
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel salenCo(III) photoinitiators and their application for cycloaddition of carbon dioxide†
Daoqing Chen,
LongChao Du * and
Jie Yang
* and
Jie Yang
School of Chemistry and Chemical Engineering & the Key Laboratory of Environment-friendly Polymer Materials of Anhui Province, Key Laboratory of Structure and Functional Regulation of Hybrid Materials, Ministry of Education, Anhui University, Hefei, 230601, P. R. China. E-mail: dulongchao@sina.com
First published on 2nd June 2023
Abstract
Carbon dioxide (CO2) is a renewable carbon resource that can be effectively used in the production of polycarbonate (PPC) and cyclic carbonate (CPC) through open-loop copolymerization with epoxides and CO2. SalenCo(III) can successfully break the carbon–oxygen link between propylene oxide (PO) and CO2. On this basis, we prepared four different types of photosensitive salenCo(III) complexes and investigated their catalytic copolymerization of CO2 and PO. The results show that the catalytic performance of 1,2-cyclohexamediamine complexes is better than that of 1,2-o-phenylenediamine complexes. The catalytic efficiency of salenCo(III) catalyst increases with the expansion of the photosensitive conjugate system. In addition, the introduction of light can improve the catalytic efficiency. When we increased the power of the external light source from 100 W to 200 W, the TON of the catalyst [C4] increased by nearly 50%.
1 Introduction
Carbon dioxide is currently considered as the most important greenhouse gas, and at the same time also regarded as a component of C1 resources in today's world.1–4 How to convert carbon dioxide has been put on the research agenda. Since Inoue and his colleagues first reported that carbon dioxide and epoxide can be converted into polycarbonate (PPC) in 1969, rapid progress has been made in catalyzing the copolymerization of carbon dioxide and PO.5 PPC as one of the products has excellent properties, such as good tensile strength, low density, durability, good permeability, good heat resistance and insulation properties. In addition, it is easy to process and color. Therefore, PPC is usually used in electronics, optical media, glass and films, the automobile industry, the medical industry and many other consumer goods.6–9 As another product, cyclic carbonate (CPC) can be used as a solvent for degreasing, pickling and cleaning due to its low odor and toxicity, high boiling point and flash point.10–12 During the copolymerization of carbon dioxide with epoxy compounds, not only polycarbonate (PPC) and cyclic carbonate (CPC), but also some polyether (PPO) are produced because of the successive ring opening of epoxy compounds (Scheme 1).13–15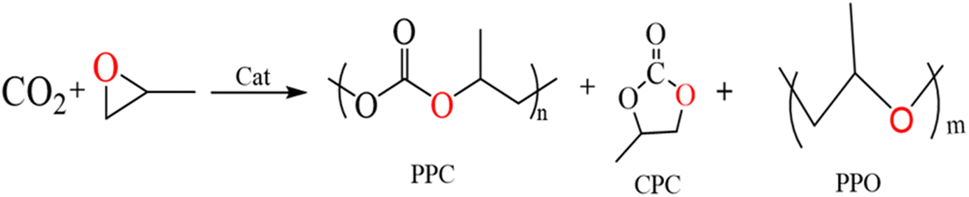 | ||
| Scheme 1 Copolymerization route: CO2 and PO copolymerization by catalyst. | ||
Among these copolymerization, various catalysts have been developed,16–19 in which homogeneous and heterogeneous metal complex catalysts are also included. For the heterogeneous catalytic systems, Sebastian Joby et al. prepared Co–Zn bimetallic cyanide (DMCs) catalysts in 2015, the catalyst has good catalytic activity and selectivity for carbon dioxide copolymerization with epoxide. The amount of base ions in the DMCs catalyst determines the activity of the copolymerization reaction and the selectivity for PPC.20 Zhang et al.21 prepared a nanolamellar [Zn–Co(III) DMCC] catalytic system in 2016. The carbon dioxide copolymerization with epoxide was carried out by free radical initiator. When the “Fe” metal in the center of the catalyst is replaced by “Co” metal, the catalytic activity was greatly improved. The lanthanum isoscorpionate complexes catalyze the copolymerization of carbon dioxide and epoxide with high selectivity for cyclic carbonate when the reaction conditions are milder.22 Among heterogeneous nonmetallic catalysts, the porous PILs catalyst prepared by Li Guoqing et al.23 can greatly promote the cycloaddition of carbon dioxide and epoxide to generate cyclocarbonate under relatively mild conditions. And the value of TOF will decrease with the extension of reaction time. Guo Zengjing et al.24 synthesized phenol hydroxyl imidazole salt from phenol and used it in the cycloaddition reaction of carbon dioxide. By adjusting the position of the hydroxyl group, they improve the anion's superior nucleophilic ability to leave, thus improving the catalytic activity of the catalyst. Wu Yue et al.25 synthesized a series of novel linear phenol hydroxyl functional imidazole dazolium-PILs to realize PIL species with ionic parts and surface group space satisfaction to achieve synergistic CO2 epoxide cycloaddition transformation. The catalyst has high yield and stable recovery. The catalyst is an efficient metal-free catalyst used to fix carbon dioxide.
Schiff base first appeared in 1864 and was synthesized by Hugo26 Schiff using salicylaldehyde, aniline and copper ions. This kind of compound with imine group was named Schiff base. Schiff bases themselves can be used as catalysts for cycloaddition reactions of carbon dioxide with epoxy compounds. Many metal complexes can improve the efficiency of the copolymerization of carbon dioxide with epoxides, for example, transition state metals such as Cr, Co and Mg.27–29 These catalysts has a very high selectivity for PPC. In addition, they can be used with water to produce efficiently polycarbonate polyols.30 Charlotte K. Williams et al.31 designed bimetallic heteronuclear catalysts in 2018. Comparison with related mononuclear analogues, among the heteronuclear complexes, Zn/Co catalysts has high catalytic efficiency, good stability and selectivity.
Cyclic carbonate (CPC) is a class of widely used compounds that can take the greenhouse gas carbon dioxide as a raw material, which can be used as a green solvent and a sustainable alternative to the toxic reactants currently used in the chemical industry.32 Therefore, CPC has great potential to become an important raw material for future chemical products. Dimethyl carbonate has proven to be an excellent oxidation additive for gasoline and jet fuel, and cyclic carbonate is the main raw material for the synthesis of dimethyl carbonate. Metal complex catalysts have very high selectivity for CPC in catalyzing the reaction of carbon dioxide with epoxides.33–35 In 2011, Dr Carl Young36 used the bimetallic aluminum complex [(salen)Al]2O or [(acen)Al]2O to catalyze the copolymerization of carbon dioxide and epoxides under very mild reaction conditions, and it promotes the formation of small molecular CPC by linking the individual carbonate chains first and last. This shows that metal (salen)–cobalt complexes exhibit significantly higher catalytic activity in the synthesis of CPC. In 2022, Lamia A. Siddig et al.37 prepared Bi-gallate as a powerful photocatalytic material that uses solar energy under mild conditions to form annular carbonates through cycloaddition of epoxides with carbon dioxide. Luping Zhang et al.38 synthesized a kind of photothermal catalyst containing organic ligand (TPA) of bimetallic oxides (Fe and Co). With the addition of TPA, the oxygen vacancy of bimetallic oxides (Fe and Co) could be increased, and the optic performance of the catalyst was bettered greatly. Under the condition of photothermal catalysis and high pressure, the catalyst can reply CO2 with PO to form annular carbonate.
In this paper, several kinds of salenCo(III) complex containing photosensitive groups s were designed and applied to the copolymerization of CO2 and PO. First, the obtained Schiff bases were complexed with cobalt acetate to form salenCo(II), and then reacted with azophenol or imidazolol to obtain salenCo(III) complex. Finally, the maximum absorption wavelength of salenCo(III) complex were measured, and the catalytic efficiency on the copolymerization was discussed.
2 Experimental process
2.1 Materials
1H NMR (400 MHz, chloroform-d) δ 14.10 (s, 2H), 8.63 (s, 2H), 7.37 (d, J = 7.8, 1.7 Hz, 2H), 7.35–7.29 (m, 2H), 7.23 (d, J = 6.9 Hz, 2H), 7.13–6.99 (m, 2H), 6.90–6.67 (m, 2H), 1.42 (s, 18H). IR(KBr) cm−1: 3075 (OH), 1615 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) (Fig. S2†).
O) (Fig. S2†).
1H NMR (400 MHz, DMSO-d6) δ 14.10 (s, 2H), 8.44 (s, 2H), 7.17 (d, J = 7.8, 1.7 Hz, 2H), 7.10 (d, 2H), 6.69 (t, J = 7.6 Hz, 2H), 5.10 (t, 2H), 1.88 (d, J = 12.8 Hz, 1H), 1.77 (d, J = 9.0 Hz, 1H), 1.68–1.42 (m, 2H), 1.28 (s, 18H). IR(KBr) cm−1: 3060 (OH), 1634 (CO) (Fig. S4†).
1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 2H), 7.51 (d, 2H), 7.45 (d, J = 14.2, 1.7 Hz, 2H), 7.39 (d, 1H), 7.31 (d, 2H), 7.25–7.10 (m, 2H), 6.93 (t, 2H), 6.73 (d, 2H), 6.53–6.37 (m, 2H), 1.47 (s, 18H). MS: m/z 644.22 for C37H34N4O3Co1, found 645.25.
1H NMR (400 MHz, DMSO-d6) δ 9.03 (d, J = 14.2 Hz, 2H), 8.77 (s, 2H), 8.41 (dt, J = 26.8, 5.1 Hz, 2H), 7.69 (d, J = 7.3 Hz, 2H), 7.64 (d, J = 6.9 Hz, 2H), 7.57 (dt, 2H), 7.53–7.42 (m, 2H), 7.37 (s, 1H), 6.96 (d, J = 9.3 Hz, 2H), 6.71 (t, J = 7.5 Hz, 2H), 2.55 (t, J = 7.7 Hz, 2H), 1.74 (t, J = 7.3 Hz, 2H), 1.55 (s, 19H), 0.80 (q, J = 7.5 Hz, 3H). MS: m/z 725.28 for C42H37N5O3Co1, found 725.96.
1H NMR (400 MHz, DMSO-d6) 8.16 (s, 2H), 7.51 (d, J = 8.6 Hz, 2H), 7.42 (d, J = 4.8 Hz, 1H), 7.25 (d, J = 6.7 Hz, 2H), 7.15 (s, 1H), 6.92 (t, J = 7.2 Hz, 2H), 6.49 (d, J = 7.5 Hz, 2H), 6.32 (ddd, J = 18.9, 14.1, 7.7 Hz, 2H), 2.92 (d, J = 12.8 Hz, 2H), 1.71(d, J = 24.5, 7.6 Hz, 4H), 1.56 (m, 4H), 1.41 (s, 9H), 1.33 (s, 9H). MS: m/z 650.27 for C37H40N4O3Co1, found 651.19.
1H NMR (400 MHz, DMSO-d6) δ 8.72 (d, J = 6.5 Hz, 2H), 8.24 (s, 2H), 8.06 (d, J = 1.9 Hz, 2H), 7.72 (d, J = 8.1 Hz, 2H), 7.43 (m, 2H), 7.34 (d, J = 7.5, 1.7 Hz, 1H), 6.92 (t, J = 7.2 Hz, 2H), 6.77 (d, J = 6.7 Hz, 1H), 6.60 (dt, J = 7.6 Hz, 1H), 2.79 (d, J = 11.1 Hz, 2H), 2.24 (d, J = 7.5 Hz, 2H), 1.87 (dt, J = 11.1 Hz, 2H), 1.79 (d, J = 7.6 Hz, 2H), 1.73–1.55 (m, 4H), 1.55 (s, 9H), 1.40 (s, 9H), 0.89 (t, J = 7.3 Hz, 2H), 0.80 (t, 3H). MS: m/z 731.32 for C42H43N5O3Co1, found 731.73.
2.2 Structural analysis
By comparing the FT-IR spectra of compound 1 and catalyst [C1], we found that only the –OH peak at 3075 cm−1 disappeared in the FT-IR spectra of catalyst [C1], while the peaks at other positions still existed. In addition, the infrared spectrum changes of other catalysts are the same. From this, we can infer that the ‘Co’ atom is reacting with the ‘O’ atom.Because our four catalysts containing photosensitive groups have similar structures (as shown in Fig. 1), we took out the catalyst [C3] and analyzed its structure with XPS.39 As shown in Fig. 2, in the full spectrum scan of X-ray photoelectron spectroscopy (XPS) of catalyst [C3], we can see the presence of Co, C, N, and O elements respectively, and the analysis of the Co spectral peak of elements in Fig. 8(c) shows that, the peaks of Co–N and Co–O at 796.08 eV and 780.68 eV, respectively, indicate that Co is combined with N and O in the catalyst. The spectral peak analysis of element N in Fig. 8(d) shows that the peaks of N-(C3) and C–NC at 401.18 eV and 399.28 eV respectively. From this, we can infer that the prepared product is [C3] (Table 1).
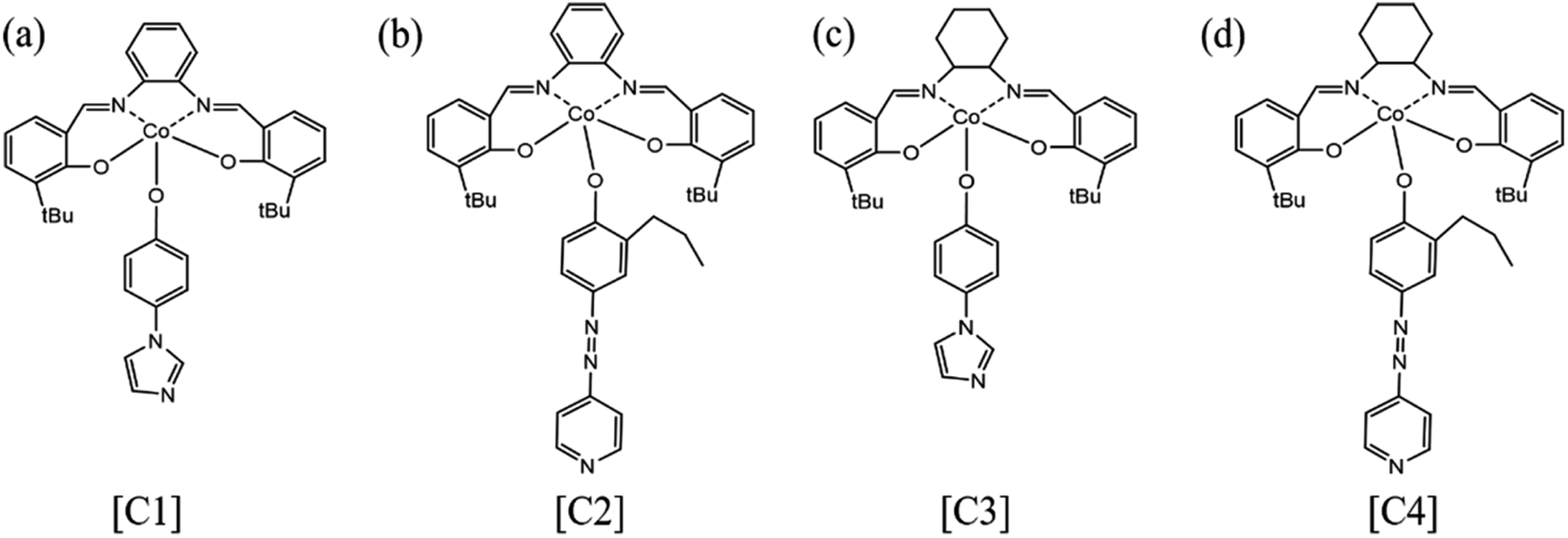 | ||
| Fig. 1 Catalyst structure formula (a) [C1]; (b) [C2]; (c) [C3]; (d) [C4]. | ||
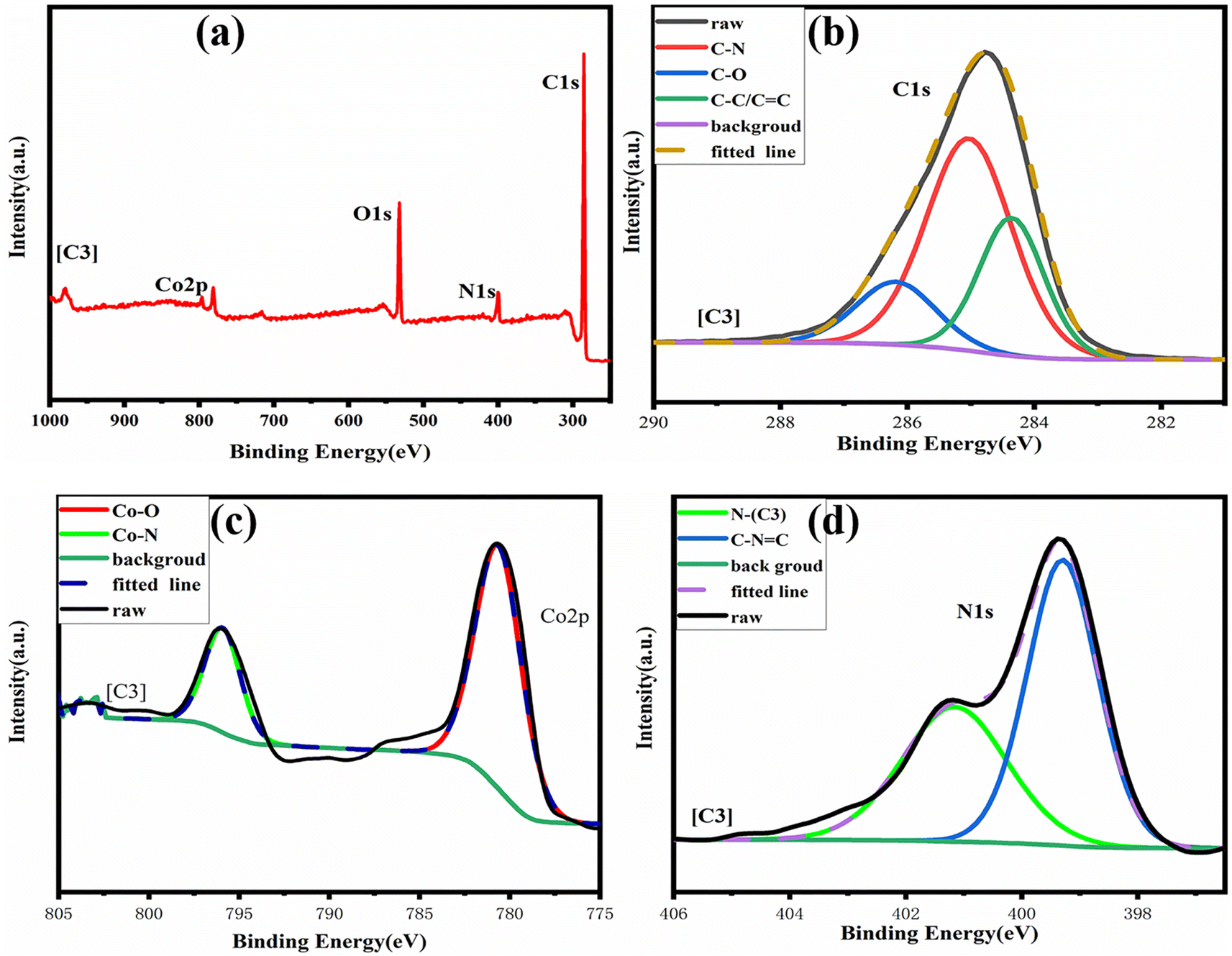 | ||
| Fig. 2 (a) XPS spectra of [C3] catalyst; (b–d) XPS narrow spectrum of characteristic elements. | ||
| Type (catalyst) | Synthetic route | Name | |
|---|---|---|---|
| Cat1 | SalenCo(III)-1 | Scheme S1 | [C1] |
| Cat2 | SalenCo(III)-2 | Scheme S1 | [C2] |
| Cat3 | SalenCo(III)-3 | Scheme S2 | [C3] |
| Cat4 | SalenCo(III)-4 | Scheme S2 | [C4] |
2.3 CO2 and PO copolymerization
The autoclave was dried in vacuum at 60 °C for 12 hours and then cooled naturally to room temperature. Add the catalyst and PO to the autoclave and bring the autoclave to the set temperature, and then CO2 was passed through to the set pressure. When the reaction reaches the set time, the copolymerization was stopped, and the autoclave was cooled to room temperature naturally to discharge CO2 slowly. The copolymerization products in the reactor were collected by syringe. For shading catalytic copolymerization, the quartz glass of the autoclave was covered with an opaque black cloth. In the experiment of photopolymerization, an external light source was introduced and an incandescent lamp was used for illumination. The distance between the light source and the reactor quartz glass is 20 cm. Light of other wavelengths is filtered by the filter to obtain the required wavelength (see Table 2). Each filter has a filter range (±18 nm; transmittance > 90%, Fig. S14–S18†).| Entry | Maximum UV visible light absorption wavelength (nm) |
|---|---|
| Compound 1 | 340 |
| Compound 2 | 325 |
| 4-(Imidazole-1-yl)phenol | 240 |
| 2-Propyl-4-(4-pyridylazo)phenol | 360 |
| Compound 1 + Co2+ | 380 |
| Compound 2 + Co2+ | 356 |
| [C1] | 474 |
| [C2] | 553 |
| [C3] | 410 |
| [C4] | 542 |
3 Results and analysis
3.1 Synthesis & characterization
Compound 1 was synthesized from o-phenylenediamine (OPD) and 3-tert-butyl-2-hydroxybenzaldehyde. Firstly, o-phenylenediamine was added to the three-necked flask, followed by the addition of an appropriate amount of anhydrous ethanol to the three-necked flask and the passage of nitrogen to remove the air inside, and the reaction lasted for 36 hours. Then, the reactants were filtered and washed to obtain a yellow solid and dried under vacuum, and the compound 1 was obtained. Finally, compound 1 was dissolved by dichloromethane and added to a three-necked flask. Meanwhile, the ethanol solution of anhydrous cobalt acetate was added to the three-necked flask using a constant pressure funnel. When the above substances were added, a large amount of red precipitate appeared, and then the reaction system was lowered to 0–5 °C and kept for 30 minutes. Filter pressing under nitrogen protection, the product was washed using anhydrous ethanol and dried, and the salenCo(II) complex was obtained as a red solid. Finally, a dichloromethane solution of 2-propyl-4-(4-pyridylazo)phenol or 4-(imidazole-1-yl)phenol was added to the three-necked flask with the red solid salenCo(II) complex and then O2 was passed to oxidize the salenCo(II) complex to get the salenCo(III) complex [C1] or [C2] (Scheme S1†).For the catalyst [C3] or [C4], firstly, 3-tert-butyl-2-hydroxybenzaldehyde is reacted with 1,2-diaminocyclohexane to obtain compound 2. The ligand reacts with cobalt acetate, and the “Co” metal coordinates with the ligand to form a complex of Co(II), and then a CH2Cl2 solution of 2-propyl-4-(4-pyridazinyl)phenol or 4-(imidazole-1-yl)phenol is dropped into the salenCo(II) complex and O2 is passed through; the salenCo(II) complex is oxidized to get the salenCo(III) complex. The desired catalyst [C3] or [C4] was obtained (Scheme S2†).
3.2 UV visible light absorption test
In order to study the maximum UV-visible absorption wavelength of Schiff base, photosensitive group and salenCo(III) catalyst, their UV-visible absorption wavelength were measured. Table 2 lists their UV-visible maximum absorption peaks. As can be seen in Fig. 3(a) that the UV-visible maximum absorption peak of compound 1 and compound 2 are 340 nm and 325 nm respectively. From Fig. 3(b), it can be seen that the UV-visible maximum absorption peak of 4-(imidazole-1-yl)phenol and 2-propyl-4-(4-pyridinazo)phenol are 240 nm and 360 nm, respectively. As can be seen from Fig. 2(c), the maximum UV absorption of salenCo(II) of 1,2-o-phenylenediamine and salenCo(II) of 1,2-cyclohexylenediamine are 380 nm and 356 nm, respectively. From Fig. 1(d), it can be seen that the UV-visible maximum absorption peaks of [C1], [C2], [C3] and [C4] are 474 nm, 553 nm, 410 nm and 542 nm, respectively. Due to the addition of the photosensitive group, for [C1] and [C3], the UV-visible maximum absorption peaks increase by 134 nm and 213 nm when the 4-(imidazole-1-yl)phenol and 2-propyl-4-(4-pyridinazo)phenol is added. For [C2] and [C4], the maximum absorption peak increases by 85 nm and 217 nm due to the addition of 4-(imidazole-1-yl)phenol and 2-propyl-4-(4-pyridinazo)phenol.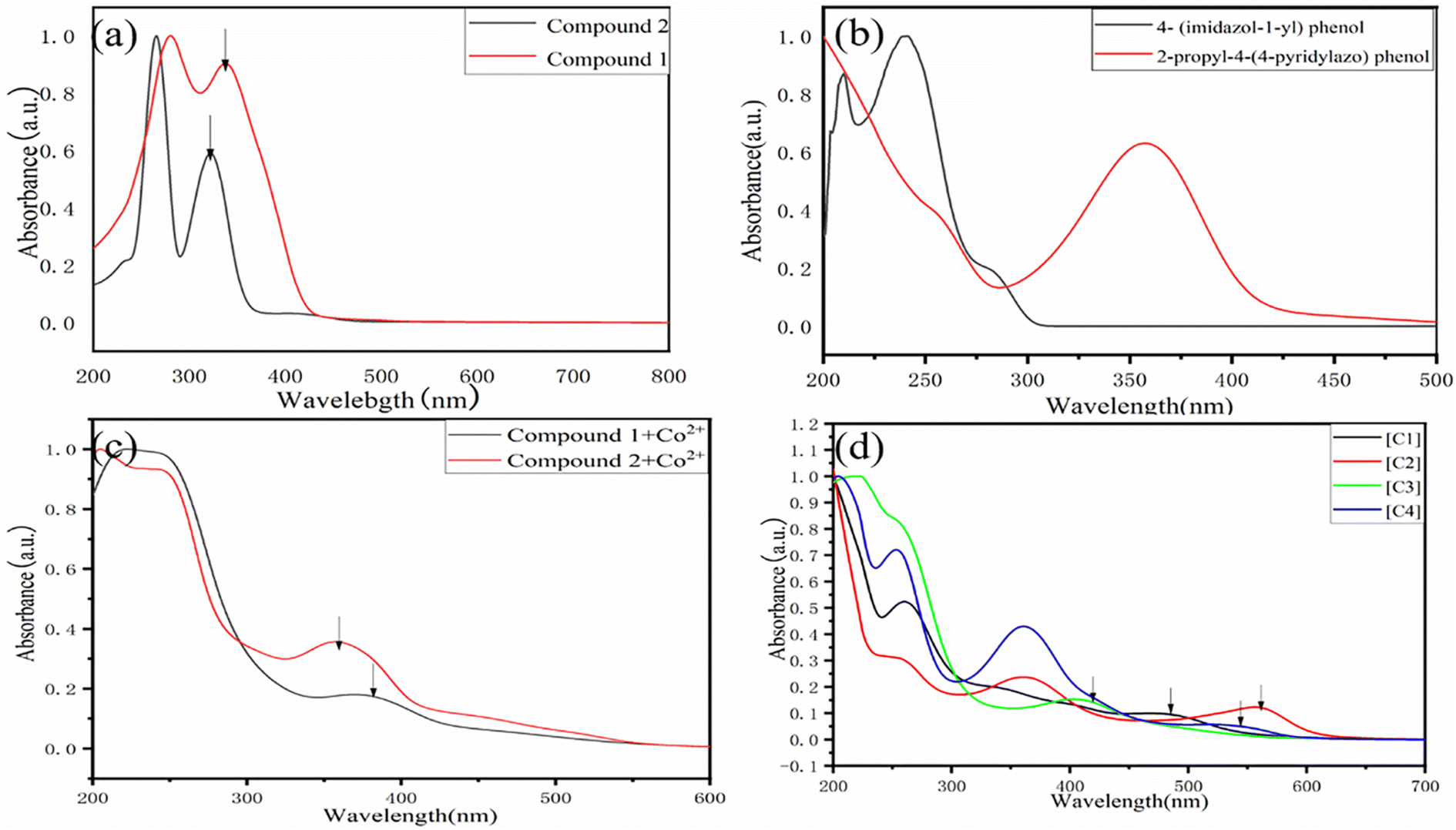 | ||
| Fig. 3 UV test (a) dichloromethane solution with a compound concentration of 0.01 mmol L−1. (b) Using methanol as solvent, 4-(imidazole-1-yl)phenol and 2,4-(4-pyridinazo)phenol with concentration of 0.001 mmol L−1. (c) Using methanol as solvent, salenCo(II) with concentration of 0.001 mmol L−1 (d) catalyst concentration is 0.01 mmol L−1, epoxy propane solvent: 1,2-dichloroethane (v/v = 3; 1). | ||
As can be seen in Fig. 2, in the Schiff base without metal coordination, the π→π* leap at 250–280 nm corresponds to the π→π* leap of the CN group and the n→π* leap of the CN group at 300–340 nm. In the catalysts after metal coordination, 360–400 nm correspond to the d→d leap of the metal complex, which also indicates the successful coordination of the metal to the ligand. The position of the absorption peak is significantly red-shifted in the catalyst with the metal Co bound compared to the catalyst without the metal ligand. The addition of the photosensitive group enhances the ligand conjugation and effectively improves the light absorption capacity of the catalyst, which explains to a certain extent the superior photocatalytic activity of the salenCo(III) catalyst containing the photosensitive group.
3.3 Copolymerization of CO2 and PO
The TON value of the catalytic product was calculated by the following formula: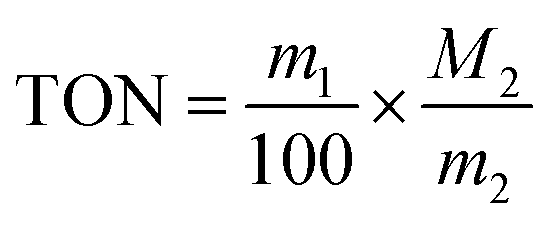
m1: copolymer quality after copolymerization; m2: catalyst quality; M2: molar mass of catalyst; 100 is the molar mass of a copolymerization unit (per mole of copolymerization unit has 1 mol of PO); h: time.
From the copolymerization results in Table 3, it can be seen that the sum of CPC/PPC contents for these four salenCo(III) catalysts does not exceed 20% of the total products in the light-free catalytic copolymerization, which indicates that the selectivity of light-free copolymerization for CPC/PPC is poor. The lowest catalytic efficiency of [C1] was only TON 82.3, and the PPC content in the product was 10.1%; the highest catalytic efficiency of [C4] reached TON 128.4, and the CPC content in the product also reached 13.8%. This is similar to the study results of R. Duan et al.40 We compared [C1] with [C2] or [C3] with [C4], and found that the catalytic efficiency of catalysts containing 2-propyl-4-(4-pyridinazo)phenol was significantly higher than that of catalysts containing 4-(imidazole-1-yl)phenol. The introduction of photosensitive groups with different conjugate lengths, the catalytic activity and selectivity of salenCo(III) catalyst will have different effects, and the activity and catalytic ability of the catalyst will be enhanced with the longer the conjugate system of the photosensitive groups introduced.
| Cat. | Wb | TOF−h | TONc | PPC (%) | PPO (%) | CPC (%) |
|---|---|---|---|---|---|---|
| a Polymerization conditions: PO 10 g, [cat] 3 × 10−5 mol, CO2 2 MPa, temperature 60 °C.b External light source power: 0 W indicates no external light source; 100 W indicates that there is an external light source and the power is 100 W.c Turnover number: moles of PO consumed per mole of catalyst. | ||||||
| [C1] | 0 | 13.7 | 82.3 | 9.8 | 9.8 | 48.0 |
| [C2] | 0 | 14.7 | 88.4 | 7.3 | 7.3 | 45.4 |
| [C3] | 0 | 17.5 | 105.4 | 1.1 | 6.1 | 46.4 |
| [C4] | 0 | 21.4 | 128.4 | 5.2 | 5.2 | 43.1 |
| [C1] | 100 | 17.1 | 102.8 | 1.1 | 15.2 | 83.7 |
| [C2] | 100 | 19.3 | 115.7 | 0.7 | 12.4 | 86.9 |
| [C3] | 100 | 25.9 | 155.7 | 0.6 | 13.1 | 86.3 |
| [C4] | 100 | 31.5 | 189.3 | 0.7 | 10.1 | 89.2 |
| [C4] | 200 | 51.4 | 308.2 | 0.2 | 15.5 | 84.3 |
When light was used in the polymerization experiment, we found that the TON of catalyst [C1]–[C4] increased by 20.5, 27.3, 50.3 and 60.9 (as in Fig. 4), respectively. This shows that the specific wavelength of light can enhance the activity of the catalyst, the longer the conjugate system of photosensitive groups and catalysts, the higher the catalytic activity. At the same time, we increased the lighting power of [C4] catalyst to 200 W for specific wavelength, and the catalytic efficiency was also improved, with TON increased by nearly 50%. Compared with the salenCo(III) catalyst with quaternary ammonium swab prepared by Hu Yang et al.41 the catalytic effectiveness (TOF−h) of the salenCo(III) catalyst prepared by us is 2–3 times that of the salenCo(III) catalyst containing photosensitive groups. In the 1H-NMR spectra of copolymer, the characteristic peaks of polycarbonate (PPC) are 1H-NMR (CDCl3), δ (ppm) 1.34 (3H; –CH3), 4.17 (2H; –CH2) and 5.00 (1H; –CH). The peak values of PPO were δ (ppm) 1.16 (3H; –CH3), 3.58 (2H; –CH2) and 3.45 (1H; –CH). The peak values of cyclic carbonate (PPC) were δ (ppm) 1.49 (3H; –CH3); 4.03 (1H; –CH2); 4.56 (1H; –CH2) and 4.86 (1H; –CH) (Fig. 5).41
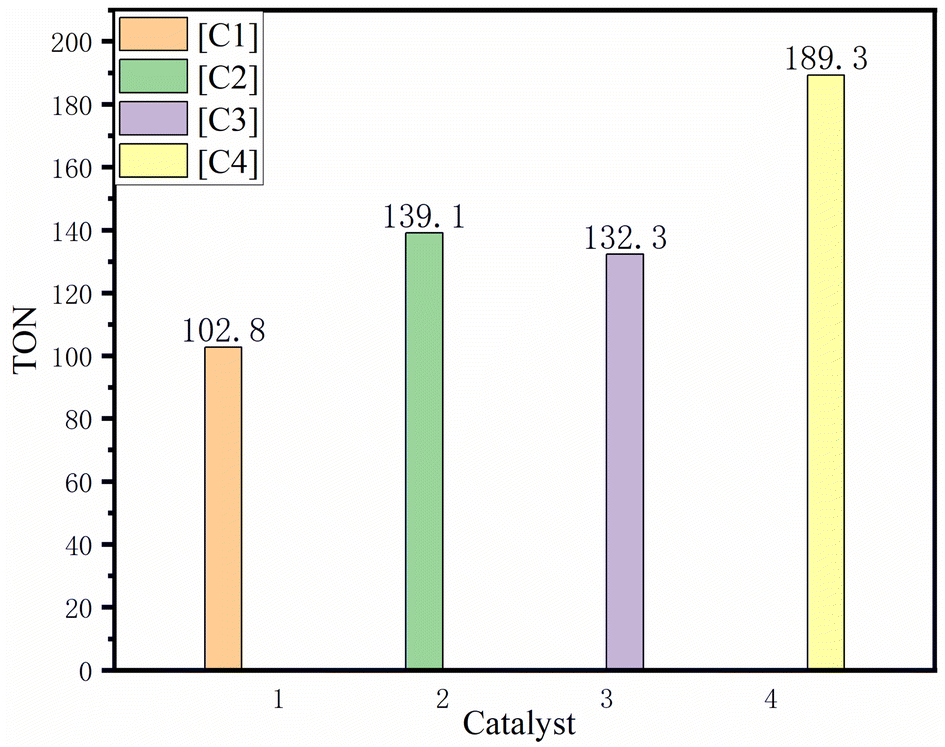 | ||
| Fig. 4 The TON for [C1]–[C4] at propylene oxide (10 g), catalyst (3 × 10−5 mol), 60 °C, 6 h, 2 MPa CO2 and 100 W light. | ||
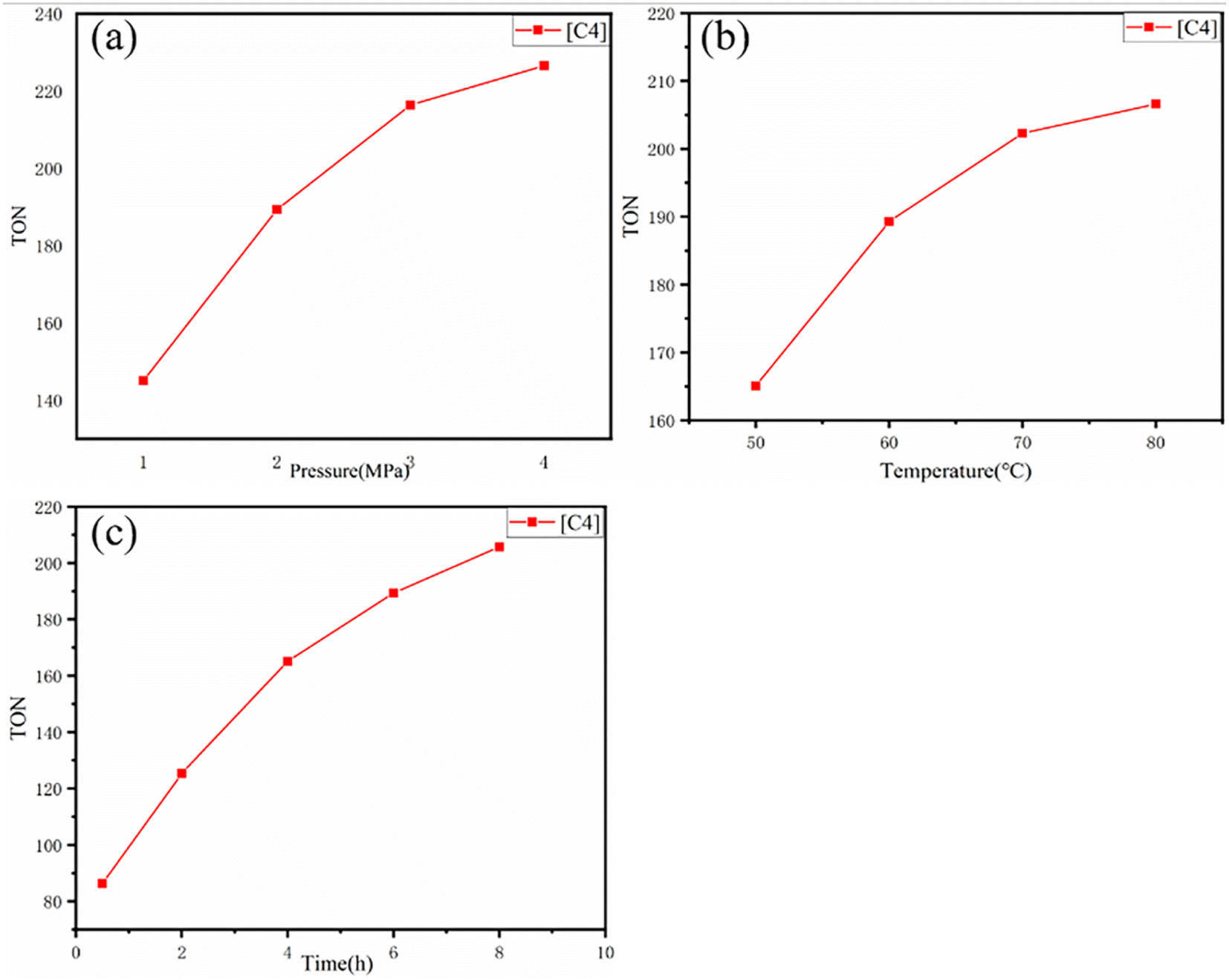 | ||
| Fig. 5 The relationship between TON and (a) carbon dioxide pressure, (b) temperature and (c) reaction time during the cycloaddition of carbon dioxide and propylene oxide catalyzed by [C4] catalyst. Reaction conditions: propylene oxide (10 g), catalyst (3 × 10−5 mol). There is only one change in the reaction condition for each graph (pressure, temperature and time). | ||
The catalytic activity of [C4] and the change of TON under different CO2 pressure, temperature and time were further evaluated. It can be observed from (Fig. 4 and Table S2†) that with the change of CO2 pressure, TON increases with the increase of pressure at the beginning, but its increase rate decreases somewhat. In addition, by changing the reaction temperature and time, the TON also has a similar rule. This result was similar to Degong Jia et al.42 In order to explore the reusability of the catalyst, we collected the catalyst after each catalytic copolymerization.43 Since the catalyst is undoable in ethyl acetate, the copolymer is soluble in ethyl acetate. The copolymer was dissolved in ethyl acetate and the catalyst was collected by filtering. As can be seen from Fig. 6(a), when [C4] catalyst is used constantly for catalysis, its TON value changes little, and from Fig. 6(b) and (c), we find that the infrared and XRD of [C4] hardly change. Thus, we can infer that the catalyst can be used again and again.
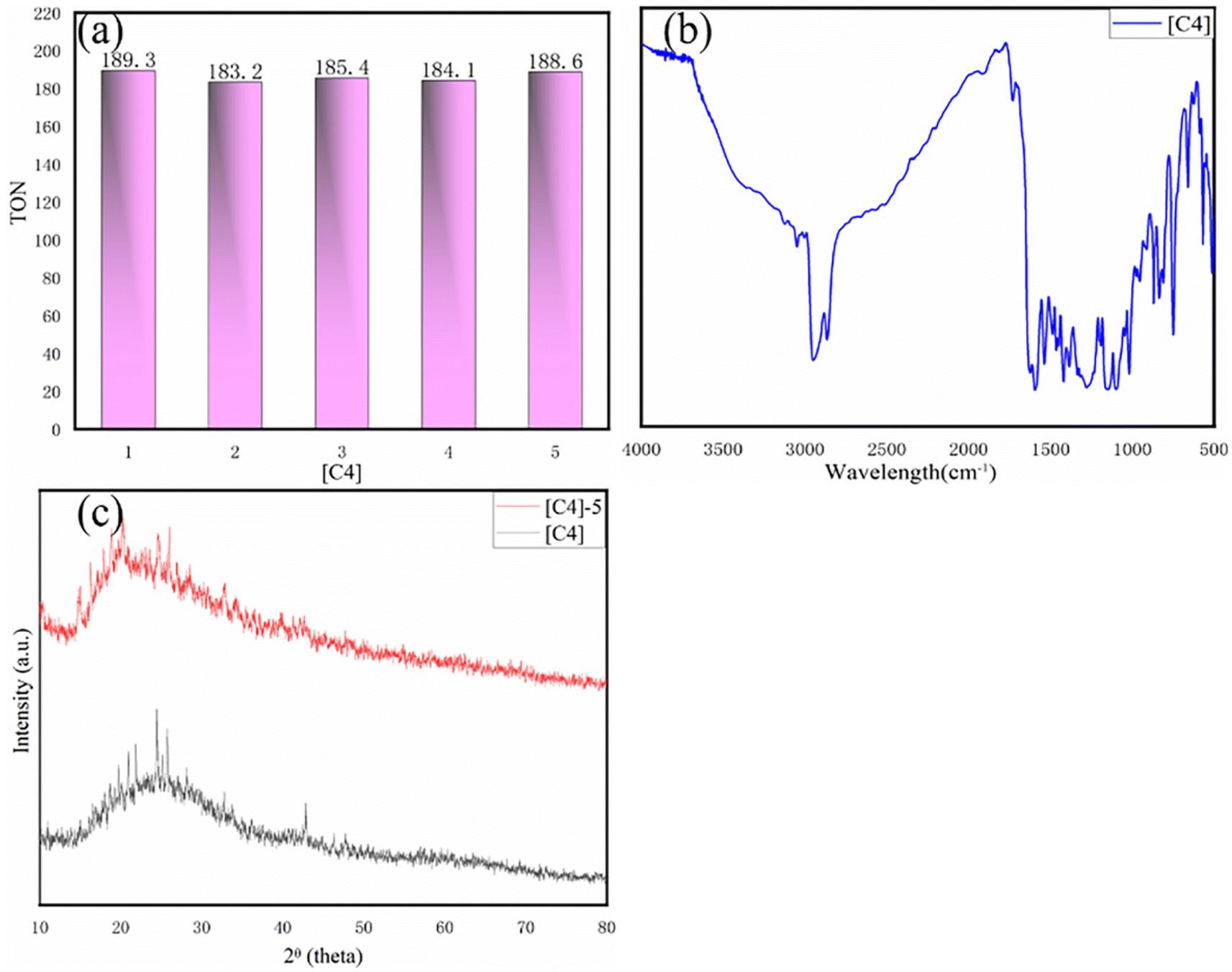 | ||
| Fig. 6 (a) [C4] catalytic reuse. (b) IR spectra of catalyst [C4] after the 5th catalysis. (c) XRD pattern [C4] refers to before the first catalysis; [C4]-5 refers to after the 5th catalysis. Reaction conditions: PO (10 g), catalyst (3 × 10−5 mol), CO2 (2 MPa), 60 °C, 6 h. | ||
3.4 Mechanistic explanation of [C3]
We investigated the feasibility of the reaction between compound 2 and Co-4-(imidazole-1-yl)phenol to form [C3], and the energy changes of compound 2 and [C3] were calculated by Gaussian software using density flooding theory (DFT).43,44 As shown in Fig. 7(a), from the simulation model of compound 2 and the [C3], it can be seen that the “Co” atom in the catalyst [C3] is coordinated to the “N” atom of the two imines and the “O” atom of the phenolic hydroxyl groups in compound 2 to form the salenCo(II) complex; subsequently, the 4-(imidazole-1-yl)phenol reacts with the salenCo(II) complex by the “Co” atom to form [C3]. As shown in Fig. 7(b), the LUMO of compound 2 is (1.3737 eV), HOMO is (−2.4362 eV), and Δ E = 3.8099 eV. As can be seen from Fig. 7(c), after complexation, LUMO of [C3] is (0.3691 eV), HOMO is (−2.3856 eV) and ΔE = 3.0247 eV. From the results of the DFT calculation, it can be inferred that compound 2 and Co-4-(iminazol-1-yl)phenol formed a stoichiometric 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex. From the view angle of ΔE, the ΔE 3.0247 eV of [C3] is less than that of compound 2 (3.8099 eV), while means the [C3] catalyst has a stable structure. This also supports the phenomenon that the maximum UV absorption of [C3] after complexation is significantly enhanced. The same is true for the rest of the catalysts.
:1 complex. From the view angle of ΔE, the ΔE 3.0247 eV of [C3] is less than that of compound 2 (3.8099 eV), while means the [C3] catalyst has a stable structure. This also supports the phenomenon that the maximum UV absorption of [C3] after complexation is significantly enhanced. The same is true for the rest of the catalysts.
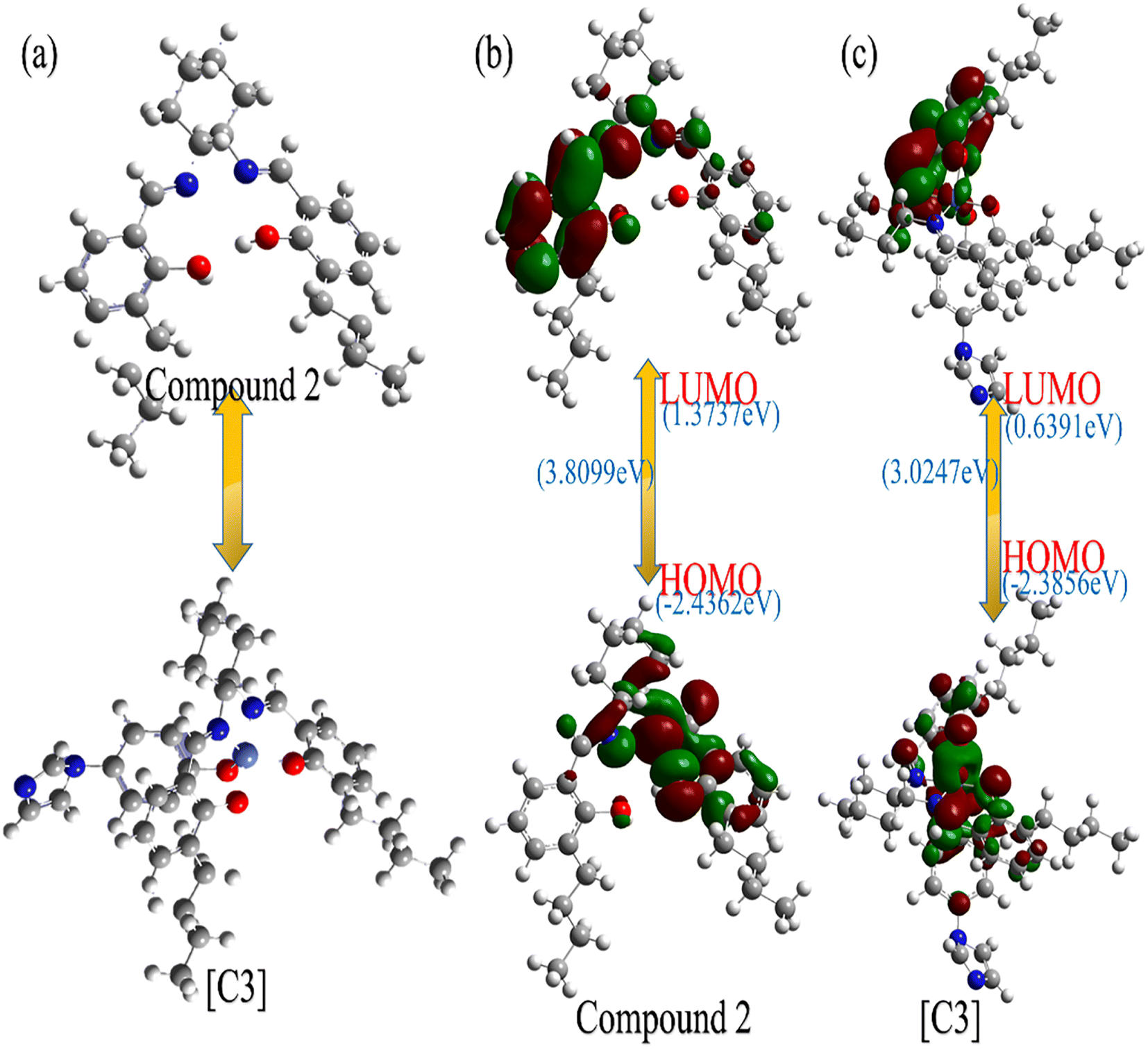 | ||
| Fig. 7 (a) Optimized structures of compounds 2 and [C3], color coding: C (black), N (blue), H (white), O (red), Co (light blue). (b) The frontier orbital shape and energy level of compound 2. (c) The frontal orbit shape of [C3] and its energy levels. Color coding: C (black), N (blue), H (white), O (red), Co (light blue). | ||
3.5 Catalytic mechanism
Based on previous salenCo(III) catalyst experience in catalyzing carbon dioxide and epoxides. Therefore, we can deduce the reaction process during copolymerization (as in Fig. 8). Firstly, the catalyst active substance attacks the C–O bond of PO to form a ring-opening intermediate; then, the negatively charged oxygen atom of this intermediate attacks the carbon atom of CO2 to form a ring-opening carbonate; finally, the negatively charged oxygen atom within the molecule attacks the carbon atom on the other side of the ring opening to release the desired products and active substances.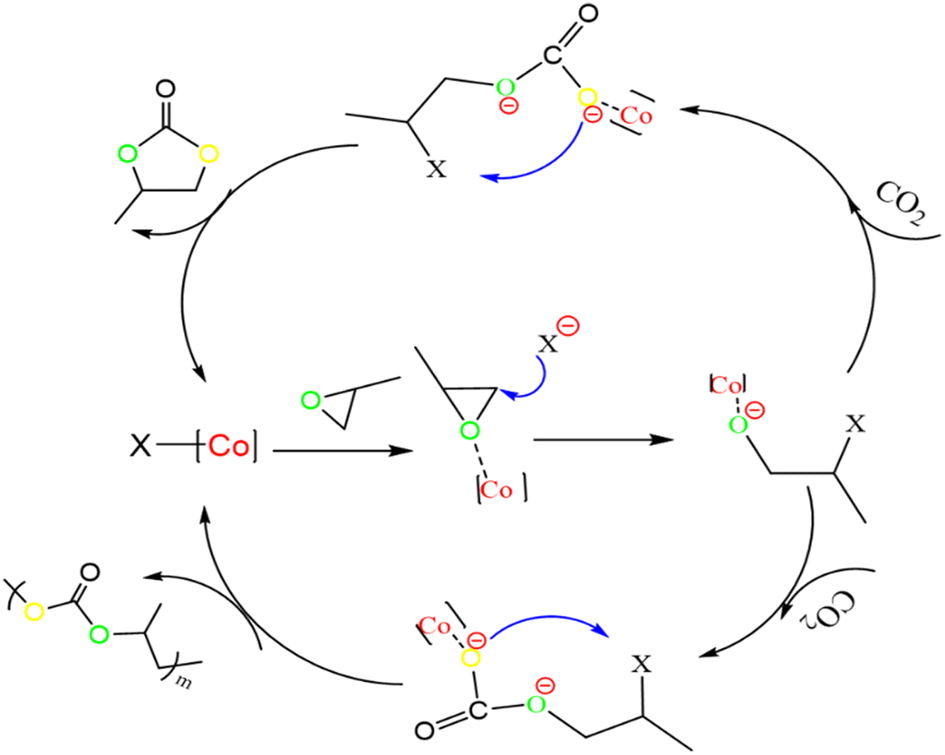 | ||
| Fig. 8 [C3] catalyzed the copolymerization of CO2 and PO. | ||
After irradiation, the catalyst can absorb visible light to form a state of charge separation, which promotes the transfer of electrons from the photosensitive group to the “Co” atom of salenCo(III) ligand and promotes the cycloaddition reaction of PO and CO2, as shown in the EPR data in Fig. 9. According to EPR measured under dark conditions, it can be seen that under the condition of no light, only a small part of electrons on the photosensitive group transfer to the “Co” atom. Therefore, after the catalyst is irradiated, a large number of electrons of the photosensitive group are transferred to the “Co” atom (as shown in Scheme 2), which greatly enhances its activity and is more conducive to the ring-opening of PO. This in turn significantly improves the efficiency of the catalyst.
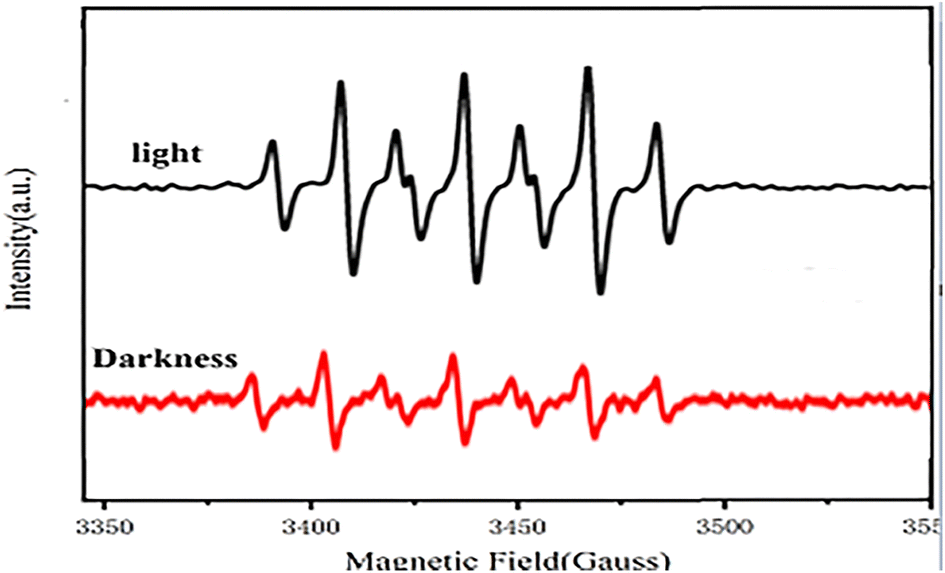 | ||
| Fig. 9 EPR spectra of [C4] catalyst before and after light exposure. | ||
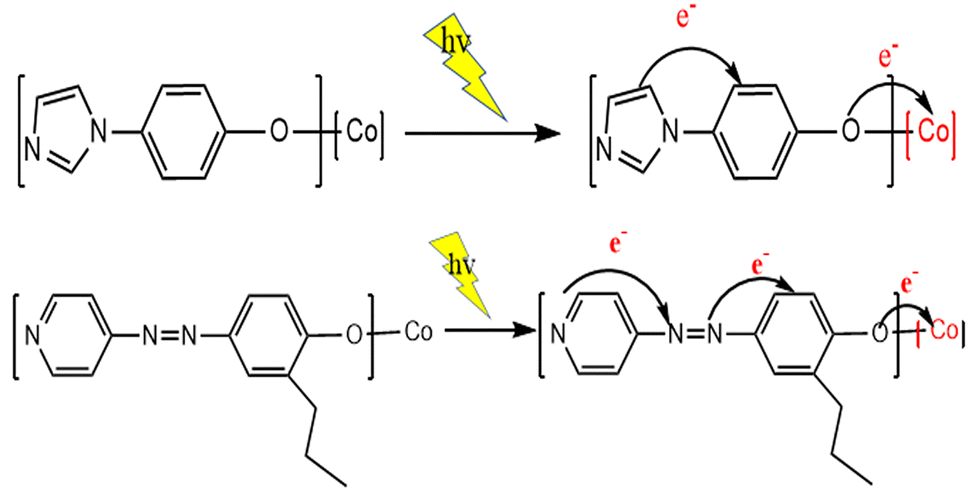 | ||
| Scheme 2 Electron transfer of photosensitive groups in catalyst under light. | ||
4 Conclusion
In summary, the structure of salenCo(III) and the CO2/PO copolymerization catalyzed by the photosensitive groups were studied in this paper. The catalytic effectiveness of [C3] or [C4] is higher than [C1] or [C2] because the electron transfer between the π series of Schiff base of 1,2-cyclohexylenediamine and Co atoms is stronger than that of o-phenylenediamine. Compared with [C3], the catalytic efficiency of [C4] is much higher than that of [C3], because the conjugated system of 2-propyl-4-(4-pyridylazo)phenol is much longer than that of 4-(imidazole-1-yl)phenol. It can be inferred that light can greatly ameliorate the catalytic exertion of carbon dioxide and epoxide, and the catalytic effect increases with the increase of light power.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author thanks the Scientific45 Research Foundation of Education Department of Anhui Province of China (2022AH050103), Anhui Natural Science Foundation (No. 1908085MB51), Anhui Key Laboratory of Environmentally Friendly Polymer Materials and Anhui University Key Laboratory of Hybrid Materials Structure and Function Regulation (Anhui University) for their support; Thank you for your guidance from Qiong Zhang, School of Chemistry and Chemical Engineering, Anhui University. Address: 230601, Hefei, China.References
- D. J. Darensbourg, R. Mackiewicz and J. L. Rodgers, J. Am. Chem. Soc., 2005, 127, 14026–14038 CrossRef CAS PubMed
.
- D. J. Darensbourg and D. R. Billodeaux, Inorg. Chem., 2005, 44, 1433–1442 CrossRef CAS PubMed
- Z. Yu, L. Xu, Y. Wei, Y. Wang, Y. He, Q. Xia, X. Zhang and Z. Liu, Chem. Commun., 2009, 3934 RSC
- D. J. Darensbourg and G.-P. Wu, Angew. Chem., Int. Ed., 2013, 52, 10602–10606 CrossRef CAS PubMed
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS
- S. D. Thorat, P. J. Phillips, V. Semenov and A. Gakh, J. Appl. Polym. Sci., 2003, 89, 1163–1176 CrossRef CAS
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed
- C. J. Whiteoak, N. Kielland, V. Laserna, F. Castro-Gómez, E. Martin, E. C. Escudero-Adán, C. Bo and A. W. Kleij, Chem.–Eur. J., 2014, 20, 2264–2275 CrossRef CAS PubMed
- M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC
- Y. Hu, J. Steinbauer, V. Stefanow, A. Spannenberg and T. Werner, ACS Sustainable Chem. Eng., 2019, 7, 13257–13269 CrossRef CAS
- K. A. Andrea and F. M. Kerton, ACS Catal., 2019, 9, 1799–1809 CrossRef CAS
- C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS PubMed
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed
- P. K. Saini, C. Romain and C. K. Williams, Chem. Commun., 2014, 50, 4164–4167 RSC
- P. Wang, Y. Fei, Y. Long and Y. Deng, J. CO2 Util., 2018, 28, 403–407 CrossRef CAS
- J. Marbach, T. Höfer, N. Bornholdt and G. A. Luinstra, ChemistryOpen, 2019, 8, 828–839 CrossRef CAS PubMed
- R. Liu, X. Liu, K. Ouyang and Q. Yan, ACS Macro Lett., 2019, 8, 200–204 CrossRef CAS PubMed
- J. Sebastian and D. Srinivas, Appl. Catal., A, 2015, 506, 163–172 CrossRef CAS
- B. Liu, Y.-Y. Zhang, X.-H. Zhang, B.-Y. Du and Z.-Q. Fan, Polym. Chem., 2016, 7, 3731–3739 RSC
- J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, J. A. Castro-Osma, A. Lara-Sánchez and A. Otero, ChemSusChem, 2017, 10, 2886–2890 CrossRef PubMed
- G. Li, S. Dong, P. Fu, Q. Yue, Y. Zhou and J. Wang, Green Chem., 2022, 24, 3433–3460 RSC
- Z. Guo, Y. Hu, S. Dong, L. Chen, L. Ma, Y. Zhou, L. Wang and J. Wang, Chem. Catal., 2022, 2, 519–530 CrossRef CAS
- Y. Wu, L. Ma, Z. Song, S. Dong, Z. Guo, J. Wang and Y. Zhou, Carb. Neutrality, 2023, 2, 1 CrossRef
- Y. Niu, W. Zhang, H. Li, X. Chen, J. Sun, X. Zhuang and X. Jing, Polymer, 2009, 50, 441–446 CrossRef CAS
- M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef CAS
- Y. Niu and H. Li, Colloid Polym. Sci., 2013, 291, 2181–2189 CrossRef CAS PubMed
- C.-H. Ho, H.-J. Chuang, P.-H. Lin and B.-T. Ko, J. Polym. Sci., Part A-1: Polym. Chem., 2017, 55, 321–328 CrossRef CAS
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed
- A. C. Deacy, C. B. Durr, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2018, 57, 15575–15583 CrossRef CAS PubMed
- P. P. Pescarmona, Curr. Opin. Green Sustainable Chem., 2021, 29, 100457 CrossRef CAS
- M. North, S. C. Z. Quek, N. E. Pridmore, A. C. Whitwood and X. Wu, ACS Catal., 2015, 5, 3398–3402 CrossRef CAS
- M. North, B. Wang and C. Young, Energy Environ. Sci., 2011, 4, 4163 RSC
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC
- M. North and C. Young, ChemSusChem, 2011, 4, 1685–1693 CrossRef CAS PubMed
- L. A. Siddig, R. H. Alzard, H. L. Nguyen and A. Alzamly, Inorg. Chem. Commun., 2022, 142, 109672 CrossRef CAS
- T. Yan, H. Liu, Z. X. Zeng and W. G. Pan, J. CO2 Util., 2023, 68, 102355 CrossRef CAS
- D. Jia, L. Ma, Y. Wang, W. Zhang, J. Li, Y. Zhou and J. Wang, Chem. Eng. J., 2020, 390, 124652 CrossRef CAS
- R. Duan, C. Hu, Z. Sun, H. Zhang, X. Pang and X. Chen, Green Chem., 2019, 21, 4723–4731 RSC
- L. Du, C. Wang, W. Zhu and J. Zhang, J. Chin. Chem. Soc., 2020, 67, 72–79 CrossRef CAS
- D. Jia, L. Ma, Y. Wang, W. Zhang, J. Li, Y. Zhou and J. Wang, Chem. Eng. J., 2020, 390, 124652 CrossRef CAS
- Z. Guo, Q. Jiang, Y. Shi, J. Li, X. Yang, W. Hou, Y. Zhou and J. Wang, ACS Catal., 2017, 7, 6770–6780 CrossRef CAS
- P. Xu, X. Liu, X. Zhao, W. Zhu, M. Fang, Z. Wu, L. Du and C. Li, J. Chin. Chem. Soc., 2020, 67, 298–305 CrossRef CAS
- L. A. Siddig, R. H. Alzard, H. L. Nguyen and A. Alzamly, Inorg. Chem. Commun., 2022, 142, 109672 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02370b |
| This journal is © The Royal Society of Chemistry 2023 |