
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCurrent status of N-, O-, S-heterocycles as potential alkaline phosphatase inhibitors: a medicinal chemistry overview
Rabab S. Jassasa,
Nafeesa Naeem b,
Amina Sadiqc,
Rabia Mehmoodc,
Noof A. Alenazid,
Munirah M. Al-Rooqie,
Ehsan Ullah Mughal
*b,
Reem I. Alsantalif and
Saleh A. Ahmed
*e
b,
Amina Sadiqc,
Rabia Mehmoodc,
Noof A. Alenazid,
Munirah M. Al-Rooqie,
Ehsan Ullah Mughal
*b,
Reem I. Alsantalif and
Saleh A. Ahmed
*e
aDepartment of Chemistry, Jamoum University College, Umm Al-Qura University, Makkah 21955, Saudi Arabia
bDepartment of Chemistry, University of Gujrat, Gujrat 50700, Pakistan. E-mail: ehsan.ullah@uog.edu.pk
cDepartment of Chemistry, Govt. College Women University, Sialkot 51300, Pakistan
dDepartment of Chemistry, College of Science and Humanities in Al-Kharj, Prince Sattam Bin Abdulaziz University, Al-kharj 11942, Saudi Arabia
eDepartment of Chemistry, Faculty of Applied Sciences, Umm Al-Qura University, 21955 Makkah, Saudi Arabia. E-mail: sahmed@uqu.edu.sa
fDepartment of Pharmaceutical Chemistry, College of Pharmacy, Taif University, P.O. Box 11099, Taif 21944, Saudi Arabia
First published on 1st June 2023
Abstract
Heterocycles are a class of compounds that have been found to be potent inhibitors of alkaline phosphatase (AP), an enzyme that plays a critical role in various physiological processes such as bone metabolism, cell growth and differentiation, and has been linked to several diseases such as cancer and osteoporosis. AP is a widely distributed enzyme, and its inhibition has been considered as a therapeutic strategy for the treatment of these diseases. Heterocyclic compounds have been found to inhibit AP by binding to the active site of the enzyme, thereby inhibiting its activity. Heterocyclic compounds such as imidazoles, pyrazoles, and pyridines have been found to be potent AP inhibitors and have been studied as potential therapeutics for the treatment of cancer, osteoporosis, and other diseases. However, the development of more potent and selective inhibitors that can be used as therapeutics for the treatment of various diseases is an ongoing area of research. Additionally, the study of the mechanism of action of heterocyclic AP inhibitors is an ongoing area of research, which could lead to the identification of new targets and new therapeutic strategies. The enzyme known as AP has various physiological functions and is present in multiple tissues and organs throughout the body. This article presents an overview of the different types of AP isoforms, their distribution, and physiological roles. It also discusses the structure and mechanism of AP, including the hydrolysis of phosphate groups. Furthermore, the importance of AP as a clinical marker for liver disease, bone disorders, and cancer is emphasized, as well as its use in the diagnosis of rare inherited disorders such as hypophosphatasia. The potential therapeutic applications of AP inhibitors for different diseases are also explored. The objective of this literature review is to examine the function of alkaline phosphatase in various physiological conditions and diseases, as well as analyze the structure–activity relationships of recently reported inhibitors. The present review summarizes the structure–activity relationship (SAR) of various heterocyclic compounds as AP inhibitors. The SAR studies of these compounds have revealed that the presence of a heterocyclic ring, particularly a pyridine, pyrimidine, or pyrazole ring, in the molecule is essential for inhibitory activity. Additionally, the substitution pattern and stereochemistry of the heterocyclic ring also play a crucial role in determining the potency of the inhibitor.
 Ehsan Ullah Mughal | Ehsan Ullah Mughal is a Tenured Associate Professor of Organic Chemistry at the Department of Chemistry in University of Gujrat, Gujrat, Pakistan. He obtained his PhD from Bielefeld University, Germany in 2013 under the supervision of Prof. Dr Dietmar Kuck. For postdoctoral studies, he joined the group of Prof. Dr Xinliang Feng, Max-Planck-Institute for Polymer Research, Mainz, Germany. His current research interests include the design and synthesis of bioactive heterocycles, synthetic flavonoids, transition metal-based terpyridine complexes and their uses in the fabrication of DSSCs and as efficient photo-catalysts, and organic functional materials for the applications in organic electronics. |
 Saleh A. Ahmed | Prof. Dr Saleh A. Ahmed received his PhD in photochemistry (photochromism) under the supervision of Prof. Heinz Dürr at Saarland University, Germany. He has more than 20 years of experience as a postdoctoral fellow, senior researcher and visiting professor in France, Japan, Germany, Italy and USA. Since 2010 he promoted to full professor of organic photochemistry. He published more than 235 publications in high ranked journals and more than 10 US-patents. His research interests include synthesis and photophysical properties of novel organic compounds, and developments of synthetic methodologies for the synthesis of compounds with unique theoretical and biological applications. |
1. Introduction
Heterocycles are a class of molecule that exist in many biological compounds in the human body and include atoms other than carbon and hydrogen in a ring structure. This natural presence, combined with their complexity and ability to participate in hydrogen bonding, makes heterocycles highly sought after for use as drugs. Medicinal chemists create drug structures that resemble the heterocycles found in the body with the aim of either enhancing or restricting their function. Modern advancements have enabled the synthesis of numerous heterocyclic scaffolds, aimed at facilitating drug discovery. These compounds have received significant attention in recent years due to their potential as therapeutic agents, as they can interact with enzymes and modulate their activity.1–6 Enzymes are biological molecules that catalyze chemical reactions and are essential for various physiological processes. They have become attractive targets for drug discovery and development due to their central role in cellular metabolism and signaling pathways.4,5,7–12 Heterocyclic compounds can interact with enzymes by binding to their active sites, thereby altering their activity and inhibiting their function.13–16 One particular area of interest for the use of heterocyclic compounds as enzyme inhibitors is the modulation of alkaline phosphatase (AP) activity.Alkaline phosphatases (APs; EC 3.1.3.1) are ubiquitous membrane-bound glycoproteins that distribute broadly in nature, and thus are found in prokaryotes to eukaryotes, with the exception of fewer higher plants.17–19 AP belongs to a broad family of dimeric enzymes that are often found only on the surface of cells. It hydrolyzes different monophosphate esters at high pH, releasing inorganic phosphate in the process.20–22
Numerous varieties of APs have been identified and characterized23–26 since their discovery in the 1920s,27 allowing for a better knowledge of their evolution. Extensive and in-depth research has been conducted on the crystal structure and catalytic characteristics of AP from Escherichia coli, which has been the prototype for all APs since the 1950s.28 In the 1980s, numerous mammalian AP complementary DNAs had been cloned, and matching enzymes had been characterized. Numerous biological uses for AP were suggested in the 1990s.29 The discovery of the human placental AP's, three-dimensional structure in 2001,30 allowed for a deeper understanding of the enzyme's active site and key residues, as well as useful information for the design of enzyme inhibitors, substrates and prodrugs for biochemical applications, the cure of AP-related diseases, and drug delivery.31
APs in mammals are ectonucleotidases that belong to the zinc-containing metalloenzyme family. These dimeric entities are encoded by a multigene family and require three metal ions, two Zn2+ and one Mg2+, for their enzymatic activity. Besides their role in catalysis, these metal ions also play a significant role in shaping the AP monomer and controlling the interactions between subunits.32
APs catalyze a wide range of chemical reactions, including the conversion of nucleotides to nucleosides, by removing phosphate groups from molecules hydrolytically.33 Adenosine monophosphate (AMP) is hydrolyzed by APs to produce adenosine, a chemical involved in cell signaling. Additionally, they can be seen as significant regulators of purinergic cell signaling.34,35 The differentiation of adipocytes and osteoblasts, as well as their maturation processes, is another significant function of APs. Numerous adipocyte tissues have been found to include APs, which are significant participants in the adipogenesis process.36,37
The focus of researchers has been on synthesizing lead compounds that can act as inhibitors of various APs. This review article delves into the relationship between these disease states and APs, as well as the inhibitors that have been published from 2009 to 2023.
2. Alkaline phosphatase isoenzymes, their distribution and physiological functions
Human APs can be divided into four tissue-specific variants or isozymes, primarily based on how specifically a tissue could express them (Fig. 1).38,39 Three of them are known as tissue-specific alkaline phosphatases enzymes, and they are called placental alkaline phosphatase (PLAP or Regan isozyme), intestinal alkaline phosphatase (IAP), and germ cell alkaline phosphatase (GCAP or NAGAO isozyme).40,41 The fourth enzyme is known as tissue-nonspecific alkaline phosphatase (TNAP). Each of these enzymes has a great deal of homology.42–44 PLAP is highly expressed in the placenta and is overexpressed in ovarian and testicular malignancies, while being under-expressed in pregnant Chagas disease patients.38,45–47 TNAP is primarily found in the kidney, liver, and bone.37,48,49 The gastrointestinal tract (GIT), particularly the duodenum, contains a significant amount of IAP, which makes it a potential target for drug therapy aimed at treating sepsis, antibiotic-associated diarrhea and inflammatory bowel syndrome.50–52 New research indicates that an increase in serum AP levels could lead to various disease conditions. Additionally, these studies have found a link between elevated AP levels and an increased likelihood of developing Alzheimer's disease (AD) and other related conditions.53,54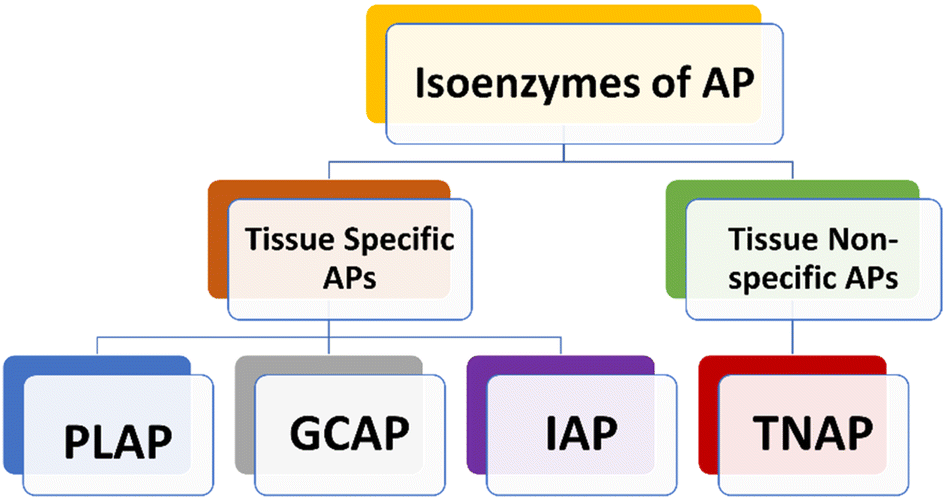 | ||
| Fig. 1 Classification of Alkaline Phosphatase (AP) isozymes. | ||
2.1 Placental alkaline phosphatases
PLAP is primarily expressed at high levels in the placenta (Table 1 and Fig. 2). Though, lower concentrations of PLAP can be seen in the serum.17 In addition, it is found in trace amounts in the ovaries, type 1 pneumocytes, and the cervix of non-pregnant women.55 As opposed to the other isoenzymes, PLAP has a low catalytic constant, making it a less effective enzyme. This inefficiency is linked to the pH of the placenta, which is approximately 7, and the PLAP's ideal pH, which is 11.38 Due to its heat stability property, PLAP is an exceptional isoenzyme because it does not inactivate in the presence of 10−2 M magnesium at temperatures below 75 °C. Because PLAP lacks protein and comes from a source that makes it stable to heat, PLAP has a high level of heat resistance.56 Regan is a variation of the PLAP isoenzyme that is specifically found in the plasma of roughly 15% of individuals with liver, gut, and lung cancer.17| Organ/tissue | Isoenzymes | Functions of the isoenzyme | ALP activity (IU g−1 tissue) | References |
|---|---|---|---|---|
| Intestine | IAP | • Lipids absorption | 38 ± 14 | 50, 66, 86–88 |
| • Removal of bacterial lipopolysaccharides and free nucleotides | ||||
| • Control of the bacterial transmucosal passage | ||||
| • Reduced gut inflammatory response | ||||
| • Dephosphorylation of tricellular adenosine triphosphate (ATP) to adenosine | ||||
| Colon | IAP, colonic TNAP | • Reduction of inflammation and oxidative stress | 2.3 ± 0.8 | 89 and 90 |
| Testis | GCAP | • Sperm glycolytic reactions and fructose production | 0.5 ± 0.1 | 91 |
| Placenta | PLAP | • Tumor marker | 69 ± 44 | 30, 91 and 92 |
| • Removal of bacterial endotoxins from the body | ||||
| Bone | TNAP | • Bone development and mineralization | — | 91 |
| • Sign of osteoblastic activity | ||||
| Liver | TNAP | • Breakdown of phosphorylcholine | 2.6 ± 1.4 | 93 |
| Kidney | TNAP | • Dephosphorylation and detoxification of ATP | 2.1 ± 0.7 | 94 |
| Lung | TNAP | • Repair of damaged tissue and the clearance of mucus | 2.1 ± 0.5 | 95 |
| • Formation of new blood vessels and the repair of damaged tissue | ||||
| Vagina, endometrium | TNAP | • Maintenance of acidic pH | — | 96 and 97 |
| • Maintenance of vaginal flora | ||||
| • Wound healing | ||||
| • Endometrial development | ||||
| • Menstrual cycle | ||||
| • Pregnancy | ||||
| Eyes | TNAP | • Ocular development | — | 98 and 99 |
| • Maintenance of the retinal pigment epithelium | ||||
| • Removal of toxic waste products | ||||
| • Regulation of cell growth and differentiation | ||||
| • Age-related macular degeneration | ||||
| • Retinitis pigmentosa |
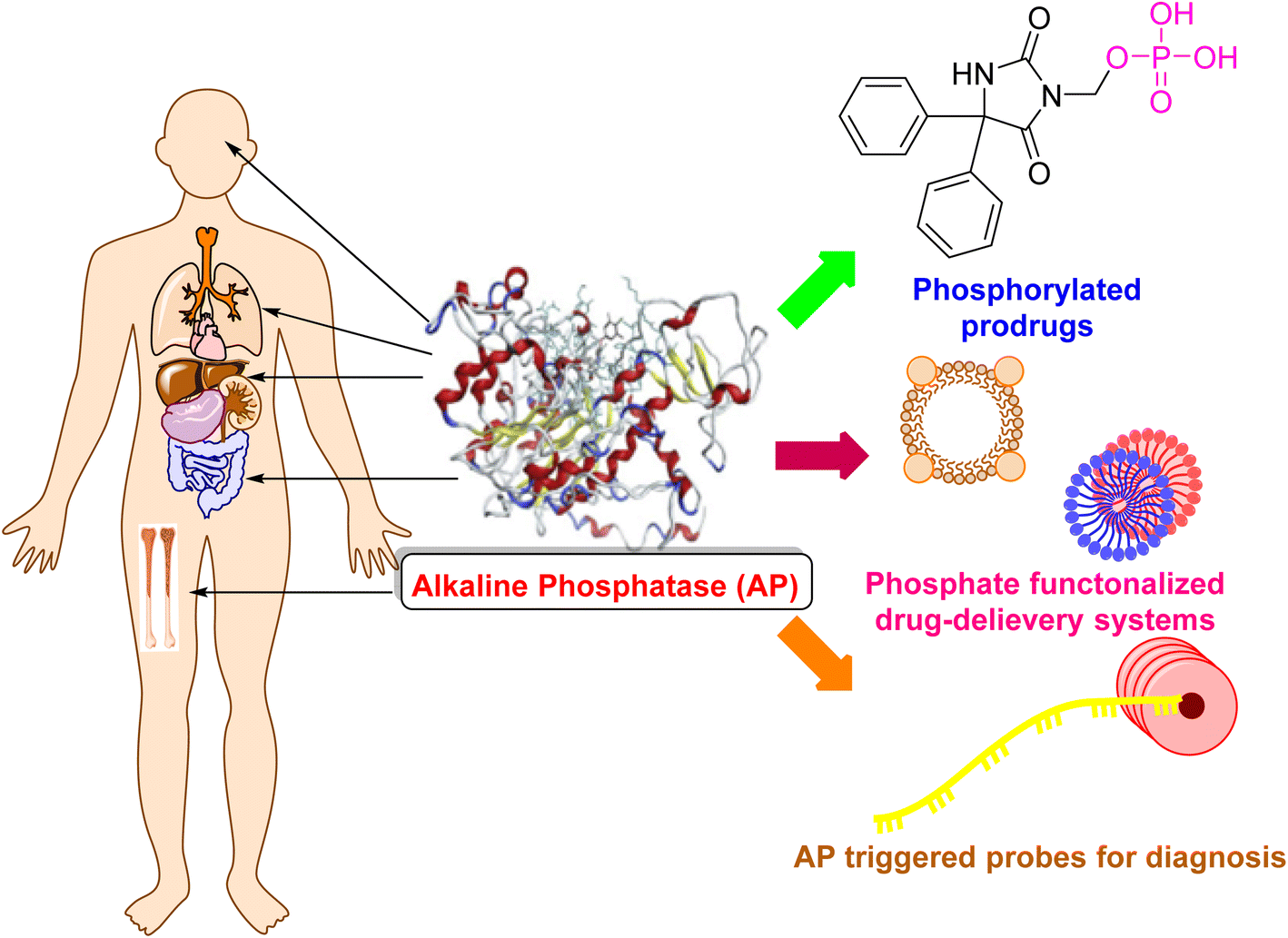 | ||
| Fig. 2 Distribution of alkaline phosphatase that is a target for drug delivery and diagnosis. | ||
2.2 Intestinal alkaline phosphatase
Intestinal alkaline phosphatase (IAP) is a hydrolase enzyme that is produced by the enterocytes lining the small intestine (Table 1 and Fig. 2). IAP is responsible for the removal of any potential toxins or pathogens that may be present in the gut lumen by hydrolyzing the phosphates from the surfaces of these molecules, rendering them unable to attach to the gut epithelium.59 IAP is specifically expressed in the brush border of the enterocytes in the small intestine and is secreted into the lumen, where it acts on luminal antigens, such as lipopolysaccharides (LPS) and flagellin, which are components of Gram-negative bacteria. This function of IAP is important for the maintenance of gut integrity and barrier function.60,61 IAP has also been shown to have anti-inflammatory properties, which are thought to be mediated by its ability to inactivate LPS and other pro-inflammatory molecules.62 In addition, IAP has been found to play a role in the regulation of the gut microbiome and the maintenance of gut immune homeostasis. Moreover, IAP has been found to be involved in the regulation of various physiological processes such as glucose metabolism, lipid metabolism, and the gut–brain axis. It has also been linked to several diseases such as inflammatory bowel disease (IBD), obesity and cancer.63,64In summary, IAP is an enzyme that is produced by the enterocytes lining the small intestine and plays a critical role in gut integrity and barrier function, as well as in the maintenance of gut immune homeostasis. It also has anti-inflammatory properties and is involved in the regulation of various physiological processes such as glucose metabolism, lipid metabolism, and the gut–brain axis. Studies have also linked IAP to several diseases such as inflammatory bowel disease (IBD), obesity, and cancer, making it a potential therapeutic target for these conditions.65
2.3 Germ cell alkaline phosphatase
Germ cell alkaline phosphatase (GCAP) is an enzyme that is expressed in the germ cells of the testis and ovary17 (Table 1 and Fig. 2). GCAP is essential for the proper development and maturation of germ cells in the testis and ovary, and it plays a critical role in the formation of sperm and ova. GCAP is expressed in the early stages of germ cell development, and its expression decreases as germ cells differentiate and mature.70 In the testis, GCAP is expressed in the spermatogonia, which are the precursor cells that give rise to sperm, and in the primary spermatocytes, which are the cells that undergo meiosis to form sperm. In the ovary, GCAP is expressed in the oogonia, which are the precursor cells that give rise to ova, and in the primary oocytes, which are the cells that undergo meiosis to form ova.71 GCAP has been found to be involved in the regulation of various physiological processes such as DNA repair, cell cycle progression, and apoptosis. GCAP is also involved in the regulation of the sex steroid hormone biosynthesis, and it has been found to be essential for the proper development and maturation of germ cells.72 GCAP deficiency has been linked to several diseases such as male infertility, and in some cases, female infertility. GCAP has also been shown to be involved in the onset and progression of certain cancers, such as testicular cancer, and it has been proposed as a potential therapeutic target for these conditions.73In summary, GCAP is an enzyme that is expressed in the germ cells of the testis and ovary, and it plays a critical role in the development and maturation of germ cells. GCAP is involved in the regulation of various physiological processes such as DNA repair, cell cycle progression, and apoptosis. GCAP deficiency has been linked to several diseases such as male and female infertility and certain cancers, making it a potential therapeutic target for these conditions.74
In summary, GCAP plays a critical role in the development and maturation of germ cells, by regulating DNA repair, cell cycle progression, apoptosis, sex steroid hormone biosynthesis and spermatogenesis and oogenesis. Additionally, GCAP deficiency has been linked to several diseases such as male and female infertility, and certain cancers, making it a potential therapeutic target for these conditions.79
2.4 Liver/bone/kidney alkaline phosphatase
Tissue nonspecific alkaline phosphatase (TNAP) is an enzyme that belongs to the family of APs.80 It is also known as liver/bone/kidney alkaline phosphatase (LBK-AP) because it occurs in various tissues such as the liver, bone, and kidney (Table 1, Fig. 2 and 3).81 TNAP is responsible for the hydrolysis of phosphomonoesters, and it plays a critical role in several physiological processes. TNAP is expressed in bone and plays a critical role in bone mineralization by hydrolyzing inorganic pyrophosphate (PPi) which is a potent inhibitor of mineralization.82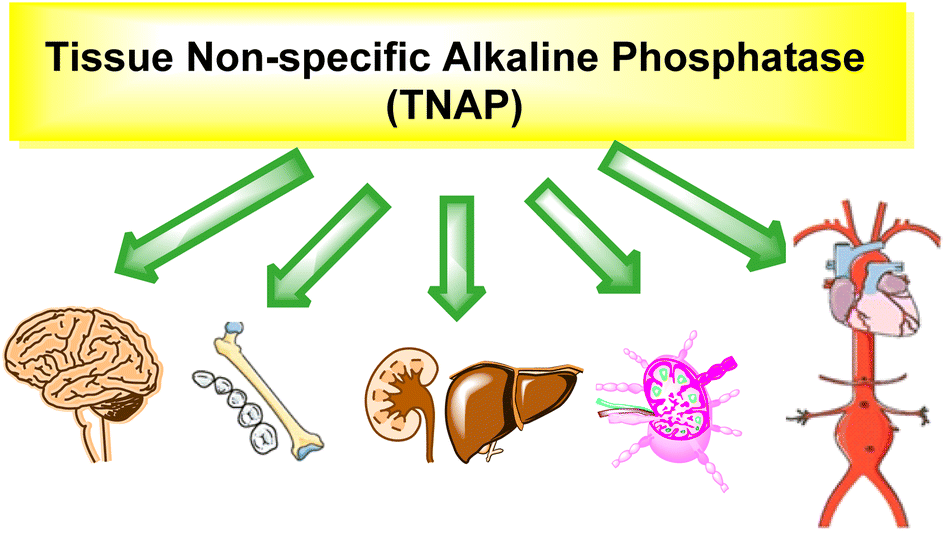 | ||
| Fig. 3 Distribution of TNAP in various body organs. | ||
TNAP spreads in the proximal tubule of the kidney and thus plays a role in the regulation of phosphate homeostasis.83 It is expressed in the liver and plays a role in the regulation of bile acid metabolism and detoxification of xenobiotics. TNAP has been found to be involved in the regulation of vascular calcification, cardiovascular disease and the progression of certain cancers, such as prostate cancer.84,85
3. Structure of alkaline phosphatases
APs are a group of enzymes that belong to the phosphatase family. They are known for their ability to hydrolyze phosphomonoesters, and they have a common structure that is composed of several domains. The catalytic domain contains the active site of the enzyme, where the phosphomonoester substrate binds and is hydrolyzed. The active site typically contains a metal ion, such as zinc or magnesium, which plays a critical role in the catalytic activity of the enzyme.100 The regulatory domain is responsible for regulating the activity of the enzyme, typically through the binding of specific inhibitors or activators. The membrane-binding domain is accountable for the attachment of the enzyme to the cell membrane. This domain is found only in the membrane-bound forms of the enzyme. The signal peptide domain is liable for directing the enzyme to the correct location within the cell, typically to the cell membrane or secretory pathway.101The specific structure of alkaline phosphatases varies depending on the type of the enzyme, for instance, the structure of the Intestinal alkaline phosphatase (IAP) is composed of six domains, the catalytic, dimerization, membrane-binding, regulatory, carbohydrate-binding, and signal peptide domains, while in the Germ cell alkaline phosphatase (GCAP) the structure is composed of four domains, the catalytic, regulatory, transmembrane, and signal peptide domains.102–106
The homodimeric metal-containing enzymes, known as Aps, are substrate non-specific in nature. In addition to five cysteine residues, each monomer contains three metal ions: two Zn2+ and one Mg2+ (Cys474, Cys467, Cys183, Cys121, and Cys101 in PLAP). While the Cys101 residue is left in a free form, two disulfide linkages are created between Cys121–Cys183 and Cys467–Cys474. The catalytic mechanism of PLAP is mediated by a number of specific amino acid residues, including Asp42, Glu311, His153, Ser92, Ser155, Asp357, Asp316, His432, His360, and His358.107 The active site of PLAP contains two Zn2+ ions, one of which is directly attached by three bonds to various amino acids (Asp316, His320 and His432), while the other is with a water molecule, which can be replaced by a suitable substrate. Even though there are four connections between the second Zn2+ ion and the residues of the amino acids Asp42, Ser92, Asp357, and His358, and using water or another suitable substrate, the fifth bond is created. The Mg2+ ion is hexacoordinated, which means that it establishes six bonds: three with Ser155, Glu311and Asp42 amino acid residues, and three more with three water molecules, which can be swapped out for substrate binding. Additionally, the presence of the Ca2+ ion is essential for the proper operation of the enzyme in all mammalian APs.108
4. Mechanism of action of alkaline phosphatases
All species have APs, and interestingly Escherichia coli and humans have about a 25–30% similarity in terms of their AP composition.109 The transphosphorylation and hydrolysis of different phosphate monoesters are an example of the APs catalytic function. A covalent phosphoserine intermediate is formed by APs to release inorganic phosphate. The general catalytic mechanism of APs two reaction stages is shown in Fig. 4. The initial alkaline phosphatase (E) catalyzed reaction includes a substrate (DO-Pi) binding phase, phosphate-moiety transfer to Ser-93 (in its active site's TNAP sequence), and product alcohol (DOH) release.110 Phosphate is liberated in the second stage of the reaction by hydrolysis of the covalent intermediate (E-Pi) and non-covalent complex (EPi) of inorganic phosphate in the active site. Phosphate is released via a transphosphorylation event in the presence of nitrogen-containing alcohol molecules (AOH), such as the buffer diethanolamine (DEA).111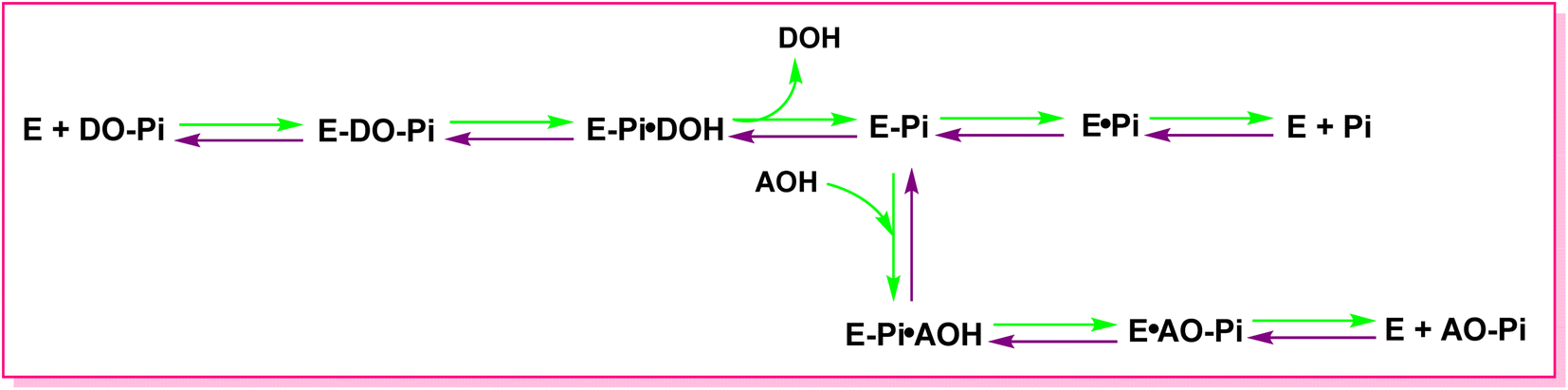 | ||
| Fig. 4 The catalytic mechanism of AP reaction. DO-Pi, substrate molecule; E, alkaline phosphatase enzyme molecule; E-Pi, phosphoenzyme. | ||
5. Significance of alkaline phosphatase in several physiological conditions
5.1 Dialysis
Dialysis is a medical process that involves removing excess water, solutes, and toxins from the blood of patients whose kidneys are not working correctly. Studies have shown that patients who undergo dialysis and have elevated levels of AP serum face a substantial mortality risk and an increased likelihood of cardiovascular disease. When AP concentrations are high, patients may develop coronary calcification due to the promotion of mineral deposition in their veins and arteries, as well as the production of polyphosphoric acid.1125.2 Cerebral small vessels disease (CSVD)
Stroke and dementia are neurological conditions brought on by CSVD. Blood circulation in this area of the brain declines when the brain's arterial vessels stiffen, leading to ischemic stroke. The activation of AP is caused by the vessel walls becoming calcified. The idea that AP may be used as a biomarker for CSVD was reinforced by the link between the serum levels of APs and the disease.1135.3 Periodontitis
The soft tissues encircling the teeth are attacked and harmed by the gum infection known as periodontitis. The AP enzyme is secreted during periodontitis, contributing to gingival inflammation, and serving as a biomarker. Patients with diabetes mellitus who have periodontitis have even greater levels of AP because their immune systems are weakened, rendering them more prone to infection.1145.4 Cancer
5.5 Relation between bone diseases and AP level
The amount of APs in the body is influenced by a variety of variables, including age, sex, and blood type. The level of APs will change in the presence of numerous pathological states in different tissues, making APs a valuable diagnostic tool for bone illnesses such as osteomalacia, osteoporosis and rickets.123 The AP level test will therefore be an excellent indicator of osteoporosis illness.124 Additionally, the AP level test can be used to assess the effectiveness of treatment for several disorders, such as osteoporosis (by administering alendronate), where a drop in the AP level denotes effective treatment. Because of this, it is essential to administer the AP tests accurately in order to prevent any changes in the findings that could have an impact on the diagnosis of disorders.1256. Alkaline phosphatase inhibitors
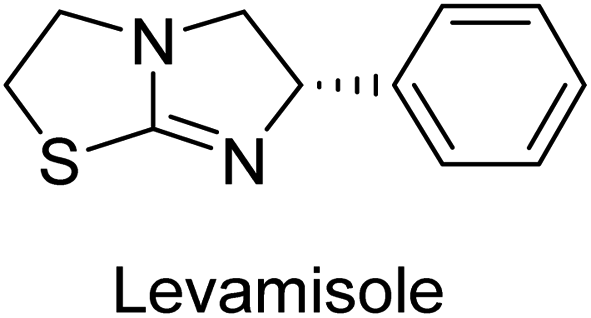
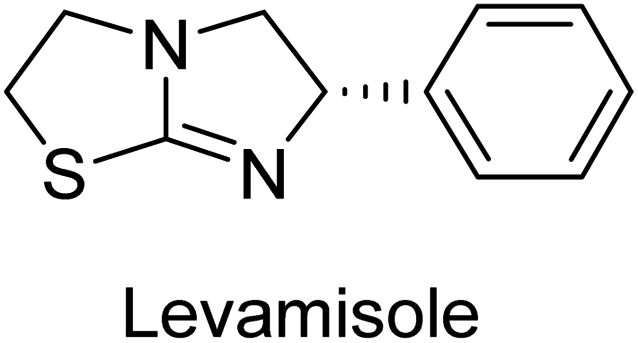
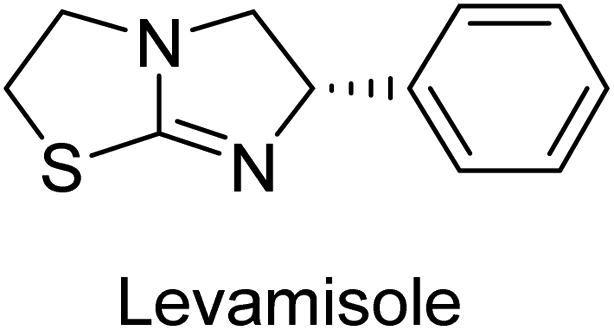
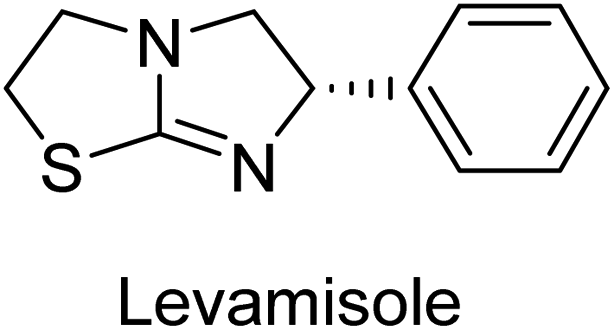
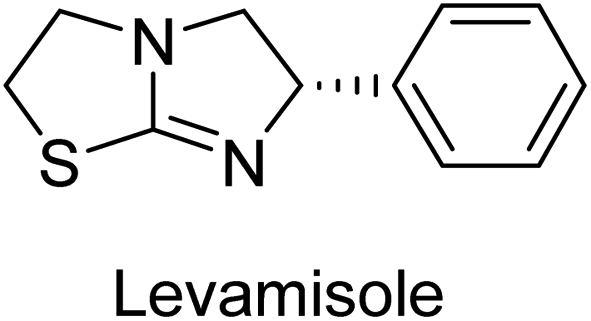
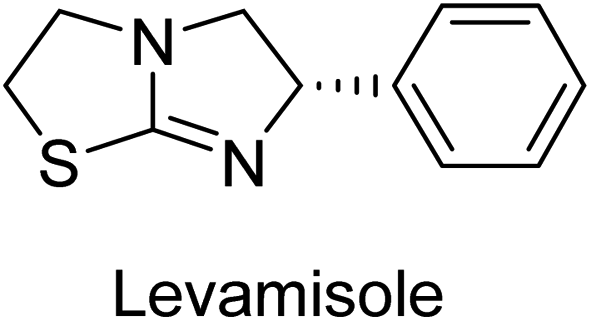
Millan et al. (2009) have discovered three new inhibitors of alkaline phosphatase's natural pyrophosphatase activity and demonstrated their potential in reducing vascular calcification in two different models. Specifically, these compounds were effective in inhibiting the in vitro calcification of cultured Enpp1−/− VSMCs and in suppressing the increased pyrophosphatase activity in a rat aortic model. The inhibitors 1, 2 and 3 possess highly aromatic molecular structures and were found to inhibit TNAP via an uncompetitive mechanism, both at physiological pH and at a pH of 9.8. The small variations in Ki values observed for two of the three inhibitors between pH 9.8 and pH 7.5 suggest that their positioning is non-ionic, which is consistent with the abundance of aromatic nitrogen atoms in their backbone structure as compared to the standard Levamisole (Fig. 5). The most potent inhibitor 3 demonstrated even greater potency at pH 7.5 and could be successfully modeled within the active site pocket of TNAP, spanning the enzyme's large pocket. These findings suggest that these inhibitors have potential as therapeutic agents for treating TNAP-related conditions and warrant further investigation.126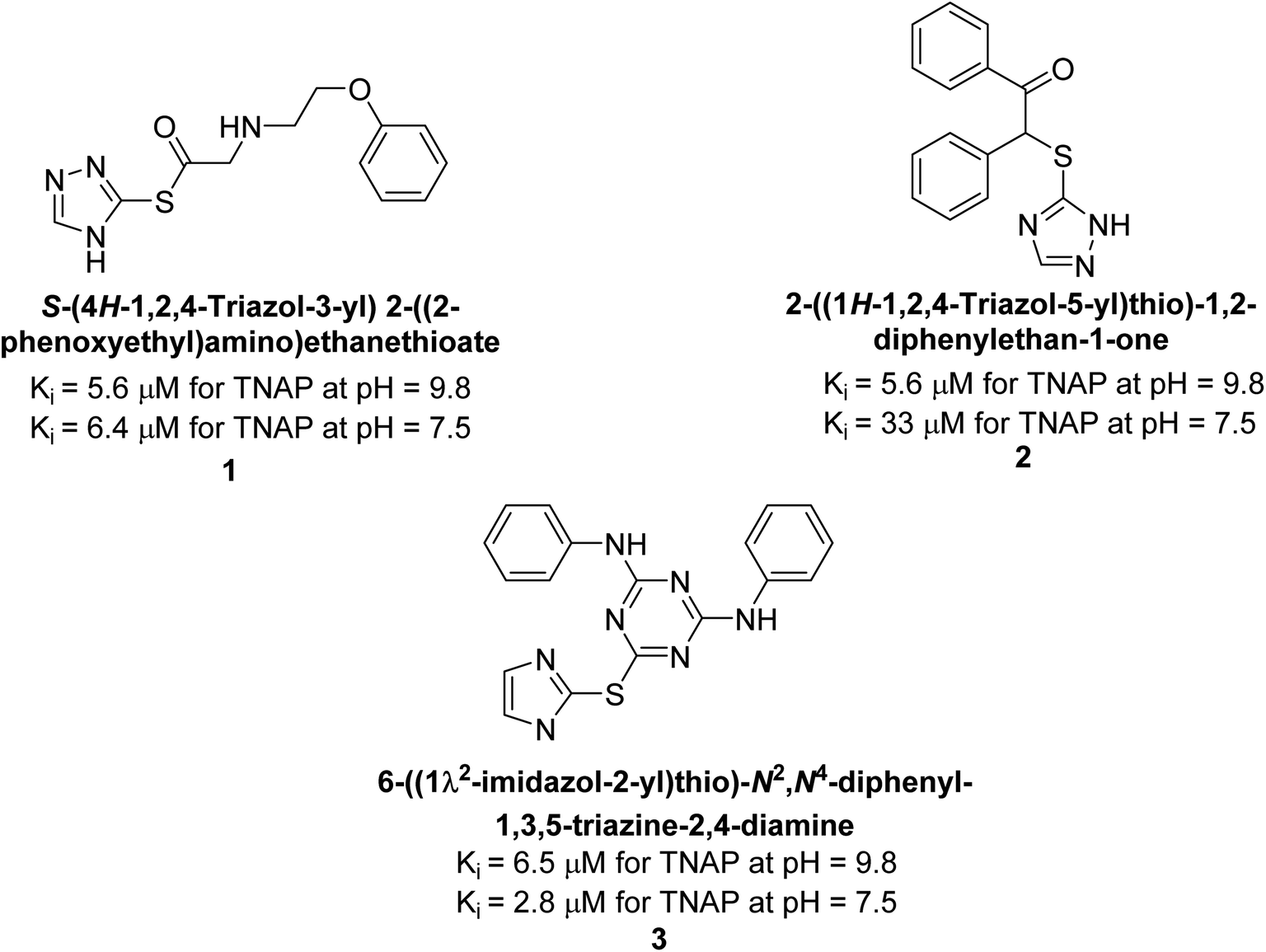 | ||
| Fig. 5 Chemical structure of compounds 1, 2 & 3 and its TNAP inhibition. | ||
Cosford et al. (2009) reported the design and synthesis of pyrazole derivatives as potent and selective inhibitors of TNAP. The 2,3,4-trichlorophenyl analog 4 was the most powerful compound in the series. The in vitro IC50 of 4, which showed excellent activity, was 0.005 μM (Fig. 6). Furthermore, compound 4 had selectivity for TNAP of at least 2000-fold because it was inert (IC50 > 10 μM) against the related PLAP isozyme. By performing the kinetic study, it was demonstrated that compound 4 behaved as a competitive inhibitor. Compound 4 is almost 200 times more effective against TNAP and exhibits a high degree of selectivity against the associated PLAP isozyme. The study has proved that the pyrazole derivatives with specific substitutions at the 5-position of the pyrazole ring, such as N-substitution and C-substitution, show potent and selective inhibition of TNAP. Moreover, the studies also highlighted the importance of optimizing the lipophilicity and hydrogen-bonding potential of the pyrazole derivatives for their activity.127
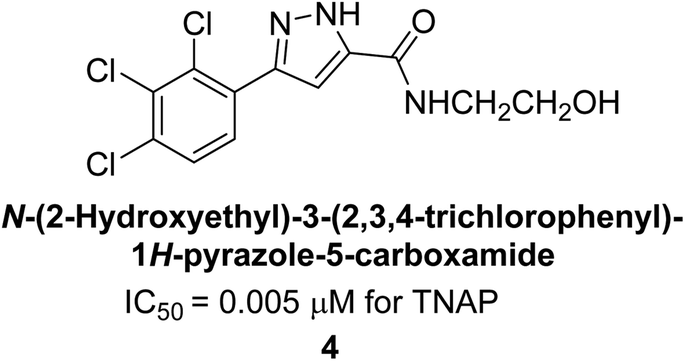 | ||
| Fig. 6 Chemical structure of compound 4 and its TNAP inhibition. | ||
Lemaire et al. (2009) reported the synthesis and evaluation of benzo[b]thiophene derivatives as inhibitors of AP. The benzo[b]thiophene derivatives were synthesized and assessed against AP enzymes as inhibitors. The results showed that some of the derivatives exhibited potent inhibitory activity, with IC50 values in the low micromolar range. The protocol describes the synthesis and TNAP inhibitory activity of benzo[b]thiophene derivatives, which may be used in osteoarthritis medication therapy. It was discovered that there exists water-soluble racemic 2-phenyl-benzo[b]thiophene-3-carbaldehyde 5 with apparent inhibition constants Ki = 85 μM, which are equivalent to the enantiomeric levamisole's 93 μM (Fig. 7). The findings suggest that benzo[b]thiophene derivatives have the potential to act as lead inhibitors of AP and warrant further investigation for their therapeutic potential in diseases associated with abnormal alkaline phosphatase activity.128
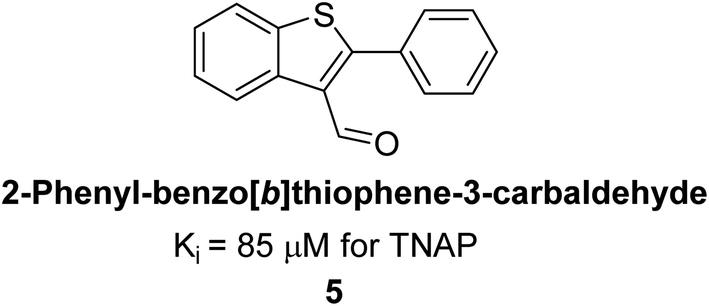 | ||
| Fig. 7 Chemical structure of compound 5 and its TNAP inhibition. | ||
Lanier et al. (2010) reported that the design and synthesis of selective inhibitors of PLAP were carried out using molecular modeling and chemical synthesis techniques. The activity of the inhibitors was evaluated using in vitro assays. The results showed that the synthesized inhibitors were effective and selective towards PLAP as compared to other related enzymes. Compound 6 has IC50 = 14.8 μM in the IAP assay, whereas compound 7 2-methylated counterpart did not exhibit any measurable inhibition (IC50 > 100 μM), resulting in a selectivity of more than 80-fold for IAP over PLAP (IC50 = 2.5 μM) (Fig. 8). These catechol compounds may be helpful as tools to comprehend the physiological significance of PLAP in biology and pharmacology due to their inhibitory specificity. The study provides a valuable contribution to the development of PLAP inhibitors for therapeutic use.129
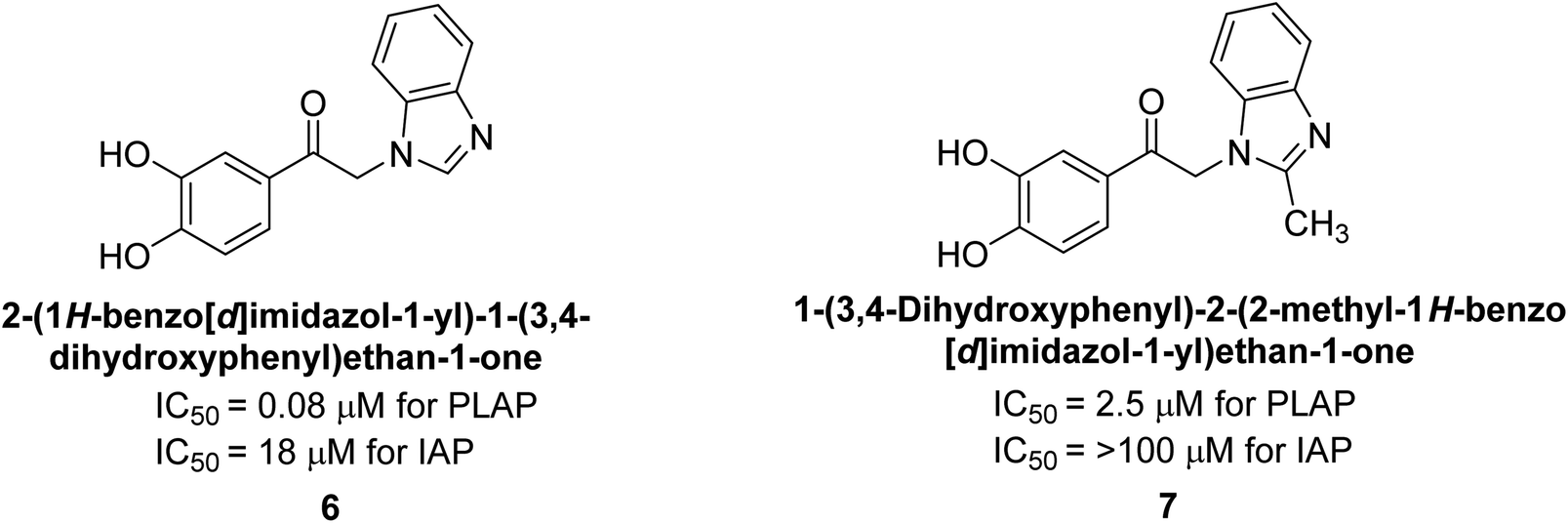 | ||
| Fig. 8 Chemical structures of compounds 6 and 7 and their PLAP and IAP inhibitions. | ||
Iqbal et al. (2012) reported the synthesis of polyoxometalates (POMs) and evaluated them as inhibitors of AP. These enzymes play a key role in various biological processes, including bone metabolism and tumor cell growth. All substances had nanomolar levels of AP inhibitory activity. The structure Na10[H2W12O42]·27H2O 8 was discovered with Ki = 313 nM as the effective inhibitor for TNAP, while the most effective inhibitor for TNAP was Na33[H7P8W48O184]·92H2O 9, with a Ki value of 135 nM. These findings highlight the therapeutic potential of POMs as inhibitors of alkaline phosphatases, offering a new approach to the treatment of cancer and other diseases associated with these enzymes.130
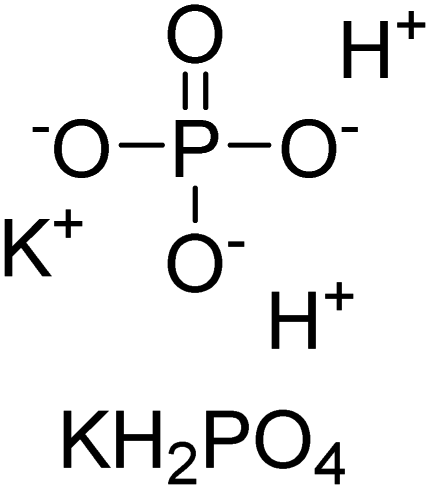
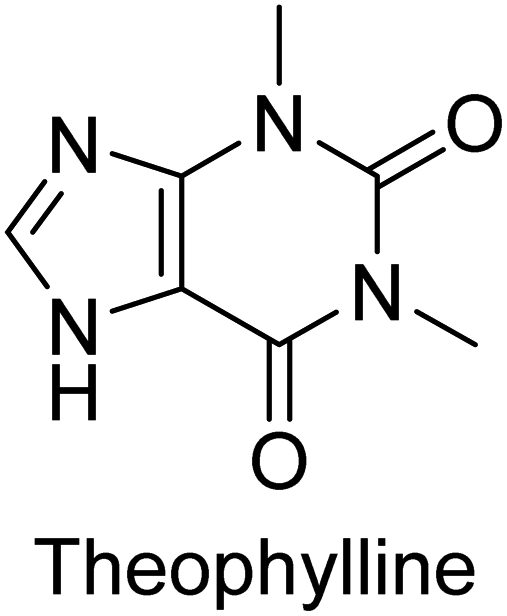
Iqbal et al. (2012) investigated the synthesis of new 1,2,4-triazole and 1,3,4-thiadiazole derivatives and their potential as inhibitors of AP. Among the synthesized series, only four of the synthetic compounds were effective inhibitors of AP in comparison to the standards KH2PO4 (IC50 = 3.11 μM; Ki = 1.5 μM) and theophylline (IC50 = 47 μM; Ki = 91 μM). Others either precipitated out or exhibited weak inhibition. In the triazole series, compound 10 with a chloro group at the meta position showed the greatest effectiveness with IC50 = 0.061 μM and Ki value 0.044 μM, however for compound 11, the inhibition declines (IC50 = 6.7 μM) upon transferring the chloro substituent to the para position. In contrast to 12, which has chloro functionality at the meta position and an IC50 of 20 μM, the powerful molecule 13 in the thiadiazole series has an IC50 of 0.15 μM. Accordingly, the pattern implies that inhibition rises when the chloro group is substituted at the para position (Table 2). Overall, the results showed that the synthesized compounds were moderate to good inhibitors against AP enzyme, indicating their potential as drug candidates for the treatment of related diseases.131
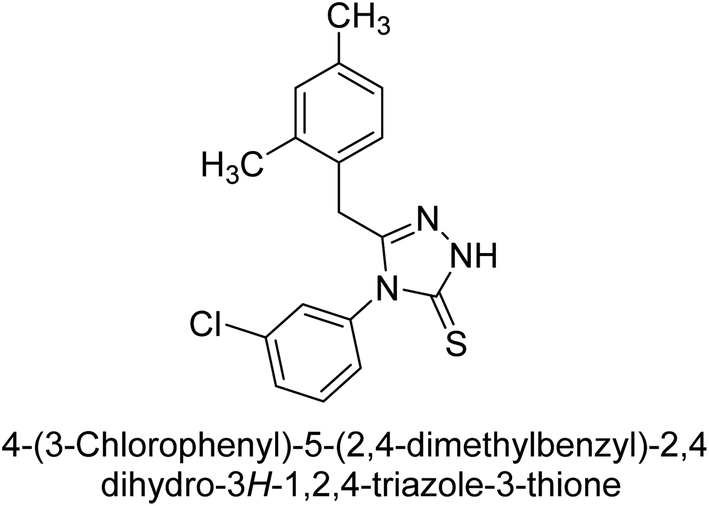
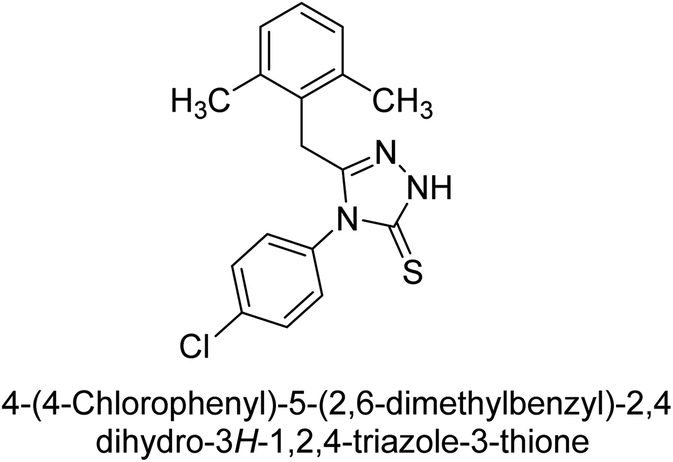
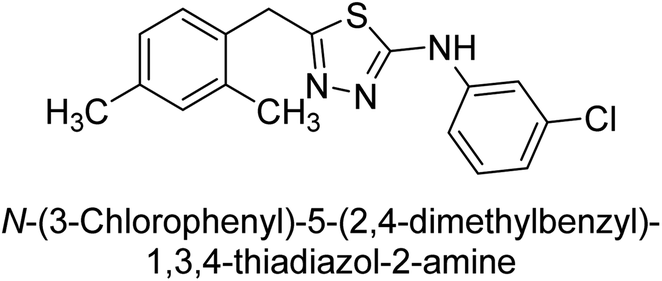
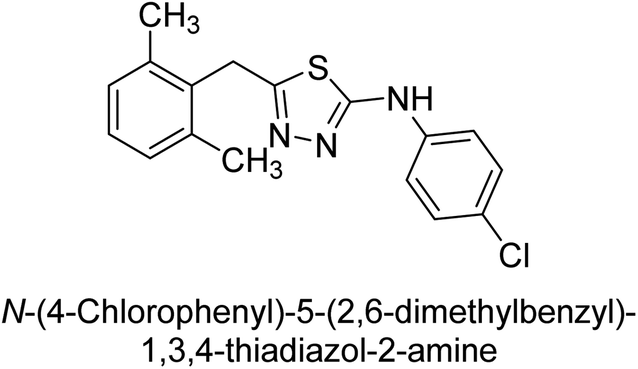
Buchet et al. (2013) reported inhibitors of TNAP have been an active area of research in recent years. Organic small molecules, including bisphosphonates, polyphenols, and quinoline derivatives, have shown great promise as potential therapeutic agents for TNAP-related disorders. These compounds 14 and 15 have demonstrated effective inhibition of TNAP both in vitro and in vivo and have shown the ability to reduce calcification in cell culture models (Fig. 9). Kinetic studies have provided insights into the mechanism of action and structure–activity relationships of these TNAP inhibitors. Furthermore, biomineralization assays have been used to evaluate their effects on mineralization processes, while cellular tests have demonstrated their potential in treating TNAP-related disorders.132
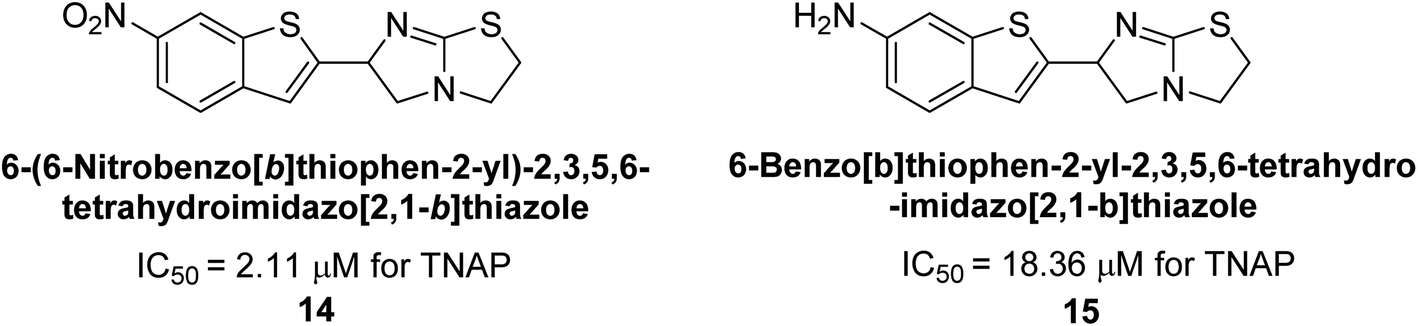 | ||
| Fig. 9 Chemical structures of compounds 14 and 15 and their TNAP inhibition. | ||
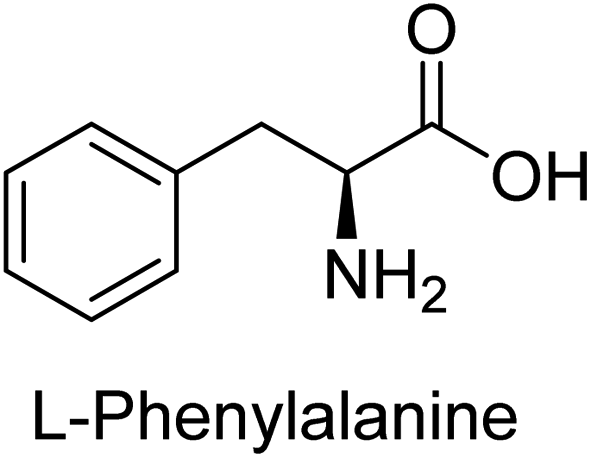
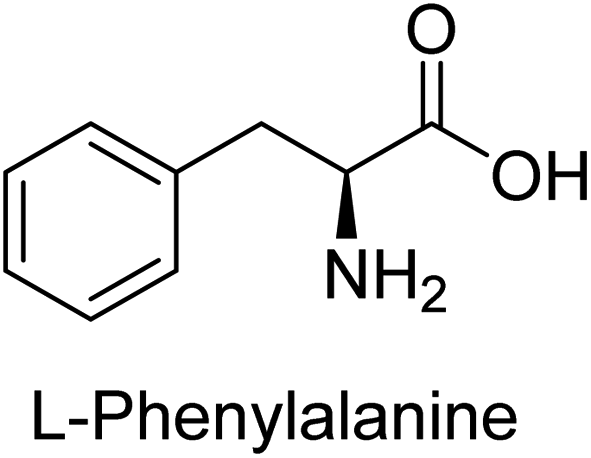
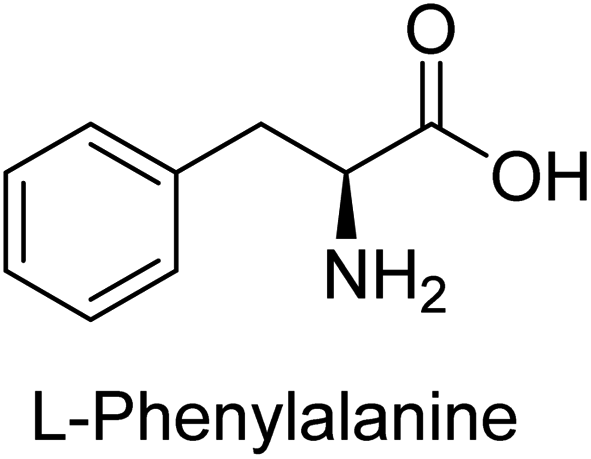
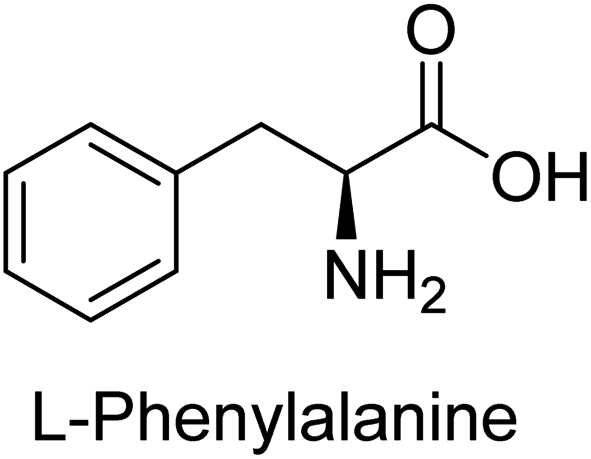
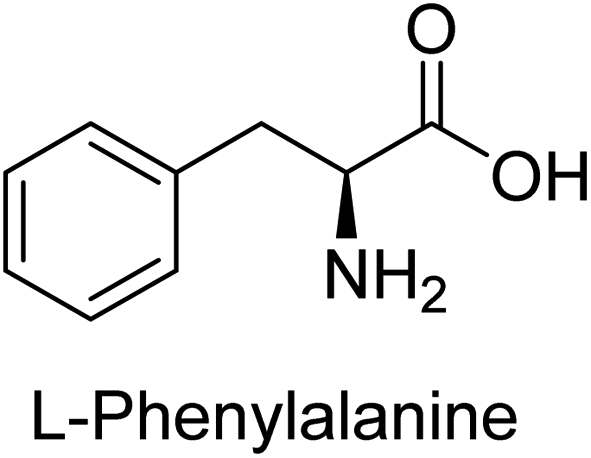
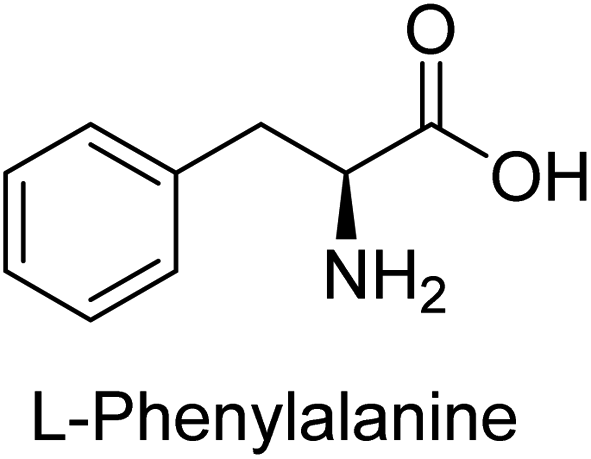
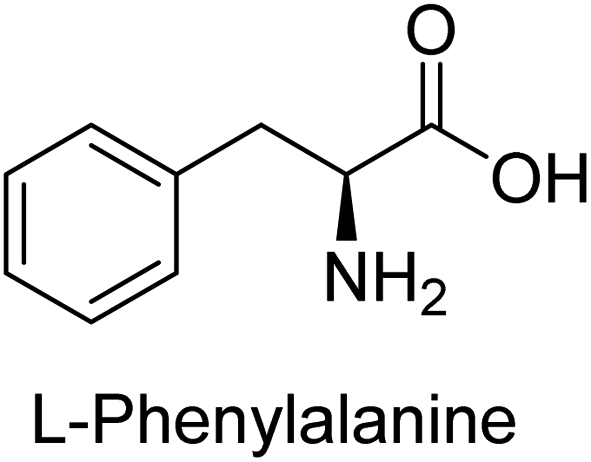
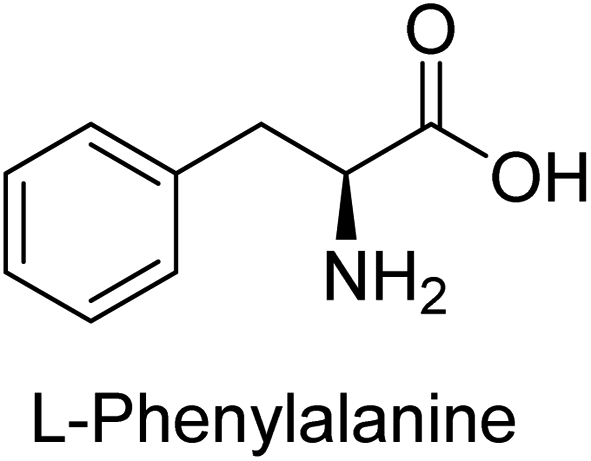
Hameed et al. (2015) studied the influence of structural variations on the imine functionality of 4-bromophenylacetic acid derived hydrazones in relation to their inhibition of AP through synthesis and molecular modeling. Several compounds from the examined series were recognized as lead candidates, with IC50 values ranging from micro to nanomolar. Against h-IAP, compound 16 p-allylated hydroxyl substituent exhibited ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold strong inhibition strength, and thus found as the most active inhibitor (IC50 = 10 μM). This inhibitory effect has a potency that is about 10000 times greater than that of the standard L-phenylalanine (IC50 = 300 μM). The compounds 17 (IC50 = 0.026 μM; ∼24-fold strong inhibition), 18 (m-acetylated hydroxyl group displayed ∼312-fold strong inhibition strength; IC50 = 0.32 μM) and 19 (o-benzylated hydroxyl group; ∼405-fold strong inhibition; IC50 = 0.74 μM) were effective inhibitors of TNAP, PLAP, and GCAP, respectively (Table 3). The results show the impact of diversified structural variations on the inhibition of AP.133
000-fold strong inhibition strength, and thus found as the most active inhibitor (IC50 = 10 μM). This inhibitory effect has a potency that is about 10000 times greater than that of the standard L-phenylalanine (IC50 = 300 μM). The compounds 17 (IC50 = 0.026 μM; ∼24-fold strong inhibition), 18 (m-acetylated hydroxyl group displayed ∼312-fold strong inhibition strength; IC50 = 0.32 μM) and 19 (o-benzylated hydroxyl group; ∼405-fold strong inhibition; IC50 = 0.74 μM) were effective inhibitors of TNAP, PLAP, and GCAP, respectively (Table 3). The results show the impact of diversified structural variations on the inhibition of AP.133
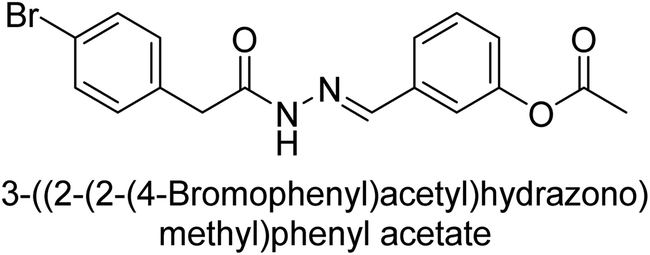
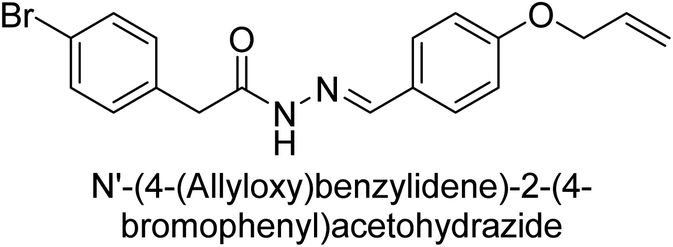
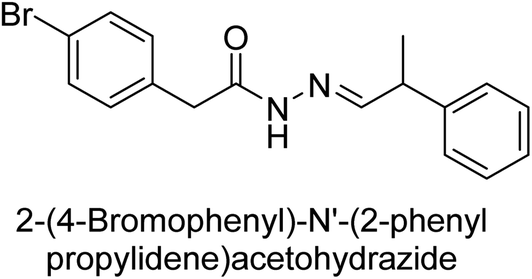
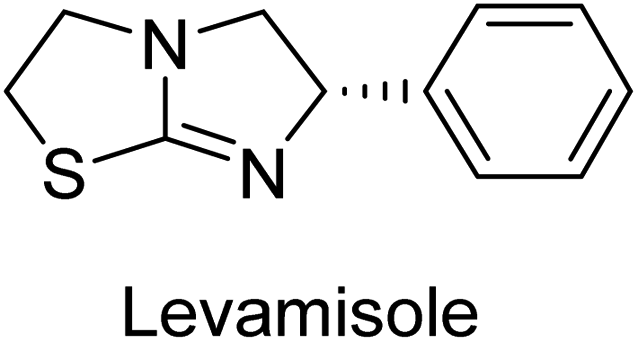
Iqbal et al. (2015) explored the inhibitory properties of diarylsulfonamides and their bioisosteres towards AP enzyme. Some of these substances were shown to be extremely effective and specific AP inhibitors. The synthesized compounds were reported to be selective b-TNAP and b-IAP inhibitors. The most potent compound among the series was 20 with (IC50=0.035 μM) for TNAP and 0.14 μM for IAP as compared to standards levamisole (IC50 = 19 μM) and L-phenylalanine (IC50 = 80 μM) (Fig. 10). Detailed kinetic tests for the most active AP inhibitor 20 revealed a competitive mechanism of inhibition against TNAP and a non-competitive method of inhibition against IAP. The study focused on understanding the SAR of these compounds and the molecular mechanisms behind their inhibitory activity. The results of the study provide insights into the design and development of novel dual inhibitors of AP enzyme.134
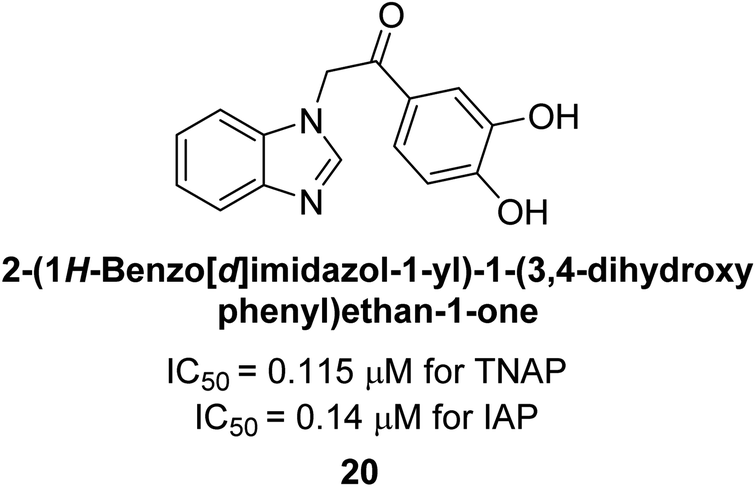 | ||
| Fig. 10 Chemical structure of compound 20 and its TNAP and IAP inhibitions. | ||
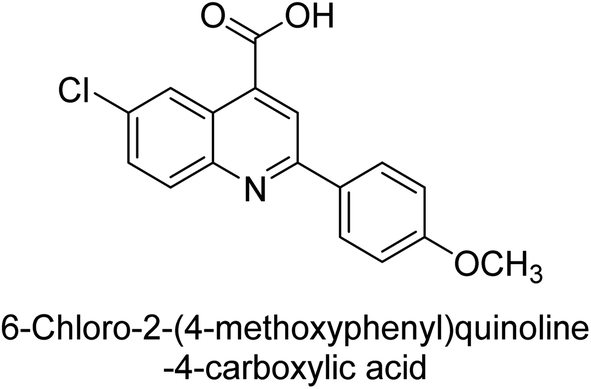
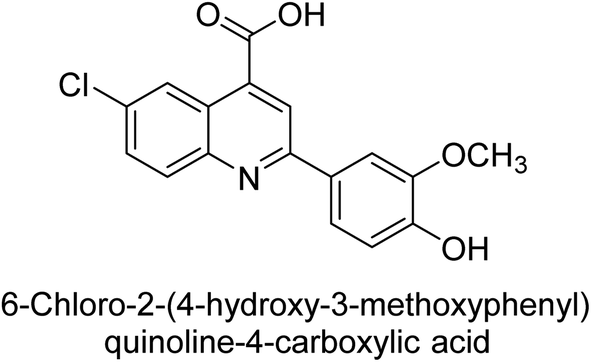
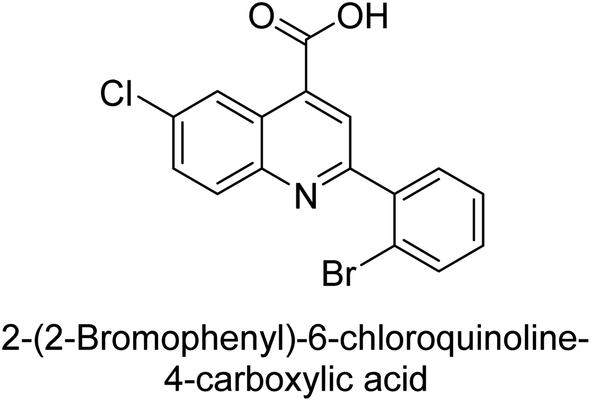
Iqbal et al. (2015) investigated quinoline-4-carboxylic acid as a promising scaffold for developing inhibitors of AP. The majority of the examined substances significantly inhibited h-TNAP, h-IAP and h-PLAP. While 21 appeared as a leading contender against h-IAP and h-PLAP with IC50 values of 34 μM and 82 μM, respectively, 22 was discovered as a powerful inhibitor of h-TNAP with an IC50 value of 22 μM. With an IC50 value of 150 μM, 23 was a strong inhibitor of h-GCAP (Table 4). Molecular modeling studies provided insights into the interactions between the inhibitors and AP.135
Saeed et al. (2015) reported the synthesis of bis-coumarin-iminothiazole hybrids and their inhibitory activity against AP. When the synthetic analogs were tested against AP, compound 24 emerged as a powerful inhibitor with an IC50 value of 1.38 μM. In contrast to the standard inhibitor KH2PO4 (IC50 = 2.43 μM), this inhibitor's efficacy is two times higher (Fig. 11). The two methyl groups that are present at the 3- and 5-positions of the aryl ring that is immediately linked to the thiazole core may be responsible for this two-fold strong inhibitory effect. Molecular modeling was used to gain insights into the molecular interactions of the hybrids with the target proteins.136
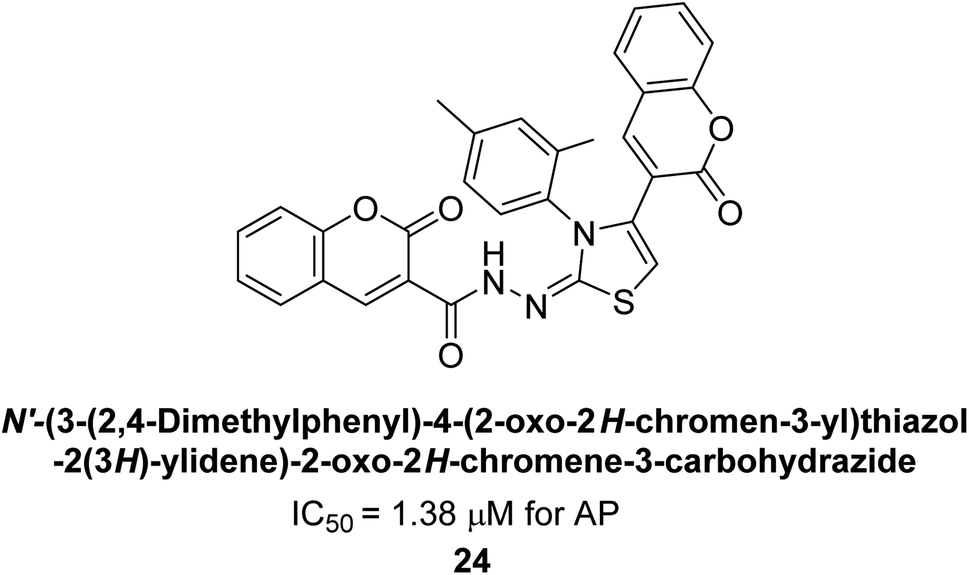 | ||
| Fig. 11 Chemical structure of compound 24 and its AP inhibition. | ||
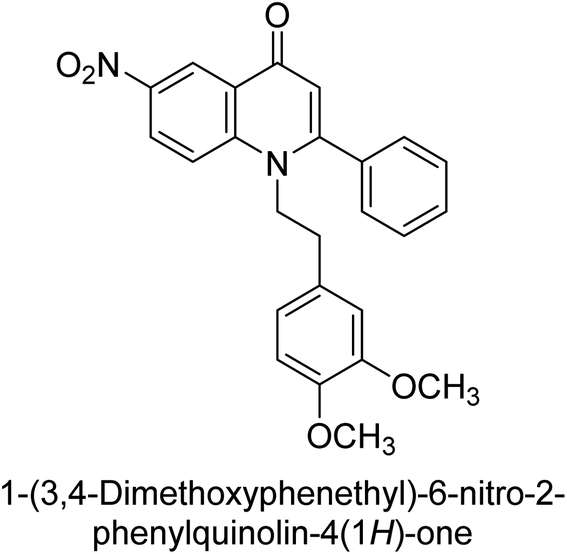
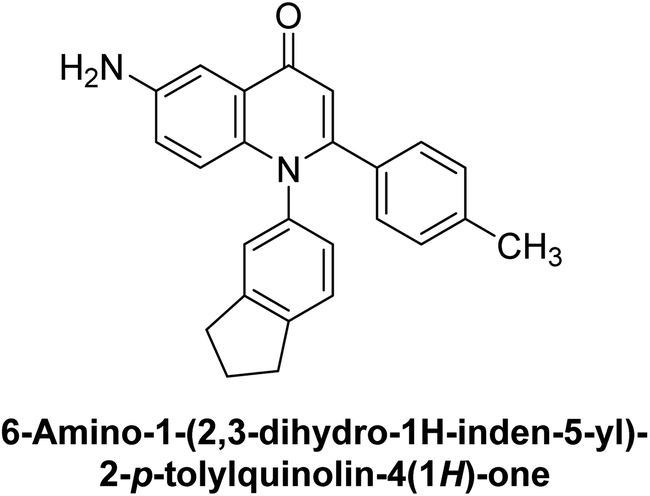
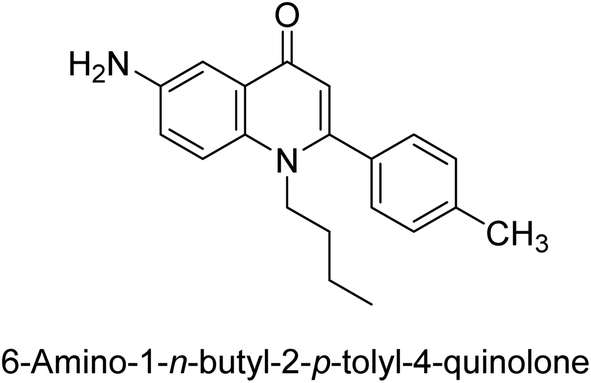
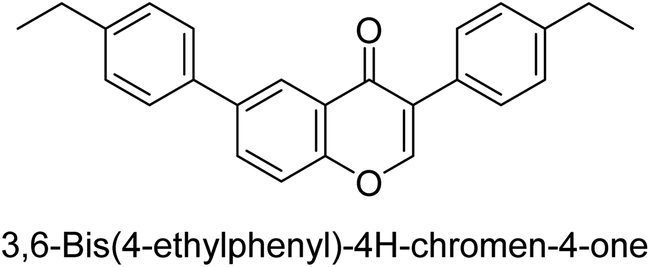
Langer et al. (2015) synthesized a series of 6-nitro- and 6-aminoquinolones and evaluated them for their inhibitory activity against AP. They investigated the ability of the 6-nitroquinolones and the 6-aminoquinolones to inhibit the b-TNAP and c-IAP, respectively. All nitroquinolones were capable of inhibiting TNAP, with IC50 values ranging from 1.43 to 134.1 μM for each compound. A particularly effective and specific inhibitor of b-TNAP with an inhibitory value of IC50 = 6.34 μM was discovered to be 25 among them. Comparing this substance to the assay's reference standard, Levamisole, which had an IC50 value of 19.21 μM, revealed a threefold increase in potential. All aminoquinolones showed strong anti-b-TNAP action, with IC50 values ranging from 1.14 to 78.1 μM. Although some of these compounds were also effective against c-IAP, it was discovered that most of these compounds were selective inhibitors of b-TNAP. The range of the activity against c-IAP was IC50 = 0.443 to 176.4 μM. The most effective derivative was discovered to be compound 26, with an IC50 value of 0.443 μM. Among the series, compounds 27 and 28 are the only two to have a chromene substructure. It was discovered that these substances were more effective against c-IAP than b-TNAP. The IC50 values for the inhibitory effects on c-IAP were 0.797 and 1.41 μM, respectively. Contrarily, these compounds displayed an IC50 range of 5.84 to 40.9 μM against b-TNAP (Table 5). The results showed that some of the compounds exhibited potent inhibitory activity, making them promising candidates for further development as AP inhibitors and potential therapeutic agents.137
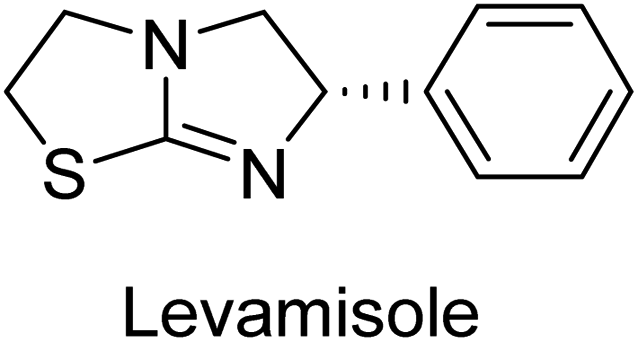
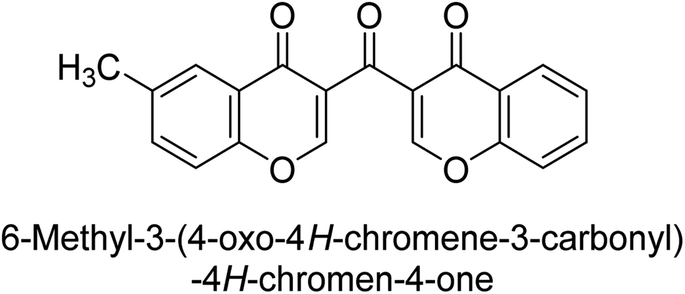
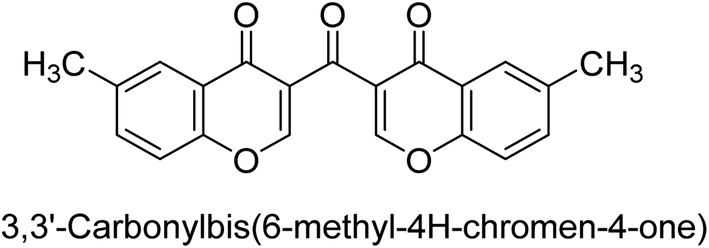
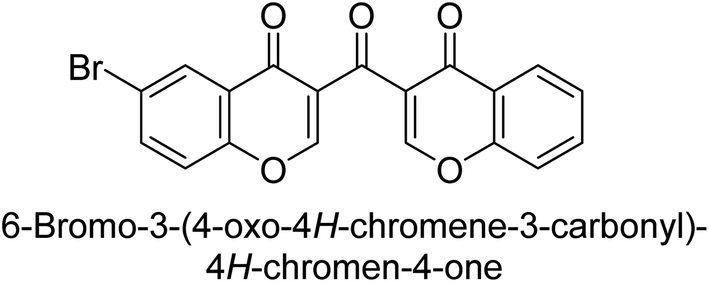
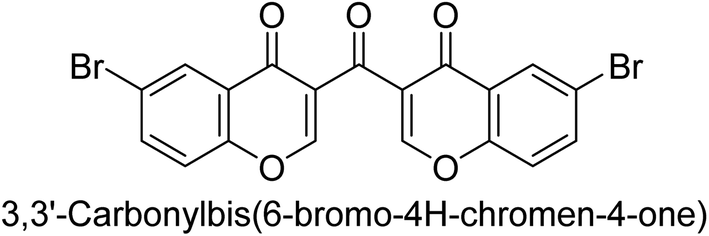
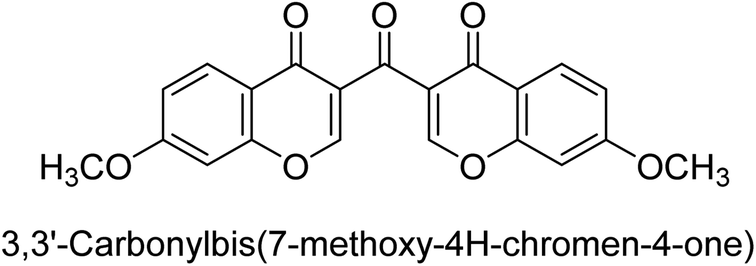
Langer et al. (2016) synthesized 3,3′-carbonyl-bis(chromones) and evaluated them for their inhibitory activity against mammalian AP, and some compounds showed promising inhibitory effects, with some having IC50 values in the low micromolar range. The molecular mechanisms of inhibition were also studied using molecular docking simulations, which revealed that the target compounds form hydrogen bonds with the active site residues of AP, thereby inhibiting its activity. The inhibitory potential of all synthesized compounds was evaluated against two types of alkaline phosphatases, b-TNAP and c-IAP. The results demonstrated that the investigated derivatives showed significant inhibition against both enzymes. The potency of compounds 29, 30, 31, and 32 on b-TNAP and c-IAP was shown to be equal. The most effective inhibitor of b-TNAP among all the compounds studied was 30, with an inhibitory value of IC50 = 2.47 μM. It demonstrated a 9-fold more inhibitory capacity than the standard drug Levamisole, with an IC50 value of 19.21 μM. However, compound 33 was discovered to be a selective b-TNAP inhibitor, with an inhibitory value of IC50 = 6.61 μM. Compared to the reference compound, it demonstrated three times the potential (Table 6). With an inhibitory value of IC50 = 0.653 μM, compound 34 was discovered to be the most effective inhibitor against c-IAP.138
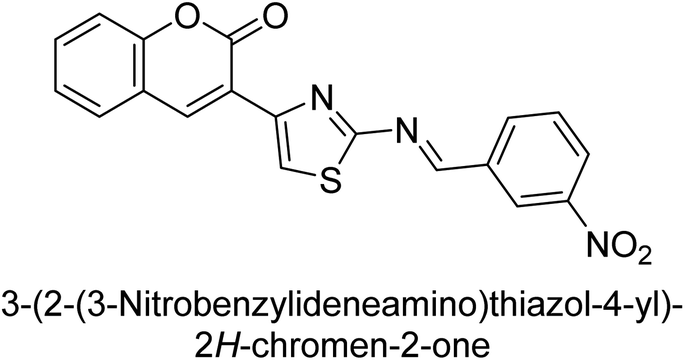
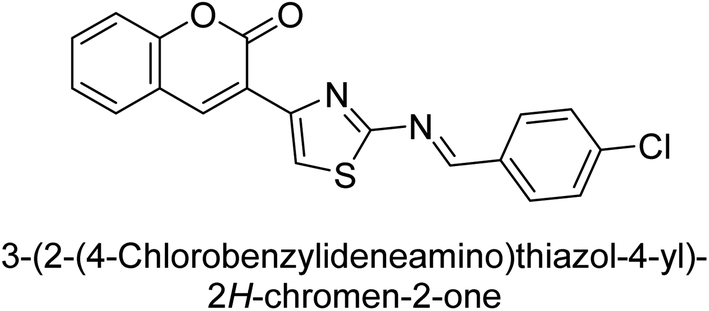
Saeed et al. (2016) described the discovery of a new class of compounds, 3-(5-(Benzylideneamino)thiozol-3-yl)-2H-chromen-2-ones, which have been found to inhibit both AP and ecto-5′-nucleotidase. The ability of each substance to inhibit human recombinant ecto-nucleotidases, such as h-TNAP, h-IAP, and human and rat ecto-5′-nucleotidase (h-e5′NT & r-e5′NT), was examined. All compounds were shown to be effective and selective inhibitors of h-e5′NT, while compounds 35 and 36 were the most effective (IC50 = 0.25 μM and 0.28 μM, respectively). Most of the compounds were found to have inhibitory effects in the lower micromolar range and to preferentially inhibit h-TNAP over h-IAP. Compounds 37 and 38 exhibited the most potent h-TNAP inhibition (IC50 = 0.21 μM and 0.22 μM, respectively), which is approximately 91 times more potent than the inhibitory action of the benchmark drug levamisole. Compound 39, which showed approximately 11 times better selectivity for h-IAP over h-TNAP, was discovered to be the most effective h-IAP inhibitor (IC50 = 0.05 μM) (Table 7). These inhibitors have been evaluated for their potency and efficacy, showing their potential as therapeutic agents for various medical conditions related to phosphatase and nucleotidase overactivity.139
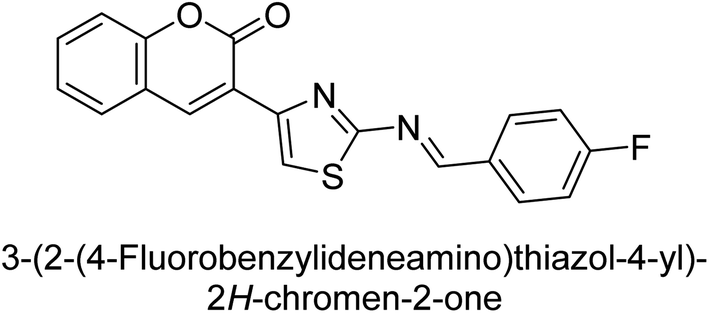
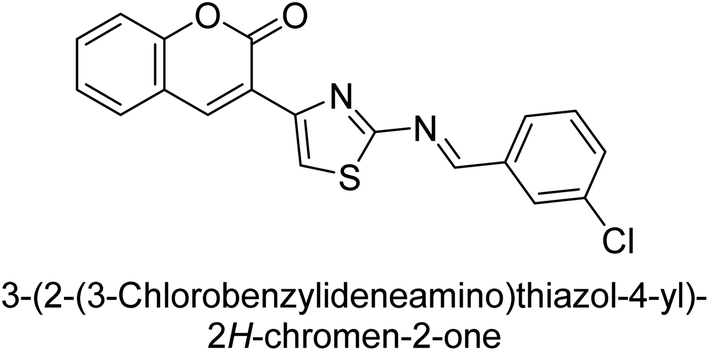
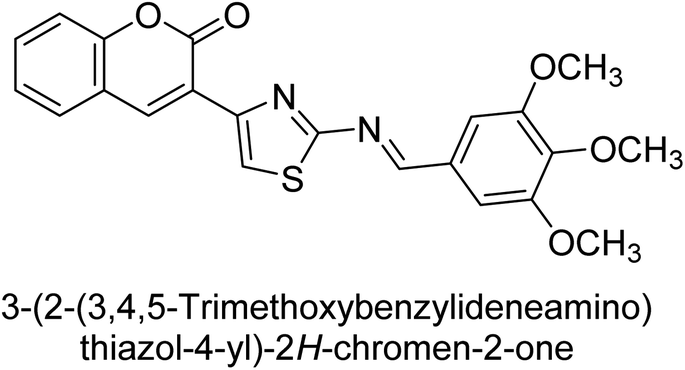
Saeed et al. (2016) also synthesized a series of coumarin-triazolothiadiazine hybrid analogs and evaluated them against AP. The synthesis of these hybrids was achieved through a combination of chemical reactions and the results were characterized through various analytical techniques. Compound 40, which has bis-coumarinyl motifs at the heteroaromatic core's 3- and 6-positions, proved to be a powerful inhibitor with an IC50 value of 1.15 μM as compared to KH2PO4 (IC50 = 2.41 μM) (Fig. 12). The molecular docking analysis was performed to study the binding modes and interactions of the hybrids with the target enzymes, providing insights into the nature of their inhibitory effects.140
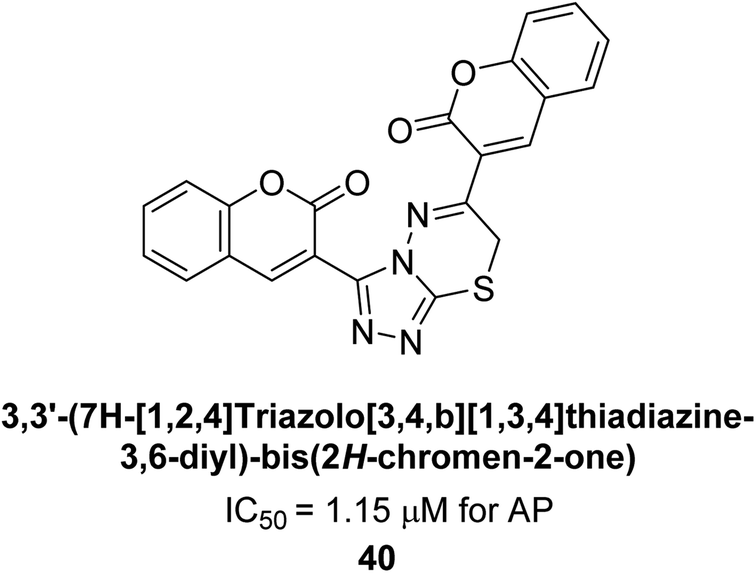 | ||
| Fig. 12 Chemical structure of compound 40 and its AP inhibition. | ||
Iqbal et al. (2017) investigated novel chalcone sulfonamide hybrids as inhibitors of intestinal IAP. The synthesized compounds were tested for their ability to block the activity of the c-IAP isozyme. The target series displayed magnificent inhibition activity with IC50 values in the range of 0.12 to 2.57 μM as compared to the standard L-phenylalanine (IC50 = 80.2 μM). Compound 41 showed maximum inhibition of c-IAP with an IC50 = 0.12 μM (Fig. 13). The SAR analysis of these compounds revealed that the presence of a substituent on the phenyl ring and the nature of the sulfonamide moiety are important for IAP inhibition. According to the docking experiment of compound 41 into the crystal structure of c-IAP, the sulfonamide group interacts with the Ser111 and Arg185 residues of the active site by hydrogen bonds, and the sulfonamide nitrogen atom interacts with two zinc metal ions. Additionally, the chlorine atom at the benzene ring interacts electrostatically with the amino acid residue Arg127. Furthermore contributing to the stability of compound 41, the benzene ring with the sulfonamide group forms a π–π stacking with His339 (Fig. 13).141
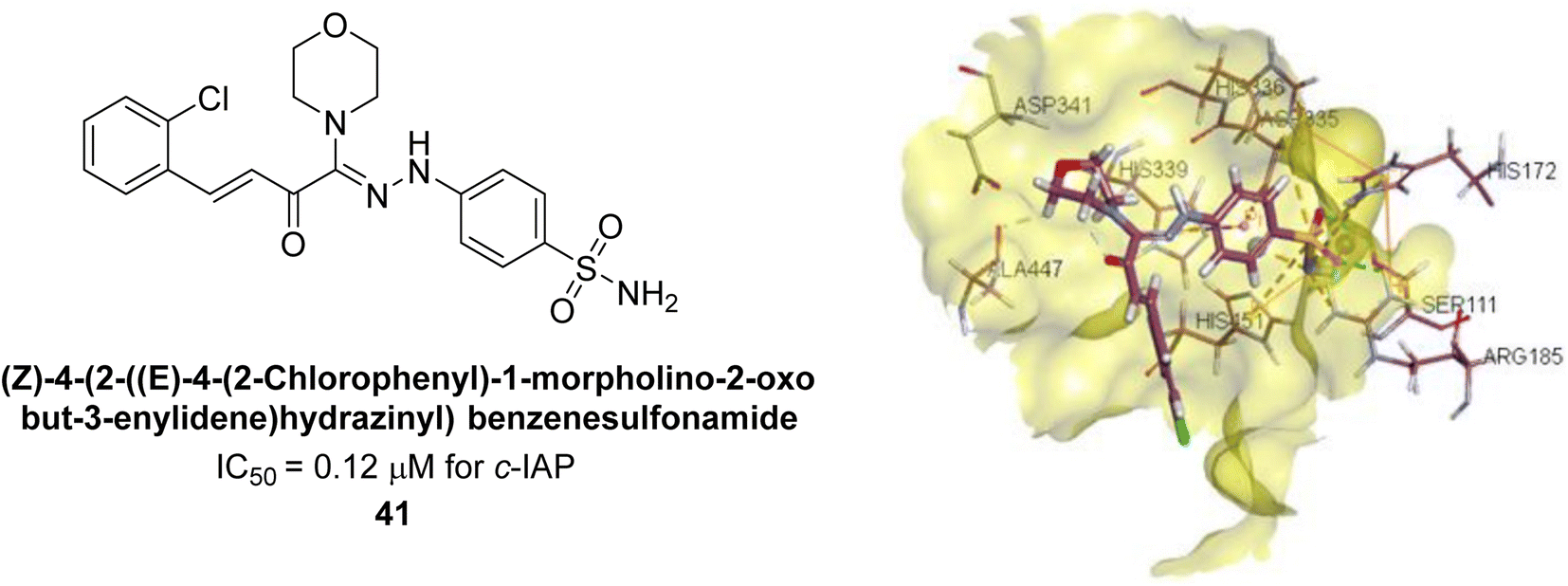 | ||
| Fig. 13 Chemical structure and docking image of 41 and its c-IAP inhibition. | ||
Iqbal et al. (2017) described the synthesis and evaluation of cyclic sulfonamides as AP inhibitors. The trigger for the synthesis was the introduction of dimethyl amino groups. A facile synthesis was used to produce the compounds, which were then evaluated for their inhibitory activity against AP. The potential of all compounds to inhibit AP (b-TNAP and b-IAP) was evaluated. The results showed that all compounds were excellent inhibitors of b-TNAP with IC50 values in the low micromolar range (0.11–6.63 μM). The majority of the compounds were selective towards b-TNAP over b-IAP, with only six compounds being active against b-IAP with IC50 values ranging from 0.38–3.48 μM, furthermore, the most potent inhibitor was 42 (IC50 = 0.38 μM). Compound 43, with a p-NO2 substituent, was found to be the most potent inhibitor of b-TNAP (IC50 = 0.11 μM), followed by compounds 44 (IC50 = 0.19 μM) and 45 (IC50 = 0.20 μM). 43 was not only the most active inhibitor but also a highly selective one for b-TNAP, showing only 40% inhibition against the other isozyme, b-IAP (Table 8). The results revealed promising potential as inhibitors, indicating a possible future application in the treatment of diseases related to overactivity of AP.142
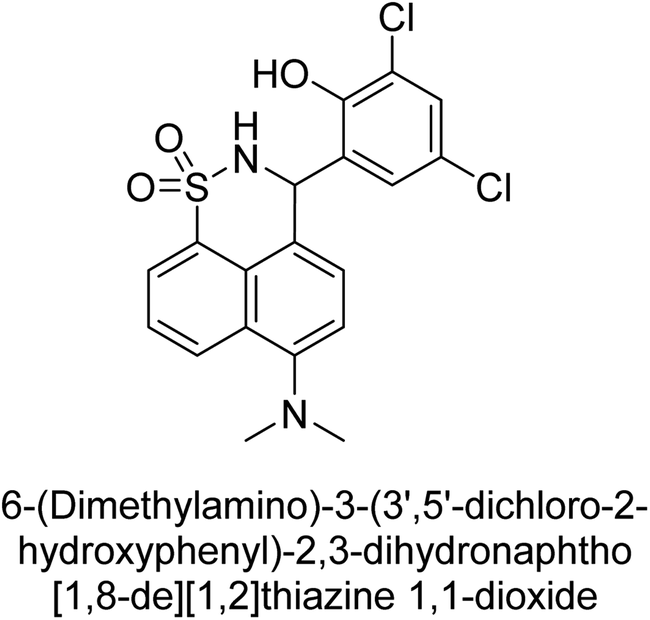
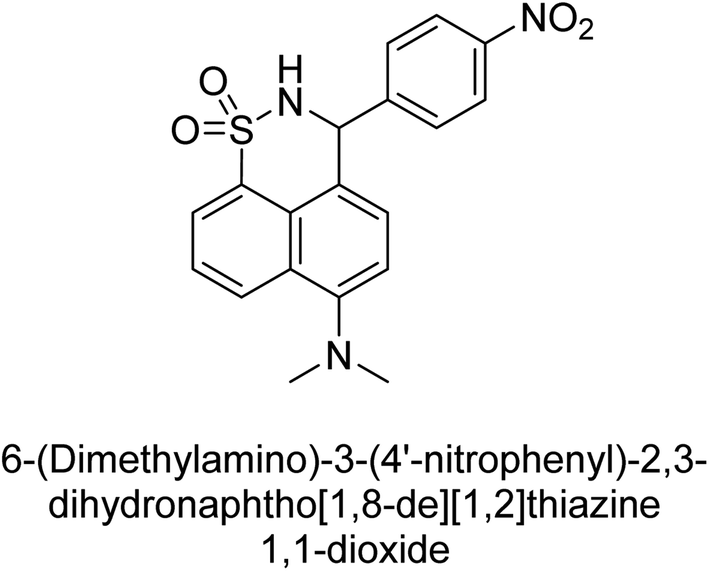
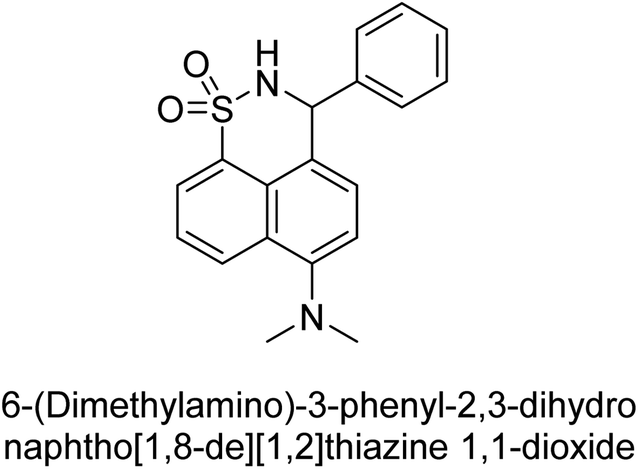
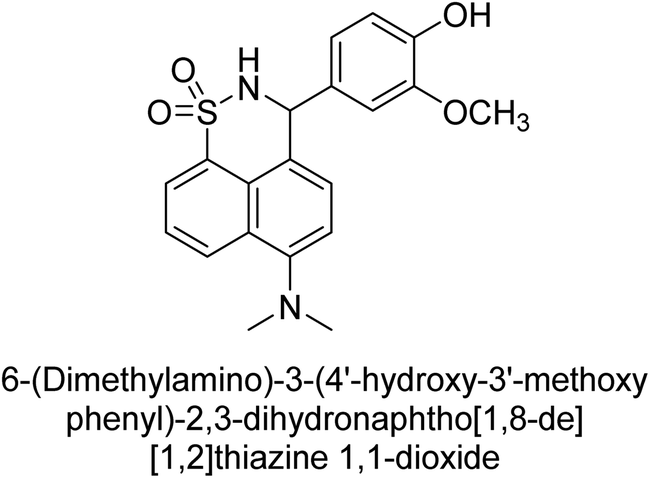
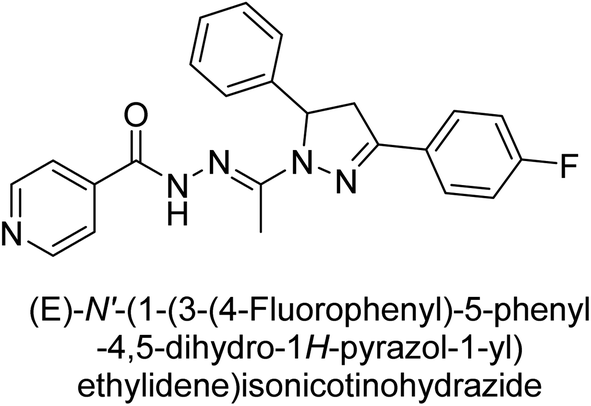
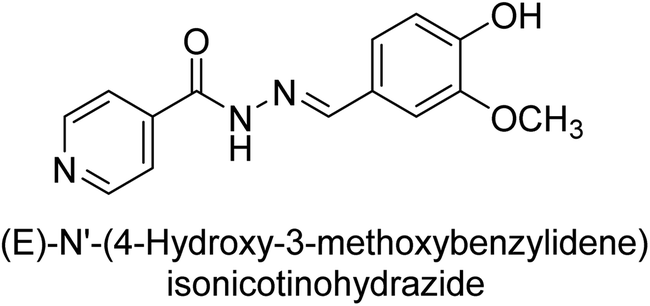
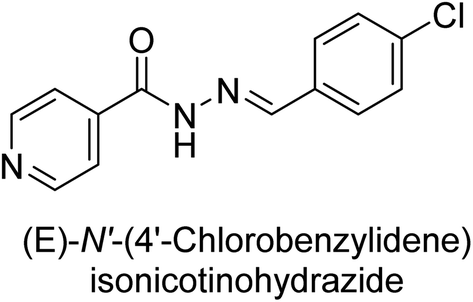
Iqbal et al. (2017) synthesized and tested a series of isonicotinohydrazide derivatives as inhibitors of recombinant h-e5′NT, r-e5′NT and AP isozymes, such as b-TNAP and c-IAP. These enzymes play a role in various medical conditions, including vascular calcifications, solid tumors, and cancers. The results showed that all tested derivatives were active against both enzymes, with the most potent inhibitors being 46 (IC50 = 0.19 μM) for h-e5′NT, 47 (IC50 = 0.14 μM) for r-e5′NT, 48 (IC50 = 0.67 μM) for c-IAP, and 47 (IC50 = 0.35 μM) for b-TNAP (Table 9). The study also included computational determination of the binding modes of the most potent inhibitors against the target enzymes.143
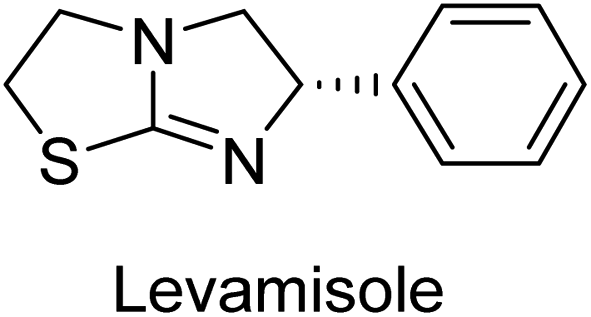
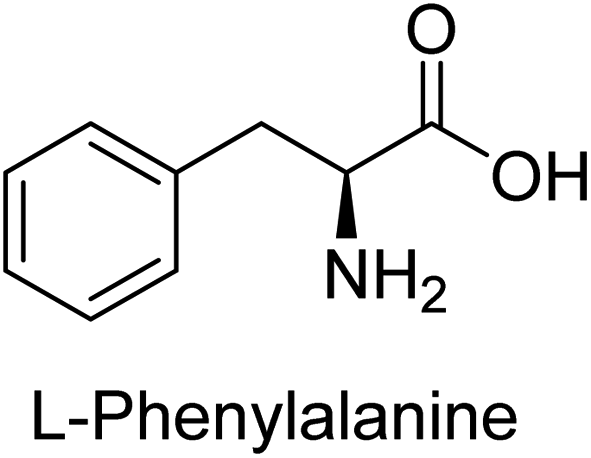
Khan et al. (2017) focused on the evaluation of coumarin sulfonates as potential inhibitors of AP enzymes. The study employs both in vitro and in silico techniques to examine the inhibition properties of these compounds. The ability of each substance to reduce alkaline phosphatase activity was examined (h-TNAP and h-IAP). The majority of the substances were discovered to be AP inhibitors. Compound 49 was found to be the most potent h-IAP inhibitor, with an IC50 value of 1.11 μM, whereas compound 50 was found to be the most potent h-TNAP inhibitor, with an IC50 value of 0.58 μM as compared to the standards levamisole (IC50 = 20.2 μM) and L-phenylalanine (IC50 = 100 μM) (Fig. 14). The results suggested that coumarin sulfonates have potential as AP inhibitors, with certain compounds showing strong inhibition activity in vitro. The in silico analysis provides further insight into the molecular interactions and mechanisms of inhibition. The findings of this study contribute to the understanding of coumarin sulfonates as potential therapeutic agents for AP-related disorders.144
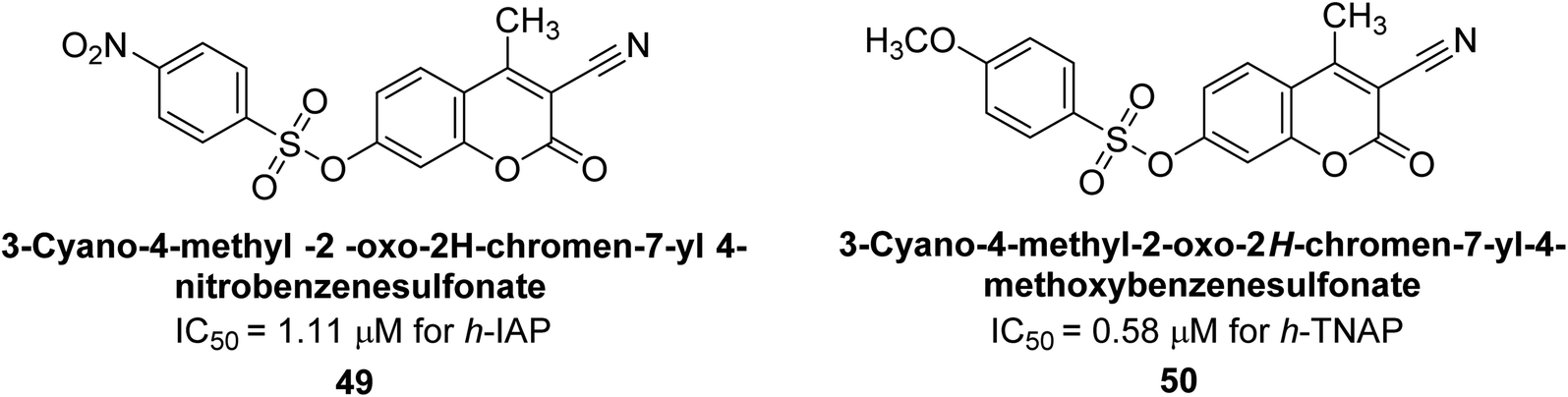 | ||
| Fig. 14 Chemical structures of compounds 49 and 50 along with their TNAP and IAP inhibitions. | ||
Langer et al. (2017) synthesized and identified new furan-2(3H)-one analog as AP inhibitors. Many of the analogs were discovered to be powerful and selective inhibitors of TNAP. Inhibition of b-TNAP by compound 51 (IC50 = 1.34 μM) was 14 times larger than that of levamisole (the reference standard), whilst that of c-IAP by compound 52 (IC50 = 5.37 μM) was 104 times more than that of reference L-phenylalanine. One molecule was shown to be the most effective b-TNAP inhibitor among the derivatives examined, having a strong inhibitory value and a 6-fold higher level of b-TNAP specificity compared to c-IAP (Fig. 15). Due to the presence of two furan rings, this molecule has a stronger inhibitory action towards b-TNAP as compared to levamisole and L-phenylalanine. The most effective inhibitor of c-IAP was determined to be another derivative with a furan ring, which had a reduced inhibitory capacity towards b-TNAP.145
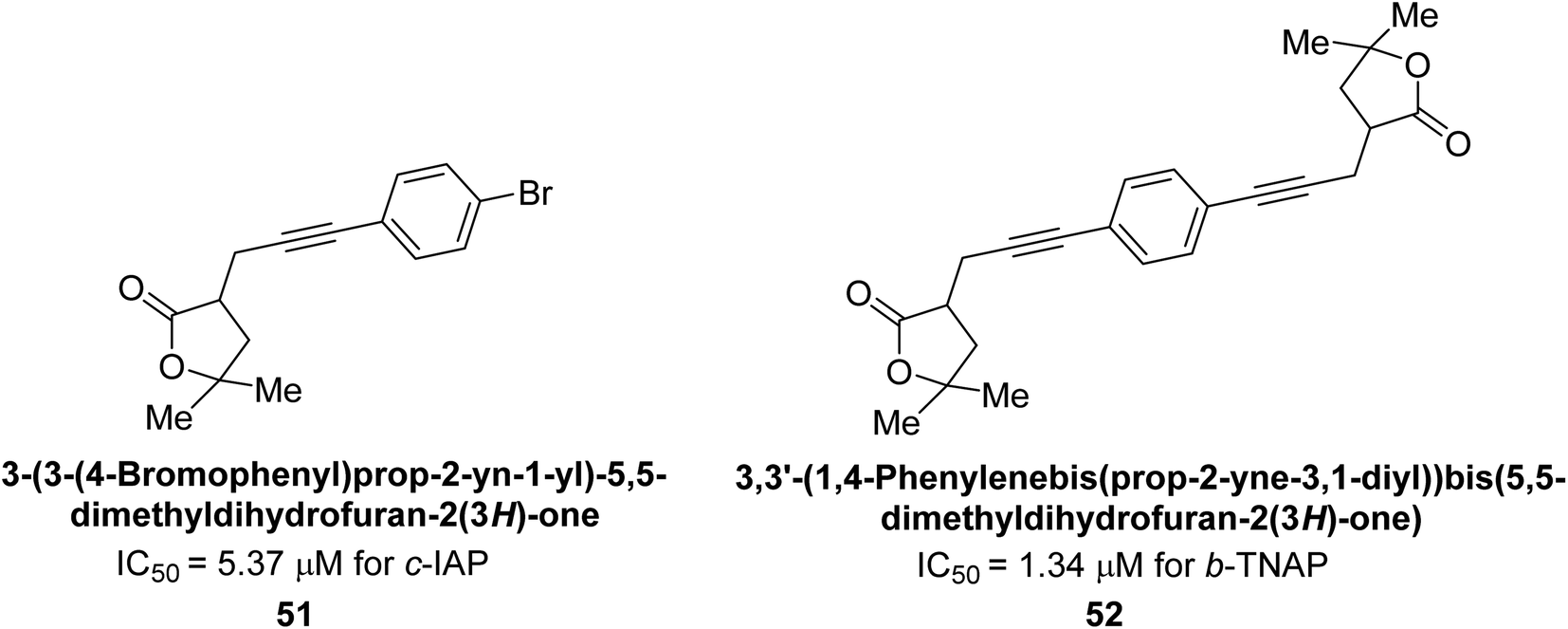 | ||
| Fig. 15 Chemical structures of compounds 51 and 52 and their TNAP and IAP inhibitions. | ||
Langer et al. (2017) described the synthesis and characterization of novel derivatives of 4-quinolones, and their inhibitory effects on the enzyme alkaline phosphatase, including in vitro and molecular docking to predict the interactions between the compounds and the enzyme. Superior inhibitory efficacy and moderate selectivity are characteristics shared by the majority of the compounds. The range of the IC50 for IAP was 1.06 to 192.10 μM, whereas the range for TNAP was 1.34 to 44.80 μM. In comparison to TNAP, the most active derivative 53 (Fig. 16) (IC50 = 14.80 μM for TNAP; 1.06 μM for IAP) has a ∼14-fold greater selectivity and a powerful inhibition on IAP than standards L-phenylalanine (80.21 μM) or levamisole (19.21 μM), respectively. The study found that compound 53 binds to the active site of IAP with high binding affinity, and the binding mode is in agreement with the known structure of IAP inhibitors. The compound forms hydrogen bonds with the key residues in the active site and interacts with other residues through hydrophobic interactions. The docking study suggests that 53 is a potential IAP inhibitor, and it may be useful as a lead compound for the development of new drugs against cancer (Fig. 16). The study provides valuable information for further optimization of the compound to improve its binding affinity and specificity, and for the design of in vitro and in vivo studies to evaluate its potential as a therapeutic agent. The results of the study suggested that these 4-quinolone derivatives have potential as new inhibitors of AP and might be useful in the treatment of diseases related to this enzyme.146
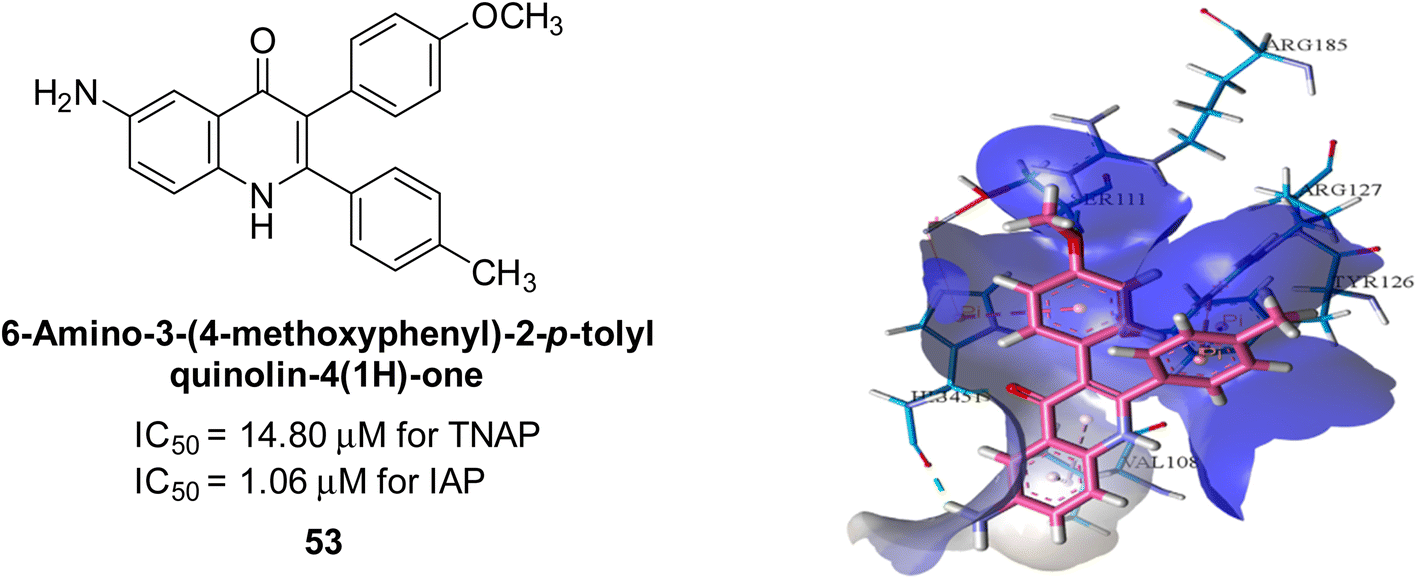 | ||
| Fig. 16 Chemical structure and docking image of 53 and its IAP inhibition. Putative binding interactions of compound 53 (colored light pink) inside calf IAP active site (colored sky blue). | ||
Iqbal et al. (2018) reported the synthesis of tricyclic coumarin sulfonate derivatives and evaluated their inhibitory effects on AP in vitro. Here, the ability of a group of tricyclic coumarin sulphonates inhibited AP against h-TNAP and h-IAP. The most effective h-TNAP inhibitor was discovered to be the methylbenzenesulphonate derivative 54 (IC50 = 0.38 μM) as compared to levamisole (IC50 = 20.2 μM). The most effective inhibitor of h-IAP was discovered to be a different 4-fluorobenzenesulphonate derivative, 55 (IC50 = 14 μM) as compared to L-phenylalanine (IC50 = 100 μM). Results showed that these derivatives exhibited potent inhibitory effects on AP with IC50 values ranging from 1.2 to 10.6 μM (Fig. 17). Docking simulations were performed to gain insights into the interactions between the tricyclic coumarin sulfonates and AP, providing evidence for the binding modes and key amino acid residues involved in the inhibitory process. These findings provide a basis for the further development of tricyclic coumarin sulfonates as potential inhibitors of AP.147
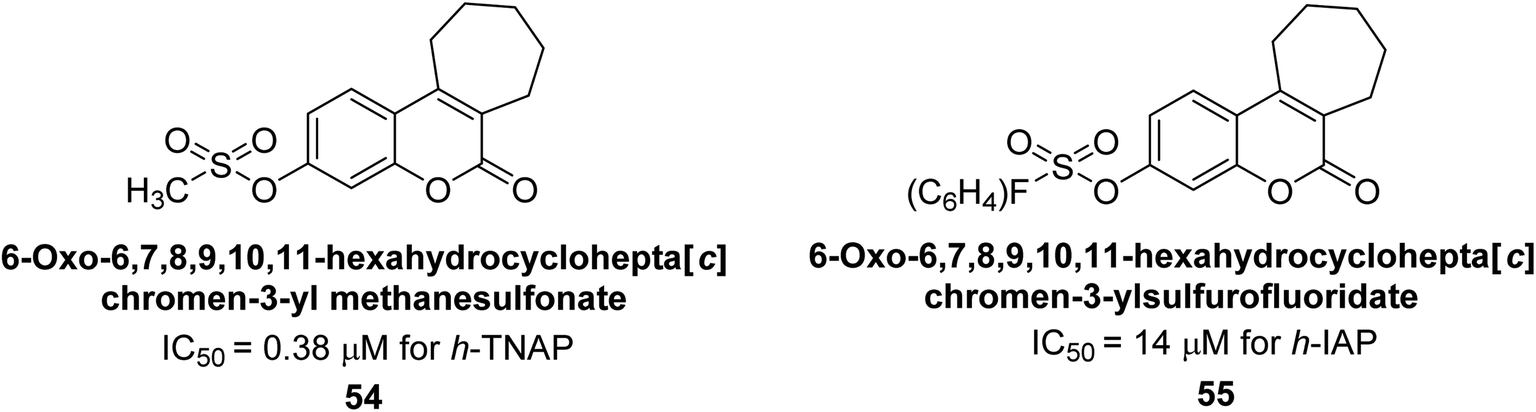 | ||
| Fig. 17 Chemical structures of compounds 54 and 55 and their TNAP and IAP inhibition. | ||
Iqbal et al. (2018) reported the synthesis and AP inhibition studies of carboxy pyrazole derivatives. The synthetic compounds inhibited h-TNAP between IC50 values of 0.91 to 42.0 μM and h-IAP between IC50 values of 0.39 to 22.3 μM. Compounds 56 (IC50 = 0.91 μM) and 57 (IC50 = 5.91 μM) with electronegative substitution were found to be more efficient against h-TNAP as compared to levamisole (IC50 = 20.21 μM) (Fig. 18). Furthermore, molecular docking and in silico research were used to elucidate the binding method of interactions.148
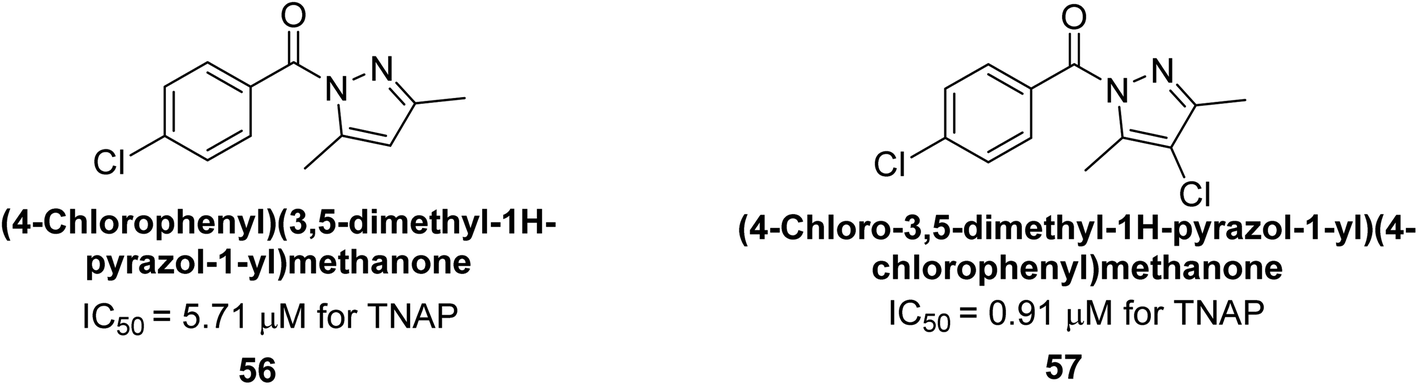 | ||
| Fig. 18 Chemical structures of compounds 56 and 57 and their TNAP inhibition. | ||
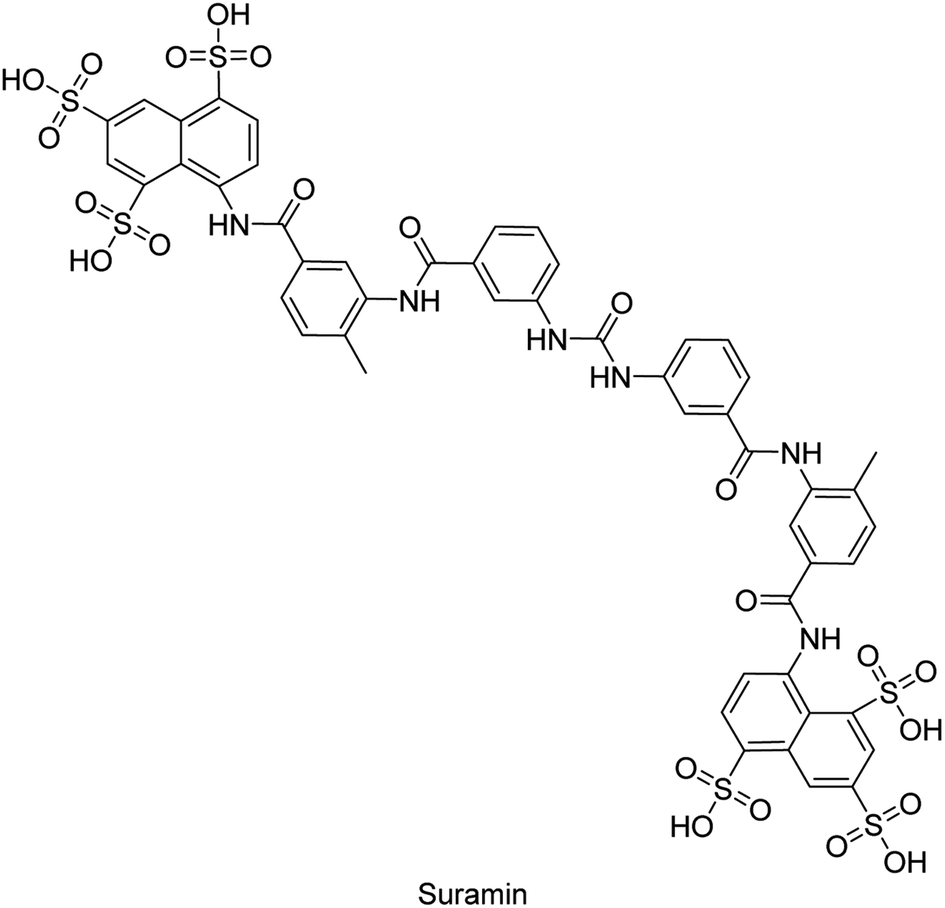
Iqbal et al. (2018) reported design, synthesis, and biological evaluation of 4-aminopyridine-based amide derivatives as potent inhibitors of h-TNAP. A series of compounds were synthesized by the condensation reaction of 4-aminopyridine and various amide derivatives. The synthesized compounds were found to possess good inhibition potential having IC50 values ranging between 0.05 and 0.82 μM as compared to the standard levamisole (IC50 = 19.2 μM). It was discovered that compound 58 was more effective than the previously known inhibitor suramin (IC50 = 42.1 μM), which showed considerable inhibition (IC50 = 0.25 μM) (Fig. 19). The SAR analysis of these compounds revealed that the presence of a substituent on the pyridine ring and the nature of the amide moiety are important for TNAP inhibition.149
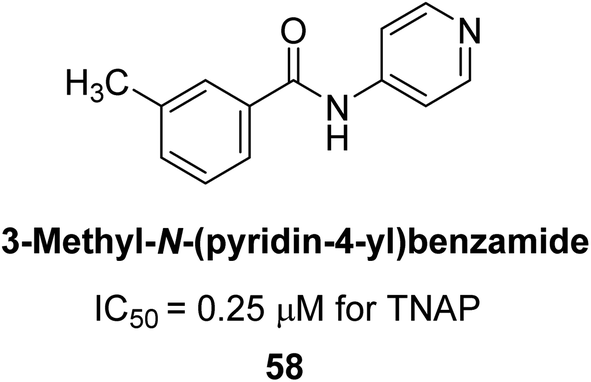 | ||
| Fig. 19 Chemical structure of compound 58 and its TNAP inhibition. | ||
Hanif et al. (2018) disclosed several hybrid compounds generated from 1,2-benzothiazine cores and chalcones. Compound 59 was the most potent inhibitor of h-IAP with an IC50 value of 1.04 μM and compound 60 was a selective inhibitor of h-TNAP with an IC50 value of 0.25 μM. According to molecular docking experiments, compounds 59 and 60 interact differently with the active sites of the two AP isoforms, h-IAP and h-TNAP, respectively (Fig. 20). The compounds did not create H bonds with the isoform in which they displayed lesser activity, despite the fact that they interacted with the Zn ions in both isoforms.150
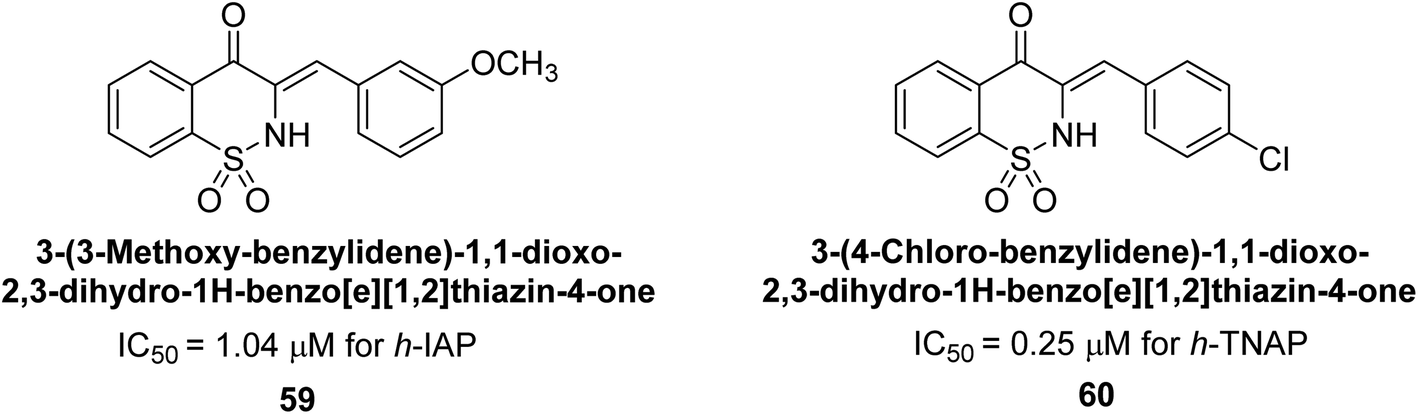 | ||
| Fig. 20 Chemical structures of compounds 59 and 60 and their TNAP and IAP inhibitions. | ||
Langer et al. (2018) reported the synthesis of novel deazapurine analogs featuring a 1H-pyrazolo[3,4-b]pyridin-3(2H)-one core and their biological activity. These analogs were synthesized through a multi-step process, including the reaction of 4-chloro-1H-pyrazolo[3,4-b]pyridine with different aldehydes, followed by cyclization and deamination. The most potent and effective inhibitor of h-TNAP was discovered to be compound 61 (IC50 = 0.31 μM) as compared to levamisole (IC50 = 19.21 μM). The significant inhibition of h-TNAP may be due to the presence of the chlorodifluoromethyl and methyl groups at positions 4 and 2 of the pyridine ring in compound 61. The most effective h-IAP inhibitor, compound 62, has an inhibitory concentration of 0.52 μM as compared to L-phenylalanine (IC50 = 80.21 μM) (Fig. 21).151
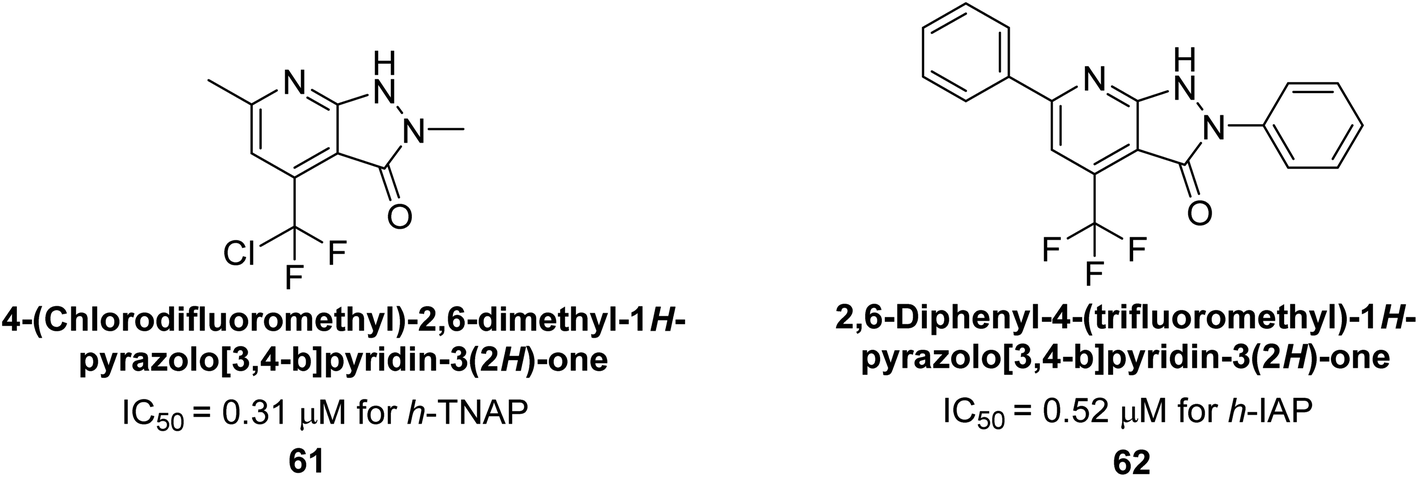 | ||
| Fig. 21 Chemical structure of compounds 61 and 62 and their TNAP and IAP inhibitions. | ||
Langer et al. (2018) reported the synthesis and evaluation of 2-substituted 7-trifluoromethyl-thiadiazolopyrimidones as inhibitors of AP. SAR and molecular docking studies were conducted to understand the relationship between chemical structure and inhibitory activity. Results showed that the 2-substituted derivatives exhibited strong AP inhibitory activity, with some compounds displaying high potency. An enhanced inhibitory value against h-IAP was obtained from the mono-substituted methyl derivative 63 (IC50 = 0.36 μM of h-IAP; 1.06 μM of TNAP) (Fig. 22). The molecular docking results suggest that the inhibitors bind to the active site of the enzyme in a specific manner. These findings provide new insight into the development of 2-substituted 7-trifluoromethyl-thiadiazolopyrimidones as potential inhibitors of AP for therapeutic applications.152
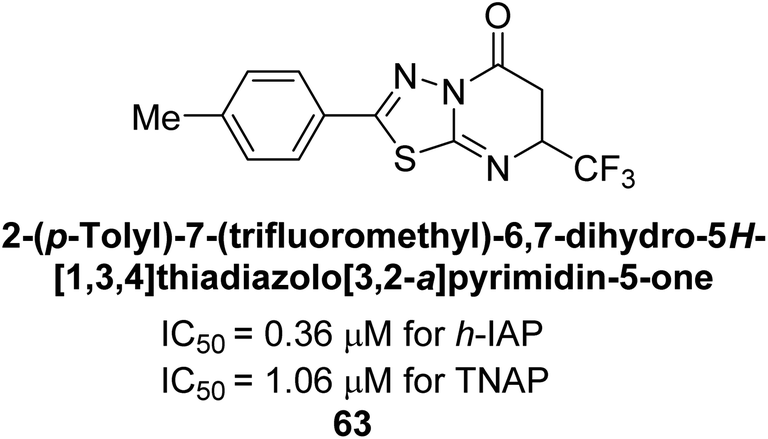 | ||
| Fig. 22 Chemical structure of compound 63 and its TNAP and IAP inhibition. | ||
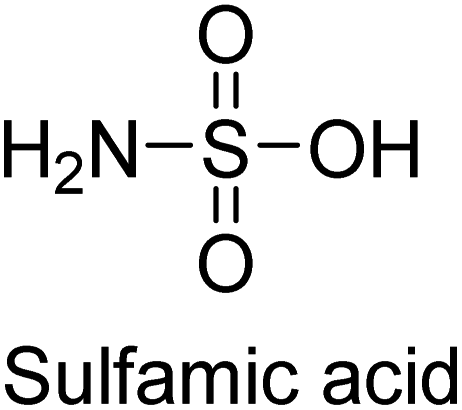
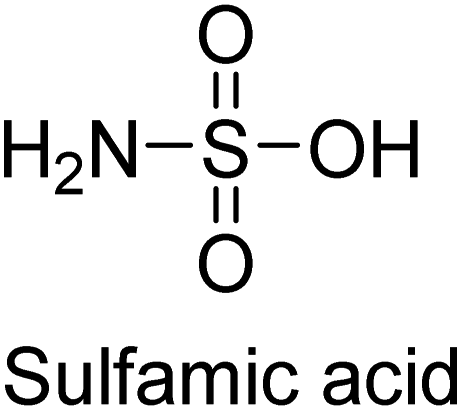
Langer et al. (2018) reported the synthesis of new pyrazolopyridines and benzofuropyridines through a domino reaction of 3-chlorochromones with aminoheterocycles and demonstrated their inhibitory activity against ecto-5′-nucleotidase, making them potential lead compounds for further drug development. The pyrazolo[3,4-b]pyridines showed intriguing results in their interaction with h-e5′NT. Among these derivatives, 64 stood out as a selective and highly potent inhibitor of h-e5′NT when compared to the other derivatives. With an inhibitory value of 0.32 μM, it was approximately 132 times more effective than the reference standard, sulfamic acid, which had an IC50 value of 42.1 μM. The derivative of pyrrolo[2,3-b]pyridine, compound 65 (IC50 = 0.67 μM), was found to be a dual inhibitor of both enzymes, with a higher degree of selectivity towards r-e5′NT. It was the most potent inhibitor of r-e5′NT, exhibiting approximately 16 times higher selectivity over h-e5′NT and 115 times higher selectivity than sulfamic acid (IC50 = 77.3 μM) (Fig. 23). The optical and ecto-5′-nucleotidase inhibitory effects of the resulting compounds were evaluated, and it was found that some of the compounds exhibited potent inhibitory activity.153
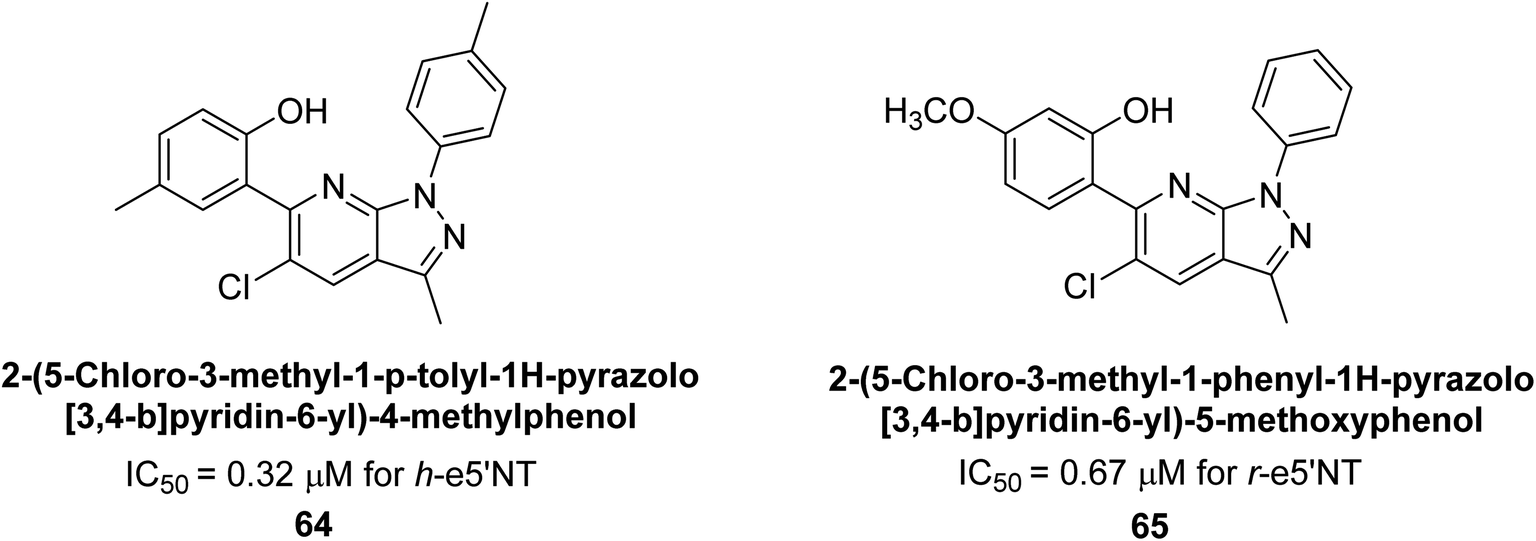 | ||
| Fig. 23 Chemical structures of compounds 64 and 65 and their AP inhibition. | ||
Larik et al. (2018) synthesized acridine analogs and evaluated their inhibitory activity on AP, and some of the analogs showed significant inhibition. The ability of all the produced compounds to inhibit c-IAP was examined. The nitrogen-containing analog 66 displayed the highest potential in the series with an IC50 of 0.0102 μM (Fig. 24) (standard KH2PO4 4.317 μM). This study highlights the potential of using ionic liquids in the synthesis of acridine analogs and their potential as inhibitors of enzymes such as AP.154
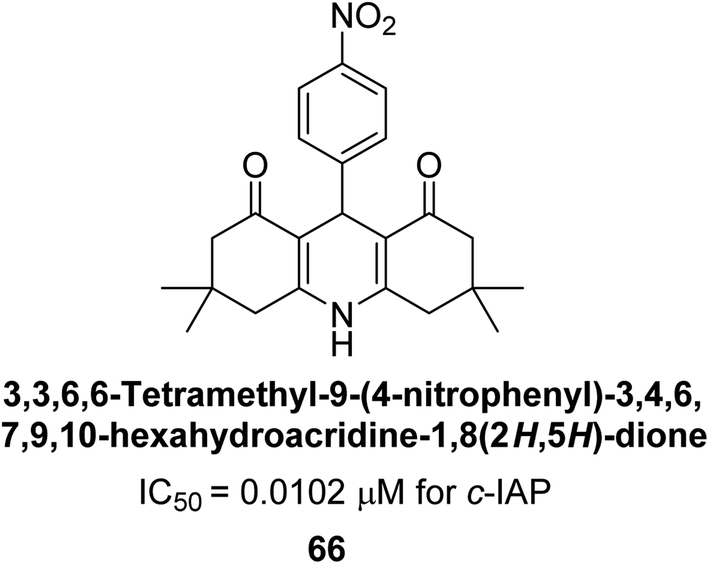 | ||
| Fig. 24 Chemical structure of compound 66 and its c-IAP inhibition. | ||
Saeed et al. (2018) synthesized sulfadiazinyl acyl/aryl thiourea derivatives as inhibitors of c-IAP. The results showed that the sulfadiazinyl acyl/aryl thiourea derivatives had good inhibitory activity against c-IAP, promising pharmacokinetic properties, and strong binding affinity. When compared to conventional monopotassium phosphate, the synthesized derivatives demonstrated more inhibitory potential. Compound 67 with an IC50 of 0.251 μM showed the highest potential in the series as compared to KH2PO4 (IC50 = 4.317 μM) (Fig. 25). The most effective derivative's ability to inhibit c-IAP via a mixed-type pathway was revealed by Lineweaver–Burk plots. According to a molecular docking investigation, chemical 67 interacts with the amino acid residues Asp273, His317, and Arg166. The findings of this study provide important information for further optimization and development of sulfadiazinyl acyl/aryl thiourea derivatives as c-IAP inhibitors.155
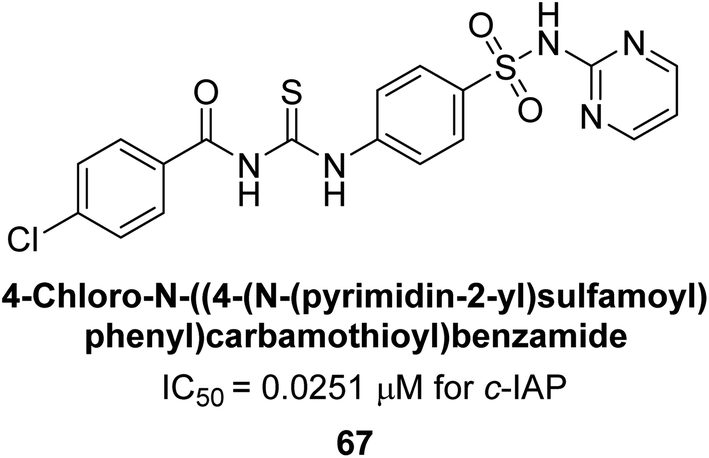 | ||
| Fig. 25 Chemical structure of compound 67 and its c-IAP inhibition. | ||
Seo et al. (2018) reported the synthesis of benzamides and evaluated them against AP. When the inhibitory properties of bi-heterocyclic benzamides were assessed against AP, all of the synthesized compounds were shown to be highly potent inhibitors in comparison to the reference standard KH2PO4 (IC50 = 5.2421 μM). Analyzing the Lineweaver–Burk plots revealed that compound 68 inhibited AP non-competitively to create an enzyme–inhibitor complex, which was the kinetics mechanism attributed. The molecule 68 (IC50 = 0.0427 μM) with a p-ethoxy group was identified as the most potent in the synthetic series (Fig. 26). The compounds having medium-sized polar groups in the aryl portion exhibited excellent inhibitory potential. Dixon plots were used to compute the compound's inhibitory constant Ki, which came out at 1.15 μM.156
 | ||
| Fig. 26 Chemical structure of compound 68 and its AP inhibition. | ||
Ashraf et al. (2019) reported the synthesis and molecular docking of N-(5-(alkylthio)-1,3,4-oxadiazol-2-yl)methylbenzamide analogs as potential inhibitors of the AP. The aim was to evaluate the effect of alkylthio substitution on the inhibitory activity and binding affinity of these compounds towards AP. The synthesized compounds were characterized, and their inhibitory potency was determined through in vitro assays. According to the results of the bioassay, target compounds had good to excellent AP inhibitory activity. Compound 69 had the highest level of activity, with an IC50 value of 0.420 μM compared to 2.80 μM for the standard (KH2PO4) (Fig. 27). The molecular docking studies were performed to predict the binding mode and affinity of the compounds towards the target enzyme. The amino acid His265 with a binding distance of 2.13 Å interacted with the nitrogen on the oxadiazole ring in compound 69. The results suggest that the alkylthio substitution has a significant effect on the inhibitory activity and binding affinity of the compounds towards AP.157
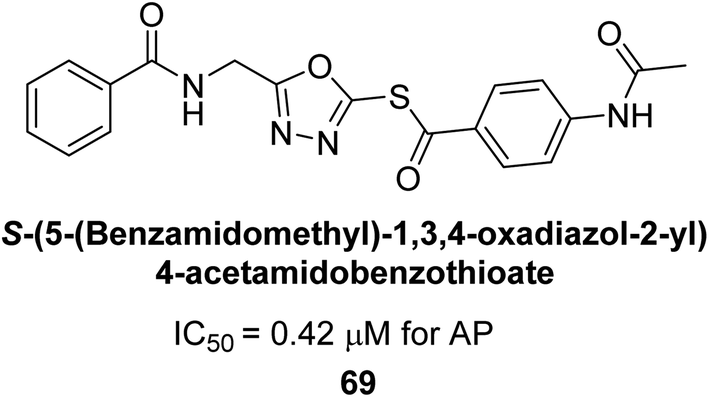 | ||
| Fig. 27 Chemical structure of compound 69 and its AP inhibition. | ||
Ashraf et al. (2019) synthesized and evaluated substituted acetamides as AP inhibitors. The inhibitory potential of each of the title compounds was assessed against h-AP. The subjected compounds showed a significant inhibition potential having activity in the range of 0.420 to 5.012 μM, which was superior to the standard KH2PO4 (IC50 = 2.80 μM) (Fig. 28). Compound 70 shows particularly powerful activity with an IC50 value of 0.420 μM compared to the standard's KH2PO4. Lineweaver–Burk plots depicting the non-competitive mode of binding with enzyme were used to assess the enzyme inhibitory kinetics of the most potent inhibitor 70. In comparison to other derivatives, molecule 70 displayed a good binding affinity with a binding energy value of around 7.90 kcal mol−1.158
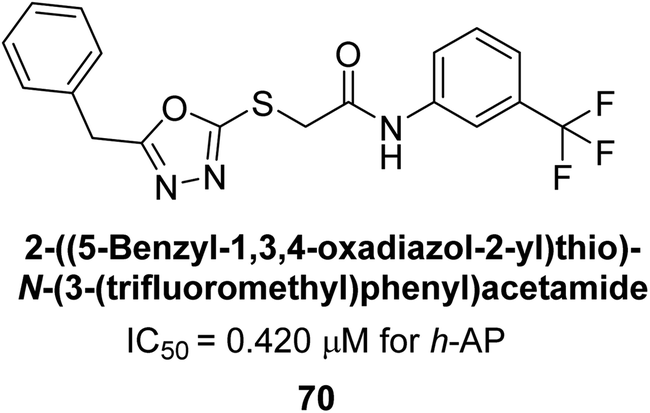 | ||
| Fig. 28 Chemical structure of compound 70 and its h-AP inhibition. | ||
Hameed et al. (2019) reported on the discovery of two new classes of compounds, pyrazolyl pyrimidinetriones (PPTs) and thioxopyrimidinediones (PTPs), as selective and efficient inhibitors of recombinant human ectonucleotidases. (h-TNAP, h-IAP) and ectonucleotidase (h-NPP1 and h-NPP3) enzymes were used to test the synthetic hybrid drugs' ability to inhibit them. With varying degrees of inhibition based on the functionalized hybrid structure, the majority of the tested analogs were extremely effective. According to SAR of synthesized derivatives, compound 71 from the PPT series led to the potent and selective inhibition of h-TNAP (IC50 = 0.33 μM), while compound 72 selectively inhibited h-IAP isozyme (IC50 = 0.86 μM). As lead scaffolds against h-NPP1 and h-NPP3, respectively, compounds 73 (IC50 = 0.61 μM) and 74 (IC50 = 0.57 μM) were also discovered (Table 10). These compounds were found to have high potency in inhibiting the enzymes and were shown to be non-nucleotide inhibitors. The study provides evidence for the potential of these inhibitors as therapeutic agents for medical conditions related to ectonucleotidase overactivity.159
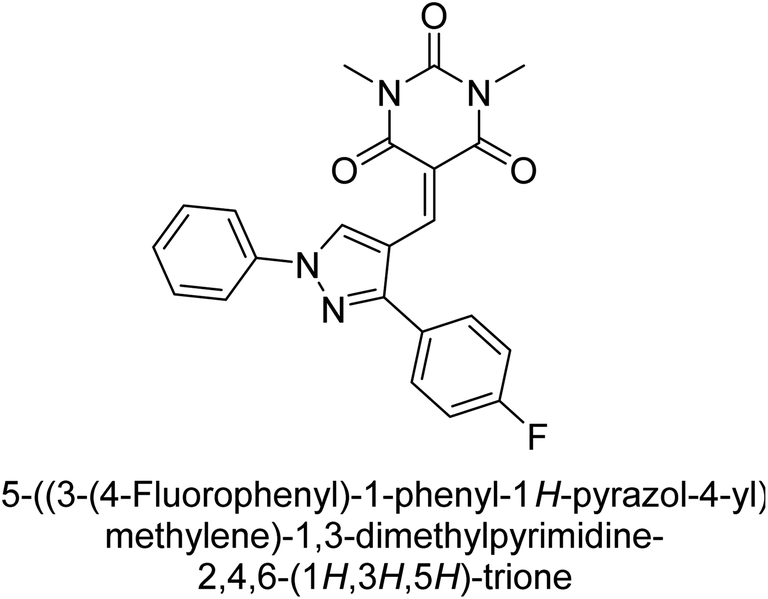
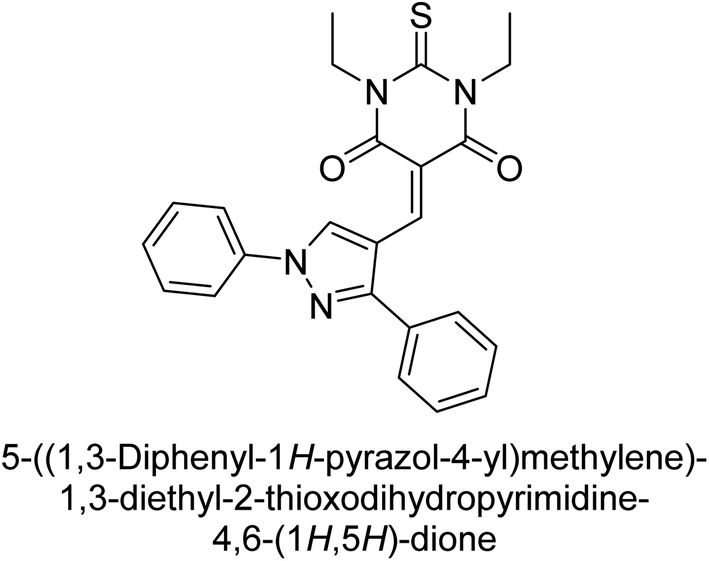
Saeed et al. (2019) reported the design, synthesis, and biological evaluation of trinary benzocoumarinthiazoles-azomethines derivatives as inhibitors of h-TNAP and h-IAP. A series of compounds were synthesized by the condensation reaction of benzocoumarin, thiosemicarbazide, and various aromatic aldehydes. Some of the compounds may act as h-IAP inhibitors, whereas the majority of them were selective for h-TNAP. While 75 (IC50 = 1.02 μM) was identified as the possible inhibitor of h-IAP, 76 (IC50 = 0.76 μM) was shown to be the most effective h-TNAP inhibitor (Table 11). The IC50 values for the other compounds in the same series ranged from 1.04 to 2.90 μM, indicating that they had good inhibitory activity against h-TNAP. The remaining compounds inhibited h-IAP between 1.17 and 2.28 μM. It was shown that these analogs had substantially more capacity to inhibit the corresponding isozyme when their IC50 values were compared to the inhibitory values of the reference standards, levamisole (IC50 = 19.2 μM) and L-phenylalanine (IC50 = 80.1 μM). The SAR analysis of these compounds revealed that the presence of a substituent on the phenyl ring, the length of the alkyl chain, and the substitution of the thiosemicarbazide nitrogen atom are important for AP inhibition. The results of this study demonstrated that trinary benzocoumarinthiazoles-azomethines derivatives are a promising new class of AP inhibitors with potential therapeutic applications.160
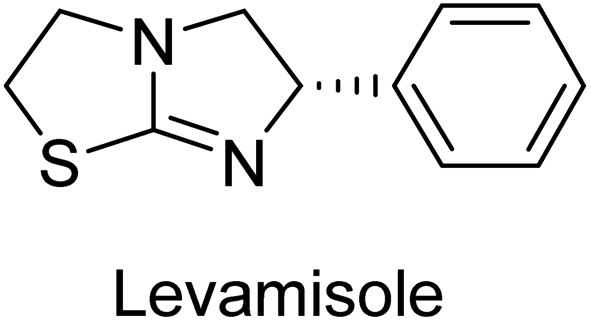
Hameed et al. (2020) reported on the synthesis and computational studies of highly selective inhibitors of human recombinant h-TNAP, with a focus on developing new therapies against vascular calcification. The results of the study show that the inhibitors exhibit high selectivity for h-TNAP, offering promising potential as a therapeutic target against this debilitating condition. Compound 77 was shown to be a highly effective inhibitor of h-TNAP (IC50 = 0.16 μM), with 127-fold more inhibition than levamisole. Compound 77, which has a 127-fold increase in inhibition relative to levamisole, inhibits h-TNAP with great potency. It contains an o-chlorophenyl moiety at the triazole ring and a p-tolyl ring at the pyrazole skeleton. Contrarily, compound 78 (IC50 = 1.59 μM) was discovered to be the most selective inhibitor against the studied AP (Fig. 29). The computational studies provide a deeper understanding of the molecular mechanisms of inhibition and help to guide future optimization efforts.161
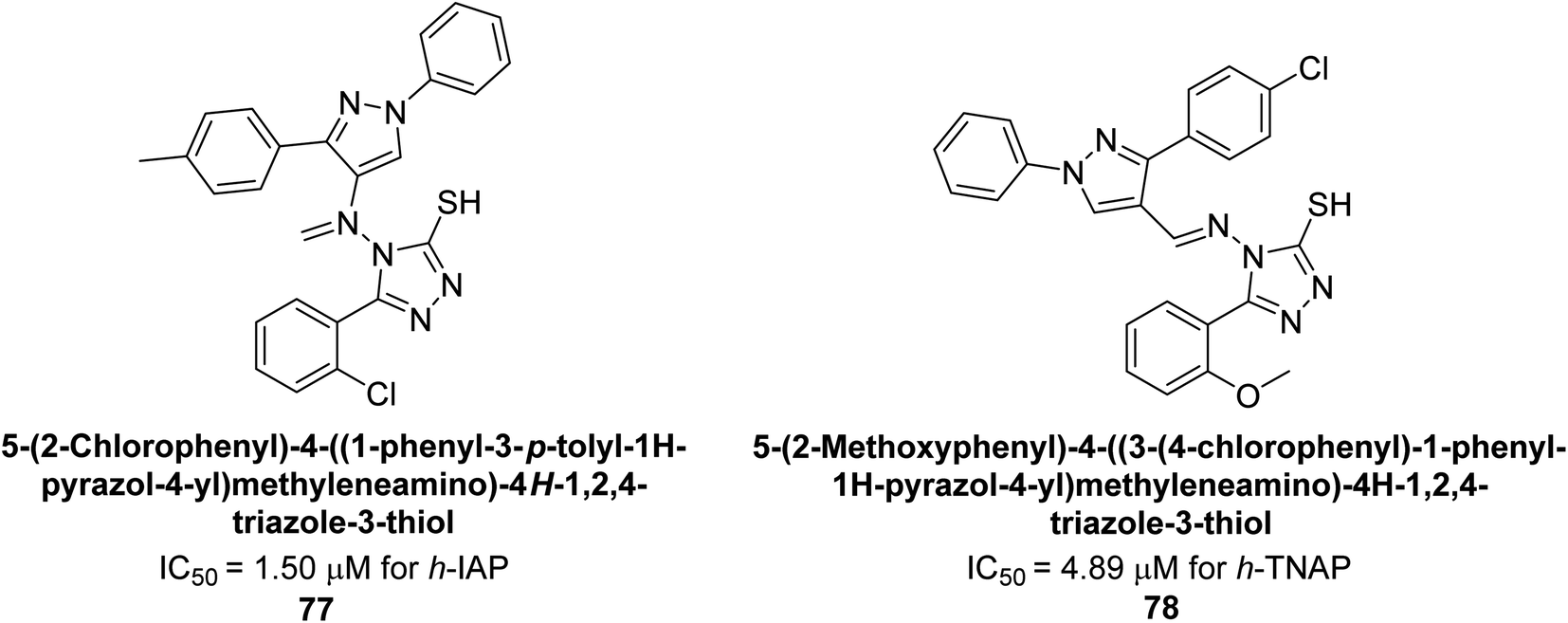 | ||
| Fig. 29 Chemical structures of compounds 77 and 78 and their h-TNAP and h-IAP inhibitions. | ||
Iqbal et al. (2020) described the synthesis of bisthioureas of pimelic acid and 4-methylsalicylic acid derivatives and evaluated them as selective inhibitors of h-TNAP and h-IAP. The inhibitory activity of compound 79 for h-TNAP was the highest of the bis(thiourea) derivatives, with an IC50 value of 4.63 μM, or almost four times that of the positive control levamisole (IC50 = 19.2 μM). With an IC50 value of 1.50 μM compared to the positive control (L-phenylalanine: 80.1 μM), compound 80 was shown to have the maximum efficacy and selectivity for h-IAP. Compound 81 demonstrated inhibition for h-TNAP with an IC50 value of 4.89 μM, which is equivalent to the inhibitory potential of a standard inhibitor levamisole (Fig. 30). Molecular docking studies were then conducted to analyze the binding interactions of the compounds with TNAP and IAP. The results showed that the bisthioureas of pimelic acid and 4-methylsalicylic acid derivatives exhibited high binding affinities and selectivity for h-TNAP and h-IAP, suggesting their potential as inhibitors of these enzymes. Compound 80 displayed a significant number of hydrogen bonds between Ser92 and the 4-nitrobenzyl group and Arg150 with the benzamide group. Additionally, other significant interactions were noted, including π-stacked with His317 and π–π T-shaped with His153. For compound 79, Glu 321 with the heptanediamide moiety, Ser 92 with the nitro phenyl group, Tyr 276 and Arg 150 with the oxygen atom all displayed hydrogen bonds. The compound's His153 sulfur atom observed sulfur interactions. Tyr276 also shows π–π T-shaped interactions.162
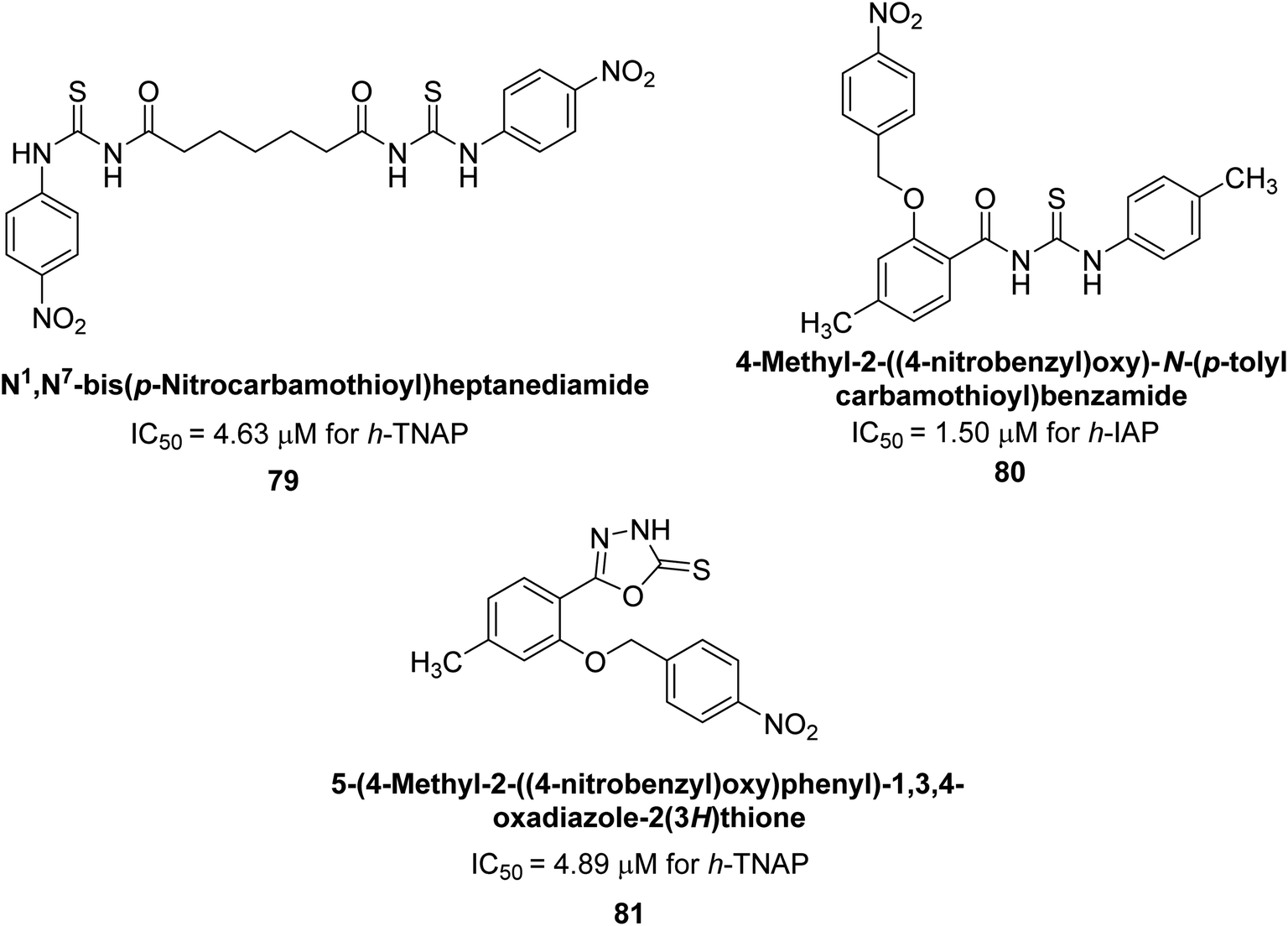 | ||
| Fig. 30 Chemical structures of compounds 79, 80 and 81 and their TNAP and IAP inhibitions. | ||
Khurshid et al. (2020) described the synthetic approach toward the preparation of 1,3-oxathiol-2-ylidenes which involves the use of thiocarbonyl ylides, which are generated by the reaction of dithioacetals with lithium diisopropylamide (LDA). This approach has been shown to be efficient in the synthesis of a variety of 1,3-oxathiol-2-ylidenes with good yields. In terms of biological activity, 1,3-oxathiol-2-ylidenes have been found to inhibit AP in a dose-dependent manner. Inhibition of AP was investigated for the synthesized compounds. The results indicated that nearly every compound has a good percentage of inhibition against both enzymes (Kidney bovine-TNAP and calf-IAP), with compound 82 (IC50 = 2.41 μM for b-TNAP and 0.67 μM for c-IAP) demonstrating dual inhibition and 83 (p-NO2 substituted derivative; IC50 = 0.37 μM) and 84 (m-Cl substituted derivative; IC50 = 2.90 μM) being strong and selective inhibitors of TNAP and c-IAP, respectively as compared to the standards Levamisole (IC50 = 19.21 ± 0.01 μM for b-TNAP) and L-phenylalanine (IC50 = 80.21 ± 0.01 μM for c-IAP) (Table 12). Docking experiments corroborated the dual inhibition of 82 because it participates in H-bonding at numerous sites with the amino acid residues His154 and Glu174, as well as π–π interactions. Molecular docking experiments have been used to determine the structure–activity correlation for the series' active members. The outcome of the SAR demonstrates the participation of active inhibitors in secondary metal ion interactions with Zn ions inside the active pocket of the enzyme, as well as H-bonding at multiple places with distinct amino acid residues (Fig. 31).163
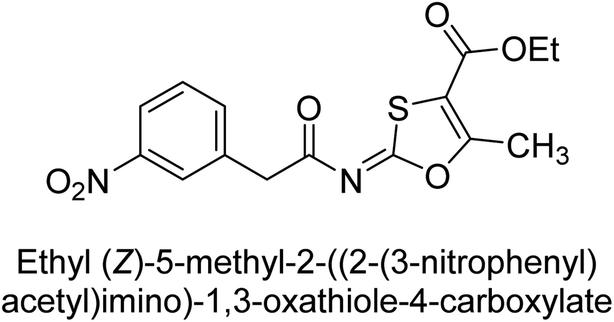
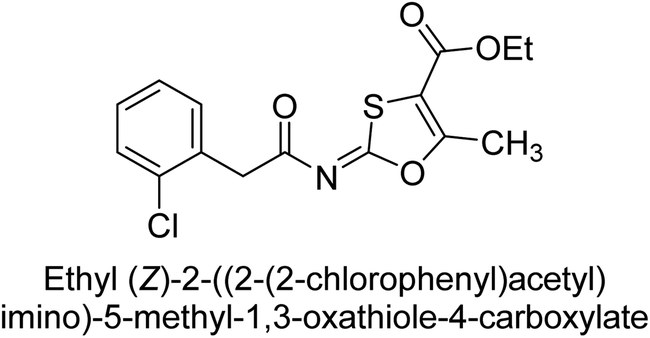
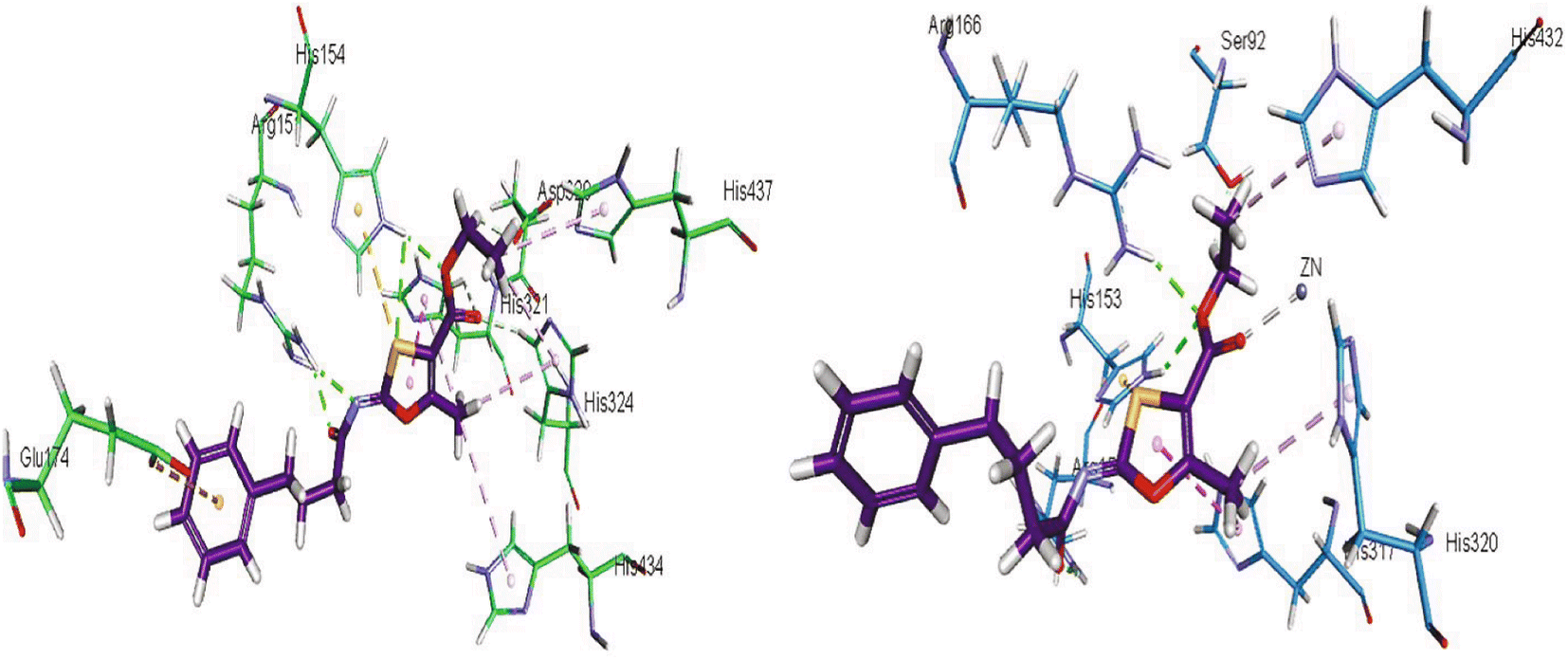 | ||
| Fig. 31 Docking images of 82 inside the bovine tissue-TNAP and calf-IAP. | ||
Rashida et al. (2020) synthesized a series of sulfonylhydrazones and evaluated them against AP. Among the chromen-2-one scaffold-based sulfonylhydrazones, compound 85 was discovered to be the most effective inhibitor for h-TNAP and h-IAP, with IC50 values of 1.02 and 0.32 μM, respectively, in comparison to levamisole (IC50 = 25.2 μM). However, compound 86 was discovered to be the most effective against h-TNAP and h-IAP among the series of phenyl ring-based sulfonylhydrazones, with IC50 values of 0.85 and 0.52 μM, respectively (Fig. 32). This pattern suggests that for h-TNAP inhibitory action, a nitro substituent, as well as a 2-hydroxy substituent at the phenyl ring, are required. Compounds 85 and 86 demonstrated uncompetitive inhibition against h-IAP while exhibiting competitive inhibition against h-TNAP.164
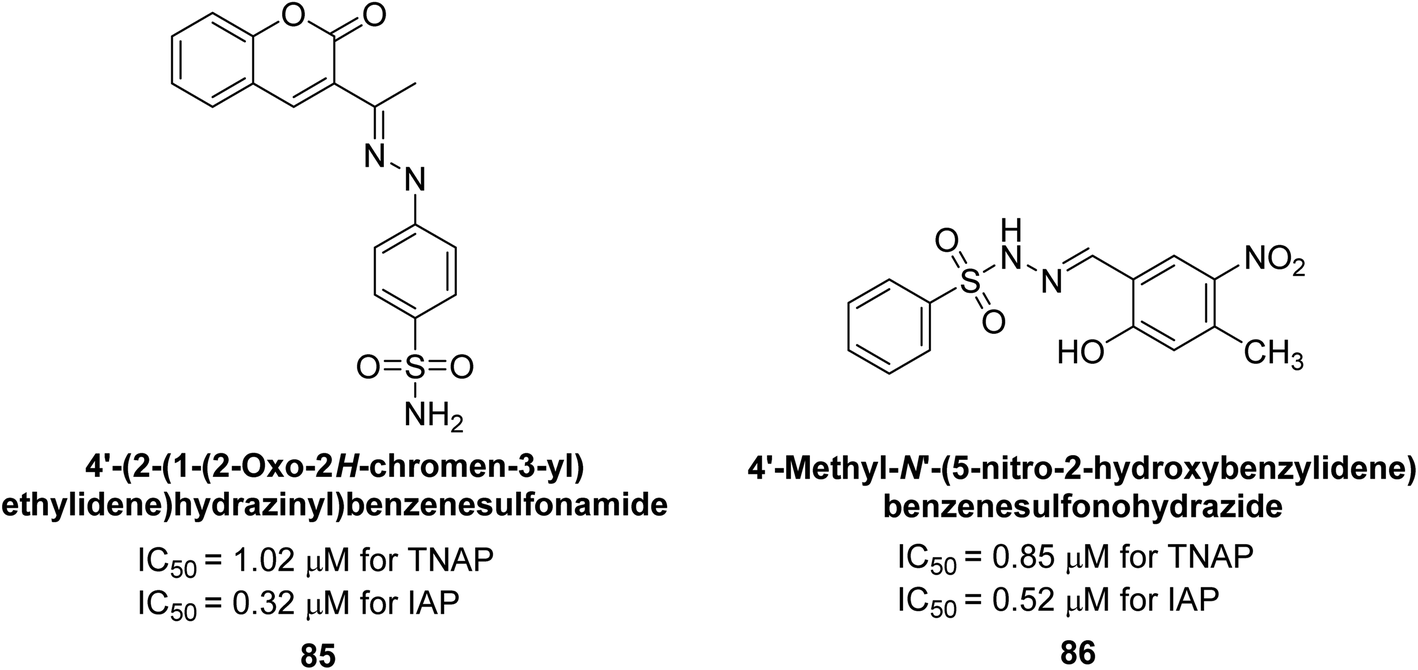 | ||
| Fig. 32 Chemical structures and IC50 values of compounds 85 and 86. | ||
Abbasi et al. (2021) described the synthesis of N-(substituted-phenyl)-3-(4-phenyl-1-piperazinyl)propanamides and evaluated their in vitro AP inhibitory activity. Compound 87 with p-ethyl group had a better activity (IC50 = 0.531 μM) as compared to the standard KH2PO4 (IC50 = 5.242 μM) (Fig. 33). The kinetic studies of compound 87 showed its non-competitive behavior. The substance 87 inhibited the catalytically active site for the zinc and magnesium ions and demonstrated hydrogen bonding with side chains of Arg166 via the nitrogen atom of the piperazinyl moiety. The ligand's ethyl phenyl moiety is tightly encapsulated in a hydrophobic pocket and interacts with His432 by stacking on top of itself. The His320, His317, and His153 are interacted with by the phenyl piperazinyl moiety, which is located outside the pocket. The authors have also performed in silico studies to provide molecular insights into the interaction of these compounds with AP. The results suggest that these compounds are potent inhibitors of AP and could have potential therapeutic applications.165
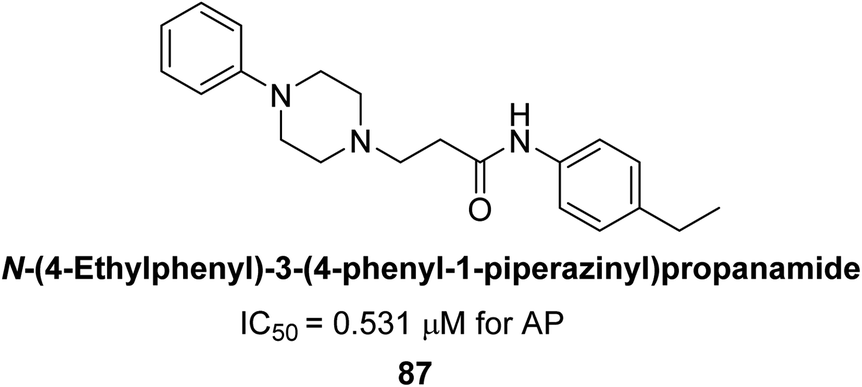 | ||
| Fig. 33 Chemical structure of compound 87 and its AP inhibition. | ||
Ashraf et al. (2021) rationally reported the synthesis of 2-benzylidenebenzofuran-3(2H)-ones using various methods, including condensation reactions and cyclization reactions and their inhibitory potential against AP, was evaluated. The compounds showed excellent bioactivity with IC50 values in the range of 1.055 to 5.786 μM as compared to the standard KH2PO4 (IC50 = 2.80 μM). Compound 88 exhibited the best inhibition potential towards AP having an IC50 value of 1.055 μM and a docking score of 5.211 kcal mol−1 (Fig. 34). The SAR analysis of target compounds has shown that the presence of a substituent on the phenyl ring and the length of the alkyl chain in the substituent are important for AP inhibition. Compound 88 with a p-methoxy group on ring B of the aurone scaffold showed exceptional inhibitory potential against human AP, which was 2.6-fold higher than KH2PO4. The nucleophilic character of the methoxy group at the para of the phenyl ring (ring B), which interacts with the enzyme active site, may account for the strong inhibitory potential. The –NO2 group of 88 interacts via a hydrogen bond with Lys87, while its methyl group interacts via a hydrogen bond with HOH2338, the water molecule of the biological specimen 2GLQ (Fig. 34). Kinetic analysis of 88 exhibited non-competitive inhibition of AP, with Ki values in the low micromolar range and Km is 0.027. These studies have provided insights into the structural features of the inhibitors that are important for binding and inhibition, and have helped to guide the design of new and more potent inhibitors.166
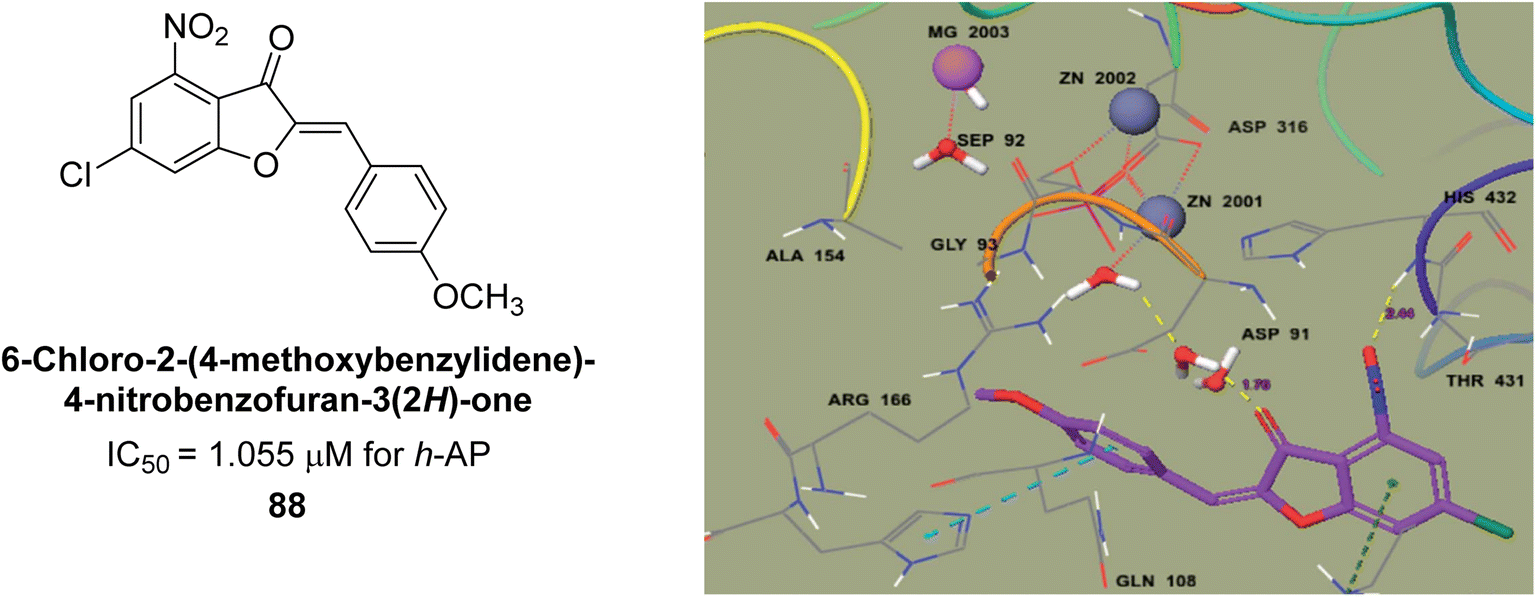 | ||
| Fig. 34 Chemical structure and IC50 value of compound 88 and its docking image inside the active pocket of AP enzyme. | ||
Neri et al. (2021) identified a specific inhibitor of PLAP. The identified inhibitor, ML228 (IC50 = 32 nM), demonstrated high potency and selectivity towards PLAP and was shown to inhibit the growth of PLAP-expressing cancer cell lines in vitro and in vivo. Moreover, ML228 was found to exhibit minimal toxicity towards normal cells, suggesting its potential as a safe and effective therapeutic agent for PLAP-associated cancers.167
Saeed et al. (2021) studied the inhibition of the IAP enzyme by aryl thioureas derived from aminophenazone using computational molecular dynamics simulations. The lead member 89 has an IC50 value of 0.420 μM (Fig. 35), which is significantly better than the reference standard used (KH2PO4 IC50 = 2.8 μM and L-phenylalanine IC50 = 100 μM), as a result of the screening of synthesized target compounds for their anti-inflammatory potential against c-IAP. Kinetic studies revealed that potent inhibitor 89 binds to AP via a non-competitive fashion.168
 | ||
| Fig. 35 Chemical structure of compound 89 and its c-IAP inhibition. | ||
Saeed et al. (2021) described the synthesis and characterization of 1-benzylidene-2-(4-tertbutylthiazol-2-yl)hydrazines as potential inhibitors of AP. An inhibition assay was performed to evaluate the ability of the compounds to inhibit AP activity. Compound 90 was discovered to be the most effective inhibitor of h-TNAP in this set of compounds, with an IC50 value of 1.09 μM. Compound 91 demonstrated selectivity and potency for h-IAP with an IC50 value of 0.71 μM. For h-TNAP and h-IAP, respectively, 90 showed competitive and uncompetitive modes of inhibition, whereas 91 was discovered to have a non-competitive mode of inhibition (Fig. 36). Additionally, molecular modeling was employed to gain insights into the interaction of the compounds with the AP enzyme. The results of the study showed that some of the synthesized compounds showed promising inhibitory activity against AP and provided useful information for the design of more potent inhibitors.169
 | ||
| Fig. 36 Chemical structures of compounds 90 and 91 and their TNAP and IAP inhibitions. | ||
Ejaz et al. (2022) explored the use of thiazole-linked thioureas as potential inhibitors of AP activity. The authors conducted a comprehensive study of these compounds using a combination of biochemical evaluation, computational analysis, and SAR studies. All of the synthetic compounds had high AP inhibitory activity, although 92 and 93 have the lowest IC50 values at 0.057 μM and 0.019 μM, respectively as compared to KH2PO4 (IC50 = 4.611 μM). The greatest docking energy of all the compounds is those of 92 and 93, measuring 32.18 kJ mol−1 and 30.09 kJ mol−1, respectively (Fig. 37). The results show that the thiazole-linked thioureas exhibit inhibitory activity towards AP, with the efficacy of inhibition dependent on various structural features of the compounds. The findings of this study provide a basis for further optimization and development of thiazole-linked thioureas as potential inhibitors of AP and have potential implications for the treatment of various diseases associated with elevated AP activity.170
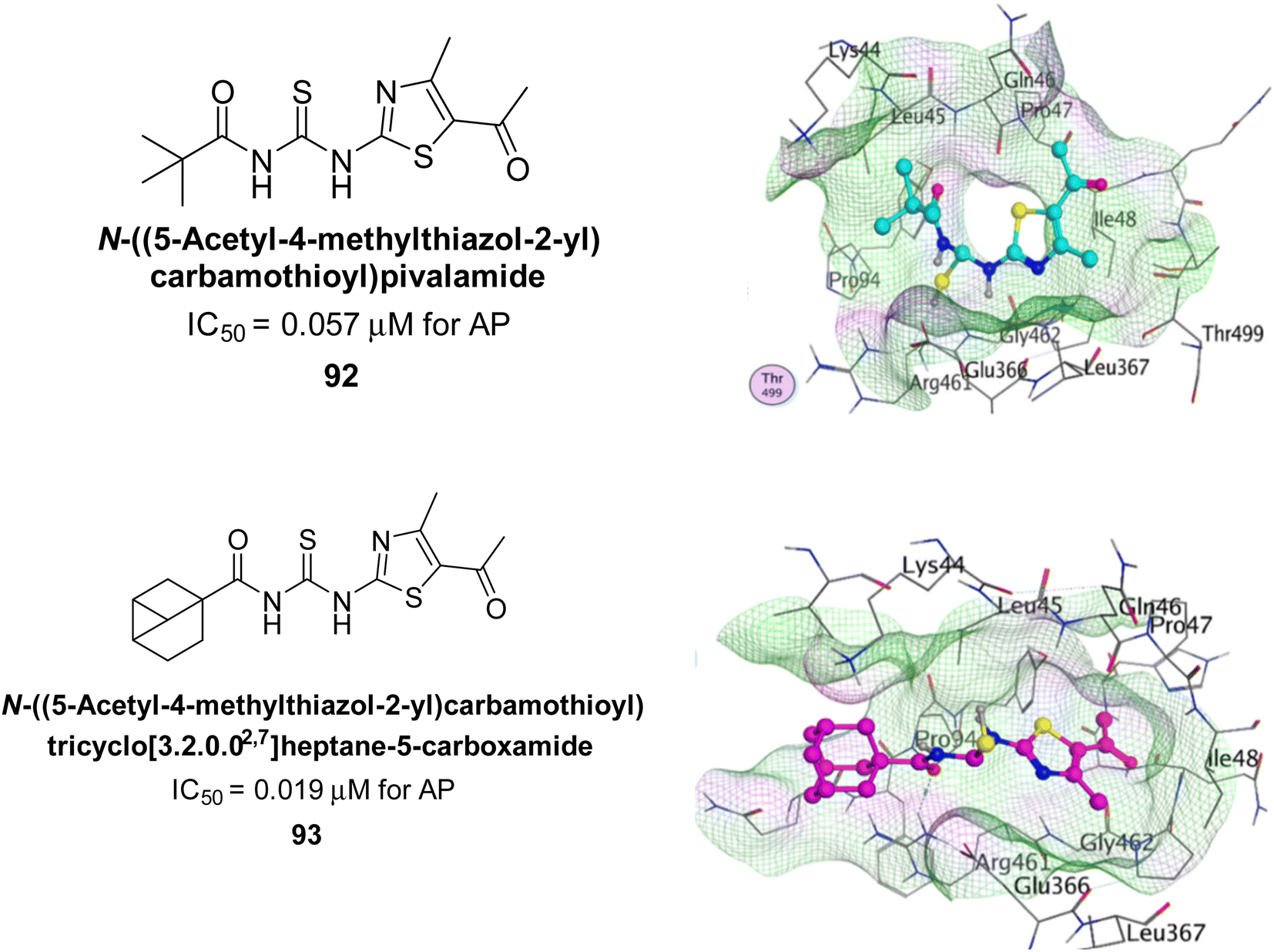 | ||
| Fig. 37 Chemical structures of compounds 92 and 93 and their AP inhibition. | ||
Ejaz et al. (2022) explored 2-tetradecanoylimino-3-aryl-4-methyl-1,3-thiazolines derivatives as potential inhibitors of AP, an enzyme that plays a role in various biological processes. The authors conducted a biochemical evaluation of these compounds and analyzed the results through computational analysis. The ability of each produced compound to suppress c-IAP was assessed. KH2PO4 was utilized as the benchmark. The IC50 values for compounds 94 (IC50 = 0.019 μM), 95 (IC50 = 0.032 μM) and 96 (IC50 = 0.052 μM) each indicated a better and more effective inhibition potential (Fig. 38). The findings suggest that these derivatives have potential as inhibitors of AP and could be further studied for their potential use in various medical applications.171
 | ||
| Fig. 38 Chemical structures of compounds 94, 95 and 96 and their c-IAP inhibition. | ||
Kim et al. (2022) described the synthesis of a number of pyrazolo-oxothiazolidines and biologically tested them in order to create AP inhibitors. The results revealed that each of the created compounds strongly inhibited AP. The most potent inhibitory activity was specifically shown by compound 97 (IC50 = 0.045 μM), which is 116-fold more potent than KH2PO4 (IC50 = 5.242 μM) as a standard reference. When the series' most potent molecule 97 was examined for its method of enzyme binding, it was discovered that it bound to the target enzyme non-competitively. When interacting with His-153 and His-317, the oxygen atom of 92 creates two hydrogen bonds with each (Fig. 39).172
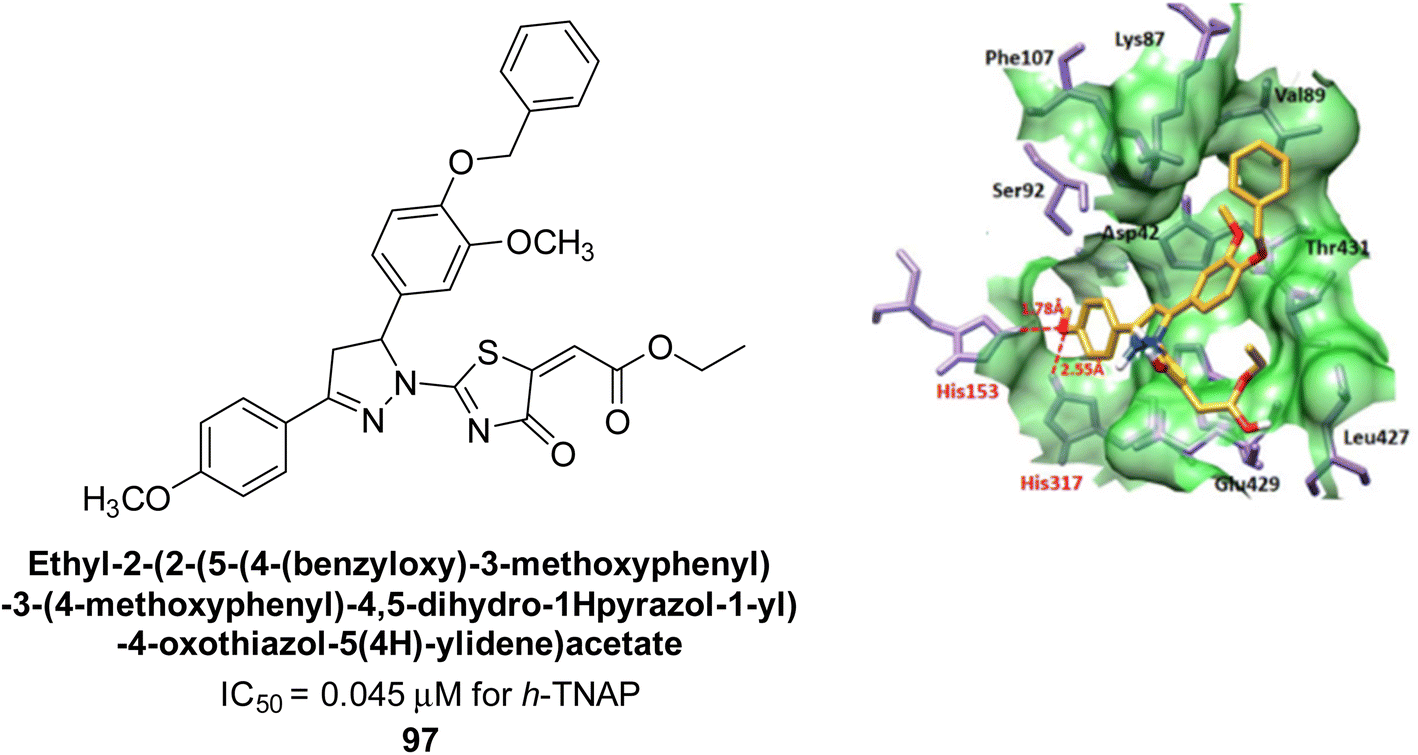 | ||
| Fig. 39 Chemical structure of compound 97, its TNAP inhibition and docking image. | ||
Shahzad et al. (2022) described the synthesis of new pyridine and dihydropyridine analogs with various pharmacophores. The enzyme h-TNAP was used to investigate the effects of each target compound. The majority of the compounds demonstrated excellent inhibition against h-TNAP with IC50 values ranging from 0.49 to 8.8 μM, multi-fold greater than the typical inhibitor (levamisole, 22.65 μM) of the h-TNAP enzyme. Among all the synthesized analogs, compound 98 (IC50 = 1.32 μM) (Fig. 40) exhibited excellent inhibitory efficacy and was approximately 17 times more effective than the reference medication levamisole. The docking result of 98 indicated that the naphthyl moieties' orientation and position are key factors in the suppression of h-TNAP activity.173
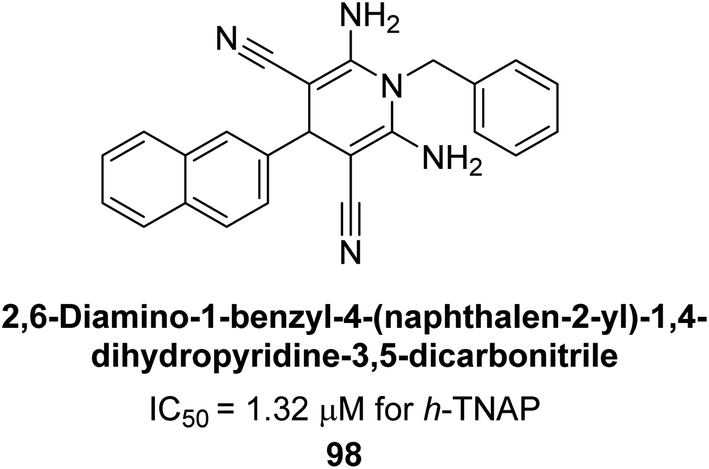 | ||
| Fig. 40 Chemical structure of compound 98 and its h-TNAP inhibition. | ||
Saeed et al. (2023) described the synthesis, kinetic characterization, and in silico investigation of a series of novel quinolinyl-iminothiazolines as AP inhibitors. Most of the synthetic compounds revealed promising inhibitory activity against TNAP with IC50 values in the range of 0.337 to 7.247 μM as compared to the standard KH2PO4 (IC50 = 5.245 μM). Additionally, most of the compounds demonstrated superior inhibitory activity in comparison to KH2PO4. Among the synthesized series, compound 99 (IC50 = 0.337 μM; Docking score = −28.86 kJ mol−1) was found to be a potent TNAP inhibitor. Compound99 (Ki = 0.47 μM) inhibits AP non-competitively to generate an enzyme inhibitor complex. Compound 99 (Fig. 41) with an unsubstituted benzene ring was shown to be the most powerful inhibitor, which may be related to the reduced steric crowding of benzoyl moiety around the iminothiazoline component. These subjected compounds have a unique structural feature, a quinoline moiety, which is known to have a high binding affinity towards enzymes. In addition, the thiazoline moiety is known to have a high binding affinity towards enzymes and proteins. Therefore, the combination of these two moieties is expected to result in compounds with high binding affinity and specificity towards AP. The results of this study provided new insights into the potential of quinolinyl-iminothiazolines as AP inhibitors and may lead to the development of more potent and specific inhibitors for the treatment of diseases associated with abnormal AP activity.174
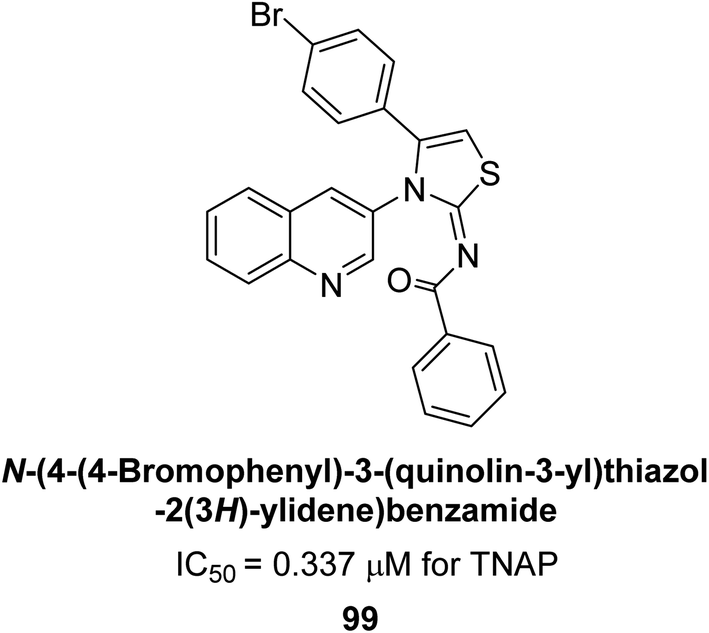 | ||
| Fig. 41 Chemical structure and IC50 value of compound 99. | ||
Iqbal et al., (2023) explored the potential of chromone sulfonamides and sulfonylhydrazones as selective inhibitors of ectonucleotidases, which are enzymes that regulate purinergic signaling. The research team synthesized a series of compounds and evaluated their biological activity in vitro against ectonucleotidases. Compound 100 demonstrated the highest activity (h-IAP IC50 = 0.51 μM; h-TNAP = 36.5%) among the inhibitors of h-IAP, being the most potent and selective. On the other hand, compound 101 showed the highest activity and selective inhibition against h-TNAP inhibitor (h-TNAP IC50 = 1.41 μM; h-IAP = 43.1%). Additionally, these compounds were tested against rat and human ecto-5′-nucleotidases (r-e5′NT and h-e5′NT), and some of them exhibited remarkable inhibitory activity against ecto-5′-nucleotidases. Among these, compound 102 demonstrated the highest inhibition against h-e5′NT (IC50 = 0.18 μM) (Fig. 42). The most promising compounds were then subjected to in silico studies to gain insight into their binding interactions with the target enzyme. The results showed that several chromone sulfonamides and sulfonylhydrazones exhibited significant inhibition of ectonucleotidases with high selectivity and low cytotoxicity. These compounds could serve as lead candidates for the development of novel drugs for the treatment of various purinergic signaling-related disorders.175
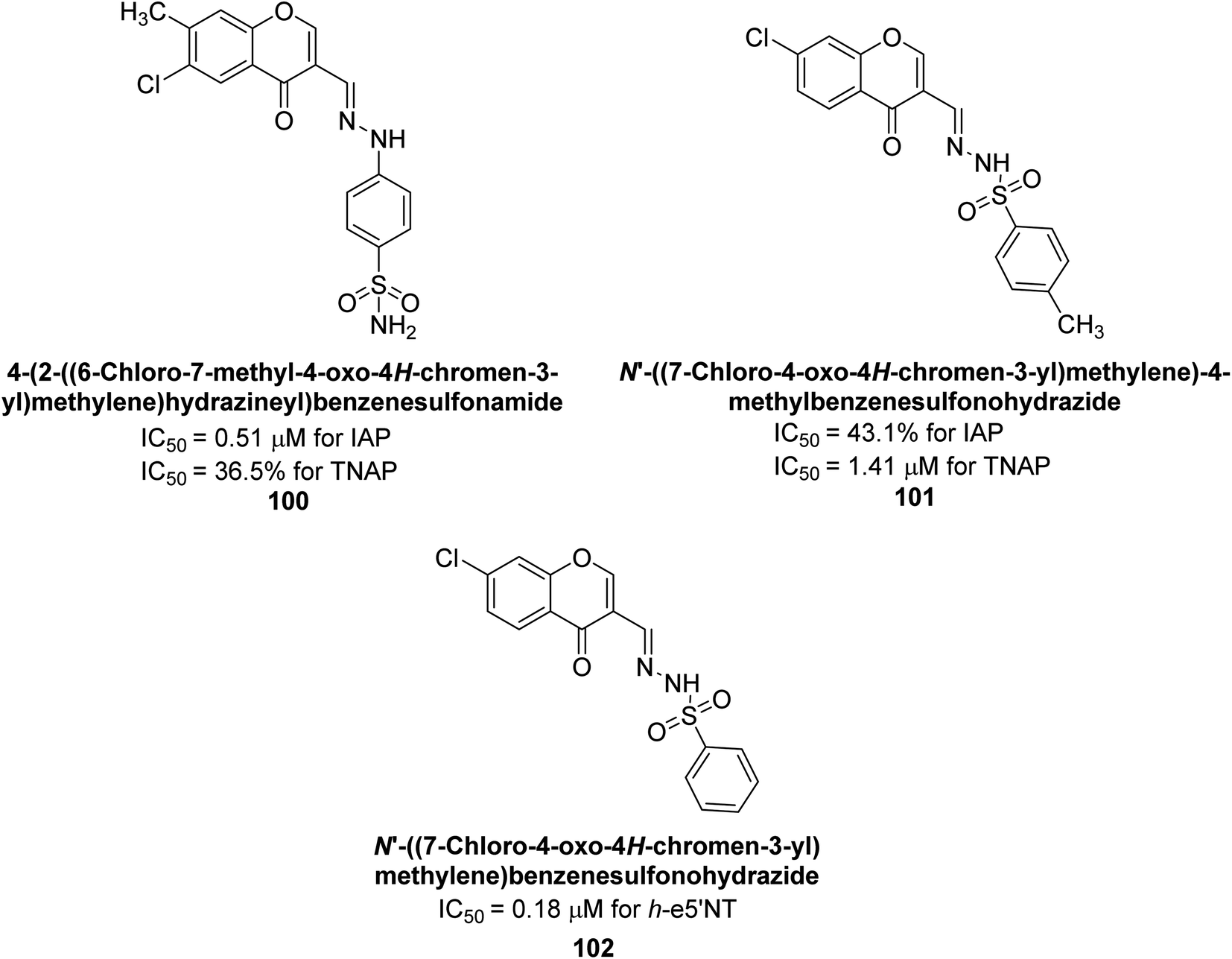 | ||
| Fig. 42 Chemical structures and IC50 values of compounds 100, 101 and 102. | ||
Saeed et al. (2023) reported the design, synthesis, and characterization of a series of novel quinolinyl-iminothiazoline compounds as potential AP inhibitors. Kinetic studies revealed that the synthesized compounds exhibit effective inhibition of AP, with inhibition constants ranging from 0.3 to 3.4 μM. Among the synthesized compounds, compound 103 exhibited a higher inhibitory value (TNAP, IC50 = 0.337 μM) as compared to the standard KH2PO4 (IC50 = 5.245 μM) (Fig. 43). In silico investigations provided valuable insights into the binding modes of the inhibitors within the active site of AP, highlighting the importance of key interactions with specific amino acid residues. Biomineralization assays demonstrated the ability of the synthesized compounds to reduce mineralization in cell culture models, indicating their potential for the treatment of AP-related disorders.174
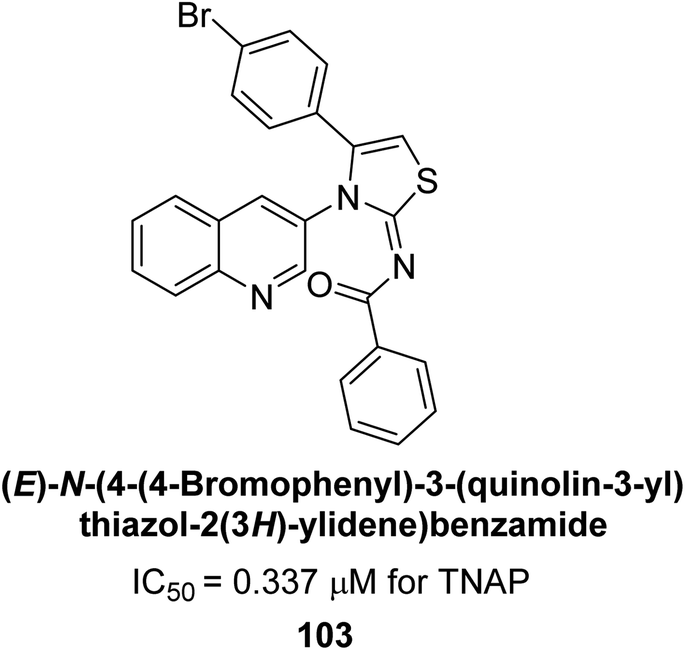 | ||
| Fig. 43 Chemical structure and IC50 value of compound 103. | ||
7. Structure–activity relationship (SAR)
The structure–activity relationship (SAR) of heterocyclic compounds as potent AP inhibitors has been extensively studied in the literature. Some of the key SAR findings include:(1) Heterocyclic ring size: smaller heterocyclic rings, such as pyridine, have been found to have higher inhibitory activity compared to larger rings, such as pyrrolidine.
(2) Ring substitution: substituents on the heterocyclic ring, such as chlorine or bromine, can increase the inhibitory activity. The presence of a hydroxyl group has also been found to enhance the activity.
(3) Hydrophobic moieties: hydrophobic moieties, such as alkyl chains, can increase the potency of the inhibitors by enhancing the lipophilicity of the compounds.
(4) Heteroatom: the presence of nitrogen or sulfur atoms in the heterocyclic ring can increase the inhibitory activity.
(5) Conformation: compounds with rigid and planar structures have been found to have higher inhibitory activity compared to compounds with flexible or non-planar structures.
Electron-donating groups, such as alkyl, alkoxy, and amino groups, increase the electron density on the heterocyclic ring, making it more nucleophilic and therefore more reactive towards electrophiles. On the other hand, electron-withdrawing groups, such as nitro, carbonyl, and cyano groups, decrease the electron density on the heterocyclic ring, making it more electrophilic and therefore more reactive towards nucleophiles. The effect of these groups on the biological and pharmacological activities of heterocyclic compounds can be explained using structure–activity relationships (SARs). SARs involve the systematic modification of the structure of a molecule and the measurement of its activity, allowing for the identification of structural features that are important for activity (Fig. 44).
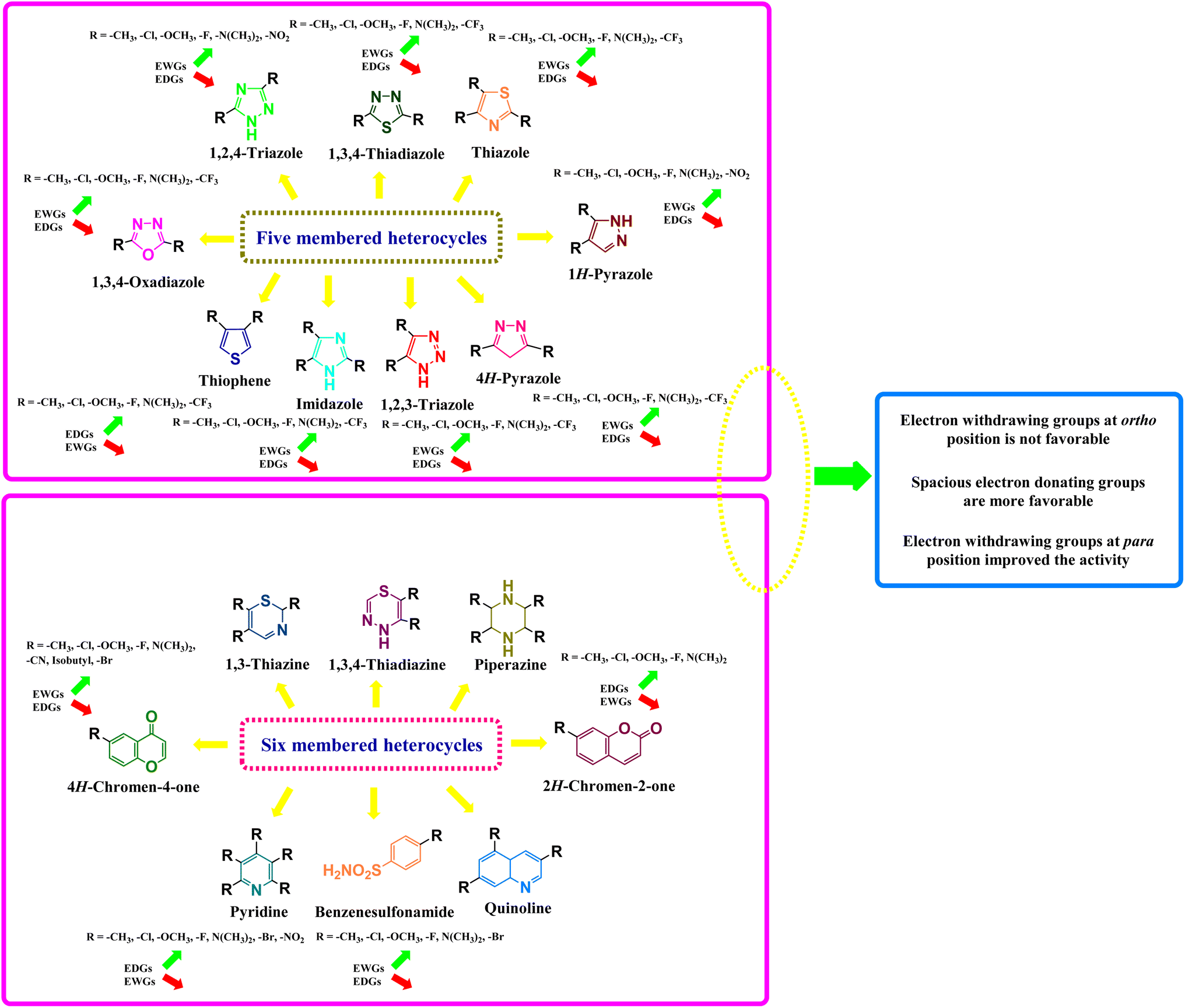 | ||
| Fig. 44 Structure–activity relationship of different heterocyclic scaffolds against AP inhibitors. | ||
Pyridine is a commonly studied heterocycle for its AP inhibitory activity. Pyridine derivatives with electron-withdrawing groups, such as nitro and cyano groups, have been shown to increase inhibitory activity. The position of the substituent also plays a role, with the 2-position being the most common site of substitution. Pyridine derivatives with electron-donating groups, such as amino and hydroxyl groups, have shown mixed results in terms of inhibitory activity. However, the addition of an amino group at the 3-position of a pyridine ring has been shown to enhance inhibitory activity.
Pyrimidine is another commonly studied heterocycle for its AP inhibitory activity. Like pyridine derivatives, pyrimidine derivatives with electron-withdrawing groups, such as nitro and cyano groups, have been shown to increase inhibitory activity. The position of the substituent also plays a role, with the 2- and 4-positions being the most common sites of substitution. Pyrimidine derivatives with electron-donating groups, such as amino and hydroxyl groups, have shown mixed results in terms of inhibitory activity.
Pyrazine derivatives have also been studied for their AP inhibitory activity. Pyrazine derivatives with electron-withdrawing groups, such as nitro and cyano groups, have been shown to increase inhibitory activity. The position of the substituent also plays a role, with the 2- and 4-positions being the most common sites of substitution. Pyrazine derivatives with electron-donating groups, such as amino and hydroxyl groups, have shown mixed results in terms of inhibitory activity.
Similarly, the SAR of imidazole derivatives has shown that electron-donating groups at the 2-position increase the activity of the compound, while electron-withdrawing groups decrease the activity. This is because the 2-position is the site of protonation in many biological systems, and the presence of electron-donating groups makes this position more basic, increasing the compound's activity.
Indole derivatives have shown promising inhibitory activity against alkaline phosphatase. Indole derivatives with electron-withdrawing groups, such as nitro and carbonyl groups, have been shown to increase inhibitory activity. The position of the substituent also plays a role, with the 2- and 3-positions being the most common sites of substitution. Indole derivatives with electron-donating groups, such as amino and hydroxyl groups, have shown mixed results in terms of inhibitory activity.
Quinoline derivatives have also been studied for their alkaline phosphatase inhibitory activity. Quinoline derivatives with electron-withdrawing groups, such as nitro and carbonyl groups, have been shown to increase inhibitory activity. The position of the substituent also plays a role, with the 2- and 4-positions being the most common sites of substitution. Quinoline derivatives with electron-donating groups, such as amino and hydroxyl groups, have shown mixed results in terms of inhibitory activity.
Overall, SAR studies have displayed that a combination of small heterocyclic rings, ring substitution, hydrophobic moieties, and the presence of heteroatoms can lead to potent AP inhibitors.
8. Conclusions and future perspectives
In conclusion, heterocycles have emerged as a promising class of compounds as potent inhibitors of AP. AP is a widely distributed enzyme, involved in many physiological processes such as bone metabolism, cell growth and differentiation, and has been linked to several diseases such as cancer and osteoporosis. Heterocyclic compounds have been found to inhibit AP by binding to the active site of the enzyme, thereby inhibiting its activity. Heterocyclic compounds such as imidazoles, pyrazoles, and pyridines, among others, have been found to be potent AP inhibitors and have been studied as potential therapeutics for the treatment of cancer, osteoporosis, and other diseases. However, further research is needed to fully understand the mechanism of action of these compounds, as well as their potential side effects. The development of more potent and selective inhibitors that can be used as therapeutics for the treatment of various diseases is also an ongoing area of research.Heterocycles constitute a class of compounds that act as potent inhibitors of AP, and thus have shown great potential as therapeutics for the treatment of various diseases. As research in this field continues to advance, several areas of focus hold potential for future development. One area of focus is the development of more potent and selective inhibitors. Current heterocyclic AP inhibitors have varying degrees of potency and selectivity, and there is still a need for the development of compounds with higher efficacy and specificity for AP. Another area of focus is the development of new heterocyclic scaffolds. The structural diversity of heterocycles allows for the generation of a wide range of compounds, and the discovery of new scaffolds could lead to the identification of new and more potent inhibitors. Additionally, the combination of different strategies, such as the use of heterocycles as AP inhibitors in combination with other therapeutic approaches, could lead to more effective treatments for diseases such as cancer and osteoporosis. Moreover, the study of the mechanism of action of heterocyclic AP inhibitors is an ongoing area of research, which could lead to the identification of new targets and new therapeutic strategies. Future research in this field will focus on the development of more potent and selective inhibitors, new heterocyclic scaffolds, and the study of the mechanism of action.
Among the different heterocyclic compounds tested, it has been found that quinoline shows the most promising inhibitory activity against AP. This discovery provides a potential avenue for the development of novel therapeutics that can effectively target AP and prevent or treat various diseases. Further research is warranted to fully understand the mechanism of inhibition of this heterocyclic compound and to optimize its potency and selectivity. Nevertheless, this study highlights the potential of heterocycles as a valuable source of lead compounds for drug discovery in the field of AP inhibition.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Deanship of Scientific Research at Umm Al-Qura University, for supporting this work by Grant code: (23UQU4320545DSR004). The financial support by the Higher Education Commission of Pakistan (HEC) under Project No. (NRPU-15800) is gratefully acknowledged.References
- R. J. Obaid, E. U. Mughal, N. Naeem, M. M. Al-Rooqi, A. Sadiq, R. S. Jassas, Z. Moussa and S. A. Ahmed, Process Biochem., 2022, 120, 250–259 CrossRef CAS.
- R. J. Obaid, N. Naeem, E. U. Mughal, M. M. Al-Rooqi, A. Sadiq, R. S. Jassas, Z. Moussa and S. A. Ahmed, RSC Adv., 2022, 12, 19764–19855 RSC.
- J. Ashraf, E. U. Mughal, A. Sadiq, M. Bibi, N. Naeem, A. Ali, A. Massadaq, N. Fatima, A. Javid and M. N. Zafar, J. Biomol. Struct. Dyn., 2021, 39, 7107–7122 CrossRef CAS PubMed.
- A. Naseer, F. A. Osra, A. N. Awan, A. Imran, A. Hameed, S. A. Ali Shah, J. Iqbal and Z. A. Zakaria, Pharmaceuticals, 2022, 15, 1288 CrossRef CAS PubMed.
- H. A. Ezelarab, S. H. Abbas, M. A. Abourehab, M. Badr, S. Sureram, P. Hongmanee, P. Kittakoop, G. E.-D. A. Abuo-Rahma and H. A. Hassan, Med. Chem. Res., 2021, 30, 2168–2183 CrossRef CAS.
- F. M. Alkhatib, T. A. Farghaly, M. F. Harras and H. A. El-Ghamry, Inorg. Nano-Met. Chem., 2021, 1–12 Search PubMed.
- R. Mehmood, A. Sadiq, R. I. Alsantali, E. U. Mughal, M. A. Alsharif, N. Naeem, A. Javid, M. M. Al-Rooqi, G.-e.-S. Chaudhry and S. A. Ahmed, ACS Omega, 2022, 7, 3775–3795 CrossRef CAS PubMed.
- R. Mehmood, E. U. Mughal, E. B. Elkaeed, R. J. Obaid, Y. Nazir, H. A. Al-Ghulikah, N. Naeem, M. M. Al-Rooqi, S. A. Ahmed and S. W. A. Shah, ACS Omega, 2022, 7, 30215–30232 CrossRef CAS PubMed.
- H. A. Al-ghulikah, E. U. Mughal, E. B. Elkaeed, N. Naeem, Y. Nazir, A. Y. A. Alzahrani, A. Sadiq and S. W. A. Shah, J. Mol. Struct., 2023, 1275, 134658 CrossRef CAS.
- E. U. Mughal, A. Sadiq, M. Ayub, N. Naeem, A. Javid, S. H. Sumrra, M. N. Zafar, B. A. Khan, F. P. Malik and I. Ahmed, J. Biomol. Struct. Dyn., 2021, 39, 6154–6167 CrossRef CAS PubMed.
- R. J. Obaid, E. U. Mughal, N. Naeem, A. Sadiq, R. I. Alsantali, R. S. Jassas, Z. Moussa and S. A. Ahmed, RSC Adv., 2021, 11, 22159–22198 RSC.
- M. Samir, M. Ramadan, M. H. Abdelrahman, M. S. Abdelbaset, M. A. Abourehab, M. Abdel-Aziz and G. E.-D. A. Abuo-Rahma, Bioorg. Chem., 2021, 110, 104698 CrossRef CAS PubMed.
- J. Ashraf, E. U. Mughal, R. I. Alsantali, R. J. Obaid, A. Sadiq, N. Naeem, A. Ali, A. Massadaq, Q. Javed and A. Javid, Bioorg. Med. Chem., 2021, 35, 116057 CrossRef CAS PubMed.
- R. I. Alsantali, E. U. Mughal, N. Naeem, M. A. Alsharif, A. Sadiq, A. Ali, R. S. Jassas, Q. Javed, A. Javid and S. H. Sumrra, J. Mol. Struct., 2022, 1251, 131933 CrossRef CAS.
- E. U. Mughal, J. Ashraf, E. M. Hussein, Y. Nazir, A. S. Alwuthaynani, N. Naeem, A. Sadiq, R. I. Alsantali and S. A. Ahmed, ACS Omega, 2022, 7, 17444–17461 CrossRef CAS PubMed.
- T. Al-Warhi, D. M. Elimam, Z. M. Elsayed, M. M. Abdel-Aziz, R. M. Maklad, A. A. Al-Karmalawy, K. Afarinkia, M. A. Abourehab, H. A. Abdel-Aziz and W. M. Eldehna, RSC Adv., 2022, 12, 31466–31477 RSC.
- U. Sharma, D. Pal and R. Prasad, Indian J. Clin. Biochem., 2014, 29, 269–278 CrossRef CAS PubMed.
- K. Rani, S. Datt and R. Rana, Int. J. Biol. Sci., 2012, 2, 1–4 Search PubMed.
- R. B. McComb, G. N. Bowers Jr and S. Posen, Alkaline phosphatase, Springer Science & Business Media, 2013 Search PubMed.
- S. M. Shaban, S. B. Jo, E. Hafez, J. H. Cho and D.-H. Kim, Coord. Chem. Rev., 2022, 465, 214567 CrossRef CAS.
- T.-C. Chang, J.-K. Wang, M.-W. Hung, C.-H. Chiao, L.-C. Tsai and G. Chang, Biochem. J., 1994, 303, 199–205 CrossRef CAS PubMed.
- A. Sadeghirizi and R. Yazdanparast, Acta Biochim. Pol., 2007, 54, 323–329 CrossRef CAS PubMed.
- S. Sharifian, A. Homaei, S.-K. Kim and M. Satari, Process Biochem., 2018, 64, 103–115 CrossRef CAS.
- S. Zappa, J.-L. Rolland, D. Flament, Y. Gueguen, J. Boudrant and J. Dietrich, Appl. Environ. Microbiol., 2001, 67, 4504–4511 CrossRef CAS PubMed.
- J. Kieleczawa, S. J. Coughlan and G. Hind, Plant Physiol., 1992, 99, 1029–1036 CrossRef CAS PubMed.
- M. Eguchi, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1995, 111, 151–162 CrossRef CAS PubMed.
- R. McComb, G. Bowers and S. Posen, Alkaline Phosphatase Plenum, New York, 1979, pp. 133–134 Search PubMed.
- A. Torriani, The alkaline phosphatase of Escherichia coli, Bacteria, Bacteriophages and Fungi, Springer, 1974, vol. 1, pp. 173–181 Search PubMed.
- J. P. Bilezikian, L. G. Raisz and T. J. Martin, Principles of bone biology, Academic press, 2008 Search PubMed.
- M. H. l. n. Le Du, T. Stigbrand, M. J. Taussig, A. Ménez and E. A. Stura, J. Biol. Chem., 2001, 276, 9158–9165 CrossRef CAS PubMed.
- B. Le-Vinh, Z. B. Akkuş-Dağdeviren, N. M. N. Le, I. Nazir and A. Bernkop-Schnürch, Adv. Thermoelectr., 2022, 5, 2100219 CAS.
- M. F. Hoylaerts, T. Manes and J. L. Millán, J. Biol. Chem., 1997, 272, 22781–22787 CrossRef CAS PubMed.
- D. M. Zaher, M. I. El-Gamal, H. A. Omar, S. N. Aljareh, S. A. Al-Shamma, A. J. Ali, S. Zaib and J. Iqbal, Arch. Pharm., 2020, 353, e2000011 CrossRef CAS PubMed.
- M. al-Rashida and J. Iqbal, Med. Res. Rev., 2014, 34, 703–743 CrossRef CAS PubMed.
- O. A. Azher, A. Hossan, R. A. Pashameah, A. Alsoliemy, A. Alharbi, T. M. Habeebullah and N. M. El-Metwaly, Arabian J. Chem., 2023, 16, 104437 CrossRef CAS.
- A. T. Ali, C. B. Penny, J. E. Paiker, G. Psaras, F. Ikram and N. J. Crowther, Ann. Clin. Biochem., 2006, 43, 207–213 CrossRef CAS PubMed.
- S. Wang, P. Su, J. Huang, J. Wu and Y. Yang, J. Mater. Chem. B, 2013, 1, 1749–1754 RSC.
- P. Llinas, E. A. Stura, A. Ménez, Z. Kiss, T. Stigbrand, J. L. Millán and M. H. Le Du, J. Mol. Biol., 2005, 350, 441–451 CrossRef CAS PubMed.
- H. Harris, Clin. Chim. Acta, 1990, 186, 133–150 CrossRef CAS PubMed.
- T. Stigbrand, Human alkaline phosphatases, 1984 Search PubMed.
- D. Lowe, T. Sanvictores, M. Zubair and S. John, in StatPearls, StatPearls Publishing, 2022 Search PubMed.
- M. Hanschkow, N. Boulet, E. Kempf, A. Bouloumié, W. Kiess, R. Stein, A. Körner and K. Landgraf, J. Clin. Endocrinol. Metab., 2022, 107, e836–e851 CrossRef PubMed.
- S. Vimalraj, Gene, 2020, 754, 144855 CrossRef CAS PubMed.
- M. P. Whyte, Bone, 2017, 102, 15–25 CrossRef CAS PubMed.
- M. Orsaria, A. P. Londero, S. Marzinotto, C. Di Loreto, D. Marchesoni and L. Mariuzzi, Cancer Biomarkers, 2016, 17, 479–486 CAS.
- M. Sartori, L. Mezzano, S. Lin, S. Munoz and S. De Fabro, Trop. Med. Int. Health, 2003, 8, 832–839 CrossRef CAS PubMed.
- S. Lin, M. Sartori, L. Mezzano and S. de Fabro, Placenta, 2005, 26, 789–795 CrossRef CAS PubMed.
- J. L. Millán and M. P. Whyte, Calcif. Tissue Int., 2016, 98, 398–416 CrossRef PubMed.
- C. Halling Linder, B. Ek-Rylander, M. Krumpel, M. Norgård, S. Narisawa, J. L. Millán, G. Andersson and P. Magnusson, Calcif. Tissue Int., 2017, 101, 92–101 CrossRef CAS PubMed.
- A. Tuin, K. Poelstra, A. de Jager-Krikken, L. Bok, W. Raaben, M. P. Velders and G. Dijkstra, Gut, 2009, 58, 379–387 CrossRef CAS PubMed.
- F. Su, R. Brands, Z. Wang, C. Verdant, A. Bruhn, Y. Cai, W. Raaben, M. Wulferink and J.-L. Vincent, Crit. Care Med., 2006, 34, 2182–2187 CrossRef CAS PubMed.
- S. N. Alam, H. Yammine, O. Moaven, R. Ahmed, A. K. Moss, B. Biswas, N. Muhammad, R. Biswas, A. Raychowdhury and K. Kaliannan, Ann. Surg., 2014, 259, 715–722 CrossRef PubMed.
- T.-R. Kuo and C.-H. Chen, Biomark. Res., 2017, 5, 1–9 CrossRef PubMed.
- S. Rao, A. Snaith, D. Marino, X. Cheng, S. Lwin, I. Orriss, F. Hamdy and C. Edwards, Br. J. Cancer, 2017, 116, 227–236 CrossRef CAS PubMed.
- S. Jeftic, Interfacial binding surface of PI-specific phospholipase C from Bacillus thuringiensis, Doctoral dissertation, University of Guelph, 2007 Search PubMed.
- F. C. Neale, J. S. Clubb, D. Hotchkis and S. Posen, J. Clin. Pathol., 1965, 18, 359–363 CrossRef CAS PubMed.
- G. A. P. Boechat, S. T. Stinghen, G. Custódio, M. A. D. Pianovski, F. R. de Oliveira Figueiredo, J. Jenkins, G. P. Zambetti, R. C. Ribeiro and B. C. Figueiredo, J. Pediatr. Hematol./Oncol., 2011, 33, e149–e153 CrossRef CAS PubMed.
- N. Ravenni, M. Weber and D. Neri, mAbs, 2014, 6, 86–94 CrossRef PubMed.
- J. Fawley and D. M. Gourlay, J. Surg. Res., 2016, 202, 225–234 CrossRef CAS PubMed.
- K. Kaliannan, S. R. Hamarneh, K. P. Economopoulos, S. Nasrin Alam, O. Moaven, P. Patel, N. S. Malo, M. Ray, S. M. Abtahi and N. Muhammad, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7003–7008 CrossRef CAS PubMed.
- M. M. Al-Rooqi, E. U. Mughal, Q. A. Raja, E. M. Hussein, N. Naeem, A. Sadiq, B. H. Asghar, Z. Moussa and S. A. Ahmed, RSC Adv., 2023, 13, 3210–3233 RSC.
- J. Bilski, A. Mazur-Bialy, D. Wojcik, J. Zahradnik-Bilska, B. Brzozowski, M. Magierowski, T. Mach, K. Magierowska and T. Brzozowski, Mediators Inflammation, 2017, 2017, 1–9 CrossRef PubMed.
- S. Narisawa, L. Huang, A. Iwasaki, H. Hasegawa, D. H. Alpers and J. L. Millán, Mol. Cell. Microbiol., 2003, 23, 7525–7530 CAS.
- M. Mizumori, M. Ham, P. H. Guth, E. Engel, J. D. Kaunitz and Y. Akiba, J. Physiol., 2009, 587, 3651–3663 CrossRef CAS PubMed.
- K. S. Munawar, S. Ali, M. N. Tahir, N. Khalid, Q. Abbas, I. Z. Qureshi, S. Hussain and M. Ashfaq, J. Coord. Chem., 2020, 73, 2275–2300 CrossRef CAS.
- J.-P. Lallès, Nutr. Rev., 2010, 68, 323–332 CrossRef PubMed.
- P. S. Henthorn, M. Raducha, T. Kadesch, M. Weiss and H. Harris, J. Biol. Chem., 1988, 263, 12011–12019 CrossRef CAS PubMed.
- J.-P. Lallès, Nutr. Rev., 2019, 77, 710–724 CrossRef PubMed.
- M. Malo, S. N. Alam, G. Mostafa, S. Zeller, P. Johnson, N. Mohammad, K. Chen, A. Moss, S. Ramasamy and A. Faruqui, Gut, 2010, 59, 1476–1484 CrossRef CAS PubMed.
- J. H. Beckstead, Am. J. Clin. Pathol., 1983, 7, 341–350 CAS.
- J. L. Millan and T. Manes, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 3024–3028 CrossRef CAS PubMed.
- H. Merchant-Larios, F. Mendlovic and A. Alvarez-Buylla, Differentiation, 1985, 29, 145–151 CrossRef CAS PubMed.
- M. Okamoto, S. Yamaguchi, Y. Ishi, H. Motegi, T. Mori, T. Hashimoto, Y. Terashita, S. Hirabayashi, M. Sugiyama and A. Iguchi, Oncology, 2021, 99, 23–31 CrossRef CAS PubMed.
- M. Al-Rashida and J. Iqbal, Mini-Rev. Med. Chem., 2015, 15, 41–51 CrossRef CAS PubMed.
- K. Koshida, A. Nishino, H. Yamamoto, T. Uchibayashi, K. Naito, H. Hisazumi, K. Hirano, Y. Hayashi, B. Wahren and L. Andersson, J. Urol., 1991, 146, 57–60 CrossRef CAS PubMed.
- D. Lewis-Jones, P. Johnson, A. Desmond and P. McLaughlin, Br. J. Urol., 1992, 69, 418–420 CAS.
- W. H. Fishman, Clin. Biochem., 1990, 23, 99–104 CrossRef CAS PubMed.
- C. M. Povinelli and B. J. Knoll, Placenta, 1991, 12, 663–668 CrossRef CAS PubMed.
- H. van Belle, Biochim. Biophys. Acta, Enzymol., 1972, 289, 158–168 CrossRef CAS PubMed.
- M. Schoppet and C. Shanahan, Kidney Int., 2008, 73, 989–991 CrossRef CAS PubMed.
- M. J. Weiss, K. Ray, P. Henthorn, B. Lamb, T. Kadesch and H. Harris, J. Biol. Chem., 1988, 263, 12002–12010 CrossRef CAS PubMed.
- N. J. Fernandez and B. A. Kidney, Vet. Clin. Pathol., 2007, 36, 223–233 CrossRef PubMed.
- E. E. Golub and K. Boesze-Battaglia, Curr. opin. orthop., 2007, 18, 444–448 CrossRef.
- C. Zucchini, M. Bianchini, L. Valvassori, S. Perdichizzi, S. Benini, M. C. Manara, R. Solmi, P. Strippoli, P. Picci and P. Carinci, Bone, 2004, 34, 672–679 CrossRef CAS PubMed.
- A. K. Moss, S. R. Hamarneh, M. M. R. Mohamed, S. Ramasamy, H. Yammine, P. Patel, K. Kaliannan, S. N. Alam, N. Muhammad and O. Moaven, Am. J. Physiol.: Gastrointest. Liver Physiol., 2013, 304, 597–604 CrossRef PubMed.
- J.-P. Lallès, Nutr. Rev., 2014, 72, 82–94 CrossRef PubMed.
- V. Vallon, B. Muhlbauer and H. Osswald, Physiol. Rev., 2006, 86, 901–940 CrossRef CAS PubMed.
- K. Hirano, H. Matsumoto, T. Tanaka, Y. Hayashi, S. Lino, U. Domar and T. Stigbrand, Clin. Chim. Acta, 1987, 166, 265–273 CrossRef CAS PubMed.
- R. López-Posadas, R. Gonzalez, I. Ballester, P. Martínez-Moya, I. Romero-Calvo, M. D. Suárez, A. Zarzuelo, O. Martínez-Augustin and F. Sanchez de Medina, Inflammatory Bowel Dis., 2011, 17, 543–556 CrossRef PubMed.
- T. Harada, I. Koyama, A. Shimoi, D. H. Alpers and T. Komoda, Cell Tissue Res., 2002, 307, 69–77 CrossRef CAS PubMed.
- J. L. Millán, W. H. Fishman and R. Stinson, Crit. Rev. Clin. Lab. Sci., 1995, 32, 1–39 CrossRef.
- H. G. Wada, J. E. Shindelman, A. E. Ortmeyer and H. H. Sussman, Int. J. Cancer, 1979, 23, 781–787 CrossRef CAS PubMed.
- J. M. Pekarthy, J. Short, A. I. Lansing and I. Lieberman, J. Biol. Chem., 1972, 247, 1767–1774 CrossRef CAS.
- E. Peters, S. Heemskerk, R. Masereeuw and P. Pickkers, Am. J. Kidney Dis., 2014, 63, 1038–1048 CrossRef CAS PubMed.
- A. Capelli, M. Lusuardi, C. G. Cerutti and C. F. Donner, Am. J. Respir. Crit. Care Med., 1997, 155, 249–253 CrossRef CAS PubMed.
- D. Smith, W. Hunter and L. Spadoni, Fertil. Steril., 1970, 21, 549–554 CrossRef CAS PubMed.
- J. Gautray, P. Couderg, M. Colomb, H. Sibut and C. Maurel, Am. J. Obstet. Gynecol., 1969, 104, 818–822 CrossRef CAS PubMed.
- S. Pandey, A. K. Sharma, K. H. Sharma, Y. Nerthigan, M. S. Khan, D.-R. Hang and H.-F. Wu, Sens. Actuators, B, 2018, 254, 514–518 CrossRef CAS.
- J. Reis, Br. J. Ophthalmol., 1951, 35, 149 CrossRef CAS PubMed.
- M.-H. Le Du and J. L. Millán, J. Biol. Chem., 2002, 277, 49808–49814 CrossRef CAS PubMed.
- M. Al-Rashida, S. U. Qazi, N. Batool, A. Hameed and J. Iqbal, Expert Opin. Ther. Pat., 2017, 27, 1291–1304 CrossRef CAS PubMed.
- B. Le-Vinh, N.-M. N. Le, I. Nazir, B. Matuszczak and A. Bernkop-Schnürch, Int. J. Biol. Macromol., 2019, 133, 647–655 CrossRef CAS PubMed.
- J. M. Motion, J. Nguyen and F. C. Szoka, Angew. Chem., 2012, 124, 9181–9185 CrossRef.
- F. Prüfert, S. Bonengel, S. Köllner, J. Griesser, M. D. Wilcox, P. I. Chater, J. P. Pearson and A. Bernkop-Schnürch, Nanomedicine, 2017, 12, 2713–2724 CrossRef PubMed.
- W. Suchaoin, I. P. de Sousa, K. Netsomboon, H. T. Lam, F. Laffleur and A. Bernkop-Schnürch, Int. J. Pharm., 2016, 510, 255–262 CrossRef CAS PubMed.
- J. Li, Y. Gao, Y. Kuang, J. Shi, X. Du, J. Zhou, H. Wang, Z. Yang and B. Xu, J. Am. Chem. Soc., 2013, 135, 9907–9914 CrossRef CAS PubMed.
- K. M. Holtz and E. R. Kantrowitz, FEBS Lett., 1999, 462, 7–11 CrossRef CAS PubMed.
- E. E. Kim and H. W. Wyckoff, J. Mol. Biol., 1991, 218, 449–464 CrossRef CAS PubMed.
- J. Silvent, B. Gasse, E. Mornet and J.-Y. Sire, J. Biol. Chem., 2014, 289, 24168–24179 CrossRef CAS PubMed.
- G. L. Borosky, J. Phys. Chem. B, 2014, 118, 14302–14313 CrossRef CAS PubMed.
- K. M. Holtz, B. Stec and E. R. Kantrowitz, J. Biol. Chem., 1999, 274, 8351–8354 CrossRef CAS PubMed.
- A. Owaki, D. Inaguma, A. Tanaka, H. Shinjo, S. Inaba and K. Kurata, Nephron Extra, 2017, 7, 78–88 CrossRef PubMed.
- P. Xiangyu, J. Zhao and Y. Wu, Clin. Invest. Med., 2019, 42, E47–E52 CrossRef PubMed.
- A. De, R. Puttannavar, F. Rahman, A. Adak, R. Sahoo and B. R. Prakash, J. Oral Maxillofac. Pathol., 2018, 22, 445 CrossRef PubMed.
- R. Gu and Y. Sun, J. Cancer Res. Ther., 2018, 14, S468–S472 CAS.
- J. Zhu, Y. Luan and H. Li, Medicine, 2018, 97, 1–4 CrossRef PubMed.
- M. Khosravifarsani, M. Bahadoram, S. M. Kazem Nourbakhsh, R. Nikkhah and M. Davoodi, J. Nephropathol., 2018, 7, 1–5 Search PubMed.
- M. F. Tolba, H. A. Omar, F. Hersi, A. C. Nunes and A. M. Noreddin, Mol. Cell. Endocrinol., 2019, 488, 79–88 CrossRef CAS PubMed.
- A. R. Metwalli, I. L. Rosner, J. Cullen, Y. Chen, T. Brand, S. A. Brassell, J. Lesperance, C. Porter, J. Sterbis and D. G. McLeod, Urol. Oncol.: Semin. Orig. Invest., 2014, 32, 761–768 CrossRef CAS PubMed.
- S. M. Abdin, D. M. Zaher, E.-S. A. Arafa and H. A. Omar, Cancers, 2018, 10, 32 CrossRef PubMed.
- M. A. Abdelgawad, A. Belal, H. A. Omar, L. Hegazy and M. E. Rateb, Arch. Pharm., 2013, 346, 534–541 CrossRef CAS PubMed.
- W.-X. Du, S.-F. Duan, J.-J. Chen, J.-F. Huang, L.-M. Yin and P.-J. Tong, J. Cancer Res. Ther., 2014, 10, C140–C143 CrossRef PubMed.
- J. Compston, D. Judd, E. Crawley, W. Evans, C. Evans, H. Church, E. Reid and J. Rhodes, Gut, 1987, 28, 410–415 CrossRef CAS PubMed.
- H. A. Fink, S. Litwack-Harrison, B. C. Taylor, D. C. Bauer, E. S. Orwoll, C. G. Lee, E. Barrett-Connor, J. T. Schousboe, D. M. Kado and P. S. Garimella, Osteoporosis Int., 2016, 27, 331–338 CrossRef CAS PubMed.
- S. S. Hegde, A. V. Revankar and A. K. Patil, Indian J. Dent. Res., 2018, 29, 721 CrossRef PubMed.
- S. Narisawa, D. Harmey, M. C. Yadav, W. C. O'Neill, M. F. Hoylaerts and J. L. Millán, J. Bone Miner. Res., 2007, 22, 1700–1710 CrossRef CAS PubMed.
- S. Sidique, R. Ardecky, Y. Su, S. Narisawa, B. Brown, J. L. Millán, E. Sergienko and N. D. Cosford, Bioorg. Med. Chem. Lett., 2009, 19, 222–225 CrossRef CAS PubMed.
- L. Li, L. Chang, S. Pellet-Rostaing, F. Liger, M. Lemaire, R. Buchet and Y. Wu, Bioorg. Med. Chem., 2009, 17, 7290–7300 CrossRef CAS PubMed.
- M. Lanier, E. Sergienko, A. M. Simão, Y. Su, T. Chung, J. L. Millán and J. R. Cashman, Bioorg. Med. Chem., 2010, 18, 573–579 CrossRef CAS PubMed.
- R. Raza, A. Matin, S. Sarwar, M. Barsukova-Stuckart, M. Ibrahim, U. Kortz and J. Iqbal, Dalton Trans., 2012, 41, 14329–14336 RSC.
- I. Khan, M. Hanif, M. T. Hussain, A. A. Khan, M. A. S. Aslam, N. H. Rama and J. Iqbal, Aust. J. Chem., 2012, 65, 1413–1419 CrossRef CAS.
- J. Debray, L. Chang, S. Marquès, S. Pellet-Rostaing, S. Mebarek, R. Buchet, D. Magne, F. Popowycz and M. Lemaire, Bioorg. Med. Chem., 2013, 21, 7981–7987 CrossRef CAS PubMed.
- I. Khan, A. Ibrar, S. A. Ejaz, S. U. Khan, S. J. A. Shah, S. Hameed, J. Simpson, J. Lecka, J. Sévigny and J. Iqbal, RSC Adv., 2015, 5, 90806–90818 RSC.
- M. Al-Rashida, S. A. Ejaz, S. Ali, A. Shaukat, M. Hamayoun, M. Ahmed and J. Iqbal, Bioorg. Med. Chem., 2015, 23, 2435–2444 CrossRef CAS PubMed.
- I. Khan, S. J. A. Shah, S. A. Ejaz, A. Ibrar, S. Hameed, J. Lecka, J. L. Millán, J. Sévigny and J. Iqbal, RSC Adv., 2015, 5, 64404–64413 RSC.
- A. Ibrar, S. Zaib, I. Khan, F. Jabeen, J. Iqbal and A. Saeed, RSC Adv., 2015, 5, 89919–89931 RSC.
- M. Miliutina, A. Ivanov, S. A. Ejaz, J. Iqbal, A. Villinger, V. O. Iaroshenko and P. Langer, RSC Adv., 2015, 5, 60054–60078 RSC.
- M. Miliutina, S. A. Ejaz, V. O. Iaroshenko, A. Villinger, J. Iqbal and P. Langer, Org. Biomol. Chem., 2016, 14, 495–502 RSC.
- A. Saeed, S. A. Ejaz, M. Shehzad, S. Hassan, M. al-Rashida, J. Lecka, J. Sévigny and J. Iqbal, RSC Adv., 2016, 6, 21026–21036 RSC.
- A. Ibrar, S. Zaib, F. Jabeen, J. Iqbal and A. Saeed, Arch. Pharm., 2016, 349, 553–565 CrossRef CAS PubMed.
- S. A. Ejaz, A. Saeed, M. N. Siddique, Z. un Nisa, S. Khan, J. Lecka, J. Sévigny and J. Iqbal, Bioorg. Chem., 2017, 70, 229–236 CrossRef CAS PubMed.
- H. A. Bhatti, M. Khatoon, M. Al-Rashida, H. Bano, N. Iqbal, S. Yousuf, K. M. Khan, A. Hameed and J. Iqbal, Bioorg. Chem., 2017, 71, 10–18 CrossRef CAS PubMed.
- P. A. Channar, S. J. A. Shah, S. Hassan, Z. u. Nisa, J. Lecka, J. Sévigny, J. Bajorath, A. Saeed and J. Iqbal, Chem. Biol. Drug Des., 2017, 89, 365–370 CrossRef CAS PubMed.
- U. Salar, K. M. Khan, J. Iqbal, S. A. Ejaz, A. Hameed, M. Al-Rashida, S. Perveen and M. N. Tahir, Eur. J. Med. Chem., 2017, 131, 29–47 CrossRef CAS PubMed.
- A. Petrosyan, T. V. Ghochikyan, S. A. Ejaz, Z. Z. Mardiyan, S. U. Khan, T. Grigoryan, A. Gevorgyan, M. A. Samvelyan, A. S. Galstyan, S. Parpart, Q. Rahman, J. Iqbal and P. Langer, ChemistrySelect, 2017, 2, 5677–5683 CrossRef CAS.
- M. Miliutina, S. A. Ejaz, S. U. Khan, V. O. Iaroshenko, A. Villinger, J. Iqbal and P. Langer, Eur. J. Med. Chem., 2017, 126, 408–420 CrossRef CAS PubMed.
- J. Iqbal, M. I. El-Gamal, S. A. Ejaz, J. Lecka, J. Sévigny and C.-H. Oh, J. Enzyme Inhib. Med. Chem., 2018, 33, 479–484 CrossRef CAS PubMed.
- P. A. Channar, S. Afzal, S. A. Ejaz, A. Saeed, F. A. Larik, P. A. Mahesar, J. Lecka, J. Sévigny, M. F. Erben and J. Iqbal, Eur. J. Med. Chem., 2018, 156, 461–478 CrossRef CAS PubMed.
- S. Hassan, S. A. Ejaz, A. Saeed, M. Shehzad, S. U. Khan, J. Lecka, J. Sévigny, G. Shabir and J. Iqbal, Bioorg. Chem., 2018, 76, 237–248 CrossRef CAS PubMed.
- A. Ashraf, S. A. Ejaz, S. U. Rahman, W. A. Siddiqui, M. N. Arshad, J. Lecka, J. Sévigny, M. E. M. Zayed, A. M. Asiri, J. Iqbal, C. G. Hartinger and M. Hanif, Eur. J. Med. Chem., 2018, 159, 282–291 CrossRef CAS PubMed.
- L. Supe, S. Afzal, A. Mahmood, S. A. Ejaz, M. Hein, V. O. Iaroshenko, A. Villinger, J. Lecka, J. Sévigny, J. Iqbal and P. Langer, Eur. J. Org. Chem., 2018, 2018, 2629–2644 CrossRef CAS.
- B. Jafari, M. Ospanov, S. A. Ejaz, N. Yelibayeva, S. U. Khan, S. T. Amjad, S. Safarov, Z. A. Abilov, M. Z. Turmukhanova, S. N. Kalugin, P. Ehlers, J. Lecka, J. Sévigny, J. Iqbal and P. Langer, Eur. J. Med. Chem., 2018, 144, 116–127 CrossRef CAS PubMed.
- M. Miliutina, J. Janke, S. Hassan, S. Zaib, J. Iqbal, J. Lecka, J. Sévigny, A. Villinger, A. Friedrich, S. Lochbrunner and P. Langer, Org. Biomol. Chem., 2018, 16, 717–732 RSC.
- M. Faisal, S. Shahid, S. A. Ghumro, A. Saeed, F. A. Larik, Z. Shaheen, P. A. Channar, T. A. Fattah, S. Rasheed and P. A. Mahesar, Synth. Commun., 2018, 48, 462–472 CrossRef CAS.
- A. Saeed, G. Saddique, P. A. Channar, F. A. Larik, Q. Abbas, M. Hassan, H. Raza, T. A. Fattah and S.-Y. Seo, Bioorg. Med. Chem., 2018, 26, 3707–3715 CrossRef PubMed.
- M. A. Abbasi, M. Nazir, A. ur-Rehman, S. Z. Siddiqui, M. Hassan, H. Raza, S. A. Shah, M. Shahid and S. Y. Seo, Arch. Pharm., 2019, 352, 1800278 CrossRef PubMed.
- Z. Iqbal, A. Iqbal, Z. Ashraf, M. Latif, M. Hassan and H. Nadeem, Drug Dev. Res., 2019, 80, 646–654 CAS.
- Z. Iqbal, Z. Ashraf, M. Hassan, Q. Abbas and E. Jabeen, Bioorg. Chem., 2019, 90, 103108 CrossRef CAS PubMed.
- H. Andleeb, S. Hameed, S. A. Ejaz, I. Khan, S. Zaib, J. Lecka, J. Sevigny and J. Iqbal, Bioorg. Chem., 2019, 88, 102893 CrossRef CAS PubMed.
- P. A. Channar, H. Irum, A. Mahmood, G. Shabir, S. Zaib, A. Saeed, Z. Ashraf, F. A. Larik, J. Lecka and J. Sevigny, Bioorg. Chem., 2019, 91, 103137 CrossRef PubMed.
- H. Andleeb, M. Hussain, S. A. Ejaz, J. Sevigny, M. Farman, M. Yasinzai, J. Zhang, J. Iqbal and S. Hameed, Bioorg. Chem., 2020, 101, 103999 CrossRef CAS PubMed.
- A. Mumtaz, K. Saeed, A. Mahmood, S. Zaib, A. Saeed, J. Pelletier, J. Sévigny and J. Iqbal, Bioorg. Chem., 2020, 101, 103996 CrossRef CAS PubMed.
- A. Saeed, A. Khurshid, G. Shabir, A. Mahmood, S. Zaib and J. Iqbal, Bioorg. Med. Chem. Lett., 2020, 30, 127238 CrossRef CAS PubMed.
- H. A. Younus, A. Hameed, A. Mahmood, M. S. Khan, M. Saeed, F. Batool, A. Asari, H. Mohamad, J. Pelletier, J. Sévigny, J. Iqbal and M. A. Rashida, Bioorg. Chem., 2020, 100, 103827 CrossRef CAS PubMed.
- M. Abbasi, M. Nazir, S. Siddiqui, H. Raza, A. Zafar, S. A. Shah and M. Shahid, Russ. J. Bioorg. Chem., 2021, 47, 1086–1096 CrossRef CAS.
- J. Ashraf, E. U. Mughal, R. I. Alsantali, A. Sadiq, R. S. Jassas, N. Naeem, Z. Ashraf, Y. Nazir, M. N. Zafar and A. Mumtaz, RSC Adv., 2021, 11, 35077–35092 RSC.
- G. Bassi, N. Favalli, C. Pellegrino, Y. Onda, J. r. Scheuermann, S. Cazzamalli, M. G. Manz and D. Neri, J. Med. Chem., 2021, 64, 15799–15809 CrossRef CAS PubMed.
- A. Khurshid, A. Saeed, Z. Ashraf, Q. Abbas and M. Hassan, Mol. Diversity, 2021, 25, 1701–1715 CrossRef CAS PubMed.
- H. Aziz, A. Mahmood, S. Zaib, A. Saeed, H. R. El-Seedi, J. Pelletier, J. Sévigny and J. Iqbal, J. Biomol. Struct. Dyn., 2021, 39, 6140–6153 CrossRef CAS PubMed.
- S. A. Channar, P. A. Channar, A. Saeed, A. A. Alsfouk, S. A. Ejaz, R. Ujan, R. Noor, M. S. Bilal, Q. Abbas and Z. Hussain, Med. Chem. Res., 2022, 31, 1792–1802 CrossRef CAS.
- A. Ahmed, S.-u. Rehman, S. A. Ejaz, A. Saeed, R. Ujan, P. A. Channar, K. Mahar, R. Sahito, S. M. Albogami and Q. Abbas, Molecules, 2022, 27, 6766 CrossRef CAS PubMed.
- N. Hosseini Nasab, H. Raza, R. S. Shim, M. Hassan, A. Kloczkowski and S. J. Kim, Int. J. Mol. Sci., 2022, 23, 13262 CrossRef CAS PubMed.
- N. A. Khan, F. Rashid, M. S. K. Jadoon, S. Jalil, Z. A. Khan, R. Orfali, S. Perveen, A. Al-Taweel, J. Iqbal and S. A. Shahzad, Molecules, 2022, 27, 6235 CrossRef CAS PubMed.
- M. N. Mustafa, P. A. Channar, M. Sarfraz, A. Saeed, S. A. Ejaz, M. Aziz, F. A. Alasmary, H. Y. Alsoqair, H. Raza and S. J. Kim, J. Enzyme Inhib. Med. Chem., 2023, 38, 2163394 CrossRef PubMed.
- H. A. Younus, M. Saeed, A. Mahmood, M. S. K. Jadoon, A. Hameed, A. Asari, H. Mohamad, J. Pelletier, J. Sévigny and J. Iqbal, Bioorg. Chem., 2023, 106450 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |