
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular crankshaft-type rearrangement in a photoisomerised glycoconjugate†
Michal Hricovínia,
James R. Asherbc and
Miloš Hricovíni *d
*d
aInstitute of Chemistry, Slovak Academy of Sciences, Dúbravská cesta 9, 845 38 Bratislava, Slovak Republic
bInstitute of Inorganic Chemistry, Slovak Academy of Sciences, Dúbravská cesta 9, 845 36 Bratislava, Slovak Republic
cFaculty of Natural Sciences, Department of Inorganic Chemistry, Comenius University, Mlynská Dolina, CH2, 84215, Bratislava, Slovak Republic
dInstitute of Chemistry, Slovak Academy of Sciences, Dúbravská cesta 9, 845 38 Bratislava, Slovak Republic. E-mail: milos.hricovini@savba.sk; Fax: +421-2-5941-0222; Tel: +421-2-5941-0323
First published on 22nd March 2023
Abstract
High-resolution NMR spectroscopy revealed that a novel glycoconjugate, consisting of two β-glucopyranoses attached to a quinazolinone-like structure, exhibited photoisomerization around the –N–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) and CH–C– bonds of the –N–NCH–C– linkage in the same timeframe (the so-called “crankshaft rotation”) upon exposure to UV light. Experimental NMR data combined with DFT calculations discovered that the attachment of carbohydrate residues to photoactive compounds significantly changed the isomerization process and intramolecular rearrangement compared to the unglycosylated system, while the overall molecular structure remained virtually unchanged.
and CH–C– bonds of the –N–NCH–C– linkage in the same timeframe (the so-called “crankshaft rotation”) upon exposure to UV light. Experimental NMR data combined with DFT calculations discovered that the attachment of carbohydrate residues to photoactive compounds significantly changed the isomerization process and intramolecular rearrangement compared to the unglycosylated system, while the overall molecular structure remained virtually unchanged.
1 Introduction
Many biological systems1,2 are subject to radiation-induced isomerisation processes, most commonly in the near-ultraviolet or visible range. These processes are very often accompanied by bond rotation,3,4 in which the more energetically favourable conformation changes to a lower energy conformer. The progress and nature of the isomerization depends mainly on the structure of the compound, which must be photochemically active; usually it involves conjugated systems with multiple bonds that may have other functional groups attached.5,6 The excitation and relaxation processes vary depending mainly on the arrangement of these individual substituents in these systems.7,8 The incorporation of substituents of organic nature into the structure of conjugated systems may lead to interesting properties of these systems. Conjugated compounds with various bulky substituents, showing the photochemical activity, have been recently investigated.9–11 Incorporation of glycoconjugate moiety into photoactive structures result in the synthesis of a new class of photoactive glycoconjugates that can exhibit such interesting properties as controlled and reversible conformational switching. Glycoconjugates are important not only from the biophysical point of view12 (for e.g. biomimetic properties) but in nanoscience and nanotechnology as well.13 To date, most photochemical studies have focussed on chemical systems with double bonds, typically azobenzene and its derivatives,14,15 where photoisomerisation proceeds at the NN bond.16,17 Photochemistry of glycoconjugates has not been studied in detail so far, and their photochemical properties are poorly understood. Because of their complicated structure, one may expect these molecules' response to UV irradiation to be similarly complex, and difficult to predict.
In this paper we present results of analysis of photoisomerisation of a glycoconjugated molecule (Fig. 1). Unlike the more widely-studied –NN– moiety,18,19 studies of molecular structures containing an –N–NCH– array were performed in details only recently20,21 and showed that this array of atoms exhibited interesting photochemical properties. The role of two glucose residues upon photoisomerization at the –N–N bond in a quinazolinone-like structure is analysed by means of high-resolution NMR spectroscopy and DFT analysis.
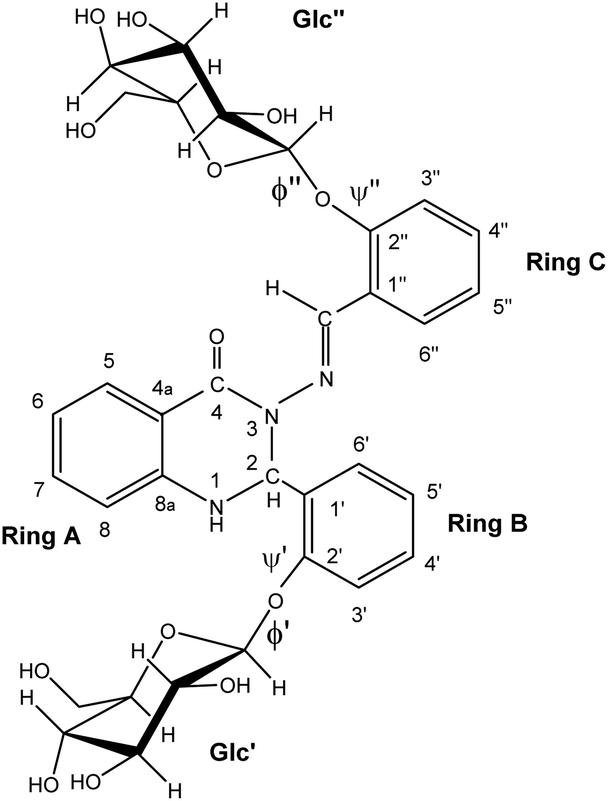 | ||
| Fig. 1 The structure of the studied compound 1. | ||
2 Experimental section
2.1 Instruments
The stock solution of compound 1 (c ∼ 1 mM)22 in DMSO (SeccoSolv, Merck, Germany) was freshly prepared directly before the measurements. High-resolution NMR spectra were recorded in a 5 mm cryoprobe on a Bruker Avance III HD spectrometer at 14 T. One-dimensional 600 MHz 1H and 150 MHz 13C NMR spectra, together with two-dimensional COSY, NOESY, HSQC, and HMBC, enabled determination of the 1H and 13C chemical shifts (referenced to internal TMS) and 1H–1H intramolecular NOEs at 25 °C. Sample were then exposed to high-powered (500 μW cm−2 at a distance of 5 cm) UV irradiation (365 nm) using a UV/vis lamp (Krüss Optronics, Germany) equipped with VIS filters which effectively filter the visible light from the tubes. The possible irradiation times were 5, 15, 30, 45, 60 and 90 minutes, 1D and 2D NMR spectra were then measured at 14 T under the same experimental conditions as before exposure to UV light.2.2 Computational details
The calculations were performed on the studied compound 1 (shown in Fig. 1) using Gaussian 16 software23 employing the MN15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 functional and the 6-311++G(2d,p) basis set. The polarizable continuum model (PCM), using the integral equation formalism variant (IEF-PCM),25 was used to approximate the solvent (DMSO) environment. The convergence criteria were set to tight, using an ultrafine integration grid. Exploratory calculations to examine crankshaft rearrangement were performed on a small model system, HCO–N(Me)–NCH–Ph, at the level of theory just outlined (ωB97X-D26/6-311++G(2d,p) with IEF-PCM). The energy barrier for crankshaft motion was examined by restricted optimisation, in which two dihedrals – D(Ccarbonyl–N–NC) and D(NC–Cipso–Cortho) – were fixed to the same value, which was varied to find the energy maximum. Variation with only one of those dihedrals fixed was also used to find the energy barriers for simple rotation around the relevant bonds (N–N and Cimine–Cipso, respectively).
24 functional and the 6-311++G(2d,p) basis set. The polarizable continuum model (PCM), using the integral equation formalism variant (IEF-PCM),25 was used to approximate the solvent (DMSO) environment. The convergence criteria were set to tight, using an ultrafine integration grid. Exploratory calculations to examine crankshaft rearrangement were performed on a small model system, HCO–N(Me)–NCH–Ph, at the level of theory just outlined (ωB97X-D26/6-311++G(2d,p) with IEF-PCM). The energy barrier for crankshaft motion was examined by restricted optimisation, in which two dihedrals – D(Ccarbonyl–N–NC) and D(NC–Cipso–Cortho) – were fixed to the same value, which was varied to find the energy maximum. Variation with only one of those dihedrals fixed was also used to find the energy barriers for simple rotation around the relevant bonds (N–N and Cimine–Cipso, respectively).
3 Results and discussion
3.1 NMR spectroscopy
High-resolution 600 MHz 1H NMR spectra of glycoconjugate 1 are shown in Fig. 2A, D and Table S1 (see ESI†). Analysis of data revealed that two conformations (labelled as a and b, a:b ∼ 1:0.9) are present in DMSO solution at room temperature. NMR and DFT analysis showed that these two conformations differ in the ϕ′ (O5Glc′–C1Glc′–O1Glc′–C2C-ring′) and ψ′ (C1Glc′–O1Glc′–C2C-ring′–C1C-ring′) angles at the glycosidic linkage connecting ring B and the Glc′ residue (Fig. 3).
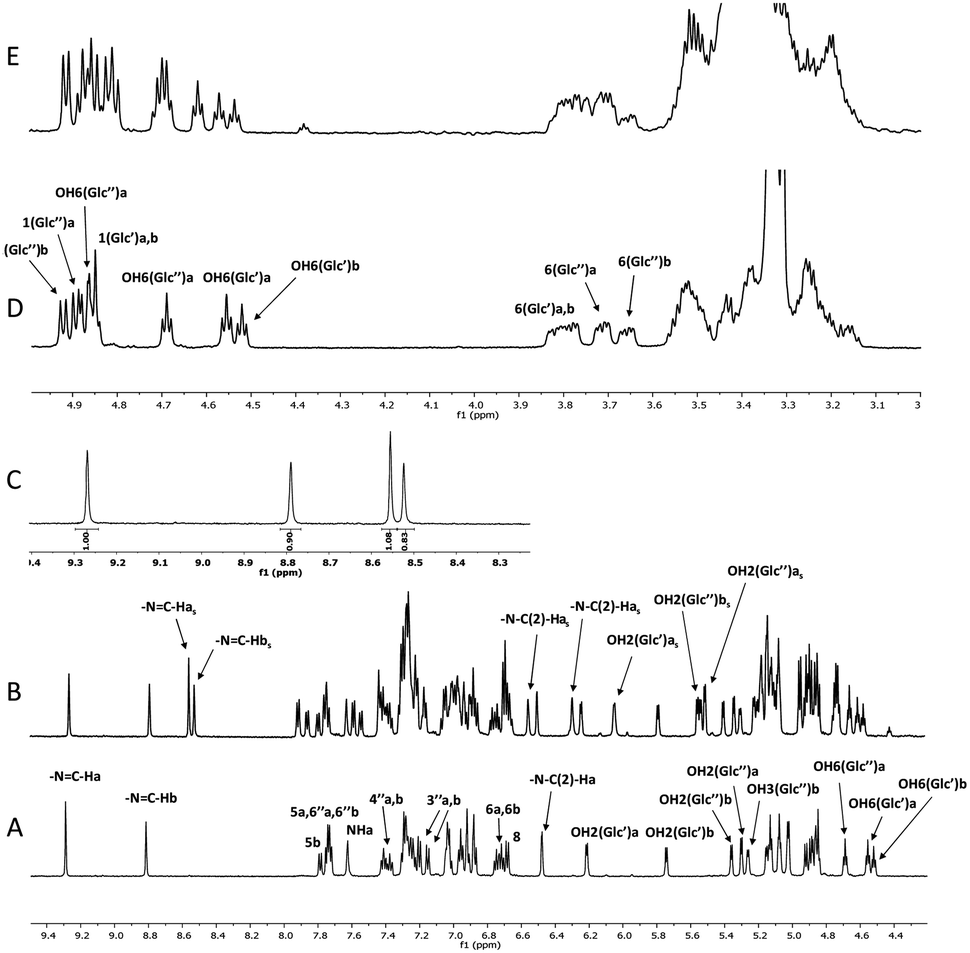 | ||
| Fig. 2 High-resolution 600 MHz 1H NMR spectrum of 1 in DMSO at 25 °C (expanded into two parts (A): 4.2–9.5 ppm (D): 3.0–5 ppm). 1H spectrum obtained after 60 min. UV irradiation (365 nm) ((B): 4.2–9.5 ppm; (E): 3.0–5 ppm). Expansion of the 1H spectrum showing the integral intensities of the azomethine group resonances originating from the two conformers (a and b) for anti- and syn-forms (C). Signals marked with “s” belong to the syn-isomer which formed under irradiation. | ||
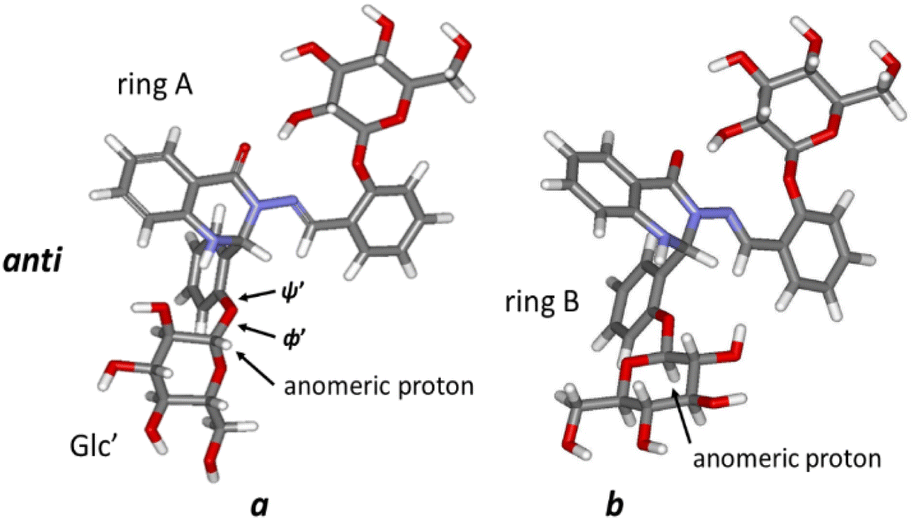 | ||
| Fig. 3 The optimised structures of both conformers (a and b) of 1 (anti-form). | ||
DFT calculations, using the MN15/6-311++G(2d,p) level of theory, yielded values of ϕ′ ∼ 74°; ψ′ ∼ 148° for the prevalent conformer (a, Fig. 3) and ϕ′ ∼ −76°; ψ′ ∼ −172° for the less populated conformer b form (Fig. 3). The main difference between these two forms is thus the orientation of the Glc′ residue with respect to rings A and B: the anomeric proton is oriented towards ring A in the a conformation, whereas the Glc′ residue is flipped around O5Glc′–C1Glc′ and O1Glc′–C2C-ring′ bonds in the b conformation, and the anomeric proton is close to ring B. It should be noted that a strong hydrogen bond between OH(2)′′ in the Glc′′ and carbonyl oxygen (ring A) was obtained (d–OH(2)′′…CO = 186 pm in both forms a and b), keeping ring C and the attached Glc′′ residue in approximately the same conformation regardless of the Glc′ position relative to ring B.
DFT studies of various possible conformers predicted the a and b conformers (with anti-conformation around N–N) to be the most stable and hence abundant (Table 1). However, the calculations predicted slightly different relative abundance of these two conformers to that seen in experiment: the conformer designated b above had the lower energy (ΔE ∼ 2.22 kJ), with a computed abundance of 71%, compared to the 47% inferred from the NMR experimental data (Fig. 2A). This difference is partly due to the use of the IEF-PCM solvent model, but it should also be taken into account that the results are at the accuracy level of the MN15/6-311++G(2d,p) approach.
| anti-form | syn-form | ΔE/(anti-/syn-) | |
|---|---|---|---|
| Conformer a | −2420.004931 | −2420.002568 | 6.20 |
| Conformer b | −2420.005776 | −2420.003759 | 5.29 |
| ΔE/(a/b) | 2.22 | 3.13 |
Next we investigated the influence of glucose residues on photoisomerisation in this glycoconjugate 1. Compound 1 displayed photoinduced switching on UV irradiation (365 nm) and NMR data showed that both forms (a and b; Fig. 3, 4A) photoisomerised around the –N–N and CH–C– bonds of the –N–NCH–C– array in the same timeframe. The original and the photoisomerised forms (for both a and b) were then present in a ∼1:1 ratio in DMSO at room temperature (Fig. 2B and C). We identify the old form as anti- and the new as syn- on the basis of prior work on similar molecules,20,21 supported by changes in the magnitudes of 1JC–H (174.1 Hz (a) and 171.1 Hz (b) for anti-, vs. 180.9 Hz (a) and 180.0 Hz (b) for syn-; see Fig. S1, ESI†) in the azomethine group (CH–C–), an indicator of conformational change in the –N–NCH–C– chain. 2D NOESY spectra (see Fig. S2, ESI†) showed dipolar interactions between the azomethine protons and the hydroxyl (OH(2)′′) protons at the Glc′′ unit linked to ring C for both a and b forms of the syn-isomer (Fig. 4B). These NOEs are compatible with a structure where the Glc′′ residue has a similar position as in the anti-isomer (Fig. 4A). For the molecule to retain such a structural feature on photoisomerisation requires that the intramolecular rearrangement of the –N–NCH–C– array proceed around the –N–N and the CH–C– bonds simultaneously instead of around –N–N only. It should also be noted that the ratio between the two forms a and b was 1:0.77 (a:b) (Fig. 1C) in the new isomer produced by UV-irradiation, indicating a small shift towards the a form due to photoisomerisation (DFT calculations predicted abundances of 22% for a and 78% for b, compared to 29:71 in the anti-form, Table 1). The theoretical results obtained fit well with the experimental data (including the presence of anti- and syn-forms during photoisomerisation), although they are not completely quantitative. As mentioned above, the accuracy level of the MN15/6-311++G(2d,p) approach should be taken into account.
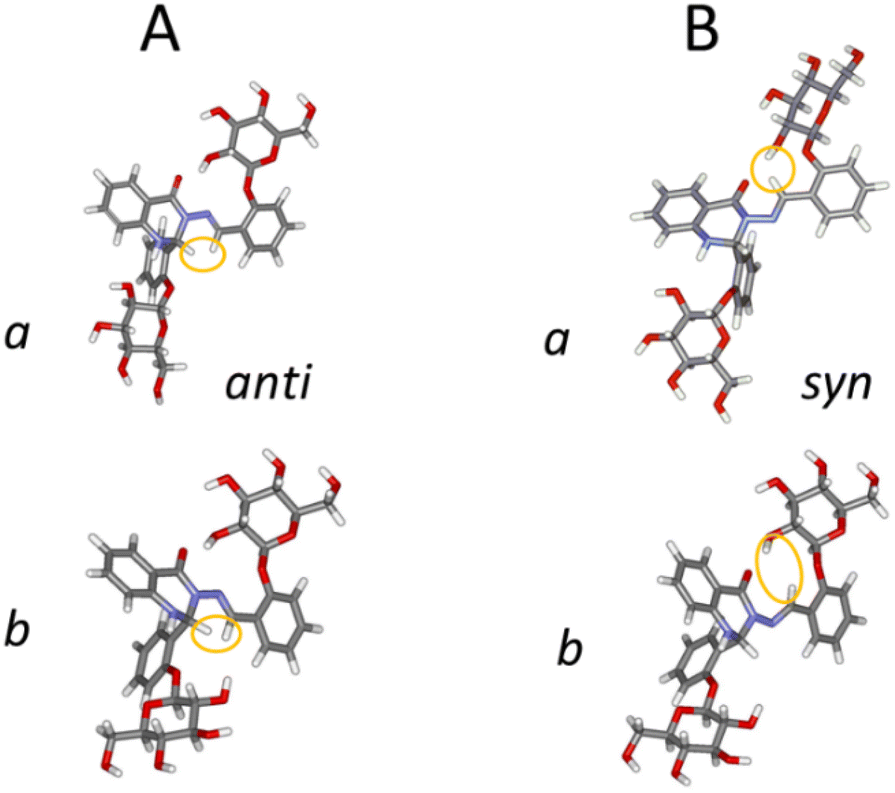 | ||
| Fig. 4 The optimised structures of both conformers (a and b) of 1 for anti- and syn-forms. We have highlighted the azomethine (NCH) hydrogen and the hydrogens to which it shows distinctive dipolar coupling in the 2D NOESY spectrum: for anti-, the tertiary H on the heterocycle; for syn-, 2-OH′′ of the glucose residue. | ||
The presence of the conjugated π-system spanning from ring A to ring C did not prevent instantaneous and concurrent isomerisation at the –N–N and C(H)–C– bonds, both having partial double-bond character (d–N–N = 137 pm and dC(H)–C– = 146 pm). In addition, the spatial orientation of the Glc′′ residue adopted the same conformation in the syn-isomer as in the anti-form, with the OH(2)′′ groups and the carbonyl oxygens remaining in close enough proximity for strong hydrogen bonds to be present: d–OH(2)′′…CO = 195 pm in the a and d–OH(2)′′…CO = 194 pm in the b forms. These conformations were stabilised by several hydrogen bonds, including a bifurcated hydrogen bond between the azomethine proton (CH–) and the glycosidic oxygen (Glc′′ residue) and OH(2)′′ oxygen. Some weak hydrogen bonds were present in both anti- and syn-isomers, i.e. unchanged by isomerisation: e.g. between the ring oxygen in Glc′′ and H-3′′ on the aromatic ring C. This well-defined H-bond network may influence the re-establishment of thermodynamic equilibrium, i.e. the reversion from the syn- to the anti-isomer, which took three months at room temperature (Table S2, see ESI†). This is notably longer than the unglycosylated compound with ortho-OH instead of o-Glc on rings B and C (compound 8 in our previous work21), which took 15 minutes to revert; this is in line with our hypothesis that o-OH permits fast reïsomerisation by a different mechanism (tautomerisation).
Previously, we used NMR data supplemented by DFT calculations to examine the anti-/syn- isomerisation of structurally-related quinazolinone-derived Schiff bases, including one which was the same as 1 but with –OH groups instead of –OGlc.20,21 That system showed 25% conversion on photoirradiation in DMSO, and reversion to the anti-form was several orders of magnitude faster: thus, attaching the Glc residue roughly doubles the yield, and greatly increases the stability, of the syn-form. We concluded that the faster reversion of the original –OH derivative (and others with an ortho-OH group) is probably due to a special mechanism involving tautomerisation – known to occur for other Schiff bases with intramolecular H-bonding to the imine nitrogen27,28 – and subsequent rotation around the –N–N moiety; this exact mechanism would not apply to 1. The mechanism in the slower-relaxing systems was assumed to be simple rotation around the N–N bond – no evidence to the contrary being seen in the NMR data – and our calculations thus focussed on that.
However, the NMR evidence discussed above indicates that glycoconjugate 1 isomerises instead by a concerted rotation (crankshaft rotation) around both the N–N and the C–C bonds. To examine this mechanism, we thus carried out exploratory calculations (ωB97XD/6-311++G(2d,p), IEF-PCM: DMSO as before) on crankshaft rotation in a small model system, HCO–N(Me)–NCH–Ph. This model system has planar symmetry, making it easy to define a reaction coordinate for crankshaft rotation – namely, setting the dihedrals Ccarbonyl–N–NCH and NCH–CAr–CArH to be equal and varying them between 0 and 180°. (Larger systems have C–N–NCH dihedrals other than 0 and 180° at their local minima, making definitions less obvious.) These calculations show that crankshaft rotation has a large energy barrier slightly less than the sum of the energy barriers for rotation around N–N and rotation around C–Ph (relative to syn, Eb,crankshaft = 10.1 mH; Eb,N–N = 2.6 mH; Eb,C–C = 8.2 mH). Thus, crankshaft rotation should be about exp(7.5/kT) ≅ 4000 times slower than N–N rotation – raising the question of why simple N–N rotation does not occur instead. We hypothesise that, because crankshaft motion maintains the relative positions of the heterocycle and ring C, it involves far less solvent friction than N–N rotation; it also preserves the intramolecular hydrogen bond from Glc′′ to CO, which as noted would be broken by simple rotation around N–N, leading to conformers which we do not see in the NMR spectra. This solvent friction obstructs simple rotation around the N–N bond enough that crankshaft rotation takes place instead.
4 Conclusions
In summary, experimental high-resolution NMR spectroscopy and DFT calculations have found that the attachment of saccharide residues to photoactive compounds can significantly change their isomerisation processes. In this case, a glycoconjugate consisting of two glucose residues linked by a quinazolinone-like structure exhibited an unusual crankshaft-type intramolecular rearrangement from anti- to syn-forms in DMSO solution. Interestingly, the overall molecular structure remained virtually the same in both anti- and syn-isomers due to a strong intramolecular hydrogen bond between Glc′′ and the carbonyl group. Thus, crankshaft motion maintains the relative positions of the heterocycle and ring C, leading to far less solvent friction than N–N rotation, and preserving the aforementioned intramolecular hydrogen bond. Other intramolecular H-bonds are also seen between Glc′ and the rest of the molecule, but are not changed by syn-/anti-isomerisation.Author contributions
Michal Hricovíni: formal analysis, investigation, writing – original draft, visualisation. James Asher: formal analysis, writing – original draft, review & editing. Miloš Hricovíni: conceptualisation, funding acquisition, investigation, writing – original draft, review & editing. All authors co-wrote the paper and extensively discussed interpretation of the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Slovak Grant Agency VEGA grants no. 2/0071/22, 2/0135/21 and grant APVV-19-0516. Calculations were performed at the Computing Centre of the SAS using the supercomputing infrastructure acquired in projects ITMS 26230120002 and 26210120002, both supported by the Research & Development Operational Program funded by the ERDF.References
- H. V. Kiefer, E. Gruber, J. Langeland, P. A. Kusochek, A. V. Bochenkova and L. H. Andersen, Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
- J. Chang, M. G. Romei and S. G. Boxer, J. Am. Chem. Soc., 2019, 141, 15504–15508 CrossRef CAS PubMed.
- C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475–2532 CrossRef CAS PubMed.
- T. Kumpulainen, B. Lang, A. Rosspeintner and E. Vauthey, Chem. Rev., 2017, 117, 10826–10939 CrossRef CAS PubMed.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- A. J. Pepino, M. A. Burgos Paci, W. J. Peláez and G. A. Argüello, Phys. Chem. Chem. Phys., 2015, 17, 12927–12934 RSC.
- L. Vuković, C. F. Burmeister, P. Král and G. Groenhof, J. Phys. Chem. Lett., 2013, 4, 1005–1011 CrossRef PubMed.
- Y. T. Wang, X. Y. Liu, G. Cui, W. H. Fang and W. Thiel, Angew. Chem., Int. Ed., 2016, 55, 14009–14013 CrossRef CAS PubMed.
- I. G. Ovchinnikova, G. A. Kim, E. G. Matochkina, M. I. Kodess, P. A. Slepukhin, I. S. Kovalev, E. V. Nosova, G. L. Rusinov and V. N. Charushin, J. Photochem. Photobiol., A, 2018, 351, 16–28 CrossRef CAS.
- J. Pang, Z. Gao, H. Tan, X. Mao, H. Wang and X. Hu, Front. Chem., 2019, 7, 1–11 CrossRef PubMed.
- E. Léonard and A. Fayeulle, Molecules, 2021, 26, 3063–3085 CrossRef PubMed.
- C. I. C. Crucho, P. Correia-Da-Silva, K. T. Petrova and M. T. Barros, Carbohydr. Res., 2015, 402, 124–132 CrossRef CAS PubMed.
- M. Kalita, M. M. Payne and S. H. Bossmann, Nanomedicine, 2022, 42, 102542 CrossRef CAS PubMed.
- A. Georgiev, A. Kostadinov, D. Ivanov, D. Dimov, S. Stoyanov, L. Nedelchev, D. Nazarova and D. Yancheva, Spectrochim. Acta, Part A, 2018, 192, 263–274 CrossRef CAS PubMed.
- K. Bujak, H. Orlikowska, J. G. Małecki, E. Schab-Balcerzak, S. Bartkiewicz, J. Bogucki, A. Sobolewska and J. Konieczkowska, Dyes Pigm., 2019, 160, 654–662 CrossRef CAS.
- J. García-Amorós and D. Velasco, Beilstein J. Org. Chem., 2012, 8, 1003–1017 CrossRef PubMed.
- J. Volarić, J. Buter, A. M. Schulte, K. O. Van Den Berg, E. Santamaría-Aranda, W. Szymanski and B. L. Feringa, J. Org. Chem., 2022, 87, 14319–14333 CrossRef PubMed.
- V. Chandrasekaran and T. K. Lindhorst, Chem. Commun., 2012, 48, 7519–7521 RSC.
- A. Müller, H. Kobarg, V. Chandrasekaran, J. Gronow, F. D. Sönnichsen and T. K. Lindhorst, Chem.–Eur. J., 2015, 21, 13723–13731 CrossRef PubMed.
- M. Hricovíni, J. R. Asher and M. Hricovíni, RSC Adv., 2020, 10, 5540–5550 RSC.
- M. Hricovíni, J. R. Asher and M. Hricovíni, RSC Adv., 2022, 12, 27442–27452 RSC.
- Z. Hricovíniová, M. Hricovíni and K. Kozics, Chem. Pap., 2018, 72, 1041–1053 CrossRef.
- M. J. Frisch, G. W. Trucks, B. H. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, Ö. Farkas, J. V. Foresman and D. J. Fox, Gaussian 16, Rev. B.01, Wallingford, CT, 2016 Search PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- B. R. Muhyedeen, S. R. Salman and A. G. Petros, Iraqi J. Sci., 1995, 36, 451–459 Search PubMed.
- D. Yordanov, V. Deneva, A. Georgiev, A. Crochet, K. M. Fromm and L. Antonov, Spectrochim. Acta, Part A, 2020, 237, 118416 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01678a |
| This journal is © The Royal Society of Chemistry 2023 |