
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The mechanism of water pollutant photodegradation by mixed and core–shell WO3/TiO2 nanocomposites†
Abdisa Habtamu and
Masaki Ujihara*
and
Masaki Ujihara*
Graduate Institute of Applied Science and Technology, National Taiwan University of Science and Technology, 43 Keelung Road, 10607, Taipei, Taiwan. E-mail: masaki.ujihara@mail.ntust.edu.tw
First published on 25th April 2023
Abstract
Environmental pollution is one of the biggest concerns in the world today, and solar energy-driven photocatalysis is a promising method for decomposing pollutants in aqueous systems. In this study, the photocatalytic efficiency and catalytic mechanism of WO3-loaded TiO2 nanocomposites of various structures were analyzed. The nanocomposites were synthesized via sol–gel reactions using mixtures of precursors at various ratios (5%, 8%, and 10 wt% WO3 in the nanocomposites) and via core–shell approaches (TiO2@WO3 and WO3@TiO2 in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of TiO2:WO3). After calcination at 450 °C, the nanocomposites were characterized and used as photocatalysts. The kinetics of photocatalysis with these nanocomposites for the degradation of methylene blue (MB+) and methyl orange (MO−) under UV light (365 nm) were analyzed as pseudo-first-order reactions. The decomposition rate of MB+ was much higher than that of MO−, and the adsorption behavior of the dyes in the dark suggested that the negatively charged surface of WO3 played an important role in adsorbing the cationic dye. Scavengers were used to quench the active species (superoxide, hole, and hydroxyl radicals), and the results indicated that hydroxyl radicals were the most active species; however, the active species were generated more evenly on the mixed surfaces of WO3 and TiO2 than on the core–shell structures. This finding shows that the photoreaction mechanisms could be controlled through adjustments to the nanocomposite structure. These results can guide the design and preparation of photocatalysts with improved and controlled activities for environmental remediation.
:1 ratio of TiO2:WO3). After calcination at 450 °C, the nanocomposites were characterized and used as photocatalysts. The kinetics of photocatalysis with these nanocomposites for the degradation of methylene blue (MB+) and methyl orange (MO−) under UV light (365 nm) were analyzed as pseudo-first-order reactions. The decomposition rate of MB+ was much higher than that of MO−, and the adsorption behavior of the dyes in the dark suggested that the negatively charged surface of WO3 played an important role in adsorbing the cationic dye. Scavengers were used to quench the active species (superoxide, hole, and hydroxyl radicals), and the results indicated that hydroxyl radicals were the most active species; however, the active species were generated more evenly on the mixed surfaces of WO3 and TiO2 than on the core–shell structures. This finding shows that the photoreaction mechanisms could be controlled through adjustments to the nanocomposite structure. These results can guide the design and preparation of photocatalysts with improved and controlled activities for environmental remediation.
1. Introduction
Increasing wastewater discharges from various sources pose enormous environmental challenges worldwide.1 Due to rapid industrial growth, the environment has become highly contaminated with various organic and inorganic pollutants.2,3 Dyes are common hazardous organic contaminants in wastewater.4 They also impart color to the water and can produce harmful byproducts through chemical reactions.5 The long-term consumption of water containing dyes could harm the liver, central nervous, and digestive systems of humans.6 For this reason, many researchers are engaged in developing techniques to eliminate organic dyes from water systems.Recently, various techniques, including using a green biochar/iron oxide composite,7 coated membranes,8 surfactant-modified biomass,9 coagulation,10 modified magnetic nanosorbents,11,12 hydrochar adsorption,13,14 and photocatalytic degradation,15,16 have been applied to remove both cationic and anionic dyes from water. Among these methods, semiconductor-based photocatalysis is thought to be the most promising method because it can convert a broad range of organic contaminants into less toxic compounds, including CO2, and H2O, without the use of expensive oxidants. With the aim of developing effective photocatalysts, various semiconductors have been examined individually or in combination with other materials. The modification of photocatalyst surfaces with other materials can improve the efficiency of photocatalysis.17 Combining semiconductors with metals can enhance charge separation.18–21 Elemental doping and combining different semiconductors22,23 can change the band gap of the resulting materials and induce charge separation. Among the photocatalysts, TiO2 has been widely investigated as a typical semiconductor photocatalyst24,25 due to its high photocatalytic activity, low price, physicochemical stability, nontoxicity, and environmental friendliness.26 Despite these advantages, the wide bandgap of TiO2 (3.20–3.35 eV) limits the use of light to the UV range, shows rapid charge recombination and has limited efficiency.27 To address these limitations, doping and combining TiO2 photocatalysts with narrow bandgap semiconductors are viable options. Semiconductors such as MoO3,28 Ag2CO3,29 ZnO,30 and WO3 (ref. 27) have been coupled with TiO2 to improve its photocatalytic efficiency under UV light. Among them, WO3 has attracted considerable amounts of attention due to its ability to absorb visible light (typically wavelengths <500 nm (ref. 31 and 32) and extended to >500 nm by the effects of oxygen vacancies32) WO3 is also stable in oxidative and acidic environments and has low cost and low toxicity.33 The crystal ionic radius of W6+ is close to that of T4+; therefore, W6+ can be easily introduced into the lattice of titania to replace Ti4+ and form W–O–Ti links, or it can be positioned at interstitial locations,34,35 which effectively induces lattice defects and increases the surface area of WO3-coupled TiO2. Moreover, WO3-coupled TiO2 shows enhanced O2 chemisorption on its surfaces,36 and this adsorbed oxygen improves charge separation. Thus, WO3-coupled TiO2 has emerged as a promising adsorbent and catalyst. However, based on the preparation methods and the nature of pollutants to be decomposed, different behaviors have been reported.34,36–40 Some studies have stated that WO3 doping boosted TiO2 photocatalytic activity, whereas others reported that it had the reverse effect. Various factors, such as the nature of the dopants and their concentrations, the nature of pollutants, the intensity of light and irradiation time, dissolved oxygen concentration, reaction temperature, pH, surface area, the quantity of catalyst, and the surface morphology of the catalysts,41 are now considered to have an impact on photocatalytic decomposition. For effective photocatalysts, WO3/TiO2 core–shell nanorods were developed.42,43 Mixed WO3/TiO2 composites were utilized.27,36,40,44–48 However, the comparative study among the core–shell and co-mixed structures, and the effects of structures on reaction mechanisms were not strongly reported. Therefore, we focused on the structural dependence for the dye decomposition.
During the photodecomposition process, adsorption of target compounds is a key first step to be considered.49–51 In this study, cationic and anionic dyes (MB+ and MO−) were used to analyze the adsorption process of target compounds on the surface of WO3-loaded TiO2. Then, 3 different types of WO3–TiO2 nanocomposites (a mixture of TiO2 and WO3 formed by the sol–gel reaction and core–shell structures of TiO2@WO3 and WO3@TiO2 prepared by a hydrothermal method) were examined as photocatalysts, in addition to the single-component photocatalysts (TiO2 and WO3). The photodegradation of model target compounds was analyzed in terms of both the adsorption kinetics and reaction mechanism. The results can help guide the further design of photocatalysts consisting of semiconductor nanocomposites.
2. Experimental section
2.1 Materials
Titanium(IV) isopropoxide (TTIP, ACROS ORGANICS, CHINA), isopropanol (≥99.8% (GC), Honeywell|Riedel-de Haën™, France), sodium tungstate oxide dihydrate (Na2WO4·2H2O, 99.0–101.0%, Thermo Fisher Scientific.), ethanol (≥99.9%, Honeywell|Fluka™, Germany), nitric acid (65% w/w), hydrochloric acid (37%, reagent grade), ethylene glycol (99.5%, ACROS ORGANICS), methylene blue (ACROS ORGANICS, INDIA), and methyl orange (ACROS ORGANICS, INDIA) were used without further purification. Para-benzoquinone (p-BQ) and ethylenediaminetetraacetic acid disodium salt (Na2-EDTA) were purchased from Sigma-Aldrich and Fisher Chemical, respectively. Ultrapure water (resistivity of 18.2 mΩ cm, Yamato, Japan) was used throughout the experiments.2.2 Sol–gel synthesis of TiO2 nanoparticles
The TiO2 nanoparticles were prepared via a sol–gel approach.52 First, 6.0 mL of TTIP was mixed with 11.6 mL of isopropanol. The mixture was vigorously stirred for 1 h, and 14.6 mL of water was added with vigorous stirring. After aging for 24 h, the white precipitate that formed was filtered and thoroughly washed with water. Then, the residue was dried at 80 °C for 12 h and calcined at 450 °C for 2 h. The obtained white mass was ground into a powder with a mortar and pestle.2.3 Hydrothermal synthesis of TiO2 nanoparticles
For comparison, TiO2 nanoparticles were synthesized by a hydrothermal method.53 First, 5.9 mL of TTIP was dissolved in 9.0 mL of ethylene glycol and stirred for 2 h. Then, the solution was transferred into a Teflon-lined autoclave, and 30.3 mL of water was added. The white slurry formed was heated at 220 °C for 6 h, and the resulting white precipitate was washed three times with water and twice with ethanol using centrifugation. Then, the white paste was dried at 80 °C for 12 h and calcined at 450 °C for 2 h. The obtained white mass was ground to a powder with a mortar and pestle. This powder was labeled hyd-TiO2.2.4 Synthesis of WO3 nanoparticles by the sol–gel method
The sol–gel procedure for WO3 synthesis was adapted from a previous report.23 A powder of Na2WO4·2H2O (1.0 g) was dissolved in 15 mL of water with stirring. To the solution, 7 mL of 1.0 M HCl was slowly added under vigorous stirring. Then, the obtained light yellowish solution was heated to 80 °C for 1 h. After the suspension was cooled to ambient temperature, the light yellowish precipitate was separated using centrifugation and washed three times with water to remove residual NaCl and HCl. Then, the yellow paste was dried at 80 °C for 12 h and ground into a powder with a mortar and pestle.2.5 Hydrothermal synthesis of WO3 nanoparticles
WO3 nanoparticles were also prepared by a hydrothermal method. A powder of Na2WO4·2H2O (2.0 g) was dissolved in 30.0 mL of water. Then, 10 mL of 5 N HNO3 was added to the solution with vigorous stirring at ambient temperature. The mixture was moved to an autoclave and heated to 220 °C for 6 h. After the mixture was cooled to room temperature, the precipitates were collected by centrifugation, washed 3 times with water followed by ethanol, and dried at 80 °C for 12 h. A yellowish mass was obtained after calcination at 450 °C for 2 h and was ground to a powder with a mortar and pestle. This powder was labeled hyd-WO3.2.6 Sol–gel synthesis of WO3/TiO2 nanocomposites
Coprecipitation of WO3 and TiO2 was carried out for the preparation of the WO3/TiO2 nanocomposites. First, 6.0 mL of TTIP was mixed with 11.6 mL of isopropanol, and the mixture was vigorously stirred for 1 h. To control the ratio of WO3 in the nanocomposites, a powder of Na2WO4·2H2O (0.243 g for 5 wt%, 0.401 g for 8 wt%, and 0.512 g for 10 wt%) was dissolved in 14.6 mL of water and added to the TTIP solution with vigorous stirring. After the mixture was aged for 24 h, the precipitate was filtered and washed 3 times with water followed by ethyl alcohol. Finally, nanocomposites were obtained after drying at 80 °C for 12 h and calcination at 450 °C for 4 h, followed by crushing in a mortar. The single-component metal oxides (TiO2 and WO3) were denoted sol-TiO2 and sol-WO3, respectively.2.7 Synthesis of core–shell TiO2@WO3
The core–shell nanocomposite TiO2@WO3 was produced with a hydrothermal method.54 The hydrothermally synthesized TiO2 (hereafter, called hyd-TiO2, 632.5 mg) was dispersed in a solution of Na2WO4·2H2O (100 mg of Na2WO4·2H2O dissolved in 30 mL of water) with stirring for 60 min; the final mass ratio of TiO2:WO3 = 9:1. The resulting white suspension was treated with dropwise additions of 5 N HNO3 with vigorous stirring. Then, the suspension was transferred to a 50 mL Teflon-lined autoclave and heated at 220 °C for 6 h. The precipitate was washed using water and ethanol by centrifugation and dried at 80 °C for 12 h. Then, the TiO2@WO3 composite was obtained after calcination in air at 450 °C for 2 h, followed by crushing in a mortar.
2.8 Synthesis of core–shell WO3@TiO2
The reverse core–shell structure of TiO2@WO3 was also synthesized. First, 2.4 mL of TTIP and 10 mL of ethylene glycol were mixed and stirred for 2 h at ambient temperature. Next, a suspension of WO3 was prepared by sonicating 70.3 mg of hyd-WO3 in 28.9 mL of water for 60 min. These solutions and suspensions were mixed in a 50 mL Teflon-lined autoclave to make the final mass ratio of TiO2:WO3 9:1. Then, the obtained yellowish to white gel was heated at 220 °C for 6 h. After cooling, the product was washed using water and ethanol by centrifugation and dried for 12 h at 80 °C. Finally, a yellowish to white mass was calcined at 450 °C for 2 h, followed by crushing in a mortar.
2.9 Characterization
The morphology of the nanocomposites was observed with a field-emission scanning electron microscope (FESEM, JSM-7900F, JEOL LTD, Japan) at an acceleration voltage of 15 kV. Before SEM inspection, all samples were sputtered with Pt using a JEC-3000FC Auto Fine Coater (JEOL LTD, Japan). Elemental analysis was carried out using an energy dispersive X-ray (EDX) spectrometer equipped with an FESEM. The crystal structures were characterized using XRD (X-ray diffractometer, 2nd Gen D2 PHASER, Bruker) with Cu Kα radiation at an acceleration voltage of 30 kV and a current of 10 mA within the 2θ range from 10° to 80°. The presence and oxidation state of each element in the nanocomposites were determined using X-ray photoelectron spectroscopy (XPS, ULVAC PHI 5000 Versa Probe) using Al Kα monochromator (1486.6 eV) X-rays. A UV-VIS spectrophotometer (V-670, JASCO, Japan) was used to measure the absorption spectra of the organic dye solutions. UV-VIS diffuse reflectance spectroscopy (DRS) was conducted at a 45° irradiation angle with a UV-VIS spectrometer (SEC2000, ALS, Japan) with a light source from Ocean Optics DH-2000-BAL.2.10 Adsorption analyses
The adsorption properties of the nanocomposites were analyzed by a batch process. A powder of the nanocomposite (4 mg) was dispersed in dye solutions (65 mL) of varying concentrations (1.0, 2.0, 4.0, 6.0, and 8.0 mg L−1). Then, the suspension was stirred at ambient temperature under dark conditions for 30 min to achieve adsorption equilibrium. After adsorption, the suspension was separated into a supernatant and precipitate (the nanocomposite absorbed some of the dye) by centrifugation (for 60 s at 6000 rpm at neutral pH and room temperature), and the free dye concentration was determined from the UV-VIS absorption spectra of the supernatant: absorbance at the λmax of the dye (662 nm for MB+ and 464 nm for MO−) was compared to that of the original dye solution (see Fig. S1 in the ESI†). The quantity of dye adsorbed on each nanocomposite (qe) in mg g−1 was estimated according to eqn (1).
 | (1) |
2.11 Photocatalytic activity analyses
To evaluate photocatalytic activity, a powder of the nanocomposite (4 mg) was dispersed in 65 mL of a dye solution (2.0 ppm) under continuous stirring at room temperature. After 30 min in the dark, the dispersion was irradiated at 365 nm using an LED light source (LLS-365, Ocean Optics, Tokyo, Japan). Then, 2.0 mL of the dispersion was sampled at 10 min intervals and centrifuged for solid–liquid separation. The dye concentration of the supernatant was then measured at the λmax of the dye. To confirm the reactive species, scavenger solutions (1 ppm of p-BQ, Na2-EDTA, and IPA) were used to scavenge superoxide radicals, holes, and hydroxyl radicals, respectively. The quantity (Qd) of dye degraded was estimated in mg g−1 by subtracting the free dye concentration at time t (Ct, mg L−1) from the dye concentration before light irradiation. Then, the decomposed quantity of the dye was calculated using eqn (2).
 | (2) |
The dye decomposition efficiency (%D) was also assessed using eqn (3).
 | (3) |
The decomposition kinetics were analyzed as pseudo first-order reactions using the Langmuir–Hinshelwood model55 and were plotted as ln(C0/Ct) vs. the photoirradiation time (t, min), as in eqn (4).
 | (4) |
3. Results and discussion
3.1 Structure of nanocomposites
The XRD patterns for the obtained nanocomposites are shown in Fig. 1. The sol-TiO2 showed peaks at 2θ = 25.4°, 38.0°, 48.1°, 54.1°, 55.2°, 62.8°, and 75.3°, which correspond to the (101), (004), (200), (105), (211), (204), and (215) planes of the hexagonal crystal lattice of the TiO2 anatase phase (JCPDS PDF 89-4921), while the sole diffraction peak at 31.0° (marked by an asterisk in Fig. 1A(a)) corresponds to the (121) plane of the orthorhombic crystal lattice of brookite phase of TiO2 (JCPDS PDF 29-1360). | ||
| Fig. 1 XRD results of (A) the sol–gel prepared (a) sol-TiO2, (b) sol-WO3, (c–e) 5 wt%, 8 wt%, and 10 wt% WO3/TiO2; (B) hydrothermally prepared (a) hyd-TiO2, (b) hyd-WO3, (c) TiO2@WO3 and (d) WO3@TiO2. | ||
The sol-WO3 showed peaks at 2θ = 14.2°, 23.1°, 24.5°, 28.4°, 33.8°, 36.8°, 50.0°, 52.3°, 55.6°, 58.2°, and 63.5°, which were, respectively assigned to the (100), (002), (110), (200), (112), (202), (220), (213), (222), (312), and (402) planes of the hexagonal crystal lattice of WO3 (JCPDS PDF 85-2459). In the 5 wt% WO3/TiO2 nanocomposite, the presence of WO3 was not well confirmed, which was probably due to the low concentration of WO3.
The 8 wt% and 10 wt% WO3/TiO2 nanocomposites showed diffraction peaks for WO3; however, some peaks (marked by ♦) significantly shifted from those of sol-WO3 and were identified as a monoclinic phase of the tungsten oxide W18O49 (JCPDS PDF 05-0392). In contrast, the XRD peaks for TiO2 in these nanocomposites appeared at the same positions as those in sol-TiO2. These results suggested that the W ions were minor components in the nanocomposites (only 8 wt% and 10 wt%) and formed new crystal structures under the influence of Ti compounds.
The hyd-TiO2 exhibited diffraction peaks at 2θ = 25.4°, 38.0°, 48.1°, 54.1°, 55.2°, 62.8°, and 75.2°, which correspond to the (101), (112), (200), (105), (211), (204), and (215) planes, respectively (Fig. 1B(a)). All the observed diffraction peaks belonged to anatase TiO2 (JCPDS PDF 89-4921), and the peak for the (121) plane of the brookite phase was not observed. The hyd-WO3 showed diffraction patterns at 2θ = 23.4°, 23.9°, 24.6°, 26.8°, 28.9°, 33.5°, 34.4°, 35.7°, 42.1°, 47.5°, 48.5°, 50.1°, 53.6°, 56.1°, 62.4°, and 76.6° corresponding to the (001), (020), (200), (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 20), (11), (
20), (11), (![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 01), (220), (121), (221), (002), (040), (140), (022), (
01), (220), (121), (221), (002), (040), (140), (022), (![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 20), (
20), (![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 40), and (422) planes, respectively, which indicated a monoclinic WO3 crystal (JCPDS PDF 05-0363). Thus, in contrast with the sol–gel method, the hydrothermal synthesis process could be adjusted to result in the monoclinic crystalline phase. Both the hydrothermally produced core–shell TiO2@WO3 and WO3@TiO2 showed diffraction peaks for anatase TiO2, and peaks for WO3 were not observed. The weakness of the XRD peak intensity of WO3 suggested that the WO3 shell was very thin and that the WO3 core was covered with a thick TiO2 shell.
40), and (422) planes, respectively, which indicated a monoclinic WO3 crystal (JCPDS PDF 05-0363). Thus, in contrast with the sol–gel method, the hydrothermal synthesis process could be adjusted to result in the monoclinic crystalline phase. Both the hydrothermally produced core–shell TiO2@WO3 and WO3@TiO2 showed diffraction peaks for anatase TiO2, and peaks for WO3 were not observed. The weakness of the XRD peak intensity of WO3 suggested that the WO3 shell was very thin and that the WO3 core was covered with a thick TiO2 shell.
The crystallite size (D) was computed from Debye–Scherrer's equation (eqn (5)).52
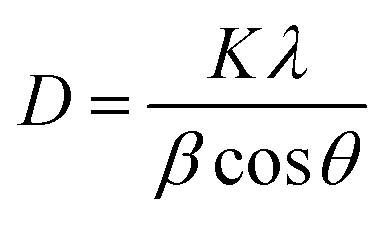 | (5) |
3.2 Optical properties of nanocomposites
The optical properties of the nanomaterials were studied with UV-VIS DRS (Fig. 2). Both sol-TiO2 and hyd-TiO2 exhibited high reflectance in the range greater than 350 nm, and with the addition of WO3, the material reflectance decreased in this range (Fig. 2a and b). The reflectance of TiO2@WO3 was slightly lower than that of WO3@TiO2. Since WO3 can absorb light of wavelengths shorter than 500 nm,31,32,56 the lower reflectance could be partially explained by the photoexcitation of WO3 in the short wavelength region. At longer wavelengths (>500 nm), the WO3 and WO3–TiO2 nanocomposites showed lower reflectance than TiO2. This could be explained by the existence of WOx (2 < x < 3), which could absorb light, and by the structures of the film specimens.32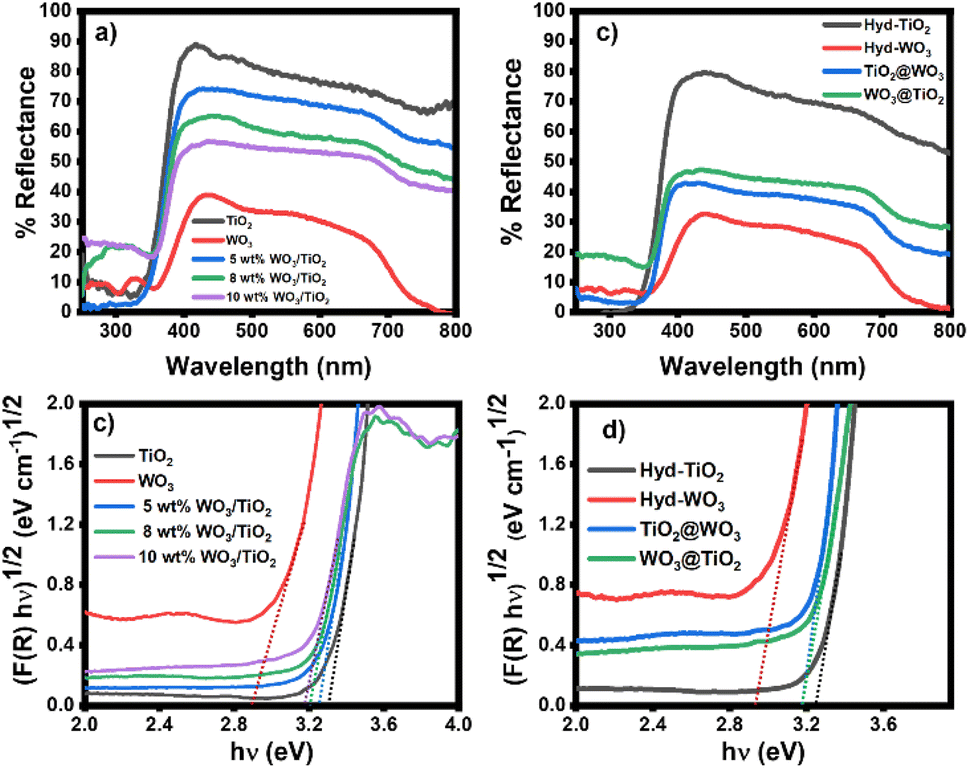 | ||
| Fig. 2 (a and b) UV-VIS DRS spectra of the sol–gel and hydrothermally prepared samples and (c and d) bandgap energy analyses of nanocomposites. | ||
The bandgap energy (Eg) of the metal oxides was determined using the Kubelka–Munk function and Tauc plots (Fig. 2c and d).57 The Eg values were evaluated from the intercepts of the energy axis. The Eg values of TiO2 and WO3 were 3.2–3.3 eV and 2.8–2.9 eV, respectively, and compared with the materials prepared by the sol–gel method, hyd-TiO2 and hyd-WO3 showed slightly decreased Eg values, which was attributed to their larger crystal sizes.58 The nanocomposites had Eg values between those of TiO2 and WO3: the sol–gel method resulted in Eg values of 3.25, 3.20, and 3.18 eV, corresponding to 5 wt%, 8 wt%, and 10 wt% WO3/TiO2, respectively (Fig. 2c). For core–shell TiO2@WO3 and WO3@TiO2, the Eg values were nearly identical at 3.18 eV (Fig. 2d). The intermediate Eg values (in between those of TiO2 and WO3) suggested interactions between TiO2 and WO3 in both the mixed and core–shell structures. Mutual effects in the nanocomposites can decrease the electron–hole recombination rate at the interface for TiO2 and WO3.
3.3 Morphological analyses of nanocomposites
The morphologies of the nanocomposites were observed using SEM (Fig. 3). The materials prepared by the sol–gel method consisted of aggregated spherical nanoparticles, and no systematic difference was observed. The hyd-TiO2 consisted of aggregates of small nanoparticles, while the hyd-WO3 showed large crystalline structures, as indicated by the XRD analysis. However, the surface morphology of the core–shell nanocomposites did not show a significant difference. This suggests that the WO3 shell in the TiO2@WO3 nanocomposite could not develop large crystals because it grew from small TiO2 particles.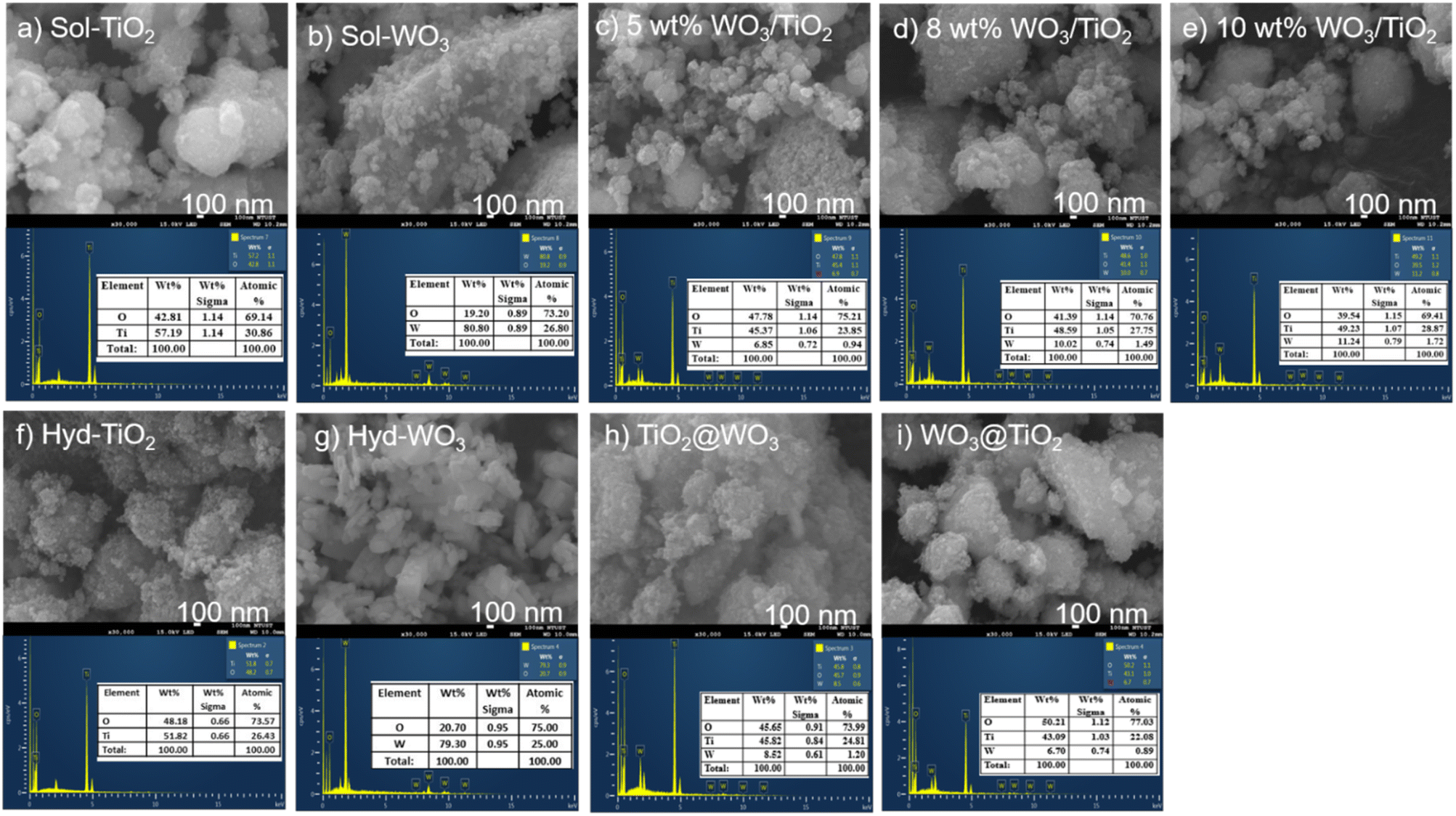 | ||
| Fig. 3 SEM images (upper) and EDX (lower) results for sol–gel (a–e) and hydrothermally (f–i) synthesized materials. | ||
The EDX results identified the presence of titanium (Ti) and oxygen (O) in TiO2, tungsten (W) and O in the WO3 oxide, as well as the presence of Ti, W, and O in the nanocomposites obtained from both the sol–gel and hydrothermal methods. The weight ratios of W to Ti in the sol–gel and core–shell nanocomposites are shown in Table 1. The experimentally obtained W ratios in the nanocomposites prepared by the sol–gel method were significantly higher than the corresponding theoretical values, which suggests that a significant amount of Ti compounds were not recovered during the preparation process relative to the W compounds. In contrast, the hydrothermally prepared nanocomposites showed W ratios that agreed well with the theoretical values. This suggests that the hydrothermal process immobilized the Ti compounds in the nanocomposites during crystal growth. The higher W ratio in TiO2@WO3 relative to WO3@TiO2 could be explained by the fact that the TiO2 core was covered by the WO3 shell.
| Nanocomposite | Synthesis method | EDS result | XPS result | Theoretical ratio |
|---|---|---|---|---|
| TiO2 (Ti:O) |
Sol–gel process | 1.3 | 1.4 | |
| WO3 (W:O) |
4.2 | 3.8 | ||
| 5 wt% WO3/TiO2 (W:Ti) |
0.15 | 0.22 | 0.07 | |
| 8 wt% WO3/TiO2 (W:Ti) |
0.21 | 0.28 | 0.12 | |
| 10 wt% WO3/TiO2 (W:Ti) |
0.23 | 0.33 | 0.15 | |
| TiO2 (Ti:O) |
Hydrothermal process | 1.1 | 1.4 | |
| WO3 (W:O) |
3.8 | 3.8 | ||
| TiO2@WO3 (W:Ti) |
0.18 | 0.26 | 0.15 | |
| WO3@TiO2 (W:Ti) |
0.16 | 0.20 | 0.15 |
3.4 Chemical states of nanocomposites
XPS analysis was conducted to determine the chemical states of the sol–gel synthesized and hydrothermally synthesized nanocomposites (Fig. 4). The peak parameters are shown in the ESI (Tables S1–S3 in the ESI†). The survey XPS spectrum showed the existence of oxygen (O 1s), titanium (Ti 2p), tungsten (W 4p and 4f), and carbon (C 1s) on the material surfaces.44 The carbon contribution originated from the substrate and was used to calibrate the binding energy. | ||
| Fig. 4 XPS survey and high-resolution spectra of the sol–gel method: (A) 5 wt% WO3/TiO2, (B) 8 wt% WO3/TiO2 and (C) 10 wt% WO3/TiO2, and core–shell method: (D) TiO2@WO3 and (E) WO3@TiO2 with (a) survey, (b) O 1s, (c) Ti 2p, and (d) W 4f. | ||
The deconvoluted XPS spectra of O 1s showed three distinct peaks in all specimens. The peak at the lowest binding energy (530.4 eV for 5 wt%, 529.7 eV for 8 wt%, 530.0 eV for 10 wt% WO3/TiO2, 530.0 eV for TiO2@WO3, and 530.0 eV for WO3@TiO2) was attributed to the lattice oxygen (O2−) of O–Ti bonds in TiO2 and O–W bonds in WO3,40 whereas the middle peaks (531.8 eV for 5 wt%, 530.9 eV for 8 wt%, 531.3 eV for 10 wt% WO3/TiO2, 531.0 eV for TiO2@WO3, and 531.3 eV for WO3@TiO2) could be attributed to substoichiometric WOx (2 < x < 3)40,59,60 or hydroxide groups adsorbed on the oxide surface as W–O–H and Ti–O–H,40 which correspond to oxygen vacancies. To compensate for the charge imbalance in the oxygen-deficient state, OH groups were bound to the metal cations. Thus, the density of oxygen vacancies is indicated by the intensity of these mid-binding energy peaks.61 The third peaks, which were located at the highest binding energy (533.4 eV for 5 wt%, 532.8 eV for 8 wt%, 533.0 eV for 10 wt% WO3/TiO2, 532.7 eV for TiO2@WO3, and 532.5 eV for WO3@TiO2), could be attributed to contamination from oxygen-containing hydrocarbons,40 H2O,59 or surface-adsorbed O2.62 The peak areas (%) of the mid-binding energy peaks ranged from 11% to 18%, which indicated that significant numbers of oxygen vacancies were formed in the nanocomposites, as suggested from the UV-VIS DRS spectra (Fig. 2). These oxygen vacancies could extend the lifetime of the charge carriers and increase the photocatalytic activity of these catalysts.
The presence of only one Ti 2p doublet for Ti 2p3/2 and Ti 2p1/2 indicated that all Ti atoms shared the same oxidation state (Ti4+).40 The binding energies of Ti 2p3/2 for the sol–gel synthesized nanocomposites of 5 wt%, 8 wt%, and 10 wt% WO3/TiO2 were 459.1 eV, 458.4 eV, and 458.9 eV, respectively, while they were 459.3 eV and 458.7 eV for the hydrothermally prepared TiO2@WO3 and WO3@TiO2, respectively. The binding energies of Ti 2p1/2 for 5 wt%, 8 wt%, and 10 wt% WO3/TiO2 were 464.8 eV, 464.0 eV, and 464.6 eV, respectively, whereas they were 464.9 eV and 464.4 eV for TiO2@WO3 and WO3@TiO2, respectively. The minor change in energy in the nanocomposites could be attributed to the interactions of W–O–Ti bonds; however, the changes were not significantly or systematic.
The peaks of W 4f appeared as two doublets. The first pair (peaks 1 and 3) might have arisen from W5+ in substoichiometric WOx (2 < x < 3),40 which corresponds to an oxygen vacancy. The second pair (peaks 2 and 4) was ascribed to W6+ in WO3.61 The ratio of W 4f to Ti 2p determined from the peak area in the nanocomposites (Tables 1 and S3 in the ESI†) was 0.22, 0.28, 0.33, 0.26, and 0.20 for 5 wt%, 8 wt%, 10 wt% WO3/TiO2, TiO2@WO3, and WO3@TiO2, respectively. These values were larger than those obtained by the EDX method, which suggested that the W component existed more on the surface than in the bulk phase of the nanocomposites. The detection of W in the core–shell WO3@TiO2 nanocomposite and Ti in the core–shell TiO2@WO3 nanocomposite suggested that the core–shell structures were imperfect, although the compositions were controlled to an extent.
Fig. 5 shows the ratios of the %area of XPS peaks for each element in the nanocomposites. The changes in the %area of both W5+ and W6+ in the nanocomposites indicated the high ratios of W5+ on the surface of the nanocomposites. The existence of W5+ can extend the light absorption range, and W5+ can provide an electron to molecular oxygen to form superoxide radicals (O2˙−) under light irradiation. Therefore, a higher ratio of W5+ on the surface could be an advantage for photocatalysis. On the other hand, the low positive charge on the surface induces low attractive interactions with negatively charged dyes, which is disadvantageous for photocatalysis.
 | ||
| Fig. 5 Ratios of chemical states for nanocomposites prepared by the sol–gel method ((a) O 1s, (b) Ti 2p, and (c) W 4f) and the core–shell method ((d) O 1s, (e) Ti 2p, and (f) W 4f). | ||
3.5 Adsorption process of dyes
The adsorption behaviors of cationic MB+ and anionic MO− onto the nanocomposites were investigated in the range of 1–8 mg L−1 initial dye concentrations (the data and the fitting curves are shown in Fig. S2 and S3 in the ESI†). The adsorption performances of the pure oxides and composite materials were evaluated by the Langmuir isotherm adsorption models.63–65 The Langmuir adsorption isotherms of the dyes are depicted in eqn (6) below:
 | (6) |
| Nanocomposite | qm (mg g−1) | KL (×105 L mol−1) | R2 | ΔG (K J mol−1) | |
|---|---|---|---|---|---|
| MB+ | Sol-TiO2 | 6.6 | 3.64 | 0.9038 | −31.7 |
| hyd-TiO2 | 13.1 | 2.76 | 0.9887 | −31.0 | |
| Sol-WO3 | 31.2 | 31.82 | 0.9679 | −37.1 | |
| hyd-WO3 | 35.6 | 27.91 | 0.9279 | −36.8 | |
| 5 wt% WO3/TiO2 | 38.7 | 5.55 | 0.9798 | −32.9 | |
| 8 wt% WO3/TiO2 | 40.8 | 6.41 | 0.9684 | −33.1 | |
| 10 wt% WO3/TiO2 | 38.5 | 7.63 | 0.9768 | −33.6 | |
| TiO2@WO3 | 55.8 | 5.94 | 0.9533 | −33.0 | |
| WO3@TiO2 | 39.0 | 4.34 | 0.9643 | −32.2 | |
| MO− | Sol-TiO2 | 3.04 | 2.67 | 0.349 | −31.0 |
| hyd-TiO2 | 1.62 | 2.85 | 0.7081 | −31.1 | |
| Sol-WO3 | 0.24 | 0.77 | 0.7724 | −27.9 | |
| hyd-WO3 | 0.23 | 0.79 | 0.7303 | −27.9 | |
| 5 wt% WO3/TiO2 | 0.55 | 2.07 | 0.6907 | −30.3 | |
| 8 wt% WO3/TiO2 | 0.39 | 1.09 | 0.8428 | −28.7 | |
| 10 wt% WO3/TiO2 | 0.46 | 0.82 | 0.6463 | −28.0 | |
| TiO2@WO3 | 0.27 | 1.21 | 0.6739 | −29.0 | |
| WO3@TiO2 | 0.73 | 2.34 | 0.5965 | −30.6 |
The adsorption of MB+ from different initial concentrations was explored at pH 7.4 without pH control. The adsorption of different MO− initial concentrations was probed at pH 6.7 without pH control. As observed in Fig. 6a, the amount of adsorbed MB+ (qe) rose as the initial concentration increased and became saturated at high concentrations (6–8 ppm). The amount of adsorbed MO− (qe) also increased as the initial concentration increased in the low concentration range (1–2 ppm) and became almost saturated at higher concentrations (2–8 ppm), except for a slight increase and decline for TiO2 (Fig. 6b). For the adsorption of MB+, the Langmuir model fitted well (R2 > 0.9), indicating that the adsorption of MB+ on the nanocomposites followed a monolayer adsorption process. However, the Langmuir model did not yield good fits for adsorption of MO− (R2 was in the range of 0.3490 to 0.8428), which could be explained by the low adsorption amounts of MO− (two orders of magnitude smaller than those of MB+, except for TiO2), leading to large errors.
 | ||
| Fig. 6 (a & b) Adsorbed amounts of MB+ and MO− on TiO2, WO3, and nanocomposites at various concentrations, respectively; (c &d) MB+ and MO− decomposition in dark; (e &f) degradation rates for MB+ and MO− under UV light; (g & h) pseudo-first-order kinetic model for decomposition of MB+ and MO− under UV light. Left column is for MB+ and right column is for MO−, respectively. | ||
Then, dye adsorption on the nanocomposites was analyzed with eqn (7).5
|
ΔG = −RTlnKL
| (7) |
In terms of the adsorption behavior of MO−, it should be noted that the ΔG of MO− adsorption could not be precisely estimated because of the poor correlation coefficients (<0.9) obtained. However, TiO2 showed a higher adsorption capacity than the others for MO− adsorption, and the nanocomposites also exhibited stronger interactions due to the higher compositional ratio of titanium, as expected from the surface charge of the nanocomposites. The KL and ΔG of TiO2 for MO− were comparable to those for MB+ adsorption. This finding suggests that the TiO2 nanoparticles provided binding sites for both the cationic and anionic dyes. The lower adsorption capacity of hyd-TiO2 for MO− compared with MB+ suggests that the number of cationic binding sites was lower in hyd-TiO2, which could be due to the difference in crystallinity and the crystal structures shown by the XRD measurements (Fig. 1). The tungsten enhanced the adsorption of MB+ and weakened the adsorption of MO− on the nanocomposite surfaces.68 However, the KL and ΔG values indicated that some nanocomposites also provided effective binding sites for MO−, especially the 5 wt% WO3/TiO2 and WO3@TiO2 nanocomposites.
3.6 Photocatalysis of dyes
The photocatalytic activity of the synthesized materials was evaluated by dye decomposition. The resulting absorption spectra are shown in Fig. S4 and S5 in the ESI.† The decomposition curves and their kinetic analyses for MB+ and MO− are shown in Fig. 6.The MB+ was irradiated with UV light in an aqueous solution of pH 7.4. Measurements were also performed under dark conditions, and the decrease in concentration was in the range of 1–8% of the initial concentration, which could have been caused by the disaggregation of the nanocomposites induced by stirring. In the absence of nanocomposite materials, the degradation of MB+ was 14.4% under UV irradiation, which was lower than that in the presence of nanocomposites. Compared with TiO2 and WO3, the nanocomposites exhibited better photocatalytic activity under UV light irradiation. The photocatalytic activity of 8 wt% WO3/TiO2 was highest among the sol–gel nanocomposites, with 94.9% decomposition after 2 h, while 5 wt% WO3/TiO2 achieved the lowest degradation (84.7%) during the same irradiation time. The core–shell TiO2@WO3 demonstrated superior photocatalytic activity, with 95.8% decomposition after 1 h of UV irradiation, while WO3@TiO2 demonstrated lower activity (decomposition of 82.5%) after 2 h than 5 wt% TiO2@WO3. The reaction rates were analyzed as pseudo first-order reactions using eqn (4) and the Langmuir–Hinshelwood model.65 The rate constant  for each nanocomposite and dye is summarized in Table 3.
for each nanocomposite and dye is summarized in Table 3.
| Photocatalysts | MB+ | MO− | ||||
|---|---|---|---|---|---|---|
|
R2 |
|
|
R2 |
|
|
| Sol-TiO2 | 0.23 | 0.9904 | 9.57 | 0.1 | 0.9816 | 12.32 |
| Sol-WO3 | 0.86 | 0.9927 | 0.87 | 0.01 | 0.9335 | 54.11 |
| 5 wt% WO3/TiO2 | 1.59 | 0.9974 | 7.40 | 0.04 | 0.9634 | 35.12 |
| 8 wt% WO3/TiO2 | 2.48 | 0.9967 | 9.48 | 0.02 | 0.9905 | 47.05 |
| 10 wt% WO3/TiO2 | 2.05 | 0.9959 | 6.98 | 0.03 | 0.9930 | 79.53 |
| Hyd-TiO2 | 0.46 | 0.9978 | 12.72 | 0.07 | 0.9605 | 15.16 |
| Hyd-WO3 | 1.04 | 0.9925 | 1.05 | 0.008 | 0.9409 | 44.03 |
| TiO2@WO3 | 5.33 | 0.9986 | 16.08 | 0.02 | 0.9778 | 61.22 |
| WO3@TiO2 | 1.41 | 0.9930 | 8.33 | 0.05 | 0.9962 | 29.27 |
| Without catalyst | 0.13 | 0.9954 | — | 0.007 | 0.9067 | — |

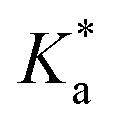

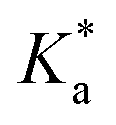
These activities were inconsistent with the orders of both the adsorption rate (KL) and the adsorption capacity (qm) of the nanocomposites (Table 2). To some extent, the magnitude of the reaction rates could be explained by several factors: (1) the tungsten in the nanocomposites provided higher reaction rates due to its strong interaction with MB+, (2) the absorbance of TiO2 was low at an excitation wavelength of 365 nm, (3) among the sol–gel nanocomposites, the adsorption capacities determined the order of the reaction rates, and (4) TiO2@WO3 showed highest rate constant, which could because it also had the highest adsorption capacity. However, these explanations were not sufficient for explaining the lower rate constant of WO3, which had a high adsorption constant and moderate adsorption capacity. Upon dividing  by KL and qm, WO3 exhibited the lowest rate constant per (adsorption rate x adsorption mass), while that of TiO2@WO3 was the highest, followed by that of hyd-TiO2. Therefore, the photocatalytic activity was not determined only by the adsorption amount and the adsorption rate.
by KL and qm, WO3 exhibited the lowest rate constant per (adsorption rate x adsorption mass), while that of TiO2@WO3 was the highest, followed by that of hyd-TiO2. Therefore, the photocatalytic activity was not determined only by the adsorption amount and the adsorption rate.
For comparison, the anionic MO− was also degraded at pH 6.7 under UV light irradiation. The reaction rate was much lower than that of MB+, which could be expected from the adsorption parameters discussed above (Table 2). Among the nanocomposites, WO3@TiO2 demonstrated the highest efficiency (degradation of ∼6% MO−), whereas TiO2@WO3 demonstrated the lowest efficiency (degradation of ∼2% MO−). The negative surface charge from the tungsten oxides had adverse effects on the photocatalysis of MO−. Upon dividing  by KL and qm, the nanocomposites demonstrated a clear tendency: a higher composition of tungsten resulted in a higher rate constant. This could be explained by the stronger light absorption at 365 nm by tungsten components. However, the higher rate constants per (adsorption rate × adsorption mass) of 10 wt% WO3/TiO2 and TiO2@WO3 suggested that the nanocomposite decomposed MO− more effectively than TiO2 and WO3.
by KL and qm, the nanocomposites demonstrated a clear tendency: a higher composition of tungsten resulted in a higher rate constant. This could be explained by the stronger light absorption at 365 nm by tungsten components. However, the higher rate constants per (adsorption rate × adsorption mass) of 10 wt% WO3/TiO2 and TiO2@WO3 suggested that the nanocomposite decomposed MO− more effectively than TiO2 and WO3.
Compared with other studies for the MB+ degradation performance (Table S4 in the ESI†), the activity of 8 wt% WO3/TiO2 was 1.34 times higher than that of 25 wt% mixed WO3/TiO2.46 The activity of the core–shell TiO2@WO3 of the current study was 3.59 times higher than that of 36 wt% core–shell WO3/TiO2.43 These results indicate that the large amount of WO3 is not essential for the effective photocatalyst.
3.7 Dye decomposition mechanism
To study the active species present during photocatalysis with the nanocomposites, dye photocatalysis was analyzed in the presence of active species scavengers. The active species generated by TiO2 and WO3 are considered to be superoxide anions (O2−), holes (h+), and hydroxyl radicals (HO˙), which can be scavenged by p-BQ, Na2-EDTA, and IPA, respectively.38,48,69,70 The degradation curves and the kinetic analyses are shown in Fig. 7 and Table 4.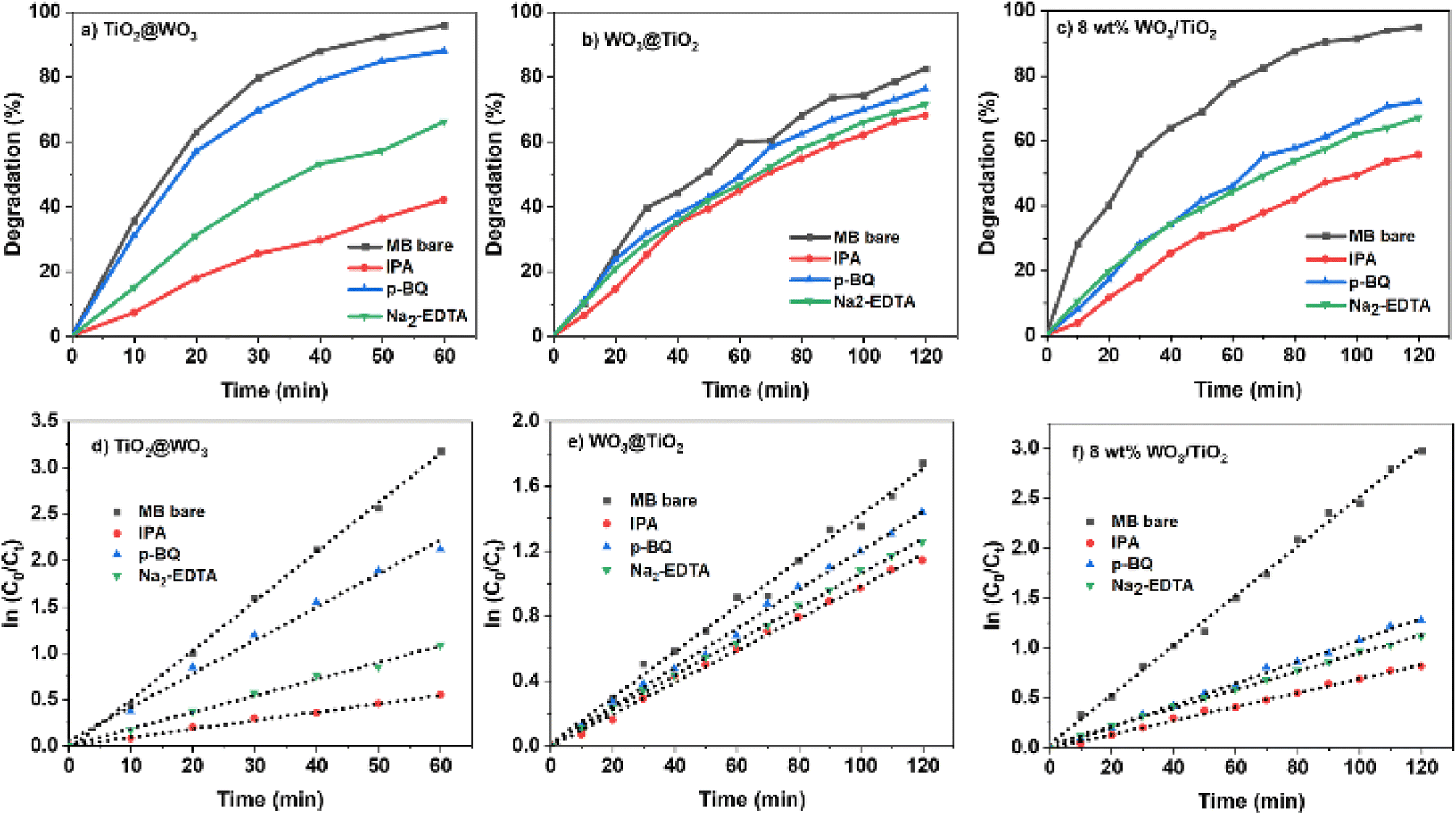 | ||
| Fig. 7 (a–c) Photocatalytic decomposition of MB+ over core–shell TiO2@WO3, WO3@TiO2, and coprecipitated 8 wt% WO3/TiO2 with various scavengers under UV light irradiation; (d–f) kinetic analysis of MB+ decomposition. | ||
| Sample type | 8 wt% WO3/TiO2 | TiO2@WO3 | WO3@TiO2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| %D | Ka (×10−2 min−1) | R2 | Inhibition | %D | Ka (×10−2 min−1) | R2 | Inhibition | %D | Ka (×10−2 min−1) | R2 | Inhibition | |
| Without scavenger | 94.9 | 2.48 | 0.9967 | — | 95.8 | 5.33 | 0.9986 | — | 82.5 | 1.41 | 0.993 | — |
| p-BQ | 72.0 | 1.09 | 0.9969 | −56% | 87.9 | 3.60 | 0.9931 | −32% | 76.2 | 1.20 | 0.9974 | −15% |
| Na2-EDTA | 67.0 | 0.92 | 0.9981 | −63% | 66.1 | 1.79 | 0.9941 | −66% | 71.5 | 1.05 | 0.9992 | −26% |
| IPA | 55.6 | 0.70 | 0.9969 | −72% | 42.1 | 0.91 | 0.9955 | −83% | 68.1 | 0.99 | 0.9968 | −30% |
The addition of scavengers decreased the decomposition rate in all cases. These results indicated that hydroxyl radicals were the most active species for all the photocatalysts examined. When IPA was used to quench hydroxyl radicals, the Ka values of the photocatalysts decreased to a similar level (0.70–0.99 × 10−2 min−1). The activity of TiO2@WO3 exhibited the greatest decrease with the use of IPA, and the effect of IPA on WO3@TiO2 was the smallest. This suggests that WO3 on the nanocomposite surface mainly provides hydroxyl radicals as the active species; the OH− groups attached to W5+/6+ could be directly oxidized by holes to generate hydroxyl radicals.
A comparison of the effects of p-BQ and Na2-EDTA show that the Ka of TiO2@WO3 decreased more with the addition of Na2-EDTA than p-BQ, while the 8 wt% WO3/TiO2 nanocomposite was similarly quenched by both scavengers. These results suggest that the activity of the core–shell TiO2@WO3 depended on the activity of holes more than photoexcited electrons, while the codeposited WO3/TiO2 used both to a similar extent. In the reversed structure, the core–shell WO3@TiO2 nanocomposite also demonstrated a stronger effect with Na2-EDTA than p-BQ; however, its activity was much lower than that of TiO2@WO3, and the decrease in the Ka value was not large. The effects of Na2-EDTA and p-BQ (i.e., the amounts of scavenged holes and electrons) were not greatly different for WO3@TiO2, and the Ka values became similar to those of 8 wt% WO3/TiO2. The weak effects of scavengers on WO3@TiO2 suggested that the active species (O2−, hole, and HO˙) were rapidly changed to other species or quenched in the nanocomposite by recombination.
In the core–shell nanocomposites, the photoinduced charges were considered to be adequately separated. Because the conduction band (CB) of TiO2 has a more negative redox potential (−0.5 V) than the CB of WO3 (+0.2 V), as reported by Escobar et al. (2020),38 the photogenerated electrons in TiO2 can easily be transferred to WO3 to reduce W6+ to W5+.48 According to Escobar et al. (2020), the valence band (VB) of WO3 has a more positive redox potential (+3.1 V) than the VB of TiO2 (+2.8 V);38 therefore, the photogenerated h+ can move from WO3 to TiO2 (Fig. 8). This charge separation extended the lifetime of the active species and improved the activity of holes on TiO2@WO3, while WO3 rapidly caused recombination and decreased photocatalytic activity, although it had the highest KL value (Tables 2 and 3). In the codeposited WO3/TiO2, similar charge separation occurred, and both holes and photoelectrons were active on the surface. In the reversed core–shell WO3@TiO2, the photoexcited electrons (and superoxide anions) were likely to be the main species, but the electron transfer process from TiO2 to WO3 was competitive with the process of superoxide anion generation. This competition also occurred for hole transfer, and therefore, the activity of WO3@TiO2 was strongly suppressed. These mechanisms are illustrated in Fig. 8.
 | ||
| Fig. 8 Dye decomposition mechanism using (A) TiO2@WO3, (B) WO3@TiO2, and (C) WO3/TiO2 nanocomposites. | ||
4. Conclusions
In this study, WO3-loaded TiO2 nanocomposites of various WO3 compositions were prepared using sol–gel (coprecipitation) and hydrothermal (core–shell) approaches. The structures of the nanocomposites were analyzed using XRD, SEM, EDX, XPS, and DRS, and adsorption-driven photocatalysis was comprehensively examined. Using cationic methylene blue (MB+) and anionic methyl orange (MO−), the adsorption behaviors of the dyes were explained by the electrostatic interaction between the dyes and the negatively charged surfaces of metal oxides, especially with respect to the tungsten component. From the Langmuir adsorption model, the adsorption rate and the adsorption capacity were analyzed for each metal oxide. The core–shell TiO2@WO3 demonstrated greater maximum adsorption capacity (qm = 55.8 mg g−1) than the sol–gel-produced nanomaterials, indicating that more active sites were available for photocatalysis. Through a kinetic study, the photocatalytic decomposition reactions of both MB+ and MO− on the sol–gel and core–shell metal oxides were analyzed. The WO3-loaded TiO2 nanocomposites showed considerably higher activity for MB+ than for MO−. The reaction rate per (adsorption rate x adsorption capacity) was calculated for each photocatalyst, and a synergistic effect was found. Using scavengers for active species, a charge separation mechanism was considered to improve the photocatalytic activity of complex metal oxides relative to simple oxides. Thus, the high efficiency of the core–shell TiO2@WO3 was explained. These results and the approaches used in this study could be useful for designing photocatalysts consisting of hybrid metal oxides.Author contributions
Abdisa Habtamu: investigation, formal analysis, writing-original draft, validation. Masaki Ujihara: conceptualization, methodology, project administration, supervision, resources, writing-review and editing, visualization, validation.Conflicts of interest
The authors disclaim any conflicts of interest.Acknowledgements
This investigation was partly supported by the Ministry of Science and Technology of the Republic of China, [MOST 109-2221-E-011-062-], and the Graduate Institute of Applied Science and Technology, National Taiwan University of Science and Technology.References
- M. Manna and S. Sen, Environ. Sci. Pollut. Res., 2022, 1–29 Search PubMed , https://www.x-mol.com/paperRedirect/1503426244822065152.
- I. Arslan-Alaton, F. G. Babuna and G. Iskender, in Advanced Oxidation Processes for Wastewater Treatment, CRC Press, 2022, pp. 39–51, DOI:10.1201/9781003165958.
- B. Hu, Y. Ai, J. Jin, T. Hayat, A. Alsaedi, L. Zhuang and X. Wang, Biochar, 2020, 2, 47–64 CrossRef.
- M. Ismail, K. Akhtar, M. Khan, T. Kamal, M. A. Khan, A. M Asiri, J. Seo and S. B. Khan, Curr. Pharm. Des., 2019, 25, 3645–3663 CrossRef CAS PubMed.
- J. Chen, Y. Xiong, M. Duan, X. Li, J. Li, S. Fang, S. Qin and R. Zhang, Langmuir, 2019, 36, 520–533 CrossRef PubMed.
- D. Vishnu, B. Dhandapani, S. Authilingam and S. V. Sivakumar, Curr. Anal. Chem., 2022, 18, 255–268 CrossRef CAS.
- P. Zhang, D. O'Connor, Y. Wang, L. Jiang, T. Xia, L. Wang, D. C. Tsang, Y. S. Ok and D. Hou, J. Hazard. Mater., 2020, 384, 121286 CrossRef CAS PubMed.
- J.-H. Shin, J. E. Yang, J. E. Park, S.-W. Jeong, S.-J. Choi, Y. J. Choi and J. Jeon, ACS Omega, 2022, 7(10), 8759–8766 CrossRef CAS PubMed.
- C. Karaman, O. Karaman, P.-L. Show, H. Karimi-Maleh and N. Zare, Chemosphere, 2022, 290, 133346 CrossRef CAS PubMed.
- P. Yushananta and M. Ahyanti, Jurnal Aisyah: Jurnal Ilmu Kesehatan, 2022, 7, 165–172 Search PubMed.
- M. Perwez, H. Fatima, M. Arshad, V. Meena and B. Ahmad, Int. J. Environ. Sci. Technol., 2022, 1–18, DOI:10.1007/s13762-022-04003-3.
- G. Sharma, A. Khosla, A. Kumar, N. Kaushal, S. Sharma, M. Naushad, D.-V. N. Vo, J. Iqbal and F. J. Stadler, Chemosphere, 2022, 289, 133100 CrossRef CAS PubMed.
- Z. Isik, M. Saleh, I. M’barek, E. Yabalak, N. Dizge and B. Deepanraj, Biomass Convers. Biorefin., 2022, 1–14, DOI:10.1007/s13399-022-02582-2.
- S. Praveen, J. Jegan, T. Bhagavathi Pushpa, R. Gokulan and L. Bulgariu, Biochar, 2022, 4, 1–16 CrossRef.
- K. Xie, J. Fang, L. Li, J. Deng and F. Chen, J. Alloys Compd., 2022, 163589, DOI:10.1016/j.jallcom.2021.163589.
- Y. Zhuang, Q. Zhu, G. Li, Z. Wang, P. Zhan, C. Ren, Z. Si, S. Li, D. Cai and P. Qin, Mater. Res. Bull., 2022, 146, 111619 CrossRef CAS.
- R. Katwal, R. Kothari and D. Pathania, Delivering Low-Carbon Biofuels with Bioproduct Recovery, 2021, pp. 195–213, DOI:10.1016/B978-0-12-821841-9.00005-0.
- A. Berhe and M. Ujihara, ChemistrySelect, 2018, 3, 10502–10508 CrossRef CAS.
- N. Kamely and M. Ujihara, J. Nanopart. Res., 2018, 20, 1–10 CrossRef CAS.
- A. L. Nigusie and M. Ujihara, Phys. Chem. Chem. Phys., 2021, 23, 16366–16375 RSC.
- I. Shown, M. Ujihara and T. Imae, J. Nanosci. Nanotechnol., 2011, 11, 3284–3290 CrossRef CAS PubMed.
- K. G. Motora, C.-M. Wu and S. Naseem, J. Ind. Eng. Chem., 2021, 102, 25–34 CrossRef CAS.
- A. F. Shojaei, A. Shams-Nateri and M. Ghomashpasand, Superlattices Microstruct., 2015, 88, 211–224 CrossRef.
- D. Kanakaraju and A. B. Chandrasekaran, Sci. Total Environ., 2023, 161525, DOI:10.1016/j.scitotenv.2023.161525.
- S. Varnagiris, M. Urbonavicius, S. Sakalauskaite, R. Daugelavicius, L. Pranevicius, M. Lelis and D. Milcius, Sci. Total Environ., 2020, 720, 137600 CrossRef CAS PubMed.
- N. M. Ainali, D. Kalaronis, E. Evgenidou, D. N. Bikiaris and D. A. Lambropoulou, Macromol, 2021, 1, 201–233 CAS.
- D. Hu, R. Li, M. Li, J. Pei, F. Guo and S. Zhang, Mater. Res. Express, 2018, 5, 095029 CrossRef.
- S. Kader, M. R. Al-Mamun, M. B. K. Suhan, S. B. Shuchi and M. S. Islam, Environ. Technol. Innovation, 2022, 27, 102476 CrossRef CAS.
- C. Yu, L. Wei, J. Chen, Y. Xie, W. Zhou and Q. Fan, Ind. Eng. Chem. Res., 2014, 53, 5759–5766 CrossRef CAS.
- N. Q. T. Ton, T. N. T. Le, S. Kim, V. A. Dao, J. Yi and T. H. T. Vu, J. Nanosci. Nanotechnol., 2020, 20, 2214–2222 CrossRef CAS PubMed.
- M. Ahmadi and M. Guinel, Microsc. Microanal., 2013, 19, 1580–1581 CrossRef.
- M. B. Johansson, B. Zietz, G. A. Niklasson and L. Österlund, J. Appl. Phys., 2014, 115, 213510 CrossRef.
- J. Murillo-Sierra, A. Hernández-Ramírez, L. Hinojosa-Reyes and J. Guzmán-Mar, Chem. Eng. J. Adv., 2021, 5, 100070 CrossRef CAS.
- F. Riboni, L. G. Bettini, D. W. Bahnemann and E. Selli, Catal. Today, 2013, 209, 28–34 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- H. Li, C.-H. Wu, Y.-C. Liu, S.-H. Yuan, Z.-X. Chiang, S. Zhang and R.-J. Wu, Sens. Actuators, B, 2021, 341, 130035 CrossRef CAS.
- T. Beata, P. Michał and M. Antoni W, Int. J. Photoenergy, 2009, 2009, 297319 Search PubMed.
- J. A. P. Escobar, E. Moctezuma and B. S. Rosales, Int. J. Chem. Reactor Eng., 2020, 18(7), 20190159 Search PubMed.
- L. F. Paula, M. Hofer, V. P. Lacerda, D. W. Bahnemann and A. O. T. Patrocinio, Photochem. Photobiol. Sci., 2019, 18, 2469–2483 CrossRef CAS PubMed.
- J. A. Pinedo-Escobar, J. Fan, E. Moctezuma, C. Gomez-Solís, C. J. Carrillo Martinez and E. Gracia-Espino, ACS Omega, 2021, 6, 11840–11848 CrossRef CAS PubMed.
- A. Kumar and G. Pandey, Mater. Sci. Eng. Int. J., 2017, 1, 1–10 Search PubMed.
- P. Guo, L. T. Meng and C. H. Wang, Adv. Mater. Res., 2014, 850, 78–81 Search PubMed.
- W. Wang, H. Fu, X. Yang and X. An, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 504, 012002 CrossRef CAS.
- L. Gao, W. Gan, Z. Qiu, X. Zhan, T. Qiang and J. Li, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- F. Giuffrida, L. Calcagno, G. Pezzotti Escobar and M. Zimbone, Crystals, 2023, 13, 372 CrossRef CAS.
- R. Wahyuono, L. Ernawati, I. K. Maharsih, N. Widiastuti and H. Widiyandari, Int. J. Eng., 2019, 32, 1345–1352 Search PubMed.
- Q. Wang, W. Zhang, X. Hu, L. Xu, G. Chen and X. Li, J. Water Process Eng., 2021, 40, 101943 CrossRef.
- L. Zhang, M. Qin, W. Yu, Q. Zhang, H. Xie, Z. Sun, Q. Shao, X. Guo, L. Hao and Y. Zheng, J. Electrochem. Soc., 2017, 164, H1086 CrossRef CAS.
- S. Adhikari, S. Mandal, D. Sarkar, D.-H. Kim and G. Madras, Appl. Surf. Sci., 2017, 420, 472–482 CrossRef CAS.
- F. Chen, Z. Liu, Y. Liu, P. Fang and Y. Dai, Chem. Eng. J., 2013, 221, 283–291 CrossRef CAS.
- B. Liu, L. Wen, K. Nakata, X. Zhao, S. Liu, T. Ochiai, T. Murakami and A. Fujishima, Chem. - Eur. J., 2012, 18, 12705–12711 CrossRef CAS PubMed.
- R. Govindaraj, M. S. Pandian, P. Ramasamy and S. Mukhopadhyay, Bull. Mater. Sci., 2015, 38, 291–296 CrossRef CAS.
- M. Fathy and H. Hamad, RSC Adv., 2016, 6, 7310–7316 RSC.
- W. Meng, L. Dai, J. Zhu, Y. Li, W. Meng, H. Zhou and L. Wang, Electrochim. Acta, 2016, 193, 302–310 CrossRef CAS.
- N. Wang, F. Zhang, Q. Mei, R. Wu and W. Wang, Water, Air, Soil Pollut., 2020, 231, 1–10 CrossRef.
- S. Komornicki, M. Radecka and P. Sobaś, Mater. Res. Bull., 2004, 39, 2007–2017 CrossRef CAS.
- J. Yang, X. Zhang, H. Liu, C. Wang, S. Liu, P. Sun, L. Wang and Y. Liu, Catal. Today, 2013, 201, 195–202 CrossRef CAS.
- R. Marotti, P. Giorgi, G. Machado and E. Dalchiele, Sol. Energy Mater. Sol. Cells, 2006, 90, 2356–2361 CrossRef CAS.
- F. Ghasempour, R. Azimirad, A. Amini and O. Akhavan, Appl. Surf. Sci., 2015, 338, 55–60 CrossRef CAS.
- T. Kunyapat, F. Xu, N. Neate, N. Wang, A. De Sanctis, S. Russo, S. Zhang, Y. Xia and Y. Zhu, Nanoscale, 2018, 10, 4718–4726 RSC.
- S. Rahimnejad, J. H. He, F. Pan, W. Chen, K. Wu and G. Q. Xu, Mater. Res. Express, 2014, 1, 045044 CrossRef.
- J. Hu, L. Wang, P. Zhang, C. Liang and G. Shao, J. Power Sources, 2016, 328, 28–36 CrossRef CAS.
- F. Amano, K. Nogami, M. Tanaka and B. Ohtani, Langmuir, 2010, 26, 7174–7180 CrossRef CAS PubMed.
- A. S. Bhatt, P. L. Sakaria, M. Vasudevan, R. R. Pawar, N. Sudheesh, H. C. Bajaj and H. M. Mody, RSC Adv., 2012, 2, 8663–8671 RSC.
- M. F. Mubarak, H. Selim and R. Elshypany, J. Environ. Health Sci. Eng., 2022, 1–16, DOI:10.1007/s40201-021-00774-y.
- F. Azeez, E. Al-Hetlani, M. Arafa, Y. Abdelmonem, A. A. Nazeer, M. O. Amin and M. Madkour, Sci. Rep., 2018, 8, 1–9 CrossRef CAS PubMed.
- T. T. Nguyen, S.-N. Nam, J. Son and J. Oh, Sci. Rep., 2019, 9, 1–18 CrossRef PubMed.
- H. Deng, J. Lu, G. Li, G. Zhang and X. Wang, Chem. Eng. J., 2011, 172, 326–334 CrossRef CAS.
- H. Huang, Y. He, R. He, X. Jiang, Z. Lin, Y. Zhang and S. Wang, Inorg. Chem. Commun., 2014, 40, 215–219 CrossRef CAS.
- K. Zhang, G.-H. Li, L.-M. Feng, N. Wang, J. Guo, K. Sun, K.-X. Yu, J.-B. Zeng, T. Li and Z. Guo, J. Mater. Chem. C, 2017, 5, 9359–9369 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The related XPS data for the prepared WO3-loaded TiO2 composites, Langmuir adsorption isotherm fitting curves for pure oxides, comixed and core–shell materials, UV-VIS spectrophotometric calibration data, and corresponding absorption spectra for the MB+ and MO− calibration solutions are provided separately in the ESI. See DOI: https://doi.org/10.1039/d3ra01582c |
| This journal is © The Royal Society of Chemistry 2023 |