
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis and super capacitive characterization of microwave-assisted highly crystalline α-Fe2O3/Fe3O4 nanoheterostructures†
Rajendra Panmand a,
Yogesh Sethia,
Animesh Jhab and
Bharat Kale*a
a,
Yogesh Sethia,
Animesh Jhab and
Bharat Kale*a
aNanocrystalline Materials Division Centre for Materials for Electronics Technology (CMET), Panchawati, Off Pashan Road, Pune 411007, India. E-mail: rpanmand@gmail.com
bSchool of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK
First published on 11th July 2023
Abstract
A facile microwave-assisted solvothermal process for the synthesis of narrow-size distributed α-Fe2O3, α-Fe2O3/Fe3O4, and Fe3O4 nanostructures was demonstrated using PVP as a surfactant. During the reaction, the influence of the reaction media, such as the mixture of ethylene glycol and water on the formation of α-Fe2O3, α-Fe2O3/Fe3O4, and Fe3O4 was systematically studied. Interestingly, pure aqueous medicated solvothermal reaction conferred phase pure rhombohedral Fe2O3 (hematite) and linearly upsurging the formation of cubic Fe3O4 (magnetite) with the increasing concentration of EG and further, in pure EG, it deliberated cubic Fe3O4. FESEM and FETEM images of α-Fe2O3/Fe3O4 nano heterostructure clearly showed the nanosized Fe3O4 particles of 4–6 nm decorated onto Fe2O3 nanoparticles. Further, the electrochemical properties of α-Fe2O3, α-Fe2O3/Fe3O4, and Fe3O4 nanoparticles were investigated with galvanostatic charge–discharge and cyclic voltammetry measurements using a 3-electrode system. The findings show that their specific capacitances are linked to the type of iron oxide. More significantly, the α-Fe2O3/Fe3O4 nanoheterostructure exhibited the utmost capacitance of 165 F g−1, which is greater than that of pristine α-Fe2O3 and Fe3O4. Enhancement in the electrochemical performance was found due to the improved charge transfer that occurred at the interface of the nanoheterostructure. The nanoparticles of Fe3O4 deposited on the Fe2O3 increased the active sites, which accelerated the process of adsorption and desorption of ions, thereby enhancing the interface-assisted charge transfer and reducing the internal resistance, which is ultimately responsible for enhanced capacitance. Such heterostructures of nano iron oxide may fulfill the requirements of electrodes in supercapacitors.
Introduction
The engineering of supercapacitors for electrical charge storage is an emerging technology where the materials used must demonstrate higher power (P) and energy density, much-enhanced reversibility, and longer cycle life when compared with batteries and common capacitors.1–4 The electrochemical capacitor was categorized as electric double-layer capacitors (ELDCs) or Faraday pseudocapacitors depending on the energy storage mechanism.5–8 As compared to EDLCs, faradaic pseudocapacitors exhibit greater energy density (E) and specific capacitance owing to their reversible multi-electron redox/faradaic reactions.9In general, transition metal oxides (TMOs) are used as electrode materials for pseudocapacitors. TMOs are promising materials as electrodes in pseudo or redox supercapacitors because of the increased E as a result of redox reactions. Amongst the TMOs, ruthenium dioxide (RuO2) is recognized as the most promising electrode material owing to its good electrical conductivity and multiple redox states.10 However, owing to its high cost and toxicity, RuO2 is limited in its practical applications, thus other metal oxides that are inexpensive and environmentally friendly have gained significance in the areas of supercapacitors. In this respect, transition metal oxides, such as MnO2,11 NiO,12 Co2O3,13 CuxO,14 V2O5,15 SnO2,16a and a few binary metal oxides, such as NiMoO4,16b,c have been identified as potential alternatives to RuO2. Recently, hematite (Fe2O3) and magnetite (Fe3O4) have emerged as promising oxide materials for supercapacitors, as both oxide forms are abundant in nature and non-toxic.17a,b Iron oxides (Fe3O4 and Fe2O3) are predicted for use as electrode materials with negative potential windows, large theoretical capacitance, and unique reversible redox reactions (Fe2+/Fe3+).18 However, the ‘P’, supercapacitance and corresponding small ‘E’ of the Fe2O3 electrode are still quite low due to its very low electrical conductivity (∼10−14 S cm−1).19 Significant research has been carried out to overcome the limitations. Notably, a number of approaches, such as the synthesis of doped Fe2O3 electrodes, nanostructured Fe2O3 electrodes, core–shell nanorod arrays, oxygen vacancy-induced Fe2O3 electrodes, and Fe2O3 composite electrodes have been investigated.20–23 Amongst these approaches, the fabrication of Fe2O3 composite electrodes with graphene,24 mesoporous carbon,25 PANI,26 and SnO2 (ref. 27) was also investigated. Although the enhanced super capacitive performance was attained, the limited electronic and ion transport remains a critical concern for Fe2O3 composites to be high-performance electrodes. Very few reports are available on the synthesis of the composite of Fe2O3 and highly conductive Fe3O4 for energy storage applications. Tang et al. reported enhanced electrochemical performance due to the improvement in the charge transport of Fe3O4/Fe2O3 composites.21
Synthesis of Fe3O4 nanoparticles using monovalent precursor is quite difficult and hence many reports on Fe3O4 preparation involve mixing Fe2+ and Fe3+ in different ratios.28 Herein, we have synthesized the nanostructured Fe2O3, Fe3O4/Fe2O3 nanoscale heterostructure, and Fe3O4 using monovalent precursor (FeCl3·6H2O) using the facile microwave method. The formation of nanoscale heterostructure was demonstrated by a microwave solvothermal method using water ethylene glycol mixture as a reaction medium. The details of the electrochemical characterization of the nanoscale heterostructure materials are also discussed alongside the capacitance measurements. The influence of nano-scale phase combination on capacitance is compared and discussed in terms of charge storage.
Experimental
All chemicals used here, namely ferric chloride (FeCl3, >99.9%), sodium hydroxide (NaOH, >99.0%), ethylene glycol (EG, >99.0%), were procured from SD Fine Chemicals Ltd, Mumbai, India and no further purification step was adopted for the synthesis of nanoparticles of iron oxides.For the synthesis of nanoparticles of iron oxide, 0.35 g (50 mM) FeCl3·6H2O, 0.3 g (150 mM) NaOH were mixed with 20 mL of ethylene glycol and water in a 100 mL beaker, as shown in Table 1. The homogenous solution was sealed in a double-walled digestion vessel lined with Teflon. A Milestone's Start D microwave digestion system oven equipped with a solution digester stage was used for heating the solutions containing FeCl3/NaOH with EG for 25 min to a controlled temperature of 150 °C. After the microwave digestion, each solution, listed in Table 1, was cooled to room temp. After centrifugal separation, the product was taken and washed in deionized water and 100% ethanol. For four hours, the reaction product was dried at 80 °C in a vacuum.
| Sample ID | Solvent | Volume ratio |
|---|---|---|
| F1 | Water | — |
| F2 | Water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EG :EG |
3:1 |
| F3 | Water:EG |
2:2 |
| F4 | Water:EG |
1:3 |
| F5 | EG | — |
The phase purity, structural, and morphological and microstructural analyses of the synthesized iron oxide phase mixtures including heterostructure were examined using an X-ray Powder Diffractometer XRD (Bruker (D8)), HR 800-Raman Spectroscopy (Horiba Joblin Yvon, France), and FESEM (FEI NOVANANOSEM 450), respectively.
Electrochemical measurements
Carbon paper was used for making electrodes for electrochemical testing. For each measurement, two carbon paper electrodes with similar surface areas were utilized. The Fe2O3/Fe3O4 nanoparticles were loaded onto the carbon paper as a substrate, as shown below. Thus, 80 wt% Fe2O3/Fe3O4 was added to 10 wt% polyvinylidene fluoride (PVDF) and acetylene black each. The viscous slurry was prepared by adding 5–7 mL of NMP (N-methyl-2-pyrrolidone) into the above mixture. The carbon paper substrate was then covered with this slurry. The electrodes were dried for 6–7 hours at 100 °C. In an aqueous 6 M KOH solution, electrochemical cyclic voltammetry experiments were performed from the −1 to 0.2 V range of potential at varied scan rates.Electrochemical impedance spectroscopy study
Electrochemical impedance (EI) spectroscopy measurements were executed for a symmetrical supercapacitor cell made up of Fe2O3/Fe3O4 nanoparticles in an aqueous 1 M Na2SO4 electrolyte. The study was performed using a frequency of 10 kHz to 10 MHz range and AC amplitude of 10 mV without the use of an external DC applied voltage.Results and discussion
The XRD patterns of the self-assembled bipyramidal-like α-Fe2O3/Fe3O4 nanoparticles are shown in Fig. 1a and b, prepared using different water-to-EG ratios as solvents. The XRD pattern of the pure aqueous mediated (F1) sample showed the presence of rhombohedral hematite without any impurity peaks (JCPDS no. 072-0469). Based on the powder diffraction data presented in Fig. 1a, the lattice constants were calculated for each sample, and it was determined that the values of lattice constants are a = b = 5.030 Å, c = 13.87 Å, which are comparable to the JCPDS data for hematite powder (a = b = 5.034 Å and c = 13.57 Å). As the amount of the EG in the solvent increased to 50% (F3) and 75% (F4), the additional peaks of cubic magnetite (JCPDS-072-0748) (Fe3O4) and (Fe2O3-JCPDS no. 072-0469) appeared. The sample prepared using pure EG solvent (F5) showed the formation of cubic magnetite (JCPDS-074-0748). The magnified pattern (29.5 to 37.5°) is shown in Fig. 1b for clarity. From Fig. 1b, it is revealed that the XRD peak intensity of α-Fe2O3 (1 1 0) and (1 0 4) diminished with an increase in EG and other planes corresponding to Fe3O4 (2 2 0) and (3 1 1) appeared, distinctly. The Fe2O3, Fe3O4@Fe2O3 heterostructure and Fe3O4 can be easily prepared using Fe(III)Cl3 and by varying the water to EG solvent ratio during the reaction.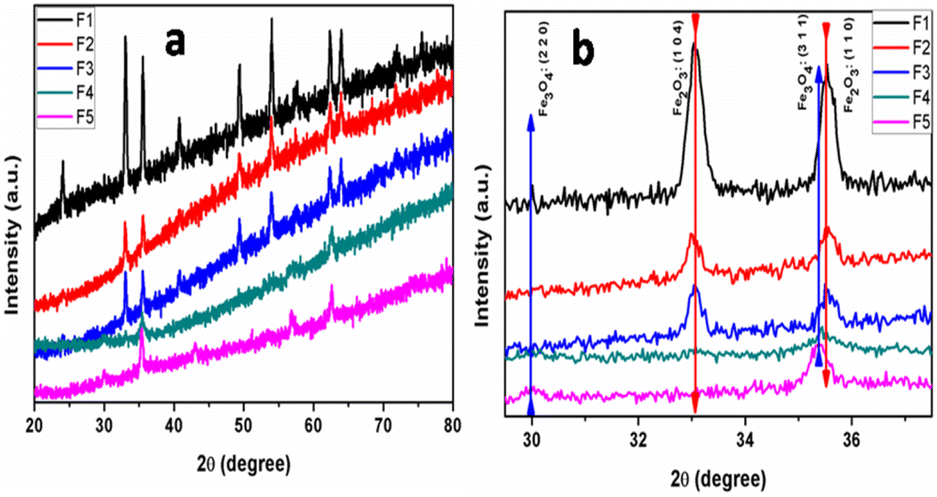 | ||
| Fig. 1 Full XRD patterns (a) and magnified XRD pattern (b) of the α-Fe2O3/Fe3O4 prepared using different water to EG ratios as a solvent. | ||
The morphological evolution of the α-Fe2O3/Fe3O4 prepared at different water:EG ratios was conducted using FESEM. Fig. 2 shows the FESEM images at different magnifications of samples obtained with different EG:water ratios (F1, F3, and F5). Uniformity and homogeneity of particles can be revealed from low magnification FESEM images. However, high-magnification images are valuable for studying the effect of the solvent ratio on morphology. Fig. 1a and b show the FESEM images of sample F1 (made purely in water medium), which revealed the uniform and monodisperse self-assembled bipyramidal α-Fe2O3 nanoparticles of size 140–150 nm in length and 100–120 nm in diameter (see Fig. 1b). The bipyramidal α-Fe2O3/Fe3O4 nano-heterostructure nanoparticles with sizes 80–90 nm in length and 50–70 nm in diameter were observed for samples prepared using 1:1 EG to water ratio (F3 see Fig. 1d). However, when the sample was prepared using pure EG as the solvent (F5), monodispersed and spherical Fe3O4 nanoparticles of diameter 30–50 nm were formed without any other phase. From FESEM, it can be concluded that the nanoparticle size of the material declined with increasing EG concentration in the reaction medium. It is obvious because of the low dielectric constant of EG (37) compared to that of water (80), the ionic mobility slowed down, which ultimately controlled the formation and growth of the nanoparticle. Hence, the lower particle size obtained in pure EG is quite justifiable. Fig. 1g and h demonstrate the images of the samples F1 and F5 dispersed in water with and without an external magnetic field. From the image, it is clear that Fe3O4 nanoparticles (sample F5) are well dispersed in water before magnetic separation. However, all Fe3O4 NPs are attracted toward the magnet, after magnetic field application. For comparison, the α-Fe2O3 nanoparticles were also dispersed in water, however, there was no change before and after magnetic separation.
 | ||
| Fig. 2 Images of FESEM at low and high magnification of samples prepared at different EG:water solvent ratios. (a and b): F1, (c and d): F3 and (e and f): F5. Photographs of α-Fe2O3 NPs (g) and Fe3O4 NPs (h) before and after magnetic separation with an external magnetic field. | ||
Being a magnetic sample, the sample was dispersed in PVA, and the film was employed for FETEM. Fig. 3 shows the TEM images of the Fe2O3/Fe3O4 nano-heterostructure prepared using a 1:3 water-to-EG solvent ratio. From TEM images, it is concluded that the tiny Fe3O4 nanoparticles of size 4–6 nm were grown on the Fe2O3 nanoparticles (see Fig. SI 1 in ESI†). TEM images clearly show that the entire Fe2O3 nanoparticles are covered with tiny Fe3O4. Fig. SF 2 from ESI† shows lattice-resolved HRTEM of Fe2O3-on-Fe2O3 nanoparticles. The interplanar spacing was measured to be about 0.367 nm, which is in good agreement with that of the (012) crystal plane. The SEAD pattern is poor due to the polymer film, which is given in the ESI.†
 | ||
| Fig. 3 TEM image of Fe2O3/Fe3O4 nanocomposite prepared at 1:3 water to EG solvent ratio. | ||
Raman spectra of iron oxide samples prepared using pure water (F1), water:EG in a 1:1 ratio (F3), and pure EG as the solvent (F5) are shown in Fig. 4. “The Raman spectra of the above-made F1 sample (α-Fe2O3 nanoparticles) do not contain the peaks belonging to Fe3O4. The peaks at 221 and 497 cm−1 are classified as A1g modes, whereas the peaks at 244, 292, 406, and 611 cm−1 are classified as Eg modes”.29,30 From Raman spectra, the peaks in the spectrum of sample F5 (Fe3O4) at “305 and 534 cm−1 are reliably allocated to the T2g mode, whereas the peak at 513 cm−1 is allocated to the Eg mode and the peak at 660 cm−1 is allocated to the A1g mode”. However, in sample F3, both iron oxide phase peaks were found, which confirmed the existence of nano-heterostructure.
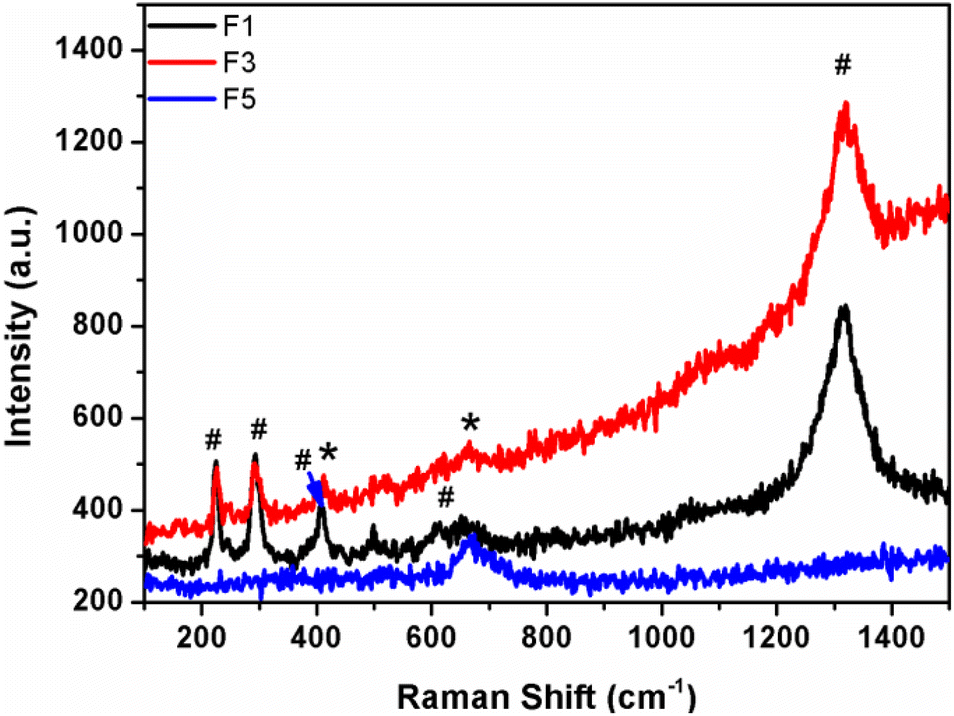 | ||
| Fig. 4 Raman spectra of samples prepared at different EG:water solvent ratios. | ||
Growth mechanism
Generally, the growth mechanism consists of two stages, i.e. nucleation of amorphous primary particles followed by slow crystallization and aggregation of primary particles.31,32 In the present case, the diluted Fe3+ ion aqueous solution was directly digested under microwave hydrothermal conditions at 150 °C. The formation of α-Fe2O3 involved two stapes.33,34a Initially, It may be that β-FeOOH is formed by hydrolysis of ([Fe(H2O)6]3+) in an aqueous solution and further, the β-FeOOH undergoes a transformation to α-Fe2O3 via the dissolution/re-precipitation mechanism.34b EG is acting as a reducing agent apart from the solvent medium. Initially, it combined with FeCl3 to form iron alkoxide nuclei, which then further grew at hydrothermal conditions and formed Fe3O4 nanoparticles of size 30–50 nm in size. However, in a mixture of EG and water solvent, the α-Fe2O3@Fe3O4 heterostructure was formed. The possible formation and growth mechanism are shown in Fig. 5. In aqueous EG, iron alkoxide formation along with FeOOH is quite understood. Preferentially, the formation of Fe2O3 from FeOOH takes place first, and then alkoxide gets reduced to Fe3O4. Hence, we can see the deposition of Fe3O4 nanoparticles on the Fe2O3 particle (Fig. 5). During the reaction, the average dielectric constant was decreased due to EG in the water, which ultimately controlled the growth of α-Fe2O3@Fe3O4 resulting in a lower nanoparticle size of α-Fe2O3/Fe3O.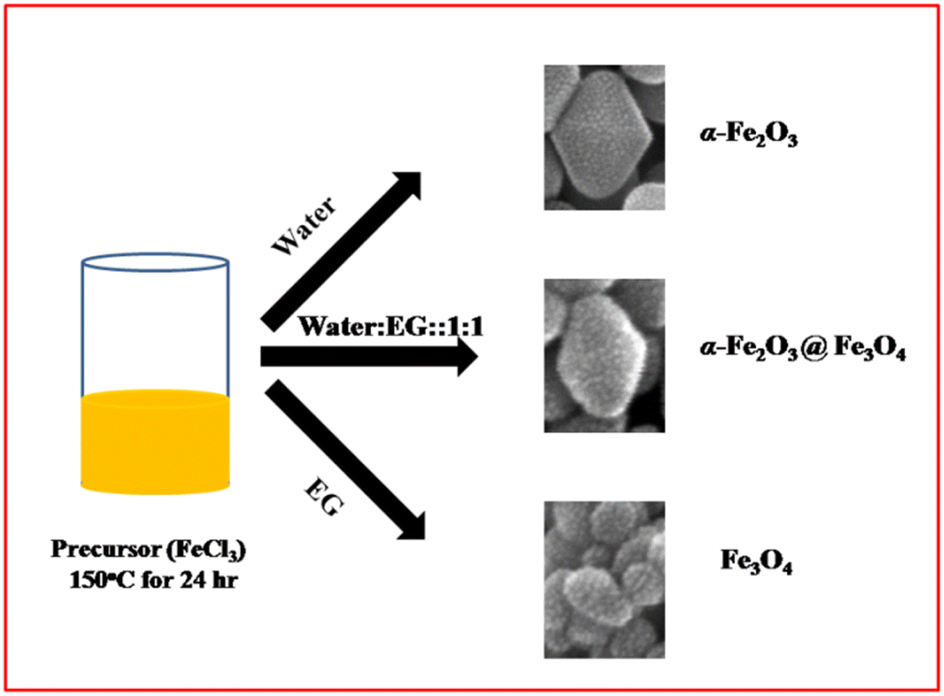 | ||
| Fig. 5 The schematic representation of the growth mechanism. | ||
Electrochemical study
Cyclic voltammetry (CV) is used to characterize the super-capacitive behavior of any material. The Csp of the cell was evaluated from the CV curve as per the following equation:
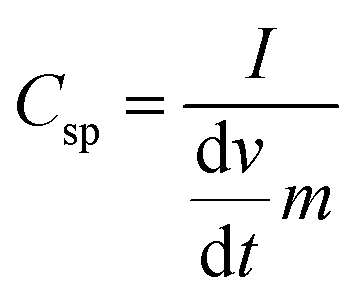 | (1) |
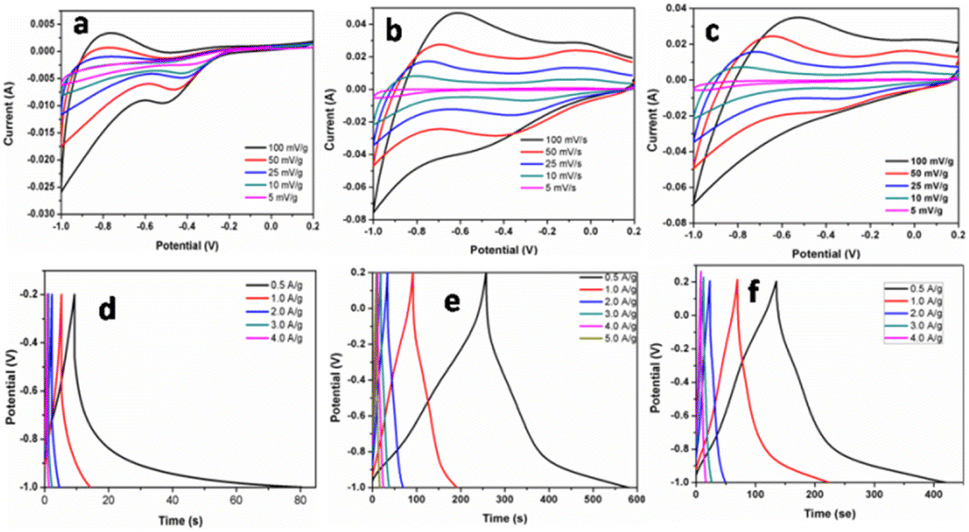 | ||
| Fig. 6 Cyclic voltammeter (potential (V) vs. saturated calomel electrode) and galvanostatic charge–discharge curve of F1 (a and d), F3 (b and e), and F5 (c and f). | ||
The galvanostatic charge–discharge curves and performance rate of the F-1, F-3, and F-5 samples with current densities ranging from 0.5 to 4 A g−1 are displayed in Fig. 6d–f. The modest charge–discharge platforms that are formed in samples F-1, F-3, and F-5, as shown in Fig. 6d–f, suggesting that tiny redox processes take place during the charge–discharge process. It is consistent with the modest redox peaks seen in the CV profile. In all three samples, there are 2 variance ranges that are present during the charge and discharge processes. The potential time dependence is linear and nonlinear below −0.4 V and above −0.4 V resp. in which the linear shows pure double-layer capacitance behaviour resulting from charge separation at the electrode–electrolyte interface35b while the nonlinear is due to the typical pseudo-capacitance behaviour. High specific capacitance values are produced at small current densities by completely accumulating charges in the electrode's inner active sites. The specific capacitances of F-1, F-2, and F-3 are 49, 165 and 157 F g−1 at 0.5 A g−1, respectively. Due to the movement of electric charges being out of phase with current rates, the specific capacitance value declines as current density increases. At very fast charge–discharge, only the outer active surface of the electrode material is used for charge storage.36 The specific capacitance change with current density is summarized in Fig. 7a and Table 2. As shown in Fig. 7a, the charge/discharge curves of the 3 electrodes collected at a 0.5 A g−1 current density showed that the sample F3 (Fe3O4@Fe2O3) electrode presented the most prolonged charge/discharge curves, confirming its outstanding capacitive performance than F1 (Fe2O3) and F5 (Fe3O4). Additionally, the internal resistance (IR) drop of 0.17 V for the F3 electrode was much smaller than 0.21 V for the F5 electrode, indicating the enhanced electrical conductivity of the F3 (Fe3O4@Fe2O3) electrode.
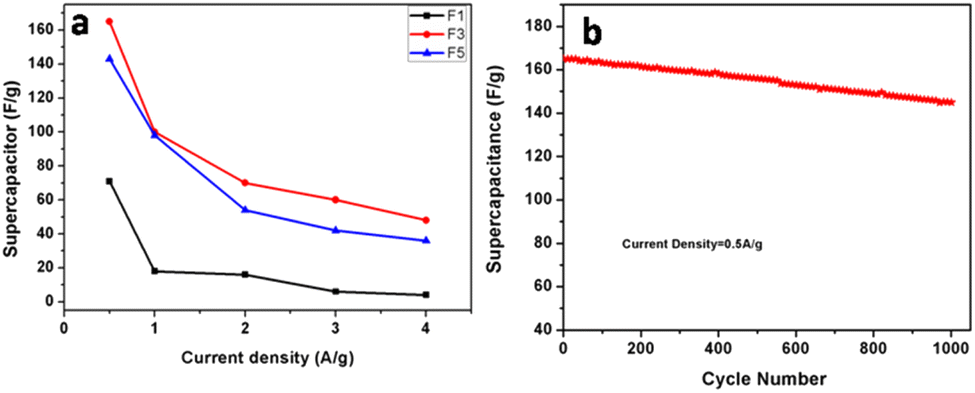 | ||
| Fig. 7 (a) Specific capacitance calculated for F1, F3, and F5 based on the charge/discharge curves as a function of current densities. (b) Cycling stability of the F3 electrode collected at a current density of 0.5 A g−1 for 1000 cycles. | ||
| Current density (A g−1) | F1 | F2 | F3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Cs (F g−1) | E (W h kg−1) | P (W kg−1) | Cs (F g−1) | E (W h kg−1) | P (W kg−1) | Cs (F g−1) | E (W h kg−1 | P (W kg−1) | |
| 0.5 | 71 | 14.2 | 716 | 165 | 33 | 364.2 | 143 | 28.6 | 358.8 |
| 1 | 18 | 3.6 | 1440 | 100 | 20 | 711.6 | 98 | 19.6 | 456.5 |
| 2 | 16 | 3.2 | 3600 | 70 | 14 | 1374.8 | 54 | 10.8 | 1160.6 |
| 3 | 6 | 1.2 | 4320 | 60 | 12 | 2168.67 | 42 | 8.4 | 2131.1 |
| 4 | 4 | 0.8 | 5760 | 48 | 9.6 | 2723.4 | 36 | 7.2 | 3014.0 |
Fig. SI 3† shows the N2 adsorption–desorption isotherms of the as-prepared F1, F2 and F3 heterostructures. The Brunauer–Emmett–Teller (BET) specific surface areas of F1, F2, and F3 heterostructures were measured to be 19.9, 34.5, and 32.14 m2 g−1, respectively. Results showed that the surface area of Fe3O4@Fe2O3 is higher than Fe3O4 and Fe2O3. Also, it has been reported that the junction formation at the Fe2O3/Fe3O4 interface can significantly improve the charge transport of Fe3O4@Fe2O3 composites.37 Hence, Due to high surface area and improved charge transport, the Fe3O4@Fe2O3 shows improved capacitance than Fe3O4 and Fe2O3.
The cycling stability of the electrode materials used in supercapacitor applications is an additional essential parameter. “Constant current charge–discharge cycling” at a 0.5 A g−1 current density was recorded in order to assess the cycling stability of F3 electrodes (Fig. 7b). The calculated specific capacitance values were plotted as a function of cycle numbers (in Fig. 7b). A slight reduction of around 12% of the initial capacitance was found for the F3 electrode after 1000 cycles. The comparison of the present supercapacitor with the reported supercapacitor is shown in Table SI 1.† The table shows that our supercapacitor is superior to the reported values.
For energy storage devices, it is very important to know the value of ‘E’ and ‘P’. The ‘E’ and ‘P’ are evaluated from the following equations:
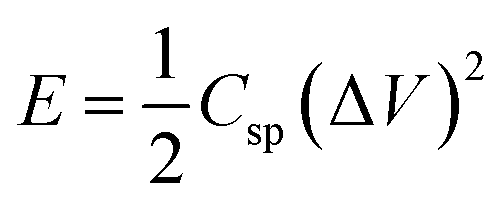 | (2) |
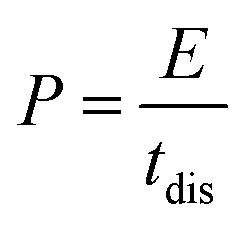 | (3) |
The relationship between ‘E’ and ‘P’, i.e., the Ragone plot for F1, F3, and F5 is illustrated in Fig. 8. According to Fig. 8, F3 obtained an ‘E’ of 33 W h kg−1 at ‘P’ of 364.20 W kg−1 and retained 9.60 W h kg−1 at 2723.40 W kg−1, both of which are greater than that of recently published Fe2O3/Fe3O4 symmetric supercapacitors in aqueous electrolyte.38 The ‘E’ observed for sample F3 at a current density 0.5 A g−1 is much higher than F1 and F5. The enhancement in the electrochemical properties in Fe2O3/Fe3O4 sample (F3) may be due to lower internal resistance and better charge transport, which arises from the junction produced at the Fe3O4/Fe2O3 interface than Fe2O3 (F1) and Fe3O4 (F5). These findings reveal that the synthesized F3 may provide an excellent electrode material for supercapacitors with high power and low cost.
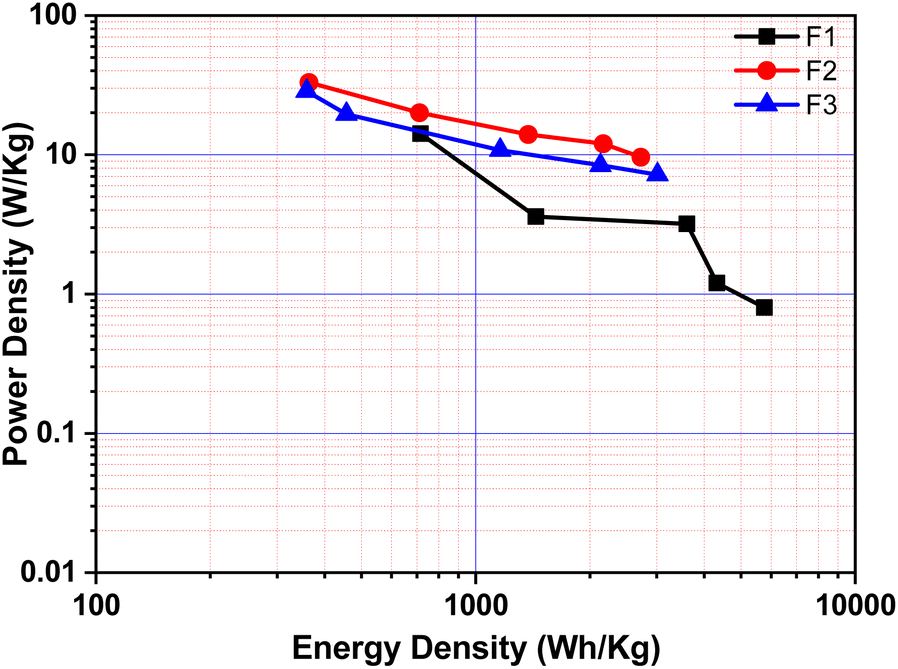 | ||
| Fig. 8 Ragone plots for F1, F3, and F5 (plot of energy density vs. power density). | ||
Conclusions
Herein, a facile microwave-assisted solvothermal route for the synthesis of Fe3O4/Fe2O3, Fe2O3, and Fe3O4 nanoparticles having narrow size distribution with FeCl3 as the iron source was investigated. The phase of the iron oxide was tuned by varying the EG to water ratio in the solvent. The prepared Fe3O4/Fe2O3, Fe2O3, and Fe3O4 nanoparticles were studied for electrochemical application. The as-prepared Fe3O4/Fe2O3 heterostructures demonstrated greater specific capacitances of 165 F g−1 at the current density of 0.5 A g−1 than Fe2O3 and Fe3O4. In a nutshell, the findings of this study indicate that hematite might be used as an electrode material for high-performance supercapacitors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Ministry of Electronics and Information Technology (MeitY), the Government of India's financial support for research, and C-MET Pune for providing research facilities. The authors would like to thank the Nanocrystalline Materials group, C-MET, Pune, and Microwave Materials Division, C-MET, Thrissur.Notes and references
- C. Hong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- D. P. Dubal, O. Ayyad, V. Ruiz and P. Gomez-Romero, Chem. Soc. Rev., 2015, 44, 1777–1790 RSC.
- D. Chen, Q. Wang, R. Wang and G. Shenb, J. Mater. Chem. A, 2015, 3, 10158–10173 RSC.
- S. Faraji and F. Nasir Ani, Renewable Sustainable Energy Rev., 2015, 42, 823–834 CrossRef CAS.
- Y. Lu, K. Fu, S. Zhang, Y. Li, C. Chen, J. Zhu, M. Yanilmaz, M. Dirican and X. Zhang, J. Power Sources, 2015, 273, 502–510 CrossRef CAS.
- D. Wang, Z. Geng, B. Li and C. Zhang, Electrochim. Acta, 2015, 173, 377–384 CrossRef CAS.
- J. Liu, J. Jiang, C. Cheng, H. Li, J. Zhang, H. Gong and H. J. Fan, Adv. Mater., 2011, 23, 2076–2081 CrossRef CAS PubMed.
- Y. Zhang, H. Feng, X. Wu, L. Wang, A. Zhang, T. Xia, H. Dong, X. Li and L. Zhang, Int. J. Hydrogen Energy, 2009, 34, 4889–4899 CrossRef CAS.
- L. Wang, H. Ji, S. Wang, L. Kong, X. Jiang and G. Yang, Nanoscale, 2013, 5, 3793–3799 RSC.
- M. Zhu, Y. Wang, D. Meng, X. Qin and G. Diao, J. Phys. Chem. C, 2012, 116, 16276–16285 CrossRef CAS.
- J. Zhi, S. Deng, Y. Wang and A. Hu, J. Phys. Chem. C, 2015, 119, 8530–8536 CrossRef CAS.
- V. Kannan, A. I. Inamdar, S. M. Pawar, H.-S. Kim, H.-C. Park, H. Kim, H. Im and Y. S. Chae, ACS Appl. Mater. Interfaces, 2016, 8(27), 17220–17225 CrossRef CAS PubMed.
- T.-Y. Wei, C.-H. Chen, K.-H. Chang, S.-Y. Lu and C.-C. Hu, Chem. Mater., 2009, 21, 3228–3233 CrossRef CAS.
- A. Lamberti, M. Fontana, S. Bianco and E. Tresso, Int. J. Hydrog. Gener., 2016, 41, 11700–11708 CrossRef CAS.
- B. Saravanakumar, K. K. Purushothaman and G. Muralidharan, ACS Appl. Mater. Interfaces, 2012, 4, 4484–4490 CrossRef CAS PubMed.
- (a) F. Li, J. Song, H. Yang, S. Gan, Q. Zhang, D. Han, A. Ivaska and L. Niu, Nanotechnology, 2009, 20, 455602 CrossRef PubMed; (b) P. Sharma, M. M. Sundaram, T. Watcharatharapong, D. W. Laird, H. Euchner and R. Ahuja, ACS Appl. Mater. Interfaces, 2020, 12(40), 44815–44829 CrossRef CAS PubMed; (c) M. Minakshi and K. Wickramaarachchi, Prog. Solid State Chem., 2023, 69, 100390 CrossRef CAS.
- (a) P. A. Lu, M. R. Manikandan, P. F. Yang, Y. L. He, F. Yang, S. T. Dang, Y. C. Shi, W. B. Lou, R. Liu, S. J. Wu, X. F. Li, Y. C. Hu, J. Shang, S. Q. Yin and X. W. Wang, J. Mater. Sci.: Mater. Electron., 2023, 34, 826 CrossRef CAS; (b) P. M. Anjana, J. F. Joe Sherin, C. Vijayakumar, S. R. Sarath Kumar, M. R. Bindhu and R. B. Rakhi, Mater. Sci. Eng., B, 2023, 290, 116313 CrossRef CAS.
- V. D. Nithya and N. Sabari Arul, J. Mater. Chem. A, 2016, 4, 10767–10778 RSC.
- Z. Song, W. Liu, W. Wei, C. Quan, N. Sun, Q. Zhou, G. Liu and X. Wen, J. Alloys Compd., 2016, 685, 355–363 CrossRef CAS.
- X. Du, C. Wang, M. Chen, Y. Jiao and J. Wang, J. Phys. Chem. C, 2009, 113, 2643–2646 CrossRef CAS.
- X. Tang, R. Jia, T. Zhai and H. Xia, ACS Appl. Mater. Interfaces, 2015, 7, 27518–27525 CrossRef CAS PubMed.
- K. Karthikeyan, S. Amaresh, S. N. Lee, V. Aravindan and Y. Sung Lee, Chem.–Asian J., 2014, 9, 852–857 CrossRef CAS PubMed.
- D. Sarkar, G. G. Khan, A. K. Singh and K. Mandal, J. Phys. Chem. C, 2013, 117(30), 15523–15531 CrossRef CAS.
- M. Saraf, K. Natarajan and M. M. Shaikh, RSC Adv., 2017, 7, 309 RSC.
- J. Hu, M. Noked, E. Gillette, F. Han, Z. Gui, C. Wang and S. Bok Lee, J. Mater. Chem. A, 2015, 3, 21501–21510 RSC.
- P. M. Padwal, S. L. Kadam, S. M. Mane and S. B. Kulkarni, J. Mater. Sci., 2016, 51, 10499 CrossRef CAS.
- R. Lei, H. Ni, R. Chen, B. Zhang, W. Zhan and L. Yang, Chem. Phys. Lett., 2017, 673, 1–6 CrossRef CAS.
- P. Panneerselvam, N. Morad and K. Aik Tan, J. Hazard. Mater., 2011, 186, 160–168 CrossRef CAS PubMed.
- A. G. Nasibulin, S. Rackauskas, H. Jiang, Y. Tian, P. Reddy Mudimela, S. D. Shandakov, L. I. Nasibulina, J. Sainio and E. I. Kauppinen, Nano Res., 2009, 2, 373–379 CrossRef CAS.
- C. Zhao, X. Shao, Y. Zhang and X. Qian, ACS Appl. Mater. Interfaces, 2016, 8(44), 30133–30142 CrossRef CAS PubMed.
- L.-S. Zhong, J.-S. Hu, H.-P. Liang, A.-M. Cao, W.-G. Song and L.-J. Wan, Adv. Mater., 2006, 18, 2426–2431 CrossRef CAS.
- X. Hu, J. C. Yu, J. Gong, Q. Li and G. Li, Adv. Mater., 2007, 19, 2324–2329 CrossRef CAS.
- Z. Pu, M. Cao, J. Yang, K. Huang and C. Hu, Nanotechnology, 2006, 17, 799–804 CrossRef CAS.
- (a) M. Lin, H. R. Tan, J. P. Ying Tan and S. Bai, J. Phys. Chem. C, 2013, 117(21), 11242–11245 CrossRef CAS; (b) C.-Y. Yin, M. Minakshi, D. E. Ralpha, Z.-T. Jiang, Z. Xie and H. Guo, J. Alloys Compd., 2011, 509, 9821–9825 CrossRef CAS.
- (a) R. Li, X. Ba, Y. Wang, W. Zuo, C. Wang, Y. Li and J. Liu, Prog. Nat. Sci.: Mater. Int., 2016, 26, 258–263 CrossRef CAS; (b) J. Wang, L. Zhang, X. Liu, X. Zhang, Y. Tian, X. Liu, J. Zhao and Y. Li, Sci. Rep., 2016, 7, 41088 CrossRef PubMed.
- R. P. Panmand, P. Patil, Y. Sethi, S. R. Kadam, M. V. Kulkarni, S. W. Gosavi, N. R. Munirathnam and B. B. Kale, Nanoscale, 2017, 9, 4801–4809 RSC.
- B. Lei, D. Xu, B. Wei, T. Xie, C. Xiao, W. Jin and L. Xu, ACS Appl. Mater. Interfaces, 2021, 13, 4785–4795 CrossRef CAS PubMed.
- X. Tang, R. Jia, T. Zhai and H. Xia, ACS Appl. Mater. Interfaces, 2015, 7(49), 27518–27525 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01967e |
| This journal is © The Royal Society of Chemistry 2023 |