
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProduction of aromatic hydrocarbons from lignin derivatives by catalytic cracking over a SiO2–Al2O3 catalyst†
Dequan Zhangac,
Xinghua Zhang *b,
Han Yinc,
Qingqi Zhenga,
Longlong Mabc,
Song Lic,
Yuchun Zhang*a and
Peng Fu*a
*b,
Han Yinc,
Qingqi Zhenga,
Longlong Mabc,
Song Lic,
Yuchun Zhang*a and
Peng Fu*a
aSchool of Agricultural Engineering and Food Science, Shandong Research Center of Engineering & Technology for Clean Energy, Shandong University of Technology, Zibo 255000, PR China. E-mail: zhangxh@seu.edu.cn
bKey Laboratory of Energy Thermal Conversion and Process Measurement and Control of Ministry of Education, School of Energy and Environment, Southeast University, Nanjing, Jiangsu 210096, PR China
cKey Laboratory of Renewable Energy, Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, PR China
First published on 5th April 2023
Abstract
Catalytic cracking of phenolic compounds to aromatic hydrocarbons is vital to the utilization of lignin. In this work, pristine amorphous SiO2–Al2O3 was used as a catalyst to produce aromatic hydrocarbons from lignin-derived phenolics by catalytic cracking using methanol as the solvent. These catalysts were characterized by various techniques (XRD, NH3-TPD, Py-IR, etc.) and evaluated on a fixed bed reactor using guaiacol as a model compound. The effects of reaction temperature, the flow of carrier gas, the molar ratio of guaiacol to methanol, and WHSV were investigated. 33-SA (SiO2–Al2O3 with the SiO2 content of 33%) exhibited the best catalytic activity due to its high content of Lewis acid sites (168.47 μmol g−1). Co-feeding with methanol promoted the removal of oxygen atoms and improved the reaction system H/Ceff. Under the optimal conditions of 400 °C, 25 mL min−1 N2, a molar ratio of methanol to guaiacol of 25, and WHSV of 8/3 h−1, the yield of aromatic hydrocarbons reached 57.93%. The deactivating species in the transformation of guaiacol into aromatic hydrocarbons on catalysts were also studied.
1. Introduction
Lignin is the only plant resource with a benzene ring structure and has been investigated as a promising alternative energy source for the production of biofuels,1–3 aromatics,4,5 monomeric phenols6,7 and other derived chemicals. Fast pyrolysis is considered one of the most promising methods for converting lignin into liquids (bio-oil). However, the application of bio-oil is limited due to its high oxygen content, poor stability, low calorific value and high corrosiveness. Therefore, pyrolysis oil must be catalytically upgraded before it can be used in the fuel industry. Guaiacol is a typical lignin derivative containing the major functional groups of lignin-derived phenols, such as hydroxyl and methoxy groups. The catalytic upgrading of guaiacol to aromatic hydrocarbons is essential to improve the quality of bio-oil.Many methods such as thermal cracking, catalytic cracking, and hydrogenation, have been developed to transform bio-oil.8,9 Direct cracking of bio-oil produces a low yield of aromatic hydrocarbons and causes significant carbon deposition. It is available to improve the quality of pyrolysis oil significantly through hydrogenation, but this requires a high-pressure operating environment.10 Catalytic cracking of bio-oil is a promising method for improving quality. However, the low yield of the target product and severe coking are the main defects in the cracking process.11–14 Zhang et al. introduced the concept of (H/Ceff) and pointed out that serious carbon deposits will occur when the raw materials H/Ceff are lower than 1.15 During pyrolysis, a large number of free radical fragments will be generated as a result of the complex structure of biomass. Without hydrogen donors, these unsaturated components will undergo polymerization and rearrangement reactions to form oligomers, and further polycondensation will generate carbon deposits, reducing aromatic hydrocarbon yield.16 Presently, hydrogen donors are provided for the cracking process in two main ways: (1) hydrotreat the feedstock before cracking.17 (2) co-cracking with some hydrogen-rich compounds during cracking.18 Valle et al. found that due to co-feeding with methanol, the conversion rate of bio-oil in raw materials was 90%, and the selectivity of aromatics was 40%.19 By changing the ratio of methanol to raw material, Zhang et al. found that it promoted the deoxidation of furan ring, thus increasing the aromatic hydrocarbon yield and reducing the generation of coke.10 Mentzel et al. diluted the small molecules of bio-oil with methanol. They observed that the conversion capacity of the catalyst could be increased by 10 times, and the presence of methanol extended the service life of the catalyst.20 Therefore, using methanol as a co-cracking reactant can increase the H/Ceff of the reactant and suppress the coke formation.
The excellent catalytic performance and economic efficiency of SiO2–Al2O3 have been widely reported in the catalytic conversion of biomass. In the reaction of diphenyl ether hydrodeoxygenation, Yang et al. found that Ni/SiO2–Al2O3 showed a better deoxygenation effect and benzene selectivity than Ni/Al2O3 (60 vs. 47%).21 The alkylation reaction of phenol and 1-octene was investigated by Mehraban et al. using SiO2–Al2O3 with different Si/Al ratios. The results indicate that SiO2–Al2O3 with a Si/Al ratio of 25 shows the highest activity and monoalkyl phenol selectivity.22 The Si/Al ratio of catalysts significantly impacts their physical properties (e.g., surface area and pore size) and acidic strength, resulting in different catalytic properties exhibited in the reactions. Considering the deoxygenation effect and alkylation ability of SiO2–Al2O3, we selected SiO2–Al2O3 with different SiO2 content and γ-Al2O3 to investigate their effects on the catalytic co-cracking of guaiacol with methanol.
This work studied the conversion of guaiacol to aromatic hydrocarbons by co-pyrolysis with methanol. The relationship between acidity and catalytic performance and the positive effect of methanol co-feeding were emphasized. The conversion of anisole, phenol, and guaiacol mixtures was studied. In addition, the possible reaction paths of guaiacol were deduced by the characterization results and product distribution of the catalysts.
2. Material and methods
2.1 Materials
Guaiacol, phenol, anisole, and 1,3,5-trimethoxybenzene, and γ-Al2O3 were purchased from Macklin Biochemical Co., Ltd (Shanghai, China). Acetone and methanol were obtained from the Guangzhou Chemical Reagent Factory. These chemicals were directly used without any pretreatment. SiO2–Al2O3 was purchased from Shanxi Juhua New Material Technology Co., Ltd, which was named x-SA (x is the SiO2 content). All catalysts were pressed and sieved to 40–60 mesh before use and calcined at 550 °C for 3 hours.2.2 Catalyst characterization
XRD analysis of the catalyst was carried out by X'Pert Pro MPD X-ray diffraction with a scanning step length of 0.02° and a scanning speed of 2° min−1. The texture structure of catalysts were measured by N2 adsorption and desorption on a QUADRASORB SI-MP-10 automatic adsorption apparatus. Before the N2 adsorption test, the catalyst samples were degassed at 300 °C for 12 hours.The acidity of the catalyst was analyzed by NH3 temperature programmed desorption (NH3-TPD) on a QuantachromeChemStar TPx chemisorption analyzer. To remove the moisture and impurities adsorbed on the surface of catalyst, a 100 mg sample was purified with helium (60 mL min−1) at 400 °C for 60 minutes, cooled to 100 °C, and exposed to 10% NH3/He (60 mL min−1) for 60 minutes. Afterwards, the sample was flushed with He gas for an hour at 100 °C. Then, the temperature was increased to 850 °C at a rate of 10°C min−1 and the desorbed NH3 was monitored on-line by thermal conductivity.
Pyridine IR analysis was conducted by Brucker's Tensor-27 FTIR. Under a high vacuum condition, the catalyst sample was pretreated at 350 °C for 2 hours. Pyridine was introduced in the IR cell when the catalyst sample was cooled to room temperature. The temperature was then raised to 150 °C and held for 5 minutes to collect the spectra.
2.3 Catalytic pyrolysis test
Catalytic pyrolysis of lignin-derived phenolics compounds was conducted on a fixed bed (see Fig. 1). In a typical procedure, equal volumes of quartz sand (40–60 mesh) and catalyst (40–60 mesh, 0.3–0.9 g) were mixed in the middle of the reaction tube. The reactor was heated to a desired temperature by an electric furnace, then carrier gas (N2, 50 mL min−1) and feedstocks (the mixtures of methanol and phenolics compounds) were quantitatively introduced into the reaction tube, and the catalytic pyrolysis experiment was started. The products and unreacted reactants were collected by double cold traps (−10 °C) using acetone as an adsorbent. | ||
| Fig. 1 Flowchart of the experimental setup. | ||
Liquid product composition was analyzed qualitatively by GC-MS (TRACE 1300ISQ) with HP-Inowax capillary column (30 m × 0.25 mm × 0.25 m). 60 °C (holding for 2 min) was set as the initial temperature. It was heated to 250 °C at a rate of 10 °C min−1 and maintained for 10 minutes. The quantitative analysis was conducted by gas chromatography (GC, Shimadzu-2014) equipped with a flame ionization detector (FID) and an HP-Inowax capillary column. The program of column temperature was consistent with the qualitative analysis of GC-MS. The spent catalyst was analyzed using an AVANCE III HD 600 MHz solid-state 13C nuclear magnetic resonance spectrometer (NMR) and a Waters SDT650 thermogravimetric analyzer (TG).
The conversion of the reactants was calculated based on eqn (1).
 | (1) |
The yield of aromatic product i can be calculated based on eqn (2).
 | (2) |
3. Result and discussion
3.1 Catalyst characterization
The XRD patterns of the aluminum silicate and γ-Al2O3 are shown in Fig. 2. The peaks centered at about 37.5°, 48.3° and 65.8° were clearly observed, which can be attributed to the characteristic peaks of γ-Al2O3.23 For the aluminum silicate, the characteristic peaks of Al2O3 disappeared while the broad dispersion peak (centered at about 22°) was observed for all samples. | ||
| Fig. 2 Wide-angle XRD spectrum. | ||
Fig. 3 presents the pore size distributions of catalysts. The diameters of these catalysts are primarily concentrated in the range of 6–50 nm. The most probable aperture distribution of catalyst sample moves to the direction of large size with the increase of SiO2 content.
 | ||
| Fig. 3 Pore size distribution. | ||
The texture structure of catalysts is exhibited in Table 1. The specific surface area, pore volume, and average pore size of SA are better than that of γ-Al2O3. This result suggests that the addition of SiO2 can improve the texture structure of γ-Al2O3. In addition, the specific surface area decreased with the increasing of SiO2 content.
| Catalyst | SBET (m2 g−1) | Vtotal (cm3 g−1) | DPore (nm) |
|---|---|---|---|
| a SBET: specific surface area; VTotal: total volume; DPore: average pore size. | |||
| 33-SA | 379.05 | 1.26 | 6.64 |
| 44-SA | 279.58 | 0.81 | 5.76 |
| 50-SA | 267.63 | 0.76 | 5.70 |
| 80-SA | 215.44 | 0.89 | 8.25 |
| γ-Al2O3 | 196.84 | 0.49 | 4.99 |
Based on the desorption peak of NH3-TPD profile shown in Fig. 4a, two prominent peaks, namely the weak acid peak in the low-temperature region (60–320 °C) and the strong acid peak in the high-temperature region (430−780 °C) were observed clearly. As the SiO2 content in the catalysts increased to 80%, the strong acid peak shifted towards the high temperature direction and the peak areas became smaller. According to the areas of the desorption peak, 33-SA had the highest number of acid sites.
 | ||
| Fig. 4 Acidity of catalysts: (a) NH3-TPD curves, (b) Py-IR curves. | ||
As shown in Table 2, the total acid sites of γ-Al2O3 is 820.7 μmol g−1, while that of 33-SA is 1290 μmol g−1. This suggests that the addition of SiO2 can increase the number of acid sites. However, the number of acid sites decreased with increasing SiO2 content. The number of acid sites of aluminum silicate is lower than that of γ-Al2O3 when the content of SiO2 increased to 80%. This is similar to the results obtained by Hernandez et al. who pointed out that the number of acid sites decreased when the Si/Al of the SiO2–Al2O3 was increased from 0.25 to 20.24 The acid sites of the catalyst originated from the unsaturated coordination of aluminum atoms. Namely, the acid sites are related to the Al–O–Al and Al–O–Si structures, while the Si–O–Si is an inactive structure. Therefore, it is plausible that the inactive Si–O–Si structure of aluminum silicate increased with the increase of SiO2 content, leading to a decrease of acid sites.
| Catalyst | (μmol NH3 g−1) | (μmol Pyrazine g−1) | |||
|---|---|---|---|---|---|
| Weak | Strong | Total | Lewis | ||
| 33-SA | 699.43 | — | 590.57 | 1290 | 168.47 |
| 44-SA | 599.26 | — | 363.14 | 962.4 | 133.42 |
| 50-SA | 548.02 | — | 308.38 | 856.4 | 124.80 |
| 80-SA | 390.95 | 190.87 | 317.88 | 709.7 | 87.64 |
| γ-Al2O3 | 366.17 | — | 454.53 | 820.7 | 146.69 |
Table 2 shows the results for the concentrations of Lewis acid calculated from FTIR bands. As shown in Fig. 4b, a major peak centered at about 1455 cm−1 was observed, suggesting the presence of Lewis acid sites of SA catalyst. The weak peak centered at about 1540 cm−1 can be assigned to the Brønsted sites. The peak centered at 1490 cm−1 belongs to the superposition peak of Brønsted acid and Lewis acid. From pyridine adsorption spectra, the increase of SiO2 decreases the amount of Lewis acid sites.25
3.2 Catalytic activity test
Pyrolysis of guaiacol was investigated over different solid acid catalysts at 400 °C. As shown in Fig. 5, 33-SA catalyst promotes the best yield of aromatic hydrocarbons and the best conversion of guaiacol, closely related to its excellent specific surface area and appropriate pore size distribution. However, the lowest yield of aromatic hydrocarbons was not correspondingly achieved by γ-Al2O3 with the lowest specific surface area but by 80-SA, suggesting that the specific surface area of the catalysts did not always correspond to the yield. As shown in Fig. 3, the pore size of each catalyst was concentrated in the range of 6–50 nm. The difference in pore size between γ-Al2O3 and 33/44-SA is small, but the catalytic performance of γ-Al2O3 is superior to that of 33/44-SA. Therefore, it was inferred that the pore size was not the main reason for the difference in aromatic hydrocarbon yield in the experiments, which was similar with the result of reported ref. 26.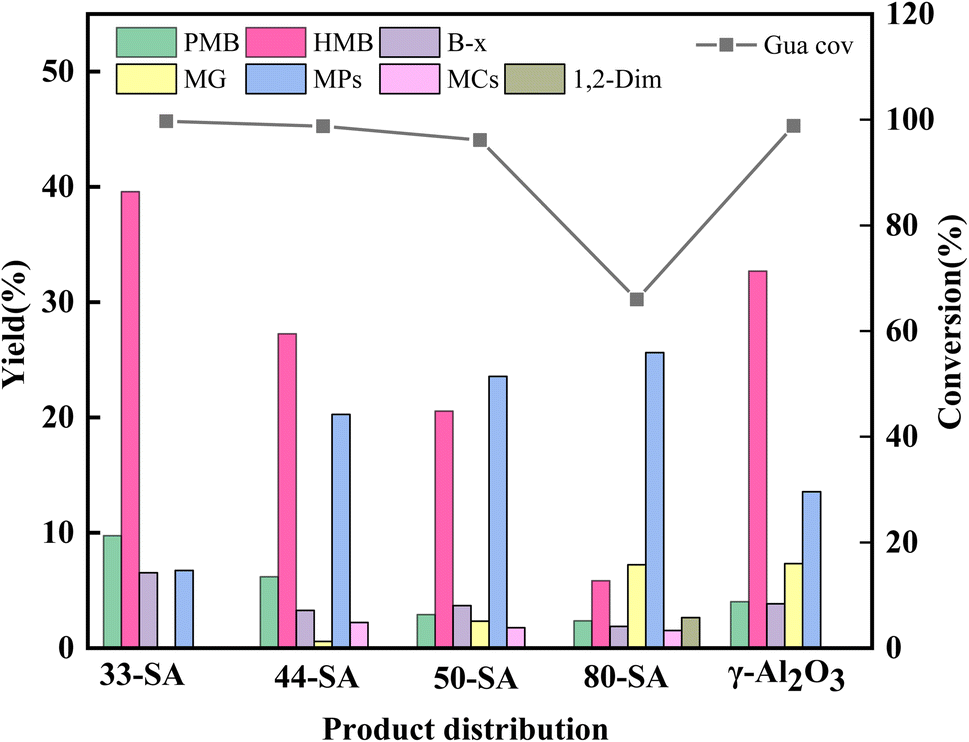 | ||
| Fig. 5 Product yield of guaiacol pyrolysis over different catalysts (PMB: pentamethylbenzene, HMB: hexamethylbenzene, MG: methylated guaiacol, MPs: methylated phenol, MCs: catechol and methylated catechol,1,2-dim: 1,2-dimethoxybenzene.). Reaction conditions: methanol to guaiacol molar ratio = 25, temperature = 400 °C, reaction time = 60 min, N2 flow rate = 50 mL min−1, WHSV = 4 h−1. | ||
According to Fig. 5, the yields of aromatic hydrocarbons over 33-SA, 44-SA, 50-SA, 80-SA, and γ-Al2O3 are 57.36%, 38.76%, 29.68%, 11.43%, and 44.46%, respectively. The 33-SA catalyst with higher Lewis acidic sites (168.47 μmol g−1) is more catalytically active. In response to an increase in SiO2 content, the Lewis acid sites decreased, and reactants conversion rates and aromatic hydrocarbons yields decreased significantly. Lewis acid is an efficient active site for cleaving C–O bonds in various lignin compounds.27 Due to the Lewis acidity of aluminum reagents, less reactive alkyl esters can be methylated, and aryl methyl ethers can be converted to valuable products after cleavage of the C–O bond and alkylation.28,29 Moreover, Ma et al. found that Ru/Nb2O5 with rich Lewis acid sites was crucial for the cleavage of cresol C–O bonds and aromatic hydrocarbons formation. The aromatic hydrocarbons yield positively correlates with the Lewis content of niobium-based supports.30 The reason for this is that Lewis acid can reduce the activation energy required for a reaction and is very effective when it comes to demethylation and transalkylation.31 Accordingly, the Lewis acid content is the main reason why 33-SA shows the best catalytic performance in the coupled co-cracking of guaiacol and methanol.
3.3 Reaction conditions optimization
As seen in Fig. 6a. It was challenging to pyrolyze guaiacol at 300 °C. The products consisted primarily of aromatic oxygenates. The highest yield of PMB and HMB were achieved at a reaction temperature of 400 °C, reaching 48.31%. By further increasing the reaction temperature to 450 °C, the yield of PMB and HMB decreased to 33.75%. Therefore, 400 °C is considered a suitable temperature for the catalytic conversion of guaiacol. | ||
| Fig. 6 Effects of different parameters on catalytic cracking. Reaction conditions: (a) N2 = 50 mL min−1, WHSV = 4 h−1, methanol to guaiacol molar ratio = 25; (b) temperature = 400 °C, WHSV = 4 h−1, methanol to guaiacol molar ratio = 25; (c) temperature = 400 °C, N2 = 50 mL min−1, methanol to guaiacol molar ratio = 25; and (d) temperature = 400 °C, N2 = 50 mL min−1, WHSV = 4 h−1. | ||
The effects of N2 flow rate and WHSV are presented in Fig. 6b and c. As the N2 flow rate gradually increased, from 25 mL min−1 to 100 mL min−1, the conversion rate of the guaiacol and the yield of HMB and PMB both declined. In the case of N2 flow rate of 25 mL min−1, the yield of PMB and HMB was 54.36%. PMB and HMB yield increased from 13.68% to 48.31% as WHSV decreased from 8 h−1 to 8/3 h−1. Reducing the WHSV did not result in a significant change in the yield of aromatic hydrocarbons. Therefore, the optimal reaction condition of the WHSV was 8/3 h−1. Generally, we believe that at lower amounts of carrier gas flow, the less gas that passes through the catalyst per unit of time, the more intermediates can be alkylated effectively.32
The effect of the molar ratio of methanol to guaiacol on catalytic performance was investigated at 400 °C with a N2 flow rate of 50 mL min−1 and a WHSV of 4 h−1. It can be seen from Fig. 6d that as the molar ratio of methanol to guaiacol increased from 10 to 25, the yield of PMB and HMB increased from 16.47% to 48.31%. It is demonstrated that the addition of methanol increases the H/Ceff in the reaction system, thus causing guaiacol to be cleaved and alkylated into aromatic hydrocarbons. However, when the molar ratio of methanol to guaiacol was increased to 30, the yield of PMB and HMB declined to 42.56%. In this case, it may be due to the increase of H/Ceff, methanol and guaiacol competed for adsorption on the active sites, and excessive dehydration reactions occurred, which inhibited the cleavage of guaiacol C–O bonds and alkylation.33
3.4 Stability of catalysts
As 33-SA showed the best catalytic activity, its stability and recoverability were further investigated. During the continuous reaction, the 33-SA catalyst was left untreated and continued to be used under the reaction conditions shown in Fig. 5. To determine the continuity of catalytic performance by measuring the yield of HMB and PMB. Based on the experimental results shown in Table S2,† the HMB and PMB yield of the catalyst decreased significantly when it was used for the second time, indicating that it had become seriously deactivated during the last continuous use. In Fig. S3,† TG curves of C1 and C2 catalysts in the air were analyzed to confirm the cause of catalyst deactivation. The weight loss rate of the catalyst after one use is 30.98%, and the weight loss rate after repeated use is 39.86%.Evaluate the recoverability of catalysts through regeneration studies. The spent catalyst was calcined at 550 °C for three hours in air atmosphere at a muffle furnace. Regeneration reactions of the catalyst are consistent with those shown in Fig. 5. A total of five cyclic regeneration experiments are conducted, numbered 1–5. It is shown in Fig. 7 that the conversion of guaiacol and the yield of the HMB and PMB did not change significantly after five cyclic regenerations of the catalyst. Consequently, 33-SA has very high reproducibility and a great deal of commercial potential.
 | ||
| Fig. 7 Renewability experiment of the 33-SA catalyst. | ||
3.5 Reaction pathway discussions
Products distribution of guaiacol pyrolysis at different temperatures is shown in Table 3, and the transformation pathway of guaiacol is proposed. According to Fig. 6a, the conversion rate of reactants is low at 300 °C, and almost no aromatic hydrocarbons are produced. Among these products are methylated guaiacol (MG), methylated phenol (MPs), catechol and methylated catechol (MCs), and 1,2-dimethoxybenzene, whose yields are respectively 15.55%, 19.39%, 4.87%, and 7.82%. Considering that the bond dissociation energy of C–O in guaiacol (Caryl–OH: 414 kJ mol −1, Caryl–OCH3: 356 kJ mol −1, and Calkyl–O: 245 kJ mol −1), the Calkyl–O is preferentially cleaved to produce free radical fragments, which are then rearranged to form catechol and 1,2-dimethoxybenzene.34 Caryl–OH requires the highest bond dissociation energy, so the yield of phenolic compounds containing Caryl–OH is the highest. As the temperature increased from 300 °C to 350 °C, the yields of MG, MCs, and 1,2-dimethoxybenzene decreased, whereas the yield of MPs increased. When the temperature was raised further, the yields of MG, MCs, 1,2-dimethoxybenzene, and MPs decreased, which indicated that MG, MCs, and 1,2-dimethoxybenzene were converted into MPs first, and MPs was then converted into PMB and HMB. The yield of PMB and HMB increased with increasing temperature, and when the temperature reached 400 °C, the yield of HMB reached 38.92%.| a Reaction condition: catalyst = 33-SA, N2 = 50 mL min−1, WHSV = 4 h−1, methanol to guaiacol molar ratio = 25. | ||||
|---|---|---|---|---|
| Reactant | Guaiacol | Guaiacol | Guaiacol | Guaiacol |
| Temperature | 300 | 350 | 400 | 450 |
| Conversion (%) | ||||
| Guaiacol | 66.03 | 86.89 | 99.86 | 98.43 |
| Yield (%) | ||||
| PMB | 3.73 | 4.66 | 9.49 | 7.79 |
| HMB | 3.05 | 16.45 | 38.92 | 25.96 |
| Methylated guaiacol | 15.55 | 8.42 | — | — |
| Benzene, 1,2-dimethoxy | 7.82 | 4.04 | — | — |
| Catechol and methylated catechol | 4.87 | 3.19 | — | — |
| Mesitylene | — | 0.34 | 1.35 | 3.37 |
| Benzene, 1,2,4,5-tetramethyl | — | 0.57 | 2.29 | 3.84 |
| Benzene, 1,2-diethyl-3,4-dimethyl | — | 0.95 | 1.35 | 2.45 |
| Phenol, 2,6-dimethyl- | 0.33 | 0.34 | 0.21 | 11.55 |
| Phenol, 2,4,6-trimethyl- | 2.07 | 3.4 | 0.72 | 3.66 |
| Phenol, 2,3,5,6-tetramethyl | 7.52 | 10.49 | 2.37 | 5.28 |
| Phenol,2-(1,1-dimethylethyl)-3-methyl | 9.47 | 15.02 | 1.98 | 4.54 |
Based on the above results, a possible reaction pathway for the coupled transformation of guaiacol and methanol is proposed. Methanol is considered a source of H-species which can provide more hydrogen for immobilizing aromatic ring fragments.35 In the presence of methanol, guaiacol can be directly methylated to MG and 1,2-dimethoxybenzene. According to Scheme 1, MCs can be generated in three ways. Since 33-SA is an active catalyst during alkylation transfers, guaiacol can be directly demethylated to catechol, as well as converted to methylated catechol via methyl transfer. Furthermore, 1,2-dimethoxybenzene can form MCs by methylation and methyl transfer. According to a study on the generation of HMB by co-pyrolyzing phenol and methanol, phenol is first converted into hydroxypentaxaethylbenzeneonium ion via hydrogen transfer. Afterward, carbon ions are converted to PMB by extracting hydride ions from methanol with one mole of water lost. Lastly, PMB is converted to HMB by methylation.36 Thus, MCs undergo the above reaction pathway and methylation to generate MPs, and then MPs are dehydroxylated and methylated again to generate polymethylbenzene. Since methanol provides sufficient methyl groups, PMB and HMB are the main aromatic products. Furthermore, methanol is cracked to CO and H2 after the hydrogen transfer reaction. Aside from the coupling conversion with guaiacol, methanol also undergoes a side reaction, which leads to the formation of DME, which can be further decomposed into CH4, CO, and H2.
 | ||
| Scheme 1 Possible reaction paths of guaiacol and methanol on 33-SA. Red: detected products, blue: reactants. | ||
Additionally, two typical phenolic compounds (phenol and anisole) were pyrolyzed over 33-SA. A summary of the reactant conversions and aromatic compounds yields is given in Table 4. Interestingly, in a reaction at 300 °C, anisole appears in the pyrolysis products of phenol, and phenol in the pyrolysis products of anisole. This is due to the fact that under the action of the Lewis acid sites, phenol can then be methylated to anisole, and anisole can be demethylated to phenol.37,38 At the same time, a large amount of MPs was detected in the product. These evidences indirectly prove the transfer path of guaiacol functional groups and the feasibility of schedule 1. As the temperature was raised to 400 °C, the conversion of phenol and anisole was close to 100%. During the pyrolysis of phenol/anisole, aromatic hydrocarbons yield was higher than that of guaiacol, suggesting that phenolic compounds with relatively simple structures are more easily converted.
| a Reaction condition: catalyst = 33-SA, N2 = 50 mL min−1, WHSV = 4 h−1, methanol to phenolic compounds molar ratio = 25. | ||||
|---|---|---|---|---|
| Reactant | Phenol | Anisole | Phenol | Anisole |
| Temperature | 300 | 300 | 400 | 400 |
| Conversion (%) | ||||
| Phenol | 63.34 | — | 99.28 | — |
| Anisole | — | 59.4 | — | 98.7 |
| Yield (%) | ||||
| PMB | 4.38 | 3.68 | 10.19 | 8.99 |
| HMB | 2.87 | 2.74 | 52.45 | 49.76 |
| Methylated anisole | — | 10.72 | — | — |
| Mesitylene | — | — | 0.71 | 0.82 |
| Benzene, 1,2,4,5-tetramethyl | — | — | 1.68 | 0.79 |
| Benzene, 1,2-diethyl-3,4-dimethyl | — | — | 2.07 | 3.32 |
| Phenol | — | 8.83 | — | — |
| Anisole | 7.6 | — | — | — |
| Guaiacol | — | 2.38 | — | — |
| Phenol, 2,6-dimethyl- | 7.12 | 3.89 | 0.76 | 1.42 |
| Phenol, 2,4,6-trimethyl- | 10.35 | 3.56 | — | 1.27 |
| Phenol, 2,3,5,6-tetramethyl | 5.54 | 2.83 | 0.52 | 0.23 |
| Phenol, 2-(1,1-dimethylethyl)-3-methyl | 8.32 | 6.54 | 1.42 | — |
3.6 Coke analysis
A TG/DTG curve for the spent catalyst is shown in Fig. 8. The temperature was programmed to 800 °C at 10°C min−1 in an air atmosphere. The TG curve indicates that 33-SA has obvious coke, whereas γ-Al2O3 exhibits less coke, which is inconsistent with NH3-TPD results. It indicates that although the active center will encourage more coke deposition, other factors (e.g., specific surface area and pore structure) also play a decisive role. | ||
| Fig. 8 TG/DTG curves of the spent catalysts (Reaction conditions of catalysts coking: methanol to guaiacol molar ratio = 25, temperature = 400 °C, reaction time = 60 min, N2 flow rate = 50 mL min−1, WHSV = 4 h−1.). | ||
According to this deduction, comparing the texture data of fresh and spent catalysts (Tables 1 and 5), the specific surface area and pore structure are indeed important factors affecting carbon accumulation.39 As a result of the abundant Lewis acid sites and the high specific surface area of 33-SA, it provides conditions for the aggregation of macromolecules during catalytic conversion.40 The maximum weight loss for SA catalysts occurred around 530 °C and a gradual broadening of the weight loss peak was observed between 33-SA and 80-SA, suggesting that a range of different coke species (e.g. hydrocarbons (CxHy), amorphous carbon and graphitic carbon) are deposited on the aluminum silicate, all of which contribute to the catalytic accumulation of carbon.39 Interestingly, the maximum weight loss temperature of γ-Al2O3 is 500 °C, which is different from that of aluminum silicate.
| Spent catalyst |
|
|
|
|---|---|---|---|
a  , ,  and and 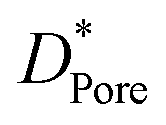 represent respectively the specific surface area, the total pore volume, and the average pore size of spent catalysts. represent respectively the specific surface area, the total pore volume, and the average pore size of spent catalysts. |
|||
| 33-SA | 217.21 | 0.58 | 10.61 |
| 44-SA | 193.38 | 0.52 | 10.67 |
| 50-SA | 175.11 | 0.49 | 11.08 |
| 80-SA | 234.61 | 0.83 | 14.22 |
| γ-Al2O3 | 162.45 | 0.38 | 9.34 |

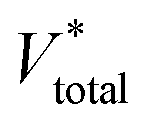

As shown in Fig. 9, the C13NMR spectra of spent catalysts indicated bands at 13 and 130 ppm corresponding to aliphatic carbonaceous nuclei, especially –CH3 groups and aromatic compounds. It has been confirmed that most of the coke formed during the co-conversion of methanol and guaiacol consists of aromatic species muti-substituted with –CH3 groups.41 The carbon bridge between aromatic rings did not appear between 130–140 ppm, which indicates that no coke product of PAHs exists. Additional bands were observed at 150 ppm and 180 ppm for γ-Al2O3, corresponding to polyalkylaromatics and carboxylic acid groups in the coke.42 This confirms that the carbon deposition in γ-Al2O3 differs from the carbon species in aluminum silicate.
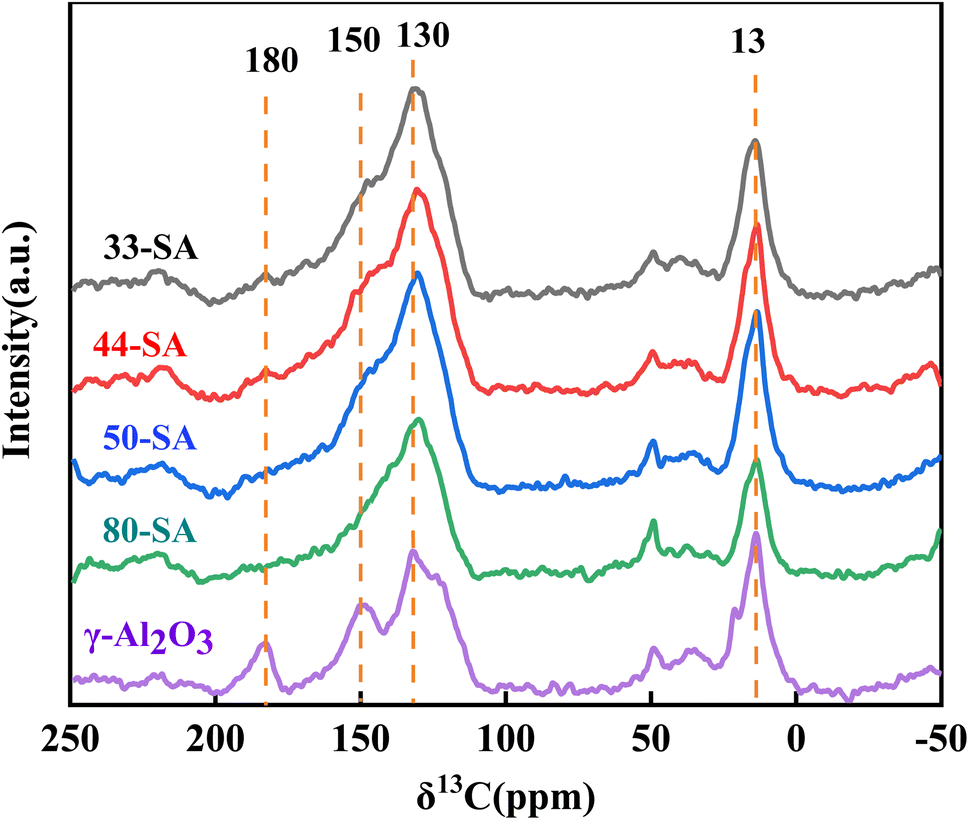 | ||
| Fig. 9 NMR spectrum of spent catalysts (Reaction conditions of catalysts coking: methanol to guaiacol molar ratio = 25, temperature = 400 °C, reaction time = 60 min, N2 flow rate = 50 mL min−1, WHSV = 4 h−1.). | ||
The degree of graphitization of coke was analyzed using Raman spectroscopy, as shown in Fig. S2.† A first observation is that all Raman spectra exhibit in-plane and out-of-plane C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond vibrational features. These bands appear at 1350 cm−1 (D) and 1600 cm−1 (G). In addition, the bands at 1260 cm−1, 1450 cm−1, and 1580 cm−1 are attributed to unique structures in coke. The five-band technique was used to fit the first-order Raman spectra of all catalysts. The distribution of the five bands is shown in Table S1.† A typical 33-SA curve fitting diagram is shown in Fig. 10.42,43 The intensity of the above-mentioned bands and the D/G band intensity ratio are shown in Table 6. The values of G band intensities increase as the content of SiO2 in aluminosilicate decreases. The results demonstrate that the proportion of coke (band G) is related to the amount of coke deposited on the catalyst.44 The 33-SA catalyst exhibits the most significant degree of graphitization.
C bond vibrational features. These bands appear at 1350 cm−1 (D) and 1600 cm−1 (G). In addition, the bands at 1260 cm−1, 1450 cm−1, and 1580 cm−1 are attributed to unique structures in coke. The five-band technique was used to fit the first-order Raman spectra of all catalysts. The distribution of the five bands is shown in Table S1.† A typical 33-SA curve fitting diagram is shown in Fig. 10.42,43 The intensity of the above-mentioned bands and the D/G band intensity ratio are shown in Table 6. The values of G band intensities increase as the content of SiO2 in aluminosilicate decreases. The results demonstrate that the proportion of coke (band G) is related to the amount of coke deposited on the catalyst.44 The 33-SA catalyst exhibits the most significant degree of graphitization.
 | ||
| Fig. 10 Raman spectrum of 33-SA. | ||
| Spent catalyst | VC–H | D | D3 | G | D2 | ID/IG |
|---|---|---|---|---|---|---|
| a Reaction condition of catalysts coking: temperature = 400 °C, N2 = 50 mL min−1, WHSV = 4 h−1, methanol to guaiacol molar ratio = 25. | ||||||
| 33-SA | 0.12 | 0.18 | 0.10 | 0.48 | 0.12 | 0.375 |
| 44-SA | 0.06 | 0.17 | 0.09 | 0.44 | 0.24 | 0.387 |
| 50-SA | 0.06 | 0.19 | 0.08 | 0.45 | 0.22 | 0.422 |
| 80-SA | 0.08 | 0.15 | 0.18 | 0.38 | 0.21 | 0.395 |
4. Conclusion
The conversion of lignin-derived phenolic compounds to aromatic hydrocarbons by catalytic cracking was investigated over aluminum silicates catalysts using methanol as a solvent. The effect of aluminum silicates with different SiO2 contents on the physical structure and catalytic performance was investigated. The 33-SA catalyst exhibited the highest catalytic activity for the conversion of phenolic compounds to aromatic hydrocarbons. Polymethylbenzenes were the dominant product. This is attributed to the alkylation of Lewis acid sites and sufficient methyl groups provided by the solvent. In addition, methanol is an effective hydrogen donor, which increases the H/Ceff ratio and inhibits carbon deposition. There is evidence that the content of SiO2 in the SiO2–Al2O3 has a remarkable effect on the catalytic activity and deactivating species. 33-SA remained active after five cyclic regenerations, demonstrating that 33-SA has excellent commercial potential.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was funded by grants from the National Natural Science Foundation of China (no. 52236010, 51976112), the Fundamental Research Funds for the Central Universities (no. 2242022R10058), China Postdoctoral Science Foundation (2022M713166).References
- S. Feng, X. H. Zhang, Q. Zhang, Y. Liang, X. L. Zhao, C. G. Wang and L. L. Ma, Fuel, 2021, 299, 120889 CrossRef CAS.
- Q. Chen, C. L. Cai, X. H. Zhang, Q. Zhang, L. G. Chen, Y. P. Li, C. G. Wang and L. L. Ma, ACS Sustainable Chem. Eng., 2020, 8, 9335–9345 CrossRef CAS.
- S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- T. Y. Guo, X. X. Ma, Z. Y. Li, L. Zheng, Q. W. Fan, X. L. Ding, S. Hu and P. Fu, J. Anal. Appl. Pyrolysis, 2022, 168, 105774 CrossRef CAS.
- J.-X. Wang, J.-P. Cao, X.-Y. Zhao, S.-N. Liu, X.-Y. Ren, L.-Y. Zhang, X.-B. Feng, Y.-P. Zhao and X.-Y. Wei, Ind. Eng. Chem. Res., 2019, 58, 22193–22201 CrossRef CAS.
- S. Song, J. Zhang and N. Yan, Fuel Process. Technol., 2020, 199, 106224 CrossRef CAS.
- X. J. Shen, Q. L. Meng, Q. Q. Mei, H. Z. Liu, J. Yan, J. L. Song, D. X. Tan, B. F. Chen, Z. R. Zhang, G. Y. Yang and B. X. Han, Chem. Sci., 2020, 11, 1347–1352 RSC.
- D.-C. Lv, K. Jiang, K. Li, Y.-Q. Liu, D. Wang and Y.-Y. Ye, Biomass Bioenergy, 2022, 159, 106425 CrossRef CAS.
- B. Shumeiko, M. Auersvald, P. Straka, P. Šimáček, D. Vrtiška and D. Kubička, ACS Sustainable Chem. Eng., 2020, 8, 15149–15167 CrossRef CAS.
- S. Zhang, J. Xu, Q. Cai and Y. Cui, Fuel Process. Technol., 2017, 161, 232–239 CrossRef CAS.
- J. M. Silva, M. F. Ribeiro, I. Graça and A. Fernandes, Microporous Mesoporous Mater., 2021, 323, 111170 CrossRef CAS.
- X. Zhang, Q. Chen, Q. Zhang, C. Wang, L. Ma and Y. Xu, J. Anal. Appl. Pyrolysis, 2018, 135, 60–66 CrossRef CAS.
- S. Wang, J. Chen, Q. Cai, F. Zhang, Y. Wang, B. Ru and Q. Wang, Int. J. Hydrogen Energy, 2016, 41, 16385–16393 CrossRef CAS.
- B. Valle, R. Palos, J. Bilbao and A. G. Gayubo, Fuel Process. Technol., 2022, 227, 107130 CrossRef CAS.
- H. Zhang, Y.-T. Cheng, T. P. Vispute, R. Xiao and G. W. Huber, Energy Environ. Sci., 2011, 4, 2297–2307 RSC.
- P. Sirous-Rezaei and Y.-K. Park, Chem. Eng. J., 2020, 386, 121348 CrossRef CAS.
- T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- B. Valle, P. Castano, M. Olazar, J. Bilbao and A. G. Gayubo, J. Catal., 2012, 285, 304–314 CrossRef CAS.
- B. Valle, A. G. Gayubo, A. T. Aguayo, M. Olazar and J. Bilbao, Energy Fuels, 2010, 24, 2060–2070 CrossRef CAS.
- U. V. Mentzel and M. S. Holm, Appl. Catal., A, 2011, 396, 59–67 CrossRef CAS.
- K. Yang, X. Chen, G. Lafaye, C. Especel, F. Epron and C. Liang, ChemistrySelect, 2018, 3, 11398–11405 CrossRef CAS.
- Z. Mehraban, F. Farzaneh, M. Ghandi and A. Abbasi, Chin. J. Catal., 2007, 28, 357–363 CrossRef CAS.
- Q. K. Tran, S. Han, H. V. Ly, S. S. Kim and J. Kim, J. Ind. Eng. Chem., 2020, 92, 243–251 CrossRef CAS.
- C. Hernandez and A. C. Pierre, J. Sol-Gel Sci. Technol., 2001, 20, 227–243 CrossRef CAS.
- Y. Matsunaga, H. Yamazaki, T. Yokoi, T. Tatsumi and J. N. Kondo, J. Phys. Chem. C, 2013, 117, 14043–14050 CrossRef CAS.
- Z. Wang, H. Zhang, S. Li, J. Wang, Q. Zhu and X. Li, J. Anal. Appl. Pyrolysis, 2017, 124, 475–485 CrossRef CAS.
- M. Asmadi, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2011, 92, 76–87 CrossRef CAS.
- X. Q. Liu, C. C. Hsiao, I. Kalvet, M. Leiendecker, L. Guo, F. Schoenebeck and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 6093–6098 CrossRef CAS PubMed.
- T. Okita, K. Muto and J. Yamaguchi, Org. Lett., 2018, 20, 3132–3135 CrossRef CAS PubMed.
- D. Ma, S. L. Lu, X. H. Liu, Y. Guo and Y. Q. Wang, Chin. J. Catal., 2019, 40, 609–617 CrossRef CAS.
- A. Saraeian, S. J. Burkhow, D. Jing, E. A. Smith and B. H. Shanks, ACS Sustainable Chem. Eng., 2021, 9, 6685–6696 CrossRef CAS.
- C. Li, L. Zhang, M. Gholizadeh, R. Westernhof, Z. Cui, B. Liu, Y. Tang, X. Jin, Z. Xu and X. Hu, Energy Fuels, 2020, 34, 11026–11040 CrossRef CAS.
- S. S. Akarmazyan, P. Panagiotopoulou, A. Kambolis, C. Papadopoulou and D. I. Kondarides, Appl. Catal., B, 2014, 145, 136–148 CrossRef CAS.
- G. Li, Z. Luo, W. Wang and J. Cen, Catalysts, 2020, 10(3), 295 CrossRef CAS.
- L. Wang, Z. Q. Wang, L. L. Zhang and C. Hu, Chem. Eng. J., 2018, 352, 664–672 CrossRef CAS.
- Z. Si, W. Lv, Z. Tian, K. Bi, X. Zhang, C. Wang, C. Pang, R. Dong and L. Ma, Fuel, 2018, 233, 113–122 CrossRef CAS.
- S. H. Jin, Z. H. Xiao, C. Li, X. Chen, L. Wang, J. C. Xing, W. Z. Li and C. H. Liang, Catal. Today, 2014, 234, 125–132 CrossRef CAS.
- J. L. Wen, S. L. Sun, B. L. Xue and R. C. Sun, Holzforschung, 2013, 67, 613–627 CrossRef CAS.
- B. S. Liu, L. Jiang, H. Sun and C. T. Au, Appl. Surf. Sci., 2007, 253, 5092–5100 CrossRef CAS.
- W. Guan, C. W. Tsang, C. S. K. Lin, C. Len, H. Hu and C. Liang, Bioresour. Technol., 2020, 298, 122432 CrossRef CAS PubMed.
- A. V. McBeath and R. J. Smernik, Org. Geochem., 2009, 40, 1161–1168 CrossRef CAS.
- B. Valle, P. Castaño, M. Olazar, J. Bilbao and A. G. Gayubo, J. Catal., 2012, 285, 304–314 CrossRef CAS.
- B. Guichard, M. Roy-Auberger, E. Devers, B. Rebours, A. A. Quoineaud and M. Digne, Appl. Catal., A, 2009, 367, 1–8 CrossRef CAS.
- Y. Echegoyen, I. Suelves, M. J. Lazaro, M. L. Sanjuan and R. Moliner, Appl. Catal., A, 2007, 333, 229–237 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00990d |
| This journal is © The Royal Society of Chemistry 2023 |