
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamics of reduced graphene oxide: synthesis and structural models
Edwin T. Mombeshora
 *a and
Edigar Muchuweni
b
*a and
Edigar Muchuweni
b
aDepartment of Chemistry and Earth Sciences, University of Zimbabwe, Mount Pleasant, Harare, MP167, Zimbabwe. E-mail: mombeshoraet@gmail.com; emombeshora@science.uz.ac.zw
bDepartment of Engineering and Physics, Bindura University of Science Education, Bindura, Zimbabwe
First published on 12th June 2023
Abstract
Technological advancements are leading to an upsurge in demand for functional materials that satisfy several of humankind's needs. In addition to this, the current global drive is to develop materials with high efficacy in intended applications whilst practising green chemistry principles to ensure sustainability. Carbon-based materials, such as reduced graphene oxide (RGO), in particular, can possibly meet this criterion because they can be derived from waste biomass (a renewable material), possibly synthesised at low temperatures without the use of hazardous chemicals, and are biodegradable (owing to their organic nature), among other characteristics. Additionally, RGO as a carbon-based material is gaining momentum in several applications due to its lightweight, nontoxicity, excellent flexibility, tuneable band gap (from reduction), higher electrical conductivity (relative to graphene oxide, GO), low cost (owing to the natural abundance of carbon), and potentially facile and scalable synthesis protocols. Despite these attributes, the possible structures of RGO are still numerous with notable critical variations and the synthesis procedures have been dynamic. Herein, we summarize the highlights from the historical breakthroughs in understanding the structure of RGO (from the perspective of GO) and the recent state-of-the-art synthesis protocols, covering the period from 2020 to 2023. These are key aspects in the realisation of the full potential of RGO materials through the tailoring of physicochemical properties and reproducibility. The reviewed work highlights the merits and prospects of the physicochemical properties of RGO toward achieving sustainable, environmentally friendly, low-cost, and high-performing materials at a large scale for use in functional devices/processes to pave the way for commercialisation. This can drive the sustainability and commercial viability aspects of RGO as a material.
 Edwin T. Mombeshora | Edwin T. Mombeshora is a Lecturer in Physical Chemistry in the Department of Chemistry and Earth Sciences, University of Zimbabwe (UZ), Harare, Zimbabwe. He has previously served as an international Technology Transfer Fellow at Swansea University, United Kingdom (2021–2022). He is currently a member of the UZ Quality Assurance Committee, Post-Doctoral Fellow member of the South African Chemical Institute and RSC Affiliate. His research interests include: Materials Chemistry and Nanotechnology. His recent research involvement focuses on the development of carbon-based materials for use as catalysts in biomass-to-platform chemicals conversion, as well as electrodes for solar energy conversion and energy storage. |
 Edigar Muchuweni | Edigar Muchuweni is a Senior Lecturer in the Department of Engineering and Physics at Bindura University of Science Education, Bindura, Zimbabwe. He has previously served as Coordinator of the Postgraduate Seminar Series at Botswana International University of Science and Technology (BIUST) (2016–2017). His research interests include: Solar Energy Materials, Nanotechnology and Materials Science. Regarding the latter, his research group works on novel nanomaterials, such as carbon nanotubes, graphene, graphene oxide and reduced graphene oxide, for several applications including energy harvesting, conversion and storage devices. |
1 Introduction
Carbon is the basic building block of biomass and is a component of the graphitic framework that constitutes most modern synthetic nanomaterials. With the well-liked potential roles that graphene (an individual sheet of carbon atoms in graphite)1 can play in the modern world and the associated current industrial scale challenges (that have hindered this progression), it is imperative to shift towards graphene derivatives as alternatives.2 The importance of graphene in recent materials with high potential in modern applications is highlighted by the fact that graphene is the basic building block of most carbon-based nanomaterials.1.1 Graphene as the building block
Graphene is a one atom thick 2-D allotrope of carbon comprising of sp2 hybrids of carbon in a hexagonal morphology.3–5 Interestingly, graphene was first isolated in 2004 by means of a mechanical “sticky tape” method3,6 though it has been around for several years as a basic building block of many carbon-based materials.7 Despite this milestone, processability and industrial scale production of pure graphene in an appreciable exfoliated form is still a challenge as it has been a problem for the longest time, hence, research has shifted to alternatives. The stacking (owing to weak van der Waals forces between sheets), zero band gap and hydrophobic nature of graphene are other essential motivations for seeking alternative materials.1,8 One closest alternative to graphene is reduced graphene oxide (RGO). Interestingly, RGO is a derivative of graphene and the scalable exfoliation pathway to this material is graphene oxide (GO). The main structural modifications of RGO from pristine graphene are the COOH functionalities that are retained from GO, on the edges, and the topological defects induced by the removal of oxygen moieties upon reduction (Scheme 1).3,9 Therefore, the current work focused on the review of the synthesis, properties and recent applications of RGO through the possible scalable GO route.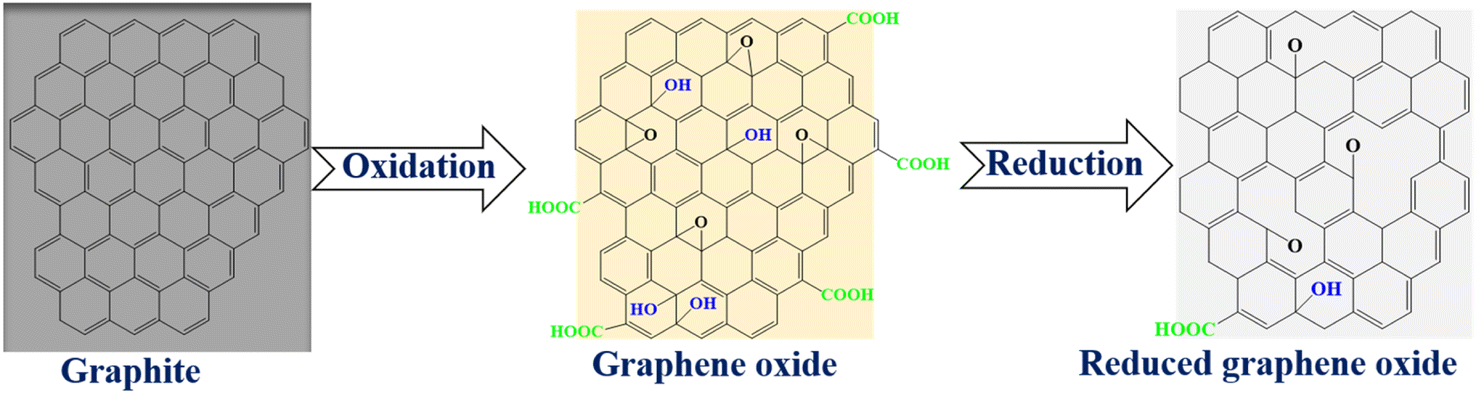 | ||
| Scheme 1 The illustration of colour changes during exfoliation and reduction to form reduced graphene oxide. | ||
2 Graphene oxide as a pathway to understanding reduced graphene oxide
The pathway to RGO is shown in Scheme 1 and a better understanding of the RGO structure is possible through models proposed for GO. The analogy here is that, since RGO is a form of GO that has some of the oxygen moieties partially removed, the various models that have been put forward to explain GO structure are assumed applicable to RGO. This approach also assumes that there are negligible structural deformations that occurs during reduction.102.1 Proposed structural models
The structure of GO has been an unresolved phenomenon for a while. Several models have been proposed, however, limitations in explaining certain issues of this family of materials has been realised over time. This is probably because the ultimate composition of RGO is influenced by the starting material, oxidation, and reduction conditions. This work reviews critical examples of key models that have been reported for GO and can be extrapolated to RGO toward future structural solutions. As a start, Matsuo (1994, Fig. 1a)11 proposed double carbon layers linked to each other by carbon sp3-bonds that are perpendicular to the carbon network.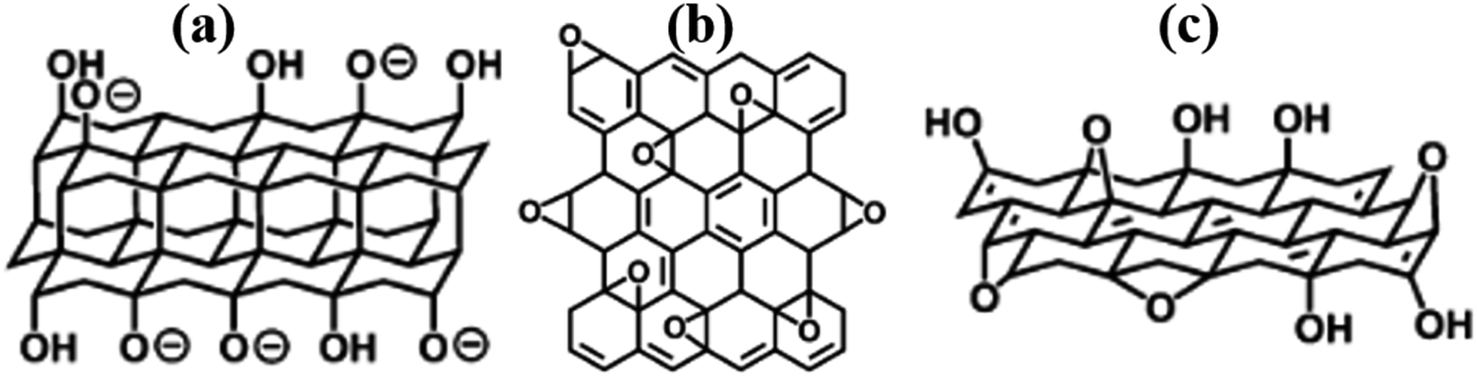 | ||
| Fig. 1 Some of the early proposed models (a) Nakajima-Matsuo,11 (b) Hofmann,12 and (c) Ruess.13 All reproduced from ref. 14 with permission from the Royal Society of Chemistry, copyright 2014. | ||
The Hofmann (1939) model proposed that GO contained only epoxy groups and that the moieties were distributed throughout the basal plane with a net molecular formula of C2O (Fig. 1b).12 The Ruess (1946) model inferred that GO consisted of a basal plane of sp3-hybridized carbon atoms in a trans-linked cyclohexane chair conformation (Fig. 1c).10,13 The key achievement of this model was the ability to account for H atoms in the GO structure. Despite this key breakthrough, the widely accepted model for GO was reported by Lerf–Klinowski in 1998.15 This model distinguishes two types of regions in the GO structure, namely, the aromatic region comprising unoxidized benzene rings and the aliphatic region containing six-membered rings with oxygen functionalities (Fig. 2). The model further proposed that the epoxy and hydroxyl groups are located in the basal plane of GO, while the carboxyl, anhydride, lactone, phenol, lactol, and pyrone groups are found in the periphery regions of GO.
 | ||
| Fig. 2 The proposed Lerf–Klinowski model. Reprinted (adapted) with permission from ref. 15. American Chemical Society, copyright 1998. | ||
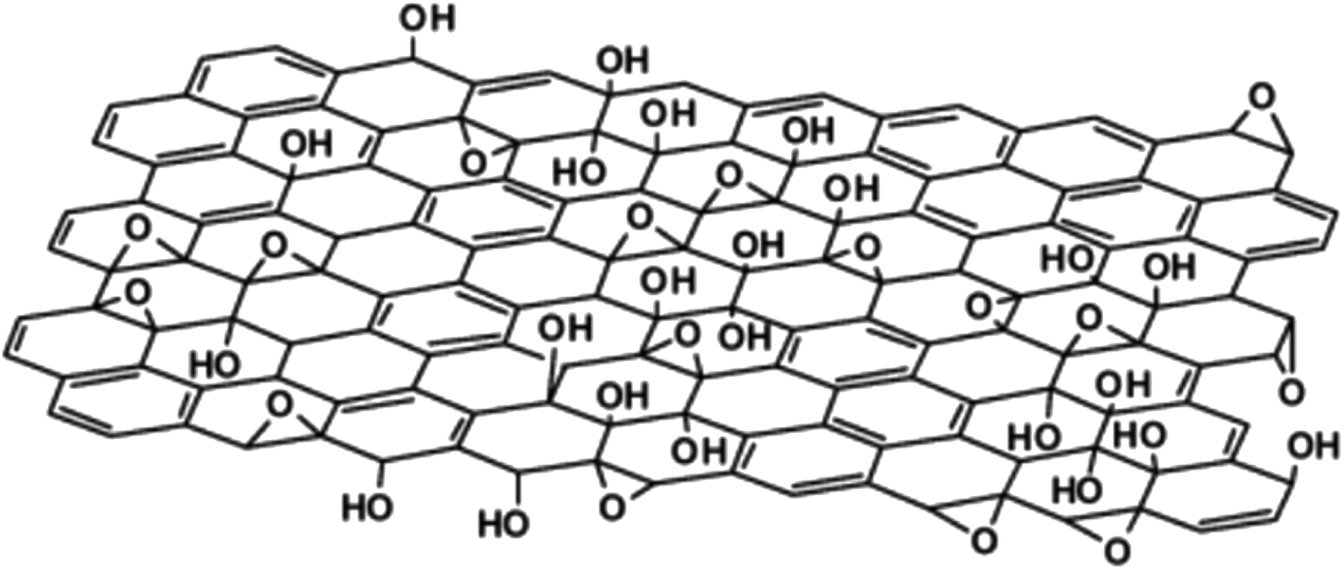
Further work in structural modelling saw Szabó et al.16 in 2006 also proposing another model consisting of two regions, namely, translinked cyclohexane chairs and ribbons of flat hexagons made up of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and functional moieties, such as 1,3-ether and tertiary hydroxyl groups (Fig. 3). The elucidation of lattice species was a key advancement from the model and provided a further understanding of the GO structure in terms of the observed planar acidity, and corroborated the idea of surface functionalisation during oxidation.
C bonds and functional moieties, such as 1,3-ether and tertiary hydroxyl groups (Fig. 3). The elucidation of lattice species was a key advancement from the model and provided a further understanding of the GO structure in terms of the observed planar acidity, and corroborated the idea of surface functionalisation during oxidation.
 | ||
| Fig. 3 The model proposed by Szabó et al., Reprinted (adapted) with permission from ref. 16. American Chemical Society, copyright 2006. | ||
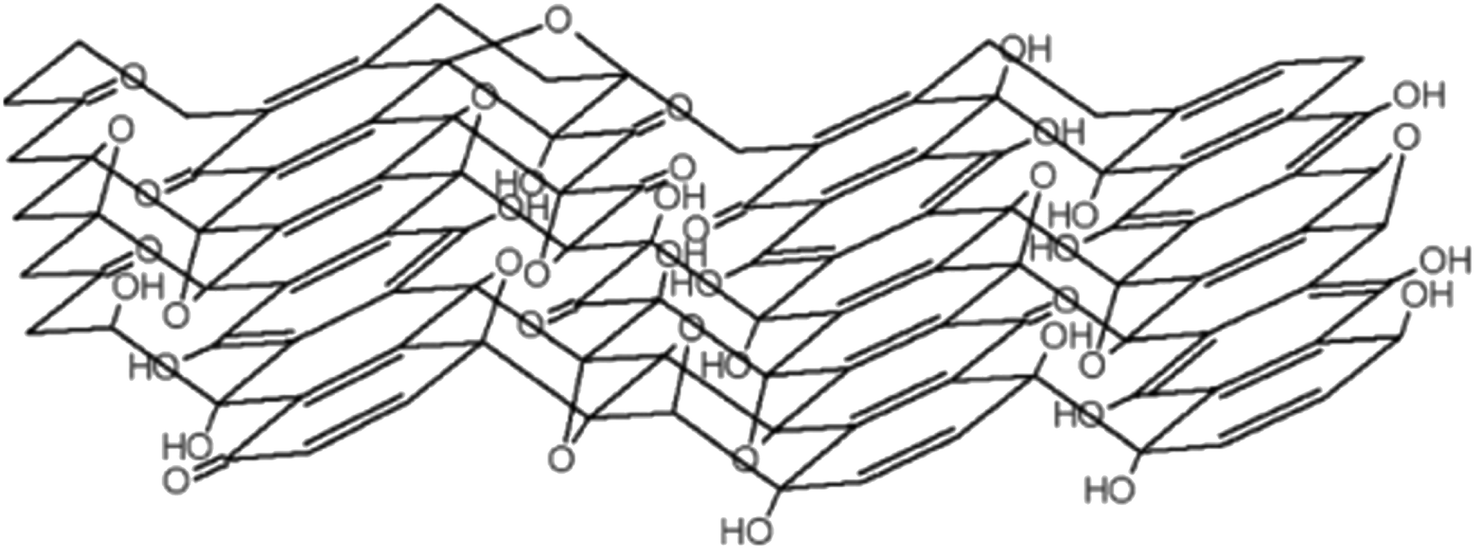
In 2010, Lee et al.,10 proposed a model that was supported by the data from several physicochemical techniques (scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction spectroscopy (XRD), Fourier transform-infra red spectroscopy (FT-IR), X-ray photoelectron spectroscopy (XPS), Carbon-13 (13C) solid-state nuclear magnetic resonance (NMR) and X-ray absorption near edge structure (XANES)) in that GO consists of amorphous carbons (sp3-hybridised carbons) and crystalline carbons (sp2-hybridised) (Fig. 4). The strength of the model was in accounting for stability in GO by suggesting the location of hydroxyl groups in opposite sites and far from epoxy moieties.10
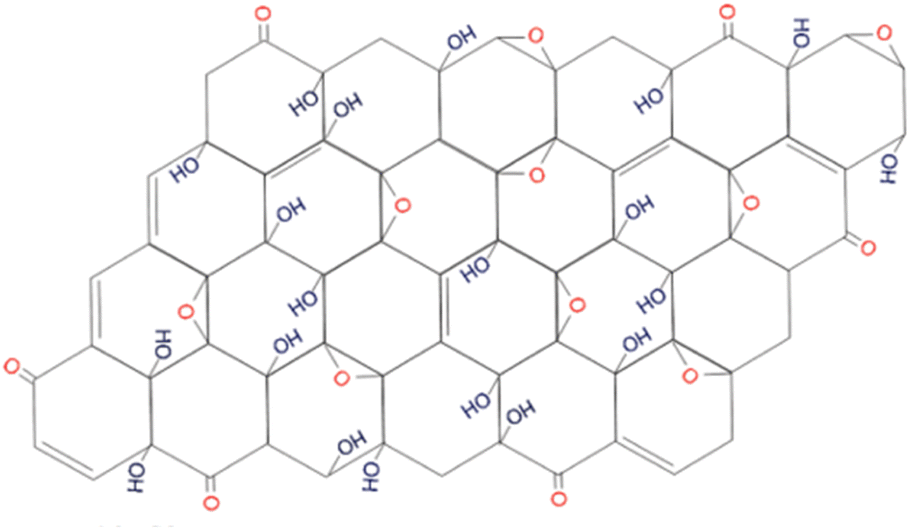 | ||
| Fig. 4 The model proposed by Lee et al., in 2010. Reprinted (adapted) with permission from ref. 10 American Chemical Society, copyright 2010. | ||
Again, in 2019 Aliyev et al.,17 proposed a model that uniquely suggested replacing C with O in GO (Fig. 5). This model supports most of the reported structures of GO.
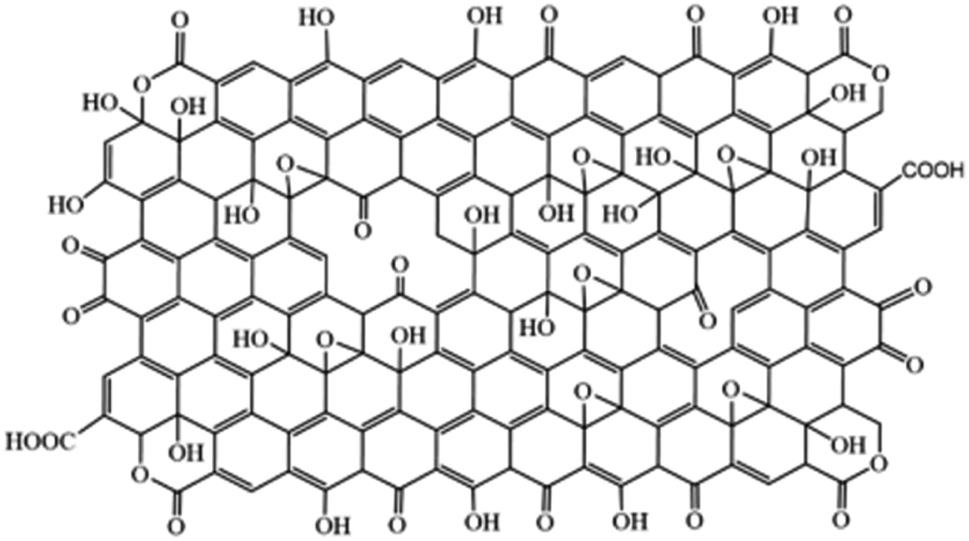 | ||
| Fig. 5 The model proposed by Aliyev et al.,17 in 2019. Reprinted (adapted) with permission from Nanomaterials, copyright 2019. | ||
Brisebois and Siaj18 proposed a model that accounted for carbon vacancies, carbon radicals, C/O ratio of ∼2, carbon esters and allylic alcohols (Fig. 6). This model would be more applicable to RGO as well.
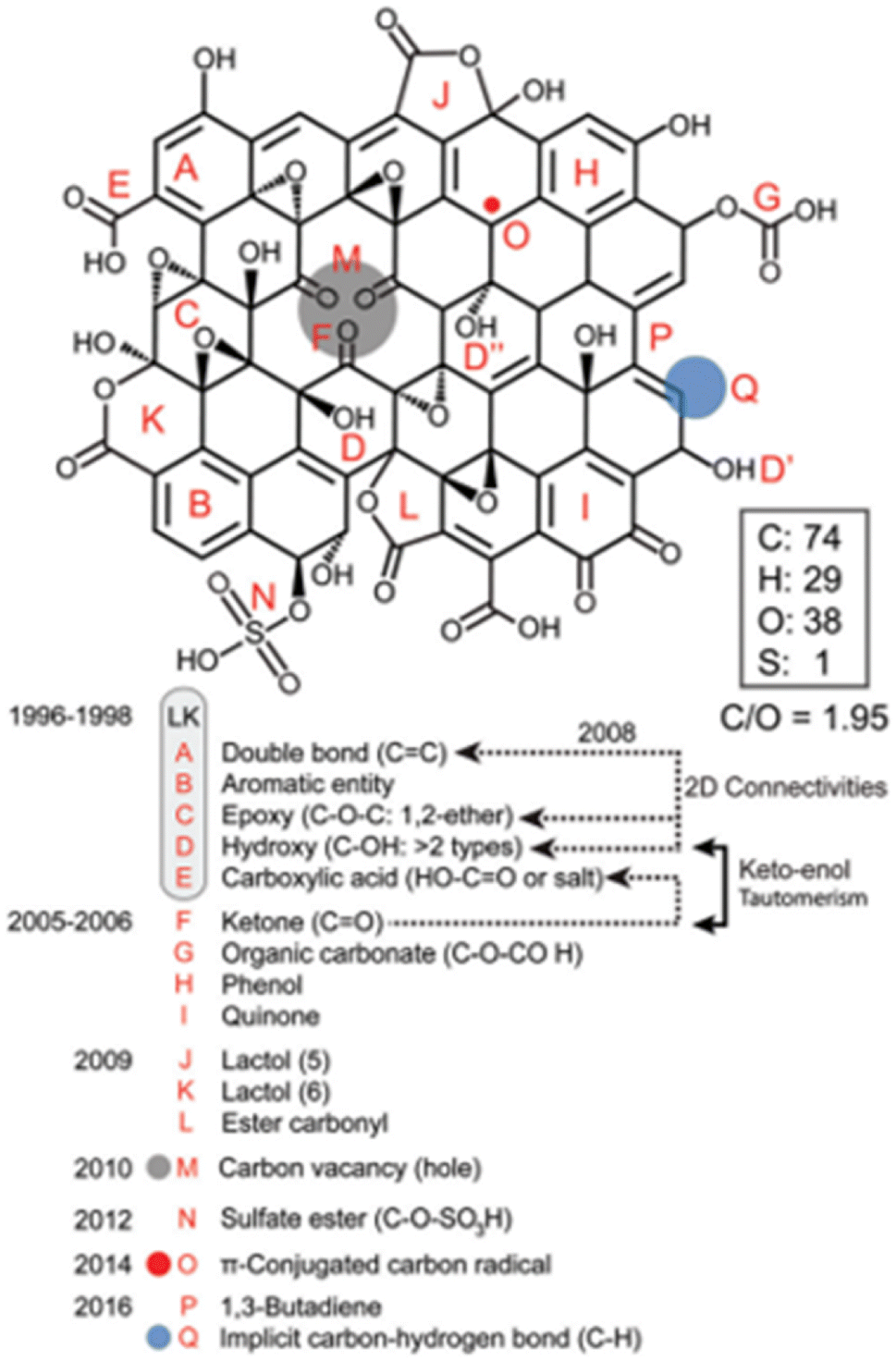 | ||
| Fig. 6 The model proposed by Brisebois and Siaj. Reproduced from ref. 18 with permission from the Royal Society of Chemistry, copyright 2020. | ||
2.2 Concerns from the existing structural models
The consensus amongst the various models and experimental data is that the GO-based structures consists of amorphous and crystalline phases. Characterization techniques have also displayed critical common inferences. For instance, GO spectra from the 13C solid-state (NMR) infer the existence of –OH and –O– moieties at 60 and 70 ppm, respectively, while the FT-IR data tend to support this deduction.10,19 The FT-IR also implies the existence of ketone functionalities which corroborate with XPS, 13C solid-state NMR and O K-edge XANES.10,20 However, there are several issues associated with each proposed model. From the early models, the Hofmann model does not support existence of other moieties since the model proposed epoxy groups only that were distributed across the basal plane with a net molecular formula of C2O with no account for H atoms. Additionally, the structure proposed in the Ruess model is not supported by characterization techniques. This is because the model suggested an sp3-hybridized basal plane structure but several techniques, such as XPS, supports the existence of an sp2-hybridized system and presence of CO which are unaccounted for in this model. On the other hand, later models also have shortfalls. For instance, the Nakajima-Matsuo model relied on the assumption of a lattice backbone similar to poly(dicarbonmonofluoride)(C2F)n but this hypothesis is yet to be proven and is most probably unstable since carbonyl functionalities with partial negative charges are feasibly stable when coordinated to three other carbon atoms.16
The latest models in an attempt to address gaps from earlier models have brought other drawbacks with them. To start with, the Lerf–Klinowski model suggests that carboxyl groups are located at the edges, however, this contradicts the NMR data which do not provide evidence that supports the existence of the moiety (absence of peak near 175 ppm).10 The Lerf–Klinowski model also lacks an account of the dependence of GO on the starting material, oxidant and oxidation conditions.21 Despite the provision of a possible explanation to the obtained 13C spectrum by Lee et al.,10 i.e., the fact that the NMR data is misleading due to cross-polarization induced by the fact that carboxyl moieties contains hydrogen, issues in their proposed model are the hydroxyl groups are located too close to each other, hence, infers a possibility of unfavourable electrical instability in the structure. The problems associated with the proposals by Szabó et al.,16 are linked to the existence of cyclohexane chair conformations which still lacks satisfactory experimental evidence and the model does not account for the existence of other moieties. Furthermore, the shortfall of the model proposed by Lee10 is the exclusion of carboxyl and lactone groups in the structure, since the solid-state 13C NMR data show the existence of both functionalities. Brisebois and Siaj18 ignored the convention that in line diagrams, carbon and hydrogen atoms may not necessarily be drawn in estimating and deducing the shortfalls of earlier models before presenting their proposed structure. To sum up, the modelling for RGO structure still needs more insights. The variations causing most shortfalls have a foundation in precursor materials, i.e., GO materials synthesised by several methods.
2.3 Summarised synthesis methods
Readers are referred to detailed reviews by Ikram et al.,22 Brisebois and Siaj.18 and Dong et al.23 on GO synthesis. In brief, synthesis methods can be classified in two broad classes. The first category can be referred to as chlorate methods and examples of this class include the Hofmann and Staudenmaier methods (additionally involving the use of HNO3),24,25 and the Brodie (based on additional use of HNO3 and H2SO4). The second class is the most popular and can be referred to as permanganate methods. This class is associated with higher levels of oxidation. The most common examples are Hummers (and several modified Hummers methods)26 and Offeman methods, which are based on H2SO4 and NaNO3 additives. Another recent example is known as the Tour method (and derivatives) and these methods involve a combination of H2SO4 and H3PO4 acids as additives. The tailoring of physicochemical properties of GO is mostly through varying the synthesis method and several critical parameters summarised in Table 1. For instance, zeta potential of GO was tuned to approximately −50, −49, −48, −43 and −33 mV (with corresponding C/O ratio of 1.47, 1.59, 1.62, 1.64 and 1.69, respectively) by adding 0, 10, 20, 30, 40 cm3 of H2O2, respectively.27 Despite the terminating role through elimination of residual MnO42− and MnO2 (conversion to MnSO4), excessive H2O2 may cause structural transformations due to creation of substantial π-conjugated carbon radicals in GO (due to reaction of ˙OH−, from the H2O2, with the CC in the disrupted π-conjugated plane of GO).27 This suggests that future modified synthesis protocols must either use small quantities or avoid adding large arbitrary volumes of H2O2 that are not systematically determined. Also, the surface area of GO was tuned from 2 to 8 and 185 m2 g−1 (with corresponding C/O ratios of 0, 1.67 and 1.48, respectively) by varying graphite (starting material): Na2NO3 ratios in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 2:1 and 1:1 fashion, respectively.28 In a similar study involving carbon nanoplates, lattice sizes of 8.72, 7.38 and 4.84 nm (with corresponding C/O ratios of 0.87, 0.95 and 0.86, respectively) were obtained by changing carbon nanoplatelets (CNP, starting material): Na2NO3 ratio of 1:0, 2:1 and 1:1, respectively.20 In the same study, CNP:KMnO4 ratios (1:0, 1:3, 1:6, 1:9, 1:12) were used to tailor the ratios (4.08, 1.48, 1.16, 0.87 and 0.78, respectively). One of the common critical weaknesses of oxidation protocols is the associated aggressiveness, which may introduce unrequired chemical functionalities and contamination from residual reagents.
:0, 2:1 and 1:1 fashion, respectively.28 In a similar study involving carbon nanoplates, lattice sizes of 8.72, 7.38 and 4.84 nm (with corresponding C/O ratios of 0.87, 0.95 and 0.86, respectively) were obtained by changing carbon nanoplatelets (CNP, starting material): Na2NO3 ratio of 1:0, 2:1 and 1:1, respectively.20 In the same study, CNP:KMnO4 ratios (1:0, 1:3, 1:6, 1:9, 1:12) were used to tailor the ratios (4.08, 1.48, 1.16, 0.87 and 0.78, respectively). One of the common critical weaknesses of oxidation protocols is the associated aggressiveness, which may introduce unrequired chemical functionalities and contamination from residual reagents.
Graphite oxidation involves an initial defect generation followed by oxidation at defect sites. The theory has predicted that the main oxygen moieties found at these defect sites are ketones and quinones.17 However, depending on the size of the defects created during oxidation and impurities introduced into the carbon framework, other chemical moieties can also be formed. For instance, sulfur impurity from the permanganate method can be chemically linked to the carbon backbone.17 These are aspects that need to be considered critically in further shaping of current models of RGO. There seems to be a consensus, based on the widely accepted model,15 on the locations of the phenolic (–OH) and epoxy (C–O–C), and acidic groups (–COOH) in the basal plane and edges, respectively.8 Hence, the basal planes of RGO sheets are assumed to be basic, whilst their edges are acidic, suggesting an amphoteric characteristic.
Possible mechanism behind popular permanganate methods:12,34
| KMnO4 + 3H2SO4 → K+ + MnO+3 + H3O+ + 3HSO−4 | (1) |
| MnO+3 + MnO−4 → Mn2O7 | (2) |
| Mn2O7 + C(graphite) + H2SO4 → C-Oxy + MnO5 + H2O + SO2−4 | (3) |
The intentional introduction of oxygen-containing moieties on the surfaces of graphite often culminate in the increment of distance between layers from the usual 0.34 nm due to exfoliation initiated by repulsions between functionalised sheets (oxygen has an electronegativity of 3.44 greater than 2.55 of carbon).10,35 The Na+, from the added NaNO3 in most versions of the Hummers' method, has been reported to also have an intercalation effect during oxidation (through the mechanism in eqn (4)–(6)), thus, further increase the d-spacing by disrupting π–π interactions.17,28
| C(graphite) + NaNO3 → CNaNO3 | (4) |
| 4KMnO4 + 2H2O → 4KOH + 4MnO2 + 3O2 | (5) |
| CNaNO3 + nO2 → GO | (6) |
3 Reduced graphene oxide
RGO can be considered an intermediate material with few oxygen-containing moieties than the parent GO and is obtained during reduction towards restoration of the original conjugated system of graphene (Scheme 1).4,36 RGO has captured interest of several researchers in wide-ranging fields due to its cost-effectiveness, large-scale production, and relatively ease of synthesis, and several other attributes discussed in the following sections.3.1 Properties
Future applications can be driven by the currently established physicochemical properties that are, in turn, influenced by defects and holes induced by missing carbon atoms, type, and distribution of residual oxygen functionalities per RGO lattice.O, reduction minimises their content but retains surface polarity and exfoliation in aqueous-media.3,39 The residual oxygen-containing functionalities on the sheets of RGO allow interaction with metal ligands, and thus allow chemical linkage of metal oxides in most composites.40 The chemical composition of RGO is controlled by precise synthesis protocols and the C/O ratio in most reports is ∼12.17 The combination of polarised RGO surfaces and hydrophobic sp2 hybridized graphitic basal planes brings amphiphilic traits to the material.41 Hence, both covalent (through oxygen moieties) and noncovalent interactions (via electrostatic and π–π interactions with cations and other conjugated backbones, respectively) are possible routes to further modify RGO.On the other hand, other studies have pointed out the decrease in mechanical properties owing to oxygen moieties.53 For example, oxygen functionalities were reported to have lower mechanical attributes in cementitious composites when compared to graphene.49 Additionally, a study through molecular dynamic simulations on the impact of wt% of oxygen groups on the mechanical properties of RGO revealed that an increase from 10 to 50% triggered severe deterioration of stress (from 110 to 55 GPa) and elastic modulus (from 0.5 to 0.3 TPa).54 The same work concluded that hydroxyl and epoxides induce easy fracturing effects on sheets due to weakened C–C σ-bonds in the diamond-like structures of RGO (relative to the CC honeycomb structure in pristine graphene).54 In a similar study, molecular dynamic simulations showed that high coverage of sheet surfaces with epoxides was associated with high ripple density and longer bond lengths that were in turn detrimental to mechanical strength.55 This common contradiction most likely suggests that RGO composition improves mechanical properties up to an optimum value and thereafter causes a deteriorating effect. Another feasible rationale for the inconsistencies is the widespread characteristics emanating from a wide range of synthesis methodologies and precursor materials. Therefore, as a future research direction, more focus on practical studies can probably clarify these differences. RGO is usually restacked and more agglomerated than GO (as often depicted by microscopy techniques (darker micrographs) and XRD).56,57 Additionally, recent studies have deduced high surface areas as one of the key features that affect mechanical properties through high dispersibility of other composites components,49,50 hence, this deduction suggests that GO is better than RGO as a mechanical reinforcement agent.
300 S m−1)8 in the reduced form. The partially restored π-system in RGO lowers the band gap.12,59 For example, Olumurewa et al.60 lowered the band gap from 2.4 to 1.5 eV by reducing GO with NaBH4. Although residual oxygen moieties, structural defects and vacancies that are generated during reduction of GO lessen conductivity of ultimate RGO relative to graphene (84500 S cm−1), the bulk conductivities of some RGO falls within the useful ranges.12,44,61 On the other hand, defects and grain boundaries in RGO raise phonon scattering and in turn lower thermal conductivity to ∼2.96 W m−1 K−1 relative to that of graphene (∼5000 W m−1 K−1), consequently, RGO performs better than graphene in thermoelectric generators.7,44,61 Also, higher electron mobility in RGO than in GO62 minimises chances of recombination of photoinduced electron–hole pairs.59,63 Additionally, RGO has an inherited high electron mobility than spherical nanomaterials by virtue of a 2-D morphology that is attributed to condensed junctions and grain boundaries.64,65 Excessive defect intensity may disrupt functionality in some applications, such as piezoelectric applications, as it interferes with electron transport. Nitrogen doping into the carbon framework of RGO has been reported to improve interfacial electron transfer in RGO, and therefore the conductivity increases as nitrogen atoms contribute to the additional electron.66,67Due to lower oxygen content and often improved crystallinity (restoration of conjugated system), RGO is generally expected to have a higher thermal stability than GO but lower than that of graphene. However, this trend is dynamic since the thermal stability of RGO-based materials depends on combination of factors such as, number of graphitic sheets, size, residual oxygen functionalities and associated defects.73 For example, on the one hand, thermal stability would be expected to decrease as the lattice domain sizes of RGO declines due to the induced tearing effect during oxygen removal and on the other hand, the low oxygen content of RGO would be anticipated to induce higher thermal stabilities.62,68,71,73 Hence, thermal analysis through thermogravimetric analysis is a potential industrial-scale technique for establishing RGO quality in future developments.73
C bond.74 Hence, UV-vis spectrophotometry is a suitable technique to determine effective reduction. Also, RGO is attractive due to its strong NIR absorption75,76 and optical attributes (excellent optical transparency of ∼98%)7,37 that enable use in optics,77–79 electronics,80 photovoltaics81 and photocatalytic reactions59 (Table 2). The transmittance of RGO decreases with an increase in number of layers,7,81 hence, reduction optimisation must be pursued as a future research direction for optical needs since reduction may induce agglomeration of layers and different structural parameters. This is imperative because ultimate structural characteristics and film thickness of RGO influence optical band gap values.39 In addition, the rich oxygen located on surfaces and edges (hydroxyl, carboxyl and epoxy), delocalized π–π electrons (due to restored aromaticity) and large surface of RGO enhance intercalation/adsorption of aromatic organic pollutants that can be photocatalytically degraded, particularly in composites with semiconductors.10,82–85 Since oxidation is known to enhance the optical band gap energy (in GO relative to graphene), reduction towards a more graphene-like material (in RGO) infers a lower optical band gap energy. This may be rationalised to mean that RGO would be more appropriate for photocatalysis than GO despite a lower content of oxygen functionalities. However, RGO may not reach its maximum adsorption capacity due to possible shrinkage of surface area induced by aggregation from the closer interplanar interaction of graphene sheets after reduction.86 Also, carbon vacancies generated during reduction of GO form carbon nanoclusters that transform the material towards semiconducting and luminescence (via promotion of percolation pathways among clusters) traits that are inclined to chain/cluster sizes/fractions.64 In this regard, the tuneable textural characteristics and large surface areas offered by RGO are critical in photodegradation, since organic pollutants principally degrade on semiconductor surfaces.64,87 RGO can be applied as catalyst supports and the 2-D π–π system on its surface facilitates activity of anchored nanoparticles by serving as electron acceptors.5 Quantum dot form of RGO (RGO-quantum dots) are 0-D materials that display unique characteristics, which emanate from the quantum confinement effect and pronounced edge effects that are inclusive of tuneable band gaps and photoluminescence (PL) with size variations.88,89
| Application | Example | Ref. |
|---|---|---|
| Smart containers | Polymer–RGO composites were used to build smart containers that controlled the desorption of inhibitors and acted as dual passive and active anticorrosive agents | 90 |
| Nanofluids | The Al2O3–RGO composite achieved a thermal conductivity of 2.076 W m K−1 at 45 °C and a heat transfer coefficient of 5462 W m−2 °C−1 at the exit of the test section that used 0.05 vol% of nanofluid at Reynolds number of 7510 | 98 |
| Anticorrosive coatings | The polydopamine nanoparticles–RGO composite improved the anticorrosion performance of a waterborne epoxy coating by triggering pH-sensitive activities upon acidic exposure. The composite also displayed self-repairing traits that improved corrosion resistance during long-term exposure by producing an impermeable barrier that delay the electrolyte penetration | 99 |
| Electrochemical capacitor electrodes | Polyaniline–N-RGO composite improved specific capacitance by 14032% (at 50 mV s−1) and 4749% (at 5 mV s−1) compared to performance of pristine N-RGO and PANI in K2SO4, respectively |
62 |
| Sodium ion battery electrodes | The Ni3S2–N-RGO composite showed stable reversible capacity (299.2 mA h g−1 for up to 100 cycles at 0.1 A g−1) and coulombic efficiency of ∼77% | 100 |
| Thermoelectric materials | WO3–RGO nanocomposite attained ∼9-, 14- and 2-fold improvement in figure of merit at 313 K, electrical conductivity and lowering of thermal conductivity relative to pristine WO3 | 80 |
| Piezoelectric energy harvesting | A 2% wt% RGO in a room temperature vulcanised silicone rubber–RGO composite lowers the tensile strength to 0.1 MP and increases the fracture strain by 100 times and electrical conductivity | 46 |
| Sensors | Polypyrrole–RGO materials were able to quickly and selectively sense NH3 linearly at room temperature | 101 |
| Nano-carrier for cancer treatments | The chitosan-Fe3O4–RGO nanocomposite was applied in the targeted delivery of anticancer drug (curcumin) for the suppression of MCF-7 breast cancer cells using the facile water-in-oil emulsification protocol | 102 |
| Antibacterial agents | The covalently conjugated peptide–RGO composite displayed improved antibacterial activity against E. coli and also achieved a decreased in hemolysis | 103 |
| Nanofiltration membranes | The RGO-based membranes achieved a water permeability of 225 L m−2 h−1 bar−1 and a selectivity of 98% in size-exclusion during separation of methyl blue separation. Size and charge are central to this technique | 104 |
| Fuel cells | MoS2–Ni3S2–RGO displayed catalytic efficiency and stability of 106 and 104% after 200 uninterrupted cyclic voltammetry cycles for CH3OH/CH3CH2OH electro-oxidation, respectively. The RGO enhanced the specific surface area and electrical conductivity of the electrocatalyst | 105 |
| Lithium-ion batteries | Ni-α-MnO2–RGO anode achieved excellent rate capability (from 0.2 to 10 C), a coulombic efficiency of ∼99% and capacity retention of 615 mA h g−1 at 830 mA g−1 after 200 cycles. The small sizes of α-MnO2 improved dispersion in the RGO matrix and consequently improved electrical-ionic conductivity, low charge transfer resistance and alleviates volume expansion | 106 |
| Lithium–sulfur batteries | Sulfur–RGO cathode attained a specific capacity of 1265 and 903 mA h g−1 after the first and hundredth cycles at 0.2 A g−1, respectively owing to improved electronic conductivity from ∼10−6 to ∼0.039 S cm−1 | 107 |
3.2 Recent applications
RGO as a material has wide-ranging applications9 and Table 2 highlights some interesting but yet to achieve commercial value. The purpose of the reviewed works in this section is to point out potential areas toward which the material can be fostered. Interestingly, ‘certain defects’ from the reduction effect and doping, as well as composites tune the material for certain applications that could not have been possible with GO and graphene. For instance, the induced hydrophobic nature is advantageous when used as an anti-corrosion material.90 Also, raised conductivity and N-doping in RGO enhances the applicability in electrochemical capacitors.71 Additionally, the restored π–π restacking structure in RGO aids high loading of hydrophobic drugs in drug carrier applications (Table 2).91 In addition to the induced properties of RGO through derivatisation,92 inherited attributes are also motivations of the recent usage of RGO. To illustrate this point, the nontoxicity and biocompatibility attributes, emanating from the main elemental component, carbon, allow the application of RGO in brain tumour treatments.93 Also, the semiconducting nature has given RGO promising prospects in sensors.94 In addition, the conductive nature of RGO and related surface roughness hinder bacterial growth, hence, can be applied for antibacterial activities.95Most of the recent applications reported with potential to achieve commercial value involve the use of RGO in composites to develop novel materials. An example of useful class composites is those in which RGO is applied as a metal catalyst support owing to the large surface area, chemical stability and thermal stability of RGO, and synergistic effect of components.96,97
The GO reduction protocol with respect to the RGO is critical to potential applications of the material. For example, Lee et al.108 established that the microwave assisted reduction (resulting in d-spacing of 0.39 nm) was more appropriate in accelerating the transport of lithium ions relative to the NH2NH2 reduced GO (culminating in d-spacing of 0.37 nm) for their lithium-ion battery application work (Table 4). The investigation of reduction protocols against specific applications is scarce in the recent literature (Tables 3–5) despite the lucrative potential in this research direction.
3.3 Review of recent reduction methods
The initial requirement was to achieve the most reductive state, closer to that of graphene but well exfoliated. However, this has evolved to the need to achieve reduction alongside physicochemical property tailoring for specific needs. This means the current thrust is not just achieve a material with characteristics closer to those of graphene, but also to develop functional materials and establish a better understanding of the nanostructural parameters. This shift is crucial towards reproducibility of the RGO-based materials and the upscaling goals. Both the parent GO (synthesis method) and reduction conditions are key in tailoring the properties of RGO.8,18 The wide-ranging precursors for GO and reduction protocols have generated vast amounts of variations between the RGO materials reported to date and this is a current challenge in standardisation. The oxygen-containing functionalities of GO are removed by three main approaches to RGO, namely, thermal,36,109 electrochemical110 and chemical methods, and a combination of methods.111,112 For illustration, Regis et al.112 employed a combination of ascorbic acid and near-ultraviolet light to enhance the reduction rate of GO (Table 3). An extremely high C/O ratio (>246) was reported from a combination of NaBH4 and thermal treatment at 1100 °C procedures.8 The reduction driving force for the thermal, electrochemical and chemical methodologies is temperature, electrical current and chemical potential, respectively, therefore, RGO materials of different characteristics are obtained.2 Distinct physicochemical properties of RGO are generated by the synthesis protocols followed.113 For example, reflux of GO in 2% aqueous green tea extract produced RGO with a large surface area (Table 3).114| Reductant | Reduction conditions | Comment | Ref. | ||||
|---|---|---|---|---|---|---|---|
| GO:H2O ratio |
GO:reductant ratio |
Mixing time (h) | Temp (°C) | Reaction time (h) | |||
| Aatriplex halimus leaves extract | 1:5 |
1:2 |
0.5 | 80 | 8 | Reduction introduced wrinkles and XRD broad peak at ∼22° | 117 |
| Azadirachta indica leaf extract | — | 2:1 |
1.33 | 90 | 2 | NaOH adjusted pH to 10 and has reducing effects. Colour change from dark brown to dark black, raised C/O ratio from 2.2 to 5.9, the (002) plane peak at 2θ ∼ 24° with corresponding d-spacing of ∼0.37 nm (decline from ∼0.77 nm) | 116 |
| Clinacanthus nutans leaf extract | 1:1 |
1:1 |
1 | 100 | — | Heating in an oil bath and also at 60 and 80 °C. The OH− removal and XRD peak 2θ ∼ 22° with a d-spacing of 0.40 nm were the signatures | 118 |
| Eclipta prostrata | 14 |
5:2 |
2 | Room | — | Surface roughness was an indicator of reduction, GO (14.29 nm) > RGO (2.051 nm) > graphite (1.784 nm) | 56 |
| Green tea extract | — | 2:1 |
0.75 | 60 | 6 | The UV-vis peak shifted from 240 nm to ∼270 nm. Phytomolecules in green tea extract also functionalised the RGO surface | 125 |
| Green tea extract | 12:5 |
6:5 |
2 | 95 | 24 | Reflux with extract at 95 °C for 24 h produced RGO with a high surface area of 973 m2 g−1 | 114 |
| Green tea extract | 1:2 |
1:100 |
— | 80 | 8 | Reduction was done at a lower temperature and in a conical flask different from the sealed autoclave in most reports | 124 |
| Chenopodium album | 1:1 |
1:10 |
— | 100 | 12 | Reductant also acts as a stabilising agent. Reflux for 12 h at 100 °C was facile and cost-effective | 120 |
| A. Cruentus extract | — | 10:1 |
— | 80 | 0.75 | The freshly prepared aqueous extract was mixed by stirring at 300 rpm. The XRD peak at 2θ ∼ 24° was ascribed to the plane (002) of the RGO in the composite | 132 |
| H2CO3 (from the coca-cola classic drink) | — | 4:25 |
— | 90 | 2 | Red-shifting of UV-vis peak for π–π* electron transition indicates restoration π-system when the soft drink was added | 133 |
| Terpenoids and polyphenols | — | 10:1 |
— | 70 | 1 | After evaporation of the solvent, the product was separated from the reductant by adding H2O2. Reducing and capping agents extracted from Vernonia amygdalina, hence, “greener” and sustainable | 35 |
| Tecoma stans | 1:1 |
1:1 |
1 | 70 | 12 | The C/O ratio decreased with increase in extract concentration | 134 |
| Olea europaea (olive extract) | 2:1 |
0.17 | — | 2 | After refluxing for 2 h, UV-vis absorption peak shift from 235 to 276 nm signalling restoration of conjugation upon reduction | 121 | |
| Na2O4S2 | — | — | — | 90 | 1.5 | The sheet d-spacing was 0.38 and 0.36 nm after treatment with Na2O4S2 and further with H2SO4, respectively | 76 |
| NaBH4 | 2:1 |
— | 1 | 100 | 1 | The lower FTIR peak intensities resulting from oxygen groups were used to confirm their removal | 38 |
| NaBH4, CH3CH2OH, NH3 | 10:1 |
50:3 |
1.5 | 150 | 18 | NH3 (25%) slowly added for pH adjustment to ∼10. Red-shifting of UV-vis peak from 300 to 260 nm indicated electronic conjugation recovery | 135 |
| NaBH4, NaOH | 2:1 |
— | 7 | 30 | 0.67 | Reaction was kept at 50 °C till NaBH4 step and under a N2 atmosphere up to NaOH addition followed by the addition of H2O2. Long method, involving a mild heating | 96 |
| NaBH4 | 1:1 |
1:1 |
1.5 | Room | — | UV-vis absorption maxima at 303 nm were ascribed to π–π* transitions due to restoration and redecorating of the aromaticity after reduction | 136 |
| NaBH4 | — | — | — | — | — | Reduction induced in a controlled N2 atmosphere at 20 Pa. The chamber was vacuumed to ∼10–2 Pa before filling with N2 | 137 |
| C8H11NO2·HCl | — | 1:2 |
— | Room | 24 | Vigorous stirring at room temperature for 24 h induced reduction without heating, hence an energy-saving approach | 138 |
| C6H8O6 | — | — | — | 95 | — | Ascorbic was added dropwise under magnetic stirring and reaction time not specified | 25 |
| C6H8O6 | — | 1:4 |
— | 95 | 3.5 | The assumption of the C2O as the chemical formula used to deduce the molar mass of GO as 40 g mol−1 in this report may not be true according to the proposed models. The XPS C/O ratio increased from 1.98 to 4.47 upon reduction | 139 |
| C6H8O6 | 1:1 |
3:10 |
— | 80 | 1 | The broad peak at 2θ ∼ 22° was attributed to the diffraction peak of RGO | 69 |
| C6H8O6 | ∼5:1 |
∼1:1 |
95 | 1 | The broadened XRD peak at 2θ = 24° and d-spacing of ∼0.37 nm (greater than 0.34 nm of graphite) indicated RGO. Water contact angle of 44° (<90°) for the RGO inferred hydrophilicity | 103 | |
| C6H8O6 | — | — | — | 50 | 12 | Reductant amount nor provided and reduction was verified optically with a dark brown/light yellow to black colour change | 140 |
| C6H8O6 | 5:1 |
1:5 |
0.17 | 90 | 2.5 | Sonication was at 600 W for 0.17 h. XRD broad peak at 2θ ∼ 25° and C/O ratio of 2.22 (increase from 1.34) signalled RGO formation with a surface area of 176 m2 g−1 | 5 |
| C6H8O6 | 5:1 |
1:8 |
1.67 | 80 | 24 | Chemical and reflux treatments led to disappearance of the XRD signature peak for GO at 2θ ∼12° and decline of d-spacing to 0.35 nm (from 0.73 nm) | 141 |
| C6H8O6, NH3 | 2:3 |
1:10 |
1.17 | 85 | 1.5 | NH3 (25%) was added dropwise to adjust pH to 12. Ascorbic acid is a green reductant and, the disappearance of IR peaks due to oxygen-containing moieties occurred after reduction | 44 |
| C6H8O6 | 15:4 |
1:67 |
— | — | 48 | Irradiation done after adding reductant at 405 nm. Radiation time effect was studied, increase in time led to increased elimination of OH− (48 h raised C/O ratio from 3.44 to 4.35) | 112 |
| NH3(aq), NH2NH2 | 1:9 |
— | 0.5 | 90 | — | NH3(aq) was added as an exfoliating agent though it also has reducing effect. The NH2NH2 amount was not given | 142 |
| NH3(aq), NH2NH2 | 1:15 |
50:1 |
2 | 90 | — | Broad (002) plane at 2θ = 24° and d-spacing of 0.44 nm were indicators of reduction | 74 |
| NH2NH2, KOH | — | — | 2 | 100 | 24 | Chemical and reflux reduction signalled by the π → π* UV-vis peak shift from 230 to the 260 nm due to reduction and restoration of the conjugated structure (increased π-electron concentration) | 43 |
| NH2NH2·H2O, NaOH | 10:1 |
200:1 |
0.5 | 60 | 4 | pH adjustment to 10 by adding either HCl (0.1 N) or NaOH (0.1 M). Highly wrinkled and irregular structured sheets with corrugated surfaces was ascribed to reduction | 83 |
| NH2NH2·H2O, Al(OH)3 | 10:1 |
500:1 |
1.15 | 90 | 0.75 | Heating was by means of a water bath. Darkened dispersions with a strong-sharp UV-vis peak at ∼258 nm signalled completion of reduction and restoration of the conjugated structure of CC |
143 |
| [CH2CH(OH)]n, NaBH4, NaOH | 5:1 |
50:7 |
0.75 | 90 | 4 | NaOH (0.1 M) was slowly added to adjust pH to 11. Combination of 3 reductants and thermal treatment. Transmission electron micrographs showed darker regions and wrinkles (induced folding and re-stacking of layers at the edges) | 63 |
The reducing and stabilizing effect of most plant extracts are due to their OH− and CO moieties.125 Ascorbic acid (C6H8O6) is mild, non-flammable, functions as a reductant in acidic as well as alkaline pH and has nontoxicity advantageous (no gaseous by-products) over other chemical reductants.5,103,126 C6H8O6 is a competing alternative to hydrazine hydrate due to ability to attain similar C/O ratios, its abundance and anti-oxidative traits. Whilst reduction is possible at room temperature, the reviewed recent articles suggest that the use of C6H8O6 as a reductant requires a mild heating of ∼95 °C (Table 3). The mild heating is necessary for achieving appreciable reduction in a short time and despite this, the low temperature requirements still qualify the use of C6H8O6 reductants as a green approach. Other reduction conditions in combination with ascorbic acid, such as stirring (rate and mode), high GO:reductant ratio, sonication, and alkaline pH, also shorten the reaction time from 48 to ∼1 h.112
The use of biologically derived reductants is an interesting progress towards the sustainability of graphene-based materials, since both GO and RGO can also be biologically derived.127–129 This is a step near the elimination of expensive, explosive, toxic and carcinogenic chemicals;122,124 such as hydrazine (and its derivatives),43,58 and sodium borohydride;130 that harm the environment. This is a highlight of the tremendous potential of biologically derived reductants not only due to their biocompatibility nature, but also because the approach is a suitable standardisation step in future research.
In general, most chemical reduction methodologies initially involve the dispersion of GO in water through ultrasonic treatment (Table 3). A common feature of the recent chemical reduction methodologies is the use of mild heating in the 80–100 °C range. This could be a decent basis for developing standardised reduction protocols that will ease comparison between methods in terms of efficacy and reproducibility. For uniformity, the GO:reductant ratios presented in Table 3 were calculated using the main reductant (where more than one possible reductant was used) assuming the density of water was 1 g cm−3. According to the reviewed works, the calculated GO:reductant ratios are arbitrary; hence, the recent chemical reduction protocols are difficult to compare in terms of efficacy (Table 3). Economic and environmental sustainability considerations are a conceivable fair stimulation to focusing more on lower GO:reductant ratios, particularly in the case of inorganic reductants, in future research.
A few of these reports presented further issues with the missing pertinent details about the amount of reductant that was used. However, satisfactory qualitative tests for RGO formation were present in most reports. More insights towards a standardised GO:reductant ratio is needed in future research to aid the progression to commercialisation of products.
Future research directions must seek a better understanding of chemical reduction mechanisms, eliminate the use of toxic chemicals, and shift towards green chemistry principles via the use of biologically derived reductants with comparable reduction efficacies and avoid inorganic reducing agents. This approach has positive prospects as a strategic value-addition exercise for agricultural waste, since high tonnage is produced globally each year. Most chemical reduction protocols involve purification steps that are not only monotonous, but also add substantial costs to material development at large-scale. The purification procedures often involve washing and neutralisation steps with deionised/ultra-pure water to remove excess chemical reductants, centrifuging and drying for ∼24 h.112,117,119,121,126,128,131 Careful and cost-effective removal of residual reductants must be sought in future research to avoid chemical contaminants that may negatively affect RGO quality and intended applications. Another drawback of chemical reductants is their selective elimination of oxygen moieties.19,30 Despite the shortfalls, chemical reduction methods have the advantage of short purification times and low energy requirements when compared to other methods; therefore, they are better suited for the principles of green chemistry and sustainability.
| Reduction method/reductant | Reduction conditions | Comment | Ref. | ||||
|---|---|---|---|---|---|---|---|
| Gas | GO:reductant ratio |
Mixing time (h) | Temp (°C) | Reaction time (h) | |||
| Thermal | Ar | — | — | 300 | — | The d-spacing decreased from 0.79 to 0.43 nm after reduction | 57 |
| Thermal | Ar | — | 350 | 1 | The method used a tube furnace and an inert atmosphere | 146 | |
| Thermal | Ar | — | — | 400 | The Ar flow rate was 0.1 L min−1 and the heating rate was 10 °C min−1. Temperatures of 250, 300 and 400 °C attained increased C/O ratios of 6.13, 6.16 and 6.46 from 2.41 of GO | 33 | |
| Thermal | Air | — | — | 50 | 168 | The notable significance of this work was the ability to thermally reduce GO at a low temperature of 50 °C. The XRD peak 2θ ∼ 24° was a signal of reduction | 144 |
| Thermal | Air/He | — | — | 500 | — | Air flow rate at 300 °C (ramped at 30 °C min−1) then He at 10 °C min−1 between 300 and 500 °C. A large surface area of 439 m2 g−1 attained | 147 |
| Thermal | N2–H2 | — | — | 400 | 1 | N2–H2 flow rate was 80/40 sccm and heating rate was 10 °C min−1. No direct determination of reduction effect | 148 |
| Thermal/CH4N2S | N2 | 15:38 |
∼12 | 800 | 2 | Ramping rate of 5 °C min−1 and reduction simultaneously done with N-doping (∼10 at%) | 100 |
| Thermal/CH3CH2OH, NH3 | — | — | 24 | 1200 | 2 | The drying was carried out at 100 °C for 24 h before thermal reduction, π–π* peak shifted from 229 nm to 254 nm | 98 |
| Thermal/CH3CH2OH | — | — | 24 | 1000 | 0.03 | CH3CH2OH (70%) was mixed by ultrasonic treatment before and after thermal treatment for 24 and 3 h, respectively, to aid exfoliation and minimise the often-enumerated agglomeration of RGO sheets | 68 |
| Reflux/L-Methionine | — | — | — | 80 | 12 | L-methionine has a reducing effect on GO | 149 |
| Spray pyrolysis | Air | — | — | 150 | Spraying setup: airbrush nozzle (aperture size – 0.4 mm) and air compressor at 4 bars. The method is based on the thermal decomposition, which facilitates reduction | 36 | |
| Pulsed laser | N2 | — | — | — | — | Nd:YAG laser source (λ = 266 nm, ν = 10 Hz), evacuated to 10–2 Pa before filling reaction chamber with N2 at 20 Pa. The π–π* peak indicated restoration aromaticity | 150 |
| Ultraviolet laser | — | — | — | 50 | 96 | Sample put in an ultraviolet chamber then irradiated with a Phillips F5-40 lamp at an intensity of 12.4 W m−2 and wavelength range of 300–320 nm. Resistance decreased from 7.05 × 103 (for GO) to 4.45 × 103 and 1.27 × 103 Ω after treatment for 48 and 96 h, respectively | 151 |
| Hydrothermal and thermal | Ar | — | 4 | 900° | 2 | GO was initially treated at 100 °C for 1 h, then kept at 180 °C for 3 h before cooling to room temperature, filtering, terminating with H2O2, neutralisation to pH 7 and final treatment. The approach was different from the usual protocol of oxidation, termination, purification, and reduction, allowed H2SO4 recycling and simplified purification via filtration | 152 |
| Hydrothermal and thermal/2-chloroethylamine hydrochloride | — | 3:20 |
4 | 500 | 3 | Hydrothermal treatment performed before the thermal step for 12 h at 180 °C (rate: 10 °C min−1). Thermal treatment was in two steps: at 200 °C for 1 h (5 °C min−1) then at 500 °C for 3 h. XRD peaks at 2θ ∼ 26° (002) and ∼44° (100) ascribed to graphite and hexagonal structure of graphite planes | 153 |
| Thermal and hydrothermal/H3BO3, CH4N2S, C2H8N2 | Ar | 1:20 |
1 | 900 | 5 | CH4N2S (4 mol) and C2H8N2 (4 mL) were added prior to hydrothermal treatment at 200 °C for 12 h. Method was sequential doping to avoid formation of B–N bonds that may cause lowered activity of the target catalyst | 154 |
| Reduction method/reductant | Reduction conditions | Comment | Ref. | ||||
|---|---|---|---|---|---|---|---|
| GO: H2O ratio |
GO:reductant ratio |
Mixing time (h) | Temp (°C) | Reaction time (h) | |||
| Hydrothermal | 1:2 |
— | 0.5 | 160 | 3 | Broad XRD peaks at 2θ ∼ 24° (002) and 43° (102), and UV-vis absorption maxima shift from 232 to 275 nm were signature peaks for RGO | 155 |
| Hydrothermal | 1:4 |
— | 2 | 180 | 27 | Stirring was at 500 rpm and unlike most hydrothermal treatments, beaker with silicon oil was use for heating. Reaction time was long and reduction was signalled by the disappearance of the sharp C–O peak at 1067 cm−1 | 60 |
| Hydrothermal | — | — | — | 120 | 4 | Method lacked direct assessment of reduction effect. Decrease in defect intensity upon reduction is a possibility, but not always, and implication of effective reduction | 106 |
| Hydrothermal | — | — | — | 150 | 6 | The amount of water added was not provided | 156 |
| Hydrothermal/NaOH | 2:1 |
— | 1.67 | 200 | 24 | The NaOH was used to adjust pH to ∼10. Reduction confirmed by FTIR spectrum that showed two unique peaks at 1565 and 1193 cm−1 | 157 |
| Hydrothermal/CH4N2S | 1:1 |
15:28 |
1.67 | 180 | 12 | Mixing was done by means of ultrasonic treatment at 750 W and 20 kHz after dispersing GO in H2O by stirring for 0.5 h | 158 |
| Hydrothermal/(CH2)3CH2O | — | 12:1 |
0.3 | 130 | 8 | Ultrasonic treatment was attributed to both GO exfoliation and the carboxylic acid-carboxylate ion moiety transformations. The C/O ratio decreased from 2.39 to 1.63 | 159 |
| Hydrothermal/NaOH | 16000:7 |
— | 0.5 | 180 | 12 | Uncoordinated oxygen moieties were removed | 160 |
| Hydrothermal | 2:1 |
— | — | 180 | 12 | Reduction generated the most stable composites with F-carbon nanofiber (the highest negative zeta potential of −45 mV) | 161 |
| Hydrothermal | — | — | — | 180 | 8 | The reduction reaction induced π–π stacking | 162 |
| Hydrothermal/H2NCH2CH2NH2 | 1:2 |
1.15 | 120 | 12 | Details of the amount of H2NCH2CH2NH2 were not provided | 163 | |
| Hydrothermal/NaOH | — | 20:1 |
1.17 | 200 | 4 | The disappearance of the C–H bonding, and declined intensities of the O–H (at 3324 cm−1) and CO bending peaks (at 1812 cm−1) were the signatures of the formation of RGO |
84 |
| Hydrothermal | — | — | — | 300 | 24 | Fluorination was done after reduction by introducing F2–N2 (1:5) and removal unbound fluorine was carried out by washing with dilute Na2CO3 and H2O |
164 |
| Hydrothermal | 10:1 |
— | 0.33 | 200 | 24 | The π–π* transition of the conjugated CC bonding was redshifted to 270 nm |
64 |
| Hydrothermal/NH2NH2 | — | — | — | 200 | 0.33 | The first treatment was at 95 °C for 2 h and then the second at 200 °C in NH2NH2 (0.2 M). RGO floated on the H2O surface and the Raman ID/IG increased from 1.05 to 1.33 after reduction due to the elimination of oxygen moieties and the formation of defects along with the recovery π-system | 165 |
| Reduction method/reductant | Reduction conditions | Comment | Ref. | ||||
|---|---|---|---|---|---|---|---|
| GO:solvent ratio |
GO:reductant ratio |
Mixing time (h) | Temp (°C) | Reaction time (h) | |||
| Solvothermal/Green tea extract | — | 1:1 |
— | 90 | 8 | The stirring was continuous at 200 rpm during the reaction. Absence of the GO typical peak at 2θ ∼ 10° confirmed complete reduction | 123 |
| Solvothermal/Murraya koenigii | — | — | 0.67 | 100 | 12 | The extract was rich in polyphenol and reduction was also ascribed to this chemical | 122 |
| Solvothermal/CH2OHCH2OH | 5:3 |
20:1 |
2.5 | 200 | 8 | The UV-vis π → π* peak at ∼270 nm was used to confirm reduction and restoration of aromatic CC bonds |
75 |
| Solvothermal/C2H4(NH2)2/CH2OHCH2OH | 15:1 |
∼1:1 |
0.5 | 180 | 10 | Thermal treatment in two reductants achieved reduction. The peak at 2θ ∼ 26° (002) plane was a signal for GO reduction | 59 |
| Solvothermal/H2/(CH3)2NC(O)H/HOCH2(CH2)2O | 1:6 |
5:6 |
1 | 180 | 8 | The dropwise addition of NH4OH adjusted the pH to >10. Hydrothermal treatment done in N2 (at 0, 5, and 10 bar) and Ar (85%)–H2 (15%) atmosphere. The injection of H2 improved crystallinity of RGO. | 166 |
| Solvothermal/C2H8N2 | — | — | 120 | 12 | C2H8N2 was a source of N2 as well as an additional reductant | 167 | |
| Solvothermal | 2:5 |
— | 1 | 140 | 12 | Nitrates are well known as oxidants; hence, minimal reduction despite thermal treatment | 168 |
| Solvothermal/NH3(aq) | — | 1:10 |
— | 160 | 8 | Reduction and N-doping achieved with low temperature and indicator was increase in the XPS C/O ratio from 2.48 (GO) to 11.36 | 71 |
| Solvothermal/CH2(C6H4NH2)/CH3CH2OH/NaOH | — | 15:1 |
1 | 180 | 10 | NaOH (0.1 M) adjusted pH to 10. Disappearing of FTIR oxygen peaks and observed weak C–H stretching vibration (2915–2935 cm−1) of the aliphatic CH2 moiety | 169 |
| Solvothermal/N2H4·H2O/NH3 | 3:25 |
1.5 | 180 | 12 | H[OCH2CH2]nOH was the solvent. N2H4·H2O and H2O was used to keep pH at 11. Combination-treatment induced pyrrolic-, pyridinic-, oxidized- and graphitic-N | 170 | |
| Solvothermal-microwave irradiation | — | — | 2 | 120 | 0.5 | Solvent: C4H9NO (DMF). The reduction was signalled by UV-vis absorption maxima at 265 nm (π–π* transitions of aromatic CC) and an increase in C/O ratio from 0.61 to 2.52 |
3 |
| Microwave-assisted solvothermal | 413:180 |
— | 0.5 | — | 0.19 | Solvent: C5H9NO | 2 |
| Treatment was in an Anton-Paar Microwave (power/time ratios: 600 W/0.19 h) (most effective), 800 W/0.14 h, and 1000 W/0.117 h (fastest) | |||||||
| Microwave-assisted solvothermal/C2H6O2 | — | — | — | 180 | 0.1 | XRD spectrum (002) plane was an indicator of reduction | 171 |
| Microwave-assisted hydrothermal/NH3 | — | — | 8 | 150 | 0.08 | NH3 used to adjust pH to 10. Simultaneous microwave rapid reduction and N-doping (NH3 was N2 source) | 172 |
| Microwave-assisted hydrothermal | — | 150 | 0.25 | Microwave irradiation was at 400 W. Raised intensity peaks of the C–C and CC signalled the reduction process |
26 | ||
| Microwave-assisted hydrothermal/NH3(aq) | — | — | 2 | 180 | 0.33 | NH3 (6%) was added dropwise to achieve a pH of 10. The NH3(aq) support the reduction of GO. | 173 |
| Reduction method/reductant | Reduction condition | Comment | Ref. | ||||
|---|---|---|---|---|---|---|---|
| GO:H2O |
GO:reductant ratio |
Mixing time (h) | Power (W) | Reaction time (h) | |||
| Microwave | — | — | — | 700 | 0.08 | Quick method | 174 |
| Microwave/NH3 | 5:1 |
500:1 |
0.67 | 500 | 0.025 | The peak of the XRD spectrum at 2θ = 26.4° was broad and a signal for reduction | 24 |
| Microwave/CH4N2S | — | — | 700 | 0.17 | Microwave frequency was 2.45 GHz. The 2θ peak ∼26° (002) plane was a reduction indicator | 175 | |
| Microwave | — | — | — | — | 0.17 | The first treatment for 0.17 h was in DMF and then in toluene for 0.083 h to complete the reduction. The wider (002) XRD peak (2θ = 23°) with a d-spacing 0.39 nm was reduction indicator | 108 |
| Microwave and thermal/NH3(aq) | ∼1:1 |
— | 2 | — | 0.17 | Freshly prepared NH3(aq) was added to maintain pH at 7. Reduction was confirmed by a broad XRD peak at 2θ = 24° assigned to the (002) plane | 176 |
| Gamma irradiation/CH3CH2OH | — | 1:1 |
0.5 | — | — | Irradiation was done with 100 kGy of γ-ray at 2 kGy h−1 from a 60Co source. The γ-rays generates reductive radical (H˙) that reduces GO and d-spacing of 38.6 nm | 177 |
| Gamma irradiation/CH3CH(OH)CH3 | — | — | — | — | — | N2 was bubbled prior to irradiation with an absorbed dose of 75 kGy at 4.5 kGy h−1 from a 60Co source under the ambient conditions. CH3CH(OH)CH3 was an ˙OH− scavenger and forms reactive radicals that were excellent reductants | 178 |
| Plasma-assisted/CO(NH2)2 | — | 2:25 |
12 | 200 | 0.25 | Radio frequency inductively coupled plasma (13.56 MHz, pressure: 15 Pa, H2 flow rate: 15 sccm) was used to simultaneously reduce and N-dope (N2 source: (CO(NH2)2)) | 66 |
Thermal reduction of GO has been reported at temperatures as low as 50 °C, however, the reaction time was one of the longest reported, that is, 1 week (Table 4).144 This suggests that a potential setback to progression can be avoided in future works through careful calculations that balance reaction time and temperature inputs, since the random lowering of one parameter could sum up to the same energy requirements. Despite achievable reduction at low temperatures that mostly remove labile oxygen groups, Coros et al.33 established that reduction increases with temperature (Table 4). This is possibly needed to meet the energy requirements for breaking chemically stable bonds in oxygen moieties under an inert environment.
However, high thermal temperatures can generate extremely toxic volatile organic hydrocarbons.58 Exfoliation through thermal means is facilitated by the evolution of gases (such as CO2 from the decomposition of oxygen moieties and H2O) and weight loss (∼30%) creates vacancies and topological defects.12,31,68 Despite the minimal contamination issues, relative to chemical means, the drawbacks that may be associated with thermal methods include the requirement of higher temperatures (mainly 400–1000 °C)30 for effective reductions and the possible build-up of explosive gas evolutions (Table 4).
Hydrothermal and solvothermal reduction of GO refers to chemical transformations that result in the removal of oxygen moieties in a supercritical environment of water and specified solvents, respectively, triggered by heating.145 Hydrothermal and solvothermal treatments can be viewed as stratagems that lower the temperature required for the reduction of GO by utilising high pressures and surface chemistry. Therefore, the hydrothermal and solvothermal reduction temperatures and durations in the recent reviewed reports were mostly in the 160–200 °C and 8–12 h ranges, respectively (Table 4). This is a direction towards ease comparison between methods and reproducibility of RGO. However, lack of ratio details in most studies may hinder this advancement. The GO:H2O ratio was commonly 1:2 or 2:1 for hydrothermal methods, while in solvothermal methods, the GO:solvent ratio was random (density of H2O was assumed to be 1 g cm−3 for the calculation of GO:H2O ratio in the Table 4).
The GO:solvent ratio in hydrothermal and solvothermal protocols still needs more work to enable comparison between solvents at given temperatures and to establish if the water/solvent should be in excess. Similarly, to the general thermal protocols, common products of both solvothermal and hydrothermal approach are CO2 and CO gases. However, the use of lower temperatures in both solvothermal and hydrothermal methods has a lower reduction efficacy compared to thermal means. The use of organic solvents in solvothermal reduction reduces energy requirements and has better capabilities to self-generate pressure relative to hydrothermal needs.30 Hence, future research could prudently use solvothermal methods, however, water as a solvent still offers affordability and safe handling advantages.
Microwave reduction of GO is possible on an industrial scale and achieves reduction at lower energy costs relative to thermal treatment (Table 4); however, reduction efficacy is low.23 Hence, microwave-assisted thermal reduction of GO is a strategy to accelerate thermal reduction since energy is transferred directly to reagents.2,145,173 In this method, reduction is triggered by both heating effect, and differences in the dielectric constants of GO and the solvent, which induces an instant increase in internal temperature, consequently, causing reduction.2,145,179 Hence, selection of an appropriate solvent system for the microwave reduction of GO is critical in this regard. An ideal solvent is one that facilitates stable GO dispersions without functionalisation of the ultimate RGO, with a high boiling point and high dielectric properties (better ability to absorb and convert microwaves into heat energy), as well as surface tension values between 40 and 50 mJ m−2.3,179–182
In addition, a study by Martins et al.2 has hinted at the need of establishing a suitable compromise between high reduction rate and efficacy, since they are often achievable through high- and low-power inputs, respectively (Table 4). The microwave technique can also be combined with other reduction protocols. For illustration, microwave-assisted hydrothermal methods are advantageous than either microwave or hydrothermal approaches in that the combination accelerates heating, effects a more sensitive reaction, and facilitates uniform heating.173
The γ-irradiation has also been reported recently as a facile, green, cost-effective method for RGO production (Table 4).177 In the presence of water or alcohol, γ-rays create H˙ (reductive) and OH˙ species (oxidative) due to water radiolysis. The role of alcohol is scavenging for oxidative radicals in order to stop additional oxidation of GO. The advantages of reduction by means of γ-irradiation over chemical means are the lower agglomeration in the RGO produced177 and the absence of contamination issues. The combination of thermal treatment with chemical reductants has gained momentum in recent times towards achieving RGO with characteristics closer to that of graphene (Table 4). Despite the general calls to utilise clean chemicals in order to preserve the environment, catastrophic toxic chemicals, such as NH2NH2, are still being used in combination with thermal treatment (Table 4). Therefore, as a future research direction, chemical selection should be inclined to only “greener” chemicals.
The reviewed literature has also shown the combinations of methods such as chemical reduction and thermal reduction in one step (solvothermal protocols that use a solvent with reducing effects, Table 4). For example, reduction was achieved through a combination of microwave irradiation and the solvothermal protocol without the use of a chemical reductant (Table 4).3 Thermal treatment can also be used in combination with alcohol to enhance reduction of GO (Table 4). Alcohols are relatively mild reductants due to the preservation of edge morphology.21
| Reagents other than GO | Reduction conditions | Remark | Ref. |
|---|---|---|---|
| H2 (reductant) | Ball milling: H2 environment for 8 h, with purging every 2 h, during dual-drive ball milling of 30 Hz | The C/O ratio of the RGO was 17.51 and the method was eco-friendly and versatile | 183 |
| H2SO4 | Electrochemical: graphite foils (1 cm apart) were electrodes in H2SO4 (0.5 M). A +4 V was the applied anodic potential at room temperature | Partial oxidation of graphene is formed through formation of radicals such as, OH− radicals | 184 |
| PTFE, KOH (reductant) | Electrochemical: GO (50 mg) was added to the paraffin oil to form a paste, then ultrasonicated at 100 W and room temperature for 0.5 h. Subsequently, this was filled into a PTFE tube prior to electrochemical treatment of −1.2 to 0 V with a scan rate of 100 mV s−1 in a cell with KOH (0.1 M, pH = 12.0) | Reduction was confirmed by a diffraction peak at 2θ ∼ 25° and weak peak at about 42° | 185 |
On the other hand, in the two-step electrochemical reduction, GO is first deposited onto the electrode surface (and attachments are possibly through van der Waals forces), then dried before electrochemical reduction. When compared to chemical means, the electrochemical reduction is faster, greener, and economically more advantageous as a future research realisation.
4 Challenges and prospects
From the reviewed works it is critical to realise that RGO is a nonstoichiometric derivative of GO since numerous ratios of carbon, hydrogen, and oxygen have been reported to date.8,32 This poses some fundamental issues in determining reproducibility of RGO by comparing atomic ratios on the basis of mass ratios per sample. The atomic ratios in RGO are influenced by synthesis methodologies which are currently wide-ranging. In addition, the precise chemical structure of GO and RGO remains obscure and still requires further modelling. The gaps in understanding RGO are due to numerous variables in oxidation methods (precursors) that lead to sample-to-sample physicochemical variations, nonstoichiometric atomic composition and unavailability of specific physicochemical characterization techniques.12 The other drawbacks associated with full utilisation of RGO are emanating from lack of standardised methodologies for reducing GO, i.e., lack of pertinent details such as reduction time, GO:reductant ratios, and use of arbitrary ratios is challenging reproducibility and adoption of methods at industrial scale.
The common use of X-ray diffraction and ultra-violet visible spectroscopies as signature techniques for confirming the formation of RGO in most recent reports is a step in the standardisation direction. Future research may also utilise the scarcely used C/O ratio from X-ray photoelectron spectroscopy, since this can enable easy comparison between methods and determination of reproducibility. With what has been reported to date, RGO may be referred to as a family of materials and not as a single composition since current proposed structures differ according to varied oxidation conditions (in each of the reported methods) and properties of starting materials.16,35 The limited understanding of the chemical structure of GO could be a major setback in understanding RGO (which is simply a derivative of GO). In addition, lack of large-scale and cost-effective production of RGO at the present time is a drawback to progression towards commercialisation despite the highlighted potentials through physicochemical properties and recent applications. Hence, this in turn is limiting breakthroughs in various potential large-scale industrial applications. In-depth understanding of reduction mechanisms, elimination of toxic reductants and the use of low temperatures (for energy-saving purposes), design of simple reduction procedures, and more studies of biologically derived reductants are critical and potentially sustainable steps as future research directions. This trajectory is promising in driving RGO towards its full potential in order to broaden and/or further advance related properties. The reviewed work highlights RGO as a potential and strategic material for achieving sustainable, environmentally friendly, low-cost, high-performing, and large-scale production and use in functional devices and/or processes to pave the way for commercialisation. This can drive future commercial viability aspects of RGO as a material.
Author contributions
ETM and EM designed the work, investigated the literature, wrote the original draft, assembled the figures and tables, and revised the manuscript.Conflicts of interest
The authors report that there are no competing interests to declare.Acknowledgements
This work was supported by the University of Zimbabwe and Bindura University of Science Education.References
- A. D. Sontakke and M. K. Purkait, Chem. Eng. J., 2021, 420, 129914 CrossRef CAS
.
- A. L. B. S. Martins, E. F. da Silva, M. F. V. Marques and W. A. Pinheiro, Mater. Res., 2022, 25, e20220140 CrossRef CAS
- R. Tarcan, M. Handrea-Dragan, O. Todor-Boer, I. Petrovai, C. Farcau, M. Rusu, A. Vulpoi, M. Todea, S. Astilean and I. Botiz, Synth. Met., 2020, 269, 116576 CrossRef CAS
- E. T. Mombeshora and A. Stark, Biomass Convers. Biorefin., 2023, 13, 4619–4638 CrossRef CAS
- N. M. Dat, P. N. B. Long, D. C. U. Nhi, N. N. Minh, L. M. Duy, L. N. Quan, H. M. Nam, M. T. Phong and N. H. Hieu, Synth. Met., 2020, 260, 116260 CrossRef CAS
- Z. Çıplak, B. Getiren, C. Gökalp, A. Yıldız and N. Yıldız, Chem. Eng. Commun., 2019, 207, 559–573 CrossRef
- F. Farjadian, S. Abbaspour, M. A. A. Sadatlu, S. Mirkiani, A. Ghasemi, M. Hoseini-Ghahfarokhi, N. Mozaffari, M. Karimi and M. R. Hamblin, ChemistrySelect, 2020, 5, 10200–10219 CrossRef CAS
- S. Mao, H. Pu and J. Chen, RSC Adv., 2012, 2, 2643 RSC
- R. Tarcan, O. Todor-Boer, I. Petrovai, C. Leordean, S. Astilean and I. Botiz, J. Mater. Chem. C, 2020, 8, 1198–1224 RSC
- D. W. Lee, L. V. De Los Santos, J. W. Seo, L. L. Felix, A. D. Bustamante, J. M. Cole and C. H. W. Barnes, J. Phys. Chem. B, 2010, 114, 5723–5728 CrossRef CAS PubMed
- S. N. a. Y. Matsuo, Carbon, 1994, 32, 469–475 CrossRef
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC
- Y. X. Li, C. Y. Sun, C. J. Yu, C. X. Wang, Y. J. Liu and Y. B. Song, Adv. Mater. Res., 2012, 476–478, 1488–1495 CAS
- D. R. Dreyer, A. D. Todd and C. W. Bielawski, Chem. Soc. Rev., 2014, 43, 5288–5301 RSC
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS
- T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef
- E. Aliyev, V. Filiz, M. M. Khan, Y. J. Lee, C. Abetz and V. Abetz, Nanomaterials, 2019, 9, 1180 CrossRef CAS PubMed
- P. P. Brisebois and M. Siaj, J. Mater. Chem. C, 2020, 8, 1517–1547 RSC
- E. T. Mombeshora, P. G. Ndungu and V. O. Nyamori, Electrochim. Acta, 2017, 258, 467–476 CrossRef CAS
- E. T. Mombeshora and A. Stark, Mater. Chem. Phys., 2022, 277, 125535 CrossRef CAS
- W. Yu, L. Sisi, Y. Haiyan and L. Jie, RSC Adv., 2020, 10, 15328–15345 RSC
- R. Ikram, B. M. Jan and W. Ahmad, J. Mater. Res. Technol., 2020, 9, 11587–11610 CrossRef CAS
- L. Dong, J. Yang, M. Chhowalla and K. P. Loh, Chem. Soc. Rev., 2017, 46, 7306–7316 RSC
- R. Kumar, S. M. Youssry, M. M. Abdel-Galeil and A. Matsuda, J. Mater. Sci.: Mater. Electron., 2020, 31, 15456–15465 CrossRef
- A. C. Lazanas, A. Katsouras, M. Spanos, G. M. Manesi, I. Moutsios, D. V. Vashurkin, D. Moschovas, C. Gioti, M. A. Karakassides, V. G. Gregoriou, D. A. Ivanov, C. L. Chochos and A. Avgeropoulos, Polymers, 2022, 14, 5292 CrossRef CAS PubMed
- S. Jangu, B. K. Satpathy, M. Raju, C. Jacob and D. Pradhan, Dalton Trans., 2021, 50, 6878–6888 RSC
- M. J. Yoo and H. B. Park, Carbon, 2019, 141, 515–522 CrossRef CAS
- E. T. Mombeshora, P. G. Ndungu and V. O. Nyamori, New Carbon Mater., 2017, 32, 174–187 CrossRef CAS
- T. Nakajima, A. Mabuchi and R. Hagiwara, Carbon, 1988, 26, 357–361 CrossRef CAS
- R. Singh, S. Ullah, N. Rao, M. Singh, I. Patra, D. A. Darko, C. P. J. Issac, K. Esmaeilzadeh-Salestani, R. Kanaoujiya and V. Vijayan, J. Nanomater., 2022, 2022, 8731429 Search PubMed
- C. Kavitha, Mater. Today: Proc., 2022, 49, 811–816 CAS
- L. Sun, Chin. J. Chem. Eng., 2019, 27, 2251–2260 CrossRef CAS
- M. Coros, F. Pogacean, A. Turza, M. Dan, C. Berghian-Grosan, I.-O. Pana and S. Pruneanu, Phys. E, 2020, 119, 113971 CrossRef CAS
- T. F. Emiru and D. W. Ayele, Egypt. J. Basic Appl. Sci., 2017, 4, 74–79 Search PubMed
- B. Meka Chufa, B. Abdisa Gonfa, T. Yohannes Anshebo, G. Adam Workneh and D.-R. Hang, Adv. Condens. Matter Phys., 2021, 2021, 6681710 Search PubMed
- A. S. AlShammari, M. M. Halim, F. K. Yam and N. H. M. Kaus, Mater. Sci. Semicond. Process., 2020, 116, 105140 CrossRef CAS
- A. Adetayo and D. Runsewe, Open J. Compos. Mater., 2019, 09, 207–229 CrossRef CAS
- H. Altaee and H. A. Alshamsi, J. Phys.: Conf. Ser., 2020, 1664, 012074 CrossRef CAS
- I. Bargaoui, N. Bitri and J. M. Menard, ACS Omega, 2022, 7, 11973–11979 CrossRef CAS PubMed
- C. Hao, T. Gao, A. Yuan and J. Xu, Chin. Chem. Lett., 2021, 32, 113–118 CrossRef CAS
- N. Maslekar, R. A. Mat Noor, R. P. Kuchel, Y. Yao, P. B. Zetterlund and V. Agarwal, Nanoscale Adv., 2020, 2, 4702–4712 RSC
- K. Ahmad, M. A. Shinde and H. Kim, Microchem. J., 2021, 169, 106583 CrossRef CAS
- M. Ebrahimi Naghani, M. Neghabi, M. Zadsar and H. Abbastabar Ahangar, Sci. Rep., 2023, 13, 1496 CrossRef CAS PubMed
- A. A. Alghyamah, S. Haider, U. Khalil, R. Khan, A. Haider, W. A. Almasry, R. Ihsan, P. Tahira, I. Wazeer and A. Chafidz, Curr. Appl. Phys., 2022, 40, 1–11, DOI:10.1016/j.cap.2020.03.002
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed
- V. Kumar, T. K. Mandal, A. Kumar, M. N. Alam, N. Parvin, S. W. Joo, D. J. Lee and S.-S. Park, eXPRESS Polym. Lett., 2022, 16, 978–995 CrossRef CAS
- N. N. Malinga and A. L. L. Jarvis, J. Nanostruct. Chem., 2020, 10, 55–68 CrossRef CAS
- C. Monteserín, M. Blanco, E. Aranzabe, A. Aranzabe, J. M. Laza, A. Larrañaga-Varga and J. L. Vilas, Polymers, 2017, 9, 449 CrossRef PubMed
- H. Sun, L. Ling, Z. Ren, S. A. Memon and F. Xing, Nanomaterials, 2020, 10, 113 CrossRef CAS PubMed
- S. Yan, P. He, D. Jia, Z. Yang, X. Duan, S. Wang and Y. Zhou, Ceram. Int., 2016, 42, 752–758 CrossRef CAS
- Y. Zhang, Q. Zhang, D. Hou and J. Zhang, Appl. Surf. Sci., 2020, 504, 144152 CrossRef
- M. V. Kiamahalleh, A. Gholampour, D. N. H. Tran, T. Ozbakkaloglu and D. Losic, Constr. Build. Mater., 2020, 250, 118832 CrossRef
- R. A. Soler-Crespo, W. Gao, P. Xiao, X. Wei, J. T. Paci, G. Henkelman and H. D. Espinosa, J. Phys. Chem. Lett., 2016, 7, 2702–2707 CrossRef CAS PubMed
- Y. Sun, X. Tang, H. Bao, Z. Yang and F. Ma, RSC Adv., 2020, 10, 29610–29617 RSC
- M. Tavakol, A. Montazeri, S. H. Aboutalebi and R. Asgari, Appl. Surf. Sci., 2020, 525, 146554 CrossRef CAS
- R. Chuah, S. C. B. Gopinath, P. Anbu, M. N. Salimi, A. R. W. Yaakub and T. Lakshmipriya, 3 Biotech, 2020, 10, 364 CrossRef PubMed
- H. Ma, X. Chen, S. Mohammed, Y. Hu, J. Lu, G. P. Simon, H. Hou and H. Wang, J. Mater. Chem. A, 2020, 8, 25951–25958 RSC
- A. Jirickova, O. Jankovsky, Z. Sofer and D. Sedmidubsky, Materials, 2022, 15, 920 CrossRef CAS PubMed
- G. Vinodhkumar, J. Wilson, S. S. R. Inbanathan, I. V. Potheher, M. Ashokkumar and A. C. Peter, Phys. B, 2020, 580, 411752 CrossRef CAS
- K. O. Olumurewa and M. A. Eleruja, Fullerenes, Nanotubes Carbon Nanostruct., 2022, 30, 913–922 CrossRef CAS
- E. Muchuweni and E. T. Mombeshora, Renew. Energy Focus, 2023, 45, 40–52 CrossRef
- E. T. Mombeshora, E. Muchuweni, M. L. Davies, B. S. Martincigh and V. O. Nyamori, New J. Chem., 2023, 47, 3502–3515 RSC
- A. Nisar, M. Saeed, M. Muneer, M. Usman and I. Khan, Environ. Sci. Pollut. Res., 2022, 29, 418–430 CrossRef CAS PubMed
- M. Ikram, A. Raza, M. Imran, A. Ul-Hamid, A. Shahbaz and S. Ali, Nanoscale Res. Lett., 2020, 15, 95 CrossRef CAS PubMed
- D. Lu, L. P. Ma, J. Zhong, J. Tong, Z. Liu, W. Ren and H. M. Cheng, ACS Nano, 2023, 17, 3587–3597 CrossRef CAS PubMed
- H. Sun, Q. Wang, T. Wu, Y. Miao and Y. Fang, Appl. Surf. Sci., 2020, 527, 146574 CrossRef CAS
- J. H. Ji, G. Lee and J. H. Koh, Sci. Rep., 2022, 12, 15583 CrossRef CAS PubMed
- L. G. P. Tienne, L. d. S. Candido, B. d. S. M. da Cruz, F. F. Gondim, M. P. Ribeiro, R. A. Simão, M. d. F. V. Marques and S. N. Monteiro, J. Mater. Res. Technol., 2022, 18, 4871–4893 CrossRef CAS
- S. Rai, R. Bhujel, M. Khadka, R. L. Chetry, B. P. Swain and J. Biswas, Mater. Today Chem., 2021, 20, 100472 CrossRef CAS
- Y. Yan, Y. Meng, H. Zhao, E. Lester, T. Wu and C. H. Pang, Bioresour. Technol., 2021, 331, 124934 CrossRef CAS PubMed
- E. T. Mombeshora, J. Mater. Sci.: Mater. Electron., 2023, 34, 521 CrossRef CAS
- Z. Lin, G. Waller, Y. Liu, M. Liu and C.-P. Wong, Adv. Energy Mater., 2012, 2, 884–888 CrossRef CAS
- F. Farivar, P. Lay Yap, R. U. Karunagaran and D. Losic, C, 2021, 7, 41 CAS
- K. Chaudhary, M. Aadil, S. Zulfiqar, S. Ullah, S. Haider, P. O. Agboola, M. F. Warsi and I. Shakir, Fullerenes, Nanotubes Carbon Nanostruct., 2021, 29, 915–928 CrossRef CAS
- F. Soysal, Z. Çıplak, B. Getiren, C. Gökalp and N. Yıldız, Mater. Res. Bull., 2020, 124, 110763 CrossRef CAS
- P. Tambe, Mater. Today: Proc., 2022, 49, 1294–1297 CAS
- J. Wu, H. Lin, D. J. Moss, K. P. Loh and B. Jia, Nat. Rev. Chem., 2023, 7, 162–183 CrossRef PubMed
- J. Wu, L. Jia, Y. Zhang, Y. Qu, B. Jia and D. J. Moss, Adv. Mater., 2021, 33, e2006415 CrossRef PubMed
- Z. Dehghani, F. Ostovari and S. Sharifi, Optik, 2023, 274, 170551 CrossRef CAS
- A. K. Gautam, M. Faraz and N. Khare, Ceram. Int., 2021, 47, 27885–27889 CrossRef CAS
- E. T. Mombeshora, E. Muchuweni, R. Garcia-Rodriguez, M. L. Davies, V. O. Nyamori and B. S. Martincigh, Nanoscale Adv., 2022, 4, 2057–2076 RSC
- R. Fatima, M. F. Warsi, M. I. Sarwar, I. Shakir, P. O. Agboola, M. F. Aly Aboud and S. Zulfiqar, Ceram. Int., 2021, 47, 7642–7652 CrossRef CAS
- D. Ranjan Rout and H. Mohan Jena, Sep. Purif. Technol., 2021, 278, 119630 CrossRef
- H. Shen, B. Jang, J. Park, H. J. Mun, H. B. Cho and Y. H. Choa, Nanomaterials, 2022, 12, 2009 CrossRef CAS PubMed
- A. M. El-Khawaga, H. Tantawy, M. A. Elsayed and A. I. A. Abd El-Mageed, Sci. Rep., 2022, 12, 17075 CrossRef CAS PubMed
- S. O. Akpotu, I. A. Lawal, P. N. Diagboya, F. M. Mtunzi and A. E. Ofomaja, ACS Omega, 2022, 7, 34054–34065 CrossRef CAS PubMed
- H. A. A. Jamjoum, K. Umar, R. Adnan, M. R. Razali and M. N. Mohamad Ibrahim, Front. Chem., 2021, 9, 752276 CrossRef CAS PubMed
- N. Buatong, C. Ruttanapun and C. Sriwong, J. Taiwan Inst. Chem. Eng., 2023, 142, 104667 CrossRef CAS
- H. Zhao, J. Wang, Z. Fan, Y. Xie, D. Zhang, S. Sun, Y. Zhang and H. Cao, Green Energy Environ., 2022, 7, 440–448 CrossRef CAS
- N. Keshmiri, P. Najmi, B. Ramezanzadeh, M. Ramezanzadeh and G. Bahlakeh, Prog. Org. Coat., 2021, 159, 106416 CrossRef CAS
- M. H. Tran, I. Booth, B. G. daFonseca, P. Berrang, J. E. Wulff and A. G. Brolo, ACS Omega, 2022, 7, 43548–43558 CrossRef CAS PubMed
- A. Umar, A. A. Ibrahim, H. Algadi, H. Albargi, M. A. Alsairi, Y. Wang and S. Akbar, Environ. Technol. Innovation, 2022, 25, 102066 CrossRef CAS
- C. J. Brown, T. Simon, C. Cilibrasi, P. J. Lynch, R. W. Harries, A. A. Graf, M. J. Large, S. P. Ogilvie, J. P. Salvage, A. B. Dalton, G. Giamas and A. A. K. King, J. Mater. Chem. B, 2022, 10, 373–383 RSC
- D. Verma, K. R. Ranjan, M. D. Mukherjee and P. R. Solanki, Biosens. Bioelectron.: X, 2022, 11, 100217 CAS
- R. Niranjan, S. Zafar, B. Lochab and R. Priyadarshini, Nanomaterials, 2022, 12, 191 CrossRef CAS PubMed
- H. Chen, M. Yang, J. Yue and G. Chen, Materials, 2022, 15, 8862 CrossRef CAS PubMed
- R. Santhosh Kumar, S. Ramakrishnan, S. Prabhakaran, A. R. Kim, D. R. Kumar, D. H. Kim and D. J. Yoo, J. Mater. Chem. A, 2022, 10, 1999–2011 RSC
- K. Singh, D. P. Barai, S. S. Chawhan, B. A. Bhanvase and V. K. Saharan, Mater. Today Commun., 2021, 26, 101986 CrossRef CAS
- L. Cheng, C. Liu, H. Wu, H. Zhao and L. Wang, J. Colloid Interface Sci., 2022, 606, 1572–1585 CrossRef CAS PubMed
- H. Sun, J. Li, W. Wang, Z. Wang and L. Pan, J. Colloid Interface Sci., 2021, 594, 35–46 CrossRef CAS PubMed
- X. Tang, J.-P. Raskin, N. Kryvutsa, S. Hermans, O. Slobodian, A. N. Nazarov and M. Debliquy, Sens. Actuators, B, 2020, 305, 127423 CrossRef CAS
- S. Kazemi, M. Pourmadadi, F. Yazdian and A. Ghadami, Int. J. Biol. Macromol., 2021, 186, 554–562 CrossRef CAS PubMed
- S. Joshi, R. Siddiqui, P. Sharma, R. Kumar, G. Verma and A. Saini, Sci. Rep., 2020, 10, 9441 CrossRef CAS PubMed
- W. Zhang, H. Xu, F. Xie, X. Ma, B. Niu, M. Chen, H. Zhang, Y. Zhang and D. Long, Nat. Commun., 2022, 13, 471 CrossRef CAS PubMed
- P. Salarizadeh, M. B. Askari and A. Di Bartolomeo, ACS Appl. Nano Mater., 2022, 5, 3361–3373 CrossRef CAS
- Y. Meng, Y. Liu, J. He, X. Sun, A. Palmieri, Y. Gu, X. Zheng, Y. Dang, X. Huang, W. Mustain and S. L. Suib, ACS Appl. Energy Mater., 2021, 4, 5424–5433 CrossRef CAS
- P. H. Wadekar, A. Ghosh, R. V. Khose, D. A. Pethsangave, S. Mitra and S. Some, Electrochim. Acta, 2020, 344, 136147 CrossRef CAS
- M. Lee and S. M. Paek, Nanomaterials, 2022, 12, 1507 CrossRef CAS PubMed
- R. Shu, Y. Wu, J. Zhang, Z. Wan and X. Li, Composites, Part B, 2020, 193, 108027 CrossRef CAS
- S. Y. Toh, K. S. Loh, S. K. Kamarudin and W. R. W. Daud, Chem. Eng. J., 2014, 251, 422–434 CrossRef CAS
- E. T. Mombeshora and V. O. Nyamori, J. Mater. Sci.: Mater. Electron., 2017, 28, 18715–18734 CrossRef CAS
- J. Regis, S. Vargas, A. Irigoyen, E. Bramasco-Rivera, J. L. Bañuelos, L. C. Delfin, A. Renteria, U. Martinez, T. Rockward and Y. Lin, Int. J. Smart Nano Mater., 2021, 12, 20–35 CrossRef
- A. K. Vivekanandan, V. Subash, S. M. Chen and S. H. Chen, Ultrason. Sonochem., 2020, 68, 105176 CrossRef CAS PubMed
- E. F. Aboelfetoh, A. H. Gemeay and R. G. El-Sharkawy, Environ. Monit. Assess., 2020, 192, 355 CrossRef CAS PubMed
- B. Sharma, S. Thakur, D. Trache, H. Yazdani Nezhad and V. K. Thakur, Nanomaterials, 2020, 10, 1616 CrossRef CAS PubMed
- K. Bansal, J. Singh and A. S. Dhaliwal, Fullerenes, Nanotubes Carbon Nanostruct., 2022, 31, 277–287 CrossRef
- A. M. Abdelfatah, N. El-Maghrabi, A. E. D. Mahmoud and M. Fawzy, Sci. Rep., 2022, 12, 19372 CrossRef CAS PubMed
- D. Perumal, E. L. Albert, N. Saad, T. Y. Y. Hin, R. M. Zawawi, H. F. Teh and C. A. Che Abdullah, Crystals, 2022, 12, 1539 CrossRef CAS
- H. C. Manjunatha, Y. S. Vidya, K. N. Sridhar, L. Seenappa, B. C. Reddy, S. Manjunatha, A. N. Santhosh, N. Dhananjaya, V. Thirunavukkarasu, B. Pavithra and P. S. D. Gupta, J. Sci.: Adv. Mater. Devices, 2022, 7, 100442 CAS
- M. F. Umar, F. Ahmad, H. Saeed, S. A. Usmani, M. Owais and M. Rafatullah, Nanomaterials, 2020, 10, 1096 CrossRef CAS PubMed
- W. Sun, Y. Hong, T. Li, H. Chu, J. Liu, L. Feng and M. Baghayeri, Chemosphere, 2023, 310, 136759 CrossRef CAS PubMed
- P. Parthipan, M. A. Al-Dosary, A. A. Al-Ghamdi and A. Subramania, J. King Saud Univ., Sci., 2021, 33, 101438 CrossRef
- P. A. Hoskeri, H. B. Muralidhara, C. R. Manjunatha and M. S. Raghu, et al., J. Electroanal. Chem., 2020, 858, 113830 CrossRef
- J. Wu, Z. Lin, X. Weng, G. Owens and Z. Chen, Chemosphere, 2020, 246, 125700 CrossRef CAS PubMed
- E. Vatandost, A. Ghorbani-HasanSaraei, F. Chekin, S. Naghizadeh Raeisi and S. A. Shahidi, Food Chem.: X, 2020, 6, 100085 CAS
- M. Buldu-Akturk, M. Toufani, A. Tufani and E. Erdem, Nanoscale, 2022, 14, 3269–3278 RSC
- S. R. Joshi, A. Sharma, G. H. Kim and J. Jang, Mater. Sci. Eng., C, 2020, 108, 110465 CrossRef CAS PubMed
- A. Ganiyat Olatoye, J. Zhang, Q. Wang, E. Cao, W. Li, E. Oluwaseyi Fagbohun and Y. Cui, Carbon Resour. Convers., 2022, 5, 222–230 CrossRef CAS
- G. S. Lekshmi, R. Tamilselvi, R. Geethalakshmi, S. D. Kirupha, O. Bazaka, I. Levchenko, K. Bazaka and M. Mandhakini, J. Colloid Interface Sci., 2022, 608, 294–305 CrossRef CAS PubMed
- A. Marinoiu, M. Andrulevicius, A. Tamuleviciene, T. Tamulevicius, M. Raceanu and M. Varlam, Appl. Surf. Sci., 2020, 504, 144511 CrossRef CAS
- E. T. Mombeshora, P. G. Ndungu, A. L. L. Jarvis and V. O. Nyamori, Mater. Chem. Phys., 2018, 213, 102–112 CrossRef CAS
- K. O. Sodeinde, S. O. Olusanya, O. S. Lawal, M. Sriariyanun and A. A. Adediran, Sci. Rep., 2022, 12, 17054 CrossRef CAS PubMed
- Y. M. Y. Albarqouni, S. P. Lee, G. A. M. Ali, A. S. Ethiraj, H. Algarni and K. F. Chong, J. Nanostruct. Chem., 2021, 12, 417–427 CrossRef
- A. E. D. Mahmoud, M. Hosny, N. El-Maghrabi and M. Fawzy, Sustainable Environ. Res., 2022, 32, 22 CrossRef CAS
- M. Handayani, B. I. Suwaji, G. Ihsantia Ning Asih, T. Kusumaningsih, Y. Kusumastuti, Rochmadi and I. Anshori, Nanocomposites, 2022, 8, 74–80 CrossRef CAS
- T. Naseem, A. Zain ul, M. Waseem, M. Hafeez, S. U. Din, S. Haq and R. Mahfoz ur, J. Inorg. Organomet. Polym. Mater., 2020, 30, 3907–3919 CrossRef CAS
- L. Lu, W. Guo, C. Chen, Q. Zhu, J. Ma, H. Wu, D. Yang, G. Yang, X. Sun and B. Han, Green Chem., 2020, 22, 6804–6808 RSC
- A. Mehdinia, S. Heydari and A. Jabbari, Mater. Chem. Phys., 2020, 239, 121964 CrossRef CAS
- J. Jagiello, A. Chlanda, M. Baran, M. Gwiazda and L. Lipinska, Nanomaterials, 2020, 10, 1846 CrossRef CAS PubMed
- J. Cifuentes, C. Munoz-Camargo and J. C. Cruz, Nanomaterials, 2022, 12, 2857 CrossRef CAS PubMed
- B. Avar and M. Panigrahi, Phys. Chem. Solid State, 2022, 23, 101–112 CrossRef
- B. Bashir, M. U. Khalid, M. Aadil, S. Zulfiqar, M. F. Warsi, P. O. Agboola and I. Shakir, Ceram. Int., 2021, 47, 3603–3613 CrossRef CAS
- M. Algethami, Mater. Res. Express, 2022, 9, 025001 CrossRef
- P. Das, A. B. Deoghare and S. R. Maity, Arabian J. Sci. Eng., 2020, 46, 5467–5475 CrossRef
- R. K. Singh, R. Kumar and D. P. Singh, RSC Adv., 2016, 6, 64993–65011 RSC
- M. Huang, Y. Wang, S. Ying, Z. Wu, W. Liu, D. Chen and C. Peng, Sensors, 2021, 21, 1958 CrossRef CAS PubMed
- J. R. do Nascimento, M. R. D'Oliveira, A. G. Veiga, C. A. Chagas and M. Schmal, ACS Omega, 2020, 5, 25568–25581 CrossRef CAS PubMed
- Z. Wu, F. Li, X. Li, Y. Yang, X. Huang and H. Li, Nanomaterials, 2022, 12, 1581 CrossRef CAS PubMed
- N. Belachew, M. H. Kahsay, A. Tadesse and K. Basavaiah, J. Environ. Chem. Eng., 2020, 8, 104106 CrossRef CAS
- P. García Lebière, A. Pérez del Pino, C. Logofatu and E. György, Appl. Surf. Sci., 2021, 563, 150234 CrossRef
- D. d. L. Alves, A. O. da Silva, S. N. Monteiro, R. P. Weber, F. J. H. T. V. Ramos and L. R. Menezes, J. Mater. Res. Technol., 2022, 20, 4241–4255 CrossRef CAS
- Y. Wang, G. Hu, Y. Cao, Z. Peng and K. Du, Mater. Chem. Phys., 2021, 265, 124523 CrossRef CAS
- M. Pinzón, O. Avilés-García, A. R. de la Osa, A. de Lucas-Consuegra, P. Sánchez and A. Romero, Sustainable Chem. Pharm., 2022, 25, 100615 CrossRef
- S. Roy Chowdhury and T. Maiyalagan, ACS Omega, 2022, 7, 19183–19192 CrossRef CAS PubMed
- S. S. Naik, S. J. Lee, T. Begildayeva, Y. Yu, H. Lee and M. Y. Choi, Environ. Pollut., 2020, 266, 115247 CrossRef CAS PubMed
- Z. Huo, H. Wu, Q. Song, Z. Zhou, T. Wang, J. Xie and H. Qu, Carbohydr. Polym., 2021, 256, 117575 CrossRef CAS PubMed
- P. Karthikeyan, S. S. D. Elanchezhiyan, H. A. T. Banu, M. Hasmath Farzana and C. M. Park, Chemosphere, 2021, 276, 130200 CrossRef CAS PubMed
- W. Peng, H. Chen, W. Wang, Y. Huang and G. Han, Curr. Appl. Phys., 2020, 20, 304–309 CrossRef
- A. Lim Teik Zheng, T. Phromsatit, S. Boonyuen and Y. Andou, FlatChem, 2020, 23, 100174 CrossRef CAS
- G. Bharath, N. Arora, A. Hai, F. Banat, D. Savariraj, H. Taher and R. V. Mangalaraja, Electrochim. Acta, 2020, 337, 135668 CrossRef CAS
- Z. Said, M. A. Abdelkareem, H. Rezk, A. M. Nassef and H. Z. Atwany, Powder Technol., 2020, 364, 795–809 CrossRef CAS
- H. Qian, J. Wang and L. Yan, J. Bioresour. Bioprod., 2020, 5, 204–210 CrossRef CAS
- R. Shu, J. Zhang, C. Guo, Y. Wu, Z. Wan, J. Shi, Y. Liu and M. Zheng, Chem. Eng. J., 2020, 384, 123266 CrossRef CAS
- Y. Wu, W. Zhao, Y. Qiang, Z. Chen, L. Wang, X. Gao and Z. Fang, Carbon, 2020, 159, 292–302 CrossRef CAS
- Y. Xie, Fullerenes, Nanotubes Carbon Nanostruct., 2021, 30, 619–625 CrossRef
- H. Nosrati, R. Sarraf-Mamoory, D. Q. S. Le, R. Zolfaghari Emameh, M. Canillas Perez and C. E. Bunger, Sci. Rep., 2020, 10, 8552 CrossRef CAS PubMed
- N. Li, R. Shu, J. Zhang and Y. Wu, J. Colloid Interface Sci., 2021, 596, 364–375 CrossRef CAS PubMed
- A. G. Ramu, A. Umar, A. A. Ibrahim, H. Algadi, Y. S. A. Ibrahim, Y. Wang, M. M. Hanafiah, P. Shanmugam and D. Choi, Environ. Res., 2021, 200, 111366 CrossRef CAS PubMed
- P. K. Kahriz, H. Mahdavi, A. Ehsani, A. A. Heidari and M. Bigdeloo, Electrochim. Acta, 2020, 354, 136736 CrossRef CAS
- R. Shu, C. Zhao, J. Zhang, W. Liu, X. Cao, Y. Li and S. Liu, J. Colloid Interface Sci., 2021, 585, 538–548 CrossRef CAS PubMed
- J. Ma, Y. Yamamoto, C. Su, S. Badhulika, C. Fukuhara and C. Y. Kong, Electrochim. Acta, 2021, 386, 138439 CrossRef CAS
- W. Geng, W. Li, L. Liu, J. Liu, L. Liu and X. Kong, Fuel, 2020, 259, 116267 CrossRef CAS
- M. Shahid, T. R. Katugampalage, M. Khalid, W. Ahmed, C. Kaewsaneha, P. Sreearunothai and P. Opaprakasit, Sci. Rep., 2022, 12, 19043 CrossRef CAS PubMed
- S. Kashyap, V. Kumar, S. Abraham, S. Umrao, S. Singh, A. Kamath, R. Ms, A. Srivastava and P. S. Saxena, Austin J. Biosens. Bioelectron., 2017, 3, 1026 Search PubMed
- P. S. Selvamani, J. J. Vijaya, L. J. Kennedy, B. Saravanakumar, N. C. S. Selvam and P. J. Sophia, J. Electroanal. Chem., 2021, 895, 115401 CrossRef CAS
- J. Zhao, J. Zhang, H. Yin, Y. Zhao, G. Xu, J. Yuan, X. Mo, J. Tang and F. Wang, Nanomaterials, 2022, 12, 1210 CrossRef CAS PubMed
- M. M. Atta, M. I. A. Abdel Maksoud, O. I. Sallam and A. S. Awed, J. Mater. Sci.: Mater. Electron., 2021, 32, 3688–3698 CrossRef CAS
- S. Kianfar, A. N. Golikand and B. ZareNezhad, J. Nanostruct. Chem., 2020, 11, 287–299 CrossRef
- R. Jakhar, J. E. Yap and R. Joshi, Carbon, 2020, 170, 277–293 CrossRef CAS
- E. Murray, S. Sayyar, B. C. Thompson, R. Gorkin III, D. L. Officer and G. G. Wallace, RSC Adv., 2015, 5, 45284–45290 RSC
- J. Kang, Y. Ko, J. P. Kim, J. Y. Kim, J. Kim, O. Kwon, K. C. Kim and D. W. Kim, Nat. Commun., 2023, 14, 901 CrossRef CAS PubMed
- V. V. Chaban and O. V. Prezhdo, Nanoscale, 2017, 9, 4024–4033 RSC
- B. B. Palei, T. Dash and S. K. Biswal, IOP Conf. Ser.: Mater. Sci. Eng., 2020, 872, 012158 CAS
- K. Kakaei and M. Rahnavardi, Renewable Energy, 2021, 163, 1277–1286 CrossRef CAS
- S. D. Bukkitgar, N. P. Shetti, K. R. Reddy, T. A. Saleh and T. M. Aminabhavi, FlatChem, 2020, 23, 100183 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2023 |