
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalyzed ring-opening reactions of γ-carbonyl-substituted ε-caprolactones†
Takayuki Ota‡
a,
Valentina Montagna‡bc,
Yuji Higuchi d,
Takashi Katoe,
Masaru Tanakaf,
Haritz Sardonc and
Kazuki Fukushima*beg
d,
Takashi Katoe,
Masaru Tanakaf,
Haritz Sardonc and
Kazuki Fukushima*beg
aGraduate School of Science and Engineering, Yamagata University, Yamagata 992-8510, Japan
bGraduate School of Organic Materials Science, Yamagata University, 4-3-16 Jonan, Yonezawa, Yamagata 992-8510, Japan
cPOLYMAT, University of the Basque Country UPV/EHU, Joxe Mari Korta Center, Avda. Tolosa 72, 20018 Donostia-San Sebastian, Spain
dResearch Institute for Information Technology, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
eDepartment of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: k_fukushima@chembio.t.u-tokyo.ac.jp
fInstitute for Materials Chemistry and Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
gJapan Science and Technology Agency (JST), PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 19th September 2023
Abstract
Side-chain-functionalized aliphatic polyesters are promising as functional biodegradable polymers. We have investigated ring-opening reactions of γ-carbonyl-substituted ε-caprolactones (gCCLs) to obtain poly(ε-caprolactone) (PCL) analogues. Organic catalysts and Sn(Oct)2 often used for the ring-opening polymerization (ROP) of ε-caprolactone (CL) have been explored to find the conditions for the formation of polymeric products of gCCLs. We confirmed the consumption of gCCLs in all catalyzed reactions. However, chain propagation hardly occurs, as the propagating species are preferentially transformed to α-substituted five-membered lactones when the substituents are linked by ester or not sterically hindered. Intramolecular cyclization to form thermodynamically stable five-membered lactones releases alcohols and amines, serving as nucleophiles for the subsequent ring opening of other gCCLs. Thus, apparent chain reactions are realized for continuous consumption of gCCLs. The reaction preference remains unchanged independent of the catalysts, although the reactions of the amide-linked gCCLs by acidic catalysts are slightly mitigated. Finally, copolymerization of CL and a gCCL catalyzed by diphenyl phosphate has been investigated, which enables the chain propagation reaction to yield the linear oligomers of PCL analogues containing up to 16 mol% of gCCL units. This study contributes to understanding the chemistry of ring-opening reactions of substituted lactones for designing functional degradable polymers.
Introduction
Organocatalysts have led to significant progress in organic and polymer syntheses.1–3 Representative organocatalyzed polymerizations are applicable to ring-opening polymerizations (ROPs) of several heterocyclic monomers such as lactones, cyclic diesters, O-carboxyanhyderides, and cyclic carbonates to produce aliphatic carbonyl-containing polymers. These have been extensively studied as biodegradable polymers for a wide range of biomedical applications.3–5 These biodegradable polymers require additional functionalities, such as cell adhesion properties, enhanced drug loading, and biocompatibility, to fit the applications in regenerative medicine and nanomedicine.6 Copolymerization and end-modification are often employed to modify and expand the chemical functionalities of these polymers.7,8 In addition, the preparation of substituted cyclic carbonyl monomers and their ROPs has been explored as a potential route to provide desired functionality to these aliphatic carbonyl polymers.4,6–8 While extensive efforts have been performed in the literature to introduce different functionalities into degradable polymers, some side-chain structures still affect the polymerizability of the monomers. Depending on the reaction conditions, the ester and amide bonds in the side functional groups have shown some competing mechanisms with the transesterification-based ROPs.9 In the last two decades, enormous efforts have been addressed to develop organocatalysts, which promote the controlled polymerization of the cyclic esters and carbonates with carbonyl side groups.5 Alicyclic guanidine and amidine and pairs of thioureas and nitrogen bases have proved controllability and selectivity in the ROPs of these functional monomers, enabling the production of a wide range of side-chain-functionalized biodegradable polymers.4,5,9Poly(ε-caprolactone) (PCL) is a commercially available polylactone with unique elastic properties and has attracted significant attention due to its marine degradable properties.10,11 In order to enhance the value of these PCL-based polymers, PCL analogues with functional side chains have also been developed.12–14 These functional PCLs were designed for applications to high-valued biomaterials, such as drug delivery vehicles and tissue engineering scaffolds.14–16 γ-Carbonyl substituted caprolactones (gCCLs) with an ester linker were first reported by Hedrick and co-workers in 2000, in which they were unable to obtain high-molecular-weight products by the ROP of the gCCLs.13 Recently, Lang and co-workers reported the successful ROP of gCCLs with amide linker, producing the materials with tunable lower critical solution temperatures17 Similarly, Stefan and co-workers have recently developed a tert-amide linked PCL analogue forming self-assembled nanoparticles.18
Synthesis of ester-linked functional PCLs appears still challenging. Nevertheless, this side chain structure remains intriguing for us because of our recent discovery. We have found that functional poly(trimethylene carbonate) (PTMC) analogues with methoxyalkyl ester side chains exhibit excellent antiplatelet properties.19,20 These results further lead to the recent developments of ether-functionalized PTMC and polydioxanone.21–23 However, biodegradation of the PTMC analogues proceeds slowly compared to that of aliphatic polyesters.20,24 We hypothesize that PCL analogues with similar side-chain structures can be of interest because they can indicate different degradation profiles while retaining the blood compatibility.
In this context, we originally designed a CL analogue with 2-methoxyethoxycarbonyl group at the γ-position (1a; Fig. 1) to obtain an ether-functionalized PCL analogue by its ROP. However, the desired polymeric product was not formed. Thus, 1b was prepared as a model compound to elucidate the mechanism of the ring-opening reaction of ester-linked gCCL. In addition, a gCCL 1c containing an amide linker was examined as another candidate to form the ether-functionalized PCL analogue. These amide-linker types of monomers have shown good polymerizability.17,18 The present study focuses on understanding the unique reactivity of gCCLs in ring-opening reactions, such as the preference for isomerization into other lactone structures. To this end, we explore several catalysts, including organobases and organoacids (Fig. 1), which have been previously applied in ROPs of cyclic esters and carbonates.25–29 The most common ROP catalyst stannous 2-ethylhexanoate (Sn(Oct)2) is also examined as a control. Additionally, we demonstrate the copolymerization of gCCL and CL as a pragmatic means to form the ether-functionalized PCL analogue from gCCL.
 | ||
| Fig. 1 Chemical structures of γ-carbonyl-substituted ε-caprolactones (gCCLs) 1a–c (prepared in this study), initiators, and catalysts for ring-opening reactions of the gCCLs. PTSA, p-toulenesulfonic acid; DPP, diphenylphosphate; TBD, 1,5,7-triazabicylclo[4.4.0]dec-5-ene; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene; TU, 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexyl-2-thiourea; Sn(Oct)2, stannous 2-ethylhexanoate. | ||
Results and discussion
Ring-opening reactions of 1a
Target gCCLs 1a–c were prepared by following the methods reported by Hedrick.13 Before studying the ROP of 1a, we evaluated the catalytic activities of the catalysts shown in Fig. 1 toward the ROP of CL. Acidic organocatalysts such as p-toluenesulfonic acid (PTSA), and diphenyl phosphate (DPP) exhibited higher catalytic activities than basic 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) (Table S1†). Our preliminary screening also verified that the ROP of CL was faster in toluene than in CH2Cl2 (entries 3 vs. 4 in Table S1†). Then, we examined the first ring-opening reaction of 1a (Fig. 2) in toluene (1 M) using DPP (run 1 in Table 1), because this condition achieved a high monomer conversion and narrow molar-mass dispersity in the ROP of CL (entry 3 in Table S1†). The reaction using 1 mol% of 1-pyrenebutanol (A1) as an initiator was monitored using a 1H NMR, showing the quantitative monomer conversion in 26 h at room temperature (Fig. S1†). The reaction was quenched by adding triethylamine. As with ROP of CL, we tried to precipitate the product in hexane. However, the insoluble fraction was hardly formed. Thus, we analyzed the product using size exclusion chromatography (SEC) by eluting tetrahydrofuran (THF) at 30 °C. | ||
| Fig. 2 Desired ring-opening reaction of 1a using A1 as an initiator. | ||
| Run | Catalyst | 1a/A1/Cat. | Solvent | [1a] (M) | Temperature (°C) | Reaction time (h) | Conversiona (%) | Peak Ab (%) |
|---|---|---|---|---|---|---|---|---|
| a Monomer conversion was determined using 1H NMR.b Determined from SEC chart (THF, 30 °C) in Fig. 3.c The reaction was quenched by triethylamine.d Room temperature.e The reaction was quenched by benzoic acid. | ||||||||
| 1c | DPP | 100/1/1 | Toluene | 1.0 | 20–25d | 26 | >99 | 48 |
| 2c | DPP | 100/1/1 | Toluene | 1.0 | 20–25d | 49 | >99 | 36 |
| 3c | DPP | 100/1/1 | CH2Cl2 | 1.0 | 20–25d | 27 | 79 | 45 |
| 4c | DPP | 100/1/1 | Bulk | 20–25d | 29 | >99 | 33 | |
| 5e | TU/DBU | 100/1/5/5 | Toluene | 1.0 | 20–25d | 31 | >99 | 41 |
| 6e | TU/DBU | 100/1/5/5 | CH2Cl2 | 1.0 | 20–25d | 27 | >99 | 63 |
| 7 | Sn(Oct)2 | 100/1/1 | Toluene | 0.5 | 110 | 24 | 32 | 86 |
| 8 | Sn(Oct)2 | 100/1/1 | Bulk | 110 | 28 | >99 | 73 | |
PCL prepared under the same conditions (entry 3 in Table S1†) shows a typical SEC trace characteristic of the polymeric products, appearing at earlier retention time (∼20 min) with relatively wide distribution, including a small shoulder around 18 min (Fig. 3). The SEC trace of run 1 in Table 1 indicates a peak A as the major fraction at 28–29 min of the retention time with several small peaks at 24–28 min. Unfortunately, the lower limit of the calibration of the molecular weights was 25 min (Fig. S2a and b†). Thus, reliable molecular weight information (number-average molecular weight: Mn and dispersity: Đ) can not be listed, although Mn value of the major fractions (peak A) would be below 1.0 × 103 g mol−1 if it could be calibrated. The peak areas of peak A as the percentage of the major fraction in the reaction product were given in Table 1, instead.
 | ||
| Fig. 3 SEC traces (THF, 30 °C) of the products of the ring-opening reaction of 1a with A1 (A1/1a = 1/100) at different runs in Table 1. PCL was prepared by DPP-catalyzed ROP of CL in toluene (entry 3 in Table S1†). Molecular weights were calibrated by polystyrene standards. | ||
Subsequently, in order to find conditions to obtain the polymeric product as depicted in Fig. 2, we examined the ring-opening reactions of 1a under different conditions listed in Table 1, that are, varying catalysts, solvents, and temperatures. Strong acids and bases may induce transesterification-based cross-linking and branching, stemming from the side-chain esters. Thus, we employed DPP as a mild acid catalyst27 and less basic TU/DBU rather than TBD. Since the basic catalyst was found to be less active than acidic ones for the ROP of CL in the preliminary screening and literature,28 the feed ratios of TU/DBU were increased to 5 mol% (runs 5 and 6 in Table 1). The reaction using Sn(Oct)2 was conducted under heating conditions as it is used in most ROPs30 (runs 7 and 8 in Table 1). As with the ROP of CL, the DPP-catalyzed reaction proceeded slowly in CH2Cl2 (run 3 in Table 1). The Sn(Oct)2-catalyzed reaction in toluene was also slow (run 7 in Table 1). At all runs, the products exhibited similar NMR spectra and SEC traces (Fig. 3). These results suggest the low polymerizability of 1a and its transformation to some deactivated species with no polymerizability. The 1H NMR spectrum of the reaction mixture of run 1 in Table 1 is different from that expected for the ring-opened structure of 1a (Fig. S1b†). Accordingly, we suspected a five-membered lactone as a major product of the ring-opening reactions of 1a (Fig. 4a).
 | ||
| Fig. 4 (a) Plausible formation of five-membered lactones 2(Ax) and 2a through a ring-opening reaction of 1a. (b–d) Plausible activation models of the ring-opened structure 1a-O by acidic catalysts (b), basic catalysts (c), and Sn(Oct)2. | ||
Until a recent study by Chen,31 five-membered lactones were recognized as non- or low-polymerizable monomers owing to their thermodynamical stability.12,32 The hydroxy group in the ring-opened species 1a-O (Fig. 4a) is located four bonds away from the ester carbonyl originally introduced at the γ-position of the CL analogue, which is favorable for the formation of a thermodynamically stable five-membered lactone via intramolecular transesterification. Polymerization as an intermolecular reaction (chain propagation in Fig. 4a) is liable to proceed at a high monomer concentration, such as under bulk conditions. However, in the ring-opening reactions of bulk 1a (runs 4 and 8 in Table 1), no significant differences in the product composition were observed (Fig. 3), suggesting that 1a-O highly prefers the intramolecular cyclization toward 2(Ax) in the reaction. The formation of 2(Ax) involves the release of 2-methoxyethanol (A2) from the side chain of 1a-O, which serves as a nucleophile to ring-open 1a. Thus, the chain reaction from 1a to 2a is realized (Fig. 4a).
The DPP-catalyzed bulk reaction (run 4 in Table 1) product was further characterized by SEC eluting DMF containing 10 mM LiBr with a refractive index (RI) and ultraviolet (UV) detectors (Fig. S2d†). The SEC trace by a RI detector showed two major peaks: fractions A and B, while that by a UV detector indicated a bimodal peak corresponding to a part of fraction B. Fraction A is UV-undetectable and thus most likely to be 2a. Since the initiator A1 is UV-detectable, fraction B includes the products conjugating A1 such as 2(A1) and oligomeric P1a (Fig. 4a) with A1 structure. It should be noted that fraction B contains UV-undetectable products. Considering the feed ratio of A1 to 1a (1/100), the composition of the UV-detectable parts in the whole product should be small. Fraction A also appears to correspond to peak A in the SEC traces in THF (Fig. 3) because its eluted time is close to that of 1a (Fig. S2c and d†). These results support the formation of 2a and the chain reaction.
The percentages of peak A in SEC traces of the products in THF vary depending on the catalyst. The DPP-catalyzed reactions (runs 1–4 in Table 1) produced several fractions corresponding to oligomers P1a and 2(A1) at the retention time between 24 and 28 min (Fig. 3). Thus, the peak A percentages become relatively low (33–48%). The prolonged and concentrated reactions may further facilitate the formation of oligomeric P1a and 2(A1) via transesterification with 2a, reducing peak A (runs 2 and 4 in Table 1). In contrast, the reaction products using TU/DBU and Sn(Oct)2 predominantly showed peak A (Fig. 3) with high percentages over 60% (runs 6–8 in Table 1), indicating the preferential formation of 2a. These differences in the product compositions depend on how the ring-opened species 1a-O is activated by the catalysts (Fig. 4b–d). The DPP-catalyzed ring-opening reaction proceeds through an activated monomer mechanism or a hydrogen-bonding activation pathway, in which the Brønsted acid activates the carbonyl oxygen atom (Fig. 4b).26,27 Pseudo-anionic mechanism via hydrogen-bonding activation is proposed for DBU,24,28 and coordination–insertion mechanism is believed the most plausible for Sn(Oct)2,30,33 respectively. The hydroxy oxygen atom of 1a-O is activated in both DBU- and Sn(Oct)2-catalyzed reactions (Fig. 4c and d).
Based on these activation modes, we performed preliminary density functional theory (DFT) calculations for the model compound of 1a-O (1a′-O in Fig. S3a†). We used the alkoxide anion form (Fig. S3b†) and the oxonium cation form (Fig. S3c†) as the extreme and simplified models of 1a-O activated by acids and bases/Sn(Oct)2 (Fig. 4b–d). Consequently, the alkoxide anion form immediately yields the intermediate structure of 2, but the oxonium cation form does not (Fig. S3b and c†). The energy difference between alkoxide anion form and the ring-closed structure in Fig. S3b† was 23.27 kcal mol−1, indicating the stability of the five-membered ring. These results support that the ring-opening reaction products using TU/DBU and Sn(Oct)2 contain high percentages of peak A (Table 1). Accordingly, the organobasic catalysts are preferred over the organoacidic ones for the formation of five-membered lactones. In addition, the results of the DFT calculations also indicate that the intramolecular cyclization occurs in a geometry-dependent manner as the six-membered lactone is not favorably formed from the analogue of 1a-O with additional methylene between the ester group and OH (Fig. S3d†). Our result is consistent with the previous quantum chemical calculation, which revealed the high stability of the five-membered ring of γ-butyrolactone compared to the six-membered ring of δ-valerolactone.32
Subsequently, we performed an equimolar reaction of 1a with A2 to verify whether 1a-O (R = CH2OCH3) or 2a could be obtained. In this reaction, TU/DBU (5% each relative to 1a) were used as the catalysts to obtain products with low contents of oligomers. The reaction was completed in 4.5 h, and the crude reaction product was almost a single component (Fig. S4a†). The crude product was purified by column chromatography using ethyl acetate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane of 1:1 and silica gels. The purified product was then characterized using NMR and Fourier-transform infrared (FT-IR) spectroscopies to verify the formation of 2a (Fig. 5 and S5–S7†). The integral ratios in the 1H NMR spectrum matched those of the structure of the five-membered lactone 2a rather than those of the linear ring-opened structure of 1a-O (Fig. 5a). In addition, the FT-IR spectrum of 2a exhibited the characteristic peaks of the five-membered-ring-strained and linear esters at 1765 and 1729 cm−1, respectively (Fig. 5b). The crude reaction product exhibited a similar 1H NMR spectrum (Fig. S4a†) to that showed in Fig. 5a and S4b,† indicating instability of 1a-O in the reaction mixture. These observations verify the preferential formation of the five-membered lactone through intramolecular transesterification.
:hexane of 1:1 and silica gels. The purified product was then characterized using NMR and Fourier-transform infrared (FT-IR) spectroscopies to verify the formation of 2a (Fig. 5 and S5–S7†). The integral ratios in the 1H NMR spectrum matched those of the structure of the five-membered lactone 2a rather than those of the linear ring-opened structure of 1a-O (Fig. 5a). In addition, the FT-IR spectrum of 2a exhibited the characteristic peaks of the five-membered-ring-strained and linear esters at 1765 and 1729 cm−1, respectively (Fig. 5b). The crude reaction product exhibited a similar 1H NMR spectrum (Fig. S4a†) to that showed in Fig. 5a and S4b,† indicating instability of 1a-O in the reaction mixture. These observations verify the preferential formation of the five-membered lactone through intramolecular transesterification.
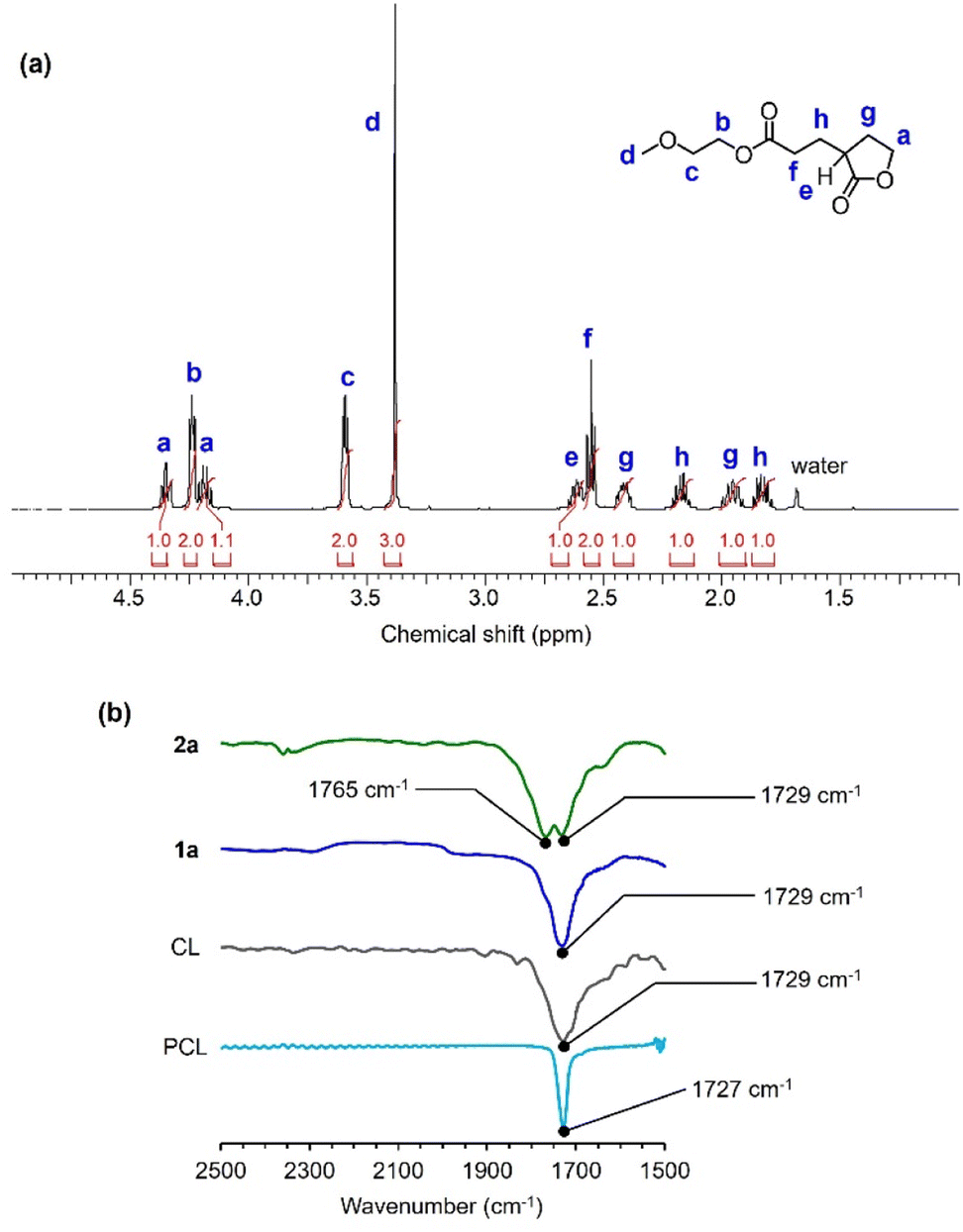 | ||
Fig. 5 Spectroscopic characterization of 2a. (a) Expanded region of 1H NMR (500 MHz) spectrum of 2a in CDCl3. (b) Expanded region of FT-IR spectra of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bands in 2a, 1a, CL, and PCL. O stretching bands in 2a, 1a, CL, and PCL. | ||
Ring-opening reactions of 1b with equimolar alcohols
The equimolar ring-opening reaction further invoked our interest regarding equilibrium between the five-membered lactone and the released alcohol (Fig. 6). Thus, we examined the equimolar ring-opening reaction of 1, using different alcohols Ax, solvents, and catalysts. Compound 1b was additionally synthesized and used because its simple structure is beneficial for spectroscopic characterizations and it is simply derived from a commercially available material in one reaction. | ||
| Fig. 6 Equimolar ring-opening reaction of 1b using alcohols Ax as initiators. | ||
Different ring-opening reactions with DPP, TU/DBU, and Sn(Oct)2 as catalysts were conducted. These three catalysts were used in the ring-opening reactions of 1a (Table 1) and thus selected as the representatives of organoacids, organobases, and organometallic catalysts. The purpose for conducting these reactions was to verify whether the released alcohol (ethanol) can compete with the initiator alcohol reagent (Ax) to form mixed five-membered lactones 2b and 2(Ax) and whether the competition depends on the catalytic mechanism and solvent (Fig. 6). The selective formation of a five-membered lactone depending on the catalyst and solvent was also verified. The DPP-catalyzed reactions produced a single component when A1 was used as the initiator (run 1 in Table 2 and Fig. S8a†). This is because 2(A1) was insoluble in toluene and precipitated once formed, avoiding competing with the released ethanol in the reaction. The products of the reactions in toluene using A2 and A3 (runs 2 and 3 in Table 2, Fig. S8b and c†) were the mixtures of 2b and 2(Ax) (1:1). These results may explain the low percentages of peak A at runs 2 and 4 in Table 1 as a result of transesterification between 2(A1) and 2a (Fig. 4). The reaction with initiator A3 in CHCl3 preferably yielded 2b with 97% of selectivity (run 4 in Table 2). Furthermore, when DBU/TU and Sn(Oct)2 were used as catalysts (runs 5–8 in Table 2 and Fig. S8d†), the same reactions yielded only 2b, irrespective of solvents.
| Runa | Alcohol | Catalyst | Loadingb (equiv.) | Solvent | 2(Ax):2bc |
|---|---|---|---|---|---|
| a All reactions were conducted for 1 h at 20–25 °C (room temperature).b Relative to 1b.c Determined using 1H NMR. | |||||
| 1 | A1 | DPP | 0.1 | Toluene | 100:0 |
| 2 | A2 | DPP | 0.1 | Toluene | 55:45 |
| 3 | A3 | DPP | 0.1 | Toluene | 55:45 |
| 4 | A3 | DPP | 0.1 | CHCl3 | 3:97 |
| 5 | A3 | TU/DBU | 0.02, 0.02 | Toluene | 0:100 |
| 6 | A3 | TU/DBU | 0.02, 0.02 | CHCl3 | 0:100 |
| 7 | A3 | Sn(Oct)2 | 0.02, 0.02 | Toluene | 0: 100 |
| 8 | A3 | Sn(Oct)2 | 0.02, 0.02 | CHCl3 | 0:100 |
DPP acts as a Lewis acid and activates the carbonyl groups through hydrogen bonding, facilitating the nucleophilic attack by the hydroxy group of ring-opened 1b and Ax. For the ring-opening reaction in toluene, the hydrogen-bonding activation by DPP is sufficiently effective for not only the lactone carbonyl of 1b but also for the linear ester of 2(Ax) to form 2b, reaching the equilibrium (runs 2 and 3 in Table 2). The predominant formation of 2b in the reactions using A3 (runs 4–8 in Table 2) suggests that the released benzyl alcohol (A3) is not involved in the substitution of the ethyl ester of 2b. It should be noted that the basicity of ethanol and benzyl alcohol is close (pKa in water: 15 vs. 14).34 Polarity of CHCl3 may enhance this slight difference in the basicity of these alcohols as nucleophilicity when DPP is used (run 4 in Table 2). In contrast, the activation of the alcohol hydroxy groups by TU/DBU and Sn(Oct)2 may differentiate their nucleophilicity over the effects of solvent polarity (runs 5–8 in Table 2). We may propose these ring-opening reactions as a way of selectively forming functional five-membered lactones, which are potentially used as precursors and building blocks of high-value chemicals and medicines,35,36 by selecting appropriate solvents and catalysts.
Ring-opening reactions of 1c
The formation of ester bonds through alcoholysis of amide is thermodynamically unfavorable. Thus, we expect to obtain PCL analogues without the formation of five-membered lactones by ROP of 1c containing the 2-methoxyethyl substituent linked through an amide bond (Fig. 7). The ring-opening reactions of 1c with different catalysts were examined and compared with those of 1a. It should be noted that these experiments are performed to find the conditions to obtain the polymeric product from 1c, not to evaluate catalytic activities and reaction rate. The monomer-initiator ratios were the same as the reactions of 1a (1c/Ax = 100/1). Compared to the reactions of 1a that were mostly completed in approximately 30 h, longer reaction times and heating (90, 100 °C) were required for high conversion of 1c (Table 3). This is partially attributable to the poor solubility of 1c in toluene. Although CH2Cl2 shows better solubility for 1c, it is not an appropriate solvent for the heating reactions due to its low boiling point. | ||
| Fig. 7 Ring-opening reaction of 1c and the estimated products. | ||
| Run | Initiator | Catalyst | 1c/Ax/Cat. | Solvent | [1c] (M) | Temperature (°C) | Time (h) | Conversiona (%) | Major fractionb (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Determined using 1H NMR.b Determined from SEC chart in Fig. 8a (THF, 30 °C).c DPP is gradually loaded up to 15 mol%.d TBD is added in the middle of the reaction to be 5 mol%. | |||||||||
| 1 | A3 | DPP | 100/1/1–15c | Toluene | 1.0 | 90 | 77 | 57 | 66 |
| 2 | A3 | TBD | 100/1/1–5d | Toluene | 1.0 | 90 | 152 | 77 | 60 |
| 3 | A1 | Sn(Oct)2 | 100/1/1 | Toluene | 1.0 | 100 | 36 | >99 | 41 |
| 4 | A1 | Sn(Oct)2 | 100/1/1.5 | Bulk | 100 | 73 | 80 | 38 | |
The incomplete monomer conversions were observed in several conditions. The DPP-catalyzed reaction with the catalyst loading of 1 mol% did not proceed. The DPP-loading was then stepwise increased up to 15 mol% to finally reach the monomer conversion of 57% for 77 h (run 1 in Table 3). Acidic catalysts are often used for the polymerization of monomers with acidic protons, such as amide NH.25,37 However, in this study, the monomer conversion appeared to stop at a certain time after loading of DPP, suggesting the catalyst deactivation by some byproducts. TBD also did not start the reaction with 1 mol% of the loading, which increased to 5 mol% to convert 77% of 1c slowly taking 152 h (run 2 in Table 3). Sn(Oct)2-catalyzed reactions facilitated high monomer conversions (runs 3 and 4 in Table 3 and Fig. S9†). Accordingly, no catalysts provided explicit polymeric products (P1c in Fig. 7) as major products under several temperatures and concentrations.
The SEC traces of the products were similar to those of the ring-opening reactions of 1a, in which the major fractions are eluted at late retention times (29–31 min; Fig. 8a). The percentages of the major fractions vary by catalysts. The reactions catalyzed by DPP and TBD produced the major fractions over 60% (runs 1 and 2 in Table 3). The Sn(Oct)2-catalyzed reactions yielded lower percentages of the major fractions (<41%; runs 3 and 4 in Table 3) with a larger area of sub-peaks at 26–28 min, suggesting more formation of oligomers. Similar results were observed in a previous report by Lang,17 which demonstrated the successful formation of several PCL analogues with amide side groups at the γ-position but a less hindered residue. The FT-IR spectra of the reaction products exhibit the characteristic CO stretching band around 1766 cm−1 (Fig. 8b), suggesting the formation of the five-membered lactone ring, such as 2(Ax) and 2c (Fig. 7). Thus, we need to consider the intramolecular cyclization through alcoholysis of the amide side chain (Fig. S10†), which is unusual and rarely observed.
 | ||
| Fig. 8 Characterization of the products of the ring-opening reactions of 1c: (a) SEC traces (THF, 30 °C) and (b) expanded regions of FT-IR spectra. | ||
The reaction pathways of the ring-opening reactions of 1c are different from those of 1a. Achieving 100% monomer conversion in the reactions of 1c was difficult, particularly when using acidic catalysts. This is probably attributed to the behavior of 2-metxhoyethylamine (2MEA) released as a byproduct during the formation of the five-membered lactone 2(Ax) from the ring-opened species 1c-O (Fig. S10†), forming salts or adducts with the acidic catalysts. The released 2MEA in the reaction of 1c can be involved in the ring-opening reaction of another 1c, leading to the chain reaction for the formation of 2c (Fig. S10†). However, the linear ester structure was remained in the products of the TBD- and Sn(Oct)2-catalyzed reactions (Fig. 8b). These results suggest that both the five-membered lactone and the ring-opened species are present in the equilibrium (Fig. S10†), differently from the case of 1a. Accordingly, the reaction products of 1c included complex structures (Fig. 7).
The equimolar reactions of 1c with benzyl alcohol (A3) were studied to understand the mechanism of the ring-opening reaction of 1c in a simpler manner. The equimolar ring-opening reaction of 1c was not completed in the presence of 0.1 equiv. of DPP (Fig. S11†). The increased loading of DPP by 1.0 equiv. resulted in full conversion of 1c (run 1 in Table 4). The product exhibited a characteristic signal for benzyl ester at 5.2 ppm and no signal around 5.9 ppm for amide NH in the 1H NMR spectrum (Fig. S11†), suggesting the predominant formation of 2(A3) (Fig. S10†). These results rationalize our assumption that the released amine forms an adduct with DPP, not attacking 2(A3) to form 2c. In contrast, the reactions catalyzed by basic organocatalysts DBU in the presence of a co-catalyst TU (run 2 in Table 4) proceeds in a different manner. The time course of the reaction mixture in the presence of TU/DBU monitored by 1H NMR (Fig. S12†) suggested that the reaction product at 2 h contained 2(A3) and 1c-O (Fig. S10†), which were eventually transformed into 2c at 24 h. In the NMR spectra, the reaction product at 2 h shows the distinct signal for benzyl ester at 5.2 ppm, which completely disappears at 24 h (Fig. S12†). Instead, the sharp and single signal for amide NH at 5.9 ppm emerges at 24 h, reminding us of the reaction between 2(A3) and the released 2MEA. These results indicate that 2MEA is active in the TU/DBU-catalyzed reaction of 1c, forming 2c. The low conversion of 1c in the reaction using water as the initiator indicates that a carboxyl group is formed on the ring-opened species 1c-O (RCH2 = H; Fig. S10†) to mitigate the basic catalyst (run 3 in Table 4).
Copolymerization of 1a and CL
The DPP-catalyzed ring-opening reactions of 1a provided more oligomeric products than those catalyzed by TU/DBU and Sn(Oct)2 (Fig. 3). Based on our analyses of various reactions mentioned earlier, reactions with acidic catalysts and at room temperature are preferable for mitigating intramolecular cyclization. In addition, incorporating other monomer units that do not induce internal cyclization would facilitate chain propagation to form a linear polymer structure. Therefore, we studied the DPP-catalyzed ring-opening copolymerization of 1a and CL in toluene at room temperature using A1 as the initiator (Fig. 9a). Two 1a/CL feed ratios of 10/90 and 25/75 were used for the reactions. All monomers were consumed for 31 h, as confirmed by 1H NNR. After precipitation in a 1:1 mixture of hexane and methanol, precipitated products were obtained. The SEC traces of the precipitates exhibited explicit broad peaks corresponding to polymeric products, although distinguished sharp peaks suggesting the presence of 2a were also confirmed (Fig. 9b). The calibrated Mns of the copolymerization products were 6.5 and 4.2 × 103 g mol−1, respectively. The dispersities (Đ = Mw/Mn) were relatively narrow, indicating the low degrees of chain transfer and transesterification in the DPP-catalyzed copolymerization. The 1H NMR spectra of the products (Fig. S13†) suggested the successful incorporation of 1a in PCL, imparting the compositions of 1a (x) in the copolymers as 0.07 and 0.16. The comonomer ratios in the copolymers of 1a and CL (1a/CL = 7/93 and 16/84) corresponded well with the original feeding ratios (10/90 and 25/75), proving that polymerization proceeded in a controlled manner. Although a five-membered lactone structure may be formed at the copolymer chain end, several emerging signals on the NMR spectra (Fig. S13†) confirm the successful formation of the ether-functional PCL analogues.
 | ||
| Fig. 9 Ring-opening copolymerization of 1a and CL: (a) synthetic scheme, (b) SEC traces of the copolymerization products (THF, 30 °C, PS standards). | ||
Conclusions
We investigated ring-opening reactions of three types of γ-substituted ε-caprolactones with the substituents linked by ester and amide bonds, using different organocatalysts. A five-membered lactone was preferentially formed in all reactions compared to the functionalized PCL analogues. Detailed studies revealed that the hydroxy groups located four bonds away from the ester carbonyl group at the ring-opened structure of gCCLs readily induced intramolecular cyclization via transesterification. The compositions of the ring-opening reaction products of gCCLs depend on the catalysts used. Basic organocatalysts facilitate the formation of five-membered lactones 2(Ax), while acidic organocatalysts provide more fractions of linear oligomeric products. Thus, DPP-catalyzed copolymerization of CL and 1a successfully provided PCL analogues bearing methoxyethyl ester side chains with molecular weights of several thousands. In this study, the five-membered lactone was also preferentially formed from a γ-amide-substituted CL, suggesting the significance of the steric effect in intramolecular alcoholytic cyclization with cleavage of the amide bond. The results of the present work will be further used in designing on-demand degradable polymers, where the equilibrium in aminolysis of five-membered lactones is engineered. In addition, our findings can be applied to prepare substituted five-membered lactones, which can be potentially used as precursors of medicines and high-value chemicals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (JP25870078, JP18K12074, JP19H05715, JP19H05716, JP19H05718, and JP19H05720), JST PRESTO (JPMJPR21N7), JST COI (JPMJCE1312), Izumi Science and Technology Foundation (H24-J-129), and Iketani Science and Technology Foundation (0301087-A). The authors are grateful to Prof. Hideharu Mori of Yamagata University for the assistance of the SEC measurements. The authors would like to thank Editage for the English language editing.Notes and references
- J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719–724 RSC.
- S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS PubMed.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- K. Fukushima, Polym. J., 2016, 48, 1103–1114 CrossRef CAS.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- K. Fukushima, Biomater. Sci., 2016, 4, 9–24 RSC.
- W. Yu, E. Maynard, V. Chiaradia, M. C. Arno and A. P. Dove, Chem. Rev., 2021, 121, 10865–10907 CrossRef CAS PubMed.
- Y. Yu, J. Zou and C. Cheng, Polym. Chem., 2014, 5, 5854–5872 RSC.
- K. Fukushima, ROP of Cyclic Carbonates, in Organic Catalysis for Polymerisation, eds., A. P. Dove, H. Sardon and S. Naumann, Royal Society of Chemistry, Cambridge, 2019, ch. 7, pp. 274–327 Search PubMed.
- M. Suzuki, Y. Tachibana and K. Kasuya, Polym. J., 2021, 53, 47–66 CrossRef CAS.
- G.-X. Wang, D. Huang, J.-H. Ji, C. Völker and F. R. Wurm, Adv. Sci., 2021, 8, 2001121 CrossRef CAS PubMed.
- K. Fukushima and T. Fujiwara, New Routes to Tailor-Made Polyesters, in Polymers for Biomedicine, ed., C. Scholz, John Wiley & Sons, Inc., Hoboken NJ, 2017, ch. 6, pp. 149–190 Search PubMed.
- M. Trollsås, V. Y. Lee, D. Mecerreyes, P. Löwenhielm, M. Möller, R. D. Miller and J. L. Hedrick, Macromolecules, 2000, 33, 4619–4627 CrossRef.
- E. A. Rainbolt, K. E. Washington, M. C. Biewer and M. C. Stefan, Polym. Chem., 2015, 6, 2369–2381 RSC.
- E. M. Pelegri-O’Day, S. J. Paluck and H. D. Maynard, J. Am. Chem. Soc., 2017, 139, 1145–1154 CrossRef PubMed.
- M. E. Prévôt, H. Andro, S. L. M. Alexander, S. Ustunel, C. Zhu, Z. Nikolov, S. T. Rafferty, M. T. Brannum, B. Kinsel, L. T. J. Korley, E. J. Freeman, J. A. McDonough, R. J. Clements and E. Hegmann, Soft Matter, 2018, 14, 354–360 RSC.
- L. Wen, S. Zhang, Y. Xiao, J. He, S. Zhu, J. Zhang, Z. Wu and M. Lang, Macromolecules, 2020, 53, 5096–5104 CrossRef CAS.
- E. L. Calubaquib, P. Soltantabar, H. Wang, H. Shin, A. Flores, M. C. Biewer and M. C. Stefan, Polym. Chem., 2021, 12, 3544–3550 RSC.
- Y. Watanabe, S. Takaoka, Y. Haga, K. Kishi, S. Hakozaki, A. Narumi, T. Kato, M. Tanaka and K. Fukushima, Polym. Chem., 2022, 13, 5193–5199 RSC.
- K. Fukushima, Y. Inoue, Y. Haga, T. Ota, K. Honda, C. Sato and M. Tanaka, Biomacromolecules, 2017, 18, 3834–3843 CrossRef CAS PubMed.
- V. Montagna, J. Takahashi, M.-Y. Tsai, T. Ota, N. Zivic, S. Kawaguchi, T. Kato, M. Tanaka, H. Sardon and K. Fukushima, ACS Biomater. Sci. Eng., 2021, 7, 472–481 CrossRef CAS PubMed.
- Y. Watanabe, R. Kato, K. Fukushima and T. Kato, Macromolecules, 2022, 55, 10285–10293 CrossRef CAS.
- K. Fukushima, Y. Ota and T. Kato, Macromol. Chem. Phys., 2022, 223, 2200192 CrossRef CAS.
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- K. Fukushima and K. Nozaki, Macromolecules, 2020, 53, 5018–5022 CrossRef CAS.
- D. J. Coady, H. W. Horn, G. O. Jones, H. Sardon, A. C. Engler, R. M. Waymouth, J. E. Rice, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2013, 2, 306–312 CrossRef CAS PubMed.
- K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999–2005 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 689–695 CrossRef CAS.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed.
- K. N. Houk, A. Jabbari, H. K. Hall and C. Alemán, J. Org. Chem., 2008, 73, 2674–2678 CrossRef CAS PubMed.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 7359–7370 CrossRef CAS.
- A. Hellqvist, Y. Hedeland and C. Pettersson, Electrophoresis, 2013, 34, 3252–3259 CrossRef CAS PubMed.
- G.-H. Pan, R.-J. Song and J.-H. Li, Org. Chem. Front., 2018, 5, 179–183 RSC.
- S. Gupta, R. Arora, N. Sinha, M. I. Alama and M. A. Haider, RSC Adv., 2016, 6, 12932–12942 RSC.
- K. Fukushima, K. Matsuzaki, M. Oji, Y. Higuchi, G. Watanabe, Y. Suzuki, M. Kikuchi, N. Fujimura, N. Shimokawa, H. Ito, T. Kato, S. Kawaguchi and M. Tanaka, Macromolecules, 2022, 55, 15–25 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01025b |
| ‡ Takayuki Ota and Valentina Montagna contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |