
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of nontriterpenoids from the rot roots of Panax notoginseng with cytotoxicity and their molecular docking study and experimental validation†
Jia-Huan Shangab,
Yi-Jun Qiaoa,
Hong-Tao Zhua,
Dong Wanga,
Chong-Ren Yanga and
Ying-Jun Zhang *a
*a
aState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, PR China. E-mail: zhangyj@mail.kib.ac.cn; Tel: +86-871-6522-3235
bUniversity of Chinese Academy of Sciences, Beijing 100049, PR China
First published on 6th April 2023
Abstract
Panax notoginseng (PN) is a well-known traditional Chinese medicine, with dammarane-type triterpenoid saponins characterized as major component and active ingredients, together with amino acids, flavonoids, polysaccharides, and polyacetylenes. The roots of PN are susceptible to root rot disease, which causes a huge loss and changes in the chemical components of this precious resource. In this study, sub-fractions of rot PN root extracts were preliminarily found to have admirable cytotoxicity on two human cancer cells. Further bioassay-guided isolation discovered nine new non-triterpenoids, including two novel N-methylacetamido-1-oxotetrahydropyrimidine alkaloids (1, 2), five 2H-furanones or 2H-pyranones (3–7), and two polyacetylenic alcohols (8, 9). Their structures were illuminated by extensive spectroscopic data, calculated ECD, and X-ray diffraction analysis. Among them, 3–7 were considered to be transformed from panaxatriol through the intermediates (8, 9). The new alkaloids (1, 2) displayed noteworthy cytotoxicity against five human cancer cells with IC50 values ranging from 14 to 24 μM. In silico target prediction and molecular docking studies showed that 1 and 2 may interact with EGFR, and were verified by the experimental inhibitory effect on EGFR tyrosine kinase.
Introduction
Panax notoginseng (Burk.) F. H. Chen (PN) is a famous medicinal herb belonging to the ginseng family (Araliaceae). The roots, possessing dammarane-type triterpenoid (DTT) as dominant chemical components with various bioactivities, such as immunostimulatory, anti-atherosclerotic, anti-hypertensive, antitumor, and CNS-protective activities, are used widely as major ingredients in many traditional Chinese medicinal preparation for treating cardiovascular diseases in the world.1 Moreover, some minor constituents are also identified from PN. For example, dencichine, a PN amino acid, plays a main hemostatic role by improving the amount and activation of platelet and inhibiting fibrinolysis.2 Polyacetylenes, found in PN and Korean ginseng, displayed cytotoxic, anti-platelet, and anti-microbial activities.3The root rot disease has become the most severe destructive problem for PN cultivation, causing a huge loss and great changes in the chemical components.4 In order to exploit PN resources further and discover a new bioactive skeleton, several highly oxidized dammarane-type triterpenes with favorable anticancer and anti-inflammatory activities were obtained from the rot PN root extracts previously by our group.5 Continuing study showed that the remaining fractions displayed significant cytotoxicity against lung cancer A-549 and hepatocellular carcinoma cancer SMMC7721 cells (inhibition rates > 96%) at a concentration of 100 μg mL−1 via MTS assay. Further bioassay-guided isolation yielded nine new nontriterpenoids, including two novel alkaloids (1, 2), five α,β-unsaturated furanones or pyranones (3–7), and two polyacetylenic alcohols (8, 9) (Fig. 1).
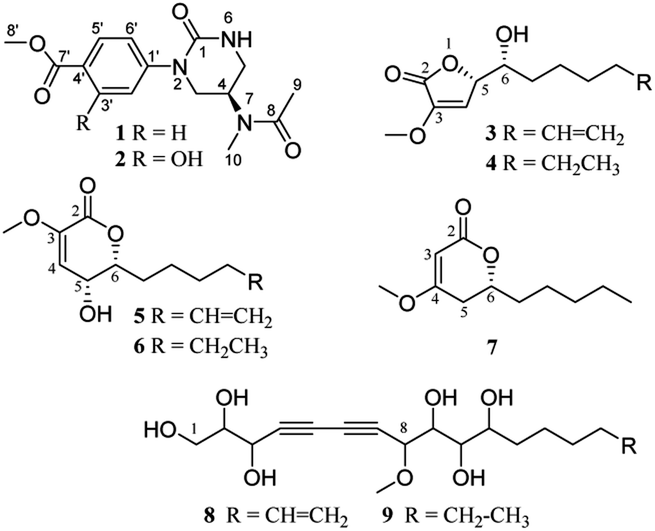 | ||
| Fig. 1 Chemical structures of compounds 1–9. | ||
The alkaloids (1, 2) contained a unique N-methylacetamido substituent on their unreported 1-oxotetrahydropyrimidine skeleton, representing the first rare non-amino acid alkaloid from ginseng plants. New lactones (3–7) were biogenetically considered to be transformed from panaxatriol (PXT), a polyacetylene naturally occurring in the genus Panax, through their intermediate products (8, 9) by a series of reactions. Herein, the structural elucidation of nine new nontriterpenoids (1–9), the plausible bio-transformed pathway for lactones, as well as their antiproliferative activity against five human cancer cell lines, were deduced in detail. Moreover, in silico target prediction and molecular docking studies were adopted to analyze the interaction of cytotoxic molecules with the predicted target protein (EGFR), which was further verified by the experimental protein inhibition assay.
Results and discussion
The methanol extract (4.0 kg) of PN rot root was fractionated into three main fractions (Fr. A–C), and Fr. C (220.8 g) was further divided into six sub-fractions (Fr. C1–C6) by an RP-18 CC. In our previous work, eight anti-inflammatory and anti-cancer triterpenes were reported from the sub-Fr. C2, C3, and C5.5b Further study showed that the remaining sub-Fr. C1 and C6 displayed significant cytotoxicities against lung cancer A-549 and hepatocellular carcinoma SMMC7721 cancer cells with inhibition rates exceeding 96%, while sub-Fr. C4 displayed no obvious inhibitory effect (Table S4†). Therefore, the present study mainly focused on the remaining bioactive fractions, resulting in the discovery of compounds 1, 2, 8, and 9 from Fr. C1, and 3–7 from Fr. C6, respectively.Structure elucidation of compounds 1–9
Compound 1 was obtained as a colorless needle crystal, and its molecular formula was determined to be C15H19N3O4 by the HRESIMS (m/z 328.1269 [M + Na]+, calcd for, 328.1273), corresponding to eight degrees of unsaturation. The 1H, 13C NMR and HSQC spectra suggested the presence of para-substituted benzene [δC 147.0, 126.8, 130.5 (2C), 124.2 (2C), δH 7.98 and 7.33 (each 2H, d, J = 8.7 Hz)], and the remaining nine carbon resonances assignable to three conjugated ketones (δC 171.6, 166.8, 155.0), one methine (δC 46.1), two methylenes (δC 50.0, 42.6), two singlet methyls (δC 32.6, 22.6) and one methoxyl (δC 52.3), as well as broad and short active hydrogen (δH 5.63) (Table 1). The 1H–1H COSY correlations (Fig. 2A) of NH (δH 5.63)/H2-5 (δH 3.53, 3.48)/H-4 (δH 5.04)/H2-3 (δH 3.08) suggested the presence of –NH–CH2–CH–CH2– fragment. The HMBC (Fig. 2A) correlations from H3-9 (δH 2.10), H3-10 (δH 3.03), and H-4 to C-8 (δC 171.6), H3-9 to C-10 (δC 32.6), H3-10 to C-4 (δC 46.1), revealed an N-methyl acetylamino group located at C-4. Likewise, the HMBC correlations of H-3'/5' (δH 7.98), H-2′/6′ (δH 7.33), and H3-8′ (δH 3.87) to C-7′ (δC 166.8), and H3-8′ to C-4′ (δC 126.8) indicated the existence of a methoxyl acyl group at C-4′. In addition, the very high field of the carbonyl group (δC 155.0) showed a correlation with H2-3/H2-5 in the HMBC spectrum, and the HMBC correlation between H2-3 and C-1′, suggested an (N-methyl acetylamino)-oxytetra-hydropyrimidine ring was attached to C-1′ (δC 147.0) of para-substituted benzene through a tertiary nitrogen atom. Finally, the structure and absolute configuration of 1 were unequivocally confirmed as (R)-2-(p-benzoate methyl)-4-(N-methylacetami-do)-1-oxotetrahydropyrimidin by X-ray single crystal diffraction [Flack parameter = 0.03 (4)] (Fig. 2B).| No. | 1 | 2 | ||
|---|---|---|---|---|
| δC | δH | δC | δH | |
| a d: doublet; m: multiplet; s: singlet; δ in ppm; J in Hz. | ||||
| 1 | 155.0 | 155.5 | ||
| 3 | 50.0 | 3.80 dd (11.8, 7.1) | 50.8 | 3.72 m |
| 4 | 46.1 | 5.04 m | 46.1 | 5.05 m |
| 5 | 42.6 | 3.53 ddd (11.8, 5.6, 2.3) | 42.9 | 3.53 ddd (11.8, 5.6, 2.7) |
| 3.48 ddd (11.8, 7.1, 1.3) | 3.49 ddd (11.8, 7.3, 1.5) | |||
| 6 | 5.63 s | 5.10 s | ||
| 8 | 171.6 | 171.6 | ||
| 9 | 22.6 | 2.10 s | 22.7 | 2.12 s |
| 10 | 32.6 | 3.03 s | 32.6 | 3.06 s |
| 1′ | 147.0 | 134.4 | ||
| 2′ | 124.2 | 7.33 d (8.7) | 127.0 | 7.70 d (2.7) |
| 3′ | 130.5 | 7.98 d (8.7) | 160.0 | |
| 4′ | 126.8 | 112.5 | ||
| 5′ | 130.5 | 7.98 d (8.7) | 118.5 | 6.95 d (8.9) |
| 6′ | 124.2 | 7.33 d (8.7) | 134.1 | 7.37 dd (8.9, 2.7) |
| 7′ | 166.8 | 170.2 | ||
| 8′ | 52.3 | 3.87 s | 52.6 | 3.91 s |
| OH | 10.67 s | |||
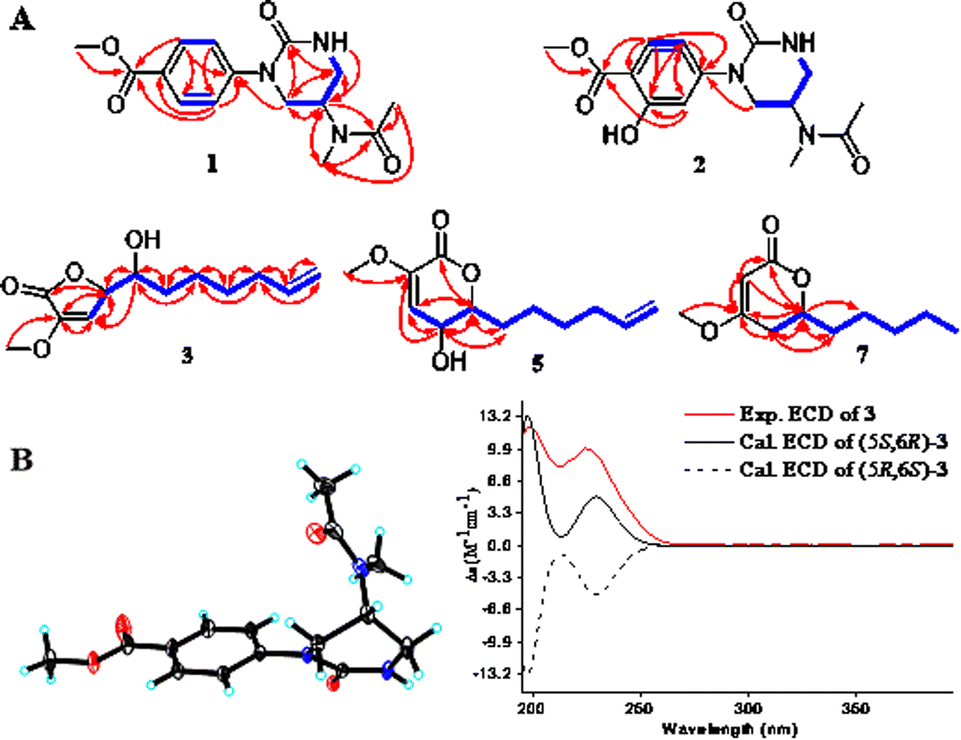 | ||
Fig. 2 (A) Key 1H–1H COSY ( ) and HMBC ( ) and HMBC ( ) correlations of 1–3, 5, 7. (B) (left) ORTEP of 1 with thermal ellipsoids shown at 30% probability. (right) Experimental ECD and calculated ECD spectra of 3. ) correlations of 1–3, 5, 7. (B) (left) ORTEP of 1 with thermal ellipsoids shown at 30% probability. (right) Experimental ECD and calculated ECD spectra of 3. | ||
Compound 2 was isolated as white amorphous powder, with a molecular formula of C15H19N3O5, as deduced by HREIMS (m/z 321.1328 [M]+), which was one more oxygen atom than that of 1. Their 1H and 13C NMR data showed some similarities, except for an additional broad active hydrogen (δH 10.67) and 1,2,4-substituted benzene ring signals in 2 (Table 1). The same chemical shifts of benzoate methyl (δC 170.2, 52.6) and (N-methylacetylamino)-oxytetrahydropyrimidine ring [δC 155.5 (C-1), 50.8 (C-3), 46.1 (C-4), 42.9 (C-5), 171,6 (C-8), 22.7 (C-9), 32.6 (C-10)] as those of 1, as well as the HMBC correlations between H-5′ (δH 6.95), H-6′ (δH 7.37) and C-1′ (δC 134.4), H-5′ and C-7' (δC 170.2)/C-4′ (δC 112.5), H3-8′ (δH 3.91) and C-7′, confirmed that two parts were connected through a tertiary nitrogen atom. The additional oxygen was assigned to a hydroxyl group at C-3′ (δC 160.0) because of H-2′ presenting as a smaller doublet signal (δH 7.70, d, 2.7 Hz) with HMBC correlation to C-1′ (Fig. 2A). On account of the only chiral carbon at C-4 in 2, the consistent positive optical ration with 1, indicated the same 4R-configuration for 2 to 1. Thus, 2 was determined as (R)-2-(3-hydroxy-4-methoxyl acyl)phenyl-4-(N-methylacetamido)-1-oxotetrahydropyrimidin.
Compound 3 was obtained as a yellow oil, possessing a molecular formula C12H18O4, as determined by the HRESIMS (m/z 271.1186 [M + HCOO]−). The 13C NMR spectrum showed 12 carbon resonances, assignable to five methylenes with a vinyl (δC 113.9), four methines with two oxygen bearings (δC 83.0, 72.0), two quaternary carbons, and one methoxyl. The 1H NMR spectrum presented a methoxy at δH 3.65 (3H, s), two oxymethines at δH 5.04 (overlap) and 4.02 (d, 3.1 Hz), two olefinic protons at δH 6.58 (d, 1.7 Hz) and 5.83 (ddt, 16.9, 10.2, 6.7 Hz), and two terminal olefinic protons at δH 5.04 and 4.99 (dd, 10.1, 0.8 Hz) (Table 2). Analysis of the 1H–1H COSY correlations (Fig. 2A) revealed a chain fragment of –CH–CH(O)–CH(O)–(CH2)4–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2. In the HMBC spectrum of 3, correlations of olefinic proton H-4 (δH 6.58) with C-3 (δC 148.7)/C-5 (δC 83.0), oxymethines H-5 (δH 5.04) with C-2 (δC 168.4)/C-3/C-4 (δC 116.0)/C-6 (δC 72.0) and H-6 (δH 4.02) with C-4/C-5/C-7 (δC 34.2)/C-8 (δC 26.2), and the methoxyl protons (δH 3.65) with C-3 were observed respectively. The aforementioned evidence constructed that the planar structure of 3 as 3-methoxy-substituted α,β-unsaturated-γ-lactone with a long chain at C-5. The absolute configurations of C-5 and C-6 were determined by combining HETLOC correlations together with ECD spectrum. The ECD spectrum showed a negative Cotton effect at 210 nm for a lactone moiety, indicating the S configuration of C-5.6 Furthermore, the small coupling constant of 3JH5–H6 (3.0 Hz) indicated the gauche conformation for H-5 and H-6, while the two small bond heteronuclear coupling constants (2JH6–C5 = 2.8 Hz, 2JH5–C6 = 2.3 Hz) in the HETLOC spectrum (Fig. S29†), suggested the relative configurations of H-5 and H-6 were threo and determined 5S,6R configurations of 3.7 Subsequent ECD calculations were conducted to further identify the absolute configuration of 3, which showed that the calculated ECD spectrum of (5S,6R)-3 was in good agreement with the experimental ECD curve of 3 (Fig. 2B). Consequently, compound 3 was determined as (5S,6R)-5-(1-hydroxy-2-pentenyl)-3-methoxy-2(5H)-furanone.
CH2. In the HMBC spectrum of 3, correlations of olefinic proton H-4 (δH 6.58) with C-3 (δC 148.7)/C-5 (δC 83.0), oxymethines H-5 (δH 5.04) with C-2 (δC 168.4)/C-3/C-4 (δC 116.0)/C-6 (δC 72.0) and H-6 (δH 4.02) with C-4/C-5/C-7 (δC 34.2)/C-8 (δC 26.2), and the methoxyl protons (δH 3.65) with C-3 were observed respectively. The aforementioned evidence constructed that the planar structure of 3 as 3-methoxy-substituted α,β-unsaturated-γ-lactone with a long chain at C-5. The absolute configurations of C-5 and C-6 were determined by combining HETLOC correlations together with ECD spectrum. The ECD spectrum showed a negative Cotton effect at 210 nm for a lactone moiety, indicating the S configuration of C-5.6 Furthermore, the small coupling constant of 3JH5–H6 (3.0 Hz) indicated the gauche conformation for H-5 and H-6, while the two small bond heteronuclear coupling constants (2JH6–C5 = 2.8 Hz, 2JH5–C6 = 2.3 Hz) in the HETLOC spectrum (Fig. S29†), suggested the relative configurations of H-5 and H-6 were threo and determined 5S,6R configurations of 3.7 Subsequent ECD calculations were conducted to further identify the absolute configuration of 3, which showed that the calculated ECD spectrum of (5S,6R)-3 was in good agreement with the experimental ECD curve of 3 (Fig. 2B). Consequently, compound 3 was determined as (5S,6R)-5-(1-hydroxy-2-pentenyl)-3-methoxy-2(5H)-furanone.
| No. | 3 | 4 | 5 | 6 | 7 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | δC | δH | δC | δH | |
| a d: doublet; m: multiplet; s: singlet; t: triplet; δ in ppm; J in Hz. | ||||||||||
| 2 | 168.4 | 168.5 | 161.0 | 161.1 | 167.6 | |||||
| 3 | 148.7 | 148.8 | 145.2 | 145.2 | 90.3 | 5.05, s | ||||
| 4 | 116.0 | 6.58, d (1.7) | 116.1 | 6.59, d (2.1) | 114.9 | 6.00, d (3.3) | 114.9 | 6.00, d (3.2) | 173.0 | |
| 5 | 83.0 | 5.04, overlap | 83.1 | 5.06, dd (4.8, 2.1) | 66.5 | 4.63, m | 66.6 | 4.65, m | 33.0 | 2.39, ddd (17.0, 12.0, 1.1) |
| 2.24, dd (17.0, 3.8) | ||||||||||
| 6 | 72.0 | 4.02, d (3.1) | 72.1 | 4.03, m | 84.7 | 4.53, td (8.3, 3.2) | 84.8 | 4.55, td (8.3, 3.3) | 76.0 | 4.29, m |
| 7 | 34.2 | 1.68, m | 34.4 | 1.71, overlap | 32.7 | 2.00, overlap | 32.9 | 2.00, m | 34.7 | 1.70, m |
| 1.77, dtd (14.0, 9.5, 4.8) | 1.78, m | 1.55, m | ||||||||
| 8 | 26.2 | 1.77, m; 1.50, m | 30.1 | 1.22, overlap | 25.5 | 1.60, m; 1.43, m | 25.9 | 1.58, m; 1.42, m | 24.5 | 1.43, m; 1.33, m |
| 9 | 29.6 | 1.39, m | 26.7 | 1.33, m | 29.4 | 1.32, m | 29.8 | 1.24, overlap | 31.6 | 1.19, overlap |
| 10 | 34.4 | 2.03, m | 32.6 | 1.77, overlap; 1.50 m | 34.3 | 1.96, dd (14.2, 7.0) | 32.3 | 1.14, overlap | 22.5 | 1.24, overlap |
| 11 | 139.7 | 5.83, ddt (16.9, 10.2, 6.7) | 23.4 | 1.25, overlap | 139.5 | 5.78, ddt (17.0, 10.2, 6.7) | 23.2 | 1.19, overlap | 14.0 | 0.81, t (7.0) |
| 12 | 115.2 | 5.04, overlap | 14.8 | 0.85, t (7.0) | 115.3 | 5.03, dd (17.0, 1.6) | 14.7 | 0.81, t (7.1) | 56.1 | 3.66, s |
| 4.99, dd (10.2, 0.8) | 4.97, m | |||||||||
| OMe | 58.4 | 3.65, s | 58.4 | 3.68, s | 55.9 | 3.57, s | 55.8 | 3.57, s | ||
Compound 4 was isolated as a colorless oil and was assigned the molecular formula C12H20O4 with three degrees of unsaturation by the HRESIMS (m/z 273.1345 [M + Na]+). Detailed comparison of NMR data between 4 and 3 indicated their structures to be quite similar, except that the terminal ethyl group (δC 139.7, 115.2) in 3 was replaced by one methylene (δC 23.4) and one methyl group (δC 14.8) in 4, as indicated by the HMBC correlations from H2-11 (δH 1.25) to C-10 (δC 32.6)/C-12 (δC 14.8) and the 1H–1H COSY correlations of H3-12 (δH 0.85)/H2-11/H2-10 (δH 1.77, 1.50)/H-9 (δH 1.33). Meanwhile, the HETLOC spectrum (Fig. S40†) showed small heteronuclear coupling constants (2JH6–C5 = 2.8 Hz, 2JH5–C6 = 2.9 Hz), and the ECD spectrum (Fig. S43†) almost overlapped with that of 3 (Fig. S32†), indicating the 5S,6R configurations of 4. Hence, compound 4 was concluded as (5S,6R)-5-(1-hydroxyheptyl)-3-methoxy-2(5H)-furanone.
Compound 5 yielded a white amorphous powder, and had the same molecular formula C12H18O4 as that of 3, evidenced by the HRESIMS data at m/z 271.1186 [M + HCOO]−. The detailed analysis of 1H and 13C NMR, DEPT, and HSQC spectra showed varied different chemical shifts with those of 3 on the two oxymethines as well as the α,β-unsaturated ketone carbons. The HMBC correlations from oxymethines H-6 (δH 4.53) to C-2 (δC 161.0)/C-4 (δC 114.9)/C-5 (δC 66.5)/C-7 (δC 32.7)/C-8 (δC 25.5) and H-5 (δH 4.63) to C-3 (δC 145.2)/C-4/C-6 (δC 84.7), as well as the 1H–1H COSY correlations of H-4/H-5/H-6/H2-7 (δH 2.00, 1.77)/H2-8 (δH 1.60, 1.43)/H2-9 (δH 1.32)/H2-10 (δH 1.96)/H-11 (δH 5.78)/H2-12 (δH 5.03, 4.97), revealed a six-membered δ-lactone in 5 replacing the γ-lactone moiety in 3. According to the small coupling constant of 3JH5–H6 (3.2 Hz), and a reliable ECD empirical rule for structure with α,β-unsaturated-δ-lactone moiety,8 6R Configuration was assigned as the ECD spectrum of 5 (Fig. S53†) showing a positive Cotton effect at 262 nm. Thus, compound 5 was elucidated as (5R,6R)-6-(1-hydroxy-5-hexenyl)-5-hydroxy-3-methoxy-2(5H)-pyranone.
Compound 6 was obtained as a yellow amorphous powder with the same molecular formula C12H20O4 as that of 4, suggested by a molecular ion peak at m/z 273.1345 [M + HCOO]−. The NMR data (Table 2) characterized an identical α,β-unsaturated-δ-lactone moiety (δC 161.1, 145.2, 114.9, 84.8, 66.6) with 5. The slight difference between them was that the terminal alkenyl (δC 139.5, 115.3) chain in 5 was reduced to an aliphatic chain at C-6, as the 1H–1H COSY spectrum showing the long coherent signals of H-3/H-4/H-5/H2-6/H2-7/H2-8/H2-9/H2-10/H3-11 in 6. Moreover, the consistency between the ECD spectra of compounds 6 and 5 and the small JH4/H5 (3.3 Hz), indicated that 6 shared the same 5R and 6R configurations with that 5. Thus, the structure of 6 was determined to be (5R,6R)-6-(1-hydroxyhextyl)-5-hydroxy-3-methoxy-2(5H)-pyranone.
Compound 7 was isolated as a colorless oil and its molecular formula C11H18O3 was deduced from HRESIMS (m/z 221.1149 [M + Na]+). The 13C NMR and DEPT spectra showed 11 carbons, which were similar to those of 6, except for a set of up and down-shifted double bond signals (δC 90.3, 173.0 for 7; δC 145.2, 114.9 for 6) and a missing oxymethine in 7. The 1H–1H COSY correlations of H2-5/H-6/H2-7/H2-8/H2-9/H2-10/H3-11 and HMBC correlations of H-6 with C-2/C-4/C-5/C-7/C-8, H2-5 with C-3/C-4/C-6/C-7, H-3 with C-2/C-4/C-6, and methoxyl protons (δH 3.66) with C-4, established the plane structure of 7. Moreover, the optical rotation of 7 ([a] −97.4) was consistent with a synthesized analogue (R)-6-hexyl-4-methoxy-6-hydroxy-2H-pyran-2-one ([a] −103.5, >99% ee)9 and opposite to the corresponding isomer ([a] +80.1, 83% ee) with the identical skeletal structure to 7 but surplus S-isomer,10 indicating R-configuration for 7. Therefore, compound 7 was identified (R)-6-(1-hydroxyhextyl)-4-methoxy-2(5H)-pyranone.
Compounds 8 and 9 were isolated as both colorless oil and had molecular formulas of C18H28O7 and C18H30O7, respectively. Their 1D NMR and HSQC spectra showed great similarities, except that an alkenyl in 8 was replaced by an aliphatic methyl in 9 (Table S1†). The 1H–1H COSY spectrum displayed clear –CH2(O)–CH(O)–CH(O)– (8 and 9)– and –(CH2)4–CHCH2 (8) or –(CH2)5–CH3 (9) correlations, and the HMBC correlations of H-8 with C-7/C-6/C-5/C-4/OCH3, H-9 with C-7, H-3 with C-4/C-5/C-6/C-7, H-2 with C-4, and methoxy proton (δH 3.46) with C-8, indicating the partial structure of C-1 to C-9 connected by the conjugate diacetylene and a methoxy group located at C-8 in both 8 and 9. Six broad active protons (OH) peaks in the downfield showed direct 1H–1H COSY correlations with H-1 to H-3, and H-9 to H-11, respectively, evidenced further the existence and location of six hydroxyls. Thus, the planar structures of 8 and 9 were identified, as shown in Fig. 1. Regrettably, the absolute configurations of 8 and 9 were undetermined because of the polyhydroxyl substitution on their flexible skeleton.
Plausible bio-transformed pathways for 3–6, 8, and 9
Panaxytriol (PXT), widely existing in Panax species, was also found in the infected PN roots in the present study, and its absolute configuration was identified as 3R,9R,10R by circular dichroism (CD) analysis11 and synthetic method.12 Based on the structure of natural PXT and the obtained intermediates (8, 9), a plausible transformation pathway from PXT to four lactones (3–6) is proposed in Scheme 1. Under the high degree of oxidization in the infected plant, the olefinic bond, C-8, C-11, and terminal fatty chain of natural PXT were selectively oxidized to intermediate products (8, 9). Their second alkynyl (C-6, C-7) was then oxidatively cracked into the carboxyl group, which was further esterified with one of the hydroxyl groups randomly to form a five- or six-member lactone ring. Finally, the hydroxyl group next to methoxy was eliminated with ortho hydrogen to form a double bond. The absolute configuration of C-10 on PXT and lactones were kept unchanged during the transformation. In turn, the identification of 6R-configuration on compounds 3–6 proved the outstanding stereoselectivity of the oxidization reaction by bio-catalysis. To the best of our knowledge, only a few studies have reported that PXT can be transformed into new metabolites by fungi.13 New lactones and their precursors discovered in this work suggested that polyacetylene in PN could also be transformed under a highly oxidized environment, and the endophyte in the rot PN roots could be a potential tool to convert PXT and ginsenosides, which was worth further investigation.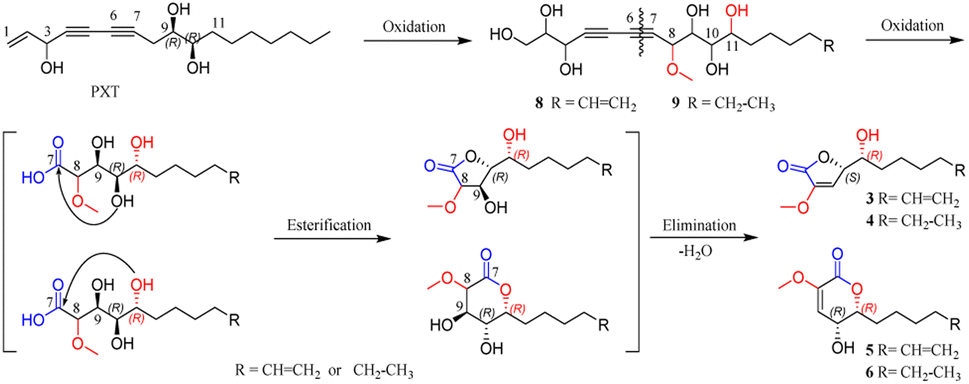 | ||
| Scheme 1 Plausible bio-transformed pathway for 3–6, 8, and 9. | ||
In vitro cytotoxic activity
All the isolates were evaluated for cytotoxicity on five human cancer cell lines (myeloid leukemia HL-60, lung cancer A-549, colon cancer SW480, breast cancer MCF-7, and hepatocellular carcinoma SMMC7721, purchased from ATCC (American Type Culture Collection), USA) by the MTS method. As shown in Fig. 3A and Table S5,† two alkaloids (1, 2) displayed a promising inhibitory activity against all five cancer cells with IC50 values ranging from 14–24 μM, which were comparable with that of positive control cisplatin (DDP, purchased from Selleck, USA). However, all the α,β-unsaturated lactones (3–7) showed no significant cytotoxicity on tested cancer cells with inhibition ratios < 50% at a concentration of 40 μM (Table S4†). This is different from the previous research on their precursor structure (PXT).3a,b The results of the in vitro bioassay suggested that endophytes in the infected PN roots may hold a detoxification mechanism, which can convert toxic metabolites into less toxic compounds to make themself survive in the rhizosphere environment.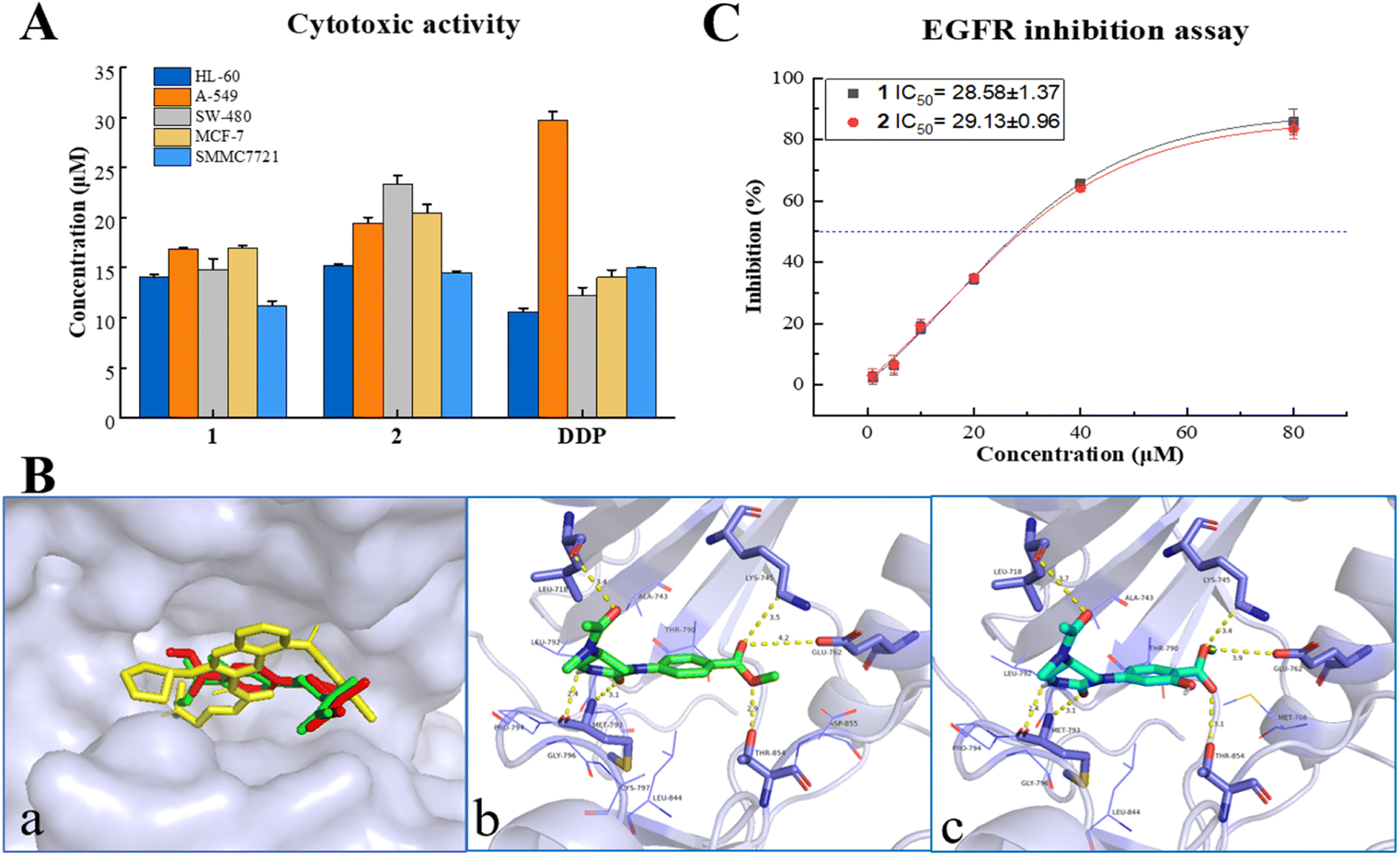 | ||
| Fig. 3 (A) Cytotoxicities of 1 and 2. (B) Molecular docking on EGFR: (a) Overlay docking positions of 1 (red), 2 (green), and afatinib (yellow) in the active site of EGFR (surface model). (b and c) The binding mode between EGFR and 1, 2, respectively. (C) IC50 values of EGFR inhibition for 1 and 2. | ||
In silico target prediction and molecular docking analysis
In silico studies nowadays play an important role in small molecule target prediction and help novel structures to find bioactive potential.14 To explore the possible target protein and obtain deeper insights into the intermolecular recognition mode, target prediction, and molecular docking simulation were performed.Of all the top 15 possible targets predicted by SwissADME for 1 and 2, epidermal growth factor receptor (EGFR), directly or indirectly related to the world's most common and deadly cancers, was investigated for molecular docking study. EGFR belongs to a family of four related receptor tyrosine kinases, which act as key mediators in cell signaling pathways involving cell proliferation, apoptosis, angiogenesis, and metastatic spread.15 As depicted in Fig. 3B, 1, 2, and the EGFR inhibitor, afatinib, were docked exactly on the ATP binding pocket of wild human EGFR tyrosine kinase (PDB: 6VHN). The EGFR/1 complex revealed a good least binding energy of −6.6 kcal mol−1, higher than that of the positive control, afatinib (−7.3 kcal mol−1). The binding profile of 1 showed that C-1, C-8 carbonyl groups interacted with MET 793 (3.1 Å) and LEU 718 (3.4 Å), 6-NH interacted with MET 793 (2.4 Å), 8′-O atom interacted with THR 854 (2.9 Å), C-7′ carbonyl group interacted with LYS 745 (3.5 Å), and GLU 762 (4.2 Å) (the same residues binding with co-crystallized ligand). Compound 2 displayed almost the same binding mode of 1 on the receptor site with a binding energy of −6.5 kcal mol−1 through several hydrogen bonds between C1O and MET 793 (3.1 Å), C8O and LEU 718 (3.7 Å), 6-NH and Met 793 (2.4 Å), 8′-O and THR 854 (3.1 Å), C7′ = O and LYS 745 (3.4 Å), GLU 762 (3.9 Å). The adopted molecular docking method was verified by redocking the co-crystallized ligand with small RMSD = 0.68 Å and a binding energy score of −7.6 kcal mol−1. In addition, physicochemical parameters for both Lipinski's and Veber's rules and pharmacokinetic properties (gastrointestinal absorption and blood–brain barrier) were determined for 1, 2, and afatinib proceeding by SwissADME.16 As shown in Table S7,† 1 and 2 obeyed the rule of five (RO5) criteria without any violations from Lipinski's Rule and passed through the Veber rule by topological polar surface area (TPSA) lower than 140 Å2. Meanwhile, compounds 1, 2, and afatinib showed high gastrointestinal absorption with no permeation to the blood–brain barrier. The new compounds 1 and 2 exhibited not only anti-proliferative activity but also promising oral bioavailability pharmacokinetic properties as the positive control, afatinib.
EGFR tyrosine kinase inhibition activity
EGFR overexpression is observed in cancers of the head and neck, ovary, cervix, bladder, oesophagus, stomach, brain, breast, endometrium, colon, and lung.17 The activation and autophosphorylation of intracellular tyrosine kinase domain of the receptors lead to several downstream signaling pathways, therewith eliciting the cascade of multiple events in the cytoplasm, as well as cell proliferation, survival, and apoptosis.18 International guidelines recommended EGFR inhibitors as the priority treatment for advanced EGFR mutation patients, regarding the higher efficacy and safety rather than standard chemotherapy.19 Due to the results of in vitro cytotoxicity and in silico simulation, the inhibition of 1 and 2 on EGFR tyrosine kinase was investigated using a commercial radio-labeled kit (Promega, China), with afatinib (Selleck, USA) as the positive control. Compounds 1 and 2 displayed a general inhibition effect on wild-type EGFR tyrosine kinase with IC50 values at 28.58 ± 1.37 and 29.13 ± 0.96 μM (Fig. 3C and Table S6†). It gave acceptable consequences coordinated with the cytotoxic and molecular docking results, and suggested that the cytotoxicity of alkaloids 1 and 2 was mediated by the inhibition on EGFR signaling. Natural alkaloids isolated from rot PN roots will provide a new scaffold candidate for developing and structural modification of novel EGFR-inhibitor.Conclusions
Further bioassay-guided phytochemical investigation of rot PN roots led to the identification of nine new non-triterpenoids including two alkaloids, and five lactones along with two biotransformed intermediates. Structurally, this study come to a consistent conclusion with our previous work that the oxidation degree was increased in the microbial-infected PN roots5a from the point of Ginseng polyalkynol. Moreover, compounds 1 and 2 revealed a noteworthy inhibition on five human cancer cell lines. The mechanism of anti-cancer action was explored by target prediction and molecular docking study, which simulated the recognition and interaction of the compounds with amino acid residues of EGFR tyrosine kinase mainly through hydrogen bonds inside the active site of the pocket. Finally, the EGFR inhibition experiment with a moderate inhibition effect verified that the EGFR signaling regulation pathway is probably involved in the anti-cancer cell lines of the active compounds. This work provided a natural candidate for probing and structural modification of novel type EGFR-inhibitor and new anti-cancer agent. Furthermore, the discovery of non-triterpenoid compounds enriched the chemical diversity of PN and reminded us that rot PN roots can be used as a special resource for bioactive components instead of being wasted or thrown away.Author contributions
Jia-Huan Shang carried out all the experiments, and prepared the manuscript; Yi-Jun Qiao reviewed and revised the manuscript; Dong Wang and Hong-Tao Zhu participated in the research; Chong-Ren Yang supervised the project; Ying-Jun Zhang designed and directed the project, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Major Science and Technique Programs in Yunnan Province, P. R. China (No. 202203AC100008, 2013FC008, 2016ZF001-01). The authors are grateful to the members of the analytical and bioactivity screening groups at the State Key Laboratory of Phytochemistry and Plant Resources in West China, KIB, CAS, for measuring the spectroscopic data, and cytotoxic and EGFR tyrosine kinase inhibition activities.Notes and references
- (a) L. P. Christensen, Adv. Food Nutr. Res., 2009, 55, 1–99 CAS; (b) L. W. Qi, C. Z. Wang and C. S. Yuan, Nat. Prod. Rep., 2011, 28, 467–495 RSC; (c) P. G. Xia, S. C. Zhang, Z. S. Liang and Z. H. Qi, Chin. Tradit. Herb. Drugs, 2014, 45, 2564–2570 CAS.
- (a) T. Wang, R. X. Guo, G. H. Zhou, X. D. Zhou, Z. Z. Kou, F. Sui, C. Li, L. Y. Tang and Z. J. Wang, J. Ethnopharmacol., 2016, 188, 234–258 CrossRef CAS PubMed; (b) L. F. Huang, H. L. Shi and B. Gao, Thromb. Res., 2014, 133, 848–854 CrossRef CAS PubMed; (c) Z. Wang, J. Y. Yang and S. J. Song, Chin. J. New Drugs, 2014, 23, 356–359 CAS.
- (a) M. Hisashi, K. Mitsuo, Y. Hiroshi, M. Masato and T. Katsumi, Chem. Pharm. Bull., 1989, 37, 1279–1281 CrossRef PubMed; (b) M. Hisashi, Cancer Chemother. Pharmacol., 1995, 4, 291–296 Search PubMed; (c) C. M. Teng, S. C. Kuo, F. N. Ko, J. C. Lee, L. G. Lee, S. C. Chen and T. F. Huang, BBA Gen. Subj., 1989, 990, 315–320 CrossRef CAS PubMed; (d) M. Kobaisy, Z. Abramowski, L. Lermer, G. Saxena, R. E. Hancock, G. H. Towers, D. Doxsee and R. W. Stokes, J. Nat. Prod., 1997, 60, 1210–1213 CrossRef CAS PubMed.
- (a) Z. S. Mao, Y. J. Long, S. S. Zhu, Z. J. Chen, F. G. Wei, Y. X. Zhu and X. H. He, J. Chin. Med. Mater., 2013, 36, 2051–2054 CAS; (b) W. F. Luo, S. F. Yu, C. F. He, Z. Y. Li, C. L. Wang and X. M. Cui, Acta Phytopathol. Sin., 1997, 27, 85–91 Search PubMed.
- (a) J. H. Shang, W. J. Sun, H. T. Zhu, D. Wang, C. R. Yang and Y. J. Zhang, J. Ginseng Res., 2020, 44, 405–412 CrossRef PubMed; (b) J. H. Shang, G. W. Xu, H. T. Zhu, D. Wang, C. R. Yang and Y. J. Zhang, Nat. Prod. Bioprospect., 2019, 9, 287–295 CrossRef CAS PubMed.
- Y. M. Shi, W. I. Xiao, J. X. Pu and H. D. Sun, Nat. Prod. Rep., 2015, 32, 367–410 RSC.
- M. Kurz, P. Schmieder and H. Kessler, Angew. Chem., 1991, 103, 1341 CrossRef CAS.
- R. F. Ma, K. Hu, W. P. Ding, B. Wang, T. B. He, X. N. Li, H. D. Sun and P. T. Puno, Phytochemistry, Elsevier, 2021, vol. 182, p. 112589 Search PubMed.
- U. Goergens and M. P. Schneider, Tetrahedron: Asymmetry, 1992, 3, 831 CrossRef CAS.
- L. L. Lin, Z. L. Chen, X. Yang, X. H. Liu and X. M. Feng, Org. Lett., 2008, 10, 1311–1314 CrossRef CAS PubMed.
- M. Kobayashi, T. Mahmud, T. Umezome, W. Wang and I. Kitagawa, Tetrahedron, 1997, 53, 15691–15700 CrossRef CAS.
- (a) W. Lu, G. R. Zheng, D. X. Gao and J. C. Cai, Tetrahedron, 1999, 55, 7157–7168 CrossRef CAS; (b) M. Satoh, M. Ishii, M. Watanabe, K. Isobe, T. Uchiyama and Y. Fujimoto, Chem. Pharm. Bull., 2002, 50, 126–128 CrossRef CAS PubMed.
- B. Y. Wang, X. Q. Yang, M. Hu, L. J. Shi, H. Y. Yin, Y. M. Wu, Y. B. Yang, H. Zhou and Z. T. Ding, J. Ginseng Res., 2020, 44, 770–774 CrossRef PubMed.
- K. Nepali, S. Sharma, M. Sharma, P. M. S. Bedi and K. L. Dhar, Eur. J. Med. Chem., 2014, 77, 422–487 CrossRef CAS PubMed.
- (a) Y. Yatabe and T. Mitsudomi, Pathol. Int., 2007, 57, 233–244 CrossRef CAS PubMed; (b) A. Ayati, S. Moghimi, S. Salarinejad, M. Safavi, B. Pouramiri and A. Foroumadi, Bioorg. Chem., 2020, 99, 103811 CrossRef CAS PubMed.
- (a) C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS; (b) D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- D. S. Krause and R. A. Van Etten, N. Engl. J. Med., 2005, 353, 172–187 CrossRef CAS PubMed.
- (a) H. H. Sedlacek, Drugs, 2000, 59, 435–476 CrossRef CAS PubMed; (b) P. A. Schwartz and B. W. Murray, Bioorg. Chem., 2011, 39, 192–210 CrossRef CAS PubMed.
- G. A. Masters, S. Temin, C. G. Azzoli, G. Giaccone, S. Baker, J. R. Brahmer, P. M. Ellis, A. Gajra, N. Rackear, J. H. Schiller, T. J. Smith, J. R. Strawn, D. Trent and D. H. Johnson, J. Clin. Oncol., 2015, 33, 3488–3515 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, experimental details, characterization of compounds, in vitro and in silico biological study, and NMR copies of new compounds. CCDC 2096666. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra00720k |
| This journal is © The Royal Society of Chemistry 2023 |