
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of high affinity broadly reactive aptamers for spike protein of multiple SARS-CoV-2 variants†
Thao T. Le *a,
Donald J. Benton*b,
Antoni G. Wrobelb and
Steven J. Gamblinb
*a,
Donald J. Benton*b,
Antoni G. Wrobelb and
Steven J. Gamblinb
aDepartment of Chemistry, Imperial College London, UK. E-mail: vtntle@gmail.com
bStructural Biology of Disease Processes Laboratory, The Francis Crick Institute, UK. E-mail: Donald.Benton@crick.ac.uk
First published on 19th May 2023
Abstract
We have developed broadly reactive aptamers against multiple variants by alternating the target between spike proteins from different SARS-CoV-2 variants during the selection process. In this process we have developed aptamers which can recognise all variants, from the original wild-type ‘Wuhan’ strain to Omicron, with high affinity (Kd values in the pM range).
Introduction
The SARS-CoV-2 virus infection is initiated by virus binding to the ACE2 cell-surface receptors, followed by fusion of the virus and cell membranes to release the virus genome into the cell.1,2 Both receptor binding and membrane fusion activities are mediated by the virus spike glycoprotein3 making it a prime candidate to be targeted for the development of vaccines as well as diagnostic and therapeutic reagents. There has been much focus on the development of both antibodies and aptamers against spike protein.4,5Aptamers are single stranded DNA or RNA molecules that are selected in vitro to bind different types of targets including proteins and small molecules.6–8 They are often regarded as nucleic acid equivalence of antibodies.9,10 Aptamers can be advantageous over antibodies due to their capability to retain or regain correct three-dimensional structures for molecular recognition11,12 in physiochemical environments where the latter are often irreversibly damaged by denaturation.13 Thus, they have increasingly been using as alternatives for or partners of antibodies in diagnostic and therapeutic applications.14,15
The spike protein of SARS-CoV-2 is a homotrimeric protein. Each monomer is post translationally cleaved into S1 and S2 subunits. The S1 subunit contains two large domains, the N-terminal domain (NTD) and the Receptor Binding Domain (RBD) as well as two smaller subdomains. Both DNA and RNA aptamers have previously been selected against the spike protein or its subunits/domains to be used as diagnostic reagents or drug candidates.16–23
The rapid emergence of large numbers of new substitutions in SARS-CoV-2 spike protein24 poses a challenge in efficacy of vaccines, as well as the reagents used for diagnostics and therapeutics which are often specifically tailored for a particular variant. We aimed to develop aptamers that bind to spike protein of multiple different strains of SARS-CoV-2 virus, which improve their ability to bind future variants. To achieve this, we alternated the selection target between the spike protein from different variants of SARS-CoV-2 virus; the wild-type ‘Wuhan’ strain and the Alpha.
Results and discussion
Two highly abundant aptamer sequences, K1 and M40, identified from the target-alternating selection showed exceptionally tight binding to spike protein of the wild-type and Alpha. Furthermore, when tested against spike protein from later emerged SARS-CoV-2 strains, i.e. Beta, Delta and Omicron, they displayed similar strong binding affinities.Both aptamers K1 and M40 were truncated to remove nucleotides in the 5′- and 3′-priming regions that showed no significant reduction in binding affinity to spike protein of SARS-CoV-2 virus in their absence. Binding affinities of truncated aptamers K1 (AATGGATCCA CATCTACGAG CTCGAACGCC CGGAGGAGAC GGGGGCAGGG CGTGTTCACT GCAGACTTGA CGAAGCT) and M40 (ATCTACGAAT TCTCCGCCAA GATAGAGGGT GGGCCGCGGA GAATTCACTGC) were determined using enzyme-linked oligonucleotide assays (ELONAs). Both aptamers showed high affinity toward all variants with dissociation constant (Kd) values of K1 to spike protein were 26 ± 6 pM to 66 ± 4 pM whilst M40 were from 155 ± 26 pM to 280 ± 63 pM. In addition, binding affinity of both aptamers to spike protein of all variants were not affected when the Tris-based buffer (20 mM Tris, 5 mM KCl, 2 mM MgCl2, 0.5 mM CaCl2 at pH 7.4) was changed to Hepes-based buffer (20 mM Hepes, 5 mM KCl, 2 mM MgCl2, 0.5 mM CaCl2 at pH 7.4). ELONA binding curves of the truncated K1 and M40 to spike protein of the wild-type, Alpha, Beta, Delta and Omicron are shown in Fig. 1. The truncated sequences and the Kd values of aptamers K1 and M40 to spike protein of the strains are presented in Table 1.
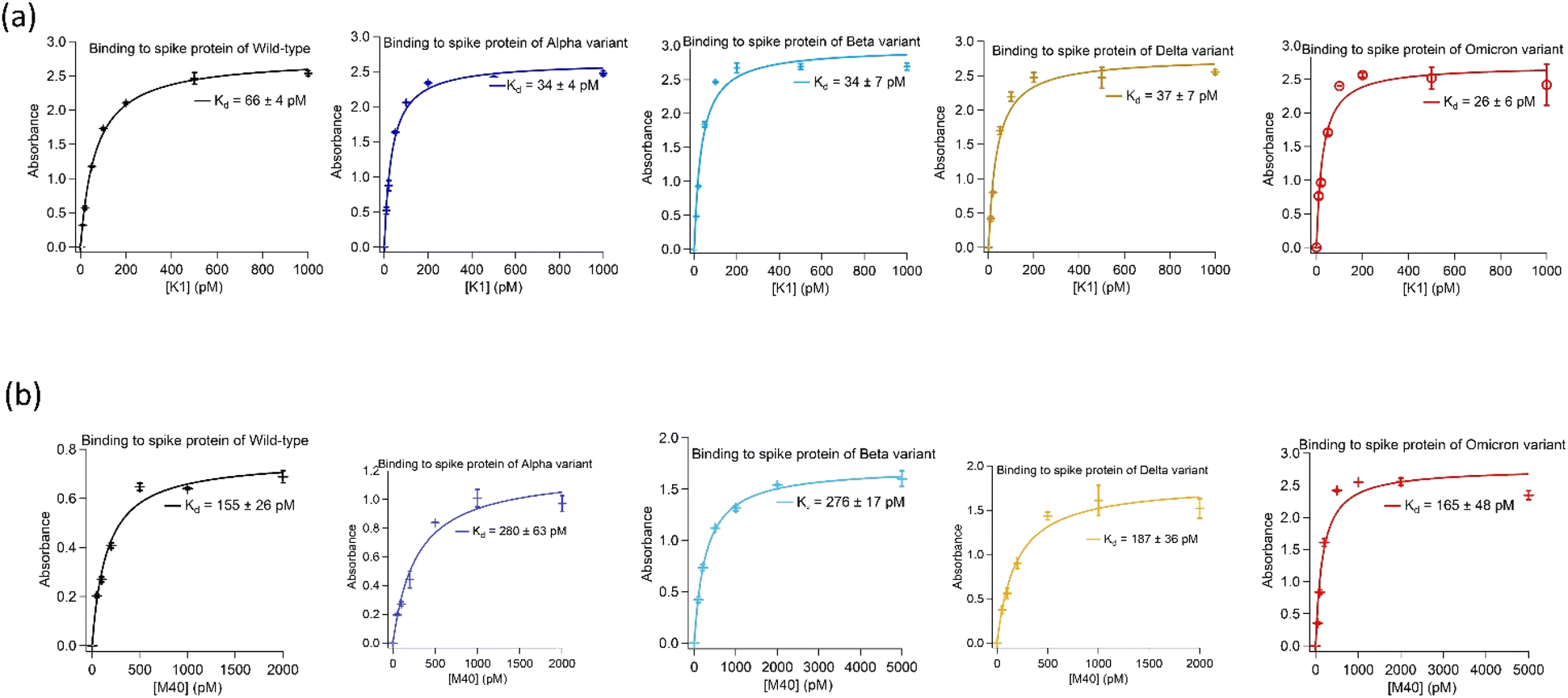 | ||
| Fig. 1 Binding affinities of the aptamers isolated using the target-alternating selection against spike protein of SARS-CoV-2 virus. (a) Binding of aptamer K1 to spike protein of wild-type, Alpha, Beta, Delta and Omicron. (b) Binding of aptamer M40 to spike protein of wild-type, Alpha, Beta, Delta and Omicron. Enzyme-linked oligonucleotide assays (ELONAs) were used to observe binding of the aptamers to spike protein. In the ELONAs, spike protein was immobilized on the microtiter plate wells and solutions of biotinylated aptamer in a range of concentrations were added. The aptamers had a biotin tag at their 3′end to facilitate enzyme (HRP) labelling through streptavidin conjugate (HRP-SA). The ELONAs were performed using buffer containing 20 mM Tris, 5 mM KCl, 2 mM MgCl2, 0.5 mM CaCl2 at pH 7.4. The Kd values obtained from fitting ELONA data to Langmuir isotherm on Igor software. Data for ELONAs were mean values obtained from two independent sets of samples performed and recorded parallelly. The error bars represent standard deviation of the values obtained from the two experiment sets. The aptamers also showed no significant changes in Kd values when replacing Tris by Hepes (20 mM Hepes, 5 mM KCl, 2 mM MgCl2, 0.5 mM CaCl2 at pH 7.4) in the assayed buffer. Binding data in Hepes buffer are presented in ESI, Fig. SI1.† | ||
| Aptamer | Sequence | Kd value (pM) | ||||
|---|---|---|---|---|---|---|
| Wild-type | Alpha | Beta | Delta | Omicron | ||
| K1 | 5'-AATGGATCCACATCTACGA GCTCGAACGCCCGGAGGAGACGGGGGCAGGGCGTG TTCACTGCAGACTTGACGAAGCT-3' | 66 ± 4 | 34 ± 4 | 34 ± 7 | 37 ± 7 | 26 ± 6 |
| M40 | 5'-ATCTACGA ATTCTCCGCCAAGATAGAGGGTGGGCCGCGGAGAA TTCACTGC-3' | 155 ± 26 | 280 ± 63 | 276 ± 17 | 187 ± 36 | 165 ± 48 |
Aptamer K1 was predicted to have one DNA G-quadruplex within its sequence with 23 possible combinations (overlaps) using QGRS Mapper.25 The overlaps are presented in Table SI1 in the ESI.† Fig. 2(a) shows a circular dichroism (CD) spectrum of aptamer K1 measured in the Tris buffer. It has been reported that DNA G-quadruplexes with the parallel topology display characteristic CD spectra of a dip at approximately 245 nm and a peak at around 265 nm whilst anti-parallel structures are expected to have a dip at about 265 nm and a peak at 295 nm.26 Aptamer K1 had a CD spectrum consisted of a dip at approximately 245 nm and a peak at around 280 nm, which is not fitted solely to the spectra of the parallel nor anti-parallel topology. However, the CD spectrum for aptamer K1 may constitute a mixture of different G-quadruplex conformations or a combination of both parallel and anti-parallel topologies.27–29 Putative G-quadruplex structures of aptamer K1 were further investigated using molecular recognition. It was reported that an antibody, BG4, bind DNA and RNA G-quadruplex structures in low nanomolar range.30 Aptamer K1 was recognised by BG4 in both free form (Fig. SI3†) or in complex (Fig. 2(b)) with spike protein. As shown in Fig. 2(b), BG4 binds aptamer K1 in complex with spike protein with Kd values of 1.77 ± 0.23 nM, which is similar to the Kd values previously reported for binding of BG4 to confirmed G-quadruplexes.30 This indicates that a G-quadruplex element is included in the three-dimensional structure of aptamer K1 in the complex formed with spike protein. It is worth noting that, this does not imply aptamer K1 retained its structure upon binding to the spike protein as confirmational changes may occur at the regions that do not involve in forming the G-quadruplex or at the G-quadruplex structure (rearrangement) or both.
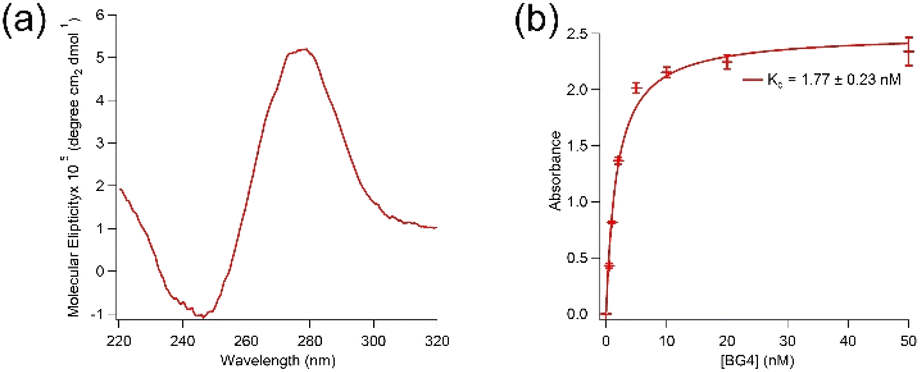 | ||
| Fig. 2 Puntative DNA G-quadruplex structures of aptamer K1 were characterised by CD and molecular recognition. (a) CD spectrum of 10 μM aptamer K1. (b) Binding of BG4, an anti G-quadruplex antibody, to aptamer K1 in complex with spike protein of wild-type CoV-2-SARS virus with a Kd value of 1.77 ± 0.23 nM, obtained from fitting ELONA data to Langmuir isotherm on Igor software. Data points were mean values obtained from two independent sets of samples. The error bars represent standard deviation of the values obtained from the two experiment sets. The CD measurements and ELONA were performed in buffer containing 20 mM Tris, 5 mM KCl, 2 mM MgCl2, 0.5 mM CaCl2 at pH 7.4. | ||
Removal of further 14 (Oligo14) and 30 (Oligo30) nucleotides from both 5′- and 3′-ends resulted in significant loss in binding affinity of the aptamer to spike protein. As shown in Fig. 3, Oligo14 and Oligo30 bind spike protein of the wild type with Kd values of 5.29 ± 0.95 nM and 245 ± 8 nM, respectively, which are several orders of magnitude bigger than the Kd value obtained with aptamer K1. This confirms that these removed nucleotides from the 5′- and 3′- ends of aptamer K1 play a role in binding of the aptamer to spike protein. Oligo14 and Oligo30 however have their DNA G-quadruplex characteristics by CD and antibody recognition retained, indicating that the DNA G-quadruplex is only one binding element of the aptamer K1 and it needs these 30 nucleotides in order to achieve high affinity for spike protein. Sequences of Oligo14 and Oligo30 are presented in Table SI2 in the ESI.†
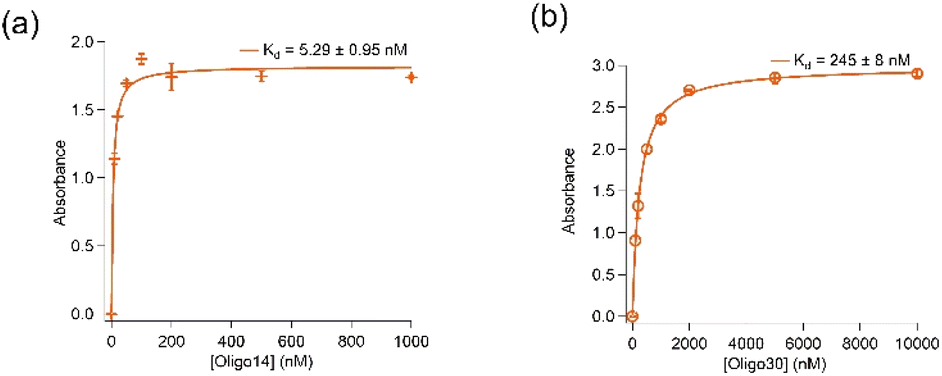 | ||
| Fig. 3 Contribution of nucleotides in binding of K1 and spike protein. (a) Oligo14 (14 nucleotides removed from both 5′- and 3′-ends) binds wild-type spike protein with a Kd value of 5.29 ± 0.95 nM. (b) Oligo30 (30 nucleotides removed from both 5′- and 3′-ends) binds wild-type spike protein with a Kd value of 245 ± 8 nM. ELONAs were performed in buffer containing 20 mM Tris, 5 mM KCl, 2 mM MgCl2, 0.5 mM CaCl2 at pH 7.4. The Kd values obtained from ELONA data used Langmuir model on Igor software. | ||
Most of the previously reported aptamers had relatively high affinity binding to the pure spike protein of SARS-CoV-2 virus with their Kd values mostly in low nanomolar range.16–19,21,23 However, it also showed that binding affinity of some of these aptamers were greatly enhanced (to picomolar range) by forming polymorphic complexes.17,18 In particular, a 'universal DNA aptamer' was recently reported that can bind spike protein of numerous variants with Kd values ranging from 2 to 10 nM.16 The aptamer was achieved by screening a library against wild-type spike protein for the first 13 rounds before finishing off by 1 round of selection for each of 5 variants. In our approach, we alternated two variants (wild-type and Alpha) from the start of the selection and kept the target rotating the whole selection process. Although the two approaches were different, they both resulted in aptamers that can bind spike proteins of multiple variants.
Experimental
Materials
Synthetic DNA libraries with a randomized region were obtained from Integrated DNA Technologies (IDT). Oligo nucleotides were made by Eurofins Scientific and IDT. Nitrocellulose membranes with 0.45 μm pore size and 13 mm diameter were from Merk Millipore. DNA polymerases (Pfu and Taq) were from New England Biolabs. dNTPs were from Promega. Streptavidin, HRP-streptavidin conjugate and TOPO™ TA Cloning sequencing kits were from Thermo Fisher Scientific. Other common chemicals were from Meck Millipore.Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Chloroform:Isoamyl Alcohol (24:24:1 v/v) in order to remove the spike protein. The remaining eluted DNA sequences in the aqueous phase of the extraction were collected for PCR amplification in order to prepare a DNA enriched library for the next round of selection. For subsequent rounds, 10 nmol of the enriched libraries were used with 1.4 μg of the spike protein. The selection target was alternated between the spike protein from the wild-type and Alpha. Every 2 cycles, the enriched library was filtered through a nitrocellulose membrane before the enriched library-target binding to eliminate the membrane-binding sequences. Selection progress was monitored through binding of enriched libraries of rounds 2nd, 6th, 10th, 15th and 19th to the targets. The selection was concluded at round 20th as binding of round 19th enriched library to spike protein had reached saturation.
:Chloroform:Isoamyl Alcohol (24:24:1 v/v) in order to remove the spike protein. The remaining eluted DNA sequences in the aqueous phase of the extraction were collected for PCR amplification in order to prepare a DNA enriched library for the next round of selection. For subsequent rounds, 10 nmol of the enriched libraries were used with 1.4 μg of the spike protein. The selection target was alternated between the spike protein from the wild-type and Alpha. Every 2 cycles, the enriched library was filtered through a nitrocellulose membrane before the enriched library-target binding to eliminate the membrane-binding sequences. Selection progress was monitored through binding of enriched libraries of rounds 2nd, 6th, 10th, 15th and 19th to the targets. The selection was concluded at round 20th as binding of round 19th enriched library to spike protein had reached saturation.Conclusions
We developed aptamers that broadly bind spike protein from multiple variants of SARS-CoV-2 virus with high affinities by alternating targets during selection. The two identified aptamers, K1 (AATGGATCCA CATCTACGAG CTCGAACGCC CGGAGGAGAC GGGGGCAGGG CGTGTTCACT GCAGACTTGA CGAAGCT) and M40 (ATCTACGAAT TCTCCGCCAA GATAGAGGGT GGGCCGCGGA GAATTCACTGC), not only bind spike protein of wild-type and Alpha, their selection targets, but also Beta, Delta and Omicron, the later emerged strains with high affinity; Kd values of both aptamers to spike protein of variants were all in pM range. In addition, aptamer K1 showed characteristic of putative DNA G-quadruplexes. This selection approach may offer a tool to develop aptamers that can cope with emergence of new variants/strains that occurs frequently as the result of a pandemic's large number of infected hosts. The aptamers can be further investigated as candidates for diagnostic and therapeutic applications. In particular, aptamer-based lateral flow tests would be suitable for mass testing that is often needed during pandemics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to thank the support from the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001078 and FC001143), the UK Medical Research Council (FC001078 and FC001143) and the Wellcome Trust (FC001078 and FC001143).References
- J. Shang, Y. Wan, C. Luo, G. Ye, Q. Geng, A. Auerbach and F. Li, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(21), 11727–11734 CrossRef CAS PubMed.
- D. J. Benton, A. G. Wrobel, P. Xu, C. Roustan, S. R. Martin, P. B. Rosenthal, J. J. Skehel and S. J. Gamblin, Nature, 2020, 588(7837), 327–330 CrossRef CAS PubMed.
- A. C. Walls, Y.-J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Cell, 2020, 181(2), 281–292.e6 CrossRef CAS PubMed.
- P. C. Taylor, A. C. Adams, M. M. Hufford, I. de la Torre, K. Winthrop and R. L. Gottlieb, Nat. Rev. Immunol., 2021, 21(6), 382–393 CrossRef CAS PubMed.
- Z. Chen, Q. Wu, J. Chen, X. Ni and J. Dai, Virol. Sin., 2020, 35(3), 351–354 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249(4968), 505–510 CrossRef CAS PubMed.
- D. L. Robertson and G. F. Joyce, Nature, 1990, 344(6265), 467–468 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346(6287), 818–822 CrossRef CAS PubMed.
- H. Sun, X. Zhu, P. Y. Lu, R. R. Rosato, W. Tan and Y. Zu, Mol. Ther.–Nucleic Acids, 2014, 3, e182 CrossRef CAS PubMed.
- S. D. Jayasena, Clin. Chem., 1999, 45(9), 1628–1650 CrossRef CAS.
- T. T. Le, A. Bruckbauer, B. Tahirbegi, A. J. Magness, L. Ying, A. D. Ellington and A. E. G. Cass, Chem. Sci., 2020, 11(17), 4467–4474 RSC.
- P. E. Burmeister, S. D. Lewis, R. F. Silva, J. R. Preiss, L. R. Horwitz, P. S. Pendergrast, T. G. McCauley, J. C. Kurz, D. M. Epstein, C. Wilson and A. D. Keefe, Chem. Biol., 2005, 12(1), 25–33 CrossRef CAS PubMed.
- A. W. P. Vermeer and W. Norde, Biophys. J., 2000, 78(1), 394–404 CrossRef CAS PubMed.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9(7), 537–550 CrossRef CAS PubMed.
- E. J. Cho, J.-W. Lee and A. D. Ellington, Annu. Rev. Anal. Chem., 2009, 2(1), 241–264 CrossRef CAS PubMed.
- Z. Zhang, J. Li, J. Gu, R. Amini, H. D. Stacey, J. C. Ang, D. White, C. D. M. Filipe, K. Mossman, M. S. Miller, B. J. Salena, D. Yamamura, P. Sen, L. Soleymani, J. D. Brennan and Y. Li, Chemistry, 2022, 28(15), e202200078 CAS.
- G. Yang, Z. Li, I. Mohammed, L. Zhao, W. Wei, H. Xiao, W. Guo, Y. Zhao, F. Qu and Y. Huang, Signal Transduction Targeted Ther., 2021, 6(1), 227 CrossRef CAS PubMed.
- J. Valero, L. Civit, D. M. Dupont, D. Selnihhin, L. S. Reinert, M. Idorn, B. A. Israels, A. M. Bednarz, C. Bus, B. Asbach, D. Peterhoff, F. S. Pedersen, V. Birkedal, R. Wagner, S. R. Paludan and J. Kjems, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(50), e2112942118 CrossRef PubMed.
- M. Svobodova, V. Skouridou, M. Jauset-Rubio, I. Viéitez, A. Fernández-Villar, J. J. Cabrera Alvargonzalez, E. Poveda, C. B. Bofill, T. Sans, A. Bashammakh, A. O. Alyoubi and C. K. O'Sullivan, ACS Omega, 2021, 6(51), 35657–35666 CrossRef CAS PubMed.
- M. Sun, S. Liu, T. Song, F. Chen, J. Zhang, J.-a. Huang, S. Wan, Y. Lu, H. Chen, W. Tan, Y. Song and C. Yang, J. Am. Chem. Soc., 2021, 143(51), 21541–21548 CrossRef CAS PubMed.
- X. Liu, Y.-l. Wang, J. Wu, J. Qi, Z. Zeng, Q. Wan, Z. Chen, P. Manandhar, V. S. Cavener, N. R. Boyle, X. Fu, E. Salazar, S. V. Kuchipudi, V. Kapur, X. Zhang, M. Umetani, M. Sen, R. C. Willson, S.-h. Chen and Y. Zu, Angew. Chem., Int. Ed., 2021, 60(18), 10273–10278 CrossRef CAS PubMed.
- A. Gupta, A. Anand, N. Jain, S. Goswami, A. Anantharaj, S. Patil, R. Singh, A. Kumar, T. Shrivastava, S. Bhatnagar, G. R. Medigeshi and T. K. Sharma, Mol. Ther.–Nucleic Acids, 2021, 26, 321–332 CrossRef CAS PubMed.
- G. Liu, W. Du, X. Sang, Q. Tong, Y. Wang, G. Chen, Y. Yuan, L. Jiang, W. Cheng, D. Liu, Y. Tian and X. Fu, Nat. Commun., 2022, 13(1), 1444 CrossRef CAS PubMed.
- W. T. Harvey, A. M. Carabelli, B. Jackson, R. K. Gupta, E. C. Thomson, E. M. Harrison, C. Ludden, R. Reeve, A. Rambaut, S. J. Peacock, D. L. Robertson and C.-G. U. Consortium, Nat. Rev. Microbiol., 2021, 19(7), 409–424 CrossRef CAS PubMed.
- O. Kikin, L. D'Antonio and P. S. Bagga, Nucleic Acids Res., 2006, 34(suppl_2), W676–W682 CrossRef CAS PubMed.
- R. del Villar-Guerra, J. O. Trent and J. B. Chaires, Angew. Chem., Int. Ed., 2018, 57(24), 7171–7175 CrossRef CAS PubMed.
- Y. Chen and D. Yang, Curr. Protoc. Nucleic Acid Chem., 2012, 50(1), 17.5.1–17.5.17 Search PubMed.
- R. Basundra, A. Kumar, S. Amrane, A. Verma, A. T. Phan and S. Chowdhury, FEBS J., 2010, 277(20), 4254–4264 CrossRef CAS PubMed.
- A. Ambrus, D. Chen, J. Dai, T. Bialis, R. A. Jones and D. Yang, Nucleic Acids Res., 2006, 34(9), 2723–2735 CrossRef CAS PubMed.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5(3), 182–186 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01382k |
| This journal is © The Royal Society of Chemistry 2023 |