of methanol and ZnSO4 (protein precipitating agent) and RMD13C6 as the internal standard (IS). The components were detected using TSQ Endura triple-quadrupole MS adjusted at positive electrospray ionization mode multiple reaction monitoring mode was utilized for data collection. The linearity range of RMD was 1–5000 μg L−1 while for GS-441524 it was 5–2500 μg L−1. LODs of RMD and GS-441524 were 0.3 and 2 μg L−1, respectively.31
van Ingen et al. developed and applied a novel bioanalytical method for the quantification of RMD and its metabolites (GS-441524 and GS-704277) in human plasma. The samples were injected on an Acquity UPLC HSS T3 column, the separation was carried out using gradient elution in a total run time of about 3.4 min. Detection was performed after optimizing the parameters to be MRM for all compounds and negative ion mode for GS-704277 while positive ion mode for GS-5734 (RMD) and GS-441524 for better sensitivity using mass spectrometer. The linearity ranges were 4–4000, 2–2000, and 2–2000 ng mL−1 for RMD, GS-441524 and GS-704277, respectively. The proposed method was found to be accurate (% Er = 11.5%) and precise (% CV = 6.6%).32
Figg et al. have developed a simple, sensitive, and selective LC-MS/MS method for the quantification of RMD in human plasma using RMD-2H5 as an internal standard (IS). Electrospray ionization (ESI) positive ion mode and MRM were applied for detection and data collection. The method was validated according to FDA guidelines and has illustrated good linearity from 0.5 to 5000 ng mL−1. It is accurate, precise, sensitive, and selective (no interference with IS or the background matrix) and could be used clinically to perform therapeutic dosing and pharmacokinetic studies.33
Kirkpatrick et al. established the paper spray mass spectrometry (PS-MS/MS) method for the determination of RMD and its metabolite in plasma. The PS technique is based on sample ionization which allows direct analysis of complex biological samples without complicated sample preparation steps. Briefly, the plasma samples were spotted on the paper substrate containing pre-made plastic cassette plates (allows the automatic analysis of 240 samples), left to dry, followed by the addition of acetonitrile:water (90:10%) with 0.1% formic acid mixture as a spraying solvent for the generation of the analyte ions under high voltage (3–5 kV). The sharp end of the paper was closed to a mass spectrometer to permit the flow of the ions (gaseous phase) into a triple quadrupole mass spectrometer utilizing automated technology with a total run time of 1.2 min only. Acceptable linearity was observed from 20–5000 ng mL−1 for RMD and 100–25000 ng mL−1 for GS-441524. Compared to other reported LC-MS/MS methods,31,34,35 PS-MS/MS sensitivity should be enhanced due to relatively high values of LOD and LLOQ of RMD and GS-441524 (ref. 36).
Ponnusamy et al. have developed UHPLC-DAD and UHPLC-MS/MS for the rapid quantification of RMD in pharmacokinetic studies and therapeutic drug monitoring in human plasma. Sample extraction was done using vortex-assisted salt-induced liquid–liquid microextraction (VA-SI-LLME) technique. Analyte solutions were prepared by dissolving the drug in DMSO:MeOH (30:70 v/v). For VA-SI-LLME 2.0 mL of 0.1 N HCl was added to the sample solution, vortexed for 30 seconds, 2.5 g of (NH4)2SO4 and 500 μL of CAN were added, vortexed for 2 minutes, centrifuged at 6000 rpm for 5 min, supernatant layer was collected, simultaneous extraction and drying by using extraction solvent under nitrogen and finally re-dissolved in methanol. Five microliters of the analyte was injected on C18 column and isocratically eluted using 0.05% (v/v) formic acid in ultrapure water: 100% ACN 52:48% at a flow rate of 0.5 mL min−1 and was detected either using DAD at 254 nm or mass spectrometer at ESI positive ion mode and multiple reaction monitoring (MRM) mode. The method was validated according to FDA guidelines, the linearity ranges were 5–5000 ng mL−1 and 1–5000 ng mL−1 for UHPLC-DAD and UHPLC-MS/MS, respectively. LOD and LOQ were 1.5 and 0.3 ng mL−1 and 5 and 1 ng mL−1 for UHPLC-DAD and UHPLC-MS/MS, respectively. Extraction recoveries for UHPLC-DAD and UHPLC-MS/MS were ranging from 90.79 to 116.74% and 85.68 to 101.34%, respectively.37
Table 1 and 2 summarizes the measurement conditions of the various analytical methods applied in the quantification of RMD in pharmaceutical dosage forms and biological matrices, respectively.
| Reference | Instrument | Chromatographic conditions | Detection | Detection wavelength (nm) | Linearity range (μg mL−1) | LOD (μg mL−1) | LOQ (μg mL−1) | |
|---|---|---|---|---|---|---|---|---|
| Stationary phase | Mobile phase | |||||||
| a HPLC = LC (high performance liquid chromatography), RP-HPLC (reversed phase high performance liquid chromatography), UV-SPM (UV-spectrophotometer), HPTLC (high performance thin layer chromatography), SF (spectrofluorimetry), ACN (acetonitrile), H2O (water), FA (formic acid), DAD = PDA (diode array detector), UV-vis (UV-visible detector), Densito (densitometer), Brij-35 (non-ionic polyoxyethylene surfactant) and SLS (sodium lauryl sulfate is an anionic surface-active agent).b (i) indicates first method; (ii) indicates second method. | ||||||||
| 24 | HPLC | Agilent Zorbax Eclipse SB-C18 column (250 × 4.6 mm, 5 μm) | ACN:distilled H2O (55:45 v/v) pH 4 |
DAD | 240 | 0.1–15 | 0.03 | 0.1 |
| 25 | (i) RP-HPLC (ii) UV-SPM | Agilent Extend C18 (4.6 mm × 250 mm, 5.0 μm particle size | 20 mM potassium dihydrogen phosphate:ACN (50:50 v/v) pH 7 |
UV-vis | (i) 247 | (i) 10–60 | (i) 3 (ii) 2.4 | (i) 9 (ii) 7.3 |
| (ii) λmax = 247 | (ii) 10–60 | |||||||
| 26 | RP-HPLC | Core–shell RP-C18 column Kinetix® (5 μm, 150 × 4.6 mm) | 0.025 M Brij-35*, 0.1 M SLS** and 0.02 M disodium hydrogen phosphate | PDA | 244 | 5–100 | 0.5 | 2 |
| 27 | SPM | — | — | 418 | 2–12 | — | — | |
| 28 | HPTLC | Precoated TLC silica gel aluminum plates 60 F254 (20 × 10 cm, 200 μm thickness) | Ethyl acetate:Ethanol (96:4 v/v) |
Densito | 245 | 6–100 | 1.67 | 5.55 |
| 29 | RP-HPLC | Zorbax Eclipse plus C18 (150 × 4.6 mm, 5 μm) | Acetonitrile and 0.1% formic acid in water (45:55, v/v) |
DAD | 245 | 2 – 100 | 0.57 | 1.73 |
| 38 | SF | — | — | — | (i) λ ex = 410 | (i) 0.050–0.50 | (i) 0.007 (ii) 0.002 | (i) 0.022 (ii) 0.007 |
| (ii) λem = 241 | (ii) 0.010–0.350 | |||||||
| Reference | Matrix | Instrument | Chromatographic conditions | Detection | Detection wavelength (nm) | Linearity range (ng mL−1) | LOD (ng mL−1) | LOQ (ng mL−1) | |
|---|---|---|---|---|---|---|---|---|---|
| Stationary phase | Mobile phase | ||||||||
| a UHPLC (ultra-high performance liquid chromatography), MS (mass spectrometer), PS (paper spray), SF (spectrofluorimetry), ACN (acetonitrile), H2O (water), FA (formic acid).b (i) indicates first method; (ii) indicates second method. | |||||||||
| 30 | Plasma | UHPLC-MS/MS | Acquity® HSS T3 1.8 μm, 2.1 × 50 mm column | Phase A: H2O:FA 0.05% |
MS | — | — | 0.24 | LLOQ: 0.98 |
| Phase B: ACN:FA 0.05% |
ULOQ: 1000 | ||||||||
| 31 | Plasma | LC-MS/MS | Kinetex® 2.6 μm Polar C18 100A LC column (100 × 2.1 mm i.d.) | 10 mM sodium formate buffer in 0.1% FA and ACN starting from 0% of ACN to 100% in 2 min | MS | — | 1–5000 | 0.3 | 1.0 |
| 32 | Plasma | UHPLC-MS/MS | Acquity UPLC HSS T3 column T3 (50 × 2.1 mm, 1.8 μm) | Mobile phase A: 10 mM ammonium formate in 5% methanol, pH 2.5 | MS | — | 4–4000 | — | LLOQ: 4 |
| Mobile phase B: 100% methanol | |||||||||
| 33 | Plasma | LC-MS/MS | Synergi™ HPLC Fusion-RP (100 × 2 mm, 4 μm) column | 1% FA in H2O (v/v, aqueous) and 1% FA in ACN (v/v, organic) | MS | — | 0.5–5000 | — | LLOQ: 0.5 |
| 36 | Plasma | PS-MS/MS | — | — | MS | — | 20–5000 | 13 | LLOQ: 43 |
| 37 | Plasma | UHPLC | ACE C18 column (150 × 4.6 mm, 3 μm) | 0.05% (v/v) formic acid in ultrapure water:100% ACN 52:48% |
(i) DAD | (i) 254 | (i) 5–5000 | (i) 1.5 | (i) 5.0 |
| (ii) MS | (ii) — | (ii) 1–5000 | (ii) 0.3 | (ii) 1.0 | |||||
| 38 | SF | — | — | — | (i) λ ex = 410 | (i) 50–500 | (i) 7.31 (ii) 2.34 | (i) 22.15 (ii) 7.10 | |
| (ii) λem = 241 | (ii) 10–350 | ||||||||
2.2 Molnupiravir (MLP)
Repudi et al. have developed a simple and economic chromatographic analytical method for routine quantification of MLP in both bulk and pharmaceutical dosage form since it does not require complex pretreatment steps like purification. Additionally, an economic mobile phase and solvent composed of methanol:phosphate buffer 35:65% v/v was used. Samples were injected on octadecylsilica HPLC column with an injection volume of 10 μL and finally detected using UV detector at 236 nm. Validation of the method has confirmed its accuracy, precision, selectivity and sensitivity with a linearity range from 20 to 100 μg mL−1, % RSD was 0.248032 and LOD & LOQ were 2.6 and 6.35 μg mL−1, respectively.40
Ramzy et al. have developed an economic green computational spectrophotometric method for the quantification of MLP in bulk and pharmaceutical dosage form by using the computational technique using Gaussian 03 software to calculate the highest binding energy (calculated using ΔE = EA–B − EA – nEB, where A is the energy of optimized MLP and B is the optimized energy) between different phenolic acid dyes (α-naphthol, β-naphthol, 8-hydroxyquinoline, resorcinol, and phloroglucinol) and the drug to reduce both cost and time. The dye 8-hydroxyquinoline was found to be the most suitable dye when added to MLP in presence of 0.5 mL of hydrochloric acid (0.5 M) and 0.5 mL sodium nitrite (4%) (diazotization of MLP) followed by 0.5 mL sodium hydroxide (2 M) and diluted with ethanol as an eco-friendly solvent as they form red complex which is detected at 515 nm. According to the ICH guidelines the method was valid, accurate, precise and green with linearity range 1–12 μg mL−1, % recovery was 99.820, % RSD was 0.623 and analytical eco-scale score was 79.41
Hasan et al. have established a novel silver-nanoparticles spectrophotometric method for ecofriendly and cost-effective assay of MLP routinely in quality control and research laboratories and have successfully applied it in studying the dissolution rate of marketed preparations. This method was based on a redox reaction between MLP (reducing agent) and silver nitrate as an (oxidizing agent), both were heated in presence of polyvinylpyrrolidone (PVP) for stabilization and sodium hydroxide then the absorbance was measured at 416 nm. Moreover, electron microscopy was used for visualizing the silver-nanoparticles. According to the ICH guidelines the method was valid, linear (100–2000 ng mL−1), accurate (Er% was 0.89), precise (% RSD was 0.60) and eco-friendly (Analytical eco-scale total score was 95).42
Abbaraju et al. have developed a RP-HPLC method for the determination of MLP in bulk form and pharmaceutical dosage forms. 2 μL of the solution was injected on C18 column, eluted at 1.5 mL min−1 using ammonium phosphate monobasic and methanol in the ratio of 47:53% v/v then detected using diode array detector at 260 nm. The method was validated according to ICH guidelines and the method was linear in the range from 25 to 150 μg mL−1. Forced degradation studies were performed on the drug under different stress conditions such as acidic, alkaline, oxidative, thermal, and photolytic conditions and the peak of the drug was well-resolved from the degradation products. Values for LOD and LOQ were as low as 0.993 μg mL−1 and 0.3 μg mL−1, respectively.43
Praharsha et al. have developed an accurate, precise, robust, selective, and sensitive RP-HPLC method for the estimation of MLP in pharmaceutical dosage forms. The mobile phase was composed of orthophosphoric acid:acetonitrile in the ratio of 60:40. Ten microliters of the sample was injected on C18 column then eluted at 1.0 mL min−1 then detected using DAD at 253 nm. The method was validated according to ICH guidelines; the method was linear at 20–60 μg mL−1, sensitive LOD and LOQ were 0.06 μg mL−1 and 0.21 μg mL−1, respectively, robust (no changes in the results were observed when slight changes were altered in the chromatographic conditions), and selective as no interference was found from the different forced degradation products (acidic, basic, hydrogen peroxide, photolytic and dry heat degradation products).44
Attia et al. have developed a RP-HPLC method for the quantification of MLP in pharmaceutical dosage forms and was applied in quality control assay of the drug. The drug was dissolved using a diluent composed of ethanol:water (50:50, v/v%). Fifty microliters of the sample was injected on a C18 column and isocratically eluted in a mobile phase consisting of 20 mM phosphate buffer pH 2.5:acetonitrile (80:20, v/v%) flowing at 1.0 mL min−1 and detected at 230 nm using DAD. Comparison of the developed method with other reported methods39,45,46 was performed and no significant differences were found between them. This method was validated according to ICH guidelines, linearity was 0.2–80.0 μg mL−1, LOD was 0.04 μg mL−1 while LOQ was 0.12 μg mL−1 indicating high sensitivity. In addition, the method was robust as no interference was observed from the excipients and precise as % RSD was less than 2%.47
Degim et al. have developed a rapid RP-HPLC method for the quantification of MLP in different matrices (pharmaceutical dosage forms, biological or non-biological) and validated it according to the ICH guidelines, the method was linear between 0.010:0.150 mg mL−1, precise as % RSD was less than 2%, selective (no interference with the excipients of dosage forms) and robust (no significant changes in the results when there was a slight changes in temperature, flow rate etc). Ten microliters of the drug solution was injected on C18 column and isocratically eluted using 10 mM phosphate buffer (pH 7):Acetonitrile mixture (80:20) with a flow rate of 1 mL min−1 and finally detected using DAD at 230 nm.48
Nemutlu et al. have developed the first electrochemical method for the quantification of MLP in pharmaceutical dosage forms and applied it in the assay of marketed capsules. The method was based on electrochemical deposition of reduced graphene oxide (rGO) on a glassy carbon electrode (GCE) using cyclic voltammetry (CV). The drug was dissolved in deionized water and Britton–Robinson buffer (BR) adjusted at pH 9 to encourage the oxidation of MLP. Analysis was done at 0.2 V by square wave voltammetry (SWV) against the Ag/AgCl reference electrode and detection was achieved using electrochemical impedance spectroscopy and cyclic voltammetry. The method was validated according to ICH guidelines and was found to be linear (0.09–4.57 μM), sensitive (LOD and LOQ were 0.03 μM and 0.09 μM, respectively), accurate and precise (% RSD was less than 2%), and robust as slight changes in the conditions led to no changes in the obtained results.49
Walker et al. have developed a novel analytical LC-MS/MS method based on the quantification of MLP and its active metabolite NHC in human plasma and saliva and have successfully applied it in pharmacokinetic studies. For plasma samples, 13C15N2-EIDD-2801 and 13C15N2-N4-hydroxycytidine were used as internal standards (IS), whereas the IS used for saliva samples were 13C15N2-EIDD-2801 and 13C15N2-N4-hydroxycytidine. Detection of the compounds was achieved using a mass spectrometer at negative ion mode using selective reaction monitoring (SRM). The method was validated according to EMA and FDA guidelines, in both plasma and saliva matrices. Good linearity was obtained in the concentration ranges 2.5–5000 ng mL−1.45
Abdel-Megied et al. developed an LC-MS/MS method for the quantification of NHC in human plasma and applied the method in studying its pharmacokinetics in Egyptian volunteers. NHC and IS (ribavirin) were mixed with plasma and proteins were precipitated using acetonitrile. A mass spectrometer was used for the detection at positive ESI and MRM modes. The method was validated according to FDA guidelines with a linearity range from 20–10000 ng mL−1. The accuracy, precision, selectivity, and sensitivity of the established method were all confirmed.51
Tables 3 and 4 summarizes the measurement conditions of the various analytical methods applied in the quantification of MLP pharmaceutical dosage forms and biological matrices, respectively.
| Reference | Instrument | Chromatographic conditions | Detection | Detection wavelength (nm) | Linearity range (μg mL−1) | LOD (μg mL−1) | LOQ (μg mL−1) | |
|---|---|---|---|---|---|---|---|---|
| Stationary & mobile phases | ||||||||
| 39 | RP-HPLC | Discovery® HS C18 column (75 × 4.6 mm, 3 μm) | ACN:H2O (20:80 v/v) |
DAD | 240 | 0.1–60.0 | 0.05 | 0.1 |
| 40 | RP-HPLC | Symmetry ODS C18 (150 × 4.6 mm, 5 μm) | Methanol:phosphate buffer (35:65% v/v) pH-4.2 adjusted with orthophosphoric acid |
UV | 236 | 20–100 | 2.6 | 6.35 |
| 41 | Spectrophotometry | — | — | UV-visible 1650 spectrophotometer | 515 | 1–12 | — | — |
| 42 | Spectrophotometry | — | — | UV-visible spectrophotometer and JEOL-1010 transmission electron microscope | 416 | 0.1–2 | 0.030 | 0.091 |
| 43 | HPLC | Waters 2695 using an Agilent Zorbax Eclipse C18 (250 mm × 4.6 mm × 5 μm) | Ammonium phosphate monobasic and methanol in the ratio of 47:53% v/v |
DAD | 260 | 25–150 | 0.3 | 0.993 |
| 44 | HPLC | Agilent C18 (250 mm × 4.6 mm, 3.6 μm) | Orthophosphoric acid:acetonitrile 60:40 |
DAD | 253 | 20–60 | 0.06 | 0.21 |
| 47 | HPLC | Inertsil C18 column (150.0 mm × 4.6 mm, 5.0 μm) | 20 mM phosphate buffer pH 2.5:acetonitrile (80:20, % v/v) |
DAD | 230 | 0.2–80.0 | 0.04 | 0.12 |
| 48 | HPLC | Phenomenex C18 column (150 × 4.6 mm, 3 μm) | 10 mM phosphate buffer pH 7/acetonitrile mixture (80:20) |
DAD | 230 | 10–150 | — | — |
| 49 | Cyclic voltammetry | — | — | Electrochemical impedance spectroscopy & cyclic voltammetry | — | 0.09–4.57 μM | 0.03 μM | 0.09 μM |
| 52 | Fluorescent polyamine quantum dots | — | — | SF | λem 504 nm after 10 min of excitation at λex 440 nm | 2–70 ng mL−1 | 0.58 ng mL−1 | 1.61 ng mL−1 |
| Reference | Matrix | Instrument | Chromatographic conditions | Detection | Detection wavelength | Linearity range (ng mL−1) | LOD (ng mL−1) | LOQ (ng mL−1) | |
|---|---|---|---|---|---|---|---|---|---|
| Stationary phases | Mobile phases | ||||||||
| a PBMC (peripheral blood mononuclear lysates). | |||||||||
| 45 | Plasma | LC-MS/MS | Waters C18 XBridge column (3.5 μm: 100 mm × 2.1 mm) | 1 mM ammonium acetate & 1 mM ammonium acetate in ACN | MS | — | 2.5–5000 | — | — |
| 50 | Plasma-PBMCa | LC-MS/MS | Zorbax Eclipse Plus C18 UPLC column, (50 mm × 2.1, 3.5 μm)-Scherzo SM-C18, column (50 mm × 3, 3 μm) | FA in H2O and FA in ACN-50 mM ammonium formate:5 mM ammonium hydroxide (MPA) and 80 mM ammonium formate:8 mM ammonium hydroxide in (80:20) H2O:ACN |
MS | — | 1–5000 | — | 1 ng mL−1–1 pmol per sample for NHC |
| 51 | Plasma | LC-MS/MS | Agilent Zorbax Eclipse plus C18, T3 (150 × 4.6 mm, 5 μm) | 0.2% acetic acid:methanol (5:95 v/v) |
MS | — | 20–10000 | — | 20.0 |
| 52 | Plasma | Fluorescent polyamine quantum dots | — | — | SF | λem 504 nm after 10 min of excitation at λex 440 nm | 2–70 | 0.58 | 1.78 |
2.3 Nirmatrelvir (NTV) and ritonavir (RTN)
Liu et al. have developed an LC-MS method for the simultaneous determination of NTV and RTN in plasma samples. Sample preparation involved protein precipitation and RMD was used as an internal standard. Mass detector was adjusted on positive polarity mode with ESI and detection parameters were then optimized. The method was linear over a range of 50–5000 ng mL−1 for NTV and 10–1000 ng mL−1 for RTN; with correlation coefficients greater than 0.99 and accurate with a % relative error Er of ±15%.17A green HPLC method was developed by Ramzy et al. for the determination of NTV and RTN combination in pharmaceutical dosage form however, the difference between the developed method and other methods that the stationary phase and mobile phases were selected using a computational software (Gauss-view software) that rely on quantum mechanics, molecular dynamics and semi empirical structure–properties relationships. Depending on the physical interaction between the drugs and the stationary phases the most appropriate stationary phase was C18 column (among C8, C18, Cyano column) and the most green and suitable mobile phase was ethanol:water (80:20, v/v) using the following equation.53
| ΔE = EA–B − EA − EB |
The drugs were dissolved in ethanol, injected on C18 column, and eluted isocratically using ethanol:water (80:20 v/v) with flow rate 1 mL min−1 and detected using DAD at 215 nm. The method was validated using ICH guidelines, the method was linear 1.0–20.0 μg mL−1, selective, accurate LOD was 0.20, 0.32 μg mL−1 while LOQ was 0.60, 0.96 μg mL−1 for NTV and RTN, respectively and green (according to analytical eco-scale, the green analytical procedure index and the AGREE evaluation method).
Aboras et al. have developed two white and novel analytical methods for the determination of NTV and RTN in pharmaceutical dosage forms and have successfully applied it in the quantification of Paxlovid®. Micellar Electrokinetic Chromatographic (MEKC) was the first method, they used 50 mM borate buffer at pH 9.2 20 with 25 mM sodium lauryl sulfate (SDS) as background electrolyte (BGE) and they injected hydrodynamically on a 21 deactivated fused silica capillary at 50 mbar pressure, for 17 s using a voltage of 30 kV. The drugs were dissolved in methanol, injected after the activation of silica wall with 0.1 M NaOH. Both drugs were detected at 210 nm using DAD detector. Second method was RP-HPLC, by 20 μL of the solution was injected on C18 column and eluted using 50 mM ammonium acetate buffer at pH 5 and acetonitrile isocratically at 1 mL min−1 flow rate then also detected using DAD at 210 nm. They have also studied the effect of various stress conditions such as acid, base, neutral, oxidative and photolytic degradation on the selectivity of the developed methods. Both methods were validated according to ICH guidelines; the two methods were linear between 10–200 μg mL−1 for NTV and 5–100 μg mL−1 RTN in both methods, white and green upon using Hexagon, AGREE and RGB12 techniques. The methods were found to be sensitive with detection limits reaching 0.83 and 1.95 μg mL−1 using MEKC and 2.22 and 1.23 μg mL−1 using HPLC for NTV and RTN, respectively, accuracy and precision were confirmed with RSD% and Er% less than 2%.54
Bode-Böger et al. have developed an accurate, selective, and sensitive (detecting concentrations at nanogram level) LC-MS/MS method for the determination of NTV and RTN in human plasma and have successfully applied it in therapeutic drug monitoring in patients who was hospitalized Covid-19 in a deteriorated condition. The drugs in methanol and D6-ritonavir was used as internal standard then human plasma was spiked, protein precipitation was performed using methanol and after processing and filtration 20 μL was injected on C18 column and eluted gradiently using 90% aqueous buffer (1 g ammoniumformate and 1 mL formic acid in 1 L water, pH 3.5) and 10% acetonitrile, after 1 min 30% buffer and 70% acetonitrile and after 9 min they used the first composition (90% buffer and 10% water). Detection was performed using the turbo-ion-spray source of the mass spectrometer without splitting after it was adjusted at the positive mode for both drugs. The method was validated according to ICH guidelines, it was linear between 10–10000 ng mL−1 and 2–2000 ng mL−1 for NTV and RTN, respectively, sensitive with LLOQ values of 10 ng mL−1 and 2 ng mL−1 for NTV and RTN, respectively, and selective since no interference was detected.55
Tables 5 and 6 summarize the measurement conditions of the various analytical methods applied in the quantification of NTV and RTN in pharmaceutical dosage forms and biological matrices, respectively.
| Reference | Instrument | Chromatographic conditions | Detection | Detection wavelength | Linearity range (μg mL−1) | LOD (μg mL−1) | LOQ (μg mL−1) | |
|---|---|---|---|---|---|---|---|---|
| Stationary phase | Mobile phase | |||||||
| a Micellar Electrokinetic Chromatographic (MEKC). | ||||||||
| 53 | HPLC | BDS Hypersil C18 column (250 × 4.6 mm, 5 μm particle size) | Ethanol:Water (80:20, v/v) |
DAD | 215 nm | 1.0–20.0 | 0.20 for NTV 0.32 for RTN | 0.60 for NTV 0.96 for RTN |
| UV-visible detector | ||||||||
| 54 | (i) MEKC | (i) Deactivated fused silica capillary (50 cm effective length × 50 μm id) | (i) 50 mM borate buffer at pH 9.2 with 25 mM sodium lauryl sulfate (SDS) | DAD | 210 nm | 10–200 for NTV | (i) 0.83 for NTV 1.95 for RTN | — |
| (ii) HPLC | 5–100 for RTN | (ii) 2.22 for NTV 1.23 for RTN | ||||||
| (ii) Zorbax-Eclipse C18 (4.6 × 250 mm, 5 μm particle size) column | (ii) 50 mM ammonium acetate buffer at pH 5 and acetonitrile | |||||||
| Reference | Matrix | Instrument | Chromatographic conditions | Detection | Linearity range (ng mL−1) | LOD (ng mL−1) | LOQ (ng mL−1) | |
|---|---|---|---|---|---|---|---|---|
| Stationary & mobile phases | ||||||||
| 17 | Plasma | LC-MS/MS | Thermo BDS Hypersil C18 column (4.6 × 100 mm, 2.4 μm) | Deionized H2O and methanol and both contained 0.1% (v/v) FA | MS | NTV: 50–5000 | — | NTV: 50 |
| RTN: 10–1000 | RTN: 10 | |||||||
| 55 | Plasma | LC-MS/MS | Zorbax XDB-C18 2.1 × 50 mm, partical size 3.5 μm | 90% aqueous buffer (1 g ammoniumformate and 1 mL formic acid in 1 L water, pH 3.5) and 10% acetonitrile for 1 min, then 30% buffer and 70% acetonitrile for 9 min, then 90% buffer and 10% water | QTRAP 4500MD mass spectrometer | NTV: 10–10000 | — | NTV: 10 |
| RTN: 2–2000 | RTN: 2 | |||||||
2.4 Drug combinations
Attia et al. developed a sensitive, simple TLC method and applied it for the simultaneous estimation of RMD and FVP in dosage forms and human plasma using TLC plates without any significant interference. Samples were spotted on normal phase TLC plates, then developed using a mobile phase composed of ethyl acetate–methanol–ammonia and visualized at 235 nm. The method was linear over a range of concentrations from 0.20–4.50 and 0.08–5.00 μg per band for RMD and FVP, respectively, sensitive with LOD of 0.04 and 0.12 ng per band for RMD and FVP, respectively. The greenness of this method was verified using the National Environmental Method Index (NEMI) and Eco scale metric systems with a score of 80.57
Eid et al. have successfully developed five simple and green spectrophotometric methods for the simultaneous determination of RMD and FVP combination in both human plasma and pharmaceutical dosage forms, namely (I) direct measurement (at λmax 244 and 323 nm), (II) derivative (RMD was second-ordered at 248 nm while FVP was first-ordered at 337 nm), (III) dual wavelength (RMD was determined at 207–244 nm but FVP was determined at 330–400 nm), (IV) ratio subtraction (272–340 nm for RMD and 222–335 nm for FVP), and (V) ratio derivative (both drugs were derivatized to first order and measured at 340 nm and 280 for FVP and RMD, respectively). All methods were validated according to the ICH guidelines and the linearity ranges were the same for the five methods for RMD it was 1–10 μg mL−1, while in the first two methods for FVP it was 1–20 μg mL−1 and for the rest of methods it was 2–20 μg/mL.58
:acetonitrile:water (15:35:50 v/v/v) adjusted at pH 4 in a run time of 3.65 min and detected at 240 nm. The linearity range for the three drugs was 0.1–10 μg mL−1. According to the FDA guidelines, the method proved be accurate, precise, selective, sensitive, and green (Eco-Scale score = 83).59Molnupiravir, favipiravir, and ritonavir in pure form and in pharmaceutical dosage forms: Ibrahim et al. have developed a simple HPTLC method for the quantification of the orally active antivirals MLP, RTN, and FVP which were used in the treatment regimens of Covid-19. Samples were spotted on Silica gel 60F254 thin layer chromatography plates and then developed using methylene chloride:ethyl acetate:methanol:25% ammonia (6:3:4:1,v/v/v/v) as a mobile phase and the chromatogram was visualized at 289 nm. The obtained linearity ranges for FVP and MLP were 3.75–100.00 μg mL−1 and 2.75–100.00 μg mL−1 for RTN. The method was proven to be sensitive (LOD = 1.12, 1.21, and 0.89 μg mL−1 for FVP, MLP, and RTN, respectively), robust, and inexpensive as it requires simple tools and recyclable reagents. Additionally, the greenness of the developed method was confirmed by the GAPI and AGREE (score of 0.62) metric systems.60
Tables 7 and 8 summarizes the measurement conditions of the various analytical methods applied in the quantification of the different drug combinations pharmaceutical dosage forms and biological matrices, respectively.
| Ref. | Chromatographic conditions | Instrument | Detection | Detection wavelength (nm) | Linearity range (μg mL−1) | LOD (μg mL−1) | LOQ (μg mL−1) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Stationary phases | Mobile phases | |||||||||||
| a PDF (pharmaceutical dosage form), Brij-35 (non-ionic polyoxyethylene surfactant) and SLS (sodium lauryl sulfate is an anionic surface-active agent), DAD PDA (diode array detector), Densito (densitometer), UV-SPM (UV-spectrophotometer).b (i) indicates first method; (ii) indicates second method. | ||||||||||||
| 56 | SF | RMD: 251 FVP: 335 Δλ: 140 | RMD: 0.02–0.10 FVP: 0.04–0.10 | RMD: 2830 FVP: 3620 | RMD: 8570 FVP: 10790 | |||||||
| 57 | HPTLC plates pre-coated silica gel aluminum plates (60 F254, 0.1 mm thickness) | Ethyl acetate:methanol:ammonia (8:2:0.2 by volume) |
TLC | Densito | 235 | RMD 0.08–5.00 μg per band FVP 0.20–4.50 μg per band | RMD 0.04 ng per band FVP 0.12 ng per band | RMD: 0.5–50 FVP: 6–22 | ||||
| 58 | — | — | Spectrophotometric | UV | RMD | FVP | RMD (I–V) 1–10 | FVP (I and II) 1–20 | RMD | FVP | RMD | FVP |
| I: 244 | I: 323 | I 0.05 | I 0.27 | I 0.16 | I 0.82 | |||||||
| II: 248 | II: 337 | II 0.14 | II 0.21 | II 0.43 | II 0.63 | |||||||
| III: 207-244 | III: 330-400 | FVP (III–V) 2–20 | III 0.20 | III0.13 | III 0.60 | III 0.38 | ||||||
| IV: 272–340 | IV: 222–335 | IV 0.26 | IV0.23 | IV 0.78 | IV 0.71 | |||||||
| V: 340 | V: 280 | V 0.26 | V 0.12 | V 0.80 | V 0.36 | |||||||
| 60 | HPTLC plates coated with silica gel 60 F254 with 250 μm thickness | Methylene chloride:ethyl acetate:methanol:25% ammonia (6:3:4:1,v/v/v/v) |
HPTLC | Densito | 289 | FVP and MLP 3.75–100 RTN 2.75–100 | FVP: 12 | FVP: 3.38 | ||||
| MLP: 1.21 | MLP: 3.66 | |||||||||||
| RTN: 0.89 | RTN: 2.68 | |||||||||||
| 61 | Kinetix® column (5 μm, 150 × 4.6 mm) | 0.1 M SDS, 0.01 M Brij-35, and 0.02 M monobasic potassium phosphate, pH 3.1 | (i)HPLC (ii)UV SPM | UV | (i) 230 (ii) 350 | (i) 0.5–50 (ii) 6–22 | FVP: 0.04 | FVP: 0.12 | ||||
| MLP: 0.02 | MLP: 0.05 | |||||||||||
| Ref | Matrix | Analytical conditions | Instrument | Detection | Detection wavelength (nm) | Linearity range | LOD | LOQ | |
|---|---|---|---|---|---|---|---|---|---|
| Stationary phase | Mobile phase | ||||||||
| a TLC = thin layer chromatography, DAD = PDA(diode array detector), Densito (densitometer), UV-SPM (UV-spectrophotometer). | |||||||||
| 56 | Plasma | — | — | SF | UV | RMD: 251 | RMD 20–100 ng mL−1 | RMD 2.83 ng mL−1 | RMD 8.57 ng mL−1 |
| FVP: 335 | |||||||||
| Δλ: 140 | FVP 40–100 ng mL−1 | FVP 10.79 ng mL−1 | |||||||
| FVP 3.62 ng mL−1 | |||||||||
| 57 | Plasma | 20 × 20 cm pre-coated silica gel aluminum plates (60 F254, 0.1 mm thickness) | Ethyl acetate:methanol: ammonia (8:2:0.2 by volume) |
TLC | Densito | 235 nm | RMD 0.20–4.50 μg per band | RMD 0.04 μg per band | RMD 0.12 μg per band |
| FVP 0.08–5.00 μg per band | FVP 0.12 μg per band | FVP 0.07 μg per band | |||||||
| 59 | Plasma | BEH C18 (150 mm × 2.1 mm, 1.7 μm) | Methanol:ACN:H2O (15:35:50, by volume) at pH 4 |
UPLC | DAD | 240 nm | 0.1–10 μg mL−1 | 0.1 μg mL−1 | |
3. Conclusion
This review encompasses the different analytical methods used for the analysis of currently used drugs and drug regimens applied in the treatment of Covid-19 patients. After literature screening, we observed that most of the developed methods were used for the analysis of RMD and most of them were dependent on using LC-MS/MS under different analysis conditions. In addition to the traditionally known method; there were novel techniques developed for the analysis of the above-mentioned drugs including quantum dots and computational chemistry. This review puts the bases for analysts who are seeking the development of more accurate, selective, and time-saving methods in both biological fluids and different pharmaceutical dosage forms for more understanding of pharmacokinetics and more efficient quality control analysis of medications used in Covid-19 therapy.Conflicts of interest
The authors declare that there are no conflicts of interest.References
- I. E. Mikhail, H. Elmansi, F. Belal and A. E. Ibrahim, Microchem. J., 2021, 165, 106189 CrossRef CAS PubMed.
- WHO, Coronavirus (COVID-19) Dashboard, https://covid19.who.int/, accessed 8/9/2022, 2022 Search PubMed.
- R. Lu, X. Zhao, J. Li, P. Niu, B. Yang, H. Wu, W. Wang, H. Song, B. Huang and N. Zhu, Lancet, 2020, 395, 565–574 CrossRef CAS PubMed.
- M. Letko, A. Marzi and V. Munster, Nat. Microbiol., 2020, 5, 562–569 CrossRef CAS PubMed.
- H. Shuheng, L. Xinghao, X. Zimu, W. Jiaquan, L. Yunxia, S. Jie, L. Yan and C. Cheng, Plasma Sci. Technol., 2018, 21, 015501 Search PubMed.
- NHS, Coronavirus (COVID-19) symptoms in adults, https://www.nhs.uk/conditions/coronavirus-covid-19/symptoms/main-symptoms/, accessed 8/9/2022, 2022 Search PubMed.
- M. Mehmandoust, Y. Khoshnavaz, M. Tuzen and N. Erk, Microchim. Acta, 2021, 188, 1–15 CrossRef PubMed.
- Y. Huang, C. Yang, X.-f. Xu, W. Xu and S.-w. Liu, Acta Pharmacol. Sin., 2020, 41, 1141–1149 CrossRef PubMed.
- WHO, Tracking SARS-CoV-2 variants, https://www.who.int/activities/tracking-SARS-CoV-2-variants, accessed 8/9/2022, 2022 Search PubMed.
- NIH, COVID-19 Treatment Guidelines, https://www.covid19treatmentguidelines.nih.gov/therapies/antiviral-therapy/remdesivir/, accessed 11/9/2022, 2022 Search PubMed.
- NIH, Covid-19 treatment guidelines, https://www.covid19treatmentguidelines.nih.gov/therapies/antiviral-therapy/remdesivir/https://www.fda.gov/drugs/emergency-preparedness-drugs/coronavirus-covid-19-drugs#:%7E:text=Veklury(Remdesivir)isapprovedfor,areathighriskfor, accessed 11/9/2022, 2022 Search PubMed.
- FDA, Coronavirus (COVID-19) | Drugs, https://www.fda.gov/drugs/emergency-preparedness-drugs/coronavirus-covid-19-drugs#:%7E:text=Veklury(Remdesivir)isapprovedfor,areathighriskfor, accessed 11/9/2022, 2022 Search PubMed.
- D. bank, Favipiravir, https://go.drugbank.com/drugs/DB12466, accessed 11/9/2022, 2022 Search PubMed.
- WHO, WHO updates its treatment guidelines to include molnupiravir, https://www.who.int/news/item/03-03-2022-molnupiravir, accessed 11/9/2022, 2022 Search PubMed.
- A. Jayk Bernal, M. M. Gomes da Silva, D. B. Musungaie, E. Kovalchuk, A. Gonzalez, V. Delos Reyes, A. Martín-Quirós, Y. Caraco, A. Williams-Diaz and M. L. Brown, N. Engl. J. Med., 2022, 386, 509–520 CrossRef PubMed.
- WHO, Administration of Nirmatrelvir-ritonavir for COVID-19, http://WHO-2019-nCoV-Therapeutics-Nirmatrelvir-ritonavir-Poster-A-2022.1-eng.pdf, accessed 11/9/2022, 2022 Search PubMed.
- C. Liu, M. Zhu, L. Cao, H. Boucetta, M. Song, T. Hang and Y. Lu, Biomed. Chromatogr., 2022, 36, e5456 CAS.
- L. H. Keith, L. U. Gron and J. L. Young, Chem. Rev., 2007, 107, 2695–2708 CrossRef CAS PubMed.
- J. Płotka-Wasylka, Talanta, 2018, 181, 204–209 CrossRef PubMed.
- F. Pena-Pereira, W. Wojnowski and M. Tobiszewski, Anal. Chem., 2020, 92, 10076–10082 CrossRef CAS PubMed.
- M. Tobiszewski, M. Marć, A. Gałuszka and J. Namieśnik, Molecules, 2015, 20, 10928–10946 CrossRef CAS PubMed.
- J. L. Young and D. E. Raynie, in Challenges in Green analytical chemistry, Royal Society of Chemistry Cambridge, 2011, pp. 44–62 Search PubMed.
- A. Gałuszka, Z. M. Migaszewski, P. Konieczka and J. Namieśnik, TrAC, Trends Anal. Chem., 2012, 37, 61–72 CrossRef.
- M. M. Hamdy, M. M. Abdel Moneim and M. F. Kamal, Biomed. Chromatogr., 2021, 35, e5212 CrossRef CAS PubMed.
- I. Bulduk and E. Akbel, J. Taibah Univ. Sci., 2021, 15, 507–513 CrossRef.
- A. E. Ibrahim, S. E. Deeb, E. M. Abdelhalim, A. Al-Harrasi and R. A. Sayed, Separations, 2021, 8, 243 CrossRef CAS.
- A. H. Abdelazim and S. Ramzy, Spectrochim. Acta, Part A, 2022, 276, 121188 CrossRef CAS PubMed.
- A. H. Abo-Gharam and D. S. El-Kafrawy, Sustainable Chem. Pharm., 2022, 29, 100744 CrossRef CAS PubMed.
- K. Thumar, M. Pandya, N. Patel and F. Rawat, Int. J. Pharm. Sci. Res., 2023, 14, 1273–1279 Search PubMed.
- V. Avataneo, A. De Nicolò, J. Cusato, M. Antonucci, A. Manca, A. Palermiti, C. Waitt, S. Walimbwa, M. Lamorde, G. Di Perri and A. D’Avolio, J. Antimicrob. Chemother., 2020, 75, 1772–1777 CrossRef CAS PubMed.
- J.-C. Alvarez, P. Moine, I. Etting, D. Annane and I. A. Larabi, Clin. Chem. Lab. Med., 2020, 58, 1461–1468 CrossRef CAS PubMed.
- D. Xiao, K. H. J. Ling, T. Tarnowski, R. Humeniuk, P. German, A. Mathias, J. Chu, Y.-S. Chen and E. van Ingen, Anal. Biochem., 2021, 617, 114118 CrossRef CAS PubMed.
- R. Nguyen, J. C. Goodell, P. S. Shankarappa, S. Zimmerman, T. Yin, C. J. Peer and W. D. Figg, J. Chromatogr. B, 2021, 1171, 122641 CrossRef CAS PubMed.
- K. Habler, M. Brügel, D. Teupser, U. Liebchen, C. Scharf, U. Schönermarck, M. Vogeser and M. Paal, J. Pharm. Biomed. Anal., 2021, 196, 113935 CrossRef CAS PubMed.
- A. Reckers, A. H. Wu, C. M. Ong, M. Gandhi, J. Metcalfe and R. Gerona, J. Antimicrob. Chemother., 2021, 76, 1865–1873 CrossRef CAS PubMed.
- C. Skaggs, H. Zimmerman, N. Manicke and L. Kirkpatrick, J. Mass Spectrom. Adv. Clin. Lab, 2022, 25, 27–35 CrossRef CAS PubMed.
- R. R. Pasupuleti, P.-C. Tsai, V. K. Ponnusamy and A. Pugazhendhi, Process Biochem., 2021, 102, 150–156 CrossRef CAS PubMed.
- T. Z. Attia, J. M. Boushra, A. F. Abdel Hakiem, A. S. Lashien and D. A. Noureldeen, Luminescence, 2022 Search PubMed.
- T. Reçber, S. S. Timur, S. E. Kablan, F. Yalçın, T. C. Karabulut, R. N. Gürsoy, H. Eroğlu, S. Kır and E. Nemutlu, J. Pharm. Biomed. Anal., 2022, 214, 114693 CrossRef PubMed.
- G. Sravanthi, K. S. Gandla and L. Repudi, Cell. Mol. Biomed. Rep., 2023, 3, 130–136 CrossRef.
- A. H. Abdelazim, M. A. Abourehab, L. M. Abd Elhalim, A. A. Almrasy and S. Ramzy, Spectrochim. Acta, Part A, 2023, 285, 121911 CrossRef CAS PubMed.
- A. R. Mohamed, E. Abolmagd, I. M. Nour, M. Badrawy and M. A. Hasan, BMC Chem., 2023, 17, 13 CrossRef CAS PubMed.
- G. R. Kumar, B. S. Babu, R. R. Rao, V. M. Vardhan and V. Abbaraju, J. Pharm. Res. Int., 2021, 33, 3026–3035 CrossRef.
- M. Bindu, K. Gandla, S. Vemireddy, S. Samuel and Y. Praharsha, World J. Adv. Res. Rev., 2022, 15, 580–590 CrossRef CAS.
- A. Amara, S. D. Penchala, L. Else, C. Hale, R. FitzGerald, L. Walker, R. Lyons, T. Fletcher and S. Khoo, J. Pharm. Biomed. Anal., 2021, 206, 114356 CrossRef CAS PubMed.
- S. Jain, S. Giri, N. Sharma and R. P. Shah, J. Liq. Chromatogr. Relat. Technol., 2021, 44, 750–759 CrossRef CAS.
- A. M. Annadi, N. M. El Zahar, N. E.-D. A. Abdel-Sattar, E. H. Mohamed, S. A. Mahmoud and M. S. Attia, RSC Adv., 2022, 12, 34512–34519 RSC.
- G. Camlik, F. Beyazaslan, E. Kara, D. Ulker, I. Albayrak and I. T. Degim, Med. Res. Arch., 2022, 10, 9 CrossRef.
- S. E. Kablan, T. Reçber, G. Tezel, S. S. Timur, C. Karabulut, T. C. Karabulut, H. Eroğlu, S. Kır and E. Nemutlu, J. Electroanal. Chem., 2022, 920, 116579 CrossRef CAS.
- T. L. Parsons, L. A. Kryszak and M. A. Marzinke, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2021, 1182, 122921 CrossRef CAS PubMed.
- A. S. Gouda, H. M. Marzouk, M. R. Rezk, A. M. Salem, M. I. Morsi, E. G. Nouman, Y. M. Abdallah, A. Y. Hassan and A. M. Abdel-Megied, J. Chromatogr. B, 2022, 1206, 123363 CrossRef CAS PubMed.
- B. I. Salman, A. E. Ibrahim, S. El Deeb and R. E. Saraya, RSC Adv., 2022, 12, 16624–16631 RSC.
- M. S. Imam, A. S. Batubara, M. Gamal, A. H. Abdelazim, A. A. Almrasy and S. Ramzy, Sci. Rep., 2023, 13, 137 CrossRef CAS PubMed.
- H. S. Elbordiny, N. Alzoman, H. Maher and S. Aboras, Validation, and Environmental Impact Studies, 2023 Search PubMed.
- J. Martens-Lobenhoffer, C. R. Böger, J. Kielstein and S. M. Bode-Böger, J. Chromatogr. B, 2022, 1212, 123510 CrossRef CAS PubMed.
- M. El-Awady, H. Elmansi, F. Belal and R. a. Shabana, J. Fluoresc., 2022, 32, 1941–1948 CrossRef CAS PubMed.
- D. A. Noureldeen, J. M. Boushra, A. S. Lashien, A. F. A. Hakiem and T. Z. Attia, Microchem. J., 2022, 174, 107101 CrossRef CAS PubMed.
- H. S. Elama, A. M. Zeid, S. M. Shalan, Y. El-Shabrawy and M. I. Eid, Spectrochim. Acta, Part A, 2023, 287, 122070 CrossRef CAS PubMed.
- A. A. Emam, E. A. Abdelaleem, E. H. Abdelmomen, R. H. Abdelmoety and R. M. Abdelfatah, Microchem. J., 2022, 179, 107580 CrossRef CAS PubMed.
- R. E. Saraya, S. E. Deeb, B. I. Salman and A. E. Ibrahim, J. Sep. Sci., 2022, 45, 2582–2590 CrossRef CAS PubMed.
- Y. A. Sharaf, S. El Deeb, A. E. Ibrahim, A. Al-Harrasi and R. A. Sayed, Molecules, 2022, 27, 2330 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |