
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafine ZnCo2O4 QD-incorporated carbon nitride mediated peroxymonosulfate activation for norfloxacin oxidation: performance, mechanisms and pathways†
Tao Zeng *ab,
Sijia Jinb,
Zhiquan Jinb,
Shuqi Libc,
Rui Zoub,
Xiaole Zhangd,
Shuang Songb and
Min Liu*a
*ab,
Sijia Jinb,
Zhiquan Jinb,
Shuqi Libc,
Rui Zoub,
Xiaole Zhangd,
Shuang Songb and
Min Liu*a
aCollege of Architecture and Environment, Sichuan University, Sichuan 610065, China. E-mail: zengtao@zjut.edu.cn; liuminscu@163.com; Tel: +86-571-88320726
bKey Laboratory of Microbial Technology for Industrial Pollution Control of Zhejiang Province, College of Environment, Zhejiang University of Technology, Hangzhou, Zhejiang 310032, P. R. China
cHangzhou Vocational & Technical College, Ecology and Health Institute, Hangzhou, 310018, P. R. China
dCollege of Life Science, North China University of Science and Technology, Tangshan, Hebei 063000, China. E-mail: smilxin@126.com
First published on 9th May 2023
Abstract
Recently, peroxymonosulfate (PMS)-based advanced oxidation processes (AOPs) are being actively investigated as a potential technology for water decontamination and many efforts have been made to improve the activation efficiency of PMS. Herein, a 0D metal oxide quantum dot (QD)–2D ultrathin g-C3N4 nanosheet (ZnCo2O4/g-C3N4) hybrid was facilely fabricated through a one-pot hydrothermal process and used as an efficient PMS activator. Benefiting from the restricted growth effect of the g-C3N4 support, ultrafine ZnCo2O4 QDs (∼3–5 nm) are uniformly and stably anchored onto the surface. The ultrafine ZnCo2O4 possesses high specific surface areas and shortened mass/electron transport route so that the internal static electric field (Einternal) formed in the interface between p-type ZnCo2O4 and the n-type g-C3N4 semiconductor could speed up the electron transfer during the catalytic reaction. This thereby induces the high-efficiency PMS activation for rapid organic pollutant removal. As expected, the ZnCo2O4/g-C3N4 hybrid catalysts significantly outperformed individual ZnCo2O4 and g-C3N4 in catalytic oxidative degradation of norfloxacin (NOR) in the presence of PMS (95.3% removal of 20 mg L−1 of NOR in 120 min). Furthermore, the ZnCo2O4/g-C3N4-mediated PMS activation system was systematically studied in terms of the identification of reactive radicals, the impact of control factors, and the recyclability of the catalyst. The results of this study demonstrated the great potential of a built-in electric field-driven catalyst as a novel PMS activator for the remediation of contaminated water.
1. Introduction
Contamination of water with antibiotics has attracted increased attention due to the environmental and public health concerns.1 As synthetic broad-spectrum antibiotics, fluoroquinolones have been utilized more frequently to manage bacterial infections in human beings, livestock, and aquaculture. Norfloxacin (NOR) is one of the most commonly discovered fluoroquinolones in drinking water, surface water, and wastewater on a worldwide scale attributable to the poor capacity of traditional sewage treatment procedures to remove antibiotics.2 Although the concentration of NOR detection varies from ng L−1 to g L−1 in aquatic environments, the negative health consequences on aquatic biota at various trophic levels owing to the continuous input and bio-accumulation make it categorized as an emerging pseudo-persistent pollutant.3 Numerous techniques, such as adsorption,4 biodegradation,5 and ultrafiltration,6 have been examined for their NOR removal potential. Nevertheless, low removal efficiency and slow reaction rate impede their practical application. By contrast, catalytic technology7–12 is an essential high-tech and green environmental protection technology, providing immense economic and social benefits while being a crucial factor in the advancement of the chemical industry and society.Recently, sulfate radical (SO4˙−)-based advanced oxidation processes (AOPs) particularly have been taken into consideration as an efficient and promising technology in terms of complete decomposition of antibiotics in a short time by producing radicals. In comparison to hydroxyl radicals (˙OH, 1.8–2.7 V), SO4˙− has higher redox potential (2.5–3.1 V) and longer lifetime with more flexible pH requirement, thus allowing the effective reaction with the target organic pollutants.13,14 Anipsitakis and Dionysiou15 found that cobalt ions (Co2+) exhibited the highest ability to activate peroxymonosulfate (PMS) for SO4˙− generation and cobalt oxide (Co3O4) was effective in heterogeneously PMS activation. However, considering that cobalt is both toxic and somewhat expensive, efforts are underway to replace Co3O4 with more environmentally benign and inexpensive alternative metals.
ZnCo2O4, which is isostructural to Co3O4, is a typical spinel with the Co3+ occupying the octahedral sites and the Zn2+ filling the tetrahedral sites in the cubic spinel structure. The replacement of Co2+ by Zn2+ may not only facilitate the PMS activation but also suppress Co leaching due to the strong Zn–Co interactions, which renders ZnCo2O4 to be a promising candidate for PMS activation.16–18 While there is still a considerable problem to be solved for substantially improving the catalytic performance and the stability of current catalysts. For instance, ultrafine nanoscale metal oxides usually provide high specific surface areas as well as reduced mass/electron transfer pathway, which are highly beneficial for catalytic processes.19–21 But individual nanoparticles (NPs) unavoidably likely to aggregate during the operation attribute to the high surface energy, leading to the decrease of the catalytic activity.22 A manageable synthesis method for ultrafine nanoscale PMS activators is therefore urgently needed.
To alleviate the agglomeration of ultrafine ZnCo2O4 NPs, support effects can be imaginably utilized to improve their dispersion over the surface of catalyst. In recent years, a variety of catalyst supports, such as metal oxides,23 graphene,24 and metal–organic frameworks,25 have all been employed to stabilize NPs,26,27 which endows the resultant hybrids with increased catalytic activity for antibiotic degradation. In addition, many researchers have also been encouraged to study the synergistic effect between the metal nanoparticles and catalyst support.28–31 Among the majority of available supports, graphitic carbon nitride (g-C3N4), with inexpensive materials and high stability for simple preparation, has prominent advantages in embedding NPs.32–35 The high affinity of the organic moieties of g-C3N4 toward transition metal species contribute to achieving the strong coupling between g-C3N4 and ZnCo2O4 NPs. Furthermore, the difference in electronegativity between N and C atoms could cause a significantly positive charge on neighboring C, facilitating PMS adsorption. More importantly, by integrating p-type semiconductor ZnCo2O4 with n-type semiconductor g-C3N4, a p–n heterojunction interface could be formed because of their favorable band alignment, which leads to the creation of an internal electric field to accelerate the interfacial electron transfer.36
With these in mind, we herein report the construction of an effective built-in electric field-driven catalyst system based on multifunctional g-C3N4 supported ultrasmall ZnCo2O4 quantum dots (QDs) for enhanced degradation of NOR in the presence of PMS. Besides the merits mentioned above, the g-C3N4 support was also able to restrict the ZnCo2O4 NPs growth, thus allowing stabilization of the smaller NPs with higher chemical reactivity. The effects of catalyst dosage, NOC and PMS concentration, and pH value on NOR removal were evaluated in details. The reaction mechanism as well as the degradation pathway of NOR was also investigated. As a green oxidation process, the activation of PMS with this built-in electric field-driven ZnCo2O4/g-C3N4 catalyst is a valuable, promising, and viable method for antibiotics-contaminated wastewater treatment.
2. Material and methods (experimental section)
2.1 Materials
Target contaminant of norfloxacin (C16H18FN3O3 98% in purity) was purchased from J&K Chemical Ltd. Other chemicals used for synthesizing ZnCo2O4 and g-C3N4, such as cobalt acetate (Co(CH3COO)3·4H2O), dicyandiamide (C2H4N4), ammonium chloride (NH4Cl) and zinc acetate (Zn(CH3COO)2·2H2O), ammonium hydroxide (NH3·H2O, 25–28% NH3), concentrated hydrochloric acid (HCl, 36–38%, w/v), and anhydrous ethanol (C2H6O) were supplied by J&K Chemical Ltd. Potassium peroxymonosulfate (2KHSO5·KHSO4·K2SO4, PMS) was purchased from Sigma-Aldrich. Sodium hydroxide (NaOH), potassium iodide (KI), and tert-butyl alcohol (TBA) were obtained from Huadong Medicine Co., Ltd. No further purification was performed on any of the compounds.2.2 Synthesis of the catalysts
The sole ZnCo2O4 NPs without g-C3N4 support was synthesized utilizing the same approach as described above except for the addition of g-C3N4.
2.3 Catalyst characterization
The crystal phases of the as-prepared samples were recorded by X-ray diffractometry (XRD, X'Pert PRO MPD, PANalytical, Netherlands) using Cu Kα radiation (λ = 1.540562 Å) over a 2θ range of 10–80°. The morphology features and microstructures of the catalysts were characterized by field-emission scanning electron microscopy (FE-SEM, S-4800, Hitachi, Japan) and high-resolution transmission electron microscopy (HRTEM, FEI Tecnai G2 F30, FEI, Holland). X-ray photoelectron spectroscopy (XPS, PHI 5000C ESCA, PerkinElmer, USA) was applied to analyze the composition and the valence state of the specimens. The specific surface area was calculated using the N2 adsorption–desorption isotherms (BET, ASAP2010, Micromeritics, USA). Fourier transform infrared spectra were recorded on a spectrophotometer (FT-IR, VERTEX 70, Bruker, Germany) in KBr pellets to recognize the functional groups. Thermo-gravimetric analysis (TGA, DTG-60h, Shimadzu, Japan) was applied within a temperature range of 25–800 °C at a rate of 10 °C per minute under static N2 atmosphere to test the thermal stability of the samples. Atomic absorption spectrophotometer (AAS, Shimadzu, Japan) was used for detecting the ion-leaching of Zn, Co from the samples.2.4 Catalytic activity test and sample analysis
The catalytic activity tests were conducted in a shaking table with a rotation speed of 250 rpm at atmospheric pressure. In a typical run, the catalysts (0.2 g L−1) and PMS (0.15 mM) were added to a 0.06 mM NOR aqueous solution without light irradiation. At preset intervals, 1.0 mL of the reaction mixture was withdrawn, quenched with excess methanol and centrifuged. In a single factor effect experiment, we keep all other conditions constant while only altering one factor condition, such as catalyst concentration, PMS concentration, pollutant solution concentration, pH, and temperature to study the impact of the change on the degradation performance. The concentrations of NOR in the supernatant were analyzed by high performance liquid chromatography (HPLC, LC-1200, Agilent, USA) equipped with a C18 column (150 mm × 4.6 mm × 5 μm, Eclipse XDB-C18, Agilent, USA) and a UV detector at λ = 278 nm. The column temperature was held at 30 °C, while the mobile phase was formic acid–water–acetonitrile (0.002![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30:70) with a flow rate of 0.3 mL min−1 and a 10 μL injection volume.
:30:70) with a flow rate of 0.3 mL min−1 and a 10 μL injection volume.
The NOR intermediates/products during reaction process were quantitated on a Waters ACQUITY UPLC-MS/MS system with triple quadrupole mass spectrometer XEVO TQ MS (Waters, Milford, MA, USA) in positive ion mode controlled by Waters Mass Lynx software. For chromatographic separation, the ACQUITY UPLC® BEH C18 column (50 mm × 2.1 mm × 1.7 μm) was used when the column temperature is 35 °C. The mobile phase was consisted of acetonitrile (A) and 0.2% formic acid (B). The solvents were set at a flow rate of 0.2 mL min−1 in the gradient mode (0 min, 5% A; 5 min, 5% A; 10 min, 10% A; 15 min, 10% A; 18 min, 15% A; 30 min, 25% A; 35 min, 25% A; 45 min, 5% A; 50 min, 5% A). MS spectra were obtained through a full scan mode (m/z 50 to 500).
The samples were collected and washed by ethanol and deionized water several times after 2 hours of reactions, and then freeze-dried in a lyophilizer for reuse. The cyclic experiment followed the same procedure mentioned above.
3. Results and discussion
3.1 Catalyst characterization
The ZnCo2O4 QDs were formed and anchored onto the g-C3N4 surface by a facile one-pot hydrothermal process for producing the hybrid catalyst ZnCo2O4/g-C3N4. The morphology and structure of g-C3N4, ZnCo2O4 and ZnCo2O4/g-C3N4 were illustrated by SEM and TEM. As displayed in Fig. 1a, individual ZnCo2O4 NPs, with a diameter of ∼20 nm, aggregate severely because of the high surface energy.37 Pure g-C3N4 appears as layered structure with wrinkled planes, suggesting the thin and flexible feature of the nanosheets (Fig. 1b). For the ZnCo2O4/g-C3N4 composites (Fig. 1c), it can be observed that the g-C3N4 nanosheet was covered by ZnCo2O4 QDs and no unbounded nanoparticles was found. It is worth noting that the particle size of ZnCo2O4 (Fig. S1†) is significantly decreased to around 3–6 nm, as compared with the individual ones, which may be originated from the support effect of g-C3N4. The high-resolution TEM image (Fig. 1d) further confirms the good anchor-hold between ZnCo2O4 QDs and g-C3N4. The lattice spacing in the heterojunction is determined to be 0.234 nm, attributing to the (222) crystal plane of ZnCo2O4,38 which can also be verified by the strongest diffraction peak of the next X-ray diffraction results. However, the lattice fringe of g-C3N4 cannot be observed probably due to its weak crystallization. More specifically, the robust coupling of N groups toward transition Zn2+ and Co2+ ions not only stabilized the highly dispersed ZnCo2O4 NPs but also controled the nucleation to form uniform QDs. The SEM image of g-C3N4 (Fig. 1e) displayed a smooth flake appearance with an average diameter of 0.5–1.0 μm, while the ZnCo2O4/g-C3N4 sample presented a relatively rougher surface (Fig. 1f), which is basically consistent with the results in TEM. To gain insights into the elemental distribution of the ZnCo2O4/g-C3N4 hybrids, EDS-elemental mapping analysis (Fig. S2†) was conducted, and the results indicated the homogeneous distribution of C, N, O, Co, and Zn elements in the sheets. | ||
| Fig. 1 TEM images of (a) ZnCo2O4, (b) g-C3N4, (c) ZnCo2O4/g-C3N4; (d) HRTEM image of ZnCo2O4/g-C3N4; and SEM images of (e) g-C3N4 and (f) ZnCo2O4/g-C3N4. | ||
XRD technique was used to dissect the structural phase of the materials. In Fig. 2a, the peaks at 2θ of 13.0° and 27.5° are demonstrated to be characteristic of the g-C3N4 (100) and (002) reflection (JCPDS no. 87-1526).39 The pattern of individual ZnCo2O4 exhibits peaks at 2θ of 18.9°, 31.2°, 36.8°, 38.5°, 44.7°, 55.6°, 59.3°, 65.2°, 77.2°, and 78.3°, corresponding to the (111), (220), (311), (222), (400), (422), (511), (440), (533), and (622) planes of ZnCo2O4 (JCPDS no. 023-1390).18 Due to the abundant ZnCo2O4 QDs covering the g-C3N4 surface in the ZnCo2O4/g-C3N4 samples, all the original diffraction peaks of g-C3N4 except the sharpest peak at 26.5° disappeared. These observations further elucidate the successful formation of ZnCo2O4/g-C3N4 materials.
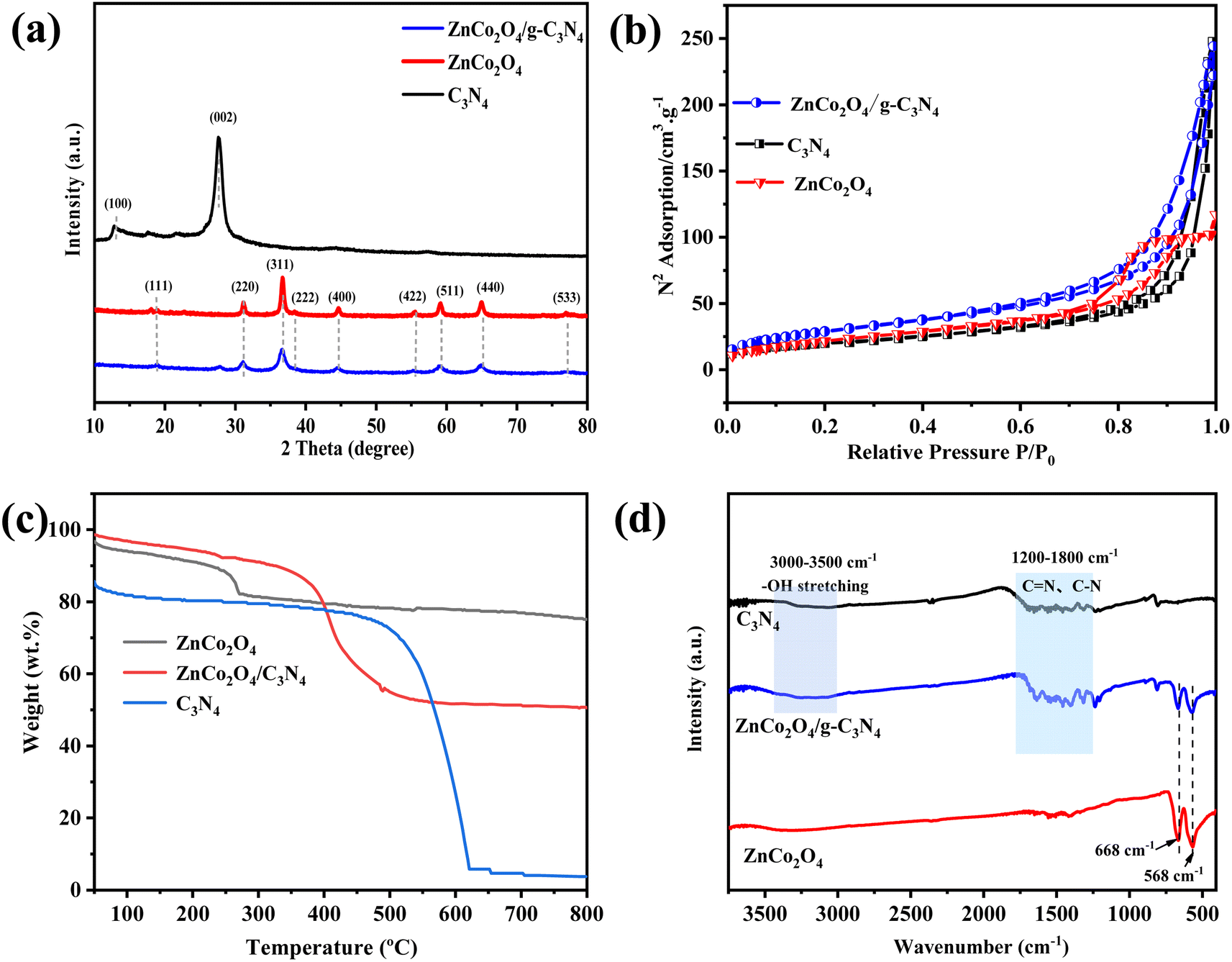 | ||
| Fig. 2 (a) XRD patterns. (b) N2 adsorption–desorption isotherms. (c) TGA, and (d) FTIR spectra of ZnCo2O4, g-C3N4 and ZnCo2O4/g-C3N4. | ||
The porous structure and surface area of the nanocomposites can be well investigated from its N2 adsorption–desorption isotherms. Fig. 2b shows that all the isotherms of g-C3N4, ZnCo2O4, and ZnCo2O4/g-C3N4 have the type IV curve with a type H3 hysteresis loop, suggesting the mesoporous structure of the catalysts. The BET specific surface area of ZnCo2O4/g-C3N4 is 92.18 m2 g−1, which is slightly higher than those of ZnCo2O4 (82.03 m2 g−1) and g-C3N4 (69.39 m2 g−1) counterparts. High surface area may endow the catalysts with more active sites and thus enhances the adsorption of reactant species. The thermal stability of the samples was examined via the TG technique. As demonstrated in Fig. 2c, adsorbed solvent molecules may have evaporated at temperatures below 100 °C, causing the first weight loss in all sample.40 In the curve of pure g-C3N4, the surface solvent loss is about 20% at first stage, followed by major weight loss between 500–650 °C reflecting the complete decomposition of g-C3N4.41 In contrast, thermal stability of pure ZnCo2O4 NPs ranges from 30 to 800 °C. Weight loss occurs at a lower temperature for ZnCo2O4/g-C3N4 than the other two materials due to the combustion of g-C3N4 under the catalysis of ZnCo2O4 on the surfaces. The final weight loss of ZnCo2O4/g-C3N4 at 800 °C reaches ∼50%, from which we conclude that the ZnCo2O4 to g-C3N4 weight ratio on the hybrid catalyst is approximate two.
Fig. 2d shows the FTIR spectra of the g-C3N4, ZnCo2O4/g-C3N4, and ZnCo2O4. The stretching vibration mode of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–N heterocycles is attributed to the broad band in the range of 1200–1800 cm−1 for pure g-C3N4. The bands at 3000–3500 cm−1 are related to the stretching vibration mode of N–H bond, and the –OH group from the surface adsorbed water.42 All of the g-C3N4 vibration bands appear in the ZnCo2O4/g-C3N4 samples, indicating that the g-C3N4 structure was preserved during the hybridization process. The peaks in the area from 525 to 725 cm−1 are originated from the typical bands of spinel metal oxide and the strong peaks at 568 cm−1 and 668 cm−1 appear in both pure ZnCo2O4 and ZnCo2O4/g-C3N4 hybrids.43
N and C–N heterocycles is attributed to the broad band in the range of 1200–1800 cm−1 for pure g-C3N4. The bands at 3000–3500 cm−1 are related to the stretching vibration mode of N–H bond, and the –OH group from the surface adsorbed water.42 All of the g-C3N4 vibration bands appear in the ZnCo2O4/g-C3N4 samples, indicating that the g-C3N4 structure was preserved during the hybridization process. The peaks in the area from 525 to 725 cm−1 are originated from the typical bands of spinel metal oxide and the strong peaks at 568 cm−1 and 668 cm−1 appear in both pure ZnCo2O4 and ZnCo2O4/g-C3N4 hybrids.43
The chemical states of the samples were investigated by XPS. According to the XPS observations (Fig. 3), C 1s and Zn 2p were found to have undergone no chemical change after loading ZnCo2O4 on g-C3N4. In the C 1s core level analysis, the peak at 284.6 eV was linked to the indefinite carbon adsorbed on the surface or the sp3 graphitic carbon generated during the polymerization, while the 288.0 eV binding energy belongs to sp2 hybrid carbon in N–CN.44 The signal at 398.4 eV in the N 1s core level analysis of g-C3N4 was attributed to the hybrid aromatic nitrogen atom (C–NC), while the peak of 400.2 eV denotes a tertiary nitrogen (N–(C)3 or C–NH–C) group. Besides, a broad peak of 404.2 eV was generated by π–π excitation between stacked interlayers. However, after ZnCo2O4 loaded, its chemical state changed to higher bonding energy, indicating that the chemical state of the hybrid aromatic N changed because of the ZnCo2O4 doping. Specifically, it was reported that the six lone pair electrons in g-C3N4 could occupy the free orbit of the center Co atoms to form Co–N, leading in reduced electron density and increased N atom binding energy.45
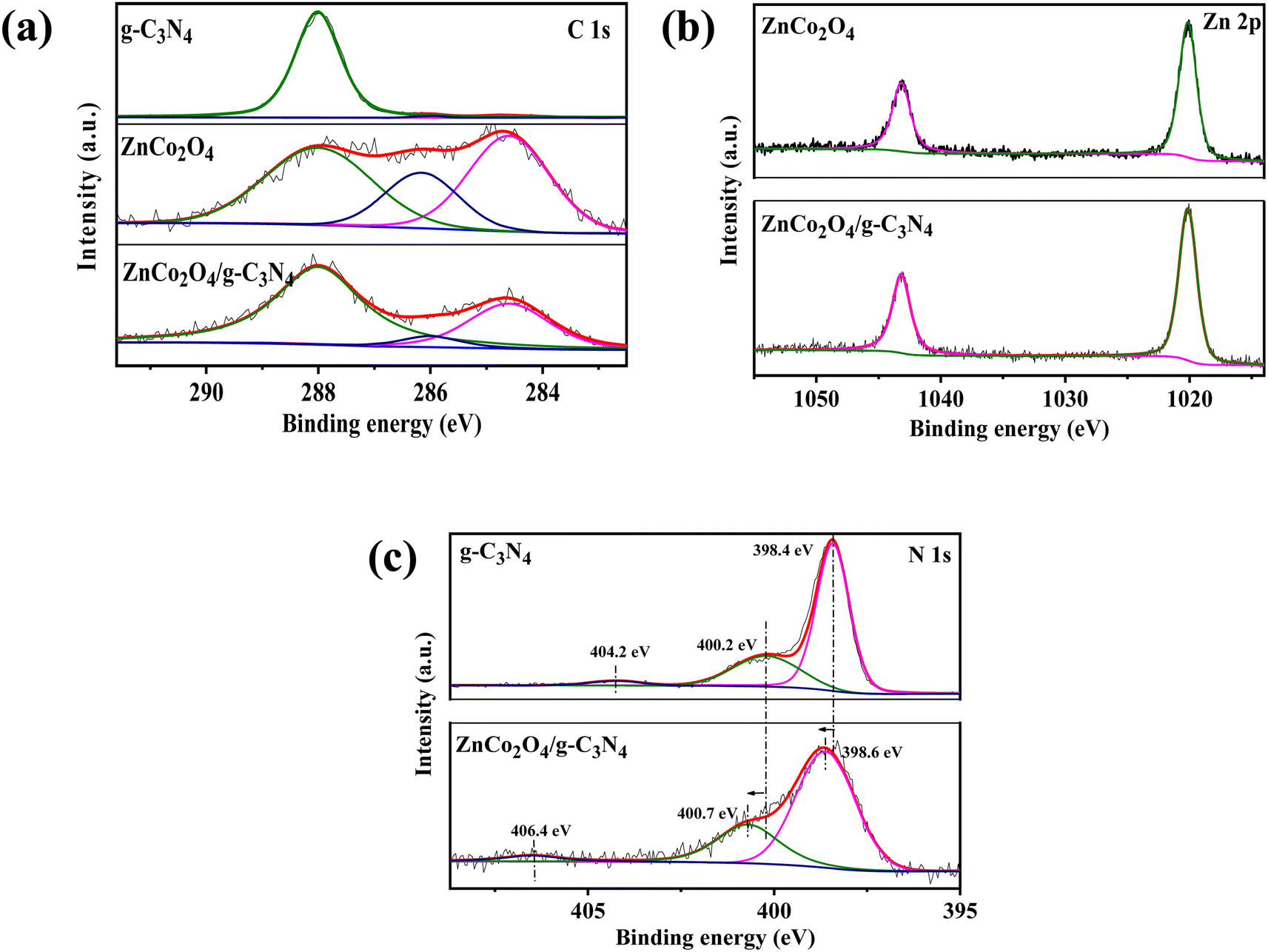 | ||
| Fig. 3 XPS spectra of (a) C 1s, (b) Zn 2p, and (c) N 1s core levels in several samples. | ||
3.2 Catalytic evaluation
The catalytic properties of g-C3N4, ZnCo2O4 and ZnCo2O4/g-C3N4 hybrids in aqueous solution were assessed by the degradation of NOR with the aid of PMS (Fig. 4). All the catalytic experiments were conducted without light irradiation, which thereby avoided the additional energy consumption. The results showed that NOR could be slowly degraded with only PMS and the addition of C3N4 could slightly accelerate the removal of NOR. However, the introduction of ZnCo2O4 can obviously increase the degradation performance of the catalytic oxidation systems. For example, ZnCo2O4 coupled with PMS could induce a NOR removal rate of 73.6% within 120 min, which is much higher than those achieved in the above two conditions. In the presence of PMS, ZnCo2O4 and g-C3N4 are physically combined in a 1:1 weight ratio, which results in similar removal efficiency of NOR to that of ZnCo2O4. The highest NOR removal rate (∼95%) in 120 min was observed for the ZnCo2O4/g-C3N4 coupled with PMS oxidation system, surpassing other counterparts. This performance can be attributed to the built-in electric field in the p–n heterojunction of ZnCo2O4/g-C3N4, in contrast to the simple physical mixture of ZnCo2O4 and g-C3N4.
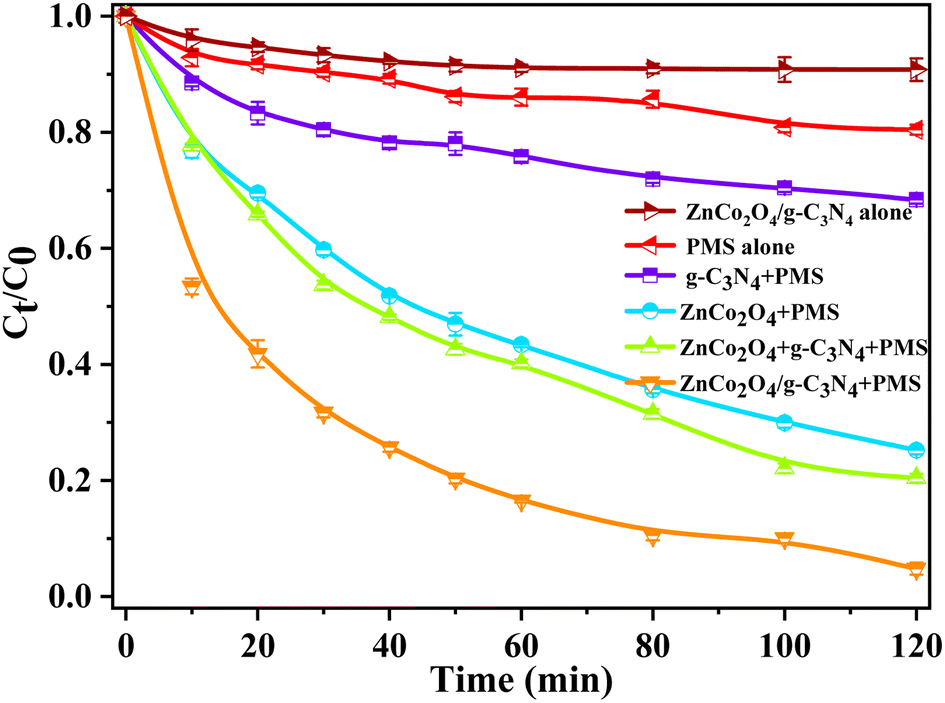 | ||
| Fig. 4 NOR degradation with different catalysts. (reaction conditions: catalyst = 0.2 g L−1, PMS dosage = 0.15 mM, initial NOR concentration = 0.06 mM, T = 25 °C, without pH adjustment). | ||
The effect of surface area on the catalytic performance of catalyst was also surveyed. As confirmed by the BET characterization, the surface area of ZnCo2O4/g-C3N4 catalyst is only somewhat higher than those of pure ZnCo2O4 and g-C3N4 and its specific activity calculated by surface area normalization is also only slightly higher than those of the other two (Table S1†). But the ZnCo2O4/g-C3N4 hybrids possess much higher catalytic activity than the other ones, which implies surface area is not the key factor affecting the catalytic activity. Therefore, the excellent catalytic performance may primarily be credited to the special combination of ZnCo2O4 and g-C3N4, which leads to not only the very stable and uniform dispersion of ZnCo2O4 QDs but also additional electronic transfer via intimate interfacial contact. These thereby enhanced the PMS activation by transition metal.
3.3 Optimization of NOR degradation
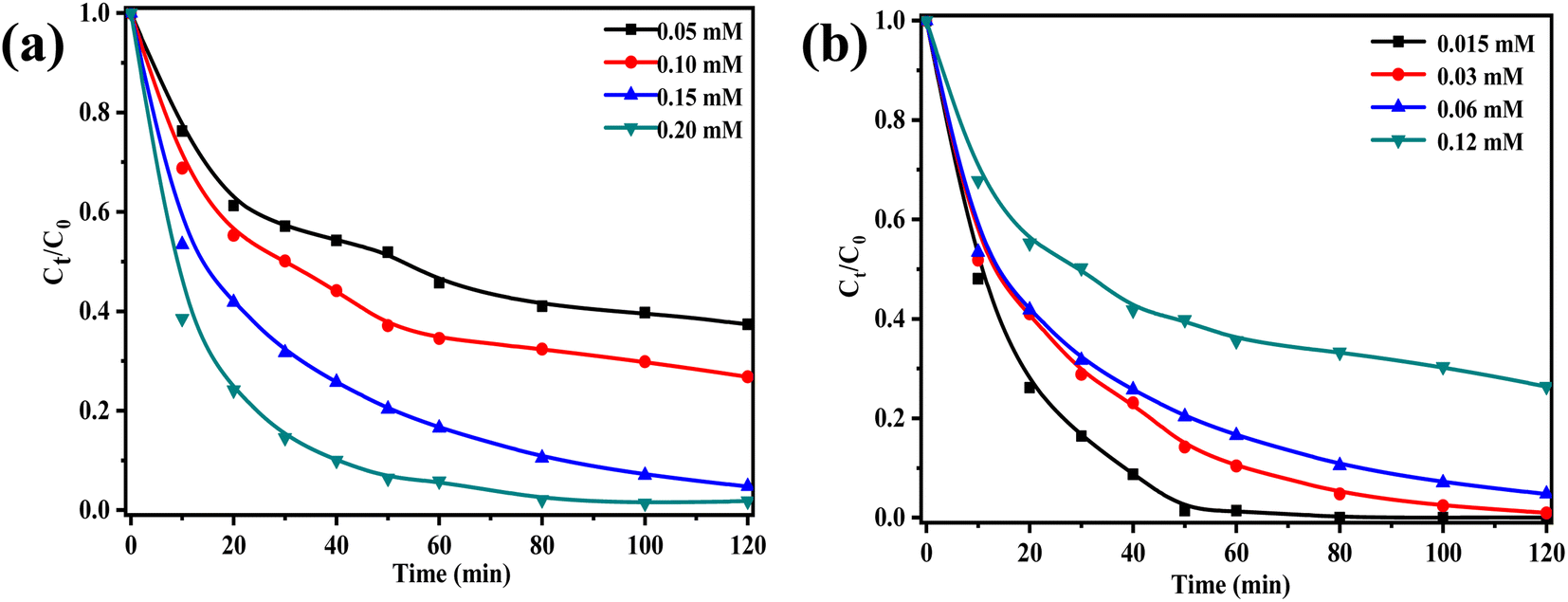 | ||
| Fig. 5 Comparison the performance of ZnCo2O4/g-C3N4 under different conditions: (a) PMS dosage, (b) initial NOR concentration. | ||
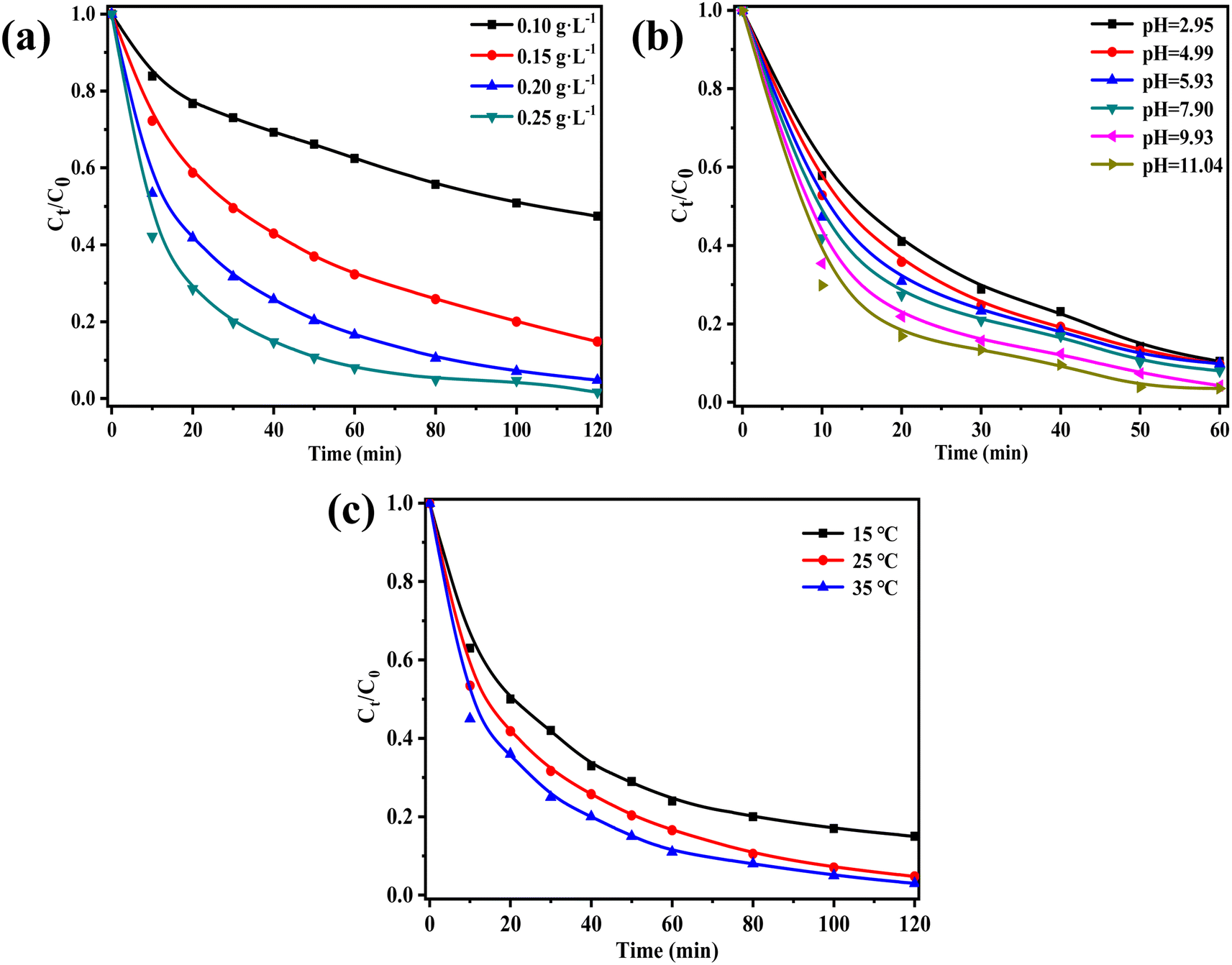 | ||
| Fig. 6 Comparison the performance of ZnCo2O4/g-C3N4 under different conditions: (a) catalyst dosage, (b) initial pH value, (c) temperature. | ||
The point of zero charge (pHpzc) of ZnCo2O4/g-C3N4 was determined to be 5.5 (Fig. S3†). At pH < 5.5, the catalyst surface becomes negatively charged, thus inhibiting the activation of PMS due to electrostatic repulsion between the catalyst and PMS. Conversely, at pH > 5.5 the catalyst becomes positively charged, allowing the adsorption of negatively charged PMS, which in turn results in ROS generation.47 Furthermore, acidic conditions such as H+ in the system forming hydrogen bonds with O–O in HSO5− can slightly inhibit the degradation effect, as it affects the activation of HSO5− into active free radicals, thus decreasing the degradation performance.
3.4 TOC reduction during reaction and reusability of the ZnCo2O4/g-C3N4 catalyst
Fig. 7a displays the TOC removal and NOR degradation efficiencies for various reaction periods. We found that NOR degradation of 95.3% were achieved after the peroxymonosulfate degradation and the mineralization degree reached 50% after 4 hours of reaction. It can be discovered that the strong oxidizing species (sulfate and hydroxyl radicals) destroyed the majority of the NOR molecules, but that the tiny organic intermediates generated during the catalytic process are not entirely degraded.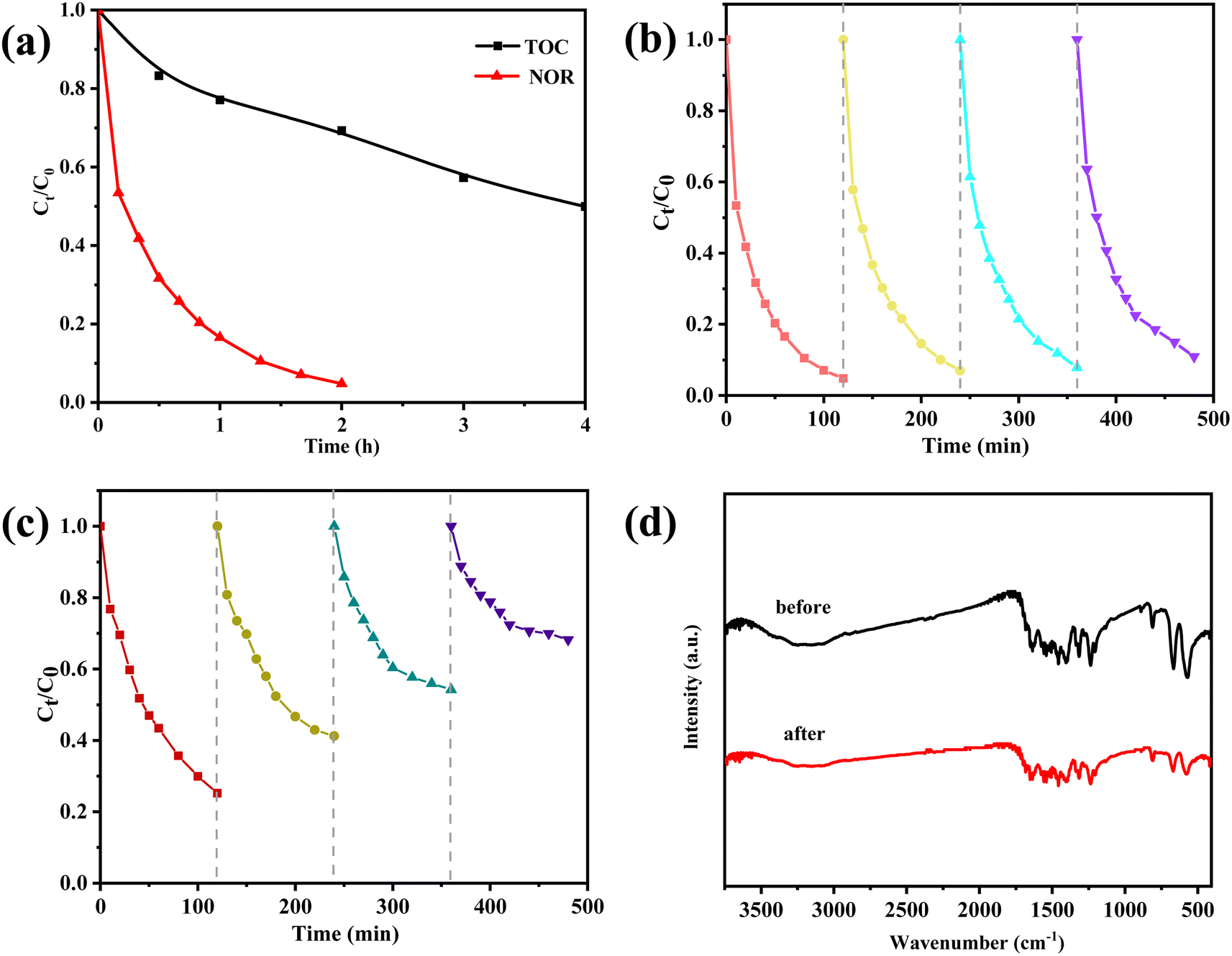 | ||
| Fig. 7 (a) TOC removal and NOR degradation during the reaction; recycle experiments of NOR removal for (b) ZnCo2O4/g-C3N4 and (c) ZnCo2O4 catalyst; (d) FTIR spectra of fresh and used ZnCo2O4/g-C3N4 catalyst. | ||
The recycled ZnCo2O4/g-C3N4 catalyst demonstrated a good degree of reusability for the degradation of NOR even after four consecutive cycles of testing (Fig. 7b), though its catalytic activity was lower than that of the freshly prepared catalyst. The ability of a catalyst to drive the reduction in activity may be affected by two factors: loss of the catalyst during the recycling process, and a decrease in the catalytic surface area due to the build-up of reaction products on the catalyst surface. In contrast, the ZnCo2O4 catalyst showed a marked reduction in NOR elimination rate from 75% to around 32% after four cycles (Fig. 7c). These findings imply that g-C3N4 support was able to enhance the durability of the ZnCo2O4 catalysts. Furthermore, the Zn and Co ions leaching concentration after reaction were detected. The Zn ion leaching in ZnCo2O4/g-C3N4 system is 0.14 mg L−1, which is lower than that of ZnCo2O4 system (0.55 mg L−1). Whereas, Co ion leaching was not significant, with 0.05 mg L−1 and 0.09 mg L−1 being measured in ZnCo2O4/g-C3N4 and ZnCo2O4, respectively. The results of the FTIR characterizations before and after use showed that ZnCo2O4/g-C3N4 has an outstanding structural durability, as there was almost no change in the results (Fig. 7d). This observation reflects the good stability of the ZnCo2O4/g-C3N4 catalyst for the activation of PMS, thus confirming its potential for practical applications.
3.5 Mechanism of PMS activation by ZnCo2O4/g-C3N4
In metal-activated PMS system, several types of radicals including SO4˙−, ˙OH, and SO5˙− are commonly generated. Both SO4˙− and ˙OH have the potential to attack organic compounds while SO5˙− cannot take part in the reaction due to its poor redox potential (1.1 V vs. NHE).50 To recognize the oxidizing radicals accountable for NOR removal in the ZnCo2O4/g-C3N4 + PMS system, different quenching agents were added to the system (tert-butyl alcohol (TBA) for ˙OH, and ethanol (EtOH) for both ˙OH and SO4˙−).51 As shown in Fig. 8a, addition of 500 mM of TBA could achieve obvious decrease of NOR removal (∼12%) within 120 min, demonstrating the generation of plenty of ˙OH in the catalytic oxidation. Moreover, the NOR removal decreased from ∼100% to 73% in 120 min as the same concentration of EtOH was added, which indicated the participation of SO4˙− into the reaction procedure. The presence of alcohols could only partially inhibit the degradation of NOR, possibly due to the fact that they were difficult to cling to the hydrophilic catalyst surface substantially to efficiently capture the surface-adsorbed radicals. Thus, KI, which may react with surface-bound radicals was utilized to another scavenger to further study the role of radicals for the oxidation degradation of NOR. On this occasion, adding 10 mM of KI significantly reduced the removal rate of NOR to 51%. Based on the above observations, we draw the conclusion that ˙OH and SO4˙− are the main reactive species in ZnCo2O4/g-C3N4 activated PMS process, which agrees with recent findings in the literature.52–54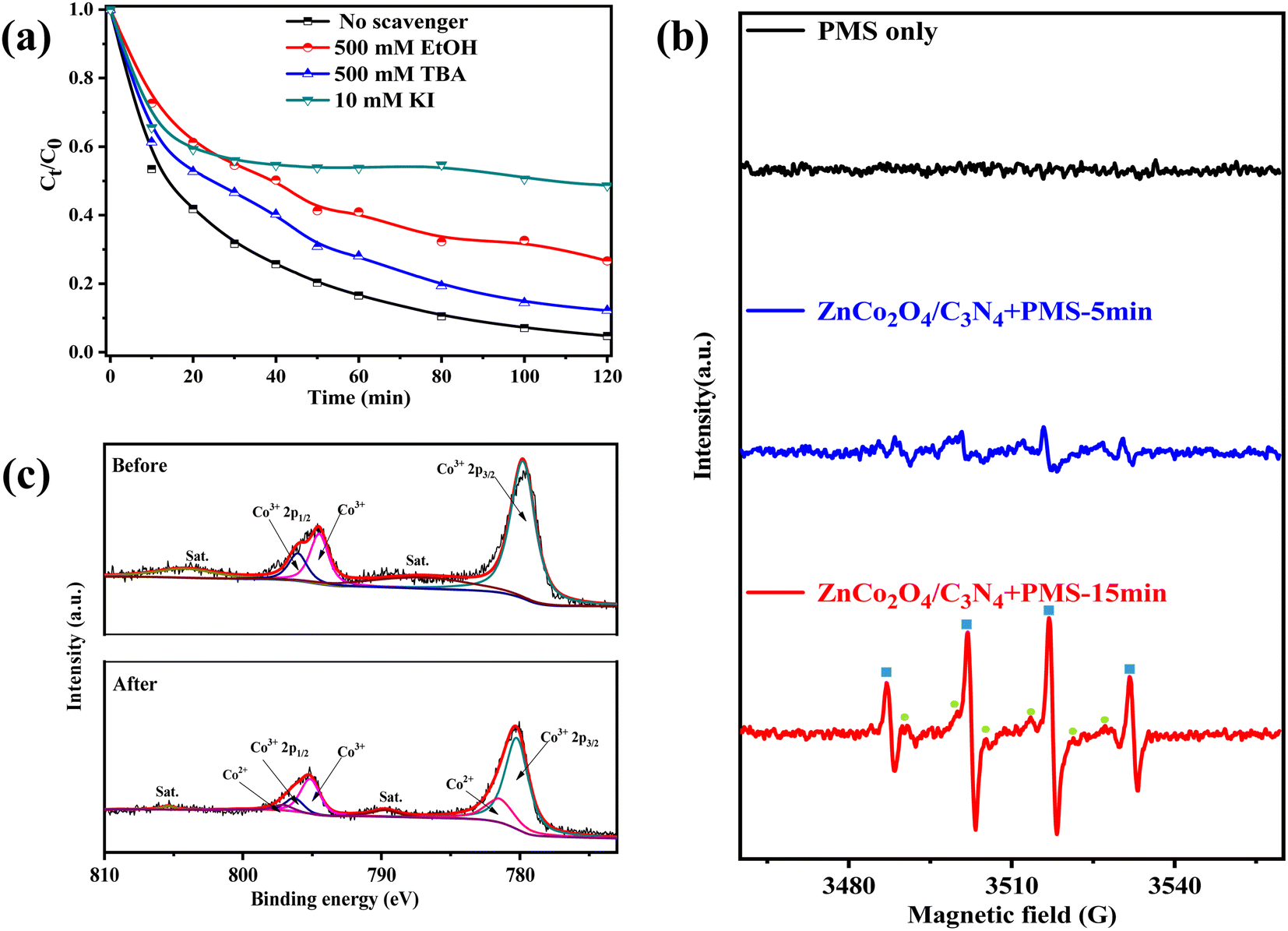 | ||
| Fig. 8 (a) The quenching experiments using EtOH, TBA, and KI. (b) DMPO–˙OH and DMPO–SO4˙− adduct in ESR spectra under different conditions. (c) Co 2p XPS spectrum of ZnCo2O4/g-C3N4 catalyst before and after reaction. | ||
Moreover, electron spin resonance (ESR) technique was employed to detect the active radicals in the ZnCo2O4/g-C3N4 + PMS reaction systems. As depicted in Fig. 8b, no DMPO–˙OH and DMPO–SO4˙− adduct was detected when PMS was present alone, indicating that these radicals could not be generated without the existence of catalysts. As expected, characteristic signals of DMPO–˙OH (quarter lines with peak strength of 1:2:2:1, refer to αN = αβ-H = 14.9 G) and DMPO–SO4˙− (refer to αN = 13.2 G, αβ-H = 9.6 G, αγ-H1 = 1.48 G, αγ-H2 = 0.78 G)55 adducts were observed in ZnCo2O4/g-C3N4 + PMS system. When reaction time was increased from 5 to 15 min, the peak signals of DMPO–˙OH and DMPO–SO4˙− adducts enhanced, indicating that the amount of reactive species increased during this period.
For the sake of understanding the catalytic reaction mechanism of the ZnCo2O4/g-C3N4 + PMS system, XPS was used to supervise the change in chemical state of the catalyst. Before reaction, XPS spectrum of Co 2p core-level shows typical peaks at Co respectively, and the peaks at Co 2p3/2 and Co 2p1/2 slight shift from 779.8 eV to 780.2 eV and from 796.0 eV to 796.3 eV,56 respectively, after reaction (Fig. 8c). Meanwhile, two new peaks with binding energy at 781.5 eV and 797.1 eV corresponding to CoII appeared, suggesting that the chemical states of some Co species on the catalyst surface were changed in the catalytic process. Three components at 397.4, 398.8 and 399.9 eV in the N 1s XPS spectrum were ascribed to the C–N–C, C–N(–C)–C, and −NHx, respectively (Fig. S4†). Indeed, the relative contribution of −NHx to the overall N intensity increased from 4% to 11% after the reaction, demonstrating that N sites may also participate in the reaction.
To comprehend the semiconducting characteristics of the catalysts, Mott–Schottky measurements were first conducted under low light conditions on FTO conductive glass. As shown in Fig. 9a, g-C3N4 displays a positive slope, which is consistent with its n-type semiconductor properties. By contrast, ZnCo2O4 exhibits a negative slope, consistent with the p-type semiconductor properties. As for ZnCo2O4/g-C3N4, a typical inverted “V-shape” plot which owns both negative and positive slopes is observed, further confirming the p–n heterojunction of the composite ZnCo2O4/g-C3N4 catalyst.
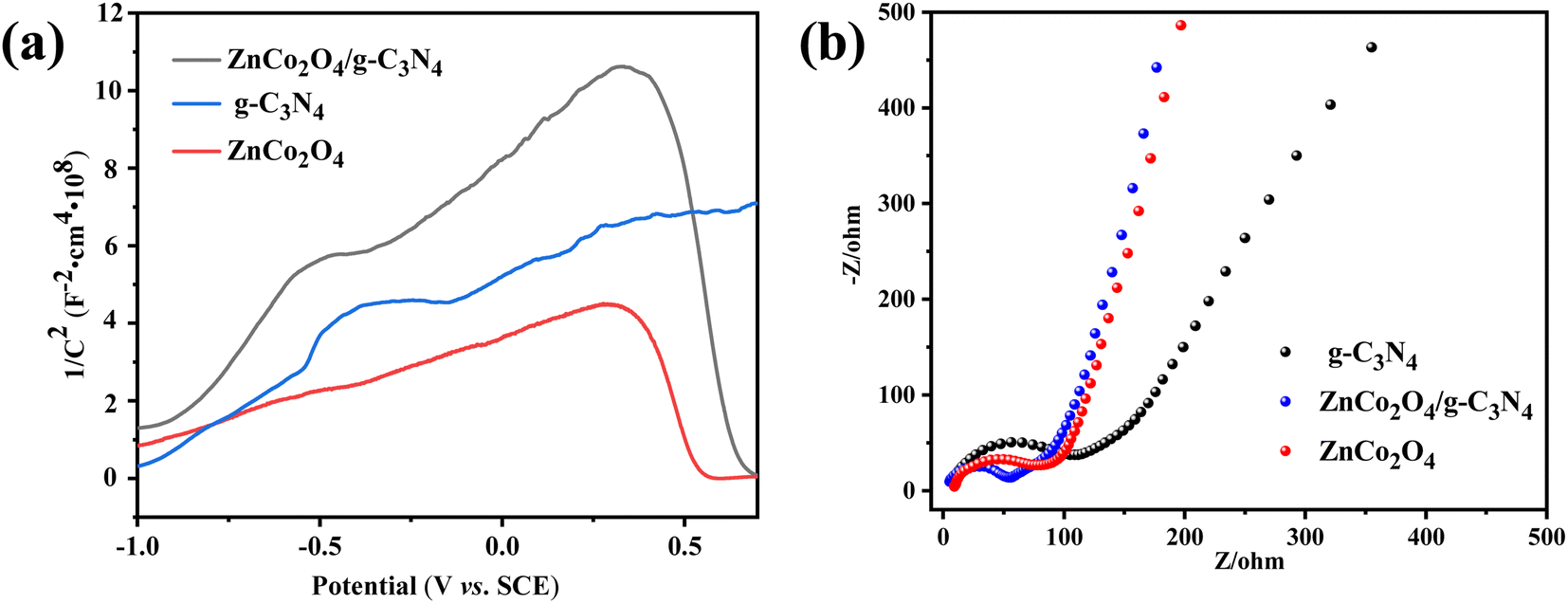 | ||
| Fig. 9 (a) Mott–Schottky and (b) electrochemical impedance spectroscopy of samples. | ||
The electrochemical impedance spectroscopy of the catalysts was also obtained under dark light conditions on the ITO conductive glass. Normally, smaller arc radii in Nyquist plots reflects stronger capacity transmit charges.57 As shown in Fig. 9b, the arc radii of ZnCo2O4/g-C3N4 is significantly smaller than those of pure ZnCo2O4 and g-C3N4, suggesting the faster interfacial charge transport. This can be originated from the existence of the internal electric field in the heterojunction interface, which provides more efficient electron transfer during the catalytic reaction.
All in all, a possible reaction mechanism for the degradation of NOR in ZnCo2O4/g-C3N4/PMS system was proposed from the above results as illustrated in Fig. 10. When a ZnCo2O4/g-C3N4 heterojunction is fabricated by tightly bounded p-type ZnCo2O4 semiconductor with n-type g-C3N4 semiconductor, ZnCo2O4 provides holes to g-C3N4 while g-C3N4 provides electrons to ZnCo2O4 in the interface to balance the Fermi level (Ef). Thus, an internal static electric field (Einternal) is fabricated at the interface with the electric field direction from g-C3N4 to ZnCo2O4. In the case of the catalytic process by ZnCo2O4/g-C3N4 hybrid, a multi-step reaction was involved for PMS activation and NOR degradation, as shown in eqn (1)–(6). According to XPS analysis, ≡CoII did not exist in ZnCo2O4/g-C3N4 and ZnCo2O4 catalysts before the reaction, but appeared after the reaction. Furthermore, the control experiment shows that pure g-C3N4 hardly exhibit catalytic activity toward the NOR oxidation with PMS and ZnCo2O4 alone also has a limit catalytic ability. Therefore, the initiation process of the degradation reaction is proposed to be the reaction of HSO5− with the variable-valent form ≡CoIII to yield ≡CoII and HSO5˙− in solution (eqn (1)). When ≡CoII loses electrons to form ≡CoIII, the e− will quickly pass through the p–n heterojunction to the g-C3N4 side due to the internal electric field (eqn (2)). Subsequently, the HSO5− reacts with e− on the side of g-C3N4 of the p–n junction interface to generate ˙OH or SO4˙− (eqn (3) and (4)). The organic molecules are then oxidized by both radicals to produce mineralization products in the last stage (eqn (5) and (6)).
| HSO5− + ≡CoIII → SO5˙− + ≡CoII + H+ | (1) |
| ≡CoII → ≡CoIII + e− | (2) |
| HSO5− + e− → SO42− + ˙OH | (3) |
| HSO5− + e− → SO4˙− + OH− | (4) |
| SO4˙− + NOR → intermediate → CO2 + H2O | (5) |
| ˙OH + NOR → intermediate → CO2 + H2O | (6) |
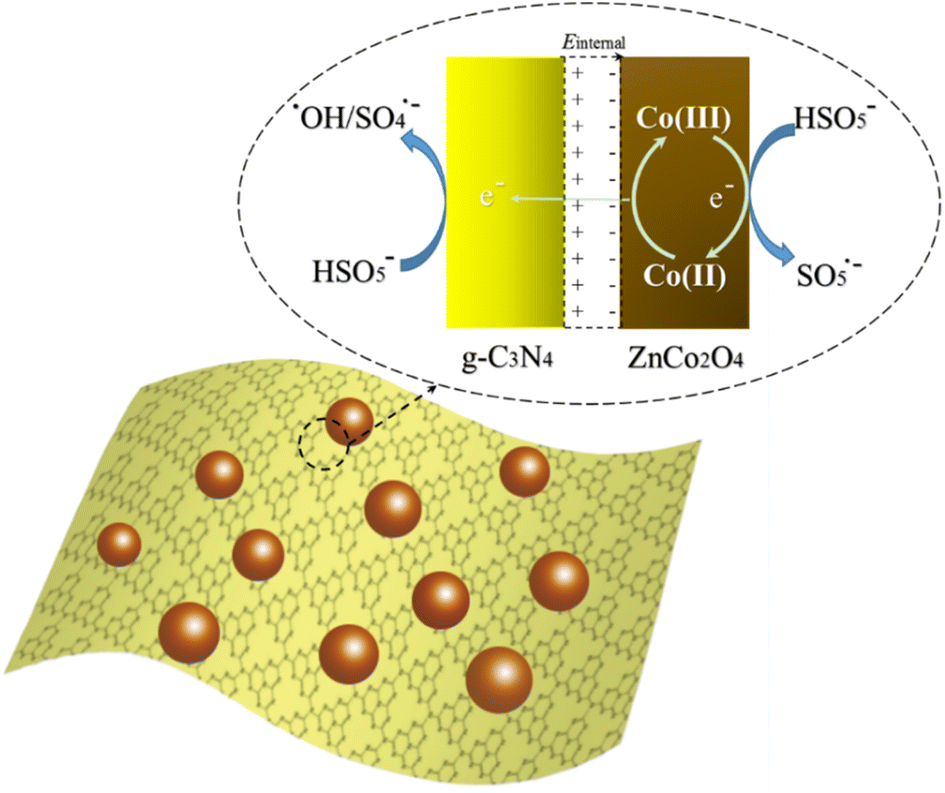 | ||
| Fig. 10 The ZnCo2O4/g-C3N4 p–n junction induced internal electric field facilitates the electron transfer during the activation of PMS. | ||
3.6 Identification of intermediates and transformation pathways
Based on the knowledge from prior literature and the fragmentation patterns acquired from liquid chromatography-mass spectrometry analysis (HPLC–QqQ–MS/MS), the structures of the intermediate products were tentatively determined.58–60 A total of 8 intermediates were detected during the degradation of NOR with ZnCo2O4/g-C3N4 + PMS system containing protonated ions ([M + H]+), as well as the proposed molecular structure and mass spectra were presented in Fig. S5.† For MS/MS analyses of these products, specific parameters are supplied in Table S2.†In reality, many of the oxidation processes, such as ozonation, photocatalysis, and thermally activated PMS, had already been identified in other AOPs. As previously indicated, when SO4˙− or ˙OH radical dots predominated the process, the degradation mechanisms were noticeably different. For example, the attack of NOR by SO4˙− would form NOR radical dots firstly through electron transfer reactions which subsequently produced (˙OH) NOR radicals by the hydroxyl abstraction or addition reaction when combined with H2O. While ˙OH would be likely to attack the carbon–carbon double bond next to the carboxylic acid group.61 When both the piperazinyl and quinolone moieties of NOR were attacked by either sulfate or hydroxyl radicals simultaneously, NOR would be degraded through multiple routes because of the concurrent oxidations of various reactive sites in the parent molecules. Despite of the complexity, prevalent degradation pathways were hypothesized based on the detected intermediates, in which defluorination, transformations of quinolone and piperazinyl groups simultaneously or successively, occurred. As shown in Fig. 11, there are three main pathways in this reaction.
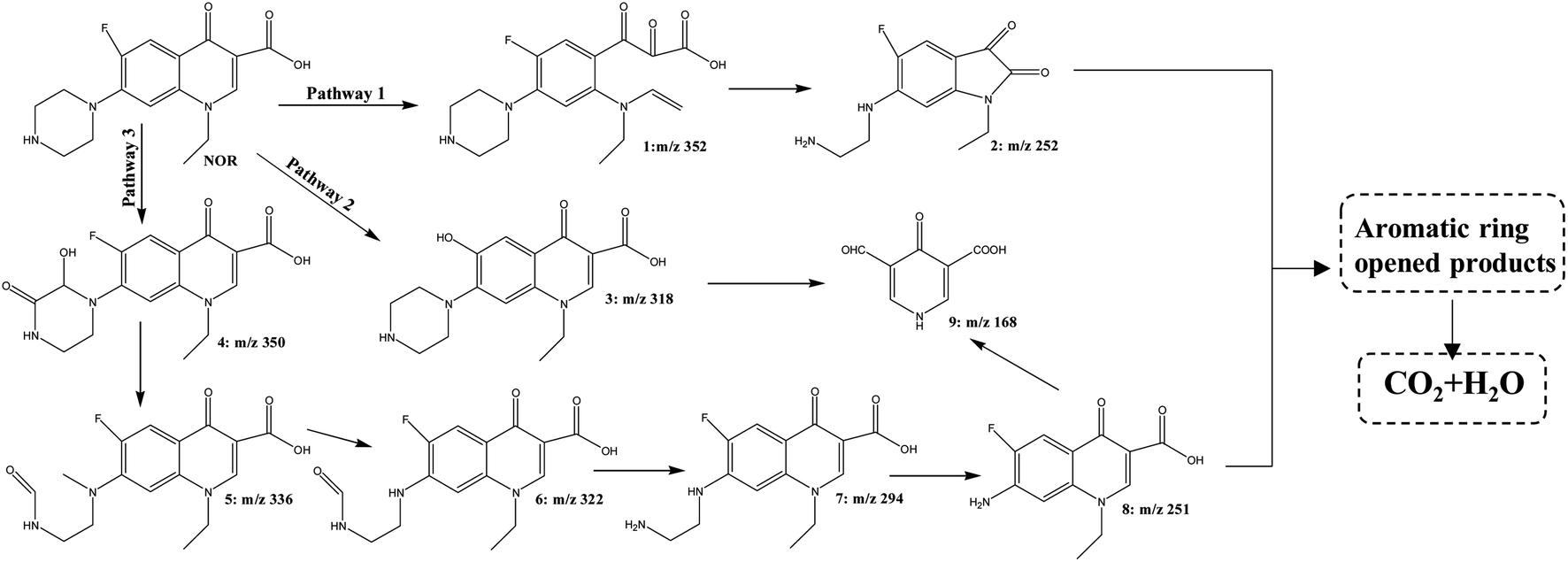 | ||
| Fig. 11 Proposed partial degradation pathway of NOR in ZnCo2O4/g-C3N4+PMS system. | ||
However, some products in this system were not detected probably owing to the weak response or the low concentration. Lack of relevant standards to determine the actual concentrations, ion counts associated with each individual degradation product were normalized according to the initial concentration of the parent antibiotics. This method was used to estimate the yields and the evolution of degradation products.
4. Conclusion
Plenty of ultrafine ZnCo2O4 QDs were anchored onto the g-C3N4 nanosheets tightly through in situ growth during a facile hydrothermal process to achieve ZnCo2O4/g-C3N4 p–n heterojunction which was employed to the PMS activation for NOR degradation. The degradation of NOR was significantly promoted due to the synergistic effect between ZnCo2O4 and g-C3N4 for PMS activation. The built-in electric field in the ZnCo2O4/g-C3N4 hybrid facilitated the electron transfer and the large specific surface area originated from the ultrafine ZnCo2O4 NPs also provided lots of catalytic activity sites and improved the diffusion of chemically active species. According to a free radical quenching experiments and ESR detection, the main oxidizing species in degradation reactions were SO4˙− and ˙OH. This proposed ZnCo2O4/g-C3N4 + PMS system can be used for the elimination of antibiotic pollution of water.Author contributions
Tao Zeng: writing – review & editing, funding acquisition. Sijia Jin: investigation, writing – original draft. Zhiquan Jin: data curation, formal analysis. Shuqi Li: methodology, formal analysis. Rui Zou: investigation. Xiaole Zhang: writing – review & editing, funding acquisition. Shuang Song: supervision. Min Liu: validation, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This study was supported by the Zhejiang Provincial Natural Science Foundation of China (LR21E080001), the National Natural Science Foundation of China (22276172 and 22076168), the Fundamental Research Funds for the Provincial Universities of Zhejiang (RF-B2022005), the Key Research and Development Plan of Zhejiang Province (2021C03176) and the Natural Science Foundation of Hebei Province of China (B2020209005).References
- Y. L. Luo, W. S. Guo, H. H. Ngo, L. D. Nghiem, F. I. Hai, J. Zhang, S. Liang and X. C. C. Wang, Sci. Total Environ., 2014, 473, 619–641 CrossRef PubMed.
- A. Wang, H. Wang, H. Deng, S. Wang, W. Shi, Z. Yi, R. Qiu and K. Yan, Appl. Catal., B, 2019, 248, 298–308 CrossRef CAS.
- C. Liu, V. Nanaboina, G. V. Korshin and W. Jiang, Water Res., 2012, 46, 5235–5246 CrossRef CAS PubMed.
- V. T. Nguyen, T. D. H. Vo, T. B. Nguyen, N. D. Dat, B. T. Huu, X. C. Nguyen, T. Tran, T. N. C. Le, T. G. H. Duong, M. H. Bui, C. D. Dong and X. T. Bui, Chemosphere, 2022, 288, 132577 CrossRef CAS PubMed.
- C. L. Amorim, A. S. Maia, R. B. R. Mesquita, A. O. S. S. Rangel, M. C. M. van Loosdrecht, M. E. Tiritan and P. M. L. Castro, Water Res., 2014, 50, 101–113 CrossRef CAS PubMed.
- H. Wu, X. Niu, J. Yang, C. Wang and M. Lu, Chem. Eng. J., 2016, 294, 410–416 CrossRef CAS.
- T. K. Das, S. Remanan, S. Ghosh and N. C. Das, J. Environ. Chem. Eng., 2021, 9, 104596 CrossRef CAS.
- T. K. Das and N. C. Das, Int. Nano Lett., 2022, 12, 223–242 CrossRef CAS.
- T. K. Das, S. Remanan, S. Ghosh, S. K. Ghosh and N. C. Das, Environ. Nanotechnol., Monit. Manage., 2021, 15, 100411 CAS.
- M. J. Hülsey, C. W. Lim and N. Yan, Chem. Sci., 2020, 11, 1456–1468 RSC.
- A. Moores and N. Yan, ACS Sustainable Chem. Eng., 2017, 5, 11124 CrossRef CAS.
- T. Wang, X. Cao and L. Jiao, Carb. Neutrality, 2022, 1, 21 CrossRef.
- W. D. Oh, Z. L. Dong and T. T. Lim, Appl. Catal., B, 2016, 194, 169–201 CrossRef CAS.
- Q. F. Wang, Y. S. Shao, N. Y. Gao, W. H. Chu, J. X. Chen, X. Lu, Y. P. Zhu and N. An, Sep. Purif. Technol., 2017, 189, 176–185 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- J. Bai, X. Li, G. Liu, Y. Qian and S. Xiong, Adv. Funct. Mater., 2014, 24, 3012–3020 CrossRef CAS.
- T. W. Kim, M. A. Woo, M. Regis and K. S. Choi, J. Phys. Chem. Lett., 2014, 5, 2370–2374 CrossRef CAS PubMed.
- Z. Q. Liu, H. Cheng, N. Li, T. Y. Ma and Y. Z. Su, Adv. Mater., 2016, 28, 3777–3784 CrossRef CAS PubMed.
- X. Li, L. Zhao, C. Shao, X. Li, W. Sun and Y. Liu, J. Colloid Interface Sci., 2018, 530, 345–352 CrossRef CAS PubMed.
- N. Liu, Z. Y. Duan, Q. Q. Zhang and J. Q. Guan, Chem. Eng. J., 2021, 419, 129567 CrossRef CAS.
- X. L. Zhang, N. Wang, L. L. Geng, J. N. Fu, H. Hu, D. S. Zhang, B. Y. Zhu, J. Carozza and H. X. Han, J. Colloid Interface Sci., 2018, 512, 844–852 CrossRef CAS PubMed.
- S. Shrestha, B. Wang and P. Dutta, Adv. Colloid Interface Sci., 2020, 279, 102162 CrossRef CAS PubMed.
- Y. P. Qiu, Q. Shi, W. Z. Wang, S. H. Xia, H. Dai, H. Yin, Z. Q. Yang and P. Wang, Small, 2022, 2106143, DOI:10.1002/smll.202106143.
- Z. P. Ma, C. Tsounis, C. Y. Toe, P. V. Kumar, B. Subhash, S. B. Xi, H. Y. Yang, S. J. Zhou, Z. H. Lin, K. H. Wu, R. J. Wong, L. Thomsen, N. M. Bedford, X. Y. Lu, Y. H. Ng, Z. J. Han and R. Amal, ACS Catal., 2022, 12, 4792–4805 CrossRef CAS.
- F. Z. Song, Q. L. Zhu, X. C. Yang, W. W. Zhan, P. Pachfule, N. Tsumori and Q. Xu, Adv. Energy Mater., 2018, 8, 1701416 CrossRef.
- W. J. Shao, C. He, M. Zhou, C. D. Yang, Y. Gao, S. Li, L. Ma, L. Qiu, C. Cheng and C. S. Zhao, J. Mater. Chem. A, 2020, 8, 3168–3179 RSC.
- F. Y. Xiao, H. Ren, H. S. Zhou, H. Z. Wang, N. Wang and D. W. Pan, ACS Appl. Nano Mater., 2019, 2, 5420–5429 CrossRef CAS.
- C. Wang, P. Shi, X. Cai, Q. Xu, X. Zhou, X. Zhou, D. Yang, J. Fan, Y. Min, H. Ge and W. Yao, J. Phys. Chem. C, 2016, 120, 336–344 CrossRef CAS.
- Y. Li, S. Ma, S. Xu, H. Fu, Z. Li, K. Li, K. Sheng, J. Du, X. Lu, X. Li and S. Liu, Chem. Eng. J., 2020, 387, 124094 CrossRef CAS.
- H. Li, C. Shan and B. Pan, Sci. Total Environ., 2019, 675, 62–72 CrossRef CAS PubMed.
- H. Liu, Z. He, J. Li and S. Zhao, Chem. Eng. J., 2023, 451, 138597 CrossRef CAS.
- N. Y. Cheng, J. Q. Tian, Q. Liu, C. J. Ge, A. H. Qusti, A. M. Asiri, A. O. Al-Youbi and X. P. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 6815–6819 CrossRef CAS PubMed.
- J. Q. Tian, Q. Liu, C. J. Ge, Z. C. Xing, A. M. Asiri, A. O. Al Youbi and X. P. Sun, Nanoscale, 2013, 5, 8921–8924 RSC.
- J. Y. Qin and H. P. Zeng, Appl. Catal., B, 2017, 209, 161–173 CrossRef CAS.
- C. Wan, L. Zhou, L. Sun, L. X. Xu, D. G. Cheng, F. Q. Chen, X. L. Zhan and Y. R. Yang, Chem. Eng. J., 2020, 396, 125229 CrossRef CAS.
- H. Wang, R. Niu, J. Liu, S. Guo, Y. Yang, Z. Liu and J. Li, Nano Res., 2022, 15, 6987–6998 CrossRef CAS.
- J. W. Liu, J. G. Jiang, Y. Meng, A. Aihemaiti, Y. W. Xu, H. L. Xiang, Y. C. Gao and X. J. Chen, J. Hazard. Mater., 2020, 388, 122026 CrossRef CAS PubMed.
- G. Huang, F. Zhang, X. Du, Y. Qin, D. Yin and L. Wang, ACS Nano, 2015, 9, 1592–1599 CrossRef CAS PubMed.
- Y. Li, K. Lv, W. Ho, F. Dong, X. Wu and Y. Xia, Appl. Catal., B, 2017, 202, 611–619 CrossRef CAS.
- X. Guo, L. Ding, K. Kanamori, K. Nakanishi and H. Yang, Microporous Mesoporous Mater., 2017, 245, 51–57 CrossRef CAS.
- Y. Su, P. Chen, F. Wang, Q. Zhang, T. Chen, Y. Wang, K. Yao, W. Lv and G. Liu, RSC Adv., 2017, 7, 34096–34103 RSC.
- X. Wang and Z. Nan, Sep. Purif. Technol., 2020, 233, 116023 CrossRef CAS.
- C. R. Mariappan, R. Kumar and G. Vijaya Prakash, RSC Adv., 2015, 5, 26843–26849 RSC.
- Z. Bao, M. Xing, Y. Zhou, J. Lv, D. Lei, Y. Zhang, J. Cai, J. Wang, Z. Sun, W. Chen, X. Gan, X. Yang, Q. Han, M. Zhang, J. Dai and Y. Wu, Adv. Sustainable Syst., 2021, 5, 2100087 CrossRef CAS.
- H. Li, C. Shan and B. Pan, Environ. Sci. Technol., 2018, 52, 2197–2205 CrossRef CAS PubMed.
- Y. Gao, Y. Rao, H. Ning, J. Chen, Q. Zeng, F. Tian and N. Gao, Sep. Purif. Technol., 2022, 297, 121555 CrossRef CAS.
- W. Zheng, Y. Sun and Y. Gu, J. Hazard. Mater., 2022, 436, 129058 CrossRef CAS PubMed.
- C. Wang, J. Kim, M. Kim, H. Lim, M. Zhang, J. You, J.-H. Yun, Y. Bando, J. Li and Y. Yamauchi, J. Mater. Chem. A, 2019, 7, 13743–13750 RSC.
- W.-C. Yun, K.-Y. A. Lin, W.-C. Tong, Y.-F. Lin and Y. Du, Chem. Eng. J., 2019, 373, 1329–1337 CrossRef CAS.
- W. D. Oh, Z. Dong and T. T. Lim, Appl. Catal., B, 2016, 194, 169–201 CrossRef CAS.
- C. Zhu, F. Zhu, D. D. Dionysiou, D. Zhou, G. Fang and J. Gao, Water Res., 2018, 139, 66–73 CrossRef PubMed.
- Z. Y. Li, Y. L. Liu, P. N. He, X. Zhang, L. Wang, H. T. Gu, H. C. Zhang and J. Ma, Chem. Eng. J., 2021, 418, 129464 CrossRef CAS.
- Z. Wang, W. Qiu, S. Pang, Y. Gao, Y. Zhou, Y. Cao and J. Jiang, Water Res., 2020, 172, 115504 CrossRef CAS PubMed.
- Z. Yang, Y. Li, X. Zhang, X. Cui, S. He, H. Liang and A. Ding, Chem. Eng. J., 2020, 384, 123319 CrossRef CAS.
- T. Liu, K. Wu, M. Wang, C. Jing, Y. Chen, S. Yang and P. Jin, Chemosphere, 2021, 262, 127845 CrossRef CAS PubMed.
- H. Y. Chen and P. C. Chen, Appl. Surf. Sci., 2020, 505, 144460 CrossRef CAS.
- X. Feng, P. Wang, J. Hou, J. Qian, C. Wang and Y. Ao, Chem. Eng. J., 2018, 352, 947–956 CrossRef CAS.
- Y. Fan, Y. R. Liu, X. Hu and Z. R. Sun, Chemosphere, 2021, 275, 130059 CrossRef CAS PubMed.
- L. W. Chen, X. Zuo, S. J. Yang, T. M. Cai and D. H. Ding, Chem. Eng. J., 2019, 359, 373–384 CrossRef CAS.
- B. M. Liu, W. B. Song, H. X. Wu, Z. Y. Liu, Y. Teng, Y. J. Sun, Y. H. Xu and H. L. Zheng, Chem. Eng. J., 2020, 398, 125498 CrossRef CAS.
- C. Liu, V. Nanaboina, G. V. Korshin and W. J. Jiang, Water Res., 2012, 46, 5235–5246 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02364h |
| This journal is © The Royal Society of Chemistry 2023 |