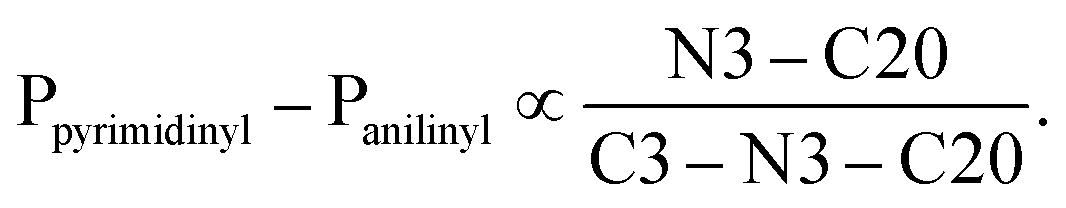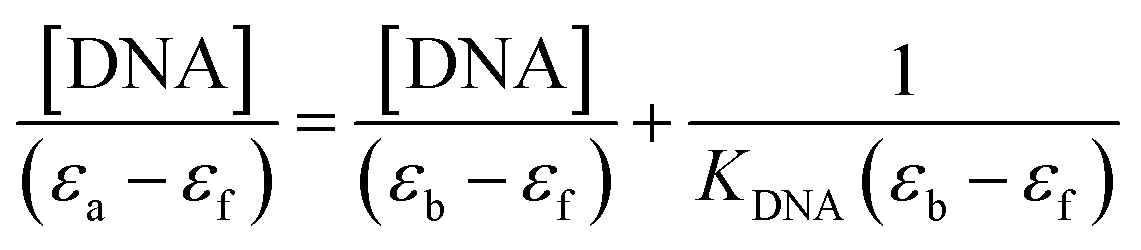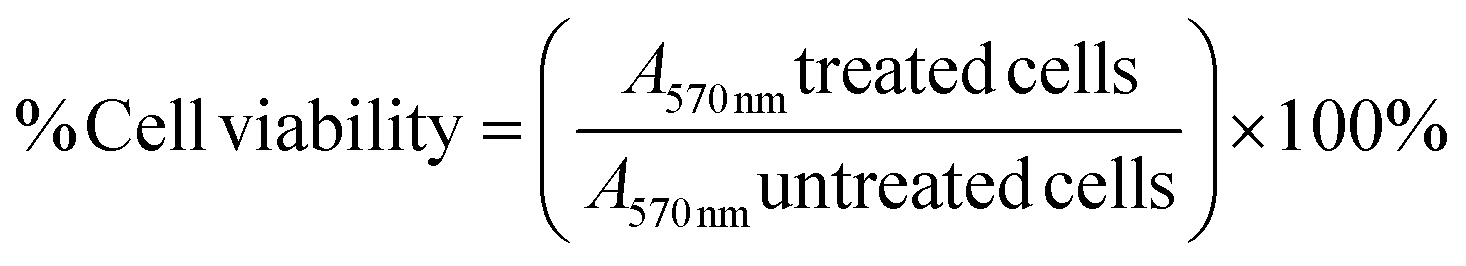DOI:
10.1039/D3RA01741A
(Paper)
RSC Adv., 2023,
13, 21820-21837
The link between relative stability constant of DNA- and BSA-chromenopyrimidine complexes and cytotoxicity towards human breast cancer cells (MCF-7)†
Received
16th March 2023
, Accepted 11th July 2023
First published on 19th July 2023
Abstract
In this study, we synthesized and characterized ten chromenopyrimidine derivatives using analytical and spectroscopic methods. Studies on DNA and albumin binding affinity, as well as cytotoxicity tests on human breast cancer (MCF-7) cells, of the chromenopyrimidines, were conducted. The natural logarithm of the relative stability constant of DNA- and BSA-chromenopyrimidine complexes [ln(KDNA/KBSA)] was used as a criterion for selecting compounds for cytotoxicity studies. We found that ln(KDNA/KBSA) was inversely related to IC50 values of the compounds in MCF-7 cells. The antiproliferative effects of the compounds were found to induce apoptosis in MCF-7 cells, which is a desired mechanism of cell death. Correlations between the DNA and albumin binding affinities of chromenopyrimidines were established. We propose that this relationship approach can, for a given set of compounds, assist in predicting the cytotoxicity of potential drug candidates towards MCF-7 cells based on their experimentally determined CT-DNA and BSA binding affinities.
1. Introduction
Breast cancer now accounts for 12.5% of the 2.3 million newly diagnosed cancer cases globally,1 surpassing lung cancer to become the most commonly diagnosed disease worldwide. In 2020, it was the most frequently diagnosed cancer in women worldwide, accounting for 15% of all female cancer cases. The prevalence of this disease is increasing in many regions, especially in developing nations.2 African nations are undergoing continuous economic and political changes however the prevalence of breast cancer which was traditionally low, has increased substantially in recent decades. This is associated with rising cases of obesity and changes in the socio-economic and cultural environments. Studies have shown that many women in resource-constrained nations either don't receive any diagnosis at all, or die because there aren't adequate facilities for early diagnosis, and treatment of breast cancers.3–7 As a result, the real rate of breast cancer in these nations is unknown. Conventional chemotherapy against breast cancer is not entirely successful due to many factors such as late diagnosis,8,9 poor compliance,10 side effects,11 treatment failures due to resistance to currently available drugs.12 For these reasons, more effective chemotherapeutic agents are continuously being sought after.13
Chromenopyrimidines are a class of organic compounds that consist of fused chromene and pyrimidine motifs. They are often synthesized by multicomponent reactions which enable the desired products to be isolated in a few steps via a single-pot operation.14–20 Chromenopyrimidine derivatives have been reported to be light emitters which can be fine-tuned by varying the substituents on the chromenopyrimidine core.14 This the class compounds as potential emitters in optoelectronic devices such as organic light-emitting diodes (OLEDs).21 They have also been studied extensively in medicinal applications as anticancer22–26 and antimicrobial24,27–30 agents. Their medicinal efficacy can be attributed to their binding affinity to biomolecules like deoxyribonucleic acid (DNA). Since pyrimidines form an integral part of the DNA, scientists have attempted to enhance the medicinal efficacy by using novel pyrimidine-based drugs.31–34 DNA-targeted drugs are intended to modulate DNA replication and transcription processes.
A drug's therapeutic potential can also be affected by its binding affinity towards plasma proteins and the stability of the resultant drug-plasma protein adduct in the body.35 Fifty-five percent of all blood proteins are serum albumins, which play an essential role in maintaining proper blood pH and colloidal osmotic pressure. Albumin carries drugs, ions, fatty acids, and hormones that aren't easily dissolved in water.36 Albumins have several pharmacological, biochemical, and therapeutic applications.37 Therefore, a better understanding of the distribution of a drug throughout the body is possible through the study of the interactions that occur between the drug and the proteins found in the plasma. In this regard, bioactive chemical binding properties of bovine serum albumin (BSA), a human serum albumin (HSA) analogue, have received the greatest attention.38
Intrinsic apoptosis (programmed cell death) is caused by intracellular signalling in response to stimuli that do not require receptors, whereas extrinsic apoptosis (programmed cell death) is induced by the attachment of an extracellular ligand to membrane-anchored death receptors.39 To prevent intrinsic pathway-induced apoptosis, the antiapoptotic B-cell lymphoma 2 (Bcl-2) protein plays a critical role by reducing caspase cascade activation.40 Bcl-2 protein expression is often high in cancer cells, which correlates with increased proliferation and reduced apoptosis.41 Scientists have discovered that fused-chromenopyrimidine derivatives' antiproliferative effects are realized through the downregulation of Bcl-2.42,43 By doing so, proapoptotic BCL2-associated X (Bax) and Bcl-2 homologous antagonist/killer (Bak) proteins are brought together in the mitochondrial membrane. Release of cytochrome complex (cyt-c) from mitochondria into the cytoplasm then activates the apoptotic peptidase activating factor (Apaf1) protein. This activates caspase 9, which in turn initiates the caspase cascade via the intrinsic pathway and, finally, promotes apoptosis.41
Developing a model that can predict the cytotoxicity of a potential antineoplastic agent could reduce time and costs for drug discovery protocols. The dawn of the fourth industrial revolution has made it possible to develop predictive models that can estimate the cytotoxicity of test compounds through machine learning and other advanced in silico techniques.44–48 However, there is a possibility of generating false positive predictions when performing virtual screening. It is important to note that the cytotoxicity of a test compound depends not only on the interaction with its targets but also on many other parameters, e.g., interaction with albumins, passage through a cell membrane, and regulation of signal and metabolic pathways, which depend on cell-line-specific cancer mutations and changes in gene expression.48 In this work, we demonstrate that the predictive models can be improved by considering the experimentally-determined binding affinities of the test compounds towards DNA and albumin.
Though there are reports containing experimental DNA- and albumin-small molecule interactions, there are no attempts to relate and predict the efficacy of potential antineoplastic agents using their binding affinity towards DNA or albumin. In our previous work, we demonstrated that the experimental DNA binding affinity of some 2-amino-3-carbonitrile derivatives could be predicted based on Hirshfeld surface and in silico techniques.49 In this work, we sought to chemically modify some of these 2-amino-3-carbonitrile derivatives into a series of chromenopyrimidines and structurally determine the molecular conformations in the solid-state via X-ray crystallography. We were hoping that the chemical modifications would enhance their DNA binding affinities. Further, we investigate the correlation between the DNA and albumin binding affinities of the chromenopyrimidines. Moreover, an approach for predicting in vitro cytotoxicity of novel chromenopyrimidine derivatives towards human breast cancer cells (MCF-7) based on their experimental binding affinity to DNA and albumin, is proposed in this work. The biosafety of selected compounds in human embryonic kidney cells (HEK-293) including the mechanism of cell death is also presented.
2. Results and discussion
2.1. Synthesis and characterization
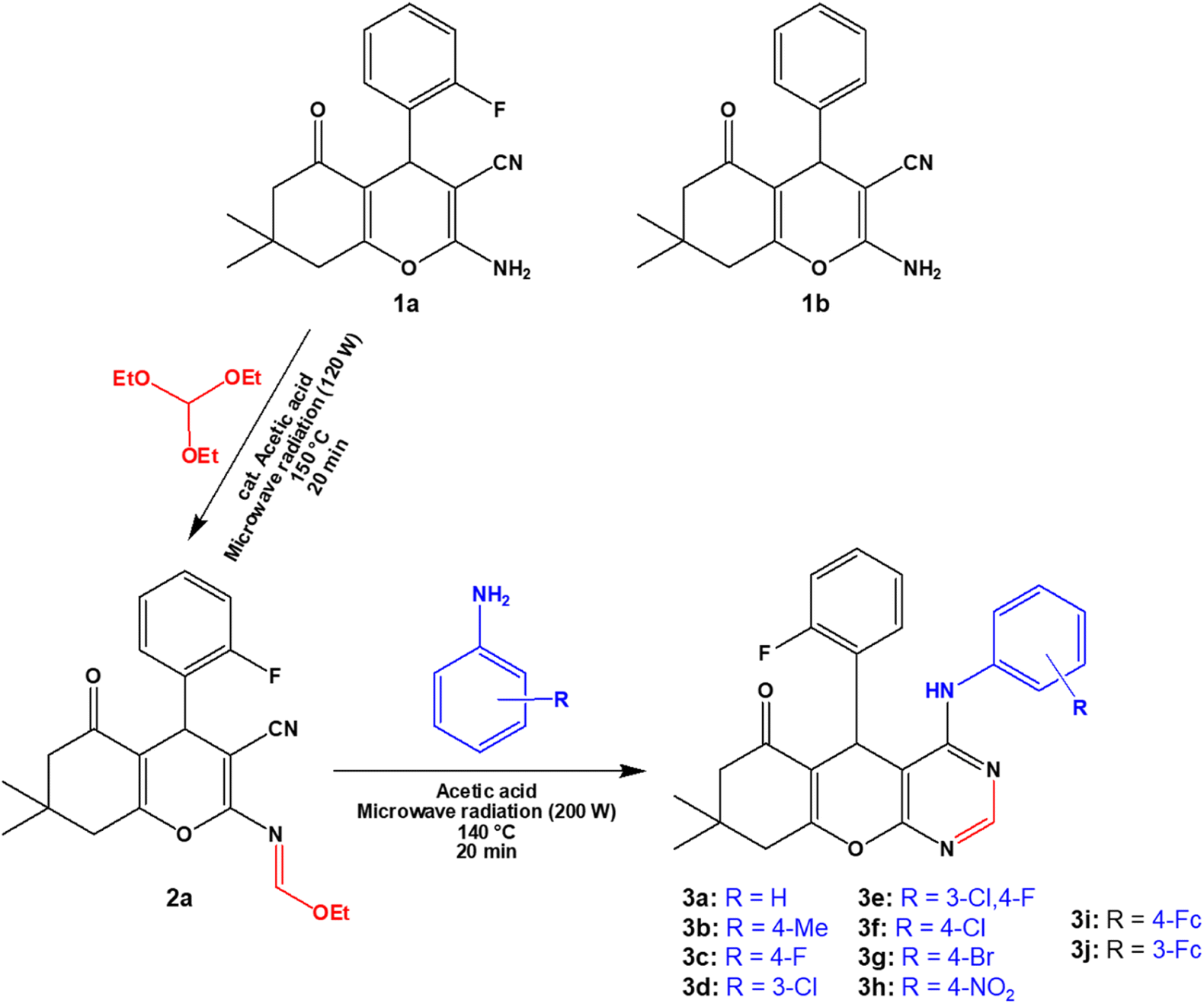
The 4H-pyran-based 2-amino-3-carbonitriles (1a and 1b) and the fluorinated 2-formimidate-3-carbonitrile intermediate (2a), were prepared according to literature.49,50 The microwave-assisted reactions of 2a with corresponding anilines in an acetic acid medium resulted in the formation of chromenopyrimidines (Scheme 1). The reactions were monitored via TLC and the desired products were precipitated from their mother liquor by the addition of water and were isolated in vacuo in moderate to high yields. The formation of compounds 3a to 3j was confirmed by the absence of the band at around 2210 cm−1 in their IR spectra, which attributed to the stretching vibration mode of the cyano functional group.50 A band in the IR spectra of 3a to 3j between 3401 and 3450 cm−1 confirmed the presence of the anilinyl moieties in 3a to 3j. In the 1H-NMR spectra of 3a to 3j, singlets between 8.37 and 9.20 ppm, and between 8.28 and 8.45 ppm attributed to the amino (NH) and pyrimidinyl (Hpyrimidinyl) protons, respectively were observed. These two singlets signified the formation of the pyrimidinyl moiety and the chemical shift of the two signals seem to be directly related (Fig. 1a). The presence of electron-donating groups on the anilinyl moiety in 3b (para-methyl), 3i (para-ferrocenyl), and 3j (meta-ferrocenyl), have a shielding effect on the N–H proton (δNH = 8.37–8.46 ppm) relative to that of the unsubstituted anilinyl derivative, 3a (δNH = 8.49 ppm). Conversely, a deshielding effect is observed in the spectra of compounds with electron-withdrawing substituents on the anilinyl moieties (para-fluoro in 3c; meta-chloro in 3d; meta-chloro, para-fluoro in 3e; para-chloro in 3f; para-bromo in 3g; and para-nitro in 3h) where the N–H proton appears between δN–H = 8.55 and 9.2 ppm. It seems like the δN–H and δC–Hpyrimidinyl values are dependent on the acid dissociation constants (pKa) of the aniline reagents used to synthesize the desired chromenopyrimidine derivatives. Table S1† lists the δN–H, δC–Hpyrimidinyl and the aniline precursors' pKa values. This is also presented in Fig. 1b where a linear inverse correlation exists between pKa and δN–H and between pKa and δC–Hpyrimidinyl. The choice of aniline precursor has a much greater effect on the chemical shift of the N–H proton than on the pyrimidinyl proton, a relation that is observed in the slopes of pKa versus δN–H and pKa versus δC–Hpyrimidinyl. This is due to the close proximity of the anilinyl ring to the amine hydrogen atom than to the pyrimidinyl hydrogen atom. A plot of pKa versus δN–H/δC–Hpyrimidinyl also reveals a linear indirect relationship between the two variables. The implication is that beyond pKa values of ∼5.98, the Hpyrimidinyl proton will be the more deshielded relative to the N–H proton. The 13C NMR spectra of chromenopyrimidines exhibited fewer signals for aliphatic carbons and more signals for the aromatic carbons than compound 1a50 further supporting the formation of the desired products.
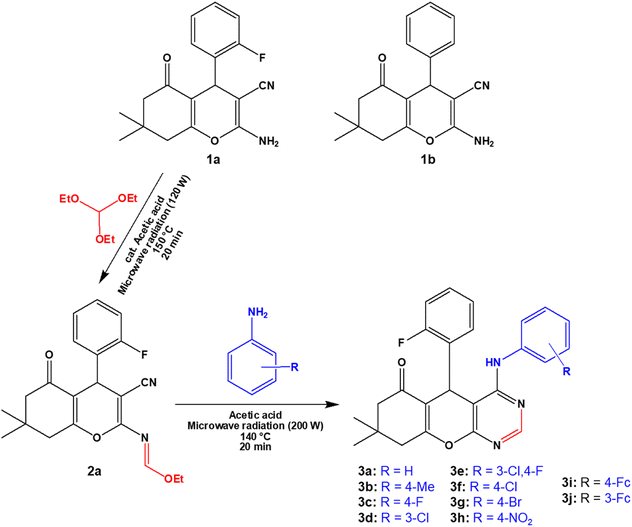 |
| | Scheme 1 Synthesis of chromenopyrimidine derivatives. | |
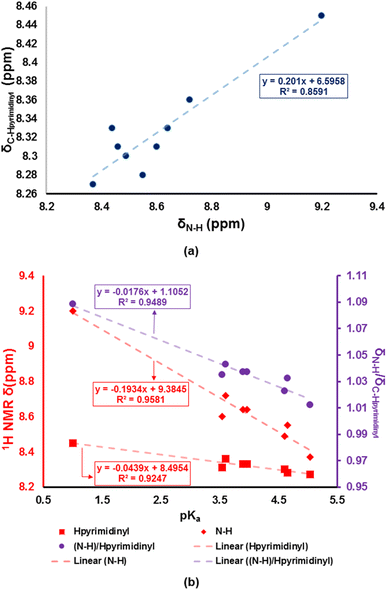 |
| | Fig. 1 Correlations between (a) δN–H and δC–Hpyrimidinyl, (b) pKa and δN–H, and δC–Hpyrimidinyl and δN–H/δC–Hpyrimidinyl. | |
2.2. Crystal structures of chromenopyrimidines
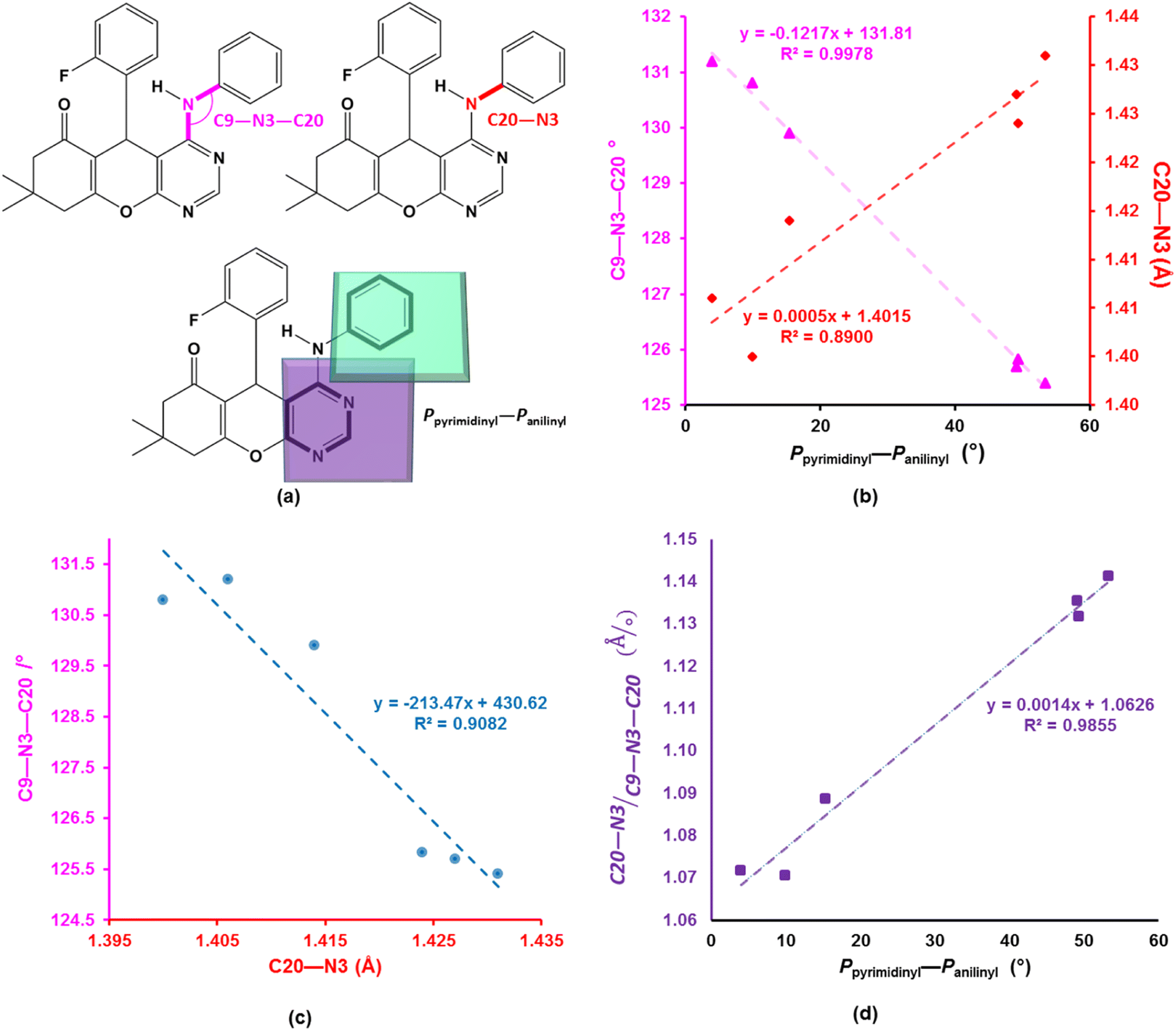
The asymmetric units of compounds 3a to 3c, 3e, and 3h all contain one molecule while that of 3j in addition, an acetone solvate molecule as depicted in Fig. 2. The molecular structures of the compounds consist of a fused ring unit comprising of chromenonyl and pyrimidinyl moieties, onto which 2-fluorophenyl and anilinyl moieties are attached on C7 and C9 atoms, respectively. The 2-fluorophenyl, pyrimidinyl and anilinyl moieties are planar in nature and are denoted P2-fluoro, Ppyridinyl and Panilinyl in this manuscript. The dihedral angles between these three planes were used to describe the molecular conformation and are listed in Table 1. The dihedral angle between the pyrimidinyl and anilinyl moieties (Ppyrimidinyl–Panilinyl) tends towards co-planarity as in 3e, 3.94(5)° and gets larger in 3a, 53.36(5)°. The presence of mono-substituted anilinyl moieties like in 3b, 3c, 3h and 3j, seems to constricts the Ppyrimidinyl–Panilinyl dihedral angle which is not observed in the unsubstituted anilinyl derivative, 3a. The 2-fluorophenyl moiety is almost orthogonal in relation to the 4H-pyran ring which is comparable to that of 1a.49
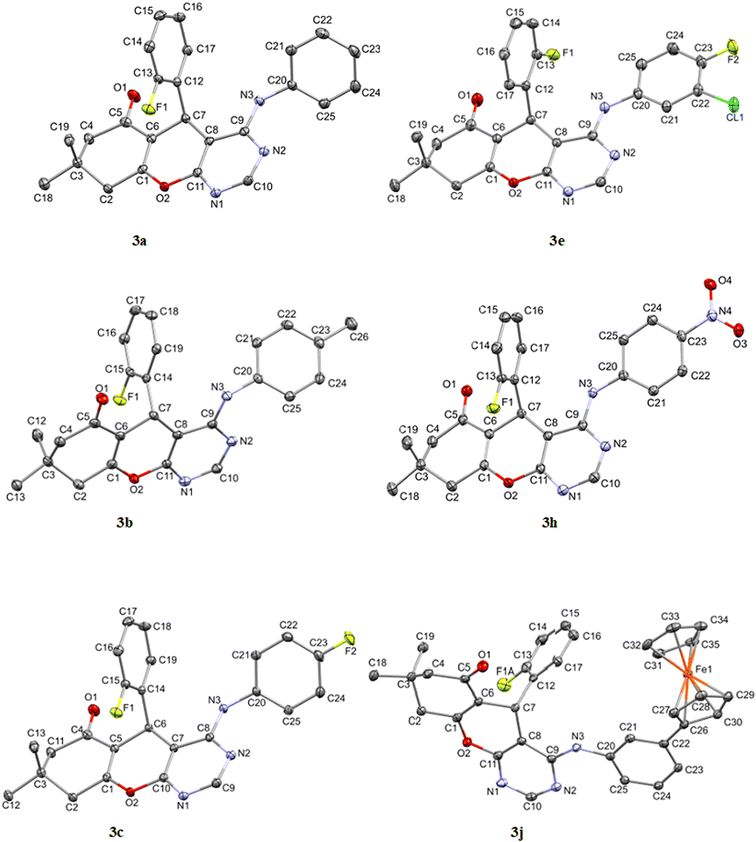 |
| | Fig. 2 Molecular structures of compounds 3a to 3c, 3e, 3h and 3j drawn at 50% thermal ellipsoid probability. All hydrogen atoms have been omitted for clarity. | |
Table 1 Selected geometric parameters observed in 3a to 3c, 3e, 3h and 3j
| Geometric parameter |
Compound |
| 3a |
3b |
3c |
3e |
3h |
3j |
| Dihedral angle |
| Panilinyl–Ppyrimidinyl/° |
53.36(5) |
49.17(5) |
49.40(4) |
3.94(5) |
10.01(4) |
15.38(6) |
| Ppyrimidinyl–P2-fluoro/° |
86.18(4) |
89.78(4) |
86.79(3) |
79.85(4) |
78.40(5) |
78.43(6) |
| Panilinyl–P2-fluoro/° |
69.58(5) |
75.45(5) |
70.53(3) |
83.69(3) |
86.97(5) |
86.33(6) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Bond distances and angles |
| N3–C9/Å |
1.367(2) |
1.363(2) |
1.365(1) |
1.370(2) |
1.366(2) |
1.367(2) |
| N3–C20/Å |
1.431(2) |
1.427(2) |
1.424(1) |
1.406(2) |
1.40(2) |
1.414(2) |
| C20–N3–C9/° |
125.4(1) |
125.7(1) |
125.83(9) |
131.2(1) |
130.1(2) |
129.9(2) |
To better understand the influence of the various anilinyl moieties on the regional molecular conformation of the chromenopyrimidines, we focused on the geometric parameters between the anilinyl and pyrimidinyl moieties. The selected geometric parameters in 3a to 3c, 3e, 3h and 3j are depicted in Fig. 3a and listed in Table 1. The N–C9 distance is significantly shorter than that of N3–C20 which suggests that the pyrimidinyl moiety tends to attract electrons more than the anilinyl moiety. This implies that the pyrimidinyl ring exhibits a greater electropositive potential relative to the anilinyl moiety. The substituents on the anilinyl moiety affect the magnitude of the N3–C20 bond, the C3–N3–C20 bond angle and the Ppyrimidinyl–Panilinyl dihedral angle. There seems to be a direct relationship between the Ppyrimidinyl–Panilinyl dihedral angle and the N3–C20 bond distance, and an inverse relationship between Ppyrimidinyl–Panilinyl and C3–N3–C20 (Fig. 3b and c). Based on Fig. 3d, these relations can all be summed up using the mathematical expression:
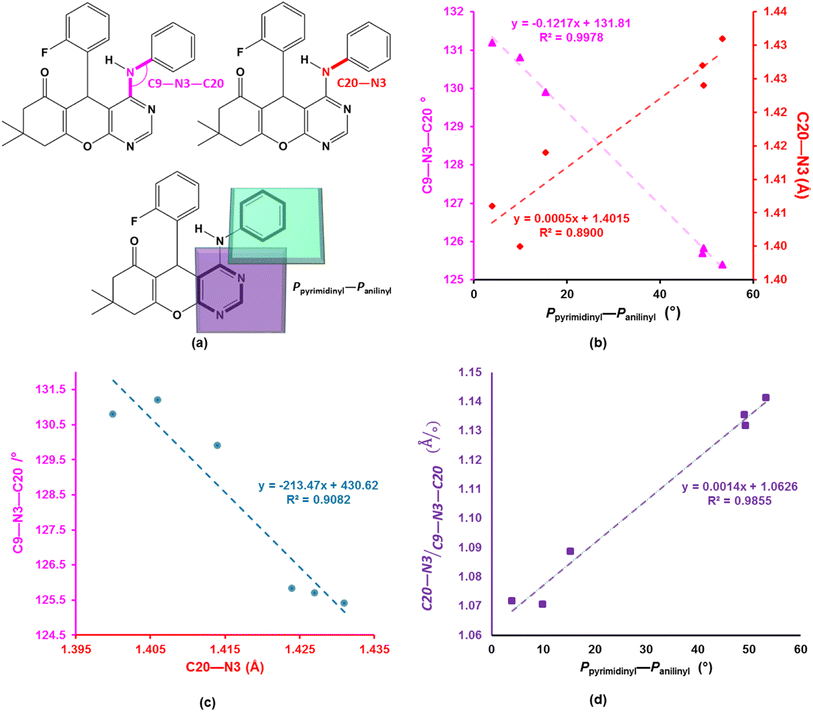 |
| | Fig. 3 (a) Depiction of the selected bond parameters. Scatterplots of (b) Ppyrimidinyl–Panilinyl versus N3–C20 and C9–N3–C20, (c) N3–C20 versus C9–N3–C20 and (d) [(N3–C20)/(C9–N3–C20)] versus Ppyrimidinyl–Panilinyl. The trendlines and equations in (b) have the colour as the corresponding y-axis. | |
The ferrocenyl unit in 3j adorpted a staggered conformation with a C26–Centroid–Centroid–C31 torsion angle of −3.65°. The intermolecular C–H⋯X (where X = N, O or F) hydrogen bonds found in the crystal packing of 3a to 3c, 3e, 3h and 3j, are depicted in Fig. S31 to S34† whilst their parameters are listed in Table S2.†
2.2.1. Molecular electrostatic potential mapping. Molecular electrostatic potential mappings were constructed over the Hirshfeld surfaces of 3a to 3c, 3e, 3h, and 3j using the CrystalExplorer21 software,51 and are illustrated in Fig. 4. Prominent red areas on the mappings were located on the oxygen, nitrogen, fluorine and chlorine atoms which signify intense electronegative potential regions. Conversely, prominent blue areas on the mappings indicated an electropositive potential which is localized around the hydrogen atoms. This further verifies our remark in Section 3.2 about the electropositive character of the pyrimidinyl moiety. On the one hand, compounds with either unsubstituted substituent as in 3a, or electron-donating substituents (as in 3b and 3j), have a lighter shade of red around the aniline moieties indicating an electronegative potential. On the other hand, the presence of electron-withdrawing substituents as in 3c, 3e and 3h, appear to have an electropositive potential on the anilinyl moiety (3e and 3h). A darker shade of blue was evident around the pyrimidinyl moiety than on the anilinyl moiety in all cases, signifying a higher electropositive potential on the pyrimidinyl moiety. These observations can directly be linked to the pKa values of the aniline precursors used (Fig. 4).
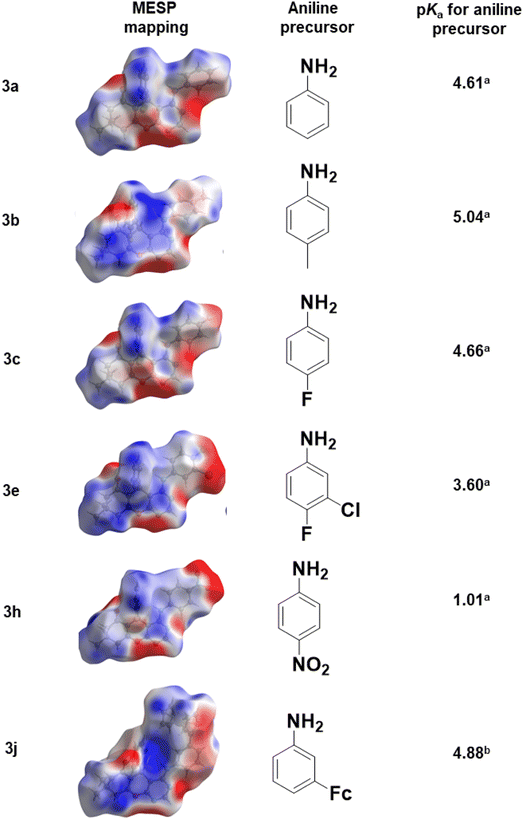 |
| | Fig. 4 Electrostatic potential mapped over the Hirshfeld surfaces of 3a to 3c, 3e, 3h and 3j. | |
2.3. CT-DNA- and albumin-small molecule binding studies
Electronic absorption spectroscopy is one of the commonly used experimental techniques for investigating biomolecule-small molecule interactions due to its availability and sensitivity.52 The changes in the electronic absorption spectral features serve as evidence of the biomolecule's electronic environment perturbation due to biomolecule-small molecule interactions. Importantly, this technique has been used and continues to be used to determine test compound's binding affinity towards DNA53–55 and BSA.53,56–58
Small-molecules are known to interact with DNA in either a covalent or non-covalent manner. The latter interaction mode is more preferred since it causes lesser toxic side effects than the former. Moreover, non-covalent DNA-small molecule interactions can be classified as either intercalation, groove or electrostatic (external) binding. In this work, the CT-DNA-small molecule binding studies were conducted via electronic absorption titration of the various small molecule (3a to 3j) solutions with incremental amounts of CT-DNA (see (a) of Fig. S36–S45 in the ESI†). The absorption spectra of 3a exhibited prominent hypochromism of the bands appearing at 240–295 nm depicting an intercalation mode of binding (Fig. 5a).59 This mode of binding can be related to the interaction between the electronic states of the compounds' chromophores and CT-DNA's nucleotides.31,49,60 The CT-DNA binding affinity of 4H-pyran-based 2-amino-3-carbonitrle derivatives (1a and 1b) compounds was determined in our earlier work.49 As for the BSA-small molecule binding studies, the electronic absorption titration spectra of BSA with incremental amounts of 3a, show distinct hyperchromism of the band at 260–300 nm which signifies the formation of BSA-small molecule complex (Fig. 5b). The rest of the compounds studied in this work, displayed similar electronic absorption titration behaviour as 3a (Fig. S44 and see (b) of Fig. S36–44†).
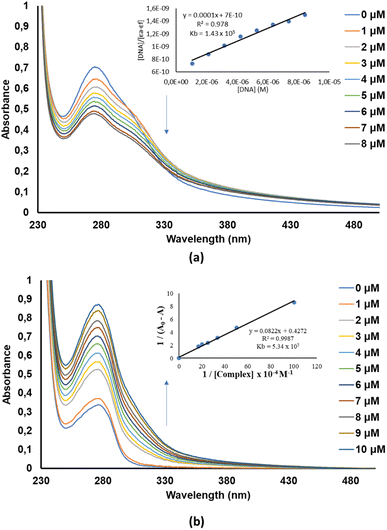 |
| | Fig. 5 (a) Electronic absorption spectrum of 3a in the absence and presence of increasing amounts of CT-DNA. (b) Electronic absorption spectrum of BSA in the absence and presence of increasing amounts of 3a. | |
The binding constants of 1a, 1b, and 3a to 3j towards CT-DNA (KDNA) and BSA (KBSA) were determined using the Wolf-shimmer and Benesi–Hildebrand equations, respectively (Fig. 6). For 4H-pyran-based 2-amino-3-carbonitrile derivatives, the presence of the 2-fluorophenyl moiety (in 1a) lowers the KDNA and KBSA values relative to the non-fluorinated derivative (1b). The electronic properties and position of the substituents on the anilinyl moiety of 3a to 3j influences the KDNA and KBSA values. Having an electron-donating methyl substituent on the anilinyl moiety (in 3b) enhances KDNA but decreases the KBSA value by three-fold in relation to the unsubstituted anilinyl derivative (3a). Replacing the substituents on the anilinyl moiety with ferrocene leads to a greater electron-donating ability due to the resonance effect which increases the KDNA value. Furthermore, the position of the ferrocenyl substituent also plays a role where being in the meta-position of the anilinyl (in 3j) leads to a higher KDNA and lower KBSA values than having it in the para-position (in 3i). For anilinyl moieties with halogen substituents, the highest KDNA value obtained was 4.50 × 105 M−1 for the para-fluoro chromenopyrimidine derivative (3c).
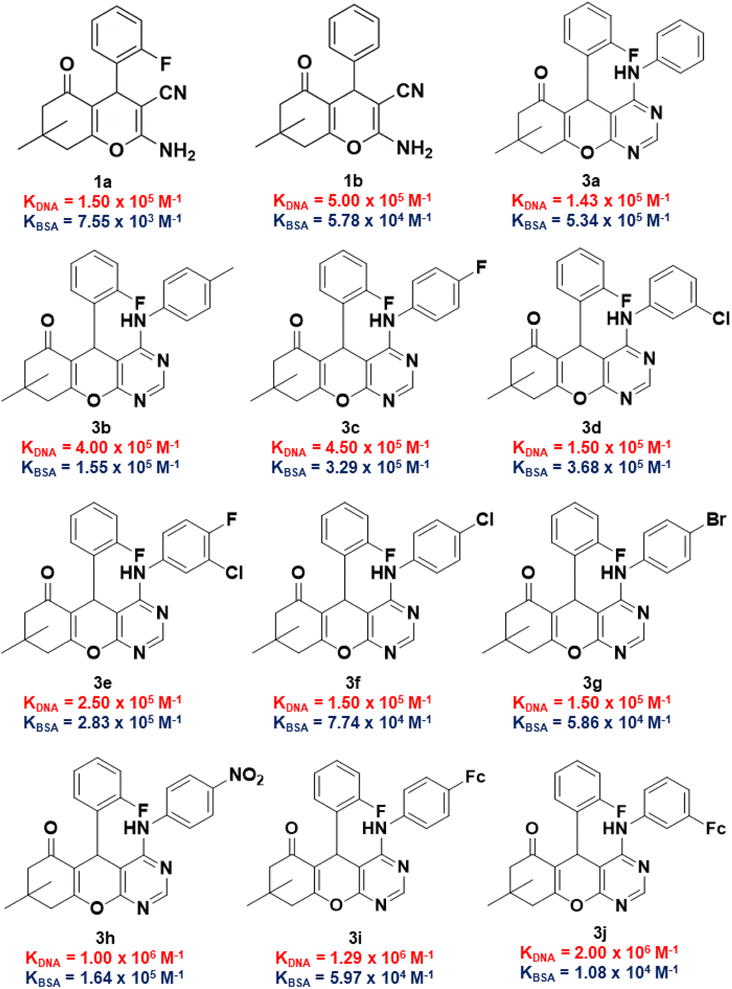 |
| | Fig. 6 KDNA and KBSA values of 2-amino-3-carbonitrile-based intermediates and fluorinated chromenopyrimidine derivatives in this work. | |
The position of the chloro-substituent (in 3d and 3f), did not seem to affect the KDNA value. Also, an increase in the atomic radii of the halogen from chloro- (in 3f) to bromo- (in 3g) and the resultant electronic properties of the corresponding anilinyl moieties did not affect the binding constants much. This could be attributed to the characteristic σ-hole of the chloro- and bromo-substituents which may exhibit halogen bonding. The presence of a para-nitro substituent on the anilinyl moiety significantly increases the KDNA value from (1.50 × 105–4.50 × 105 M−1) to 1.00 × 106 M−1 and this could be attributed to its enhanced electron-withdrawing via resonance. This observation is consistent with that of the literature,61,62 where the effect of a para-nitro substituent on high DNA binding were identified among other EWG and EDG. Although it is unknown how substituents influence DNA binding,63 their planarity due to the position of the nitro group could have led to their ease of accessing the DNA base pair pocket where intercalation with the DNA occurs.64 The KBSA values for compounds containing electron-withdrawing substituents was found to increase in the following order: 5.86 × 104 M−1 (3g) < 7.74 × 104 M−1 (3f) < 1.64 × 105 M−1 (3h) < 2.83 × 105 M−1 (3e) < 3.29 × 105 M−1 (3c) < 3.68 × 105 M−1 (3d).
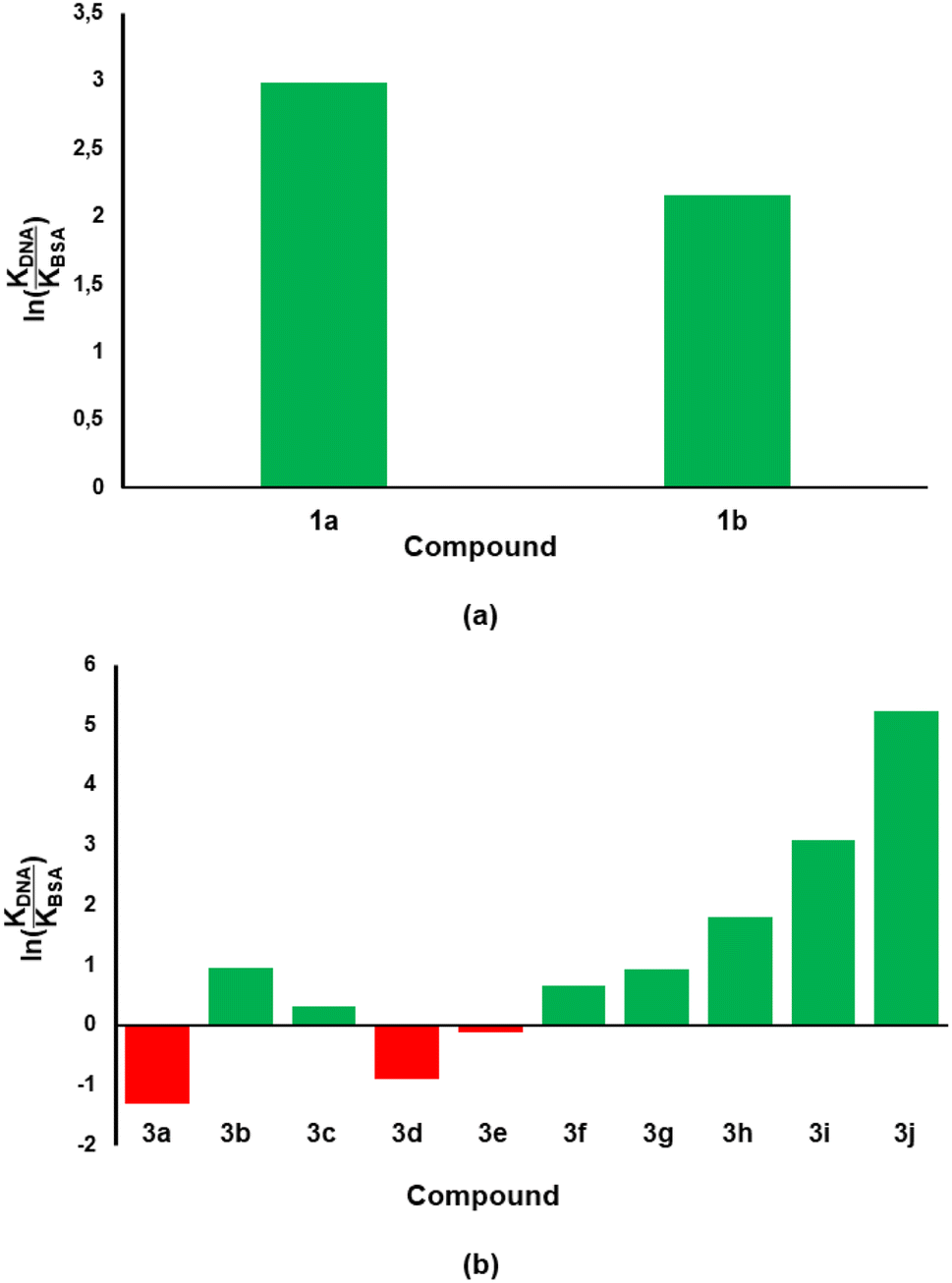
Considering that the ultimate target of our compounds is DNA, the binding constant (KDNA) needs to be higher than that of BSA (KBSA). Thus, an indicator that includes both biomolecules would be considered better with regards to biological activity and/or stability of test compounds. With this argument, we took the natural logarithm of the ratio of binding constant towards CT-DNA relative to that of BSA [ln(KDNA/KBSA)] as the indicator which defines the small molecule's relative stability towards the biomolecules. Compound 1a was shown to have a ln(KDNA/KBSA) value that is ten times lower than that of 1b (Fig. 7a). The lowest and highest ln(KDNA/KBSA) values for the chromenopyrimidines in this work (Fig. 7b), were observed in 3a and 3j, respectively.
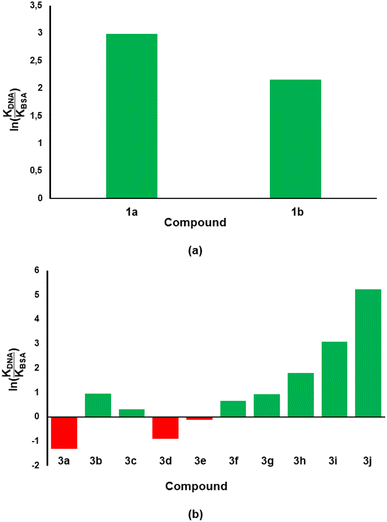 |
| | Fig. 7 The ln(KDNA/KBSA) values for (a) 2-amino-3-carbonitrile and (b) chromenopyrimidine derivatives in this work. The green- and red-coloured bars indicate positive and negative ln(KDNA/KBSA) values, respectively. | |
There seems to be a relationship between the binding constants of the 3a–3j to the DNA and BSA (Fig. S46a†). Compounds 3f and 3g with the para-chloro and para-bromo substituents are outliers possibly due to the characteristic σ-hole of the substituents which are possible involved in halogen bonding. A different way of looking at this is to also determine the Gibbs free energy of binding of the test compounds to BSA (here denoted as ΔGBSA) using eqn (1) where R is the gas constant (8.314 J mol−1 K−1) and T is the temperature at which the binding affinity studies were conducted (298 K).
| | |
ΔGBSA = −RTlnKBSA
| (1) |
An inverse trend between the two variables was observed which signifies that chromenopyrimidines with high binding affinity towards DNA, generally had low binding affinity towards BSA (Fig. S46b†).
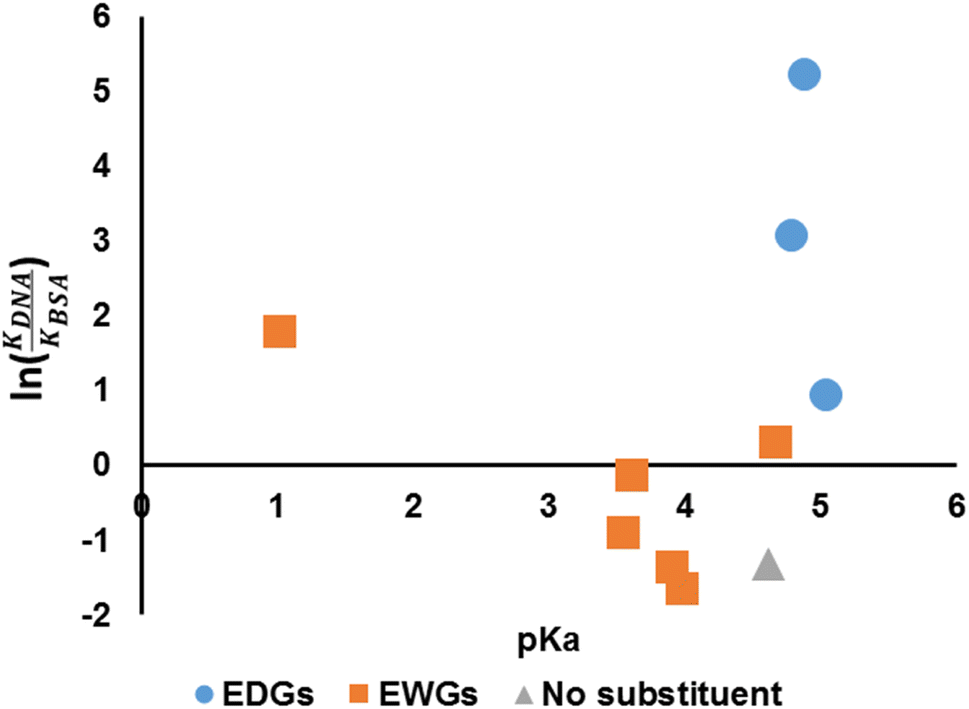
We have established that the spectroscopic behaviours of the compounds are affected by the choices of the aniline (hence pKa) and so is the binding affinity. Hence, it is of interest to investigate whether the pKa of the anilines could be related to the suggested indicator, ln(KDNA/KBSA) (Fig. 8). It would appear that the value of ln(KDNA/KBSA) rises for anilines that contain electron withdrawing groups (EWGs) whenever the acidity of the aniline precursor rises in comparison to the non-substituted. Anilines containing electron donating groups (EDGs) demonstrated greater ln(KDNA/KBSA) values when the basicity of the aniline reagent was increased.
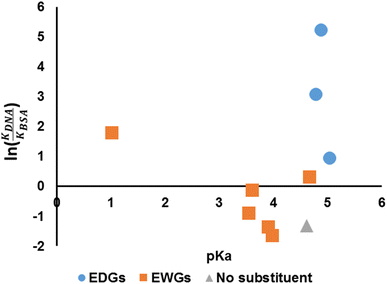 |
| | Fig. 8 pKa versus ln(KDNA/KBSA). | |
2.4. 4H-Pyran-based 2-amino-3-carbonitriles and chromenopyrimidine derivatives exhibit anticancer potential in vitro via apoptosis and biological safety to non-cancerous cells
It has previously been reported that 4H-pyran-based 2-amino-3-carbonitriles and chromenopyrimidine derivatives have antiproliferative effects on breast cancer cells-lines.65 Therefore, to determine whether chemical modifications of these derived compounds retained and enhanced antiproliferative effects, the potential of these compounds to cause significant cytotoxicity and induce programmed cell death in breast cancer cells was investigated in vitro. The 4H-pyran-based 2-amino-3-carbonitriles (1a and 1b) and two chromenopyrimidine derivatives (3a and 3j) were used for the cytotoxicity studies. The motivation for selecting 3a and 3j for performing the cytotoxicity studies is based on their binding constants towards CT-DNA and BSA using the relation, ln(KDNA/KBSA). Since 3a had the lowest ln(KDNA/KBSA) value in this work, it was hypothesized that it will have a lower cytotoxicity towards MCF-7 than 3j, which had the highest ln(KDNA/KBSA) value.
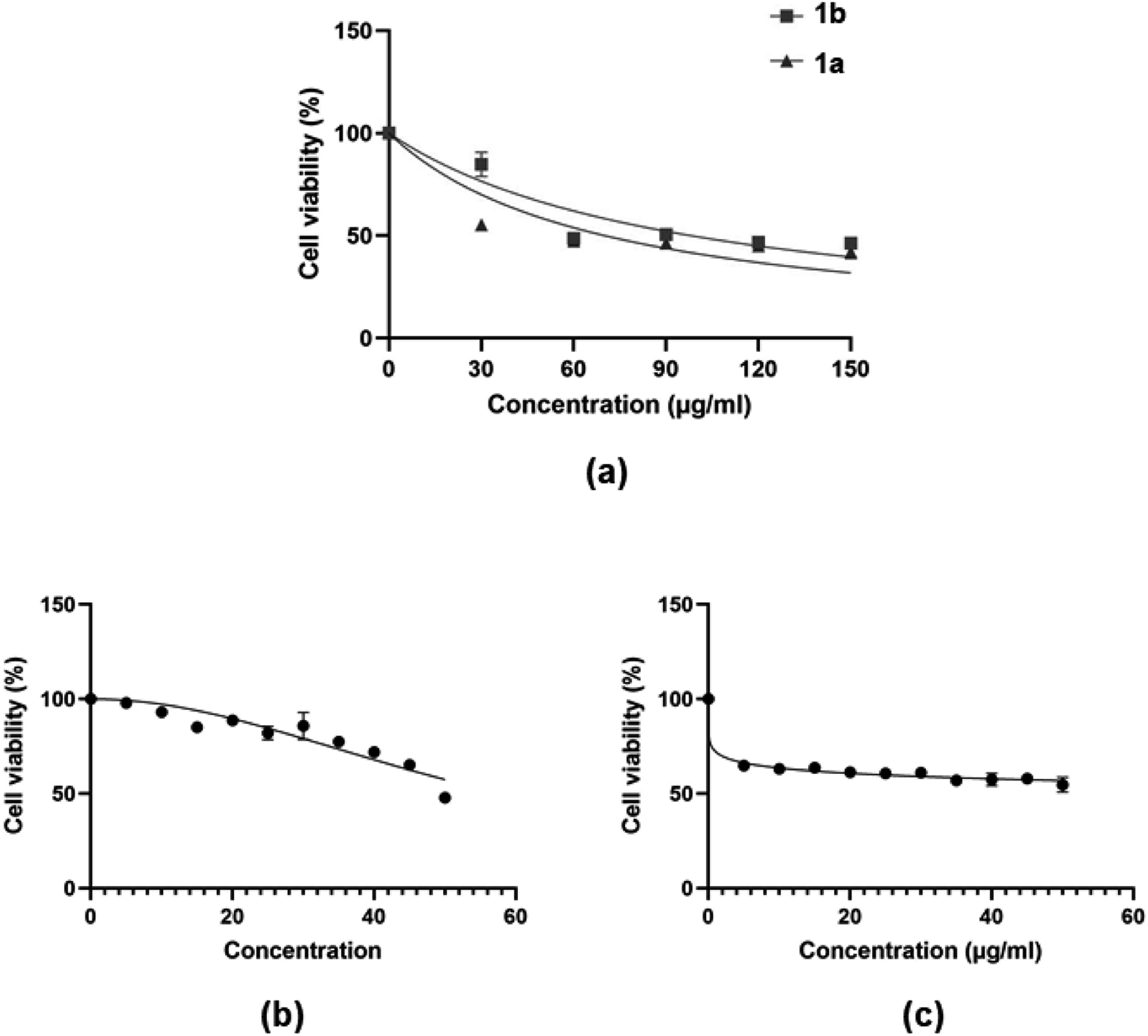
2.4.1. Cytotoxicity of compounds to MCF-7 breast cancer cell-lines. The MTT assay was used to determine MCF-7 cell viability after exposure to 4H-pyran-based 2-amino-3-carbonitriles (1a and 1b) and chromenopyrimidine derivatives (3a and 3j) (Fig. 9). The viability of MCF-7 cells were determined by calculating the IC50 values of each compound (Table 2). The assay also determined the ratios of the reducing to the oxidizing equivalents, in the cell, an indicator of the metabolic activity of the mitochondrial electron transport chain.66
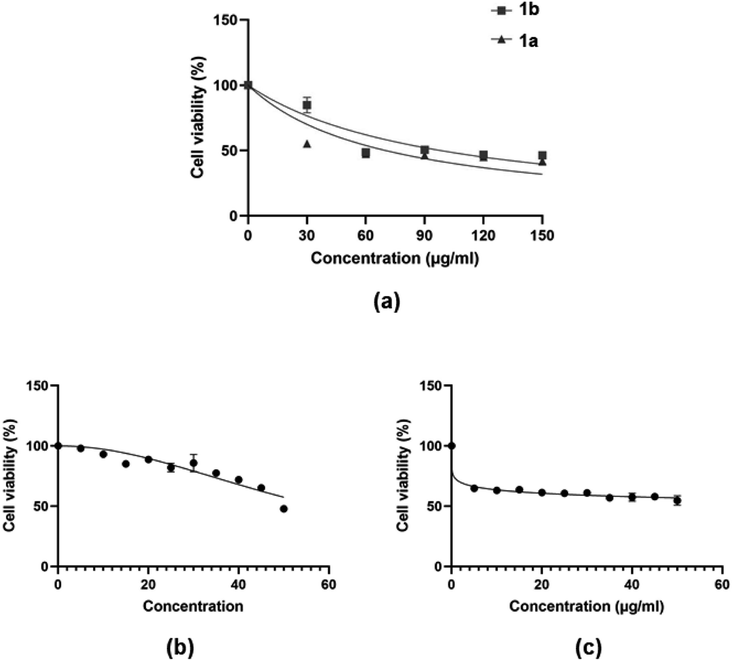 |
| | Fig. 9 Dose-dependent MCF-7 cell viability as determined MTT assay after the cells were exposed to serial concentrations of compounds (a) 1a and 1b; and (b and c) 3a and 3j. | |
Table 2 IC50 values of the test compounds towards MCF-7 cells
| Compound |
IC50 (MCF-7)/μM |
| 1a |
225 ± 4.43 |
| 1b |
334 ± 5.01 |
| 3a |
595 ± 7.14 |
| 3j |
97 ± 2.04 |
| Vinoblastine67 |
10.18 ± 0.39 |
The justification for performing cytotoxicity studies of compounds 1a and 1b was to investigate whether having a high KDNA value would translates to higher antiproliferative effect towards MCF-7 cells. Interestingly, compound 1a showed reduced viability of MCF-7 cells compared to 1b, indicated by IC50 values of 225 ± 4.43 μM and 334 ± 5.01 μM, respectively, despite its higher binding affinity towards DNA relative to 1a. This further justifies the need to factor in the BSA binding affinity when assessing the antiproliferative potential of the compounds. The chemical modifications of 1a to 3a, lead to a higher IC50 of 595 ± 7.14 μM and this suggests that the formation of the fused pyrimidine ring with an unsubstituted anilinyl moiety decreases the cytotoxicity to MCF-7 cells. However, introducing a para-ferrocenyl substituent on the anilinyl moiety significantly enhanced cytotoxicity to MCF-7 cells with an IC50 value of 97 ± 2.04 μM. Although the test compounds in this work exhibit lesser cytotoxicity than the known breast cancer antineoplastic drug, vinblastine (10.18 ± 0.39 μM),67 we established that the IC50 values of the test compounds towards MCF-7 cells exhibit an inverse relationship with respect to ln(KDNA/KBSA) values (Fig. 10); thus, validating our hypothesis.
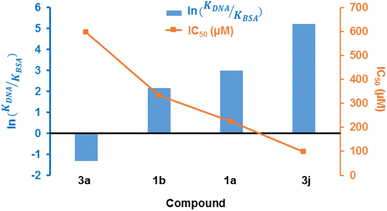 |
| | Fig. 10 Combined line and bar graphs of ln(KDNA/KBSA) and IC50 values of 1a, 1b, 3a and 3j. | |
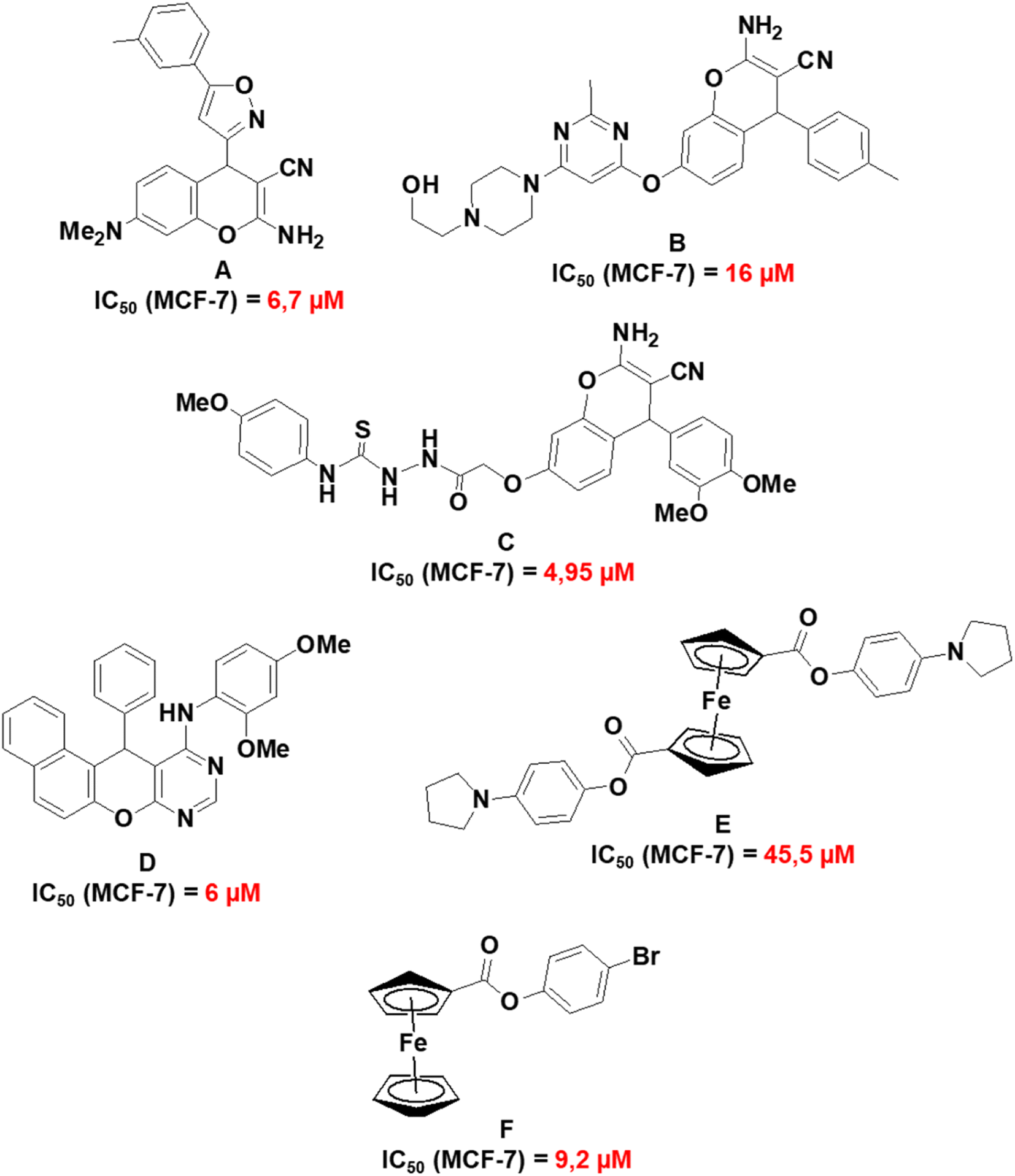
Fig. 11 presents a summary of previously reported IC50 values of related compounds which include 2-amino-7-(dimethylamino)-4-(5-m-tolylisoxazol-3-yl)-4H-chromene-3-carbonitrile (A);68 2-amino-7-(6-(4-(2-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-yloxy)-4-p-tolyl-4H-chromene-3-carbonitrile (B);69 2-(2-(2-amino-3-cyano-4-(3,4-dimethoxyphenyl)-4H-chromen-7-yloxy)acetyl)-N-(4-methoxyphenyl)hydrazinecarbothioamide (C);67 N-(2,4-dimethoxyphenyl)-12-phenyl-12H-naphtho[1′,2′:5,6]pyrano[2,3-d]pyrimidin-11-amine (D);70 1,1′-4-(1H-pyrrol-1-yl)phenyl ferrocenedicarboxylate (E)71 and 4-bromophenyl ferrocenecarboxylate (F).72 Compounds A to F exhibited the highest cytotoxicity towards MCF-7 cell lines in their respective studies and this provides a comparative perspective on the cytotoxic activities of 1a, 1b, 3a, and 3j. Compounds 1a and 1b have reduced cytotoxicity towards MCF-7 (148–159 μM) in comparison to compounds A to C (4.95–16 μM). A to C have a fused pyran-phenyl moiety, different from the fused pyran-cyclohexane moiety in 1a and 1b which has an enhanced electron delocalized system. As for the chromenopyrimidines, compound D exhibits much greater cytotoxic activity against MCF-7 cells (IC50 = 6 μM) than 3a and 3j (97–595 μM). Nevertheless, no investigations on the cytotoxic effects of compound D were conducted on any normal human cell lines.70 The selective cytotoxicity of compound D towards MCF-7 cells necessitates further investigation. It is noteworthy that compounds E and F, which are based on ferrocene, demonstrate higher IC50 concentrations against MCF-7 cells (ranging from 9.2 to 45.5 μM) in comparison to D. However, their IC50 concentrations towards the normal human mammary epithelial cell line (MCF-10A) exceed 250 μM. The observation underscores the cytotoxic selectivity exhibited by compounds E and F towards MCF-7 cells.
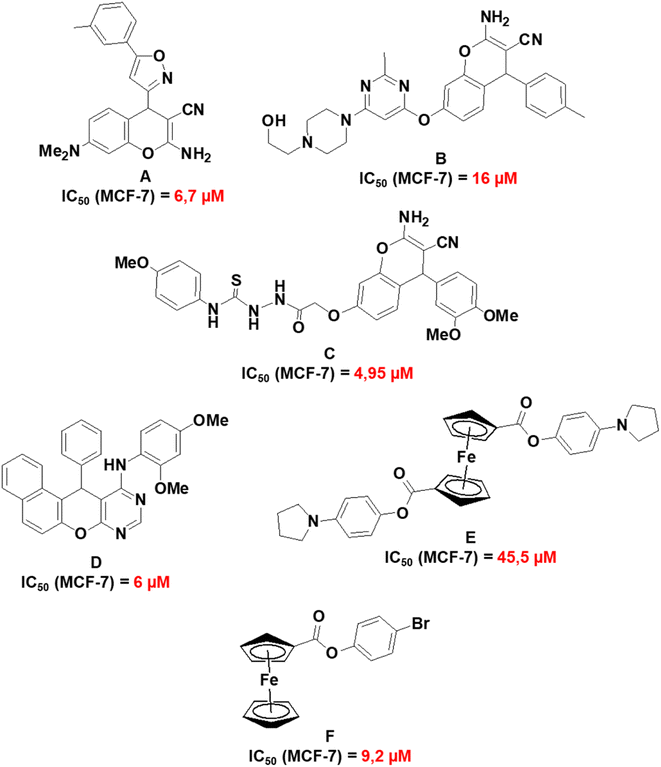 |
| | Fig. 11 Molecular structures and IC50 values of related compounds in the literature. | |
2.4.2. Cytotoxicity. Cancer cells are characterized by a loss of apoptotic control, which supports their prolonged survival and metastasis.6,39,73 Therefore, the desired mechanism of cell death induced by a proliferative compound is programmed cell death, or apoptosis.6,39,73 Therefore, after determining the antiproliferative effects of 1a, 1b, 3a and 3j on breast cancer cells via the MTT assay, apoptotic induction was assessed using the IC50 concentration of each test compound. The detection of apoptosis in MCF-7 cells were investigated by determining the activation of caspase 3/7, which is the executioner caspase of apoptosis. This study showed that 1a caused a significantly higher level of caspase 3/7 activation in MCF-7 cells (p < 0.0001), relative to 1b (Fig. 12a). Also, 3j caused a significantly higher level of caspase 3/7 activation in MCF-7 cells (p < 0.05), compared to 3a and the control, respectively (Fig. 12b). This correlates to the results obtained in the MTT assays where 1a and 3j caused lower viability compared to 1b and 3a, respectively. These results suggest that 1a and 3j halted cell proliferation through apoptosis and not necrosis.
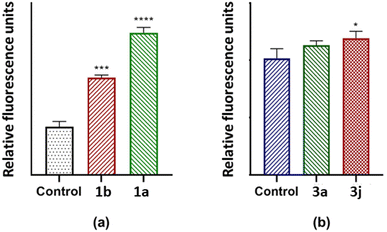 |
| | Fig. 12 Caspase 3/7 activity after MCF-7 cells were exposed to 70.21; 98.21 247.3 and 57.99 μg mL−1 for (a) 1a, 1b, (b) 3a and 3j, respectively. *p < 0.05 relative to control. ***p < 0.001 relative to control. ****p < 0.0001 relative to control. | |
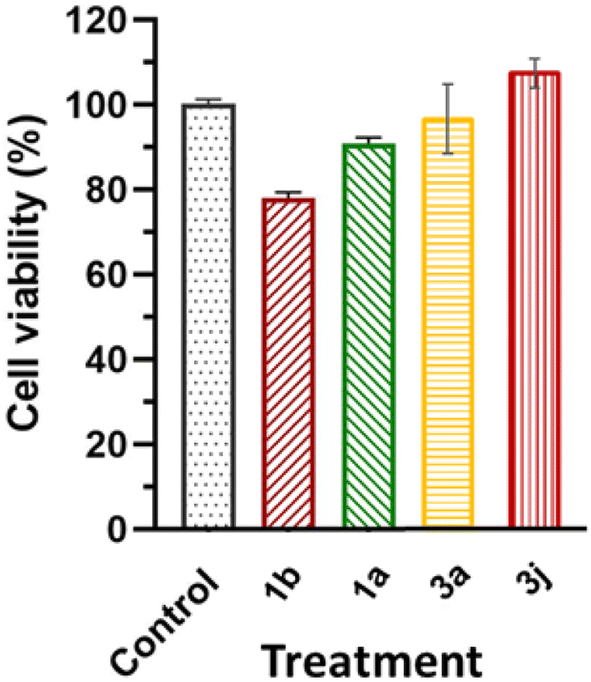
2.4.3. Selective toxicity. The effects of compounds 1a, 1b, 3a and 3j at their respective IC50 concentrations (in MCF-7 cell lines) on HEK-293 cells resulted in 89.6%, 77.6%, 96.6% and 107.43% cell viability, respectively (Fig. 13). The cell viability results reported in this study are within the limits of the International Organization for Standardization for Biomedical Material Biosafety of requires ≤30% reduction in cell viability (International Organization for Standardization, 2018).
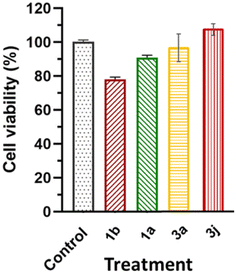 |
| | Fig. 13 The effects of 1a, 1b, 3a and 3j at their respective IC50 concentrations and a control on the cell viability of non-cancerous HEK-293 cells. | |
3. Conclusion
The microwave-assisted synthesis of chromenopyrimidine derivatives at moderate to high yields was successful. Spectroscopic IR and NMR data confirmed the formation of the pyrimidinyl moiety and the desired products. The chemical shifts of the Hpyrimidinyl and N–H protons were found to have an inverse relationship with the pKa of the corresponding aniline precursor. The molecular conformation of chromenopyrimidine derivatives in the solid-state was found to be affected by the electronic nature of the substituent on the anilinyl moiety. Upon closer inspection, correlations between the geometric parameters around the anilinyl and pyrimidinyl moieties were etablished in this work. Based on the electrostatic potential property of the Hirshfeld surface, the pyrimidinyl moiety has an electropositive potential relative to the anilinyl moiety. This resulted in shorter C9–N3 than C20–N3 distances in the crystal structures of chromenopyrimidine derivatives. The binding affinity of the 4H-pyran-based 2-amino-3-carbonitriles and chromenopyrimidine derivatives towards CT-DNA and BSA were determined via electronic absorption spectroscopy. An unprecented inverse relationship was established between KDNA and ΔGBSA for chromenopyrimidine derivatives in this work. The compound's relative stability towards the two biomolecules was defined as ln(KDNA/KBSA) and it was hypothesized that higher ln(KDNA/KBSA) values lead to greater cytotoxicity towards MCF-7 cells. So, compounds with the lowest and highest ln(KDNA/KBSA) values from the 4H-pyran-based 2-amino-3-carbonitriles (1a and 1b) and chromenopyrimidine derivatives (3a and 3j) were selected for the cytotoxicity studies. Interestingly, the pKa of the aniline precursor could be linked to the ln(KDNA/KBSA) value of the corresponding chromenopyrimidine derivative. Compounds 1a and 3j produced lower IC50 values than their respective counterpart and exhibited more potency against MCF-7 cells without presenting toxicity to non-cancerous HEK-293 cells. This suggests a potential targeted effect of the test compounds to cause selective toxicity to MCF-7 cells. Furthermore, the mechanism of cell death by the compounds in MCF-7 cells was confirmed to be apoptosis, with the 1a and 3j showing higher apoptotic activation in cancer cells than their respective counterpart. Importantly, an indirect trend was established between the ln(KDNA/KBSA) and IC50 values which validates our hypothesis. With these findings in mind, cytotoxicity of potential drug candidates towards MCF-7 can be predicted based on their experimentally determined CT-DNA and BSA binding affinities. Thus, lowers costs and improving the preclinical drug discovery protocols. Our ongoing work involves investigating the test compounds' binding site in the two biomolecules, and attempting to draw correlations with the is part of our ongoing work.
4. Experimental
4.1. Materials and methods
All chemicals used in the syntheses of target molecules were of reagent grade purchased from commercial sources. These included: ethanol, methanol, triethyl orthoformate, acetic acid, aniline, 4-fluoroaniline, 3-chloro-4-fluoroaniline, 4-chloroaniline, 4-methylaniline, 3-chloroaniline, 4-bromoaniline, 4-nitroaniline and DMSO-d6. The 2-amino-4-(aryl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (1a and 1b)49 and ethyl-N-(3-cyano-4-(2-fluorophenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromen-2-yl)formimidate precursors (2a)50 including 3-ferrocenylaniline and 4-ferrocenylaniline were prepared according to literature.74 The microwave syntheses were done using a CEM Discover system. All reactions were done in 30 mL pressurized vials fitted with “snap-on” caps. Uncorrected melting points were measured using a Stuart SMP3 melting point apparatus. Infrared spectra were recorded from 400 to 4000 cm−1 using a PerkinElmer Universal ATR Spectrum 100 spectrometer. The 1H and 13C NMR spectra were recorded using Bruker 400 MHz spectrometer. The reported chemical shift values are in parts per million (ppm) relative to the solvent residual peak which are 2.50 and 39.5 ppm for 1H and 13C NMR, respectively. Electronic absorption spectra were recorded using the Shimadzu UV-vis spectrophotometer. High-resolution mass spectra obtained on a SHIMADZU-LCMS-2020 and the elemental composition was determined done using the Vario EL cube. MCF-7 cell lines and Caspase 3/7 fluorometric assay kit were purchased from Cellonex® (Johannesburg South Africa) and Biocom® (Johannesburg, South Africa). For caspase 3/7 detection, fluorescence (excitation/emission of 535/620 nm) was measured using a microplate reader (Spectrostar Nano, Germany).
4.2. General procedure for chromenopyrimidine derivatives (3a to 3j)
A 10 mL acetic acid solution containing 2a (1 mmol) and the corresponding aniline (1.2 mmol) was added to a sealed 30 mL pressurized vial. The mixture was irradiated at 200 W in a single-mode microwave synthesis system. The reaction temperature was set at 140 °C for a duration of 20 minutes. Once the reaction was completed as confirmed using TLC, distilled water was gently added to the mixture without stirring. This formed a turbid layer over the reaction mixture which was left to stand overnight. The crude product had precipitated out of the mixture and was filtered in vacuo, washed with distilled water and recrystallized from mixture of ethanol and water.
4.2.1. 5-(2-Fluorophenyl)-8,8-dimethyl-4-(phenylamino)-5,7,8,9-tetrahydro-6H-chromeno[2,3-d]pyrimidin-6-one (3a). Aniline was used as a precursor to synthesize 3a. White solid, yield: 78% (0.32 g); m.p.: 244–246 °C, selected IR bands (cm−1): 3407 (N–H stretching), 2961 (C–H stretching), 1651 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching); 1H NMR (DMSO-d6) δ(ppm): 0.94 (s, 3H, CH3), 1.08 (s, 3H, CH3) 2.12–2.14 (d, 1H, J = 16.1 Hz, Hmethylene), 2.33–2.36 (d, 1H, J = 16.1 Hz, Hmethylene), 2.56–2.59 (d, 1H, J = 17.6 Hz, Hmethylene), 2.69–2.72 (d, H, J = 17.5 Hz, Hmethylene), 5.56 (s, 1H, Hmethine), 7.00–7.02 (t, 1H, J = 7.4 Hz, Haromatic), 7.03–7.07 (overlapping doublets, 1H, J = 8.4 and 8.5 Hz, Haromatic), 7.10–7.12 (t, 1H, J = 7.1 Hz, Haromatic) 7.19–7.20 (q, 1H, J = 6.9 Hz, Haromatic), 7.25–7.28 (t, 2H, J = 7.9 Hz, Haromatic), 7.54–7.55 (d, 2H, Haromatic, J = 7.8 Hz), 7.75–7.78 (t, 1H, Haromatic, J = 7.1 Hz), 8.30 (s, 1H, Hpyrimidinyl), 8.49 (s, 1H, –N(H)–); 13C NMR (DMSO-d6) δ(ppm): 26.7, 28.7, 29.1, 32.4, 50.4, 98.6, 112.2, 116.3, 116.4, 121.5, 123.6, 124.5, 128.9, 129.2, 129.7, 132.2, 139.6, 156.6, 159.3, 161.9, 164.9, 196.2; ESI(+)MS (m/z): 416 [M + H]+; anal. calcd (%) for [C25H22FN3O2]: C, 72.27; H, 5.34; N, 10.11; O, 7.70; found (%): C, 71.91; H, 5.31; N, 10.06; O, 7.66.
O stretching); 1H NMR (DMSO-d6) δ(ppm): 0.94 (s, 3H, CH3), 1.08 (s, 3H, CH3) 2.12–2.14 (d, 1H, J = 16.1 Hz, Hmethylene), 2.33–2.36 (d, 1H, J = 16.1 Hz, Hmethylene), 2.56–2.59 (d, 1H, J = 17.6 Hz, Hmethylene), 2.69–2.72 (d, H, J = 17.5 Hz, Hmethylene), 5.56 (s, 1H, Hmethine), 7.00–7.02 (t, 1H, J = 7.4 Hz, Haromatic), 7.03–7.07 (overlapping doublets, 1H, J = 8.4 and 8.5 Hz, Haromatic), 7.10–7.12 (t, 1H, J = 7.1 Hz, Haromatic) 7.19–7.20 (q, 1H, J = 6.9 Hz, Haromatic), 7.25–7.28 (t, 2H, J = 7.9 Hz, Haromatic), 7.54–7.55 (d, 2H, Haromatic, J = 7.8 Hz), 7.75–7.78 (t, 1H, Haromatic, J = 7.1 Hz), 8.30 (s, 1H, Hpyrimidinyl), 8.49 (s, 1H, –N(H)–); 13C NMR (DMSO-d6) δ(ppm): 26.7, 28.7, 29.1, 32.4, 50.4, 98.6, 112.2, 116.3, 116.4, 121.5, 123.6, 124.5, 128.9, 129.2, 129.7, 132.2, 139.6, 156.6, 159.3, 161.9, 164.9, 196.2; ESI(+)MS (m/z): 416 [M + H]+; anal. calcd (%) for [C25H22FN3O2]: C, 72.27; H, 5.34; N, 10.11; O, 7.70; found (%): C, 71.91; H, 5.31; N, 10.06; O, 7.66.
4.2.2. 5-(2-Fluorophenyl)-8,8-dimethyl-4-(p-tolylamino)-5,7,8,9-tetrahydro-6H-chromeno[2,3-d]pyrimidin-6-one (3b). 4-Methylaniline was used as a precursor to synthesize 3b. Pale yellow solid; yield: 88% (0.38 g); m.p.: 234–238 °C; selected IR bands (cm−1): 3410 (N–H stretching), 2964 (C–H stretching), 1653 (CO stretching); 1H NMR (DMSO-d6) δ(ppm): 0.94 (s, 3H, CH3), 1.08 (s, 3H, CH3) 2.11–2.15 (d, 1H, J = 16.2 Hz, Hmethylene), 2.23 (s, 3H, p-CH3), 2.32–2.36 (d, 1H, J = 16.2 Hz, Hmethylene), 2.55–2.59 (d, 1H, J = 17.6 Hz, Hmethylene), 2.68–2.72 (d, H, J = 17.6 Hz, Hmethylene), 5.52 (s, 1H, Hmethine), 7.02–7.12 (m, 4H, Haromatic), 7.18–7.23 (q, 1H, J = 6.8 Hz, Haromatic), 7.40–7.42 (d, 2H, J = 8.2 Hz, Haromatic), 7.45–7.78 (t, 1H, J = 7.7 Hz, Haromatic), 8.27 (s, 1H, Hpyrimidinyl), 8.37 (s, 1H, –N(H)–); 13C NMR (DMSO-d6) δ(ppm): 19.0, 20.9, 26.7, 28.7, 29.1, 32.4, 56.5, 98.2, 112.2, 116.2, 116.5, 121.7, 124.5, 129.2, 129.6, 129.7, 132.2, 132.8, 137.0, 156.6, 159.3, 161.7, 162.5, 164.9, 196.1; ESI(−)MS (m/z): 428 [M − H]−; anal. calcd (%) for [C26H24FN3O2]: C, 72.71; H, 5.63; N, 9.78; O, 7.45; found (%): C, 72.42; H, 5.60; N, 9.73; O, 7.41.
4.2.3. 5-(2-Fluorophenyl)-4-((4-fluorophenyl)amino)-8,8-dimethyl-5,7,8,9-tetrahydro-6H-chromeno[2,3-d]pyrimidin-6-one (3c). 4-Fluorolaniline was used as a precursor to synthesize 3c. Yellow solid; yield: 84% (0.36 g); m.p.: 204–206 °C; selected IR bands (cm−1): 3409 (N–H stretching), 2962 (C–H stretching), 1651 (CO stretching); 1H NMR (DMSO-d6) δ(ppm): 0.93 (s, 3H, CH3), 1.07 (s, 3H, CH3) 2.12–2.15 (d, 1H, J = 16.2 Hz, Hmethylene), 2.33–2.36 (d, 1H, J = 16.2 Hz, Hmethylene), 2.55–2.58 (d, 1H, J = 17.6 Hz, Hmethylene), 2.68–2.71 (d, H, J = 17.6 Hz, Hmethylene), 5.53 (s, 1H, Hmethine), 7.05–7.20 (m, 5H, Haromatic) 7.53 (2H, Haromatic), 7.78 (1H, Haromatic), 8.28 (s, 1H, Hpyrimidinyl), 8.55 (s, 1H, –N(H)–); 13C NMR (DMSO-d6) δ(ppm): 26.7, 28.7, 29.1, 32.4, 50.4, 98.3, 112.1, 115.6, 116.5, 123.7, 124.4, 129.1, 129.7, 132.2, 135.9, 156.5, 159.3, 159.4, 160.5, 161.9, 162.1, 165.0, 196.2; ESI(−)MS (m/z): 432 [M − H]−; anal. calcd (%) for [C25H21F2N3O2]: C, 69.27; H, 4.88; N, 9.69; O, 7.38; found (%): C, 69.06; H, 4.86; N, 9.64; O, 7.34.
4.2.4. 4-((3-Chlorophenyl)amino)-5-(2-fluorophenyl)-8,8-dimethyl-5,7,8,9-tetrahydro-6H-chromeno[2,3-d]pyrimidin-6-one (3d). 3-Chloroaniline was used as a precursor to synthesize 3d. Pale yellow solid; yield: 74% (0.33 g); m.p.: 190–194 °C; selected IR bands (cm−1): 3437 (N–H stretching), 2962 (C–H stretching), 1656 (CO stretching);1H NMR (DMSO-d6) δ(ppm): 0.94 (s, 3H, CH3), 1.08 (s, 3H, CH3) 2.12–2.15 (d, 1H, J = 16.3 Hz, Hmethylene), 2.33–2.36 (d, 1H, J = 16.1 Hz, Hmethylene), 2.56–2.59 (d, 1H, J = 17.6 Hz, Hmethylene), 2.69–2.72 (d, H, J = 17.3 Hz, Hmethylene), 5.59 (s, 1H, Hmethine), 7.03–7.06 (m, 2H, Haromatic), 7.10–7.12 (t, 1H, J = 7.5 Hz, Haromatic), 7.18–7.22 (m, 1H, Haromatic), 7.28–7.31 (t, 1H, J = 7.2 Hz, Haromatic) 7.50–7.52 (d, 1H, J = 8.3 Hz, Haromatic), 7.75–7.78 (m, 2H, Haromatic), 8.38 (s, 1H, Hpyrimidinyl), 8.69 (s, 1H, –N(H)–); 13C NMR (DMSO-d6) δ(ppm): 26.7, 28.7, 29.1, 32.4, 50.4, 99.2, 112.1, 116.5, 119.6, 120.4, 123.0, 124.5, 129.7, 130.6, 132.1, 133.2, 141.3, 156.6, 159.0, 160.5, 162.1, 165.0, 196.2; ESI(+)MS (m/z): 450 [M + H]+; anal. calcd (%) for [C25H21ClFN3O2]: C, 66.74; H, 4.70; N, 9.34; O, 7.11; found (%): C, 66.41; H, 4.68; N, 9.29; O, 7.07.
4.2.5. (3-Chloro-4-fluorophenylamino)-5-(2-fluorophenyl)-8,8-dimethyl-8,9-dihydro-5H-chromeno[2,3-d]pyrimidin-6(7H)-one (3e). 3-Chloro-4-fluoroaniline was used as a precursor to synthesize 3e. Pale yellow solid; yield: 68% (0.32 g); m.p.: 200–204 °C; selected IR bands (cm−1): 3450 (N–H stretching), 2971 (C–H stretching), 1660 (CO stretching); 1H NMR (DMSO-d6) δ(ppm): 0.93 (s, 3H, CH3), 1.07 (s, 3H, CH3) 2.12–2.15 (d, 1H, J = 16.2 Hz, Hmethylene), 2.33–2.36 (d, 1H, J = 16.2 Hz, Hmethylene), 2.56–2.59 (d, 1H, J = 17.6 Hz, Hmethylene), 2.69–2.72 (d, H, J = 17.6 Hz, Hmethylene), 5.55 (s, 1H, Hmethine), 7.03–7.06 (overlapping doublets, 1H, J = 8.3 and 8.3 Hz, Haromatic), 7.10–7.13 (t, 1H, J = 7.5 Hz, Haromatic) 7.18–7.23 (q, 1H, J = 6.9 Hz, Haromatic), 7.32–7.35 (d, 1H, Haromatic, J = 9.1 Hz), 7.50–7.52 (m, 1H, Haromatic, J = 8.8 Hz), 7.76–7.79 (t, 1H, Haromatic, J = 9.1 Hz), 7.87–7.88 (d, 1H, Haromatic, J = 9.1 Hz), 8.35 (s, 1H, Hpyrimidinyl), 8.72 (s, 1H, –N(H)–); 13C NMR (DMSO-d6) δ(ppm): 26.7, 28.7, 29.1, 32.4, 50.4, 98.9, 112.2, 116.3, 116.5, 122.8, 124.5, 127.2, 128.8, 129.2, 129.7, 132.1, 138.7, 156.5, 159.0, 160.4, 162.1, 164.9, 196.2; ESI(−)MS (m/z): 466 [M − H]−; anal. calcd (%) for [C25H20ClF2N3O2]: C, 64.17; H, 4.31; N, 8.98; O, 6.84; found (%): C, 63.91; H, 4.29; N, 8.94; O, 6.81.
4.2.6. 4-((4-Chlorophenyl)amino)-5-(2-fluorophenyl)-8,8-dimethyl-5,7,8,9-tetrahydro-6H-chromeno[2,3-d]pyrimidin-6-one (3f). 4-Chloroaniline was used as a precursor to synthesize 3f. White solid; yield: 72% (0.32 g); m.p.: 214–218 °C; selected IR bands (cm−1): 3446 (N–H stretching), 2929 (C–H stretching), 1645 (CO stretching); 1H NMR (DMSO-d6) δ(ppm): 0.94 (s, 3H, CH3), 1.08 (s, 3H, CH3) 2.12–2.15 (d, 1H, J = 16.1 Hz, Hmethylene), 2.33–2.36 (d, 1H, J = 16.2 Hz, Hmethylene), 2.56–2.59 (d, 1H, J = 17.6 Hz, Hmethylene), 2.69–2.72 (d, H, J = 17.5 Hz, Hmethylene), 5.58 (s, 1H, Hmethine), 7.02–7.06 (overlapping doublets, 1H, J = 8.3 and 8.5 Hz, Haromatic), 7.09–7.12 (t, 1H, J = 7.5 Hz, Haromatic) 7.18–7.22 (q, 1H, J = 6.9 Hz, Haromatic), 7.31–7.34 (d, 1H, Haromatic, J = 8.9 Hz), 7.6–7.63 (d, 2H, Haromatic, J = 8.8 Hz), 7.76–7.79 (t, 2H, Haromatic, J = 7.8 Hz), 8.34 (s, 1H, Hpyrimidinyl), 8.65 (s, 1H, –N(H)–); 13C NMR (DMSO-d6) δ(ppm): 26.7, 28.7, 29.1, 32.4, 50.4, 98.6, 112.2, 116.3, 116.4, 121.5, 123.6, 124.5, 128.9, 129.2, 129.7, 132.2, 139.6, 156.6, 159.3, 161.9, 164.9, 196.2; ESI(−)MS (m/z): 448 [M − H]−; anal. calcd (%) for [C25H21ClFN3O2]: C, 66.74; H, 4.70; N, 9.34; O, 7.11; found (%): C, 66.54; H, 4.68; N, 9.29; O, 7.07.
4.2.7. 4-((4-Bromophenyl)amino)-5-(2-fluorophenyl)-8,8-dimethyl-5,7,8,9-tetrahydro-6H-chromeno[2,3-d]pyrimidin-6-one (3g). 4-Bromoaniline was used as a precursor to synthesize 3g. White solid; yield: 88% (0.44 g); m.p.: 238–242 °C; selected IR bands (cm−1): 3445 (N–H stretching), 2929 (C–H stretching), 1645 (CO stretching); 1H NMR (DMSO-d6) δ(ppm): 0.93 (s, 3H, CH3), 1.07 (s, 3H, CH3) 2.12–2.14 (d, 1H, J = 16.1 Hz, Hmethylene), 2.33–2.36 (d, 1H, J = 16.2 Hz, Hmethylene), 2.56–2.59 (d, 1H, J = 17.6 Hz, Hmethylene), 2.69–2.72 (d, H, J = 17.5 Hz, Hmethylene), 5.57 (s, 1H, Hmethine), 7.02–7.06 (overlapping doublets, 1H, J = 8.2 and 8.4 Hz, Haromatic), 7.09–7.11 (t, 1H, J = 7.5 Hz, Haromatic) 7.18–7.20 (q, 1H, J = 6.3 Hz, Haromatic), 7.44–7.46 (d, 1H, Haromatic, J = 7.8 Hz), 7.54–7.56 (d, 2H, Haromatic, J = 8.9 Hz), 7.74–7.77 (t, 2H, Haromatic, J = 7.8 Hz), 8.33 (s, 1H, Hpyrimidinyl), 8.64 (s, 1H, –N(H)-); 13C NMR (DMSO-d6) δ(ppm): 26.6, 28.6, 29.1, 32.4, 50.4, 99.0, 112.1, 123.2, 124.5, 129.8, 131.7, 132.0, 139.0, 156.5, 159.0, 161.9, 165.0, 196.4; ESI(−)MS (m/z): 494 [M]−; anal. calcd (%) for [C25H21BrFN3O2]: C, 60.74; H, 4.28; N, 8.50; O, 6.47; found (%): C, 60.44; H, 4.26; N, 8.46; O, 6.44.
4.2.8. 5-(2-Fluorophenyl)-8,8-dimethyl-4-((4-nitrophenyl)amino)-5,7,8,9-tetrahydro-6H-chromeno[2,3-d]pyrimidin-6-one (3h). 4-Nitroaniline was used as a precursor to synthesize 3h. Pale yellow solid; yield: 91% (0.42 g); m.p.: 206–210 °C; selected IR bands (cm−1): 3401 (N–H stretching), 2956 (C–H stretching), 1663 (CO stretching); 1H NMR (DMSO-d6) δ(ppm): 0.92 (s, 3H, CH3), 1.06 (s, 3H, CH3) 2.10–2.14 (d, 1H, J = 16.2 Hz, Hmethylene), 2.32–2.36 (d, 1H, J = 16.2 Hz, Hmethylene), 2.54–2.58 (d, 1H, J = 17.7 Hz, Hmethylene), 2.68–2.72 (d, H, J = 17.6 Hz, Hmethylene), 5.65 (s, 1H, Hmethine), 6.98–7.03 (overlapping doublets, 1H, J = 8.2 and 7.9 Hz, Haromatic), 7.05–7.09 (t, 1H, J = 7.5 Hz, Haromatic) 7.14–7.20 (q, 1H, J = 6.9 Hz, Haromatic), 7.65–7.70 (t, 1H, Haromatic, J = 7.8 Hz), 7.81–7.83 (d, 2H, Haromatic, J = 9.1 Hz), 8.14–8.16 (d, 2H, Haromatic, J = 9.0 Hz), 8.45 (s, 1H, Hpyrimidinyl), 9.20 (s, 1H, –N(H)–); 13C NMR (DMSO-d6) δ(ppm): 26.6, 28.6, 29.0, 32.4, 50.4, 101.0, 112.1, 116.5, 119.8, 124.6, 125.2, 129.0, 129.9, 130.0, 131.8, 142.0, 156.6, 158.7, 162.4, 165.0, 196.5; ESI(−)MS (m/z): 459 [M − H]−; anal. calcd (%) for [C25H21FN4O4]: C, 65.21; H, 4.60; N, 12.17 O, 13.90; found (%): C, 64.88; H, 4.58; N, 12.11; O, 13.83.
4.2.9. 4-((4-Ferrocenylphenyl)amino)-5-(2-fluorophenyl)-8,8-dimethyl-5,7,8,9-tetrahydro-6H-chromeno[2,3-d]pyrimidin-6-one (3i). 4-Ferrocenylaniline was used as a precursor to synthesize 3i. Orange solid; yield: 85% (0.51 g); m.p.: >250 °C; selected IR bands (cm−1): 3430 (N–H stretching), 2959 (C–H stretching), 1655 (CO stretching); 1H NMR (DMSO-d6) δ(ppm): 0.94 (s, 3H, CH3), 1.08 (s, 3H, CH3) 2.12–2.15 (d, 1H, J = 16.2 Hz, Hmethylene), 2.33–2.36 (d, 1H, J = 16.1 Hz, Hmethylene), 2.56–2.59 (d, 1H, J = 17.5 Hz, Hmethylene), 2.69–2.72 (d, H, J = 17.4 Hz, Hmethylene), 4.00 (s, 5H, Hcyclopentadienyl), 4.30–4.31 (t, 2H, Hcyclopentadienyl), 4.71 (2H, Hcyclopentadienyl), 5.56 (s, 1H, Hmethine), 7.05–7.08 (overlapping doublets, 1H, J = 8.3 and 8.5 Hz, Haromatic), 7.12–7.14 (t, 1H, J = 7.1 Hz, Haromatic), 7.20–7.22 (q, 1H, J = 6.4 Hz, Haromatic), 7.43–7.45 (d, 2H, J = 8.7 Hz, Haromatic), 7.48–7.50 (d, 2H, J = 8.7 Hz, Haromatic), 7.78–7.81 (t, 2H, Haromatic, J = 7.8 Hz), 8.32 (s, 1H, Hpyrimidinyl), 8.46 (s, 1H, –N(H)–); 13C NMR (DMSO-d6) δ(ppm): 26.7, 28.7, 29.1, 32.4, 50.4, 66.3, 66.5, 69.1, 69.7, 85.3, 98.4, 112.2, 116.3, 116.5, 121.6, 124.5, 126.3, 129.2, 129.8, 132.2, 137.5, 156.6, 159.1, 160.5, 161.8, 162.1, 165.0, 196.2; ESI(−)MS (m/z): 598 [M − H]−; anal. calcd (%) for [C35H30FFeN3O2]: C, 70.12; H, 5.04; N, 7.01; O, 5.34; found (%): C, 69.77; H, 5.01; N, 6.97; O, 5.31.
4.2.10. 4-((3-Ferrocenylphenyl)amino)-5-(2-fluorophenyl)-8,8-dimethyl-5,7,8,9-tetrahydro-6H-chromeno[2,3-d]pyrimidin-6-one (3j). 3-Ferrocenylaniline was used as a precursor to synthesize 3j. Pale orange solid; yield: 86% (0.52 g); m.p.: 196–200 °C; selected IR bands (cm−1): 3431 (N–H stretching), 2953 (C–H stretching), 1647 (CO stretching); 1H NMR (DMSO-d6) δ(ppm): 0.95 (s, 3H, CH3), 1.09 (s, 3H, CH3) 2.12–2.16 (d, 1H, J = 16.2 Hz, Hmethylene), 2.34–2.38 (d, 1H, J = 16.2 Hz, Hmethylene), 2.56–2.61 (d, 1H, J = 17.6 Hz, Hmethylene), 2.70–2.74 (d, H, J = 17.6 Hz, Hmethylene), 4.05 (s, 5H, Hcyclopentadienyl), 4.34 (2H, Hcyclopentadienyl), 4.66–4.68 (d, 2H, J = 6.7 Hz, Hcyclopentadienyl), 5.58 (s, 1H, Hmethine), 7.04–7.22 (m, 5H, J = 6.9 Hz, Haromatic), 7.44 (m, 1H, Haromatic), 7.71 (s, 2H, Haromatic), 7.80–7.83 (t, 2H, Haromatic, J = 7.7 Hz), 8.33 (s, 1H, Hpyrimidinyl), 8.44 (s, 1H, –N(H)–); 13C NMR (DMSO-d6) δ(ppm): 26.7, 28.7, 29.1, 32.4, 50.4, 66.7, 67.2, 69.3, 69.9, 85.5, 98.6, 112.2, 116.3, 116.5, 119.2, 119.4, 121.5, 124.5, 128.8, 129.3, 129.8, 132.2, 139.6, 156.5, 159.3, 160.1, 161.9, 165.0, 196.2; ESI(−)MS (m/z): 598 [M − H]−; anal. calcd (%) for [C35H30FFeN3O2]: C, 70.12; H, 5.04; N, 7.01; O, 5.34; found (%): C, 69.76; H, 5.02; N, 6.98; O, 5.31.
4.3. X-ray crystal structure determinations
Single crystals suitable for X-ray diffraction studies of 3a to 3c, 3e and 3h were obtained by recrystallization from hot methanol and those of 3j were obtained via slow evaporation of acetone solution. Crystal evaluation and data collection was done on a Bruker Smart APEXII diffractometer with Mo Kα radiation source. Reflections were collected at different starting angles and the APEXII program suite was used to index the reflections.75 Data reduction was performed using the SAINT76 software and the scaling and absorption corrections were applied using the SADABS77 mult1scan technique. The structures were solved by the direct method using the SHELXS78 program and refined using SHELXL program.79 Graphics of the crystal structures were drawn using MERCURY80 and OLEX2.81 Non-hydrogen atoms were first refined isotropically and then by anisotropic refinement with the full-matrix least squares method based on F2 using SHELXL.79 All hydrogen atoms were positioned geometrically, allowed to ride on their parent atoms and refined isotropically. The fluorine atom in 3a's 2-fluorophenyl group, was found to be disordered over two positions. PART 1 and 2 instructions were used to model the disorder with the major component having a 95.0% site occupancy. In 3e, the anilinyl moiety also exhibited a two-part disorder with a 95.3% site occupancy for the major component. The fluorine atom in 3j's 2-fluorophenyl group and the acetone molecule, both exhibited a two-part disorder with the major component having 93.5% site occupancy. The crystallographic data and structure refinement details are summarized in Table S3.†
4.4. CT-DNA binding affinity determination
The interactions of the compounds with calf thymus DNA (CT-DNA) were assayed using electronic absorption titration by varying concentrations of CT-DNA (0–8 μM) in phosphate buffer saline solution against fixed concentrations (20 μM) of the compounds. The compounds-CT-DNA mixture was incubated for 10 min before measuring the absorbance using UV-vis absorption spectroscopy.82 The absorbance of CT-DNA is cancelled by adding equivalent amounts of CT-DNA to both the tested compounds and the reference solutions. In all the experimental of the interaction of the compounds with CT-DNA, the final concentration of DMSO in the incubation mixture was ∼2% (v/v).
The compounds' intrinsic binding constants, KDNA, were determined using Wolfe–Shimer equation (eqn (2))
| |
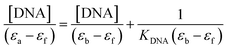 | (2) |
where [DNA],
εa,
εb, and
εf are DNA concentration, apparent, fully bound complex, and free complex extinction coefficients, respectively. The ratio of the slope to the intercept from the plot of [DNA]/(
εa −
εf) against [DNA] gave the binding constant
KDNA of the compounds.
4.5. Albumin binding affinity determination
A stock solution of BSA was prepared by dissolving an appropriate amount of BSA in phosphate-buffered saline (pH 7.2) under constant stirring for one hour at 25 °C. It was kept at 4 °C and used within four days. The BSA concentrations were determined spectrophotometrically by using ε280 nm = 44300 M−1 cm−1 absorption coefficient.83 The stock solution of the complex was prepared by dissolving 1 mmol of the complexes in DMSO. The absorption titration assay was done by adding different concentrations between 0 and 10 μM of the complexes to a constant BSA concentration (6 μM). The samples solution was incubated after each addition of the concentrations for 10 minutes at 25 °C before recording the absorbance at λmax 280 nm. Using the Benesi–Hildebrand equation (eqn (3)),84 the binding constant KBSA was calculated from the intercept to the slope ratio of 1/[A − A0] vs. 1/[complex] linear curve.| |
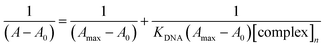 | (3) |
where A is the absorbance recorded in the presence of an added guest, A0 is the absorbance of receptor in the absence of guest, and Amax is the absorbance in the presence of added [complex]n.
4.6. Evaluation of antiproliferative potential, cytotoxicity and cell viability
4.6.1. Cell culture. To evaluate the potential of 1a, 1b, 3a and 3j to halt proliferation of MCF-7 cells, the cells were cultured in 25 cm3 flasks until 80% confluent using cell culture media comprising Dulbecco's Modified Eagles' Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 1% (v/v) penicillin–streptomycin antibiotic, 1% (v/v) L-glutamine and 2.5% (v/v) 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid (HEPES) buffer. The cell cultures were washed with sterile phosphate buffered saline (PBS), passaged using trypsin and maintained in a humidified incubator (37 °C, 5% CO2). All cell culture experiments were performed using aseptic procedures.
4.6.2. Determination of cell viability. To evaluate the cytotoxicity of 1a, 1b, 3a and 3j on breast cancer cell lines in vitro, the methylthiazol tetrazolium bromide (MTT) assay was performed. Briefly, MCF-7 cells were seeded into 96-well plates with a seeding density of 15000 cells per well and incubated for 24 hours. Thereafter, 5 mg mL−1 aliquots of the stock solutions were prepared in DMEM and DMSO (<0.1%), and cells were treated with serial concentrations of 1a and 1b (0–150 μg mL−1), 3a and 3j (0–50 μg mL−1). After 24 hour incubation, 20 μL MTT solution (5 mg mL−1 in PBS) and 100 μL cell culture media were added to the wells, respectively and incubated for another four hours at 37 °C under 5% CO2. After one hour, the MTT solution was aspirated, leaving formazan crystals at the bottom of the wells, which were solubilized using 100 μL DMSO per well. A microplate spectrophotometer (Spectrostar Nano, Germany) was used to read the absorbance of each well at 570/690 nm. This study was performed in triplicate and the concentrations of each compound that produced half the maximum inhibition (IC50) were calculated via linear extrapolation and used in all subsequent assays. Eqn (4) was used to calculate the percentage cell viability:| |
 | (4) |
4.6.3. Cytotoxicity. To determine the mechanism of cytotoxicity to MCF-7 cells following 24 hour treatment with the compounds 1a, 1b, 3a and 3j, the activation of executioner caspase 3/7 was determined. The caspase 3/7 fluorometric assay was used to determine apoptosis induction. Briefly, MCF-7 cells were seeded at a density of 10000 cells per well in a 96-well plate and incubated for 24 hours (37 °C, 5% CO2). Thereafter, cells were treated with the compounds at their IC50 values for 24 hours. Untreated cells were used as controls. Thereafter, 100 μL caspase 3/7 loading solution was added to each well and incubated for one hour at room temperature. For caspase 3/7 detection, fluorescence (excitation/emission of 535/620 nm) was measured using a microplate reader. The experiments were performed in triplicate and data were expressed as relative fluorescence units (RFU).
4.6.4. Selective toxicity. The selective toxicities of compounds 1a, 1b, 3a and 3j were determined using their IC50 values in human embryonic kidney cells (HEK-293) in vitro in pre-determined MTT assays. Briefly, HEK-293 cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS), 1% (v/v) penicillin-streptomycin antibiotic, 1% (v/v) L-glutamine and 2.5% (v/v) 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid (HEPES) buffer in an incubator. The cells were seeded in a 96-well plate at a density of 15000 cells per well and incubated for 24 hours (37 °C, 5% CO2). Thereafter, the cells were treated with the IC50 concentrations of the respective test compounds and incubated for 24 hours. Thereafter, 20 μL MTT solution (5 mg mL−1 in PBS) and 100 μL cell culture media, respectively were added to the wells and incubated for a further four hours (37 °C, 5% CO2). MTT solution was then aspirated and 100 μL DMSO was added per well. A microplate spectrophotometer was used to read the absorbance at 570/690 nm. The experiments were done in triplicate and the percentage cell viability calculated as per the formula in Section 2.6.2.
4.6.5. Statistical analysis. GraphPad Prism version 9.4.1. (GraphPad Software, San Diego, CA, USA) was used for the statistical analyses. The IC50 in the MTT experiment was analyzed utilizing the concentration-response inhibition equation with non-linear regression. We used unpaired t-tests with Welch's correction to determine whether or not the differences between treatments (1a, 1b, 3a, and 3j) and the control were statistically significant in the caspase 3/7 assay results.
Author contributions
Sizwe J. Zamisa (SJZ) and Bernard Omondi (BO) conceived the idea and designed the experiments. SJZ synthesized and characterized all the chromenopyrimidines reported in this work. Adesola A. Adeleke performed the binding affinity experiments while Nikita Devnarain, Mahasin A. Rhman conducted the cytotoxicity studies. Data curation and drafting of the manuscript was done by SJZ. BO and Peter Owira supervised the project.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
The authors greatly acknowledge support from the University of KwaZulu-Natal.
References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- R. Zheng, S. Zhang, H. Zeng, S. Wang, K. Sun, R. Chen, L. Li, W. Wei and J. He, J. Natl. Cancer Cent., 2022, 2, 1–9 CrossRef.
- W. Kadzatsa and S. Ndarukwa-Jambwa, Curr. Breast. Cancer Rep., 2019, 11, 170–174 CrossRef.
- L. E. Pace and L. N. Shulman, Oncol, 2016, 21, 739–744 CrossRef PubMed.
- O. Ginsburg, C. H. Yip, A. Brooks, A. Cabanes, M. Caleffi, J. A. Dunstan Yataco, B. Gyawali, V. McCormack, M. McLaughlin de Anderson, R. Mehrotra, A. Mohar, R. Murillo, L. E. Pace, E. D. Paskett, A. Romanoff, A. F. Rositch, J. R. Scheel, M. Schneidman, K. Unger-Saldana, V. Vanderpuye, T. Y. Wu, S. Yuma, A. Dvaladze, C. Duggan and B. O. Anderson, Cancer, 2020, 126(10), 2379–2393 CrossRef PubMed.
- F. Arvelo, F. Sojo and C. Cotte, Ecancermedicalscience, 2016, 10, 617 Search PubMed.
- B. O. Anderson, C. H. Yip, R. A. Smith, R. Shyyan, S. F. Sener, A. Eniu, R. W. Carlson, E. Azavedo and J. Harford, Cancer, 2008, 113, 2221–2243 CrossRef PubMed.
- Z. Hou, J. Liu, Z. Jin, G. Qiu, Q. Xie, S. Mi and J. Huang, Biosci. Trends, 2022, 16, 31 CrossRef CAS PubMed.
- N. Sturm, T. J. Ettrich and L. Perkhofer, Cancers, 2022, 14, 217 CrossRef CAS PubMed.
- A. Mahmood, N. Bhuva, E. Fokas and R. Glynne-Jones, Cancer Treat. Rev., 2022, 102381 CrossRef CAS PubMed.
- D. Hota and A. Tripathy, in Cancer Diagnostics and Therapeutics, Springer, 2022, pp. 287–302 Search PubMed.
- J. J. Marin, R. I. Macias, M. J. Monte, E. Herraez, A. Peleteiro-Vigil, B. S. d. Blas, P. Sanchon-Sanchez, A. G. Temprano, R. A. Espinosa-Escudero and E. Lozano, Cancers, 2020, 12, 2605 CrossRef CAS PubMed.
- R. Hong and B. Xu, Cancer Comm., 2022, 42, 913–936 CrossRef PubMed.
- A. Zonouzi, F. Hosseinzadeh, N. Karimi, R. Mirzazadeh and S. W. Ng, ACS Comb. Sci., 2013, 15, 240–246 CrossRef CAS PubMed.
- A. Zonouzi, H. A. Shahrezaee, A. Rahmani, F. Zonouzi, K. Abdi, F. T. Fadaei and K. Schenk, Org. Prep. Proced. Int., 2018, 50, 343–358 CrossRef CAS.
- L. Suresh, P. S. V. Kumar and G. V. P. Chandramouli, J. Mol. Struct., 2017, 1134, 51–58 CrossRef CAS.
- G. Keykha, M. R. Hosseini-Tabatabaei and A. Hassanabadi, J. Chem. Res., 2017, 41, 85–87 CrossRef CAS.
- S. Ameli, A. Davoodnia, M. Pordel and H. Behmadi, J. Heterocycl. Chem., 2017, 54, 1437–1441 CrossRef CAS.
- B. Umamahesh, T. R. Mandlimath and K. I. Sathiyanarayanan, RSC Adv., 2015, 5, 6578–6587 RSC.
- A. Zonouzi, M. Biniaz, R. Mirzazadeh, M. Talebi and S. Weng Ng, Heterocycles, 2010, 81, 1271 CrossRef CAS.
- S. Peng, Y. Zhao, C. Fu, X. Pu, L. Zhou, Y. Huang and Z. Lu, Chemistry, 2018, 24, 8056–8060 CrossRef CAS PubMed.
- M. R. Bhosle, D. B. Wahul, G. M. Bondle, A. Sarkate and S. V. Tiwari, Synth. Commun., 2018, 48, 2046–2060 CrossRef CAS.
- A. H. Halawa, M. M. Elaasser, A. M. El Kerdawy, A. M. A. I. Abd El-Hady, H. A. Emam and A. M. El-Agrody, Med. Chem. Res., 2017, 26, 2624–2638 CrossRef CAS.
- N. M. Sabry, H. M. Mohamed, E. S. Khattab, S. S. Motlaq and A. M. El-Agrody, Eur. J. Med. Chem., 2011, 46, 765–772 CrossRef CAS PubMed.
- H. H. Ahmed, M. F. Ahmed, M. A.-D. Al-Anood and M. E.-A. Ahmed, Lett. Drug Des. Discovery, 2016, 13, 77–88 Search PubMed.
- N. M. El-Bakhshawangy, H. B. El-Nassan, A. E. Kassab and A. T. Taher, Future Med. Chem., 2018, 10, 1465–1481 CrossRef CAS PubMed.
- R. El-Sayed and A. A. Fadda, J. Oleo Sci., 2016, 65, 929–940 CrossRef CAS PubMed.
- A. Roquia and M. Kumar, Int. J. Appl. Sci. Eng. Res., 2017, 7, 301–309 Search PubMed.
- A. Mobinikhaledi, N. Foroughifar, T. Mosleh and A. Hamtac, Iran. J. Pharm. Res., 2014, 3, 873–879 Search PubMed.
- M. Ghashang, S. S. Mansoor and K. Aswin, J. Adv. Res., 2014, 5, 209–218 CrossRef CAS PubMed.
- S. S. Chobe, B. S. Dawane, K. M. Tumbi, P. P. Nandekar and A. T. Sangamwar, Bioorg. Med. Chem. Lett., 2012, 22, 7566–7572 CrossRef CAS PubMed.
- R. S. Keri, K. M. Hosamani, R. V. Shingalapur and M. H. Hugar, Eur. J. Med. Chem., 2010, 45, 2597–2605 CrossRef CAS PubMed.
- B. Pogorelcnik, M. Brvar, B. Zegura, M. Filipic, T. Solmajer and A. Perdih, ChemMedChem, 2015, 10, 345–359 CrossRef CAS PubMed.
- S. Roy, J. A. Westmaas, K. D. Hagen, G. P. van Wezel and J. Reedijk, J. Inorg. Biochem., 2009, 103, 1288–1297 CrossRef CAS PubMed.
- P. Alam, S. K. Chaturvedi, T. Anwar, M. K. Siddiqi, M. R. Ajmal, G. Badr, M. H. Mahmoud and R. H. Khan, J. Lumin., 2015, 164, 123–130 CrossRef CAS.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- K. A. Majorek, P. J. Porebski, A. Dayal, M. D. Zimmerman, K. Jablonska, A. J. Stewart, M. Chruszcz and W. Minor, Mol. Immunol., 2012, 52, 174–182 CrossRef CAS PubMed.
- C. Tan, J. Liu, H. Li, W. Zheng, S. Shi, L. Chen and L. Ji, J. Inorg. Biochem., 2008, 102, 347–358 CrossRef CAS PubMed.
- X. Xu, Y. Lai and Z.-C. Hua, Biosci. Rep., 2019, 39, BSR20180992 CrossRef CAS PubMed.
- E. Bruckheimer, S. Cho, M. Sarkiss, J. Herrmann and T. McDonnell, Apoptosis, 1998, 75–105 CAS.
- G. Makin and J. A. Hickman, Cell Tissue Res., 2000, 301, 143–152 CrossRef CAS PubMed.
- E. Milanesi, P. Costantini, A. Gambalunga, R. Colonna, V. Petronilli, A. Cabrelle, G. Semenzato, A. M. Cesura, E. Pinard and P. Bernardi, J. Biochem., 2006, 281, 10066–10072 CAS.
- Y. Xu, M. Zhou, Y. Li, C. Li, Z. Zhang, B. Yu and R. Wang, ChemMedChem, 2013, 8, 1345–1352 CrossRef CAS PubMed.
- Z. Ji, W. Guo, E. L. Wood, J. Liu, S. Sakkiah, X. Xu, T. A. Patterson and H. Hong, Chem. Res. Toxicol., 2022, 35, 125–139 Search PubMed.
- Z. Yin, H. Ai, L. Zhang, G. Ren, Y. Wang, Q. Zhao and H. Liu, J. Appl. Toxicol., 2019, 39, 1366–1377 CrossRef CAS PubMed.
- H. Sun, Y. Wang, D. M. Cheff, M. D. Hall and M. Shen, Bioorg. Med. Chem., 2020, 28, 115422 CrossRef CAS PubMed.
- F. Stoliński, A. Rybińska-Fryca, M. Gromelski, A. Mikolajczyk and T. Puzyn, Nanotoxicology, 2022, 16, 276–289 CrossRef PubMed.
- A. A. Lagunin, V. I. Dubovskaja, A. V. Rudik, P. V. Pogodin, D. S. Druzhilovskiy, T. A. Gloriozova, D. A. Filimonov, N. G. Sastry and V. V. Poroikov, PLoS One, 2018, 13, e0191838 CrossRef PubMed.
- S. J. Zamisa, N. P. Ngubane, A. A. Adeleke, S. B. Jonnalagadda and B. Omondi, Cryst. Growth Des., 2022, 22(10), 5814 CrossRef CAS.
- S. J. Zamisa and B. Omondi, Molbank, 2022, 2022, M1364 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- N. Shahabadi and R. Farhadi, Nucleosides, Nucleotides Nucleic Acids, 2021, 40, 317–335 CrossRef CAS PubMed.
- M. A. Ragheb, R. S. Omar, M. H. Soliman, A. H. M. Elwahy and I. A. Abdelhamid, J. Mol. Struct., 2022, 1267, 133628 CrossRef CAS.
- S. J. Zamisa, N. P. Ngubane, A. A. Adeleke, S. B. Jonnalagadda and B. Omondi, Cryst. Growth Des., 2022, 22, 5814–5834 CrossRef CAS.
- A. A. Adeleke, M. S. Islam, O. Sanni, C. Mocktar, S. J. Zamisa and B. Omondi, J. Inorg. Biochem., 2021, 214, 111266 CrossRef CAS PubMed.
- A. A. Adeleke, S. J. Zamisa, M. S. Islam, K. Olofinsan, V. F. Salau, C. Mocktar and B. Omondi, Molecules, 2021, 26, 1205 CrossRef CAS PubMed.
- S. Ž. Đurić, S. Vojnovic, T. P. Andrejević, N. L. Stevanović, N. D. Savić, J. Nikodinovic-Runic, B. Đ. Glišić and M. I. Djuran, Bioinorg. Chem. Appl., 2020, 2020, 3812050 Search PubMed.
- T. H. Sanatkar, H. Hadadzadeh, Z. Jannesari, T. Khayamian, M. Ebrahimi, H. A. Rudbari, M. Torkzadeh-Mahani and M. Anjomshoa, Inorg. Chim. Acta, 2014, 423, 256–272 CrossRef CAS.
- Z. M. Lighvan, H. A. Khonakdar, A. Heydari, M. Rafiee, M. D. Jahromi, A. Derakhshani and A. A. Momtazi-Borojeni, Appl. Organomet. Chem., 2020, 34, e5839 CrossRef CAS.
- F. Auria-Luna, V. Fernández-Moreira, E. Marqués-López, M. C. Gimeno and R. P. Herrera, Sci. Rep., 2020, 10, 11594 CrossRef CAS PubMed.
- Y.-L. Bai, Y.-W. Zhang, J.-Y. Xiao, H.-W. Guo, X.-W. Liao, W.-J. Li and Y.-C. Zhang, Transition Met. Chem., 2018, 43, 171–183 CrossRef CAS.
- A. M. Gbaj, Biomed. J. Sci. Technol. Res., 2020, 24, 18318 Search PubMed.
- C. Qin, X. Hu, B. Yang, J. Liu and Y. Gao, Environ. Pollut., 2021, 268, 115798 CrossRef CAS PubMed.
- M. H. Nasir, E. Jabeen, R. Qureshi, F. L. Ansari, A. Shaukat, U. Nasir and A. Ahmed, Biophys. Chem., 2020, 258, 106316 CrossRef CAS PubMed.
- A. H. Halawa, M. M. Elaasser, A. M. El Kerdawy, A. M. A. I. Abd El-Hady, H. A. Emam and A. M. El-Agrody, Med. Chem. Res., 2017, 26, 2624–2638 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- E. K. A. Abdelall, H. A. H. Elshemy, M. B. Labib and F. E. A. Mohamed, Bioorg. Chem., 2022, 120, 105591 CrossRef CAS PubMed.
- T. Akbarzadeh, A. Rafinejad, J. M. Mollaghasem, M. Safavi, A. Fallah-Tafti, M. Pordeli, S. K. Ardestani, A. Shafiee and A. Foroumadi, Arch. Pharm., 2012, 345, 386–392 CrossRef CAS PubMed.
- I. Parveen, N. Ahmed, D. Idrees, P. Khan and M. I. Hassan, Bioorg. Med. Chem. Lett., 2017, 27, 4493–4499 CrossRef CAS PubMed.
- H. Akrami, B. F. Mirjalili, O. Firuzi, A. Hekmat, A. A. Saboury, R. Miri, O. Sabzevari, M. Pirali-Hamedani, F. Jeivad, S. Moghimi, S. Emami, A. Foroumadi and M. Khoobi, Lett. Drug Des. Discovery, 2020, 17, 640–654 CrossRef CAS.
- W. I. Pérez, Y. Soto, C. Ortíz, J. Matta and E. Meléndez, Bioorg. Med. Chem., 2015, 23, 471–479 CrossRef PubMed.
- J. L. Vera, J. Rullán, N. Santos, J. Jiménez, J. Rivera, A. Santana, J. Briggs, A. L. Rheingold, J. Matta and E. Meléndez, J. Organomet. Chem., 2014, 749, 204–214 CrossRef CAS PubMed.
- J. M. Lee and A. Bernstein, Cancer Metastasis Rev., 1995, 14, 149–161 CrossRef CAS PubMed.
- E. M. Njogu, B. Omondi and V. O. Nyamori, S. Afr. J. Chem., 2016, 69, 51 CrossRef CAS.
- Bruker, APEXII, Bruker AXS, Madison, Wisconsin, USA, 2009 Search PubMed.
- Bruker, SAINT, Bruker AXS, Madison, Wisconsin, USA, 2009 Search PubMed.
- Bruker, SADABS, Bruker AXS, Madison, Wisconsin, USA, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. A. Adeleke, S. J. Zamisa, M. Islam, K. Olofinsan, V. F. Salau, C. Mocktar and B. Omondi, Molecules, 2021, 26, 1205 CrossRef CAS PubMed.
- S. C. Gill and P. H. Von Hippel, Anal. Biochem., 1989, 182, 319–326 CrossRef CAS PubMed.
- S. Goswami, D. Sen, N. K. Das, H.-K. Fun and C. K. Quah, Chem. Commun., 2011, 47, 9101–9103 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The 1H- and 13C-NMR spectra (Fig. S1–S10), IR spectra (Fig. S11–S20), ES1MS (Fig. S21–S30) data for 3a to 3j, including the intermolecular hydrogen bonding patterns observed in the crystal packing of 3a to 3c, 3e, 3h and 3j (Fig. S31–S34), can be found in the ESI. The electronic absorption spectra for the determination of DNA/BSA binding affinity studies, are depicted in Fig. S35–S45. CCDC 2219308–219313. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra01741a |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Adesola A. Adelekea,
Nikita Devnarainb,
Mahasin Abdel Rhmanb,
Peter M. O. Owira*b and
Bernard Omondi
a,
Adesola A. Adelekea,
Nikita Devnarainb,
Mahasin Abdel Rhmanb,
Peter M. O. Owira*b and
Bernard Omondi