
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorinated saccharide-derived hard carbon as a cathode material of lithium primary batteries: effect of the polymerization degree of the starting saccharide†
Lei Chena,
Yanyan Lia,
Chao Liuab,
Feifei Guo b,
Xiaozhong Wua,
Pengfei Zhoua,
Zhiwen Fangb and
Jin Zhou*a
b,
Xiaozhong Wua,
Pengfei Zhoua,
Zhiwen Fangb and
Jin Zhou*a
aSchool of Chemistry and Chemical Engineering, Shandong University of Technology, Zibo 255000, China. E-mail: zhoujin@sdut.edu.cn
bShandong Zhongshan Photoelectric Materials Co., Ltd., Zibo 255138, China
First published on 15th May 2023
Abstract
Fluorinated hard carbon materials have been considered to be a good candidate of cathode materials of Li/CFx batteries. However, the effect of the precursor structure of the hard carbon on the structure and electrochemical performance of fluorinated carbon cathode materials has yet to be fully studied. In this paper, a series of fluorinated hard carbon (FHC) materials are prepared by gas phase fluorination using saccharides with different degrees of polymerization as a carbon source, and their structure and electrochemical properties are studied. The experimental results show that the specific surface area, pore structure, and defect degree of hard carbon (HC) are enhanced as the polymerization degree (i.e. molecular weight) of the starting saccharide increases. At the same time, the F/C ratio increases after fluorination at the same temperature, and the contents of electrochemically inactive –CF2 and –CF3 groups also become higher. At the fluorination temperature of 500 °C, the obtained fluorinated glucose pyrolytic carbon shows good electrochemical properties, with a specific capacity of 876 mA h g−1, an energy density of 1872 W kg−1, and a power density of 3740 W kg−1. This study provides valuable insights and references for selecting suitable hard carbon precursors to develop high-performance fluorinated carbon cathode materials.
Introduction
The lithium/fluorinated carbon (Li/CFx) battery boasts several advantageous characteristics, including high energy density, stable work voltage, low self-discharge rate, long shelf life, and a broad operating temperature range.1,2 The utilization of fluorinated carbon as a cathode material for lithium primary batteries dates back to 1972, and its commercialization by Panasonic Electric Co., Ltd. occurred in 1975.3 The theoretical specific capacity of Li/CFx batteries surpasses that of other lithium primary batteries, reaching a maximum of 865 mA h g−1 when x = 1, and an energy density of 2180 W kg−1. Currently, these batteries are widely used in medical and military applications.4The critical component of Li/CFx batteries is the fluorinated carbon cathode material, of which fluorinated graphite is the most prevalent one currently utilized. However, this material exhibits several limitations, such as low discharge voltage platform, initial voltage delay, and poor rate capability.5,6 Efforts have been undertaken to improve the performance of Li/CFx batteries by exploring alternative carbon sources and other means. Currently, research has been conducted on the fluorinated carbon cathode materials utilizing nanostructured carbon and hard carbon as the carbon source.7–9 Ahmad et al.10 synthesized fluorinated carbon nanodiscs which exhibited a high specific capacity of 1180 mA h g−1. The central discs' reinforcement effect and the presence of a LiF shell allow for Li+ to diffuse through the discs' edge or surface cracks, resulting in extra capacity. Li et al.11 prepared deeply fluorinated multi-walled carbon nanotubes which exhibit a conductive network of nano-scale multi-walled carbon nanotubes and the inherent rapid rate capability of one-dimensional nanostructures, enabling high discharge rates up to 5C, with a maximum power density of 7114.1 W kg−1. Damien et al.12 reported that the Li/CFx batteries utilizing fluorinated graphene with a low F/C ratio of 0.22 demonstrated excellent electrochemical performance, with a specific capacity of 767 mA h g−1 at a current density of 10 mA g−1, surpassing that of fluorinated graphite with similar fluorine content (CF0.25). Nano-CFx materials often possess the benefits of a large specific surface area, small particle size, and a porous structure, enhancing electrochemical reactivity by promoting the rapid diffusion of Li+ and reducing reaction resistance, leading to excellent electrochemical performance.13 However, the high cost and production instability of nanostructured carbon hinder their practical application as starting materials of the responding fluorinated carbon cathode in Li/CFx batteries, and thus fluorinated graphite remains the primary commercial product in the market.
Hard carbon, which is generally used as anode material for lithium/sodium ion batteries, has the advantages of low cost, wide resource, high tap density, and appropriate specific surface area.14,15 In recent years, many researchers have used hard carbon as the starting material to prepare fluorinated carbon cathode materials for high performance Li/CFx battery. Peng et al.16 synthesized fluorinated biomass-derived carbon from nut shells and found it to have a high discharge potential and specific capacity, offering an ultrahigh energy density of 2585 W kg−1. Zhou et al.17 prepared fluorinated hard carbon through the direct gas fluorination, which had an energy density of up to 2466 W kg−1 and good rate capacity with a specific capacity exceeding 600 mA h g−1 at a discharge rate of 5C. Jiang et al.18 prepared fluorinated Ketjenblack as cathode material of Li/CFx batteries, which exhibited a high specific capacity than 900 mA h g−1 and high energy density of 2544 W h kg−1, as well as a good rate discharge performance up to 20C. It is well known that the structure of the carbon starting material plays a significant role in the fluorination process and the structure of the obtained fluorinated carbon material, as well as its electrochemical performance. Moreover, the structure of the carbon material is always influenced by its precursors. However, the effect of the precursor structure of hard carbon on the structure and electrochemical performance of fluorinated carbon cathode materials has yet to be fully studied.
In this study, three hard carbon materials (HCs) are prepared through direct pyrolysis of saccharides with different degrees of polymerization (glucose, sucrose, and starch) and then subjected to fluorination by direct gas fluorination method at various temperatures. The resulting series of fluorinated carbon materials, referred to as fluorinated hard carbons (FHCs), are systematically analyzed for its micro-morphology, pore structure, crystal structure, C–F bond properties, and electrochemical properties as cathode materials. The impact of saccharides precursor structure on the structure and electrochemical performance of FHCs is explored. The fluorinated glucose pyrolytic carbon of FHC-1-500 exhibits excellent comprehensive performance, with a specific capacity of 876 mA h g−1, a maximum power density of 3740 W kg−1 and an energy density of 1872 W h kg−1, providing references for the selection of carbon sources in the preparation of fluorinated carbon materials.
Experiment
Material synthesis
Glucose, sucrose and starch from potato (hereinafter referred to as starch) were purchased directly from Shanghai Aladdin Biochemical Technology Co., Ltd. The hard carbon (HC) was prepared through the direct pyrolysis method. The saccharides were placed in corundum crucibles within a tubular furnace, and subjected to continuous Ar injection while the temperature was raised from 25 °C to 1000 °C at a rate of 2 °C min−1 and held for 2 hours. After natural cooling, the samples were labelled as HC-1, HC-2 and HC-3, respectively. Subsequently, the prepared hard carbon underwent direct fluorination in a Monel alloy tube for 4 hours, at a temperature rise rate of 2 °C min−1, within a range of 300–500 °C in an atmosphere of F2/N2 (volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) mixed gas with a temperature interval of 50 °C. Upon completion of the fluorination process, the reactor was purged with N2 to remove residual F2 and cooled to room temperature. Finally, the fluorinated product was cleaned with a dilute Na2CO3 solution and deionized water to remove free F2, and vacuum dried at 60 °C to obtain the fluorinated hard carbon. The resulting samples were labelled as FHC-X-Y, where X represents the serial number of the carbon source and Y represents the fluorination temperature.
:9) mixed gas with a temperature interval of 50 °C. Upon completion of the fluorination process, the reactor was purged with N2 to remove residual F2 and cooled to room temperature. Finally, the fluorinated product was cleaned with a dilute Na2CO3 solution and deionized water to remove free F2, and vacuum dried at 60 °C to obtain the fluorinated hard carbon. The resulting samples were labelled as FHC-X-Y, where X represents the serial number of the carbon source and Y represents the fluorination temperature.
Sample characterization
The microscopic morphology of the samples was analyzed using scanning electron microscopy (SEM, Sirion 200 FEI, Netherlands). The phase and crystal structure analysis were performed utilizing powder X-ray diffraction (XRD, Bruker D8 Advance Diffractometer) with Cu Kα radiation (λ = 0.15406 nm) and Raman spectroscopy (LabRAM HR800 JY Horiba, 325 nm). The specific surface area and porosity of the samples were determined through using the ASAP 2020 nitrogen adsorption system (Micrometrics, USA). A powder sample of about 100 mg was first degassed for 4 hours and then loaded into the system for measurement to obtain the specific surface area of the sample. The sample composition was quantitatively determined using an element analyzer (Vario EL Cube, Elemental, Germany). The surface chemical composition was analyzed using X-ray photoelectron spectroscopy (XPS, Escalab 250, USA), while the chemical properties of the C–F bond were characterized using Fourier transform infrared spectroscopy (FT-IR, Nicolet 5700 USA). The material thermal properties measured by thermogravimetric analysis (TG, STA449F5, NETZSCH, Germany).Electrochemical test
The FHC-X-Y material was employed as the cathode active material for the preparation of a working electrode. The active material (80 wt%), polyvinylidene fluoride (PVDF, 10 wt%), and Super P (10 wt%) were homogenized with N-methyl-2-pyrrolidone (NMP) to form a uniform slurry. The obtained slurry was coated onto an aluminium foil and subjected to vacuum drying at 80 °C for 12 hours. Subsequently, the material was cut into circular plates with a diameter of 12 mm and transferred to a glove box for battery assembly. Each electrode plate was estimated to contain approximately 2 mg of the active substance. The electrochemical tests were performed on CR2032 coin cells. The lithium foil served as the anode and reference electrodes, while an alumina coated film was utilized as the separator. The electrolyte was comprised of a solution of 1 mol L−1 LiClO4 in a mixed solvent of PC-DME-DOL (propylene carbonate-1,2-dimethoxyethane-1,3-dioxolane). The battery was assembled in a glove box (Mikrouna Co., Advanced 2440/750) filled with Ar, with the contents of H2O and O2 both below 1 ppm. The battery performance was evaluated using the LandCT2001 battery testing system (Wuhan Land Electronics Co., Ltd., China), utilizing galvanostatic discharge at a test temperature of 30 °C, and a cut-off voltage of 1.5 V.Results and discussion
Characterization of the saccharide-derived hard carbons
The SEM images and digital photo of three hard carbon samples are depicted in Fig. 1a–c and S1.† The results indicate that the morphology of the hard carbon samples, synthesized through pyrolysis of glucose, sucrose, and starch, are similar and characterized by massive, smooth-surfaced particles with irregular shapes and no discernible pore structure. This is because in the process of direct pyrolysis of saccharides, they will melt and foam to form irregular three-dimensional carbon foams.19 The starch-derived carbon of HC-3 exhibits the most pronounced foaming behavior, resulting in a lower yield than HC-1 and HC-2 as shown in Table S1.† Moreover, it is noted that the particle size of HC-1 and HC-2 is approximately 500 μm, while that of HC-3 is only about 100 μm (Fig. 1a–c).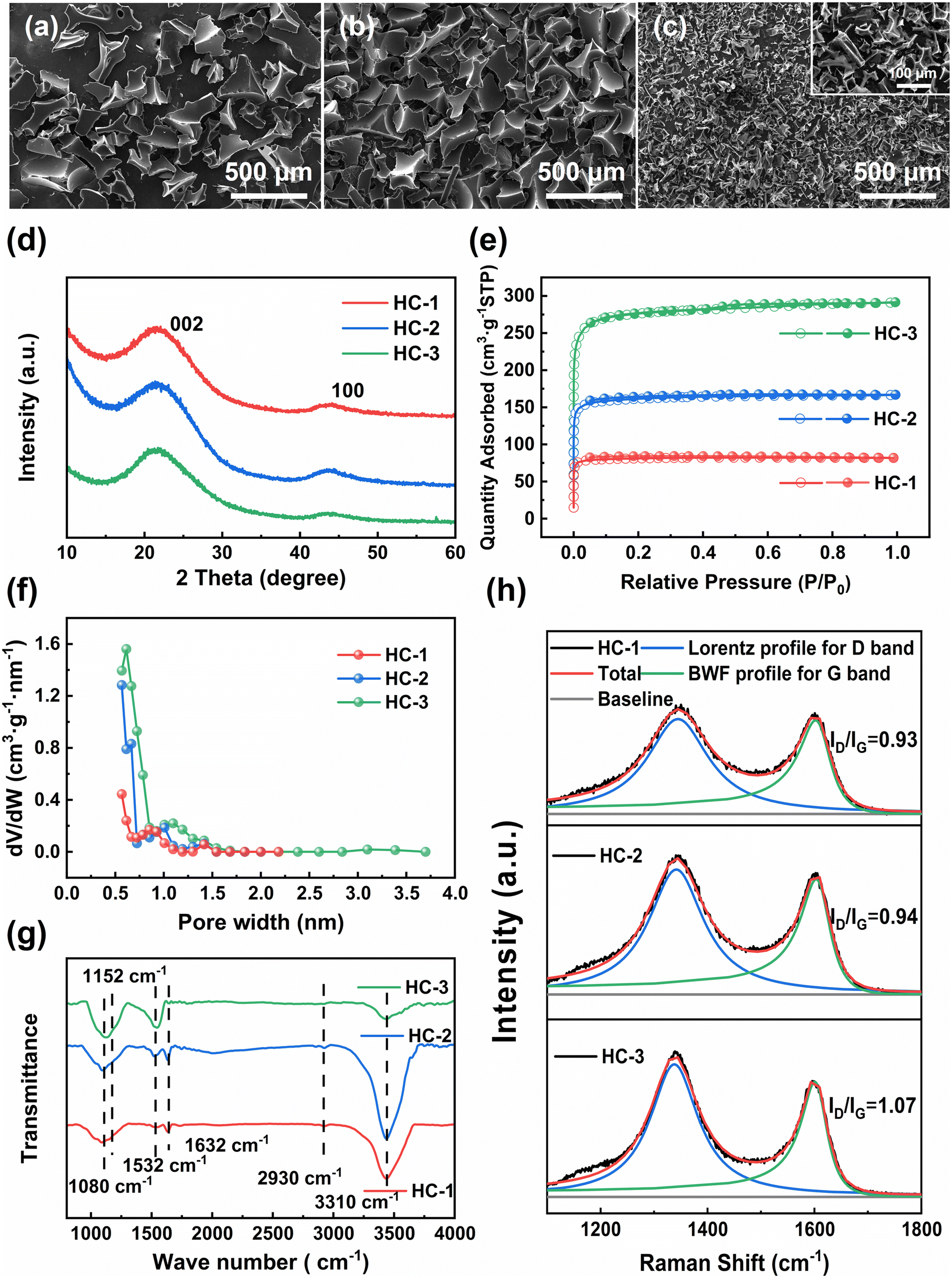 | ||
| Fig. 1 (a)–(c) SEM images of HCs; (d) XRD patterns of HCs; (e) nitrogen sorption isotherms; (f) pore size distributions of HCs; (g) FT-IR spectra of HCs; (h) Raman spectra of HCs. | ||
The crystalline structure of the HC samples is thoroughly examined via X-ray diffraction (XRD) characterization. As illustrated in Fig. 1d, all the samples display two prominent peaks at 22° and 43°, which correspond to the (002) and (100) Bragg reflections of graphite, respectively. The low and broad X-ray diffraction intensity observed suggests that the materials are highly disordered. The interlayer distance (d002) of the sp2 carbon layer is calculated by utilizing the peak position of the 002graphite peak and the Bragg formula, as expressed by the following equation:
|
2dsinθ = nλ
| (1) |
Additionally, the crystallite size along the stacking direction of the c-axis (Lc) and a-axis (La) of the carbon layer can be estimated from full-width at half maximum (FWHM) of peaks based on Scherrer formula, as follows:
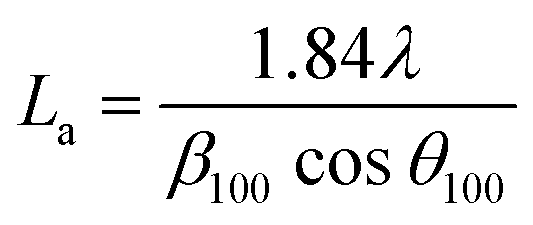 | (2) |
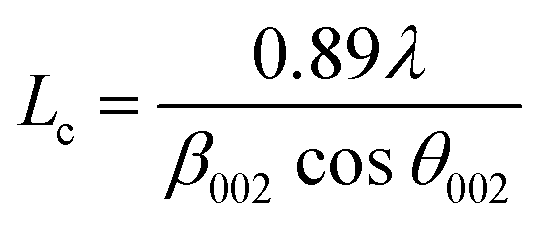 | (3) |
The pore structure and specific surface area of the carbon materials are characterized using N2 adsorption and desorption analysis (Fig. 1e and f). All the HC samples display typical isotherms of type I, exhibiting a sharp rise at low relative pressures (P/P0 < 0.1) and rapidly reaching saturation, indicating the presence of abundant micropores. The adsorption and desorption of N2 by the saccharide-derived carbon increase as the degree of polymerization increases. Additionally, a hysteresis loop is observed only for HC-3, indicating the presence of a small number of mesopores. As shown in Table S1,† both the specific surface area and average pore size increase with the polymerization degree of the saccharides increasing. Thermal gravimetric (TG) analysis is conducted from room temperature to 1000 °C in a N2 atmosphere at a heating rate of 5 °C min−1 to investigate the pyrolysis behavior of different types of saccharides. As shown in Fig. S2,† the pyrolysis curves of glucose, sucrose, and starch are found to be similar. These saccharides start to release moisture below 230 °C, with a slow pyrolysis rate, which is consistent with previous observation.22 Between 230 °C and 600 °C, these saccharides begin to decompose, undergo depolymerization and decarboxylation reactions, and release a large amount of volatile substances. The rapid escape of volatiles destroy the morphology of saccharides, resulting in the melting and foaming of the saccharide structure.23 Compared with other saccharides, starch has a higher water content and a chain-like structure, which results in more severe dehydration, depolymerization, and decarboxylation reactions. These reactions lead to an increase in the surface area and pore size of starch, but at the expense of its yield. These characteristics leads to a significant increase in structural defects for HC-3, which is beneficial for the fluorination of carbon materials.
Fig. 1g presents the FT-IR spectra of three hard carbons. The peaks at 1080, 1152, 1532, 1632, and 2930 cm−1 are attributed to the C–O stretching vibration, C–O–C asymmetric stretching vibration, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C asymmetric stretching vibration, CO asymmetric stretching vibration, and C–H asymmetric stretching vibration, respectively. Additionally, a peak cantered at 3310 cm−1 can be observed, which corresponds to the O–H stretching vibration of hydrogen bond association.19,24 It can be seen that the functional group types of the three HCs are not much different. With the growth of the carbon chains, the strength of the CO bond becomes weaker, and the strength of the CC bond increases.
C asymmetric stretching vibration, CO asymmetric stretching vibration, and C–H asymmetric stretching vibration, respectively. Additionally, a peak cantered at 3310 cm−1 can be observed, which corresponds to the O–H stretching vibration of hydrogen bond association.19,24 It can be seen that the functional group types of the three HCs are not much different. With the growth of the carbon chains, the strength of the CO bond becomes weaker, and the strength of the CC bond increases.
The surface structural defects of hard carbons are further scrutinized using Raman spectroscopy (Fig. 1h), which reveal broad bands near 1350 cm−1 and 1600 cm−1 corresponding to the D band (disordered band, A1g vibration mode of sp2 carbon ring) and the G band (graphite band, E2g vibration mode of sp2 carbon ring), respectively.25 The G peak is fitted using the Breit–Wigner–Fano (BWF) line, as expressed by the following equation:
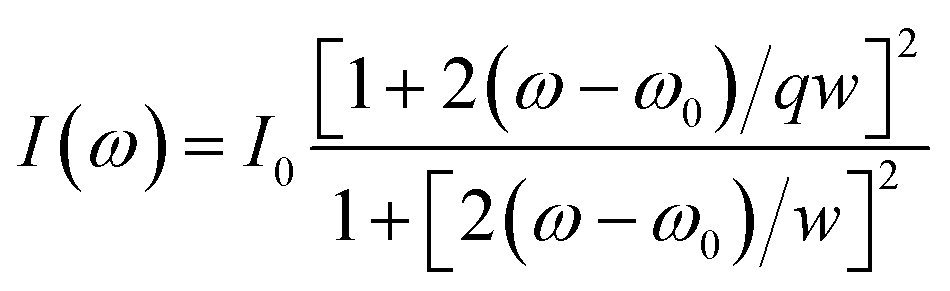 | (4) |
The surface contents of HCs and the chemical states of C and O are measured and analyzed by XPS, and the bulk contents of C and O of HCs are measured by elemental analysis. Table S2† shows that the content of O increases with the increase of polymerization degree, which may be due to the easier chemical sorption of O as the specific surface area of HC increases. As shown in Fig. S3a,† the presence of C and O is evidenced by the C 1s peak at about 285 eV and the O 1s peak at about 533 eV, respectively. The C 1s spectra of the three types of hard carbons are displayed in Fig. S3b and c,† and it is observed that peaks with binding energies of 284.8, 285.3, 286.5 and 289.8 eV correspond to C–C, C–O, CO and OC–O functional groups, respectively.27,28 Further information on the content of each functional group can be found in Table S2.†
To sum up, it can be noted that as the polymerization degree of the saccharide source increases, there appears to be no significant alteration in the crystal structure and functional groups present in saccharide-derived carbon, but the specific surface area, pore dimensions, and degree of defects all exhibit an increase.
Characterization of fluorinated hard carbons
The SEM images of the FHCs are shown in Fig. 2 and S4.† Results reveal that as the fluorination temperature increases, the surface of the fluorinated carbon undergoes fragmentation, resulting in the formation of holes, collapsing holes, and finally forming a powder with a particle size of about 20 μm. As the temperature increases, the reaction between carbon atoms and F2 is more intense, resulting in more severe erosion of the FHCs surface, leading to particle fragmentation. And at the same fluorination temperature, the higher the polymerization degree of the carbon source, the more severe the fracture of the generated fluorinated hard carbon, indicating that the increase in polymerization degree makes the hard carbon easier to fluorinate. | ||
| Fig. 2 SEM images of HC-1 and FHC-1. | ||
The crystal structure of FHCs is analyzed and characterized using X-ray diffraction (XRD). As displayed in Fig. 3a, S5a and b,† upon fluorination, a new diffraction peak appears near 14°, which is attributed to the (001) crystal plane of the fluorinated carbon material.29 As the fluorination temperature increases, the (100) reflection peak shifts towards lower angles, suggesting an increase in the length of the C–C bond and the formation of more C–F bonds.18 Additionally, the (001) reflection peak, corresponding to the hexagonal system compound with high fluorine content, increases in intensity and sharpness with increasing fluorination temperature, indicating that more layers of carbon are fluorinated. The data of d001 is shown in Table S3.† The shift towards larger angles of the (001) diffraction peak demonstrates that the interlayer spacing of the fluorinated hard carbon became narrower, potentially due to a phase transition from (C2F)n (interlayer spacing of 0.81 nm) to (CF1)n (interlayer spacing of 0.60 nm) along with the fluorination level enhancing.30
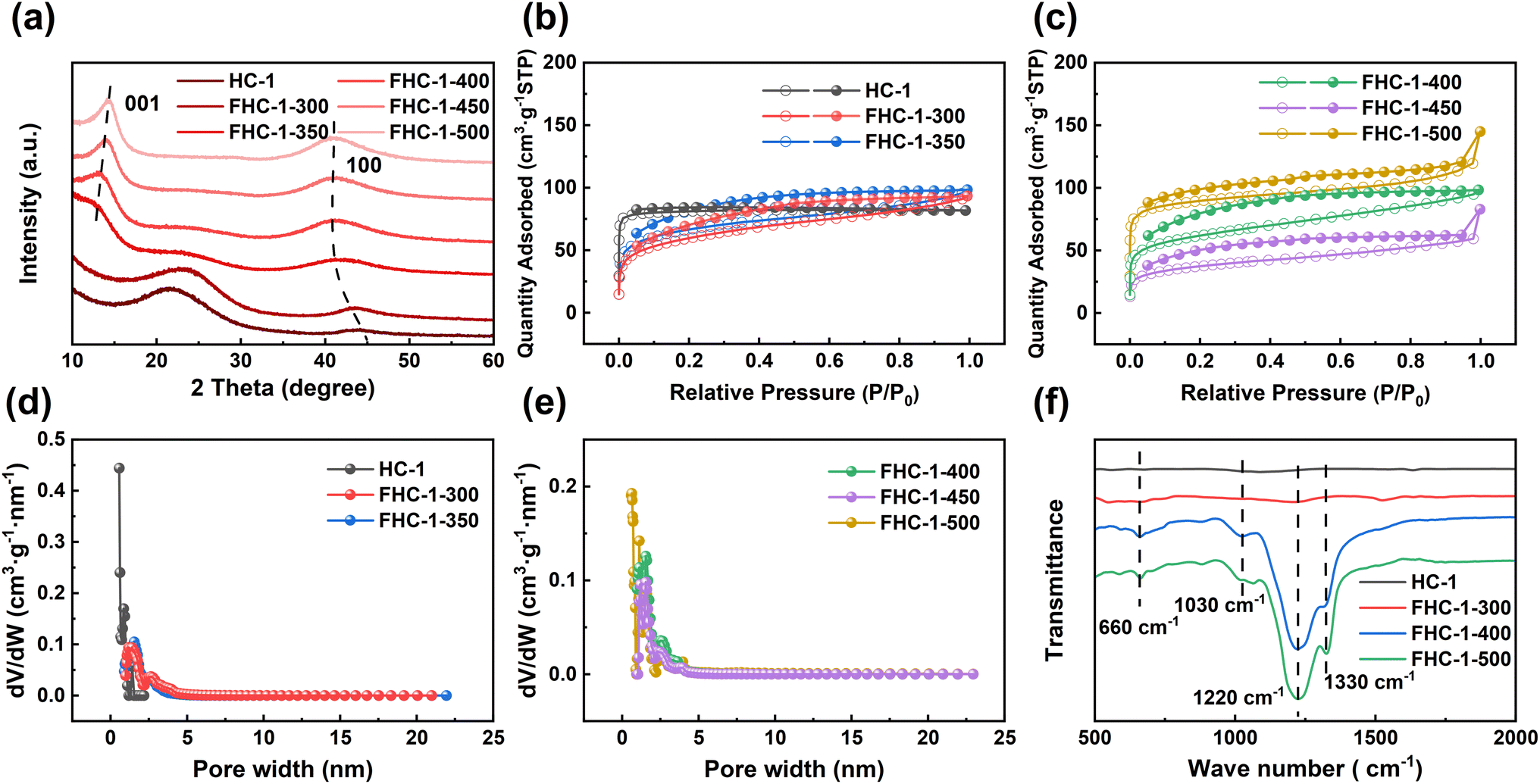 | ||
| Fig. 3 (a) XRD patterns of HC-1 and FHC-1; (b) and (c) nitrogen sorption isotherms of HC-1 and FHC-1; (d) and (e) pore size distributions of HC-1 and FHC-1; (f) FT-IR spectra of HC-1 and FHC-1. | ||
The N2 adsorption–desorption of FHC-1 is shown in the Fig. 3b and c. It can be seen that as the fluorination temperature increases, the curve gradually changes from type I to type IV, indicating that mesopores are generated in FHCs. The specific surface area and pore size of FHCs are shown in the Table S4.† After fluorination, the specific surface area of the sample decreases slightly. The reason may be that after fluorination of HC, the partially disordered structure becomes orderly and the pores merge. When the fluorination temperature is 500 °C, the specific surface area increases, probably because HC is strongly broken at higher temperature.31 The larger surface area can provide more active sites for the discharge reaction and facilitate the contact between FHCs and electrolyte. FHCs retains the porous structure, which is beneficial for the transport of Li+.32 As shown the adsorption isotherms of FHC-2 and FHC-3 are type IV, indicating that the samples are more susceptible to fluorination with increasing polymerization degree of saccharide sources. At the same fluorination temperature, the specific surface area and pore size increase with the increase of polymerization degree of the saccharide, indicating that the FHCs structures are influenced by the starting materials.
The chemical bonding properties of the FHCs are further verified through FT-IR analysis, as depicted in Fig. 3f, S5e and f.† The prominent absorption peak observed at approximately 1220 cm−1 can be attributed to the C–F covalent bond, and it is observed that with the increase in fluorination temperature, there is a shift in the FT-IR spectrum of FHCs towards higher wave numbers, accompanied by a narrowing of the peak width and an increase in peak intensity. These results demonstrate an increase in the binding strength between C and F atoms. The absorption peaks near 1320 cm−1 and 660 cm−1, corresponding to the –CF2 and –CF3 groups, respectively, display an increase in intensity with the rise in fluorination temperature.18 There is also a dispersive absorption peak at 1030 cm−1 wave number, indicative of the semi-ionic C–F bond, and when the fluorination temperature is too high, this peak disappears.33,34 Moreover, the C–F bond of FHC-3 appears earlier and has stronger strength, indicating that HC-3 is easier to be fluorinated.
The Raman spectra obtained using the UV laser (Fig. 4a–c and S6a–f†) show that the ID/IG ratio increases with the increase in fluorination temperature, indicative of an increase in the number of defects in the carbon layer. This can be attributed to the destruction of the carbon layer at high fluorination temperatures and the insertion of F atoms, which increases the number of point defects.35 At the same fluorination temperature, the ID/IG ratio also increases as the polymerization degree of saccharide increases, further demonstrating the influence of the starting materials, consistent with the results obtained from SEM, XRD, BET and FTIR analysis.
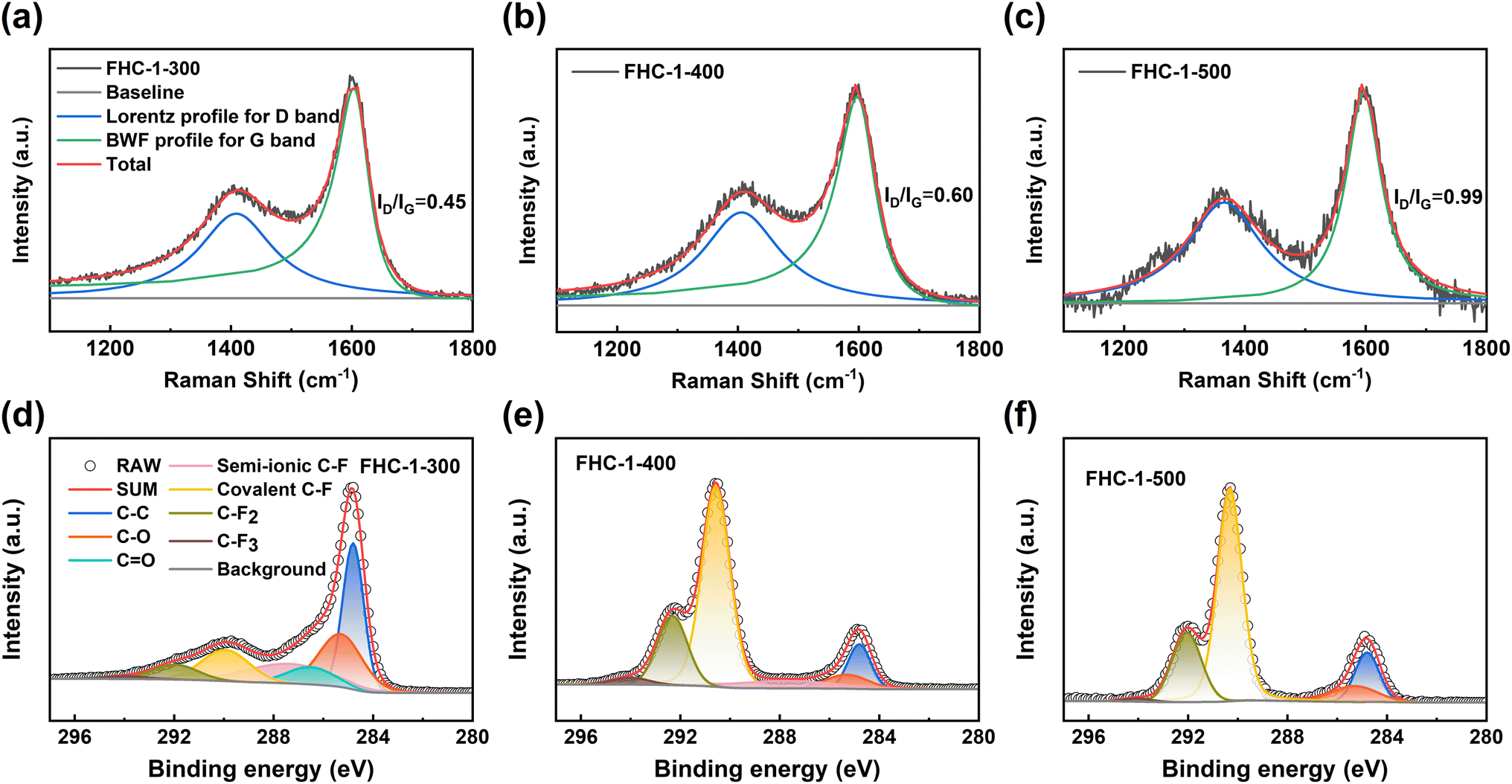 | ||
| Fig. 4 (a)–(c) Raman spectra of FHC-1; (d)–(f) C 1s spectra of the FHC-1. | ||
The composition and functional groups present in the FHCs samples are analyzed using X-ray photoelectron spectroscopy (XPS). As depicted in Fig. S7a,† the peaks observed at 285, 533, and 689 eV correspond to the presence of C, O, and F elements, respectively.36 Table S5† outlines the chemical compositions of all the FHCs. The results reveal that the fluorine content of the FHCs samples increases with an increase in the fluorination temperature. Furthermore, at the same fluorination temperature, the fluorine content increases as the degree of polymerization of the used saccharides increases, indicating an enhancement of the degree of fluorination. High-resolution XPS spectroscopy is utilized to study the chemical state of the element C. The Fig. 4d–f and S6g–l† show that four new peaks appear after fluorination. The four peaks with binding energy values of approximately 287.74, 290.26, 292, and 294 eV are attributed to semi-ionic C–F, covalent C–F, –CF2, and –CF3, respectively.37 As the fluorination temperature increases, the peak intensity of the C–F bond increases, while that of the sp2 C–C bond decreases, suggesting a conversion of C–C bond into C–F bond. Except for FHC-X-500, the other samples have semi-ionic C–F bonds on the surface. This is because FHC-X-500 has a high degree of fluorination and semi-ionic C–F is converted to other bonds. The presence of semi-ionic C–F bonds helps maintain the good conductivity of the FHCs samples, while the covalent C–F bonds significantly reduce conductivity, resulting in an insulation-prone behavior of the samples.3,38 With an increase in the fluorination temperature, the content of semi-ionic C–F bonds in FHCs decreases gradually, leading to a corresponding decrease in conductivity and an increase in ohmic polarization, which results in a decrease in the discharge voltage of the Li/CFx battery.18,39
In fact, the “semi-ionic” bonding is resulted from the incomplete fluorination of the carbon layer, which should be expressed as “weak covalent” C–F bonding due to the hyperconjugation between the un-fluorinated CC bond and the C–F bond.40–42 The interaction of fluorine gas with FHCs leads to the conversion of the ether group into C–F bond through a nucleophilic substitution reaction and the per-fluorination of the carbonyl group, dangling bond, and point defects to form –CF2 and –CF3. The binding energy of the C–F, –CF2, and –CF3 groups in the C 1s spectrum experiences a blue shift with an increase in the F/C ratio.43 This change can also be seen in the F 1s spectra of FHCs, as shown in Fig. S7b.† It can be seen that an increase in fluorination temperature results in an increase in fluorine content, a decrease in semi-ionic C–F bonds content, and an increase in covalent C–F bonds and perfluorocarbon groups. Similarly, with increasing polymerization degree of saccharide there is an increase in covalent C–F bonds and perfluorocarbon groups at the same fluorination temperature.
To summarize, it can be observed that an increase in polymerization degree of saccharide corresponds to a greater development of the specific surface and pore structure in the HC material. This, in turn, leads to an increase in the presence of defects, ultimately resulting in a higher susceptibility to fluorination. Additionally, it has been noted that under same fluorination temperatures, an elevated F/C ratio is observed, accompanied by an increase in the prevalence of –CF2 and –CF3 in the resulting C–F bonds.
Electrochemical performance test of fluorinated hard carbon
The electrochemical performance of the prepared FHCs sample as the cathode of Li/CFx battery is studied through a galvanostatic discharge test conducted at different discharge rates. Results, as shown in Fig. 5 and S8,† indicate that as the fluorination temperature increases, the maximum specific capacity of FHCs derived from the same carbon source increases, since the specific discharge capacity is directly proportional to the fluorine content. However, when the degree of fluorination is too high, such as FHC-3-500, the material cannot be discharged. Therefore, an appropriate fluorination temperature is crucial to optimize the electrochemical performance of fluorinated carbon materials. As the fluorination temperature increased, the material produced some defect structures, such as fluorinated groups such as –CF2 and –CF3 revealed by FT-IR spectroscopy and XPS C 1s spectroscopy. These perfluorinated groups on the surface are not conductive and electrochemically active, which limit the insertion and diffusion of Li+, thereby hindering the discharge reaction and reducing the discharge specific capacity and discharge voltage. In addition, it can be observed that higher current densities lead to lower specific capacities and discharge voltages, and a potential delay occurs, due to the increase of ohmic resistance.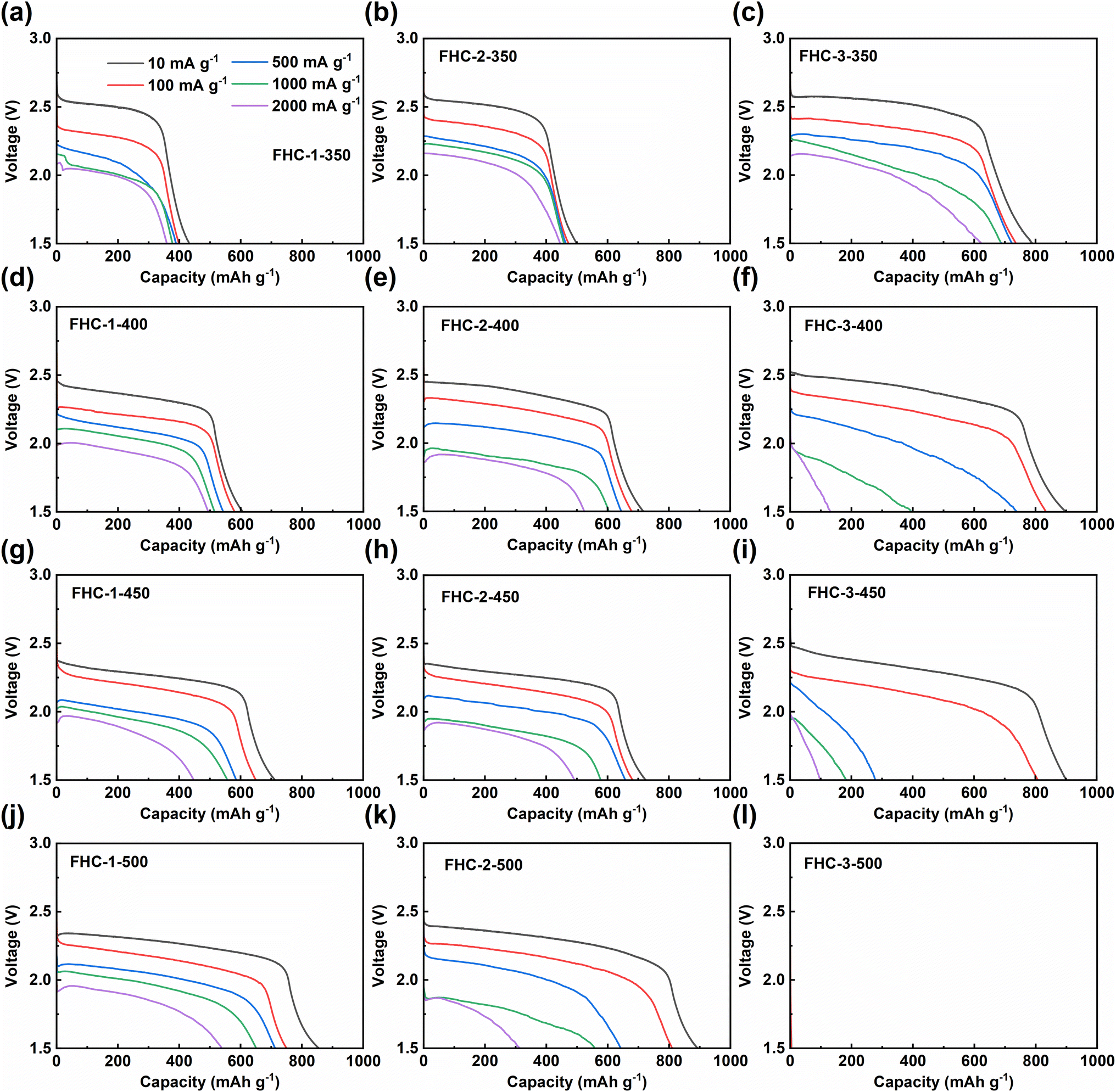 | ||
| Fig. 5 Galvanostatic discharge curves of FHCs. | ||
In order to further explore the influence of the starting saccharide sources on the discharge performance of FHCs, we compare the specific capacity, discharge voltage plateau, energy density, and power density, and the results are shown in Fig. 6 and Table 1. At low current density, the discharge specific capacity increases along with the increasing of the fluorination temperature except for FHC-3-500. This is due to that the specific capacity of CFx is proportional to its fluorine content. In same reason, for the FHCs prepared at the same fluorination temperature, the specific capacities increase when the starting materials varied from glucose to starch. However, the degree of fluorination of FHC-3-500 is too high, which makes the material unable to discharge. But the voltage plateau decreases with the increase of fluorine content, which is mainly due to the increase of ohmic polarization due to the decrease of material conductivity.44,45 As the current density increases, the specific capacity and voltage plateau decrease, and the higher the polymerization degree of the carbon source, the worse the rate performance. This is because the higher the polymerization degree of the starting saccharides, the larger the specific surface area of the generated HC, and the higher the degree of fluorination under the same conditions, resulting in less semi-ionic C–F bond content, more perfluorinated groups, and poor electrochemical performance. In addition, electrochemical polarization exacerbates the deterioration of electrochemical performance. For the same reason, with the increase of fluorination temperature, the rate performance of the material decreases. Therefore, FHC-1 series exhibited relatively high voltage and capacity retention. The Ragone plots (Fig. 6g–i) of the FHCs cathode depicts the variation of energy density with power density. It can be observed that the energy density decreases with the increase of power density. At low power density, the energy density increases with the degree of fluorination. As the power density increases, the energy density decreases, and the trend is more obvious with higher fluorination degree, which is mainly due to the decrease of output potential and discharge capacity.
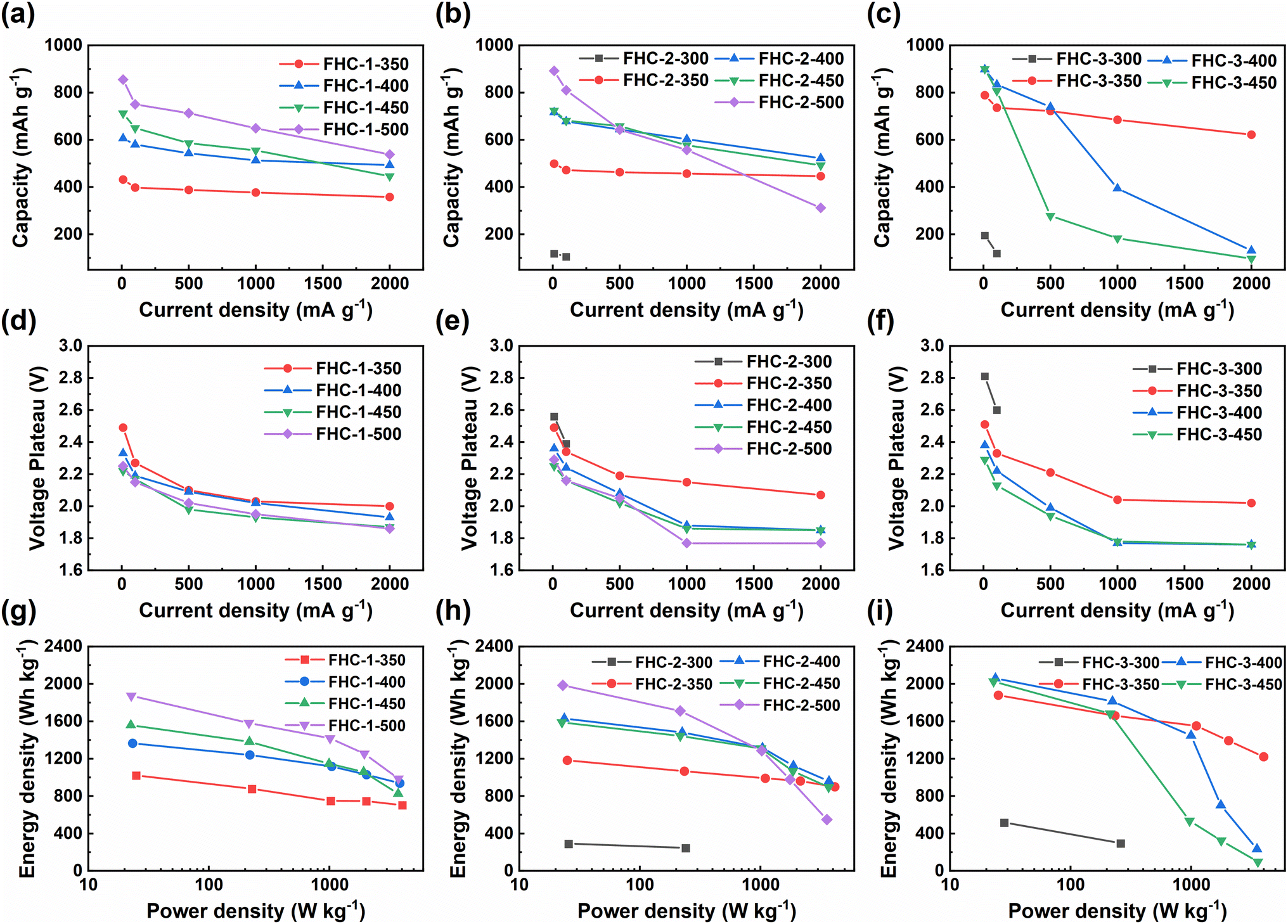 | ||
| Fig. 6 (a)–(c) Specific discharge capacities; (d)–(f) discharge voltage plateau; (g)–(i) Ragone plots of FHCs at different current densities. | ||
| Sample | Maximum capacity (mA h g−1) | Maximum energy density (W h kg−1) | Maximum power density (W kg−1) | Capacity retention at 2 A g−1 (%) |
|---|---|---|---|---|
| FHC-1-350 | 432 | 1021.1 | 4120 | 82.9 |
| FHC-1-400 | 606 | 1364.5 | 3840 | 81.3 |
| FHC-1-450 | 712 | 1558.2 | 3720 | 62.6 |
| FHC-1-500 | 876 | 1872.7 | 3740 | 61.4 |
| FHC-2-350 | 499 | 1181.8 | 4148 | 89.3 |
| FHC-2-400 | 716 | 1629.5 | 3706 | 72.9 |
| FHC-2-450 | 724 | 1588.3 | 3680 | 67.9 |
| FHC-2-500 | 892 | 1984.4 | 3560 | 34.9 |
| FHC-3-350 | 789 | 1878 | 4000 | 78.8 |
| FHC-3-400 | 898 | 2025.7 | 3580 | 14.5 |
| FHC-3-450 | 900 | 2059 | 3520 | 10.7 |
Furthermore, FHC-3-350 shows good electrochemical performance, such as an energy density of 1878 W h kg−1 and a high-rate capability with 78.8% retention of specific capacity. This is due to the large specific surface area of its precursor, which can achieve high F/C ratio even at low temperature. Although its specific capacity is slightly lower than that of commercial CFx,46,47 its moderate amount of semi-ionic bonds, large specific surface area, and rich pore structure make it have excellent capacity retention. However, the low density of FHC-3-350 derived the developed porosity will lead to a decrease in the loading capacity of the electrode sheet, more electrolyte also needs to be used. In addition, the yield of starch pyrolytic carbon is also low, and the above factors may limit the practical performance of FHC-3-350. Comparatively, FHC-1-500 has better comprehensive performance, it gives a high specific capacity of 876 mA h g−1 at 10 mA g−1 and remains 538 mA h g−1 at 2000 mA g−1, with a maximum energy density of 1872.7 W h kg−1 and a maximum power density of 3740 W kg−1. The excellent electrochemical performance of FHC-1-500 can be attributed to the small specific surface area of its precursor, which makes it difficult to be fluorinated. Therefore, even at high temperature, it maintains a certain degree of disordered structure, which provides sites for electrochemical reactions. In addition, it has a moderate specific surface area and rich pore structure, which enhance the interfacial contact between electrode materials and electrolytes, reduce the paths for Li+ diffusion, and facilitate electron transfer.
Conclusions
In summary, we use three different types of saccharides as carbon sources to prepare a series of FHCs with different properties by gas-phase fluorination, and study the effect of the polymerization degree of the starting saccharide sources on the structure and electrochemical performance of the fluorinated hard carbon. With the increase of polymerization degree, the specific surface area and pore structure of the hard carbons are more developed, and more defects will be generated at the same time, which makes HC easier to be fluorinated. At the same fluorination temperature, the resulted FHCs has a higher F/C ratio and more –CF2 and –CF3 groups in the C–F bond, which will lead to poor performance of the battery. Among them, FHC-1-500 demonstrates exceptional overall performance, exhibiting a high specific capacity of 876 mA h g−1 at 10 mA g−1, 61.4% capacity retention at 2 A g−1, a maximum power density of 3740 W h kg−1 and energy density of 1872 W kg−1. This is because the disordered structure enhances the electrochemical activity, while a certain number of semi-ionic C–F bonds and abundant pore structure facilitate the transport of electrons and Li+. These findings provide reference and guidance for preparation of high-performance CFx and carbon source selection.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22078179), Natural Science Foundation of Shandong Province (ZR2022JQ10), and Taishan Scholar Foundation (tsqn201812063).References
- L. Kong, Y. Li, C. Peng, L. Sun, K. Wang, Y. Liu and W. Feng, Nano Energy, 2022, 104, 107905 CrossRef CAS.
- L. Wang, Z. Wu, J. Zou, P. Gao, X. Niu, H. Li and L. Chen, Joule, 2019, 3, 2086–2102 CrossRef CAS.
- Y. Liu, L. Jiang, H. Wang, H. Wang, W. Jiao, G. Chen, P. Zhang, D. Hui and X. Jian, Nano. Rev., 2019, 8, 573–586 CAS.
- Y. Li, X. Wu, C. Liu, S. Wang, P. Zhou, T. Zhou, Z. Miao, W. Xing, S. Zhuo and J. Zhou, J. Mater. Chem. A, 2019, 7, 7128–7137 RSC.
- S. S. Zhang, D. Foster and J. Read, J. Power Sources, 2009, 191, 648–652 CrossRef CAS.
- Q. Zhang, S. D'Astorg, P. Xiao, X. Zhang and L. Lu, J. Power Sources, 2010, 195, 2914–2917 CrossRef CAS.
- Z. C. Liu, J. C. Lu and P. Huang, Adv. Mater. Res., 2014, 968, 16–20 CAS.
- Y. Xu, L. Zhan, Y. Wang, Y.-l. Wang and Y.-h. Shi, New Carbon Mater., 2015, 30, 79–85 CrossRef CAS.
- Z. Luo, X. Wang, D. Chen, Q. Chang, S. Xie, Z. Ma, W. Lei, J. Pan, Y. Pan and J. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 18809–18820 CrossRef CAS PubMed.
- Y. Ahmad, M. Dubois, K. Guérin, A. Hamwi and W. Zhang, Carbon, 2015, 94, 1061–1070 CrossRef CAS.
- Y. Li, Y. Feng and W. Feng, Electrochim. Acta, 2013, 107, 343–349 CrossRef CAS.
- D. Damien, P. M. Sudeep, T. N. Narayanan, M. R. Anantharaman, P. M. Ajayan and M. M. Shaijumon, RSC Adv., 2013, 3, 25702–25706 RSC.
- J. Xie, C. Li, Z. Cui and X. Guo, Adv. Funct. Mater., 2015, 25, 6519–6526 CrossRef CAS.
- K.-l. Hong, L. Qie, R. Zeng, Z.-q. Yi, W. Zhang, D. Wang, W. Yin, C. Wu, Q.-j. Fan, W.-x. Zhang and Y.-h. Huang, J. Mater. Chem. A, 2014, 2, 12733–12738 RSC.
- G. Zhao, D. Yu, H. Zhang, F. Sun, J. Li, L. Zhu, L. Sun, M. Yu, F. Besenbacher and Y. Sun, Nano Energy, 2020, 67, 104219 CrossRef CAS.
- C. Peng, Y. Li, F. Yao, H. Fu, R. Zhou, Y. Feng and W. Feng, Carbon, 2019, 153, 783–791 CrossRef CAS.
- R. Zhou, Y. Li, Y. Feng, C. Peng and W. Feng, Compos. Commun., 2020, 21, 100396 CrossRef.
- S. Jiang, P. Huang, J. Lu and Z. Liu, RSC Adv., 2021, 11, 25461–25470 RSC.
- M.-X. Song, L.-J. Xie, J.-Y. Cheng, Z.-L. Yi, G. Song, X.-Y. Jia, J.-P. Chen, Q.-G. Guo and C.-M. Chen, J. Energy Chem., 2022, 66, 448–458 CrossRef CAS.
- Y.-Y. Li, C. Liu, L. Chen, X.-Z. Wu, P.-F. Zhou, X.-Y. Shen and J. Zhou, Rare Metals, 2023, 42, 940–953 CrossRef CAS.
- K. Kubota, S. Shimadzu, N. Yabuuchi, S. Tominaka, S. Shiraishi, M. Abreu-Sepulveda, A. Manivannan, K. Gotoh, M. Fukunishi, M. Dahbi and S. Komaba, Chem. Mater., 2020, 32, 2961–2977 CrossRef CAS.
- S. Al Arni, B. Bosio and E. Arato, Renewable Energy, 2010, 35, 29–35 CrossRef CAS.
- M. Bardet, S. Hediger, G. Gerbaud, S. Gambarelli, J. F. Jacquot, M. F. Foray and A. Gadelle, Fuel, 2007, 86, 1966–1976 CrossRef CAS.
- H. Zhang, H. Ming, W. Zhang, G. Cao and Y. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 23766–23774 CrossRef CAS PubMed.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B, 2000, 61, 14095–14107 CrossRef CAS.
- Y. Liu, Y. Zhang, X. Wang, Z. Wang, W. Lai, X. Zhang and X. Liu, J. Phys. Chem. C, 2018, 122, 6357–6367 CrossRef CAS.
- D. Sun, B. Luo, H. Wang, Y. Tang, X. Ji and L. Wang, Nano Energy, 2019, 64, 103937 CrossRef CAS.
- H. Yue, W. Zhang, H. Liu, Z. Liu, G. Zhong and Y. Yang, Nanotechnology, 2013, 24, 424003 CrossRef PubMed.
- L. Wang, Y. Li, S. Wang, P. Zhou, Z. Zhao, X. Li, J. Zhou and S. Zhuo, ChemElectroChem, 2019, 6, 2201–2207 CrossRef CAS.
- S. Park, K.-S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572–578 CrossRef CAS PubMed.
- H. Touhara, H. Fujimoto, N. Watanabe and A. Tressaud, Solid State Ionics, 1984, 14, 163–170 CrossRef CAS.
- V. Mazanek, O. Jankovsky, J. Luxa, D. Sedmidubsky, Z. Janousek, F. Sembera, M. Mikulics and Z. Sofer, Nanoscale, 2015, 7, 13646–13655 RSC.
- W. Feng, P. Long, Y. Feng and Y. Li, Adv. Sci., 2016, 3, 1500413 CrossRef PubMed.
- K. Fan, J. Fu, X. Liu, Y. Liu, W. Lai, X. Liu and X. Wang, Chem. Sci., 2019, 10, 5546–5555 RSC.
- X. Wang, Y. Dai, J. Gao, J. Huang, B. Li, C. Fan, J. Yang and X. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 8294–8299 CrossRef CAS PubMed.
- S. M. Mukhopadhyay, P. Joshi, S. Datta and J. Macdaniel, Appl. Surf. Sci., 2002, 201, 219–226 CrossRef CAS.
- C. Sun, Y. Feng, Y. Li, C. Qin, Q. Zhang and W. Feng, Nanoscale, 2014, 6, 2634–2641 RSC.
- K. P. Huang, P. Lin and H. C. Shih, J. Appl. Phys., 2004, 96, 354–360 CrossRef CAS.
- Y. Sato, K. Itoh, R. Hagiwara, T. Fukunaga and Y. Ito, Carbon, 2004, 42, 3243–3249 CrossRef CAS.
- K. Yoshida, Y. Sugawara, M. Saitoh, K. Matsumoto, R. Hagiwara, Y. Matsuo, A. Kuwabara, Y. Ukyo and Y. Ikuhara, J. Power Sources, 2020, 445, 227320 CrossRef CAS.
- W. Lai, D. Xu, X. Wang, Z. Wang, Y. Liu, X. Zhang and X. Liu, Phys. Chem. Chem. Phys., 2017, 19, 19442–19451 RSC.
- W. Feng, New Carbon Mater., 2023, 38, 130–142 CrossRef.
- M. Dubois, K. Guérin, W. Zhang, Y. Ahmad, A. Hamwi, Z. Fawal, H. Kharbache and F. Masin, Electrochim. Acta, 2012, 59, 485–491 CrossRef CAS.
- W. Zhang, M. Dubois, K. Guerin, P. Bonnet, H. Kharbache, F. Masin, A. P. Kharitonov and A. Hamwi, Phys. Chem. Chem. Phys., 2010, 12, 1388–1398 RSC.
- Y. Dai, S. Cai, L. Wu, W. Yang, J. Xie, W. Wen, J.-C. Zheng and Y. Zhu, J. Mater. Chem. A, 2014, 2, 20896–20901 RSC.
- P. Zhou, J. Weng, X. Liu, Y. Li, L. Wang, X. Wu, T. Zhou, J. Zhou and S. Zhuo, J. Power Sources, 2019, 414, 210–217 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01695a |
| This journal is © The Royal Society of Chemistry 2023 |