
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective synthesis of (E)-α,β-unsaturated esters: triethylamine-catalyzed allylic rearrangement of enol phosphates†
Yulong Zhang,
Huichuang Guo,
Qian Wu,
Xiaojing Bi *,
Enxue Shi* and
Junhua Xiao*
*,
Enxue Shi* and
Junhua Xiao*
State Key Laboratory of NBC Protection for Civilian, Beijing 102205, P. R. China. E-mail: junhua@pku.edu.cn; exshi@sina.com; xiaojingbimail@yeah.net
First published on 11th May 2023
Abstract
α,β-Unsaturated esters are key structural motifs widely distributed in various biologically active molecules, and their Z/E-stereoselective synthesis has always been considered highly attractive in organic synthesis. Herein, we present a >99% (E)-stereoselective one-pot synthetic approach towards β-phosphoroxylated α,β-unsaturated esters via a mild trimethylamine-catalyzed 1,3-hydrogen migration of the corresponding unconjugated intermediates derived from the solvent-free Perkow reaction between low-cost 4-chloroacetoacetates and phosphites. Versatile β,β-disubstituted (E)-α,β-unsaturated esters were thus afforded with full (E)-stereoretentivity by cleavage of the phosphoenol linkage via Negishi cross-coupling. Moreover, a stereoretentive (E)-rich mixture of a α,β-unsaturated ester derived from 2-chloroacetoacetate was obtained and both isomers were easily afforded in one operation.
α,β-Unsaturated carbonyl motifs, such as the relevant esters, amides, and aldehydes, are widely distributed in biologically active molecules as key structural components (Fig. 1).1–4 Generally, the (Z) and (E)-isomers of those molecules possess very different living activities.5 Moreover, ubiquitous α,β-unsaturated esters are also widely employed as useful intermediates for enantioselective hydrogenation,6 allylic substitution,7 conjugate addition,8 and especially for the stereoselective generation of acyclic substituted alkenes in either (Z) or (E)-isomeric forms.9
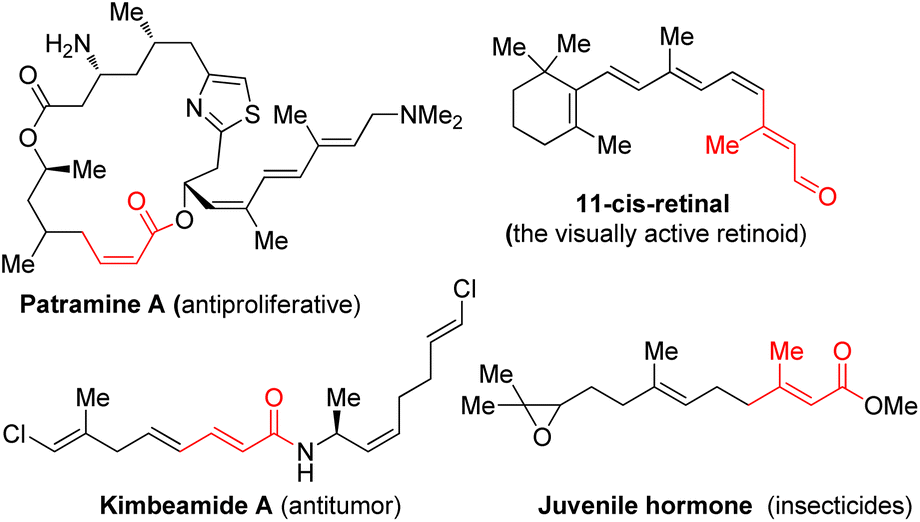 | ||
| Fig. 1 Selected bioactive α,β-unsaturated carbonyl motifs. | ||
Whilst numerous methods have been developed towards α,β-unsaturated esters,10–13 configuration-retentive transition-metal catalyzed (TMC) cross-coupling of alkenyl (pseudo)halides is universally recognized as one of the most practical methodologies.14 Among the known non-classical pseudohalides,15 diethylphosphoroxyl (DEP) functionality has been proved as a good leaving group in many organic reactions and the corresponding enol phosphates (EPs), possessing high stability and accessibility, were found to participate in various organic transformations.16 Particularly, EPs have been utilized in many types of TMC coupling reactions including Suzuki–Miyaura, Stille, Negishi, and Heck reactions by cleavage of the enol-linkage affording highly substituted alkenes.17 However, the EPs-involved (Z) and (E)-stereocomplementary synthetic method towards α,β-unsaturated esters with sufficient substrate generality is still quite limited at present. The latest impressive approach was reported by Tanabe group, which employed N-methylimidazole (NMI)-promoted phosphorylation of β-ketoesters to obtain (Z) and (E)-α,β-unsaturated esters, but which suffers from pre-activation of the unstable diphenyl phosphorochloridate (DPPCl) and usage of strong metallic tert-butoxide bases.18 Based on our recent progress in regioselective solvent-free synthesis of EPs,19 we envisioned that phosphoroxylated (Z) and/or (E)-α,β-unsaturated esters may act as the universal synthon of α,β-unsaturated esters and should be facilely obtained from the commercially available and low-cost chloroacetoacetates and phosphites via a simple metal-free Perkow reaction. Herein, we wish to present a stereoselective one-pot synthetic approach towards β-phosphoroxylated (E)-α,β-unsaturated esters, which are subsequently converted into the corresponding disubstituted α,β-unsaturated esters by Negishi cross-coupling (Scheme 1).
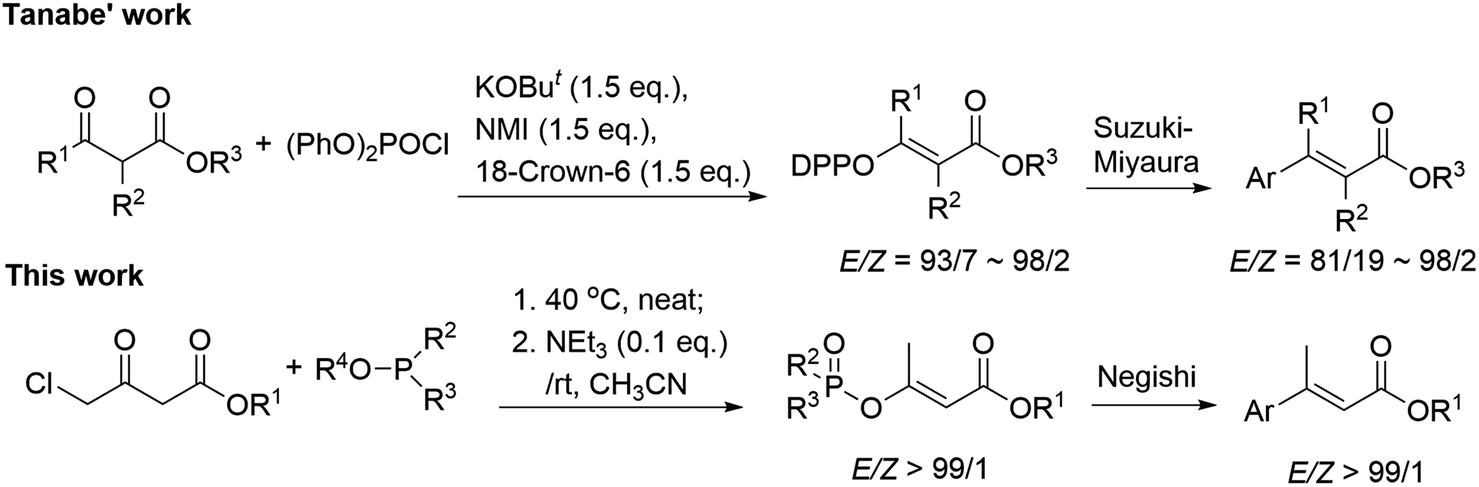 | ||
| Scheme 1 E-Stereoselective synthesis of α,β-unsaturated esters from enol phosphates. | ||
Since both 2-chloroacetoacetates and 4-chloroacetoacetates are capable of undergoing Perkow reaction with phosphites, we then took them together for comparison. Solvent-free Perkow reaction conditions were initially selected in view of high regioselectivity.19 As shown in Scheme 2, reaction between (EtO)3P and 2-chloroacetoacetate 2a gave a mixture of (E) and (Z)-isomers of β-phosphoroxylated α,β-unsaturated ester 4a in ratio of 2.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, whereas reaction between (EtO)3P and 4-chloroacetoacetate 3a gave the β-phosphoroxylated allylic ester 5a as the only product. In other words, only moderate E/Z-stereoselectivity can be achieved if using 2-chloroacetoacetate, while no conjugated EP product can be obtained if using 4-chloroacetoacetate. However, according to Seeman's report that bases, such as NaH, are supposed to be able to promote 1,3-hydrogen relocation of allyl compounds, we then suspect that the unconjugated EP product 5a may be able to be transformed into the conjugated one in a stereoselective way.20
:1, whereas reaction between (EtO)3P and 4-chloroacetoacetate 3a gave the β-phosphoroxylated allylic ester 5a as the only product. In other words, only moderate E/Z-stereoselectivity can be achieved if using 2-chloroacetoacetate, while no conjugated EP product can be obtained if using 4-chloroacetoacetate. However, according to Seeman's report that bases, such as NaH, are supposed to be able to promote 1,3-hydrogen relocation of allyl compounds, we then suspect that the unconjugated EP product 5a may be able to be transformed into the conjugated one in a stereoselective way.20
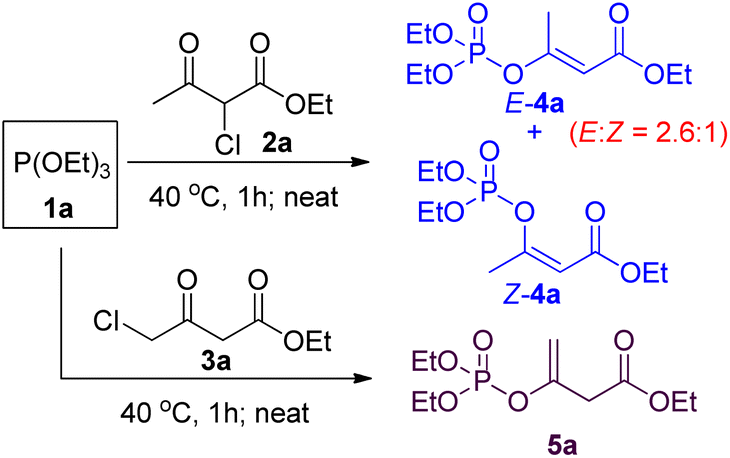 | ||
| Scheme 2 Perkow reaction of phosphite with chloroacetoacetate. | ||
Inspired by the above idea, we then turned to examine the possibility of the base-promoted 1,3-hydrogen rearrangement of 5a. As shown in Table 1, among the eight kinds of bases examined, including inorganic t-BuOK, CH3ONa, NaOH, NaH, K2CO3, and organic Et3N, Pyridine (i-Pr)2NEt, only Et3N and (i-Pr)2NEt exhibited the supposed promoting abilities, affording the desired product E-4a, but encouragely both in >99% (E)-stereoselectivity. Though only 20% yield was obtained by 1.2 equivalent (i-Pr)2NEt after 24 h reaction in THF at room temperature (Table 1, entry 8), while up to 90% yield was acquired by using Et3N (Table 1, entry 6). The following screening of solvents demonstrated that acetonitrile seemed to the best choice that the reaction could be accomplished in only 4 h and gave a higher yield of 92% (Table 1, entry 10). Further investigation about the loadage of Et3N showed that only 0.1 equivalent Et3N was sufficient to promote the rearrangement effectively, affording the comparative yield though with a few longer time of 12 h (Table 1, entry 14). Less loadage of Et3N and lower temperature both led to much longer reaction times (Table 1, entry 15&16). Though the reaction time could be shortened to 4 h at a higher temperature of 80 °C (Table 1, entry 17), we finally preferred the more benign room temperature for the following preparations.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Base | Load (x eq.) | Solvent | T (oC) | Time (h) | Yieldb (%) | E/Z (4a)c |
| a Reaction conditions: 5a (1.0 equiv.), base (x equiv.), solvent (3 ml).b Isolated yields.c Determined by NMR. | |||||||
| 1 | t-BuOK | 1.2 | THF | rt | 24 | 0 | — |
| 2 | CH3ONa | 1.2 | THF | rt | 24 | 0 | — |
| 3 | NaOH | 1.2 | THF | rt | 24 | 0 | — |
| 4 | NaH | 1.2 | THF | rt | 24 | 0 | — |
| 5 | K2CO3 | 1.2 | THF | rt | 24 | 0 | — |
| 6 | Et3N | 1.2 | THF | rt | 24 | 90 | >99:1 |
| 7 | Pyridine | 1.2 | THF | rt | 24 | 0 | — |
| 8 | (i-Pr)2NEt | 1.2 | THF | rt | 24 | 20 | >99:1 |
| 9 | Et3N | 1.2 | CH3CN | rt | 4 | 92 | >99:1 |
| 10 | Et3N | 1.2 | DCM | rt | 20 | 90 | >99:1 |
| 11 | Et3N | 1.2 | CH3OH | rt | 22 | 83 | >99:1 |
| 12 | Et3N | 1.2 | DMF | rt | 24 | 75 | >99:1 |
| 13 | Et3N | 0.5 | CH3CN | rt | 7 | 92 | >99:1 |
| 14 | Et3N | 0.1 | CH3CN | rt | 12 | 92 | >99:1 |
| 15 | Et3N | 0.05 | CH3CN | rt | 20 | 93 | >99:1 |
| 16 | Et3N | 0.1 | CH3CN | 0 | 24 | 95 | >99:1 |
| 17 | Et3N | 0.1 | CH3CN | 80 | 4 | 90 | >99:1 |
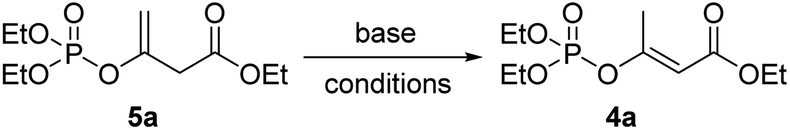
Considering the convenience of experimental operation, we then turned into the possibility of one-pot manipulation. It was found that product E-4a was afforded in 92% yield if using the crude intermediate 5a directly for the subsequent rearrangement reaction. Therefore, a mild E-stereoselective one-pot synthetic approach of β-phosphoroxylated α,β-unsaturated esters was thus established: 3 (1.0 eq.) and P(III)-reagents (1.0 eq.) react 1 h at 40 °C neatly, then added triethylamine (0.1 eq.) and acetonitrile (3 mL), and further react about 12 h at room temperature.
Having identified the optimal reaction conditions, we next set out to examine the scope of this new mild one-pot enol phosphorylation procedure (Table 2). As for the different O-alkyl 4-chloroacetoacetate substrates, all the common P(III)-reagents possessing P–O, P–C, and/or P–N bonds gave the corresponding EPs in high yields. During the preparation of compounds 4e and 4f, the rearrangement reactions were found much accelerated probably due to the higher reactivities of phosphonite and phosphinite compared to phosphites. To demonstrate the practical utility, the reaction towards product 4a was performed at the 50 mmol scale and 92% yield was obtained. The stereoscopic (E)-configuration of solid product 4f was further confirmed by single crystal X-ray analysis.

With the E-stereospecific β-phosphoroxylated α,β-unsaturated esters in hand, we then investigated their stereoretentive Negishi cross-coupling to prepare the corresponding E-stereodefined disubstituted α,β-unsaturated esters. Among the typical catalysts screened including Pd(PPh3)4, Ni(acac)2 and Pd(dppb)Cl2, the latter demonstrated the best performance in this Negishi reaction with only 0.02 equivalent loading by refluxing in acetonitrile. Various aromatic ArZnCl nucleophiles containing electron-donating and/or electron-withdrawing substituents at ortho, meta, and/or para positions were all tolerated well, affording the desired products in good to excellent yields (80–96%) without generating any stereochemical integrity (Table 3, 6a–6m). Disubstituted, condensed and hetero aromatic organometallic substrates also gave 85–92% yields of the products (Table 3, 6n–6r). However, it's regrettable that alkyl organozinc reagents was found unreactive under such conditions.
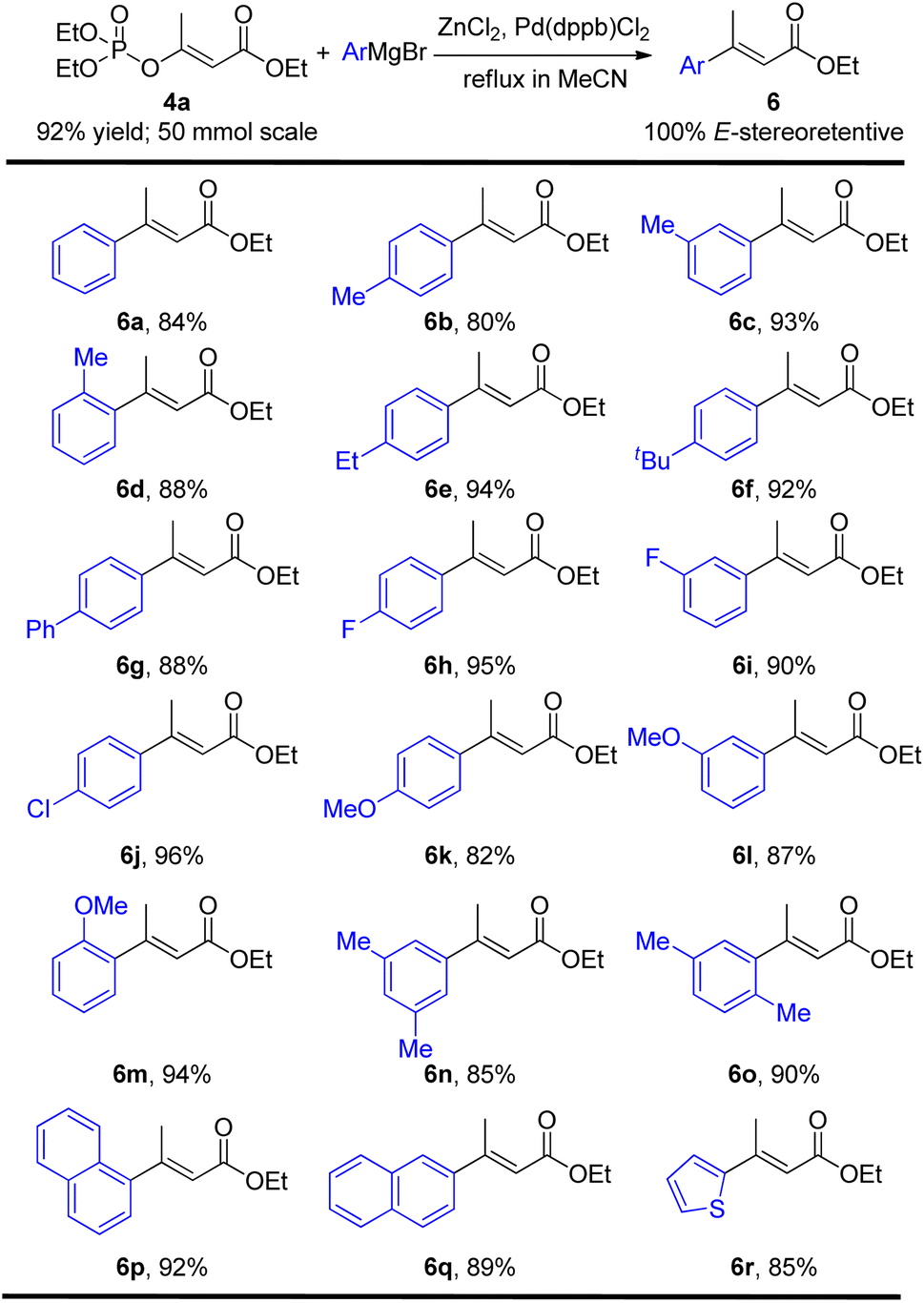
Furthermore, under the above optimal Negishi cross-coupling reaction conditions, both (Z) and (E) isomers of α,β-unsaturated esters 6a could be easily achieved, just by one operation, directly from the (Z) and (E) mixture of 4a in 22% and 70% yields respectively (Scheme 3).
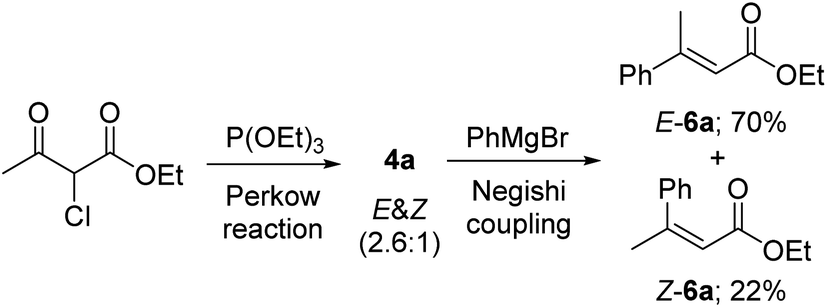 | ||
| Scheme 3 Preparation of (Z) and (E) isomers of 6a in one operation. | ||
According to the Cram's mechanistic interpretation for the allylic rearrangements, an intra-molecular pathway of the Et3N-promoted stereoselective 1,3-hydrogen rearrangement of the EPs 5a was proposed because that the degree of the observed intramolecularity depended strongly on the base and solvent used.21 As shown in Scheme 4, triethylamine firstly removes the proton from the α-carbon position of ester 5a, resulting in a coplanar anionic allylic system by three carbon atoms. The hydrogen atom of the H–Et3N ammonium then bonds to both terminal carbon atoms to form the intermediate Int, collapse of which would then give the thermodynamically favourable conjugated α,β-unsaturated ester product E-4a.
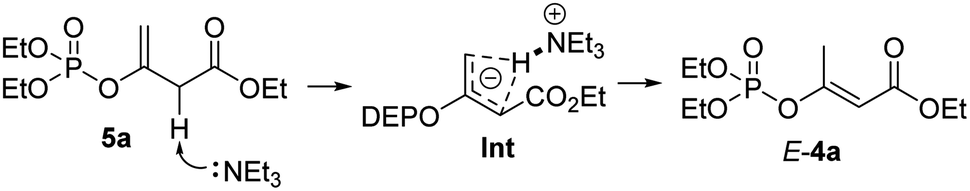 | ||
| Scheme 4 Proposed (E)-stereospecific allylic rearrangement mechanism. | ||
In summary, a mild and environmental trimethylamine-catalyzed E-stereoselective 1,3-hydrogen allylic rearrangement of enol phosphates was firstly developed to afford versatile β-phosphoroxylated (E)-α,β-unsaturated esters which can be then efficiently converted into the corresponding β,β-disubstituted (E)-α,β-unsaturated esters in high yields by a 100% stereoretentive Negishi cross-coupling reaction. Moreover, both (Z) and (E)-α,β-unsaturated esters were able to be achieved in one manipulation when just employing 2-chloroacetoacetate instead of 4-chloroacetoacetate for the solvent and metal-free Perkow reaction.
It is interesting to note that more structure-diverse α,β-unsaturated esters should be easily obtained by derivation reactions at the allylic position of α,β-unsaturated esters and/or by utilizing 2-substituted 4-chloroacetoacetates as the starting materials.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. X. Zhuo and A. Furstner, J. Am. Chem. Soc., 2018, 140, 10514 CrossRef CAS PubMed; (b) D. Romo, N. S. Choi, S. Li, I. Buchler, Z. Shi and J. O. Liu, J. Am. Chem. Soc., 2004, 126, 10582 CrossRef CAS PubMed; (c) S. D. Marco, A. Cammas, J. Pelletier and I. E. Gallouzi, Nat. Commun., 2012, 3, 8963 Search PubMed; (d) S. K. Naineni, J. Liang, B. Nagar and J. Pelletier, Cell Chem. Biol., 2021, 28, 825 CrossRef CAS PubMed.
- (a) J. K. Nunnery, N. Engene, T. Byrum, T. F. Murray and W. H. Gerwick, J. Org. Chem., 2012, 77, 4198 CrossRef CAS PubMed; (b) S. Sekharan and K. Morokuma, J. Am. Chem. Soc., 2011, 133, 19052 CrossRef CAS PubMed; (c) E. A. Zhukovsky, P. R. Robinson and D. D. Oprian, Science, 1990, 251, 558 CrossRef PubMed.
- M. Kiser and K. Golczak, Chem. Rev., 2014, 114, 194 CrossRef PubMed.
- (a) P. Guo, Y. Zhang, L. Zhang and Q. Xia, J. Biol. Chem., 2021, 297, 101234 CrossRef CAS PubMed; (b) Y. Ando, K. Matsumoto, K. Misaki, G. Mano, T. Shinada and S. G. Goto, Gen. Comp. Endocrinol., 2020, 289, 113394 CrossRef CAS PubMed; (c) M. Nouzova, C. Rivera-Pérez and F. G. Noriega, Curr. Opin. Insect. Sci., 2018, 29, 49 CrossRef PubMed; (d) K. Li, Q. Jia and S. Li, Insect Sci., 2019, 26, 600 CrossRef CAS PubMed.
- (a) D. A. Evans, P. J. Coleman, L. C. Dias and A. N. Tyler, Angew. Chem., Int. Ed. Engl., 1997, 36, 2744 CrossRef CAS; (b) D. A. Evans, D. M. Fitch, T. E. Smith and V. Cee, J. Am. Chem. Soc., 2000, 122, 10033 CrossRef CAS; (c) D. A. Evans, P. H. Carter, E. M. Carreira, A. B. Charette, J. A. Prunet and M. Lautens, J. Am. Chem. Soc., 1999, 121, 7540 CrossRef CAS; (d) I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063 CrossRef CAS PubMed.
- (a) E. Negishi, Q. Hu, Z. Huang, M. Qian and G. Wang, Aldrichimica Acta, 2005, 38, 71 CAS; (b) J. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297 CrossRef CAS PubMed; (c) V. Hornillos, M. Giannerini, C. Vila, M. F. Mastral and B. L. Feringa, Chem. Sci., 2015, 6, 1394 RSC.
- (a) K. Murakami and H. Yorimitsu, Beilstein J. Org. Chem., 2013, 9, 278 CrossRef CAS PubMed; (b) M. G. Suero, E. D. Bayle, B. S. L. Collins and M. J. Gaunt, J. Am. Chem. Soc., 2013, 135, 5332 CrossRef CAS PubMed; (c) F. Xue, J. Zhao, T. S. A. Hor and T. Hayashi, J. Am. Chem. Soc., 2015, 137, 3189 CrossRef CAS PubMed; (d) S. Wang and C. Xi, Org. Lett., 2018, 20, 4131 CrossRef CAS PubMed.
- (a) F. Guibe, Tetrahedron, 1998, 54, 2967 CrossRef CAS; (b) F. Guibe, Tetrahedron, 1997, 53, 13509 CrossRef CAS; (c) S. Escoubet, S. Gastaldi and M. Bertrand, Eur. J. Org. Chem., 2005, 3855 CrossRef CAS.
- (a) F. W. Sum and L. Weiler, Can. J. Chem., 1979, 57, 1431 CrossRef CAS; (b) M. Ide and M. Nakata, Synlett, 2001, 1511 CrossRef CAS.
- (a) A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef CAS PubMed; (b) P. Polak, H. Vanova, D. Dvorak and T. Tobrman, Tetrahedron Lett., 2016, 57, 3684 CrossRef CAS; (c) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863 CrossRef CAS; (d) Y. Ashida and Y. Tanabe, Chem. Rec., 2020, 20, 1 CrossRef.
- (a) C. Gürtler and S. L. Buckwald, Chem. - Eur. J., 1999, 5, 3107 CrossRef; (b) M. Shindo, Y. Sato, T. Yoshikawa, R. Koretsune and K. Shishido, J. Org. Chem., 2004, 69, 3912 CrossRef CAS PubMed.
- (a) A. B. Lemay, K. S. Vulic and W. W. Oglivie, J. Org. Chem., 2006, 71, 3615 CrossRef CAS PubMed; (b) J. S. Mercier, A. B. Flynn and W. W. Ogilvie, Tetrahedron, 2008, 64, 5472 CrossRef.
- (a) Y. Yamamoto, N. Kirai and Y. Harada, Chem. Commun., 2008, 2010 RSC; (b) N. Kirai and Y. Yamamoto, Org. Synth., 2010, 87, 53 CrossRef CAS; (c) Z. He, S. Kirchberg, R. Froehlich and A. Studer, Angew. Chem., Int. Ed., 2012, 124, 3759 CrossRef.
- (a) J. M. Baxter, D. Steinhuebel, M. Palucki and I. W. Davies, Org. Lett., 2005, 7, 215 CrossRef CAS PubMed; (b) A. Klapars, K. R. Campos, C. Chen and R. P. Volante, Org. Lett., 2005, 7, 1185 CrossRef CAS PubMed; (c) H. Nakatsuji, K. Ueno, T. Misaki and Y. Tanabe, Org. Lett., 2008, 10, 2131 CrossRef CAS PubMed; (d) H. Nakatsuji, H. Nishikado, K. Ueno and Y. Tanabe, Org. Lett., 2009, 11, 4258 CrossRef CAS PubMed.
- Y. Ashida, K. Nakata, D. Yoshitake, Y. Sato, Y. Miyazaki and Y. Tanabe, Asian J. Org. Chem., 2020, 9, 604 CrossRef CAS.
- (a) A. L. Hansen, J. P. Ebran, M. Ahlquist, P. O. Norrby and T. Skrydstrup, Angew. Chem., Int. Ed., 2006, 45, 3349 CrossRef CAS PubMed; (b) T. Hayashi, T. Fujiwa, Y. Okamoto, Y. Katsuro and M. Kumada, Synthesis, 1981, 1001 CrossRef CAS; (c) K. C. Nicolaou, G. Q. Shi, G. P. Grtner and Z. Yang, J. Am. Chem. Soc., 1997, 119, 5467 CrossRef CAS; (d) A. S. E. Karlstrçm, K. Itami and J. E. Bckvall, J. Org. Chem., 1999, 64, 1745 CrossRef PubMed; (e) J. Wu and Z. Yang, J. Org. Chem., 2001, 66, 7875 CrossRef CAS PubMed; (f) J. P. Ebran, A. L. Hansen, T. M. Gøgsig and T. Skrydstrup, J. Am. Chem. Soc., 2007, 129, 6931 CrossRef CAS PubMed; (g) A. L. Hansen, J.-P. Ebran, T. M. Gøgsig and T. Skrydstrup, J. Org. Chem., 2007, 72, 6464 CrossRef CAS PubMed; (h) W. You, Y. Li and M. K. Brown, Org. Lett., 2013, 15, 1610 CrossRef CAS PubMed.
- (a) T. M. Gøgsig, A. T. Lindhardt and T. Skrydstrup, Org. Lett., 2009, 11, 4886 CrossRef PubMed; (b) J. Jiang, R. DeVita, G. Doss, M. Goulet and M. Wyvratt, J. Am. Chem. Soc., 1999, 121, 593 CrossRef CAS; (c) A. L. Hansen, J. P. Ebran, T. M. Gøgsig and T. Skrydstrup, Chem. Commun., 2006, 39, 4137 RSC; (d) J. W. Coe, Org. Lett., 2000, 2, 4205 CrossRef CAS PubMed.
- H. Nakatsuji, Y. Ashida, H. Hori, Y. Sato, M. Taira and Y. Tanabe, Org. Biomol. Chem., 2015, 13, 8205 RSC.
- (a) Y. Cao, Z. Gao, J. Li, X. Bi, L. Yuan, C. Pei, Y. Guo and E. Shi, RSC Adv., 2020, 10, 29493 RSC; (b) H. Guo, Y. Zhang, Z. Li, P. Zhao, N. Li and E. Shi, RSC Adv., 2022, 12, 14844 RSC.
- S. G. Alcock, J. E. Baldwin, R. Bohlmann, L. M. Harwood and J. Seeman, J. Org. Chem., 1985, 50, 3526 CrossRef CAS.
- (a) M. Hassam, A. Taher, G. E. Arnott, I. R. Green and W. A. L. van Otterlo, Chem. Rev., 2015, 115, 5462 CrossRef CAS PubMed; (b) D. J. Cram and R. T. Uyeda, J. Am. Chem. Soc., 1964, 86, 5466 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and analytical data. CCDC 2250165. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra02430j |
| This journal is © The Royal Society of Chemistry 2023 |