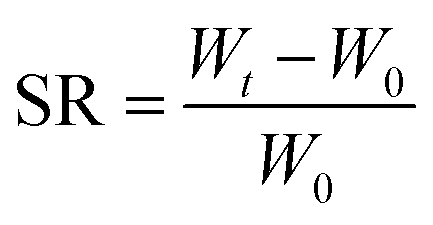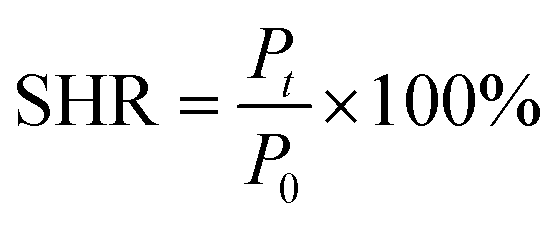DOI:
10.1039/D3RA01478A
(Paper)
RSC Adv., 2023,
13, 11530-11536
Preparation of SA/P(U-AM-ChCl) composite hydrogels by frontal polymerization and its performance study
Received
6th March 2023
, Accepted 6th April 2023
First published on 13th April 2023
Abstract
Deep eutectic solvent (DES) was synthesized from urea (U), acrylamide (AM) and choline chloride (ChCl), sodium alginate (SA) was selected as filler, and SA/P(U-AM-ChCl) composite hydrogel was prepared by thermal initiation frontal polymerization (FP). The hydrogels were characterised by Fourier transform infrared spectroscopy (FTIR) and scanning electron microscopy (SEM). The effects of SA on the swelling properties, mechanical properties and self-healing properties of the composite hydrogels were investigated. The results show that the swelling properties of the composite hydrogel with the addition of SA are greatly enhanced due to the large number of hydroxyl groups contained in the SA chain. The tensile strength of the hydrogel gradually increased with increasing SA content, with the maximum tensile strength increasing by a factor of 2.89. The self-healing efficiency of the composite hydrogel gradually increased with the increase of SA, and the healing rate of FP5 reached 94.4% after 48 h of healing. This study provides a simple and rapid method for the preparation of composite hydrogels with good mechanical properties and self-healing properties.
1 Introduction
Hydrogel is a polymer with a three-dimensional cross-linked network of internal hydrophilic groups.1 There are already applications in many fields such as drug delivery,2,3 waste water treatment,4,5 sensors,6,7 supercapacitors8,9 etc. However, conventional hydrogels have limited their widespread application due to disadvantages such as low mechanical strength and poor tensile properties. Therefore, the preparation of multifunctional composite hydrogels has become a current research hotspot. Biomass materials (e.g. sodium alginate,10 carboxymethyl cellulose,11 etc.) have attracted the attention of researchers due to their cheap price and ease of degradation.12,13
Sodium alginate (SA) is a linear anionic polysaccharide extracted from the cell wall of brown algae or Sargassum and consists of β-D-mannuronic acid and α-L-guluronic acid units in different proportions.14 SA is biocompatible, biodegradable and inexpensive, and is widely used as a food and medical material.15 It has been increasingly shown that the incorporation of SA into hydrogels can improve the water absorption and self-healing properties of hydrogels,16,17 partly because of the large number of hydrophilic groups such as hydroxyl (–OH) and carboxyl (–COOH) on the SA structure,18 and partly because SA contains a large number of –OH, which constitutes a dynamic non-covalent bond within the hydrogel.19
Frontal polymerization (FP) is a simple and rapid method of converting monomers into polymers. The FP reaction process uses external energy to initiate polymerisation to form the front-end front, after which only the front-end front needs to be maintained by its own exotherm to propagate steadily through the monomer mixture.20–22 Common FP initiation methods include thermal initiation, UV initiation, CO2 laser initiation, plasma initiation and magnetic field initiation.23 Compared to traditional polymerisation, frontal polymerisation is a new type of polymerisation. It is a polymerisation method in which low molecular weight monomers are converted into high molecular weight polymers by means of a localised reaction region and has the advantages of short reaction times and good self-propagation. The energy cost of polymer preparation and the high conversion rate provided in a short reaction time, the significant reduction in reaction time and savings in reaction costs when preparing polymers make it a new, effective and economical form of reaction.24–27
Deep eutectic solvent (DES) is composed of a hydrogen bond donor (HBD) and a hydrogen bond acceptor (HBA).28 Due to the hydrogen bonding interactions between the components, the melting point of DES is lower than that of each individual component,29 and the raw materials used to make up DES are low in toxicity, biocompatible and renewable.30,31 The ease of preparation and low cost of the synthesis process compared to conventional ionic liquids21,32 are properties that have led to the widespread use of DES in many fields.30
Based on previous research, we have developed a self-healing SA/P(U-AM-ChCl) composite hydrogel by adding sodium alginate (SA) to DES. The effect of SA content on the behaviour of FP was investigated, and the pattern of SA content on the relevant properties of the composite hydrogel was explored. Polymerisable DES was prepared by heating and mixing urea (U), acrylamide (AM) and choline chloride (ChCl) in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, and then adding different levels of SA to DES for FP, and successfully prepared SA/P(U-AM-ChCl) composite hydrogels. It was characterized by Fourier transform infrared spectroscopy (FTIR) and scanning electron microscopy (SEM), and the effects of different levels of SA on the swelling properties, mechanical properties and self-healing properties of the composite hydrogels were further investigated.
:1:1, and then adding different levels of SA to DES for FP, and successfully prepared SA/P(U-AM-ChCl) composite hydrogels. It was characterized by Fourier transform infrared spectroscopy (FTIR) and scanning electron microscopy (SEM), and the effects of different levels of SA on the swelling properties, mechanical properties and self-healing properties of the composite hydrogels were further investigated.
2 Materials and methods
2.1 Materials
Choline chloride (ChCl), acrylamide (AM), urea (U), sodium alginate (SA), N,N-methylenebisacrylamide (MBA) were purchased from Shanghai Aladdin Biochemical Technology Co; potassium persulphate (KPS) was purchased from Sinopharm Chemical Reagent Co. All raw materials are analytically pure and can be used directly; before use, choline chloride needs to be dried in a vacuum oven at 70 °C for two hours to remove the moisture from it; the water used for the experiments was all distilled water.
2.2 Preparation of DES
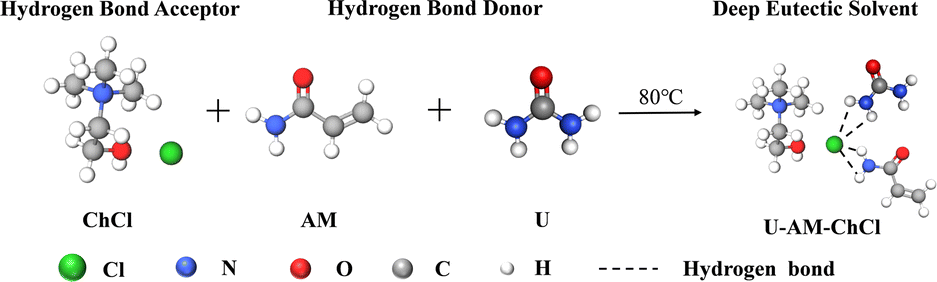
Using ChCl as HBA, U and AM as HBD, the three ingredients were mixed in a beaker in a molar ratio of 1:1:1. The beaker is immersed in an oil bath at 80 °C and stirred continuously until a clear, clarified liquid, DES, is formed. A diagram of the internal hydrogen bonding structure of DES is shown in Fig. 1.
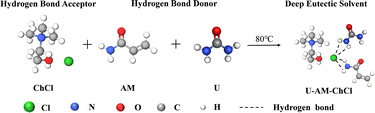 |
| | Fig. 1 Schematic diagram of the internal hydrogen bonding structure of DES. | |
2.3 Preparation of SA/P(U-AM-ChCl) composite hydrogels by frontal polymerization
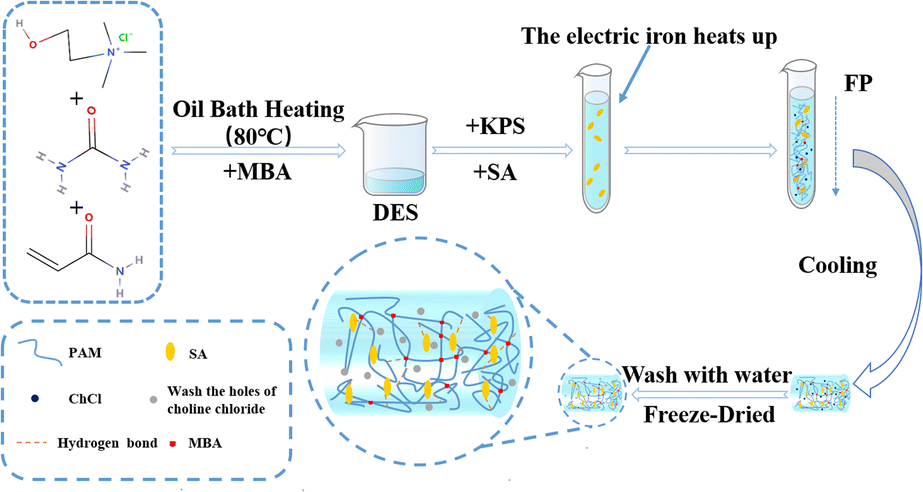
At room temperature of 25 °C, sodium alginate (SA), crosslinker (MBA) and initiator (KPS) were added to the DES in the ratio shown in Table 1 and stirred to obtain a mixed solution. After the tube has cooled to room temperature, the hydrogel sample is removed from the tube and set aside (Fig. 2).
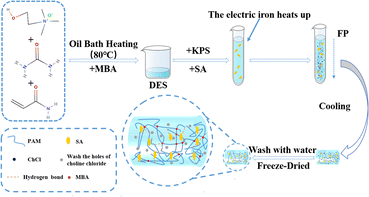 |
| | Fig. 2 Schematic diagram for the preparation of SA/P (U-AM-ChCl) composite hydrogels. | |
Table 1 SA/P(U-AM-ChCl) hydrogel sample ratios
| Samples |
AM/U/ChCl (molar ratio) |
SA (wt%) |
MBA (wt%) |
KPS (wt%) |
| FP0 |
1:1:1 |
0 |
0.7 |
0.5 |
| FP1 |
1:1:1 |
0.5 |
0.7 |
0.5 |
| FP2 |
1:1:1 |
1.0 |
0.7 |
0.5 |
| FP3 |
1:1:1 |
2.0 |
0.7 |
0.5 |
| FP4 |
1:1:1 |
4.0 |
0.7 |
0.5 |
| FP5 |
1:1:1 |
8.0 |
0.7 |
0.5 |
2.4 Measurement of front-end speed and front-end temperature
The speed of the front end can be determined by measuring the change in front end position versus time. That is, before the polymerization begins insert the K-type thermocouple 70 mm below the surface of the mixture, then trigger the top of the mixture with the soldering iron and withdraw the iron after the front-end front has been formed. The temperature displayed by the thermocouple is observed and recorded at regular intervals and the resulting data is collated to obtain a front-end temperature versus time curve.
2.5 Characterisation of SA/P(U-AM-ChCl) hydrogels
The polymer was first cut into 1 mm thick disc-shaped slices and soaked in distilled water for 7 days to remove the water-soluble ChCl. The column thin hydrogel samples were pre-frozen in a refrigerator and then placed in a freeze vacuum oven and freeze dried at −60 °C for two days to a constant weight. The dried gel samples were ground to a powder and the solid powder samples were mixed with potassium bromide, ground, pressed and then analysed for spectral characteristics using Fourier Transform Infrared Spectroscopy (FTIR). The specimens were freeze-dried in vacuum, gold sprayed on their cross-sections using a high vacuum ion sputterer, and the cross-sectional microscopic morphology was observed by SEM scanning microscopy.
2.6 SA/P(U-AM-ChCl) hydrogel performance tests
2.6.1 Hydrogel mechanical properties test. The tensile properties of the composite hydrogel samples were tested on a microcomputer-controlled electronic universal testing machine at a speed of 100 mm min−1 until the maximum tensile stress was obtained after pulling the samples. The compression properties of the composite hydrogels were tested using a TA. XTC-18 mass spectrometer with a compression head at a speed of 0.2 mm s−1 until the maximum compressive stress was obtained. The tensile and compressive strengths were calculated using the following eqn (1):| |
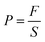 | (1) |
In eqn (1), F is the applied force and S is the cross-sectional area of the hydrogel.
2.6.2 Hydrogel swelling performance test. A disc-shaped hydrogel weighing approximately 20 mg was placed in distilled water, and at regular intervals, the hydrogel was removed and the water stains on the surface of the hydrogel were blotted out with filter paper and weighed and recorded until its weight no longer changed, and then the equilibrium swelling of the hydrogel (SR) was calculated by eqn (2):| |
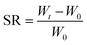 | (2) |
In eqn (2), W0 is the initial weight of the hydrogel and Wt is the weight of the hydrogel after water absorption time t.
2.6.3 Hydrogel self-healing performance test. The prepared hydrogel sample was cut into two sections and the two sections cut into the original position were closely fitted together for 30 s, left for a set period of time without any external intervention and only the sections of the sample were reunited to make them self-heal, and then the maximum tensile strength of the healed sample was tested using an electronic universal testing machine. The self-healing rate of the hydrogel is calculated using the following formula:| |
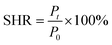 | (3) |
In eqn (3), P0 is the maximum tensile strength of the uncut hydrogel and Pt is the maximum tensile strength of the hydrogel after healing time t.
3 Results and discussion
3.1 Frontal velocity and frontal temperature measurement
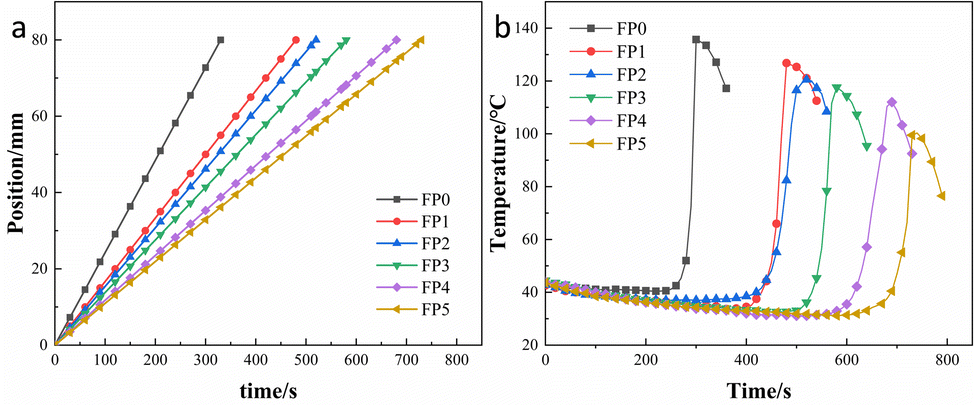
Fig. 3 shows the different patterns of effect of adding different levels of SA on frontal velocity and frontal temperature. As seen in Table 2, the frontal velocity (Vf) decreases from 1.23 cm min−1 to 0.66 cm min−1 as SA increases from 0% to 8%. As shown in Fig. 3(a), the composite hydrogel was prepared within 8 min with a good linear relationship between frontal position and time, indicating no spontaneous ontogenetic polymerization during the reaction.23,33 Fig. 3(b) shows the frontal temperature of the hydrogel as a function of time. It can be seen that the higher the amount of SA, the lower the maximum temperature (Tmax) of the hydrogel, from 135.7 °C to 100.3 °C. This is because an increase in SA leads to a decrease in AM per unit volume, which results in less heat generation per unit time of the reaction.21,28
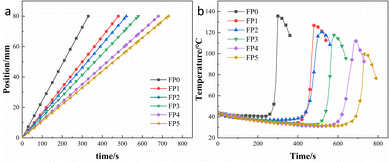 |
| | Fig. 3 (a) Position of the front end of the hydrogel as a function of time; (b) temperature of the front end of the hydrogel as a function of time. | |
Table 2 Temperature and speed parameters for frontal polymerization
| Samples |
SA (wt%) |
Vf (cm min−1) |
Tmax (°C) |
| FP0 |
0 |
1.23 |
135.7 |
| FP1 |
0.5 |
1.00 |
126.5 |
| FP2 |
1 |
0.92 |
120.5 |
| FP3 |
2 |
0.83 |
117.6 |
| FP4 |
4 |
0.71 |
112.0 |
| FP5 |
8 |
0.66 |
100.3 |
3.2 FTIR spectra of SA/P(U-AM-ChCl) hydrogels
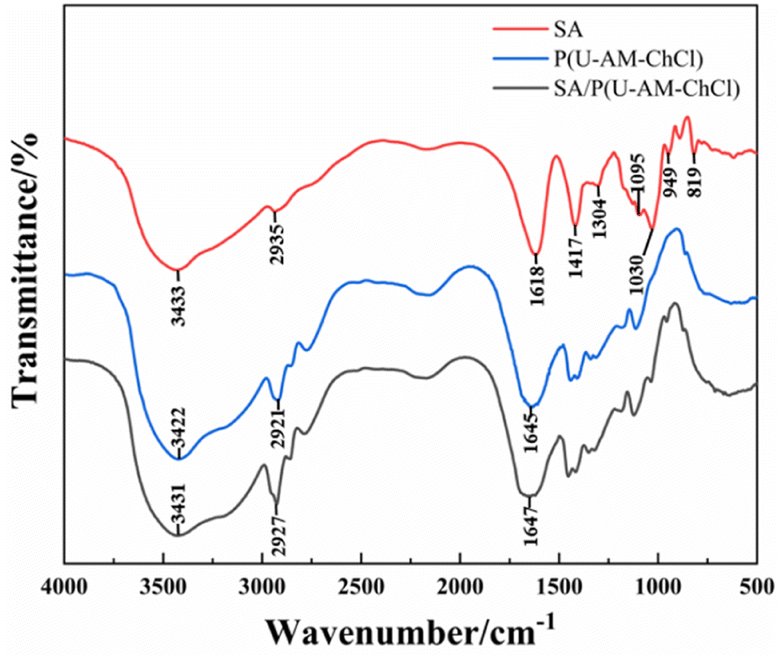
FTIR map study of SA, P(U-AM-ChCl), SA/P(U-AM-ChCl), as shown in Fig. 4. The broad absorption peak at 3433 cm−1 is attributed to the free stretching vibration of the free –OH in SA, the stretching vibration of the –CH group causes a small absorption peak at 2935 cm−1, and the three absorption peaks at 1618 cm−1, 1417 cm−1 and 1304 cm−1 are from the –COO− group, caused by the symmetric and antisymmetric stretching vibration of –COO−, respectively.16,34,35 The stretching vibration of C–O in SA causes two peaks to form at 1095 cm−1 and 1030 cm−1, and the small absorption peak at 949 cm−1 is formed by the out-of-plane bending vibration of –OH in –COOH.36 819 cm−1 corresponds to the absorption peak of Na–O in SA.37 In SA/P(U-AM-ChCl) hydrogels, the N–H stretching vibration in AM overlaps with the peak of the –OH stretching vibration in ChCl and SA, thus forming a broad absorption peak at 3431 cm−1.38 Compared to the FTIR map of the P(U-AM-ChCl) hydrogel, the SA/P(U-AM-ChCl) hydrogel showed an increase in the intensity of the absorption peak of the –CH group at 2927 cm−1. In addition, the absorption peak of –COO− at 1647 cm−1 was shifted towards high frequencies and the shape of the peak was changed, probably due to the presence of hydrogen bonds between P(U-AM-ChCl) and SA.39 In summary, SA has been successfully introduced into the P(U-AM-ChCl) hydrogel network.
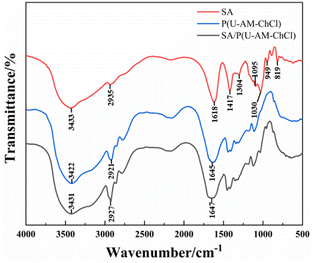 |
| | Fig. 4 FTIR maps of SA, P (U-AM-ChCl) and SA/P (U-AM-ChCl) hydrogels FTIR maps. | |
3.3 Microscopic morphological analysis of SA/P (U-AM-ChCl) hydrogels b
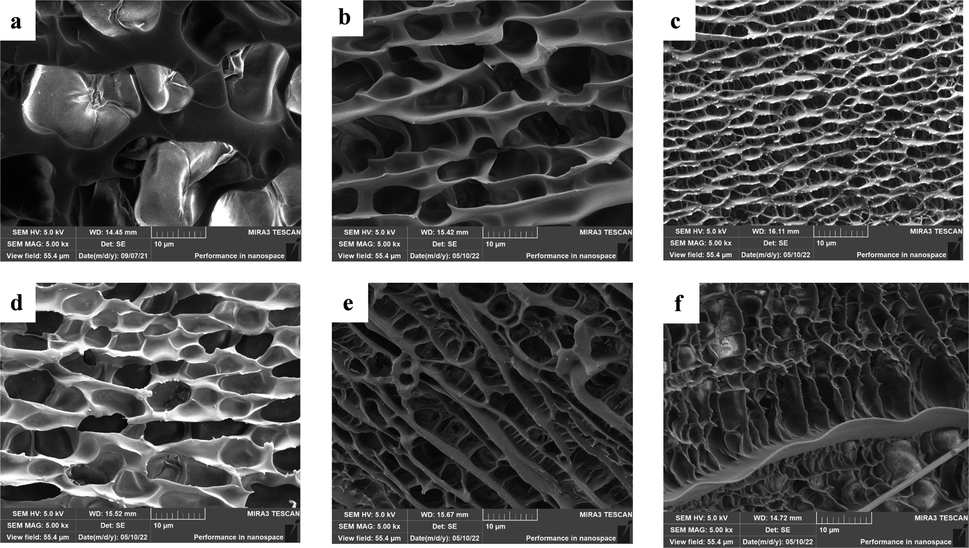
The hydrogel is soaked in deionised water for 7 days to remove soluble substances such as choline chloride. Afterwards they were pre-chilled in a refrigerator and dried in a freeze dryer at −60 °C for 48 h. The morphological structure of the hydrogels with different mass fractions of SA added was observed by scanning electron microscopy. As seen in Fig. 5(a), the cross-linked 3D network structure in the P(U-AM-ChCl) hydrogel resulted in the hydrogel without SA addition exhibiting a porous structure, while the pores appearing on its surface were observed to be sparse and smooth, caused by the sublimation of ice crystals in the interstices of the frozen hydrogel.40 However, after the addition of SA Fig. 5(b)–(f), the pores are denser and rougher, and a clear ribbon-like connecting part appears.41 The results indicate that SA can significantly change the morphology of the P(U-AM-ChCl) hydrogel from a pore structure to a ribbon structure,42 which may be due to the self-assembly of SA through hydrogen bonding, thus mosaicking and dispersing in the SA/P(U-AM-ChCl) hydrogel.
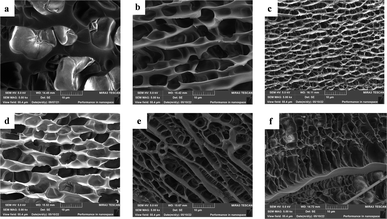 |
| | Fig. 5 SEM images of lyophilized FP0 (a), FP1 (b), FP2 (c), FP3 (d), FP4 (e), FP5 (f) hydrogels. | |
3.4 Solubilisation properties of SA/P(U-AM-ChCl) hydrogels
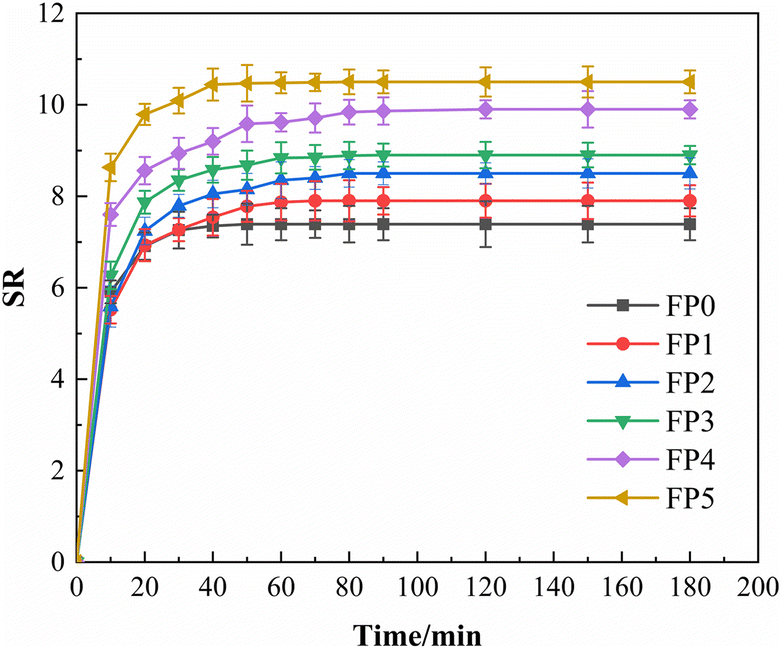
Fig. 6 reflects the swelling curves of six groups of hydrogels with different mass fractions of SA, from which it can be seen that the swelling of SA/P (U-AM-ChCl) hydrogels increases with the increase of SA content, and the swelling rate of FP5 hydrogels is the fastest and reaches the maximum equilibrium swelling, which may be due to the increase of hydrophilicity of the hydrogels due to the carboxyl and hydroxyl groups in SA.35 On the other hand, it can be shown in the SEM plots that as the SA content increases, the hydrogel forms a highly porous structure that can provide more contact surface area, thus increasing the water absorption capacity of the hydrogel (e.g. Fig. 5(b)–(f)). As can be seen in Fig. 6, the six groups of hydrogels essentially reached solubilisation equilibrium within 120 min, with maximum equilibrium solubilisation of 7.4, 7.9, 8.5, 8.9, 9.9 and 10.5 for P(U-AM-ChCl) and SA/P(U-AM-ChCl), respectively. The solubilisation of all the hydrogels was at a high level, probably due to the high content of amide groups and thus good hydrophilicity.41
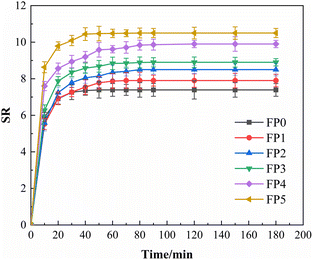 |
| | Fig. 6 Dissolution kinetic curves of SA/P (U-AM-ChCl) hydrogels. | |
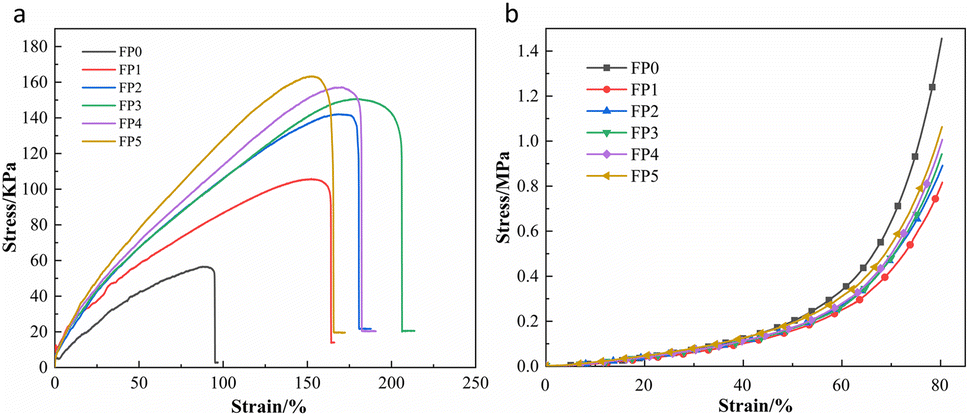
3.5 Mechanical properties of SA/P(U-AM-ChCl) hydrogels
The stress–strain curves for the stretching of SA/P(U-AM-ChCl) hydrogels are shown in Fig. 7(a). As seen in Fig. 7(a), the maximum tensile strengths of the hydrogels are 56.4 kPa, 105.6 kPa, 141.9 kPa, 150.5 kPa, 157.1 kPa and 163.3 kPa for FP0, FP1, FP2, FP3, FP4 and FP5 respectively. The maximum tensile strength of FP5 is 2.89 times that of FP0. The low density and low friction of the polymer chains formed by AM, U and ChCl were responsible for the lower tensile properties of the hydrogel FP0 without SA addition than the other experimental groups.43 It can be seen that the tensile properties of the hydrogel increase with increasing SA due to the formation of intermolecular hydrogen bonds between the –OH of the SA chain and the amide on the polymer chain.44,45 Fig. 7(b) shows the stress–strain curves for the compression of the six sets of hydrogels, setting the maximum compressive deformation variable at 80% and the maximum compressive strengths of the FP0, FP1, FP2, FP3, FP4 and FP5 hydrogels at 1.42 MPa, 0.79 MPa, 0.87 MPa, 0.92 MPa, 0.98 MPa and 1.04 MPa respectively. This shows that the hydrogel FP0 without SA is more rigid and the addition of SA increases the flexibility of the hydrogel, but the compressive strength of the SA/P(U-AM-ChCl) hydrogel increases with increasing SA, probably because the –OH on the SA chain forms hydrogen bonds with the CONH2 on the hydrogel, thus enhancing the compressive properties of the hydrogel.46
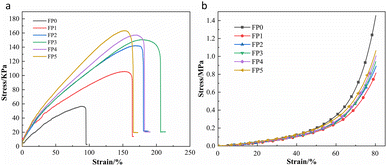 |
| | Fig. 7 (a) Tensile property test curve of SA/P(U-AM-ChCl) hydrogel; (b) compression property test curve of SA/P(U-AM-ChCl) hydrogel. | |
3.6 Self-healing properties of SA/P(U-AM-ChCl) hydrogels
To investigate the self-healing properties of SA/P(U-AM-ChCl) hydrogels, macroscopic self-healing experiments and microscopic self-healing experiments were designed respectively. The FP5 hydrogel sheet is physically divided into six equal sections of this cylinder as shown in Fig. 8(a). At room temperature, piece together in the original position to form a complete sheet, wait 10 s and then use tweezers to be able to hold it intact without breaking. As shown in Fig. 8(b), the FP5 hydrogel was cut off and self-healed for 10 min, and the FP5 hydrogel could withstand the weight of a 100 g weight. As shown in Fig. 8(d), after 24 h of self-healing, the FP4 hydrogel healing gap is almost invisible compared to the FP0 hydrogel healing gap because the dynamic hydrogen bonds inside the FP4 hydrogel are able to reorganize freely during the self-healing process (Fig. 8(c)).19,47
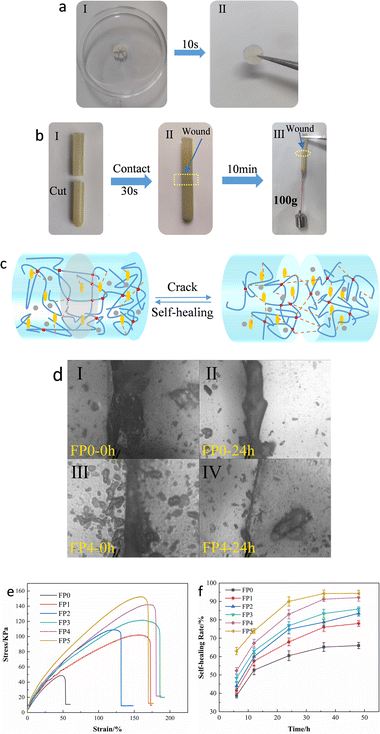 |
| | Fig. 8 (a) and (b) Macroscopic self-healing experiments of FP5 hydrogel samples; (c) schematic diagram of the self-healing mechanism of SA/P(U-AM-ChCl) hydrogels. (d) I and II are boundary control plots of FP0 after 24 h of self-healing; III and IV are boundary control experiments of FP4 after 24 h of self-healing. (e) Tensile stress–strain curves of FP0–FP5 hydrogels after 48 h of self-healing; (f) self-healing rates of SA/P(U-AM-ChCl) hydrogels at different healing times. | |
To investigate the effect of different levels of SA on the healing performance of SA/P (U-AM-ChCl) hydrogels, we explored the healing efficiency of the hydrogels after five different healing times (6 h, 12 h, 24 h, 36 h, 48 h). Fig. 8(e) shows the stress–strain curve of the composite hydrogel after fracture and self-healing for 48 h. Fig. 8(f) shows the self-healing rate of SA/P(U-AM-ChCl) hydrogels at different healing times, it can be found that the self-healing rate of the composite hydrogel gradually increases during the self-healing process from 0–48 h. At 48 h, the self-healing rates of FP0, FP1, FP2, FP3, FP4 and FP5 reached 66.0%, 78.0%, 83.5%, 85.9%, 92.1% and 94.4% respectively.48 The self-healing efficiency of P(U-AM-ChCl) hydrogels increases with increasing SA content due to the large number of hydroxyl and carboxyl groups on SA, which allows more dynamic hydrogen bonds to be formed within the hydrogel and facilitates the self-healing rate.19 The self-healing rate in SA/P(U-AM-ChCl) hydrogels is related to the hydrogen bonding between SA molecules and between SA and AM and U.49 In summary, SA/P(U-AM-ChCl) hydrogels have good self-healing properties. The self-healing rate in SA/P(U-AM-ChCl) hydrogels is related to the hydrogen bonding between SA molecules and between SA and AM and U.50
4 Conclusions
In this paper, SA was added as a filler to DES prepared from AM, U and ChCl, MBA was used as a cross-linking agent and KPS as an initiator to successfully prepare SA/P(U-AM-ChCl) composite hydrogels with good self-healing properties through frontal polymerization, and the structure and properties of the composite hydrogels were investigated, and the experimental results showed that:
(1) The experimental analysis of the swelling performance of the hydrogel showed that the swelling performance of the hydrogel increased with the increase of SA content. After 120 min swelling, the equilibrium swelling of the hydrogel reached the maximum.
(2) Tensile and compressive properties tests found that the tensile strength of the hydrogel increased with the increase of SA content, which was due to the formation of intermolecular hydrogen bonds between the –OH of SA and the amides on the polymer chain, which could obviously enhance the tensile properties of the hydrogel. The tensile strength of the hydrogel with 8% SA content was 163.3 kPa, which was 2.89 times higher than that of the hydrogel without the addition of SA. The flexibility of the hydrogel was enhanced and the compressive strength decreased after the addition of SA, but it was gradually enhanced with the increase of SA content, and the maximum compressive strength was always smaller than the initial compressive strength.
(3) The self-healing properties of the hydrogel were significantly enhanced by the addition of SA. The self-healing rate of the composite hydrogel with SA content of 8% reached 94.4% after 48 h. The self-healing rate of the composite hydrogel with SA content of 8% reached 94.4% after 48 h. The hydrogel without SA added reached 66.0% after 48 h. It can be used in flexible sensors, medical devices and other fields.
Author contributions
B. L., proposed the ideas, steps and details of the experiment, most of the experiments were done by A. L. W., W. R. H, J. Z. L., where A. L. W. was instrumental in the proper conduct of the experiments and wrote the article together with B. L., and all the authors analyzed the data, discussed the conclusions.
Conflicts of interest
The authors declare that there are no competing interests regarding the publication of this article.
Acknowledgements
The work is supported by 2022 Knowledge Innovation Dawn Special Plan Project (2022010801020393), Marine Defense Technology Innovation Center Innovation Fund (JJ-2020-719-01), Natural Science Foundation of Hubei Province (2021CFB292) and Research and Innovation Initiatives of WHPU (2022J04). This work was finished at Wuhan Polytechnic University, Wuhan.
Notes and references
- N. Yuan, L. Xu, B. Xu, J. Zhao and J. Rong, Carbohydr. Polym., 2018, 193, 259–267 CrossRef CAS PubMed.
- K. Xu, W. Shan, N. Hu, J. Wang, W. Zhou, P. Muller-Buschbaum and Q. Zhong, Colloids Surf., B, 2022, 218, 112756 CrossRef CAS PubMed.
- M. Arjama, S. Mehnath and M. Jeyaraj, Int. J. Biol. Macromol., 2022, 213, 435–446 CrossRef CAS PubMed.
- Y. Li and S. Sun, J. Ind. Eng. Chem., 2022, 114, 134–141 CrossRef CAS.
- Y. Chai, Y. Zhang, L. Wang, Y. Du, B. Wang, N. Li, M. Chen and L. Ou, Sep. Purif. Technol., 2022, 295, 121225 CrossRef CAS.
- S. Jang, S. U. Son, J. Kim, H. Kim, J. Lim, S. B. Seo, B. Kang, T. Kang, J. Jung, S. Seo and E. K. Lim, Food Chem., 2023, 403, 134317 CrossRef CAS PubMed.
- Z. Nie, K. Peng, L. Lin, J. Yang, Z. Cheng, Q. Gan, Y. Chen and C. Feng, Chem. Eng. J., 2023, 454, 139843 CrossRef CAS.
- G. Jung, H. Lee, H. Park, J. Kim, J. Wook Kim, D. Sik Kim, K. Keum, Y. Hui Lee and J. Sook Ha, Chem. Eng. J., 2022, 450, 138379 CrossRef CAS.
- Y. Wang and Y. Xie, Electrochim. Acta, 2022, 435, 141371 CrossRef CAS.
- L. Wang, L. Sun, Z. Gu, W. Li, L. Guo, S. Ma, L. Guo, W. Zhang, B. Han and J. Chang, Bioact. Mater., 2022, 15, 330–342 CrossRef CAS PubMed.
- M. Rasoulzadeh and H. Namazi, Carbohydr. Polym., 2017, 168, 320–326 CrossRef CAS PubMed.
- G. Xiao, F. Li, Y. Li, C. Chen, C. Chen, Q. Liu and W. Chen, Colloids Surf., A, 2022, 648, 129383 CrossRef CAS.
- R. Ahmad Raus, W. M. F. Wan Nawawi and R. R. Nasaruddin, Asian J. Pharm. Sci., 2021, 16, 280–306 CrossRef PubMed.
- C. B. Godiya, Y. Xiao and X. Lu, Int. J. Biol. Macromol., 2020, 144, 671–681 CrossRef CAS PubMed.
- Z. Zhang, T. Lin, S. Li, X. Chen, X. Que, L. Sheng, Y. Hu, J. Peng, H. Ma, J. Li, W. Zhang and M. Zhai, Macromol. Biosci., 2022, 22, e2100361 CrossRef PubMed.
- L. Zhu, Y. Liu, F. Wang, T. He, Y. Tang and J. Yang, Adv. Polym. Technol., 2018, 37, 2885–2893 CrossRef CAS.
- X. Hu, W. Cheng, W. Nie and Z. Shao, Polym. Adv. Technol., 2015, 26, 1340–1345 CrossRef CAS.
- S. Thakur, B. Sharma, A. Verma, J. Chaudhary, S. Tamulevicius and V. K. Thakur, J. Cleaner Prod., 2018, 198, 143–159 CrossRef CAS.
- J. Zhang, Y. Wang, Q. Wei, Y. Wang, M. Li, D. Li and L. Zhang, Int. J. Biol. Macromol., 2022, 219, 1216–1226 CrossRef CAS PubMed.
- Q. Feng, X. Chen, Y.-q. Zhao, S.-s. Hu, Z.-w. Xia and Q.-Z. Yan, Colloid Polym. Sci., 2017, 295, 883–890 CrossRef CAS.
- Y. Chen, S. Li and S. Yan, Carbohydr. Polym., 2021, 263, 117996 CrossRef CAS PubMed.
- M. Rassu, V. Alzari, D. Nuvoli, L. Nuvoli, D. Sanna, V. Sanna, G. Malucelli and A. Mariani, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1268–1274 CrossRef CAS.
- S. Li, H. Zhang, J. Feng, R. Xu and X. Liu, Desalination, 2011, 280, 95–102 CrossRef CAS.
- J. Pojman, J. Willis, D. Fortenberry, V. Ilyashenko and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 643–652 CrossRef CAS.
- K. F. Fazende, M. Phachansitthi, J. D. Mota-Morales and J. A. Pojman, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 4046–4050 CrossRef CAS.
- J. D. Mota-Morales, M. C. Gutierrez, I. C. Sanchez, G. Luna-Barcenas and F. del Monte, Chem. Commun., 2011, 47, 5328–5330 RSC.
- D. Sanna, V. Alzari, D. Nuvoli, L. Nuvoli, M. Rassu, V. Sanna and A. Mariani, Carbohydr. Polym., 2017, 166, 249–255 CrossRef CAS PubMed.
- B. Li, M. Zhou, M. Cheng, J. Liu, X. Xu and X. Xie, RSC Adv., 2022, 12, 12871–12877 RSC.
- J. M. Silva, E. Silva and R. L. Reis, Sustainable Chem. Pharm., 2021, 24, 100553 CrossRef CAS.
- L. I. N. Tomé, V. Baião, W. da Silva and C. M. A. Brett, Appl. Mater. Today, 2018, 10, 30–50 CrossRef.
- X. Fang, Y. Li, Y. L. Kua, Z. L. Chew, S. Gan, K. W. Tan, T. Z. E. Lee, W. K. Cheng and H. L. N. Lau, Food Hydrocolloids, 2022, 132, 107861 CrossRef CAS.
- X. Cao, M. Liu, W. Bi, J. Lin and D. D. Y. Chen, Carbohydr. Polym. Technol. Appl., 2022, 4, 100222 CAS.
- S. Li and S. Yan, RSC Adv., 2016, 6, 33426–33432 RSC.
- D. Zheng, K. Wang, B. Bai, N. Hu and H. Wang, Carbohydr. Polym., 2022, 282, 119113 CrossRef CAS PubMed.
- A. S. Montaser, M. Rehan and M. E. El-Naggar, Int. J. Biol. Macromol., 2019, 124, 1016–1024 CrossRef CAS PubMed.
- X. Ren, X. Hu, W. Cheng, S. Bian, Y. Zhao, M. Wu, D. Xue, Y. Li, W. Lu and P. Wang, Fuel, 2020, 267, 117261 CrossRef CAS.
- E. G. Arafa, M. W. Sabaa, R. R. Mohamed, E. M. Kamel, A. M. Elzanaty, A. M. Mahmoud and O. F. Abdel-Gawad, Carbohydr. Polym., 2022, 291, 119555 CrossRef CAS PubMed.
- H. S. Samanta and S. K. Ray, Carbohydr. Polym., 2014, 99, 666–678 CrossRef CAS PubMed.
- L. L. Zhou, D. P. Sun, L. Y. Hu, Y. W. Li and J. Z. Yang, J. Ind.
Microbiol. Biotechnol., 2007, 34, 483–489 CrossRef CAS PubMed.
- B. Li, J. Liu, D. Fu, Y. Li, X. Xu and M. Cheng, RSC Adv., 2021, 11, 35268–35273 RSC.
- Z. Cao, Y. Zhang, K. Luo, Y. Wu, H. Gao, J. Cheng, C. Liu, G. Tao, Q. Guan and L. Zhang, J. Renewable Mater., 2021, 9, 1447–1462 CAS.
- D. Zhao, M. Feng, L. Zhang, B. He, X. Chen and J. Sun, Carbohydr. Polym., 2021, 256, 117580 CrossRef CAS PubMed.
- J. P. Gong, Soft Matter, 2010, 6, 2583 RSC.
- W. Du, Z. Zhao and X. Zhang, Carbohydr. Polym., 2022, 285, 119232 CrossRef CAS PubMed.
- N. El Miri, K. Abdelouahdi, A. Barakat, M. Zahouily, A. Fihri, A. Solhy and M. El Achaby, Carbohydr. Polym., 2015, 129, 156–167 CrossRef CAS PubMed.
- M. Ding, H. Su, Y. Li, K. Yang, L. Dang, F. Li and B. Xue, Mater. Today Chem., 2022, 26, 101049 CrossRef CAS.
- G. Cai, J. Wang, K. Qian, J. Chen, S. Li and P. S. Lee, Adv. Sci., 2017, 4, 1600190 CrossRef PubMed.
- L. Zhao, Z. Ren, X. Liu, Q. Ling, Z. Li and H. Gu, ACS Appl. Mater. Interfaces, 2021, 13, 11344–11355 CrossRef CAS PubMed.
- Q. Zheng, L. Zhao, J. Wang, S. Wang, Y. Liu and X. Liu, Colloids Surf., A, 2020, 589, 124402 CrossRef CAS.
- G. Li, J. Wu, B. Wang, S. Yan, K. Zhang, J. Ding and J. Yin, Biomacromolecules, 2015, 16, 3508–3518 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Aolin Wua,
Wenrui Haoa,
Jizhen Liu
*a,
Aolin Wua,
Wenrui Haoa,
Jizhen Liu