
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA library of new organofunctional silanes obtained by thiol-(meth)acrylate Michael addition reaction†
Agnieszka Przybylska a,
Anna Szymańska*b and
Hieronim Maciejewskiab
a,
Anna Szymańska*b and
Hieronim Maciejewskiab
aFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland
bPoznań Science and Technology Park, Adam Mickiewicz University Foundation, Rubież 46, 61-612 Poznań, Poland. E-mail: anna.szymanska@ppnt.poznan.pl
First published on 9th May 2023
Abstract
A simple and efficient method for the synthesis of organofunctional silanes by the thiol-(meth)acrylate addition reaction is presented. At first, systematic studies were carried out to select an optimum initiator/catalyst of the addition reaction for the model reaction involving 3-mercaptopropyltrimethoxysilane (MPTMS) and hexyl acrylate. Photoinitiators (in the presence of UV light energy), thermal initiators (such as aza compound and peroxide) as well as catalysts (primary and tertiary amines, phosphines and Lewis acid) were studied. After selecting an effective catalytic system and optimizing the reaction conditions, reactions between the thiol group (i.e. 3-mercaptopropyltrimethoxysilane) and (meth)acrylates containing various functional groups were carried out. All derivatives obtained were characterized by 1H, 13C, 29Si NMR and FT-IR analysis. In reactions carried out at room temperature, in an air atmosphere and in the presence of dimethylphenylphosphine (DMPP) as a catalyst, quantitative conversions of both substrates were obtained within a few minutes. The library of organofunctional silanes was expanded by compounds (containing various functional groups, i.e. alkenyl, epoxy, amino, ether, alkyl, aralkyl, fluoroalkyl) which were obtained in the thiol-Michael addition of 3-mercaptopropyltrimethoxysilane to a group of organofunctional (meth)acrylic acid esters.
Introduction
Organofunctional silanes play an important role in many sectors. They combine a reactive organic group and the inorganic functionality of an alkoxysilane into a single molecule acting as a coupling agent.1 The annual global market for silane coupling agents (SCAs) is growing year by year and is estimated to be worth more than USD 529 million by 2026.2 However, organofunctional silanes can also be used as cross-linking agents, surface modifiers, substrates for the synthesis of organically modified silicas (ORMOSIL's) and many others. The interest in these compounds continues to grow and new directions for their applications are constantly emerging, which means that there is still a need for new and simple methods for their synthesis. Hydrosilylation is still one of the most popular methods for the synthesis of organosilicon compounds.3,4 Despite its versatility and many advantages, the hydrosilylation reaction also has some disadvantages.5–7 The most important include: (i) often exothermic course (increased temperature may adversely affect some functional groups), (ii) the need to use expensive catalysts (platinum and rhodium complexes are commonly used), (iii) high susceptibility of the above catalysts to poisoning (with sulfur, nitrogen and phosphorus derivatives), (iv) significant influence of reaction conditions and type of catalyst on the selectivity of the process and the occurrence of side reactions. An alternative reaction to hydrosilylation that can be used in the synthesis of organosilicon compounds is hydrothiolation.8 Hydrothiolation is an addition reaction of sulfur and hydrogen from the thiol group to unsaturated carbon–carbon bonds, commonly known as thiol–ene(yne) coupling. As in the case of hydrosilylation, the reaction may lead to two adducts, corresponding either to the Markovnikov addition (α-adduct) or the anti-Markovnikov addition (β-adduct).9,10 What distinguishes hydrothiolation from hydrosilylation is a very low contribution (less than 5%) or no formation of α-adducts, high tolerance to various functional groups, short synthesis time and mild conditions.11 In addition, from the application point of view, the products obtained in the hydrothiolation process have the same and sometimes even better properties (as adhesion promoters or modifiers) than the analogous products obtained in the hydrosilylation process. No negative effect of the presence of sulfur in these compounds is observed. The thiol–ene reaction can proceed in one of two ways: (i) radical addition or (ii) base- or nucleophile-catalyzed thiol-Michael addition. There are two ways to generate free radicals: (i) by heating – thermal initiation or (ii) by irradiation with UV or visible light photoinitiation. Photoinitiators are divided into two types, according to the type of the process of free radicals generation.12,13 According to literature data, type I photoinitiators (mainly 2,2-dimethoxy-2-phenylacetophenone, DMPA) are most often more efficient than type II ones (benzophenone or thioxanthen-9-one).12,14 The two most common classes of thermal initiators aliphatic azo-derivatives and peroxides which are very efficient and widely used.15 Thiol-Michael addition reactions usually takes place in the presence of a base or nucleophile.16–18 The reaction can also be initiated by metals, organometallic compounds, Lewis acids and others, however, bases and nucleophiles are the most effective and tend to minimize side reactions.19 Despite such a large variety of methods used, their effectiveness is largely dependent on the type of reagents.11Hydrothiolation reactions were also carried out with the participation of silanes. Commercially available 3-mercaptopropyltrialkoxysilanes are used for modification of a wide range of compounds, i.e. castor oil,20 polysiloxanes,21–23 silsesquioxanes,24–26 polyphosphasenes,27 cyclic siloxanes,28,29 polyethylene,30,31 ionic liquids,32,33 cellulose.34,35 Reactions of alkenes with 3-mercaptopropyltrialkoxysilane or thiols with allyltrialkoxysilanes in the presence of a photoinitiator (DMPA) were used to synthesize compounds that were used to modify superparamagnetic iron oxide nanoparticles.36 A similar methodology has been used in the synthesis of various derivatives of alkoxysilanes. In the next stage, the obtained compounds were precursors for the synthesis of functional polysiloxanes.23 Esquivel et al.37 have used radical hydrothiolation in the synthesis of bis-trialkoxysilyl compounds as substrates for mesoporous organosilicates. Rissing and Son8,38 have synthesized branched silanes via a photoinitiated thiol–ene reaction.
As mentioned above, the course of the hydrothiolation reaction depends on the type of reagent. The Michael thiol–ene addition is the preferred reaction between nucleophilic species and olefins under basic conditions, for example thiols with an acrylic carbon–carbon bond.18,39
Despite the above examples of the use of the thiole–ene reaction in the synthesis of organofunctional silanes, the hydrothiolation of acrylate and methacrylate derivatives with mercaptoalkoxysilanes has not been investigated, to the best of our knowledge. In addition, the hydrosilylation reaction (the main method of synthesizing organofunctional silanes) of acrylate (or methacrylate) compounds can generate mixtures of products, namely α-adduct, β-adduct, silyl ketene acetals, silyl ethers or polyacrylates, depending on the catalyst system.3,40
Therefore, we would like to expand the library of compounds obtained by thiol-(meth)acrylate addition with alkoxysilanes containing various functional groups. The article presents comparative studies of the thiol–ene reaction performed with the use of a wide spectrum of catalysts and initiators, such as two types of photoinitiators, thermal radical initiators (azo and peroxide initiators), amines, phosphines and a metal complex catalyst. 3-Mercaptopropyltrimethoxysilane and a wide group of unsaturated acrylic and methacrylic derivatives were used as the starting materials for the thiol–ene reactions.
The main aim of the work was to determine the activity of initiators and catalysts in hydrothiolation processes, to select the most effective reaction systems, and to assess the possibility of using the hydrothiolation reaction as an alternative method to the hydrosilylation reaction for the synthesis of organofunctional silanes.
Experimental
Materials
2,2-Dimethoxy-2-phenylacetophenone (DMPA, 99%), diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO, 97%), 2,2′-azobis(2-methylpropionitrile) (AIBN, ≥98%), 2,5-bis(tert-butylperoxy)-2,5-dimethylhexane (Luperox 101, 90%), 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU, 98%), 1,4-diazabicyclo[2.2.2]octane (DABCO, ≥99%), scandium(III) trifluoromethanesulfonate (99%), n-hexylamine (HA, 99%), triethylamine (TEA, ≥99%), thioxanthen-9-one (TX, 97%), benzophenone (BP, 99%), triphenylphosphine (PPh3, ≥95%), 2-(tert-butylamino)ethyl methacrylate (stabilized with 1000 ppm MEHQ, 97%), glycidyl methacrylate (stabilized with 100 ppm MEHQ, 97%), methyl acrylate (stabilized with ≤100 ppm MEHQ, 99%), methyl methacrylate (stabilized with ≤30 ppm MEHQ, 99%) were purchased from Aldrich. Hexyl acrylate (>96%, stabilized with HQ), n-decane (>99%), polyethylene glycol monomethyl ether acrylate (stabilized with MEHQ), 2-(dimethylamino)ethyl acrylate (stabilized with MEHQ, >98%), 2-(dimethylamino)ethyl methacrylate (stabilized with MEHQ, >98.5%), n-dodecyl acrylate (stabilized with MEHQ, >98%) were obtained from TCI. Methyldiphenylphosphine (PMePh2, ≥98%), 1H,1H,2H,2H-tridecafluorooctyl methacrylate (stabilized with HQ + MEHQ, 98%), n-dodecyl methacrylate (stabilized with MEHQ, >98%) were purchased from ChemScene. Dimethylphenylphosphine (DMPP, 97%), allyl methacrylate (stabilized, 98%) were purchased from Acros Organics. 3-Mercaptopropyltrimethoxysilane (MPTMS, 95%) was purchased from ABCR. 4-(Dimethylamino)pyridine (DMAP, ≥99%) was obtained from Fluka Analytical. Benzyl methacrylate (stabilized with 50 ppm MEHQ, 98%) was purchased from AmBeed. Tert-Butyl acrylate (stabilized with 15 ppm 4-methoxyphenol, 99%) was purchased from Alfa Aesar. 1,1,1,3,3,3-Hexafluoroisopropyl acrylate (stabilized with 50 ppm 4-methoxyphenol, 98%) was obtained from Angene. Chloroform-d (99.8% + Ag (CDCl3)) was purchased from Deutero. THF was purchased from ChemPur. Stabilizers were not removed from reactants.Analytical methods
Nuclear magnetic resonance (1H, 13C, 29Si NMR) spectra were recorded at 298 K on Bruker Avance III HD spectrometers. CDCl3 was used as a solvent. Fourier transform infrared (FT-IR) spectra were taken on a Bruker spectrometer, model Tensor 27, equipped with a SPECAC Golden Gate diamond ATR (Attenuated Total Reflection) unit. In each case, 16 scans were collected for a spectrum at the resolution of 2 cm−1. Gas chromatography (GC) was performed on a Varian GC-450 equipped with TCD detector, 30 m Resteck DB-5 MEGABORE capillary column with a film thickness of 1.5 μm. Helium was used as carrier gas supplied at a constant flow rate of 2 ml min−1. Temperature program was as follows: injector 220 °C, detector 240 °C and column oven program: start temperature 60 °C for 3 min ramp 10 °C min−1 to 300 °C which was held for 10 min.Test of catalysts and initiators
General procedure for synthesis of organoalkoxysilanes catalyzed by DMPP
A mixture of MPTMS (1 equiv.), acrylate or methacrylate derivatives (1 equiv.) in the amount securing obtainment of 5 g of the final product, DMPP was placed in a round bottomed flask equipped with a magnetic stirrer and was stirred at room temperature. The course of the reaction was observed using FT-IR and the reaction was performed to full disappearance of SH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. Tetrahydrofuran (to get a 50 wt% final solution) was added to the two reaction mixtures (samples 3g and 3i). The solvent was removed under reduced pressure.
C bonds. Tetrahydrofuran (to get a 50 wt% final solution) was added to the two reaction mixtures (samples 3g and 3i). The solvent was removed under reduced pressure.
Results and discussion
The key step in our research was to establish the conditions for hydrothiolation reaction of acryl- and methacryl compounds containing various functionalities for formation of silane coupling agents.Since the hydrothiolation of acrylates and methacrylates derivatives with mercaptoalkoxysilanes has been poorly covered in literature, we started our research from the selection of the best catalyst/initiator. Chemical structure of the employed catalyst and initiators are presented in Scheme 1.
 | ||
| Scheme 1 Chemical structure of catalysts/initiators used in the tested reactions. | ||
In our study, we examined 15 commercially available catalysts/initiators in total, of which 4 were photoinitiators: DMPA and TPO (type I), BP and TX (type II), 2 were thermal initiators: azo-initiator AIBN and peroxide Luperox 101, different bases and nucleophiles such as 3 phosphines, 5 amines and Sc(OTf)3 (Lewis acid). All of them were used as catalysts for hydrothiolation reaction.
The addition of 3-mercaptopropyltrimethoxysilane (1) to hexyl acrylate (2) was systematically investigated as a representative model of hydrothiolation reaction. Results of the catalyst screening are presented in Table 1.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Type of catalyst | Entry | Catalyst/initiator | Catalyst amount | Conversiona | ||||||
| Time [min] | ||||||||||
| 5 | 10 | 15 | 30 | 60 | 180 (3 h) | 1440 (24 h) | ||||
| a Conversion yields were determined by GC analysis and calculated on the basis of MPTMS (1) and hexyl acrylate (2), using n-decane peak as a standard. 1 Conversion of 3-mercaptopropyltrimethoxysilane (1), 2 conversion of hexyl acrylate (2).b Reaction using 0.5 wt% proceeded too violently, strong heating of the mixture took place leading to side reactions. | ||||||||||
| Photoinitiator/UV | 1 | DMPA/UV | 0.5 wt% | 70.71 | — | — | — | — | — | — |
| 99.92 | — | — | — | — | — | — | ||||
| 2 | TPO/UV | 0.5 wt% | 71.11 | — | — | — | — | — | — | |
| 99.52 | — | — | — | — | — | — | ||||
| 3 | TX/UV | 0.5 wt% | 68.31 | 71.41 | — | — | — | — | — | |
| 83.12 | 99.92 | — | — | — | — | — | ||||
| 4 | BP/UV | 0.5 wt% | 61.01 | 65.11 | — | — | — | — | — | |
| 96.72 | 99.92 | — | — | — | — | — | ||||
| UV | 5 | UV | — | 25.61 | 50.91 | 71.71 | — | — | — | — |
| 34.22 | 82.42 | 99.92 | — | — | — | — | ||||
| Thermal initiator | 6 | AIBN/70 °C | 0.5 wt% | 72.31 | 76.11 | — | — | — | — | — |
| 98.92 | 99.92 | — | — | — | — | — | ||||
| 7 | Luperox 101/180 °C | 0.5 wt% | 74.81 | 75.01 | — | — | — | — | — | |
| 92.62 | 1002 | — | — | — | — | — | ||||
| Amine | 8 | DABCO | 0.5 wt% | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 3.41 |
| 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 2.92 | ||||
| 9 | DMAP | 0.5 wt% | 0.01 | 0.01 | 5.01 | 13.61 | 30.61 | 38.21 | 78.41 | |
| 0.02 | 0.02 | 7.82 | 18.42 | 39.72 | 47.62 | 82.32 | ||||
| 10 | Hexylamine | 0.5 wt% | 7.21 | 12.71 | 32.31 | 41.41 | 54.21 | 73.81 | 94.31 | |
| 7.12 | 13.62 | 27.72 | 38.52 | 59.82 | 78.02 | 95.72 | ||||
| 11 | Triethylamine | 0.5 wt% | 0.01 | 0.01 | 0.01 | 0.01 | 4.01 | 11.31 | 67.71 | |
| 0.02 | 0.02 | 0.02 | 0.02 | 5.12 | 12.82 | 72.92 | ||||
| 12 | DBU | 0.05b wt% | 72.61 | 88.81 | 89.61 | 91.81 | 94.21 | 95.91 | — | |
| 74.42 | 90.22 | 94.82 | 98.52 | 98.92 | 99.92 | — | ||||
| Phosphine | 13 | PPh3 | 0.5 wt% | 0.01 | 0.01 | 0.01 | 0.01 | 0.11 | 9.71 | 28.01 |
| 0.02 | 0.02 | 0.02 | 0.02 | 10.42 | 19.82 | 34.42 | ||||
| 14 | PMePh2 | 0.5 wt% | 68.91 | 72.81 | 78.01 | 86.71 | 91.11 | 91.51 | 95.31 | |
| 75.22 | 81.92 | 83.32 | 91.52 | 94.52 | 97.32 | 99.72 | ||||
| 15 | DMPP | 0.05b wt% | 92.21 | 97.21 | — | — | — | — | — | |
| 93.82 | 99.22 | — | — | — | — | — | ||||
| Lewis acid | 16 | Sc(OTf)3 | 0.5 wt% | 20.21 | 50.21 | 64.21 | 66.91 | 67.11 | 67.21 | 67.21 |
| 18.72 | 53.72 | 63.32 | 88.72 | 94.92 | 95.02 | 95.02 | ||||
The effect of the type of catalyst on the thiol–ene addition reaction degree was studied. The efficiencies of the catalysts used were compared in the reactions carried out using their constant amount (0.5 wt%), unless the reaction was too rapid, then the amount of the catalyst was reduced to 0.05 wt%. The first reactions (entries 1–4) were triggered by photoirradiation and initiated by two Norrish type I photoinitiators DMPA and TPO as well as two Norrish type II photoinitiators BP and TX.12 Some thiol–ene reactions run with no need of a photoinitiator only upon irradiation at the appropriate wavelength, as the use of certain thiols leads to self-initiation in the absence of a photoinitiator.41 Therefore, the reaction was also carried out without the use of an initiator, only under UV irradiation (entry 5). Full conversion of hexyl acrylate took place within 5 minutes upon UV irradiation and in the presence of type I photoinitiators. In the presence of type II photoinitiators, 10 minutes were required for full conversion of hexyl acrylate. Type II photoinitiators are slightly less effective than type I ones in thiol–ene reaction between MPTMS (1) and hexyl acrylate (2). Additionally, using DMPA, TPO and TX, the conversion of MPTMS reached 71% at the complete conversion of hexyl acrylate, while with the use of BP, the conversion of MPTMS reached only 65% at the full conversion of hexyl acrylate.
Interestingly, 5 more minutes were required for the full conversion of (2) in the photoinitiated reaction without the use of photoinitiators, however, the proportion of conversions of reactants (1) and (2) was similar to that in the reaction initiated by photoinitiators.
Reactions with AIBN and peroxide (entries 6 and 7) need maximum 10 minutes for full conversion of hexyl acrylate (conversion of MPTMS reached 76% and 75%, respectively).
In all radical-initiated reactions (entries 1–7), a significantly higher conversion of hexyl acrylate was observed versus mercaptosilane which means that side reactions occurred, leading to a reduced addition efficiency. The higher conversion of hexyl acrylate than thiol is due to its (homo)polymerization taking place under the above reaction conditions.
Next, five primary and tertiary amines, i.e. 1,4-diazabicyclo[2.2.2]octane (DABCO), 4-(dimethylamino)pyridine (DMAP), hexylamine (HA), triethylamine (TEA) and 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) (entries 8–12) were investigated. The selected amines are ranked in order of increasing pKa values: DABCO (pKa = 8.8), DMAP (pKa = 9.7), HA (pKa = 10.6), TEA (pKa = 10.8) and the strongest amine is DBU (pKa = 12).42,43 Amines were not as effective as photo- and thermal-initiators, however, they showed much better selectivity and allowed the formation of only thiolation (addition) products. No homopolymerization products were observed. In the case of DABCO, DMAP and triethylamine, induction times were observed. The best results were obtained for DBU (after 10 minutes the conversion was close to 90%, after 3 h – full conversion was reached) as well as for hexylamine for which the conversion was gradually growing and reached about 95% after 24 h. Effectiveness of DBU is not only from high basicity but also their ability to act as a nucleophile.18 Better results were obtained for n-hexylamine (primary amine) than triethylamine (tertiary amine), although their pKa values are similar. HA is N-centered nucleophile and TEA is non-nucleophilic base and mechanisms of reaction (nucleophilic-catalyzed and base-catalyzed thiol-Michael addition reaction mechanisms) are different.
Three phosphines (P-centered nucleophiles), i.e. triphenylphosphine (PPh3), methyldiphenylphosphine (PMePh2) and dimethylphenylphosphine (DMPP) were examined (entries 13–15). DMPP was the most effective catalyst for thiol-Michael reaction. Full conversion was obtained after 10 minutes with the use of only 0.05 wt% of the catalyst. In general, for these three phosphines, the more methyl substituents were used instead of phenyl, the more nucleophilic character and more effective the catalyst. In addition, a rare earth metal complex catalyst Sc(OTf)3 (Lewis acid) was examined (entry 16). In the presence of this catalyst (homo)polymerization of hexyl acrylate took place.
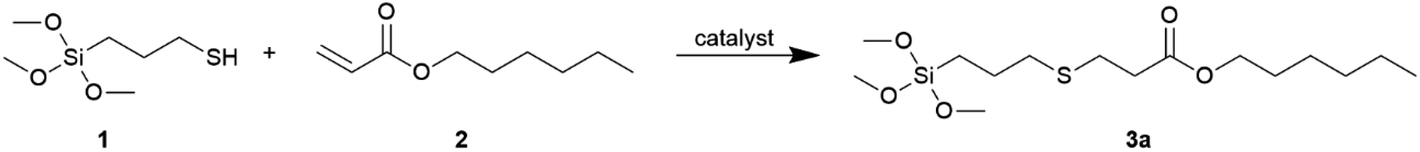
Based on the above results, DMPP was selected for further research. Having the optimized conditions at hand, we examined the addition of MPTMS to various group-containing acrylates and methacrylates, according to Scheme 2.
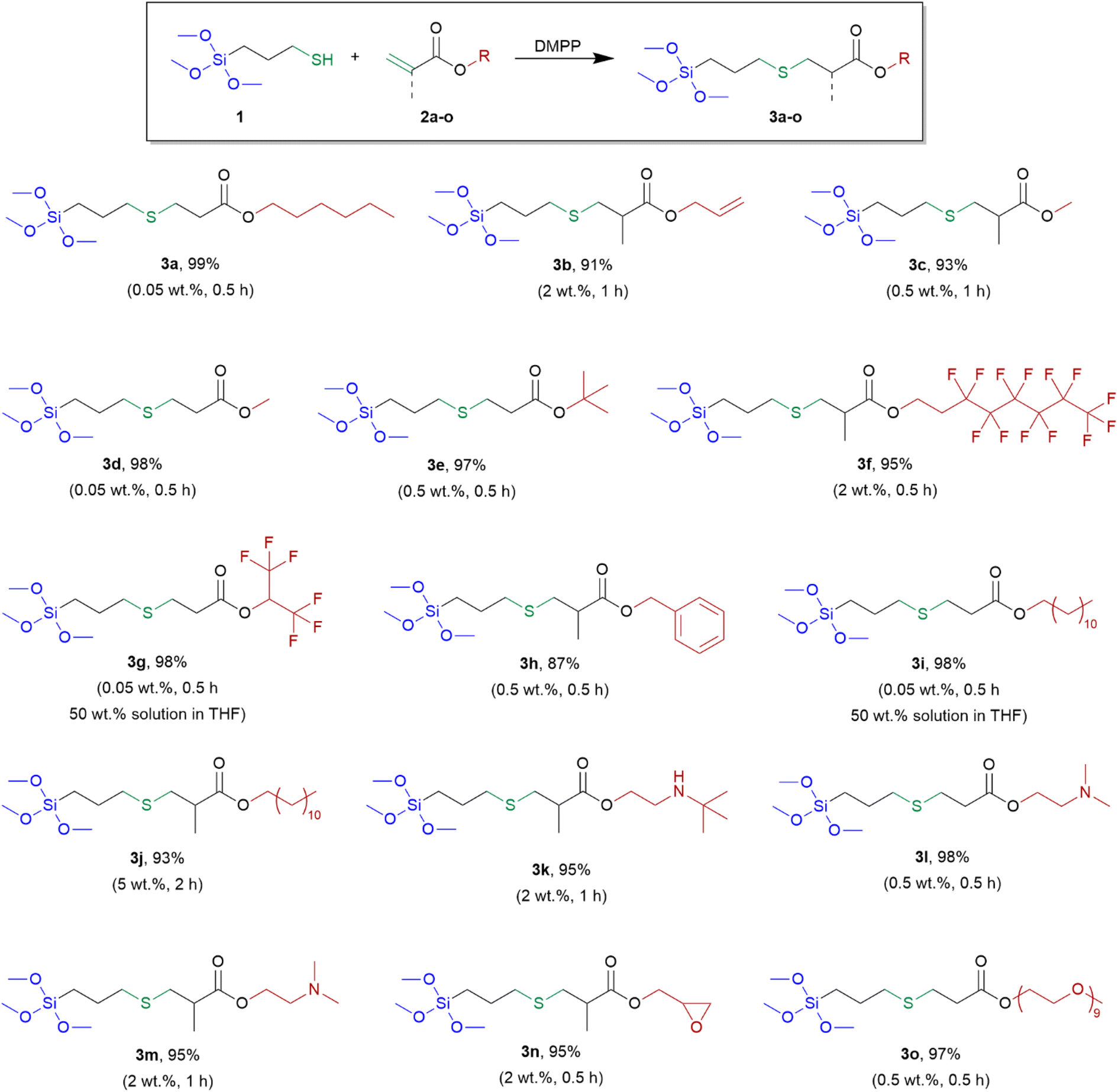 | ||
| Scheme 2 Synthesis and yields of obtained alkoxysilane derivatives (3a–o). Special reaction conditions (amount of DMPP and reaction time) are given in parentheses. | ||
In these addition reactions, 7 acrylate derivatives and 8 methacrylate derivatives containing different functional groups were used. Among them, three pairs of acrylates and the corresponding methacrylates containing the same functional groups, i.e. methyl, dodecyl and –NMe2 were investigated.
The presented method of synthesis was simple and efficient. All reactions were carried out with the substrates at a stoichiometric ratio. The catalyst was used in the amount from 0.05 to 2 wt% (5 wt% for 3j). The reactions were performed without solvent (with the exception of 3g and 3i) for 0.5 h or 1 h or (2 h for 3j) at room temperature leading to derivatives 3a–o, without the catalyst separation and further purification. The structures of the derivatives obtained are presented in Scheme 2.
Catalysis of the reaction between MPTMS (1) and various acrylates and methacrylates compounds, initiated by DMPP, resulted in quantitative formation of desired thiol-Michael products. Only the amount of MPTMS equivalent to those of acrylates and methacrylates derivatives was needed to achieve 100% conversion of double bonds. Methacrylates were less reactive than acrylates and a greater amount of the catalyst and/or longer reaction time was required when they were used. The lower reactivity of methacrylates than acrylates results from inductive effect and steric hindrance caused by the methyl group. The exception was derivative 3h (obtained from benzyl methacrylate) as the reaction to obtain it needed 0.5 wt% of the catalyst and time of 0.5 h. Comparing the counterparts of acrylates and methacrylates with the same functional groups, namely pairs 3c versus 3d and 3i versus 3j and 3l versus 3m, greater amount of catalyst and longer reaction time was required for full conversion of substrates in the case of methacrylates. No polymerization of acrylate and methacrylate derivatives was observed. When allyl acrylate was used, the thiol-Michael reaction occurred selectively to acrylate group (product 3b) and the allyl group remained unreacted, which enabled further functionalization of the obtained derivative. This course is opposite to that observed in the hydrosilylation reaction, where addition to the allyl group is preferred.
The course of the reactions was monitored using FT-IR and the reactions were performed until full disappearance of SH and CC bonds. The FT-IR spectra of the mixture of reactants (a) and the product of synthesis (derivative 3d) (b) are shown in Fig. 1.
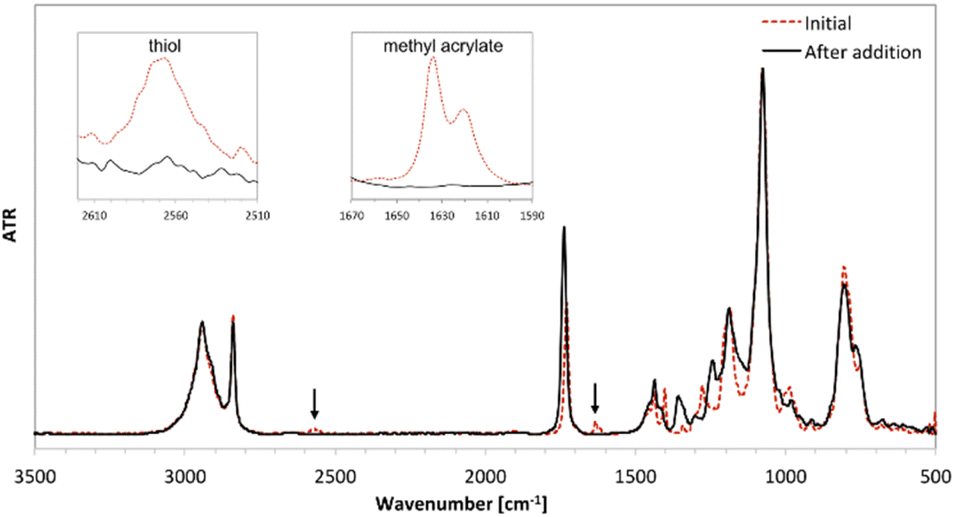 | ||
| Fig. 1 FT-IR spectra of the mixture of reactants 3-mercaptopropyltrimethoxysilane and methyl acrylate (red dashed line) and product 3d (black line). | ||
The absorption at 1634 cm−1 (corresponding to the CC stretching vibration) and 2567 cm−1 (corresponding to the –SH stretching vibration) disappeared completely after 0.5 h, which implies addition of thiol to the carbon–carbon double bond. In order to confirm that all target products were obtained, the 1H, 13C and 29Si NMR spectra were recorded (all spectra are available in ESI†).
In order to verify the structure of selected sample (derivative 3d) its 1H NMR spectrum was recorded. The 1H NMR spectra of 3-mercaptopropyltrimethoxysilane (MPTMS) (a), methyl acrylate (b) and derivative 3d (c) with peak assignments are presented in Fig. 2.
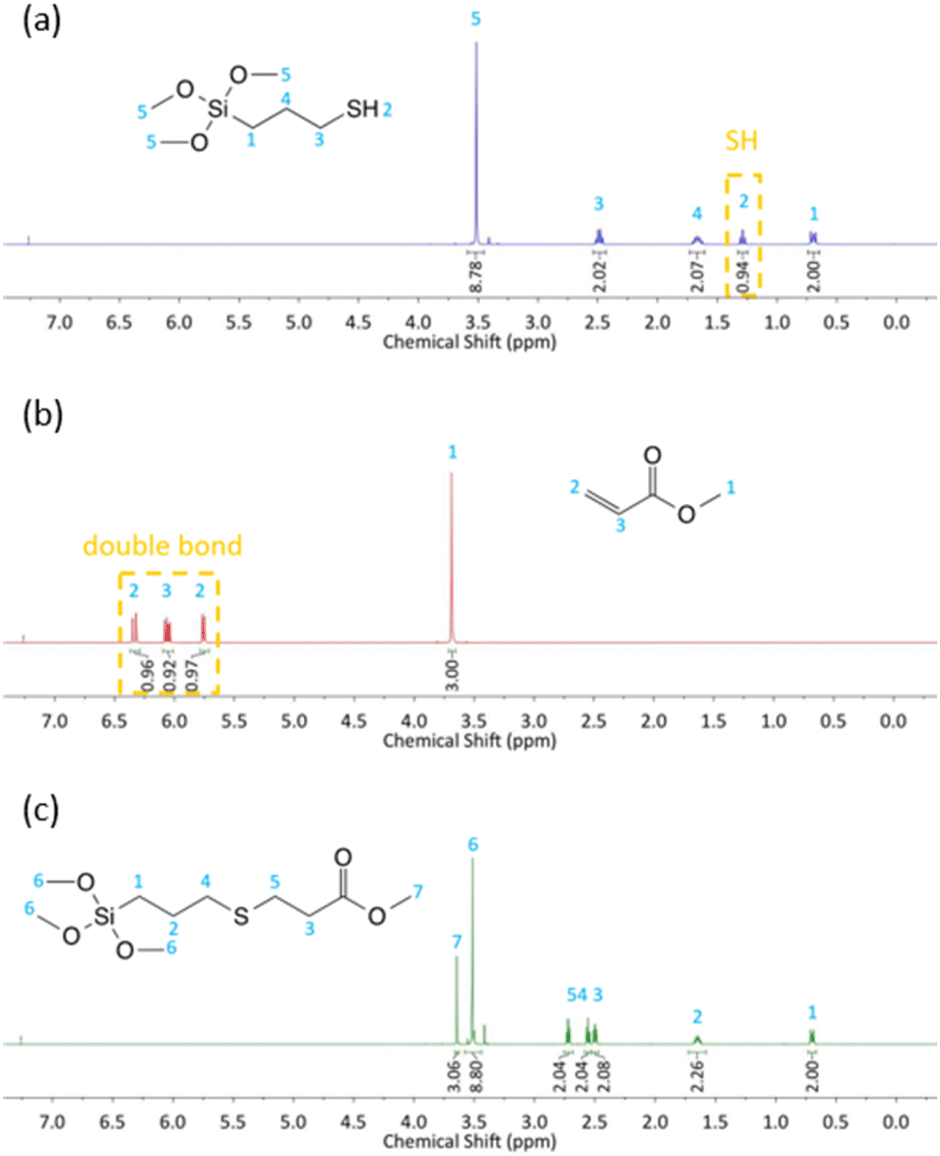 | ||
| Fig. 2 1H NMR spectra of (a) 3-mercaptopropyltrimethoxysilane (MPTMS), (b) methyl acrylate, (c) 3d. | ||
From the 1H NMR spectrum of MPTMS in Fig. 2a, the triplet at 1.29 ppm is attributed to proton 2 that represents the mercapto group SH. This peak disappeared completely in the 1H NMR spectra of the product (Fig. 2c). From the 1H NMR spectrum of methyl acrylate in Fig. 2b, the peaks at 6.35–5.74 ppm correspond to the hydrogen protons in H2CCH bond. It can be observed that the peak of the double bond totally vanished in the 1H NMR spectra of the product (Fig. 2c). CH2SCH2 bonds formation was confirmed by the appearance of two peaks at 2.73–2.54 ppm, which demonstrated that the double bonds of methyl acrylate had completely reacted with the mercapto group of MPTMS and product 3d was successfully synthesized through the thiol-Michael addition reaction.
Conclusions
The conducted screening tests of various initiators/catalysts of the hydrothiolation reaction of hexyl acrylate made it possible to select the most effective system. Of all the 15 catalytic systems tested, dimethylphenylphosphine (DMPP) turned out to be the most effective. Reactions with the participation of DMPP were very fast (even at 10 times lower concentration compared to the concentrations of other initiators/catalysts) and selectively, enabling almost quantitative conversion of both substrates. In addition, in the case of the phosphine catalysts (PPh3, PMePh2, PMe2Ph) used in the research, the influence of the electron density residing on the phosphorus center on the catalytic efficiency can be noticed. The catalytic activity of phosphines enhanced with the change of aryl substituent to methyl. The highest catalytic activity of DMPP (PMe2Ph) than PMePh2 and PPh3 results from its more nucleophilic character. Using the selected catalyst (DMPP), hydrothiolation reactions of various derivatives of acrylates and methacrylates with 3-mercaptopropyltrimethoxysilane (MPTMS) were carried out, obtaining the desired products in a selective and quantitative manner, which was confirmed by FT-IR and NMR analysis. It can be concluded that the methacrylic derivatives showed lower reactivity compared to the acrylic derivatives. Moreover, the hydrothiolation reaction of allyl methacrylate selectively adds –SH to the methacrylic moiety while the allyl group does not react. This is in contrast to the hydrosilylation reaction where the addition of the –SiH moiety occurs mainly to the allyl group. As part of the conducted research, a number of organofunctional trialkoxysilanes containing various functional groups were obtained selectively and with quantitative efficiency, the preparation of which by other methods was very troublesome. The selected catalyst (DMPP) shows high activity, thanks to which the hydrothiolation reaction is a good alternative to the hydrosilylation reaction and enables the synthesis of a whole range of organofunctional silanes.Author contributions
Agnieszka Przybylska: conceptualization, data curation, investigation. Anna Szymańska: data curation, writing – original draft, writing – review & editing, visualization. Hieronim Maciejewski: supervision, funding acquisition, writing – review & editing, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Science Centre (Poland), project OPUS no UMO-2018/29/B/ST8/00913, entitled “Synthesis and characterization of materials with defined surface properties”.References
- E. P. Plueddemann, Silane Coupling Agents, 2nd edn, Springer, Boston, MA, USA, 1991 Search PubMed.
- The Freedonia Group Report, https://www.freedoniagroup.com/World-Silicones.html/accessed November 2022.
- B. Marciniec, H. Maciejewski and P. Pawluć in Organosilicon Compounds, ed. V. Y. Lee, Academic Press, London, 2017, p. 169 Search PubMed.
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS.
- B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, Hydrosilylation. A Comprehensive Review on Recent Advances, Springer, Dordrecht, 2009 Search PubMed.
- B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, in Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook, ed. Cornils, B., Herrmann, W. A., Beller, M. and Paciello, R., Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017, vol. 8, pp. 569–620 Search PubMed.
- Z. Pan, M. Liu, C. Zheng, D. Gao and W. Huang, Chin. J. Chem., 2017, 35, 1227–1230 CrossRef CAS.
- C. Rissing and D. Y. Son, Organometallics, 2008, 27, 5394–5397 CrossRef CAS.
- M. Zhang, W. Li, Z. Zhou, S. Zhuo and Z. Su, ACS Omega, 2022, 7, 5782–5790 CrossRef CAS PubMed.
- R. Castarlenas, A. Di Giuseppe, J. J. Pérez-Torrente and L. A. Oro, Angew. Chem., Int. Ed., 2013, 52, 211–222 CrossRef CAS PubMed.
- I. Krizhanovskiy, M. Temnikov, Y. Kononevich, A. Anisimov, F. Drozdov and A. Muzafarov, Polymers, 2022, 14, 3079 CrossRef CAS PubMed.
- M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 103–110 CrossRef CAS.
- C. Mendes-Felipe, J. Oliveira, I. Etxebarria, J. L. Vilas-Vilela and S. Lanceros-Mendez, Adv. Mater. Technol., 2019, 4, 1800618 CrossRef.
- S. Dadashi-Silab, C. Aydogan and Y. Yagci, Polym. Chem., 2015, 6, 6595–6615 RSC.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed.
- P. Chakma and D. Konkolewicz, Angew. Chem., 2019, 131, 9784–9797 CrossRef.
- A. Sitterli and T. Heinze, React. Funct. Polym., 2019, 136, 66–74 CrossRef CAS.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS.
- Y. Sun, H. Liu, L. Cheng, S. Zhu, C. Cai, T. Yang, L. Yang and P. Ding, Polym. Int., 2018, 67, 25–31 CrossRef CAS.
- C. Fu, Z. Yang, Z. Zheng and L. Shen, Prog. Org. Coat., 2014, 77, 1241–1248 CrossRef CAS.
- M. Przybylak, A. Szymańska, H. Maciejewski and K. Makowska, Cellulose, 2020, 27, 8351–8367 CrossRef CAS.
- L. Xue, D. Wang, Z. Yang, Y. Liang, J. Zhang and S. Feng, Eur. Polym. J., 2013, 49, 1050–1056 CrossRef CAS.
- J. Cao, Y. Zuo, D. Wang, J. Zhang and S. Feng, New J. Chem., 2017, 41, 8546–8553 RSC.
- L. Li, L. Xue, S. Feng and H. Liu, Inorg. Chim. Acta, 2013, 407, 269–273 CrossRef CAS.
- M. Walczak, R. Januszewski, M. Dutkiewicz, A. Franczyk and B. Marciniec, New J. Chem., 2019, 43, 18141–18145 RSC.
- A. Szymańska, M. Przybylak, H. Maciejewski and A. Przybylska, Cellulose, 2022, 29, 1231–1247 CrossRef.
- V. Poscher, I. Teasdale and Y. Salinas, ACS Appl. Nano Mater., 2019, 2, 655–660 CrossRef CAS.
- K. Rózga-Wijas and J. Chojnowski, J. Inorg. Organomet. Polym. Mater., 2012, 22, 588–594 CrossRef.
- M. Rodošek, M. Koželj, L. S. Perše, R. C. Korošec, M. Gaberšček and A. K. Surca, Corros. Sci., 2017, 126, 55–68 CrossRef.
- Y. Zhang, H. Li, Z. Xu, W. Bu, C. Liu, J. H. Dong and Y. Hu, Polym. Chem., 2014, 5, 3963–3967 RSC.
- A. Szymańska, M. Dutkiewicz, H. Maciejewski and M. Palacz, Constr. Build. Mater., 2022, 315, 125624 CrossRef.
- M. S. Zayas, J. C. Gaitor, S. T. Nestor, S. Minkowicz, Y. Sheng and A. Mirjafari, Green Chem., 2016, 18, 2443–2452 RSC.
- A. Zając, A. Szpecht, A. Szymańska, D. Zieliński, O. Stolarska, M. Śmiglak and H. Maciejewski, New J. Chem., 2020, 44, 12274–12288 RSC.
- M. Beaumont, M. Bacher, M. Opietnik, W. Gindl-Altmutter, A. Potthast and T. Rosenau, Molecules, 2018, 23, 1427 CrossRef PubMed.
- P. Tingaut, R. Hauert and T. Zimmermann, J. Mater. Chem., 2011, 21, 16066–16076 RSC.
- A. K. Tucker-Schwartz, R. A. Farrell and R. L. Garrell, J. Am. Chem. Soc., 2011, 133, 11026–11029 CrossRef CAS PubMed.
- D. Esquivel, O. van den Berg, F. J. Romero-Salguero, F. Du Prez and P. Van Der Voort, Chem. Commun., 2013, 49, 2344 RSC.
- C. Rissing and D. Y. Son, Organometallics, 2009, 28, 3167–3172 CrossRef CAS.
- G. Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, R. Becker and D. M. Haddleton, Polym. Chem., 2010, 1, 1196–1204 RSC.
- J. Xiao, Z. Qiu, W. Yang, J. Qiu, T. Yang, Y. Xu, Y. Zeng, F. Wang and S. Li, Prog. Org. Coat., 2018, 116, 1–6 CrossRef CAS.
- P. Derboven, D. R. D’hooge, M. M. Stamenovic, P. Espeel, G. B. Marin, F. E. Du Prez and M. F. Reyniers, Macromolecules, 2013, 46, 1732–1742 CrossRef CAS.
- W. Xi, C. Wang, C. J. Kloxin and C. N. Bowman, ACS Macro Lett., 2012, 1, 811–814 CrossRef CAS PubMed.
- T. Kumar, S. M. Mobin and I. N. Namboothiri, Tetrahedron, 2013, 69, 4964–4972 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01583a |
| This journal is © The Royal Society of Chemistry 2023 |