
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrrolo[2,3-e]indazole as a novel chemotype for both influenza A virus and pneumococcal neuraminidase inhibitors†
Anna Egorova‡
 a,
Martina Richter‡b,
Maria Khrenovaac,
Elisabeth Dietrichb,
Andrey Tsedilina,
Elena Kazakovaa,
Alexander Lepioshkina,
Birgit Jahnb,
Vladimir Chernyshevcd,
Michaela Schmidtke*b and
Vadim Makarov*a
a,
Martina Richter‡b,
Maria Khrenovaac,
Elisabeth Dietrichb,
Andrey Tsedilina,
Elena Kazakovaa,
Alexander Lepioshkina,
Birgit Jahnb,
Vladimir Chernyshevcd,
Michaela Schmidtke*b and
Vadim Makarov*a
aFederal Research Centre “Fundamentals of Biotechnology” of the Russian Academy of Sciences (Research Centre of Biotechnology RAS), 33-2 Leninsky Prospect, 119071 Moscow, Russia. E-mail: makarov@inbi.ras.ru
bDepartment of Medical Microbiology, Section of Experimental Virology, Jena University Hospital, Hans-Knöll-Straße 2, 07745 Jena, Germany. E-mail: michaela.schmidtke@med.uni-jena.de
cChemistry Department, Lomonosov Moscow State University, 1-3 Leninskie Gory, 119991 Moscow, Russia
dFrumkin Institute of Physical Chemistry and Electrochemistry of the Russian Academy of Sciences, 31-4 Leninsky Prospect, 119071 Moscow, Russia
First published on 21st June 2023
Abstract
Influenza infections are often exacerbated by secondary bacterial infections, primarily caused by Streptococcus pneumoniae. Both respiratory pathogens have neuraminidases that support infection. Therefore, we hypothesized that dual inhibitors of viral and bacterial neuraminidases might be an advantageous strategy for treating seasonal and pandemic influenza pneumonia complicated by bacterial infections. By screening our in-house chemical library, we discovered a new chemotype that may be of interest for a further campaign to find small molecules against influenza. Our exploration of the pyrrolo[2,3-e]indazole space led to the identification of two hit compounds, 6h and 12. These molecules were well-tolerated by MDCK cells and inhibited the replication of H3N2 and H1N1 influenza A virus strains. Moreover, both compounds suppress viral and pneumococcal neuraminidases indicating their dual activity. Given its antiviral activity, pyrrolo[2,3-e]indazole has been identified as a promising scaffold for the development of novel neuraminidase inhibitors that are active against influenza A virus and S. pneumoniae.
Influenza A and B viruses cause seasonal epidemics of acute respiratory diseases with an annual death mortality of 290
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–650000 people.1 Moreover, there have been at least five influenza A virus pandemics in the past 150 years, such as the Spanish flu (1918–1920), Asian flu (1957–1958), Hong Kong flu (1968–1969), Russian flu (1977–1979) and swine flu (2009–2010), claiming millions of lives.2–4 These viral infections are often exacerbated by co/secondary bacterial infections, most commonly caused by Streptococcus pneumoniae, Staphylococcus aureus, Streptococcus pyogenes, or Haemophilus influenzae.5,6 Indeed, such a secondary infection may cause high mortality from severe pneumonia. Historical records and several systematic reviews indicate that a high number of deaths during the influenza pandemics were the result of secondary bacterial pneumonia rather than primary influenza infection.7–16 Co/secondary bacterial infection was identified in 25–95% of cases during the 1918, 1968, and 2009 pandemics, with S. pneumoniae being the most common pathogen.14–16 Murine studies have shown that influenza A virus promotes S. pneumoniae transmission and infection,17 demonstrating a lethal synergism between the virus and the bacterium.18,19 Hence, therapeutic approaches targeting both pathogens would enable the improvement of global healthcare systems against future respiratory epidemics and pandemics.
000–650000 people.1 Moreover, there have been at least five influenza A virus pandemics in the past 150 years, such as the Spanish flu (1918–1920), Asian flu (1957–1958), Hong Kong flu (1968–1969), Russian flu (1977–1979) and swine flu (2009–2010), claiming millions of lives.2–4 These viral infections are often exacerbated by co/secondary bacterial infections, most commonly caused by Streptococcus pneumoniae, Staphylococcus aureus, Streptococcus pyogenes, or Haemophilus influenzae.5,6 Indeed, such a secondary infection may cause high mortality from severe pneumonia. Historical records and several systematic reviews indicate that a high number of deaths during the influenza pandemics were the result of secondary bacterial pneumonia rather than primary influenza infection.7–16 Co/secondary bacterial infection was identified in 25–95% of cases during the 1918, 1968, and 2009 pandemics, with S. pneumoniae being the most common pathogen.14–16 Murine studies have shown that influenza A virus promotes S. pneumoniae transmission and infection,17 demonstrating a lethal synergism between the virus and the bacterium.18,19 Hence, therapeutic approaches targeting both pathogens would enable the improvement of global healthcare systems against future respiratory epidemics and pandemics.
Neuraminidase (NA) is an important target in modern anti-influenza drug discovery campaigns.20–22 Neuraminidase is a key enzyme in an influenza virus lifecycle: it facilitates viral release and spread. Its function is to catalyze the hydrolysis of sialic acid residues from host-cell glycoprotein receptors.23,24 Influenza virus neuraminidase supports S. pneumoniae infection, released sialic acids can be catabolized by these bacteria.25,26 Moreover, the resulting de-sialylated glycoproteins can be used by S. pneumoniae to bind to lung epithelial cells.5,27–29 S. pneumoniae also express neuraminidases – NanA, NanB (both found in most strains), and NanC (rarely identified).30 NanA and NanB play important roles in host colonization.31,32 The active sites of influenza virus and pneumococcal neuraminidases show structural similarity, suggesting that S. pneumoniae could be targeted by neuraminidase inhibitors.33,34 Indeed, oseltamivir is active against both viral and bacterial neuraminidases in vitro and improves survival in a mouse model of influenza A virus–S. pneumoniae synergism.35,36 Another NA inhibitor, peramivir, also showed efficacy in influenza virus-infected mice co-infected with S. pneumoniae, demonstrating prolonged survival rate and reduced bacterial burden and virus titre.37 However, no studies aimed to determine the effect of these drugs on the treatment or prevention of secondary bacterial complications following influenza infection in humans. Although resistance to these drugs occurs infrequently, resistant influenza viruses can emerge and become a major problem as seen in case of oseltamivir in the 2008 influenza season.38–40 Therefore, the search for novel compounds with activity against both influenza virus and pneumococcal neuraminidases may be an attractive way for the development of dual-acting anti-infective agents.
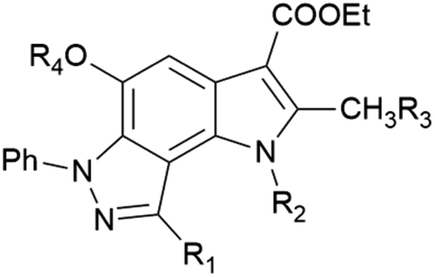
In the ongoing anti-influenza virus screening for novel dual-active neuraminidase inhibitors, we found that compounds based on a previously unexplored chemotype, pyrrolo[2,3-e]indazole, prevent virus replication by inhibiting the activity of viral neuraminidase. We then obtained a series of pyrrolo[2,3-e]indazole-core compounds with various substituents at positions R1–R4 to study structure–activity relationship. Pyrrolo[2,3-e]indazole is a heterocyclic system rarely mentioned in the scientific literature. We synthesized these compounds using the aza-Nenitzescu reaction as previously described by Lyubchanskaya and colleagues.41,42 According to Scheme 1, commercially available 1,4-benzoquinone 1 is treated with the corresponding benzaldehyde phenylhydrazones 2a–f to afford hydroquinone adducts 3a–f. Subsequent oxidation of 3a–f with an aqueous solution of potassium ferrocyanide and potassium carbonate leads to indazolequinones 4a–f.42 These intermediates are then reacted with the corresponding commercially available aminocrotonic esters 5a–d in the presence of acetic acid and acetic anhydride to cyclize into the final pyrrolo[2,3-e]indazoles 6a–k.41
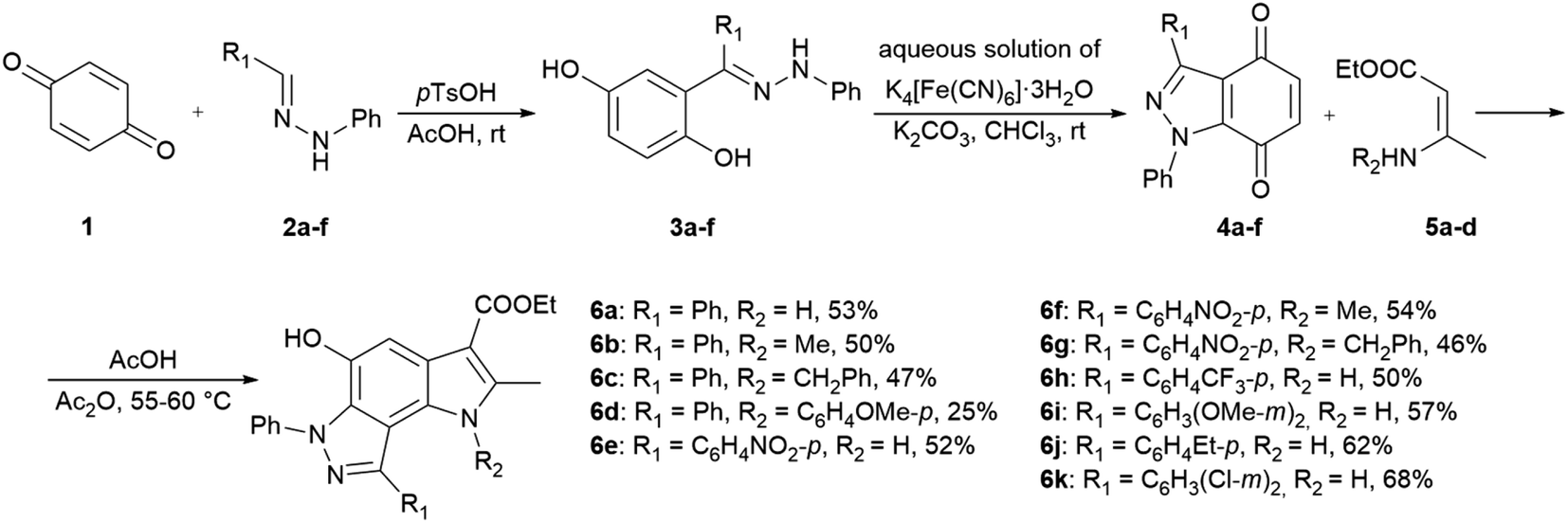 | ||
| Scheme 1 Synthesis of unsubstituted and substituted derivatives of 4-phenyl-pyrrolo[2,3-e]indazole 6a–k. | ||
2-Aminoalkyl-pyrrolo[2,3-e]indazoles 9a–c and 10a,b were synthesized according to Scheme 2 from pyrroloindazoles 6b,c. The protection of the 5-hydroxy groups of 6b,c with an acetyl group followed by bromination with bromosuccinimide (NBS) in the presence of benzoyl peroxide provides the corresponding 2-bromomethyl derivatives 8a,b. Further nucleophilic substitution of the bromine atom with piperidine, diethylamine, or morpholine results in the 2-substituted derivatives 9a–c. Deprotection of the acetyl group in 9a,c under basic condition gives 2-hydroxypyrrolo[2,3-e]indazoles 10a,b.
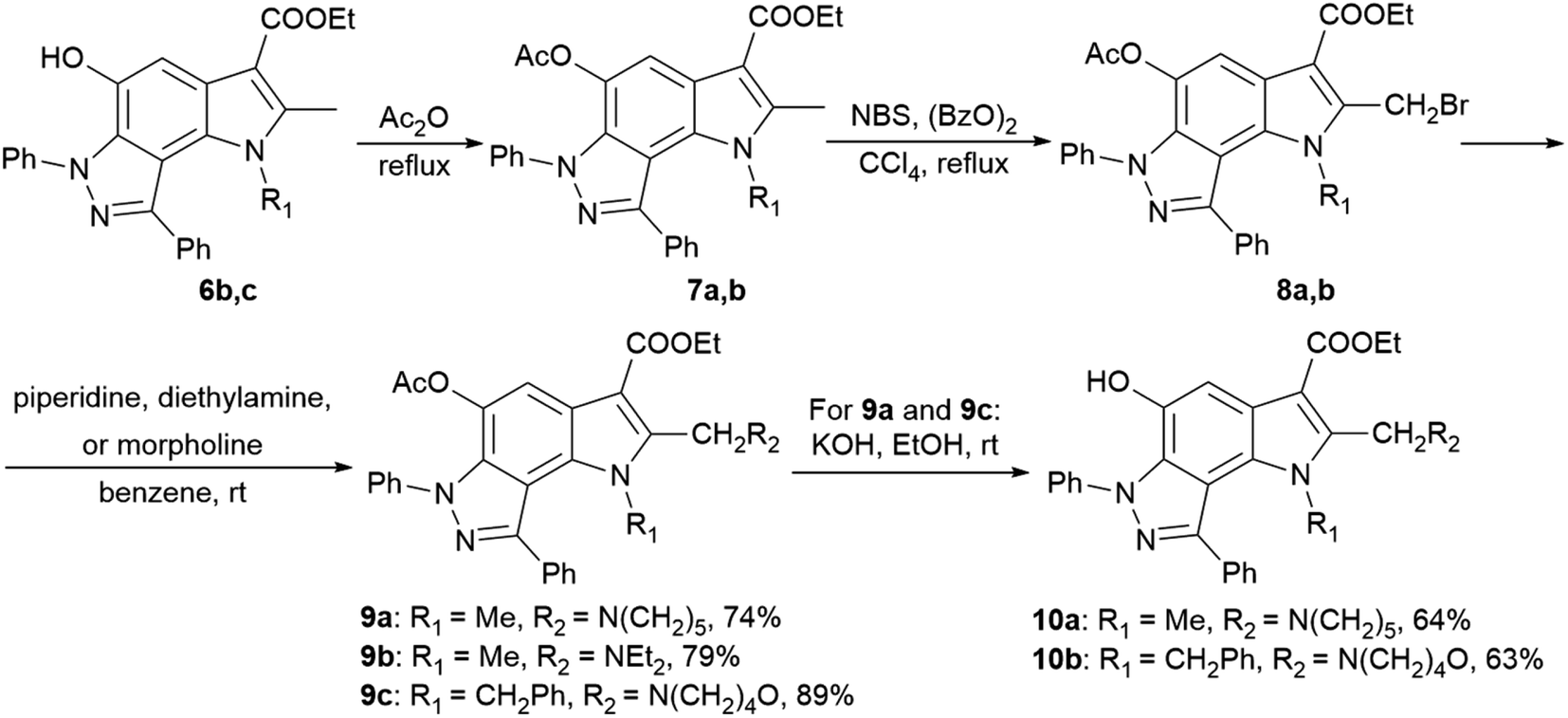 | ||
| Scheme 2 Synthesis of 2-aminoalkyl-pyrrolo[2,3-e]indazoles 9a–c and 10a,b. | ||
Using 4-nitrophenyl-pyrroloindazole 6e as a starting point, we synthesized a number of derivatives according to Scheme 3. O-Acylation with acetic anhydride of compound 6e yields 5-acetyloxy-4-nitrophenyl-pyrroloindazole 11e. Mono-(5-C) and di-substituted (1,5-C) derivatives 11a–d were synthesized via alkylation of 6e with the corresponding alkyl iodides in the presence of potassium carbonate in NMP medium. Further, we reduced the nitro group of compound 6e by catalytic hydrogenation with Pd/C to obtain 4-aminophenyl-pyrroloindazole 13. As for 6e, we performed an O-acylation with acetic anhydride for compound 12 to form 5-acetyloxy-4-aminophenyl-pyrroloindazole 13. The pyrrole derivative 14 was prepared by condensation between primary amine of 12 and 2,5-dimethoxytetrahydrofuran in the presence of acetic acid as a catalyst.
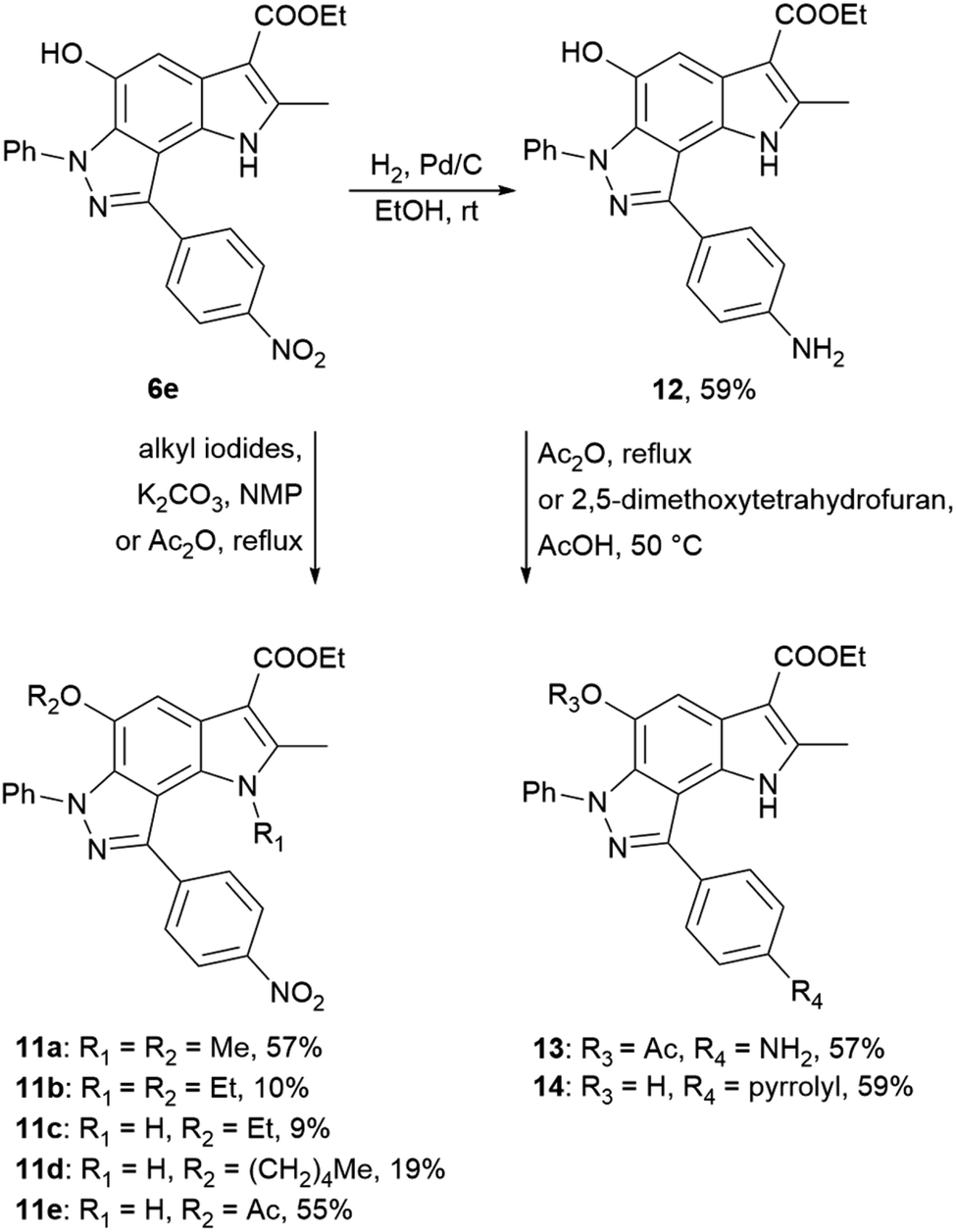 | ||
| Scheme 3 Synthesis of 4-nitrophenyl-, 4-aminophenyl-pyrrolo[2,3-e]indazole 6e, 12 and their derivatives 11a–e and 13, 14. | ||
To get a detailed insight into the structural information, X-ray diffraction analysis for compounds 6a and 12 was performed. The crystallographic data were deposited with the Cambridge Crystallographic Data Centre (CCDC ID 2246679 for compound 6a and 2249815 for compound 12).† X-ray crystal structure of 6a established its identity as a 6,8-diphenyl-pyrrolo[2,3-e]indazole (Fig. 1A and Table S1†). All bond lengths and angles in compound 12 correspond well to those observed in compound 6a and related compounds from the Cambridge Structural Database (CSD) (Fig. 1B and Table S1†). The triclinic syngony and proximity of unit cell dimensions in both compounds may indicate their crystal packing similarity. Surprisingly, both compounds form similar centrosymmetric hydrogen-bonded dimers despite the different numbers of active sites in their structures. Indeed, the hydroxy group in compound 12 participates in the formation of these dimers similar to the same group in 6a with close geometries (Fig. 1C and D). Specifically, the O1⋯O3(1 − x, 2 − y, 1 − z) distance in 6a is 2.705(4) Å, while in 12 the O1⋯O3(1 − x, −y, 1 − z) distance is 2.711(9) Å. Interestingly, the amino group of compound 12 does not involve in any hydrogen bonding formation. Instead, it participates in weak intermolecular N⋯H⋯π interactions that bind centrosymmetric dimers into ribbon-like stretches (Fig. S2†).
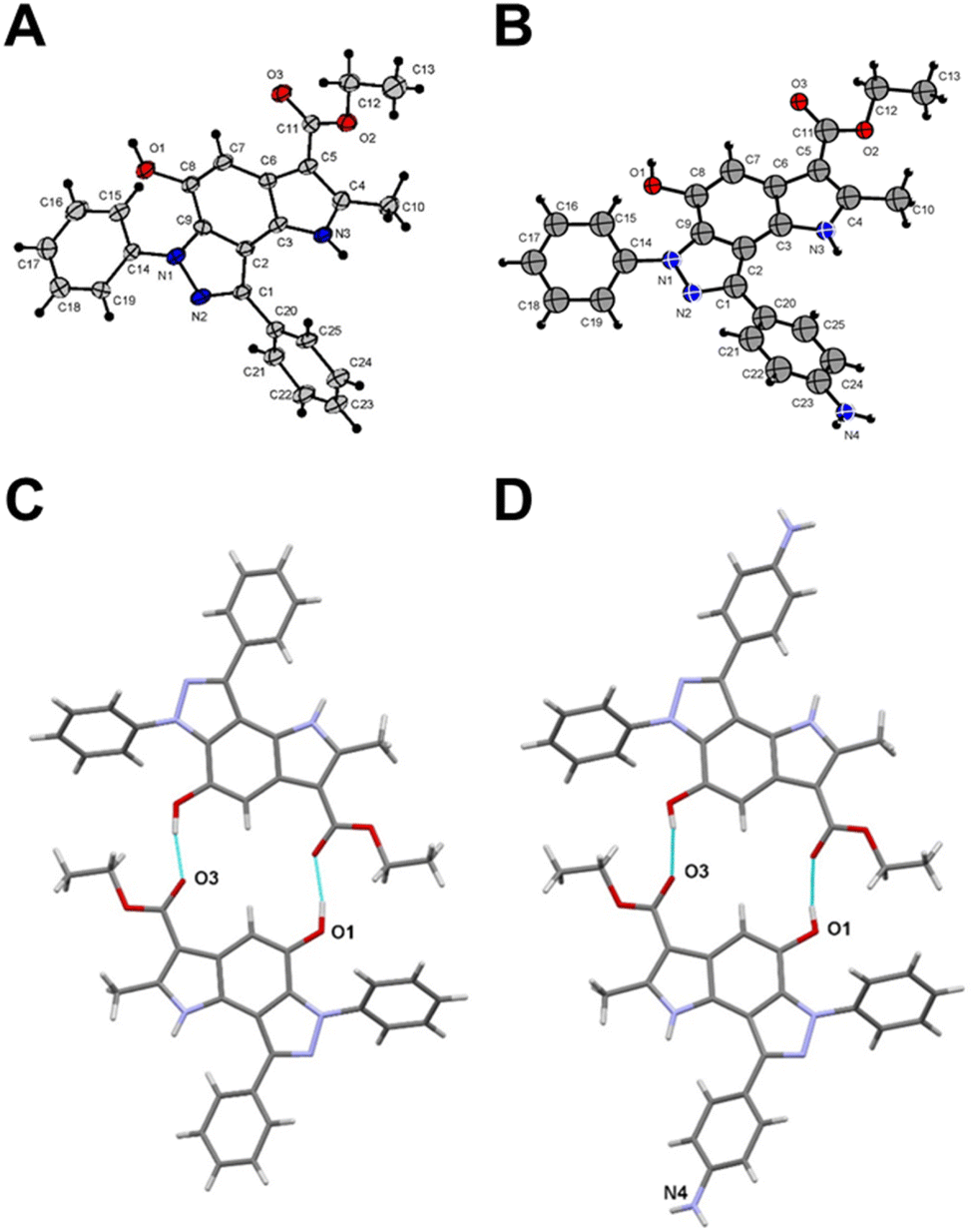 | ||
| Fig. 1 Crystal structure of compound 6a (A) and compound 12 (B). Centrosymmetric hydrogen-bonded dimers in compound 6a (C) and compound 12 (D). Thin cyan lines indicate intermolecular O⋯H⋯O hydrogen bonds. | ||
As a part of an anti-influenza screening, we evaluated synthesized compounds against influenza A virus A/HK/1/68 (subtype H3N2) and influenza A virus A/Jena/8178/09 (subtype A(H1N1)pdm09). Cytotoxicity and antiviral activity of the target compounds on MDCK cells were shown in Table 1. All pyrrolo[2,3-e]indazole-core compounds, regardless of structural fragments, showed no cytotoxicity at the maximum tested concentration, 100 μM, for MDCK cells supporting their further study for antiviral activity. The results of the structure–activity relationship study indicate a viral subtype-specific activity of the scaffold. For example, a number of compounds containing different substituents at positions R2–R4 exclusively inhibited the cytopathic effect induced by influenza virus A/Jena/8178/09 of subtype A(H1N1)pdm09 (compounds 6b–d, 7a,b, 9c, 10a,b, 11a–e, 14). In contrast, pyrroloindazoles with a small group at the R2 position (compound 6f) or without substituents at the R2–R4 positions (compounds 6j,k) exhibited activity only against influenza A virus A/HK/1/68 of subtype H3N2. At the same time, inhibition of both influenza A virus subtypes was shown for compounds without any groups at positions R2–R4 (exception for compound 13, R4 = Ac) and with a R1-phenyl ring containing para-nitro, amino or trifluoro groups (compounds 6e, 6h, 12). The control compounds oseltamivir and zanamivir acted as expected.
|
|||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | R4 | CC50 (MDCK), μM | IC50 (IAV), μM | |
| HK/1/68 | Jena/8178 | ||||||
| a n.a. – not active.b n.t. – not tested. | |||||||
| 6a | Ph | H | H | H | >100 | n.a.a | n.a. |
| 6b | Ph | Me | H | H | >100 | n.a. | 31.60 |
| 6c | Ph | CH2Ph | H | H | >100 | n.a. | 71.09 |
| 6d | Ph | C6H4OMe-p | H | H | >100 | n.a. | 38.59 |
| 6e | C6H4NO2-p | H | H | H | >100 | 16.37 | 19.54 |
| 6f | C6H4NO2-p | Me | H | H | >100 | 42.53 | n.a. |
| 6g | C6H4NO2-p | CH2Ph | H | H | >100 | n.a. | n.t.b |
| 6h | C6H4CF3-p | H | H | H | >100 | 30.48 | 24.83 |
| 6i | C6H3(OMe-m)2 | H | H | H | n.t. | n.t. | n.t. |
| 6j | C6H4Et-p | H | H | H | >100 | 52.23 | n.t. |
| 6k | C6H3(Cl-m)2 | H | H | H | >100 | 30.18 | n.t. |
| 7a | Ph | Me | H | Ac | >100 | n.a. | 83.73 |
| 7b | Ph | CH2Ph | H | Ac | >100 | n.a. | 38.66 |
| 9a | Ph | Me | N(CH2)5 | Ac | >100 | n.a. | n.a. |
| 9b | Ph | Me | NEt2 | Ac | >100 | n.a. | 53.19 |
| 9c | Ph | CH2Ph | N(CH2)4O | Ac | n.t. | n.t. | n.t. |
| 10a | Ph | Me | N(CH2)5 | H | >100 | n.a. | 86.70 |
| 10b | Ph | CH2Ph | N(CH2)4O | H | >100 | n.a. | 33.16 |
| 11a | C6H4NO2-p | Me | H | Me | >100 | n.a. | 21.52 |
| 11b | C6H4NO2-p | Et | H | Et | >100 | n.a. | 25.48 |
| 11c | C6H4NO2-p | H | H | Et | >100 | n.a. | 30.26 |
| 11d | C6H4NO2-p | H | H | Pentyl | >100 | n.a. | 26.49 |
| 11e | C6H4NO2-p | H | H | Ac | >100 | n.a. | 16.67 |
| 12 | C6H4NH2-p | H | H | H | >100 | 8.54 | 15.69 |
| 13 | C6H4NH2-p | H | H | Ac | >100 | 9.39 | 14.08 |
| 14 | C6H4-pyrrolyl | H | H | H | >100 | n.a. | 11.37 |
| Oseltamivir | n.t. | 0.004 | 0.14 | ||||
| Zanamivir | n.t. | n.t. | 0.11 | ||||
To investigate whether viral hemagglutinin or neuraminidase represent a target for pyrrolo[2,3-e]indazole-core compounds, we performed a human erythrocyte-based assay with influenza A virus A/Jena/8178/09. Viral hemagglutinin causes hemagglutination of these erythrocytes at 4 °C, and viral neuraminidase abrogates hemagglutination of human erythrocytes after incubation of the assays at 37 °C. Inhibitors targeting the viral hemagglutinin or neuraminidase might block hemagglutination and/or its abrogation, respectively. With the exception of compound 13 (hemagglutination at 31.6 μM), our compounds inhibited hemagglutination weakly or not at all (results not shown). At the same time, all small molecules were found to inhibit the viral neuraminidase in the assay (Table 2). Among them, compounds 6h, 11a, 11e, 12, and 14 were the most active in the series with MICs in the range of 14.20–24.40 μM. The exception is compound 9a, which does not inhibit either viral neuraminidase activity (Table 2) or viral replication in the CPE reduction assay (Table 1).
| Cmpd | MIC, μM | Cmpd | MIC, μM | |||
|---|---|---|---|---|---|---|
| IAV NA | S.p. NanA | IAV NA | S.p. NanA | |||
| a Published.35 | ||||||
| 6a | 70.00 | 31.60 | 9b | 77.20 | 24.40 | |
| 6b | 31.60 | 31.60 | 10a | 54.40 | 31.60 | |
| 6c | 77.20 | 24.40 | 10b | 54.40 | 10.00 | |
| 6d | 54.40 | 17.20 | 11a | 24.40 | 31.60 | |
| 6e | 54.40 | 14.32 | 11b | 77.20 | 54.40 | |
| 6f | 77.20 | 31.60 | 11c | 47.20 | 54.40 | |
| 6h | 24.40 | 10.00 | 11e | 17.20 | 10.00 | |
| 7a | 100.00 | 31.60 | 12 | 21.40 | 10.00 | |
| 7b | 100.00 | 31.60 | 13 | 14.20 | n.a. | |
| 9a | n.a. | 54.40 | 14 | 24.40 | 10.00 | |
| Oseltamivir | 1.97 | 2.08a | Zanamivir | 3.16 | n.a.a | |
Structural similarities between influenza and pneumococcal neuraminidases in their active sites assume that both neuraminidases can be targeted simultaneously by one neuraminidase small-molecule inhibitor.33,34 For example, oseltamivir, but not zanamivir, exerts such dual activity (Table 2).35,43 Inspired by the idea of developing small-molecule dual-acting neuraminidase inhibitors, we evaluated whether our pyrroloindazoles would also act on pneumococcal neuraminidase. To do this, we performed a hemagglutination-based NA inhibition assays with the neuraminidase NanA of S. pneumoniae strain DSM20566. With the exception of compound 13 all of the compounds tested were found to inhibit the enzyme at moderate concentrations (Table 2). Among them, pyrroloindazoles 6h, 10b, 11e, 12 and 14 have the highest NanA-inhibitory activity with a MIC of 10.0 μM. Taking into account the above results, we represent compounds 6h and 12 as interesting molecules with dual neuraminidase inhibitory activity. Preliminary results of the structure–activity relationship for pyrrolo[2,3-e]indazole-core compounds are shown in Fig. 2A.
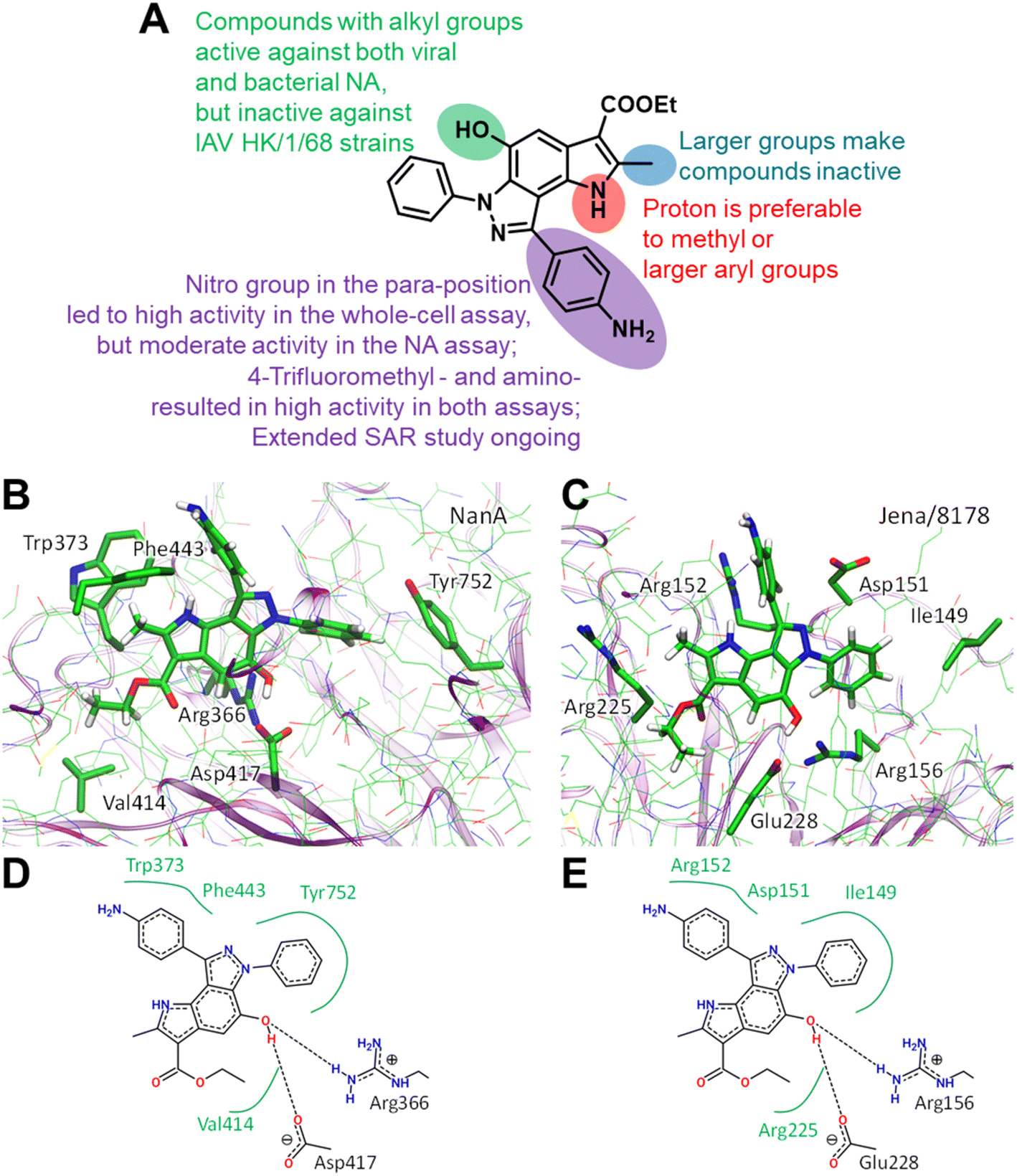 | ||
| Fig. 2 Preliminary SAR results (A). Predicted binding modes ((B and C) 3D representation; (D and E) 2D representation) for compound 12 bound to Streptococcus pneumoniae NanA (B and D) and H1N1 (C and E) neuraminidases. In 2D models hydrogen bonds are shown as dashed lines and hydrophobic interactions as green curves. Color code: carbon – green, nitrogen – blue, oxygen – red, hydrogen – white. Hydrogen atoms are shown only for compound 12. | ||
Classic neuraminidase inhibitors, zanamivir and oseltamivir, share the same structural feature, that is the carboxylate group trapping three guanidinium groups of the active site arginine residues.44–46 This structural pattern is similar to the transition state, which is formed in the NA active site during the cleavage of sialic acid from glycoconjugates.47 In contrast, our compounds do not have such carboxylate substituent in their structures. Moreover, other anionic groups are not presented either. Therefore, the search for a binding site seems challenging, especially as these compounds are active to both viral and bacterial NAs and, therefore, should form stable complexes with two proteins.
We initially performed molecular docking of pyrrolo[2,3-e]indazole 12 to the wide area around the active sites of both viral and bacterial neuraminidases. The set of complexes obtained was then studied using classical MD simulations. Most of the complexes dissociated after a short simulation time (less than 10 ns). However, we obtained another binding site that was stable for both NAs for longer than 100 ns (Fig. S1†). Despite the low amino acid sequence similarity between these proteins, we found that compound 12 binds similarly to both of them (Fig. 2). Compound 12 is anchored by two hydrogen bonds with a negatively charged carboxylate, Asp417 in NanA and Glu228 in viral NA, and a positively charged side chain of an arginine residue, Arg366 in NanA and Arg156 in viral NA. Importantly, this arginine residue is not among those that are responsible for substrate carboxylate binding. Compound 12 and its analogues have hydrophobic fragments complementary to the hydrophobic fragments of NA binding sites (Fig. 2).
Another important bacterial NA is NanB, therefore we checked whether this binding site is conserved for NanB. We reconstructed the binding mode similar to that in NanA and viral NA, and performed MD simulation. We found that the complex remains stable, and the structural patterns are the same as for NAs discussed above (Fig. S1†). Thus, we suppose that compound 12 may inhibit NanB as well. However, additional in vitro enzyme studies are needed to prove this.
Some studies reported that pneumococcal neuraminidase NanA may impact biofilm formation.48,49 So, biofilm formation of S. pneumoniae probably can be affected by neuraminidase inhibitors. We have recently observed that some neuraminidase inhibitors suppress bacterial planktonic growth and biofilm formation.50–52 In contrast, the influenza drugs oseltamivir and zanamivir do not inhibit either planktonic growth or biofilm formation (Table 3).50,51 Similarly, we observed no inhibition of planktonic growth and/or biofilm production of S. pneumoniae by most of the pyrrolo[2,3-e]indazoles studied (Table 3). Compounds 6c, 6h, and 10a active against bacterial NanA (Table 2) moderately affected S. pneumoniae planktonic growth as well as biofilm formation (Table 3). However, there is not enough evidence to conclude whether the inhibition of pneumococcal growth and biofilm formation by these molecules were the result of NanA inhibition.
| Cmpd | IC50, μM | |
|---|---|---|
| Planktonic growth | Biofilm formation | |
| 6c | 8.90 | 0.93 |
| 6h | 50.00 | 40.13 |
| 10a | 6.88 | 3.31 |
| 10b | 7.57 | n.a. |
| 14 | n.a. | 28.04 |
| Oseltamivir | n.a. | n.a. |
| Zanamivir | n.a. | n.a. |
As a result of our antiviral studies, we have discovered pyrrolo[2,3-e]indazoles as a novel class of small molecule inhibitors targeting influenza A virus neuraminidase. According to the results of structure–activity studies, the R1-phenyl ring containing para-nitro, amino or trifluoro groups of pyrrolo[2,3-e]indazoles is important for the inhibition of both circulating influenza A virus subtypes H3N2 and H1N1. Therapeutic advantage of pyrrolo[2,3-e]indazoles might represent their dual activity against the viral neuraminidase as well as the structurally related neuraminidase NanA of S. pneumoniae. Molecular dynamic simulations demonstrate that the ethyl ester moiety, hydroxy groups, and the pyrrolo[2,3-e]indazole core system are responsible for the formation of stable complexes with active sites of both viral and bacterial neuraminidases. Some pyrrolo[2,3-e]indazoles also inhibited bacterial growth. Taken together these results led us to conclude that pyrrolo[2,3-e]indazole represents novel hit compounds warranting further development.
Author contributions
AE: investigation, data curation, visualization, writing – original draft, writing – review & editing; MR: investigation; MK: investigation, data curation, visualization, writing – review & editing; ED: investigation; AT: investigation, data curation, formal analysis; EK: investigation; AL: investigation; BJ: investigation; VC: investigation, visualization, writing – review & editing; MS: conceptualization, methodology, resources, data curation, supervision, funding acquisition, writing – original draft, writing – review & editing; VM: conceptualization, methodology, resources, supervision, writing – original draft, writing – review & editing.All authors discussed the manuscript and approved the submitted version of the paper.
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported in part by the European Social Fund (ESF & TMWAT Project 2011 FGR 0137) to MR, ED, BJ, and MS.References
- World Health Organization, Influenza (Seasonal), https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal), accessed 2023-01-23 Search PubMed.
- E. D. Kilbourne, Influenza pandemics of the 20th century, Emerging Infect. Dis., 2006, 12(1), 9–14, DOI:10.3201/eid1201.051254.
- S. J. Sullivan, R. M. Jacobson, W. R. Dowdle and G. A. Poland, 2009 H1N1 influenza, Mayo Clin. Proc., 2010, 85(1), 64–76, DOI:10.4065/mcp.2009.0588.
- M. P. Girard, J. S. Tam, O. M. Assossou and M. P. Kieny, The 2009 A (H1N1) influenza virus pandemic: A review, Vaccine, 2010, 28(31), 4895–4902, DOI:10.1016/j.vaccine.2010.05.031.
- J. A. McCullers, The co-pathogenesis of influenza viruses with bacteria in the lung, Nat. Rev. Microbiol., 2014, 12, 252–262, DOI:10.1038/nrmicro3231.
- D. E. Morris, D. W. Cleary and S. C. Clarke, Secondary bacterial infections associated with influenza pandemics, Front. Microbiol., 2017, 8, 1041, DOI:10.3389/fmicb.2017.01041.
- J. F. Hers, N. Masurel and J. Mulder, Bacteriology and histopathology of the respiratory tract and lungs in fatal Asian influenza, Lancet, 1958, 2(7057), 1141–1143, DOI:10.1016/s0140-6736(58)92404-8.
- M. I. Lindsay Jr, E. C. Herrmann Jr, G. W. Morrow Jr and A. L. Brown Jr, Hong Kong influenza: clinical, microbiologic, and pathologic features in 127 cases, JAMA, J. Am. Med. Assoc., 1970, 214(10), 1825–1832, DOI:10.1001/jama.214.10.1825.
- S. W. Schwarzmann, J. L. Adler, R. J. Sullivan Jr and W. M. Marine, Bacterial pneumonia during the Hong Kong influenza epidemic of 1968-1969, Arch. Intern. Med., 1971, 127(6), 1037–1041, DOI:10.1001/archinte.1971.00310180053006.
- J. F. Brundage and G. D. Shanks, Deaths from bacterial pneumonia during 1918–1919 influenza pandemic, Emerging Infect. Dis., 2008, 14(8), 1193–1199, DOI:10.3201/eid1408.071313.
- D. M. Morens, J. K. Taubenberger and A. S. Fauci, Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness, J. Infect. Dis., 2008, 198(7), 962–970, DOI:10.1086/591708.
- K. P. Klugman, C. M. Astley and M. Lipsitch, Time from illness onset to death, 1918 influenza and pneumococcal pneumonia, Emerg. Infect. Dis., 2009, 15(2), 346–347, DOI:10.3201/eid1502.081208.
- Centers for Disease Control and Prevention (CDC), Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A (H1N1) – United States, May-August 2009, Morb. Mortal. Wkly. Rep., 2009, 58(38), 1071–1074 Search PubMed.
- C. Joseph, Y. Togawa and N. Shindo, Bacterial and viral infections associated with influenza, Influenza Other Respir. Viruses, 2013, 7(Suppl 2), 105–113, DOI:10.1111/irv.12089.
- K. R. Short, K. Kedzierska and C. E. van de Sandt, Back to the future: lessons learned from the 1918 influenza pandemic, Front. Cell. Infect. Microbiol., 2018, 8, 343, DOI:10.3389/fcimb.2018.00343.
- C. R. MacIntyre, A. A. Chughtai, M. Barnes, I. Ridda, H. Seale, R. Toms and A. Heywood, The role of pneumonia and secondary bacterial infection in fatal and serious outcomes of pandemic influenza a(H1N1)pdm09, BMC Infect. Dis., 2018, 18(1), 637, DOI:10.1186/s12879-018-3548-0.
- D. A. Diavatopoulos, K. R. Short, J. T. Price, J. J. Wilksch, L. E. Brown, D. E. Briles, R. A. Strugnell and O. L. Wijburg, Influenza A virus facilitates Streptococcus pneumoniae transmission and disease, FASEB J., 2010, 24(6), 1789–1798, DOI:10.1096/fj.09-146779.
- J. A. McCullers and J. E. Rehg, Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor, J. Infect. Dis., 2002, 186, 341–350, DOI:10.1086/341462.
- J. C. Kash, K. A. Walters, A. S. Davis, A. Sandouk, L. M. Schwartzman, B. W. Jagger, D. S. Chertow, Q. Li, R. E. Kuestner, A. Ozinsky and J. K. Taubenberger, Lethal synergism of 2009 pandemic H1N1 influenza virus and Streptococcus pneumoniae coinfection is associated with loss of murine lung repair responses, mBio, 2011, 2(5), e00172-11, DOI:10.1128/mBio.00172-11.
- X. Wu, X. Wu, Q. Sun, C. Zhang, S. Yang, L. Li and Z. Jia, Progress of small molecular inhibitors in the development of anti-influenza virus agents, Theranostics, 2017, 7(4), 826–845, DOI:10.7150/thno.17071.
- Q. Zhang, T. Liang, K. S. Nandakumar and S. Liu, Emerging and state of the art hemagglutinin-targeted influenza virus inhibitors, Expert Opin. Pharmacother., 2021, 22(6), 715–728, DOI:10.1080/14656566.2020.1856814.
- G. Li and E. De Clercq, Chapter 1: Overview of Antiviral Drug Discovery and Development: Viral Versus Host Targets, in Antiviral Discovery for Highly Pathogenic Emerging Viruses. Drug Discovery Series No. 80, ed. C. Muñoz-Fontela and R. Delgado, The Royal Society of Chemistry, Croydon, 2021, pp. 1–27, 10.1039/9781788016858-00001.
- Y. A. Shtyrya, L. V. Mochalova and N. V. Bovin, Influenza virus neuraminidase: structure and function, Acta Naturae, 2009, 1(2), 26–32, DOI:10.32607/20758251-2009-1-2-26-32.
- J. L. McAuley, B. P. Gilbertson, S. Trifkovic, L. E. Brown and J. L. McKimm-Breschkin, Influenza virus neuraminidase structure and functions, Front. Microbiol., 2019, 10, 39, DOI:10.3389/fmicb.2019.00039.
- S. J. Siegel, A. M. Roche and J. N. Weiser, Influenza promotes pneumococcal growth during coinfection by providing host sialylated substrates as a nutrient source, Cell Host Microbe, 2014, 16(1), 55–67, DOI:10.1016/j.chom.2014.06.005.
- C. Marion, A. M. Burnaugh, S. A. Woodiga and S. J. King, Sialic acid transport contributes to pneumococcal colonization, Infect. Immun., 2011, 79(3), 1262–1269, DOI:10.1128/IAI.00832-10.
- V. T. Peltola, K. G. Murti and J. A. McCullers, Influenza virus neuraminidase contributes to secondary bacterial pneumonia, J. Infect. Dis., 2005, 192(2), 249–257, DOI:10.1086/430954.
- M. Nita-Lazar, A. Banerjee, C. Feng, M. N. Amin, M. B. Frieman, W. H. Chen, A. S. Cross, L. X. Wang and G. R. Vasta, Desialylation of airway epithelial cells during influenza virus infection enhances pneumococcal adhesion via galectin binding, Mol. Immunol., 2015, 65(1), 1–16, DOI:10.1016/j.molimm.2014.12.010.
- J. A. McCullers and K. C. Bartmess, Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae, J. Infect. Dis., 2003, 187(6), 1000–1009, DOI:10.1086/368163.
- G. Xu, M. J. Kiefel, J. C. Wilson, P. W. Andrew, M. R. Oggioni and G. L. Taylor, Three Streptococcus pneumoniae sialidases: three different products, J. Am. Chem. Soc., 2011, 133(6), 1718–1721, DOI:10.1021/ja110733q.
- S. Manco, F. Hernon, H. Yesilkaya, J. C. Paton, P. W. Andrew and A. Kadioglu, Pneumococcal neuraminidases A and B both have essential roles during infection of the respiratory tract and sepsis, Infect. Immun., 2006, 74(7), 4014–4020, DOI:10.1128/IAI.01237-05.
- J. L. Brittan, T. J. Buckeridge, A. Finn, A. Kadioglu and H. F. Jenkinson, Pneumococcal neuraminidase A: an essential upper airway colonization factor for Streptococcus pneumoniae, Mol. Oral Microbiol., 2012, 27(4), 270–283, DOI:10.1111/j.2041-1014.2012.00658.x.
- S. von Grafenstein, H. G. Wallnoefer, J. Kirchmair, J. E. Fuchs, R. G. Huber, M. Schmidtke, A. Sauerbrei, J. M. Rollinger and K. R. Liedl, Interface dynamics explain assembly dependency of influenza neuraminidase catalytic activity, J. Biomol. Struct. Dyn., 2015, 33(1), 104–120, DOI:10.1080/07391102.2013.855142.
- L. Klenow, R. Elfageih, J. Gao, H. Wan, S. G. Withers, J. W. de Gier and R. Daniels, Influenza virus and pneumococcal neuraminidases enhance catalysis by similar yet distinct sialic acid-binding strategies, J. Biol. Chem., 2023, 102891, DOI:10.1016/j.jbc.2023.102891 , in press..
- M. Richter, L. Schumann, E. Walther, A. Hoffmann, H. Braun, U. Grienke, J. M. Rollinger, S. von Grafenstein, K. R. Liedl, J. Kirchmair, P. Wutzler, A. Sauerbrei and M. Schmidtke, Complementary assays helping to overcome challenges for identifying neuraminidase inhibitors, Future Virol., 2015, 10(2), 77–88, DOI:10.2217/fvl.14.97.
- J. A. McCullers and K. C. Bartmess, Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae, J. Infect. Dis., 2003, 187(6), 1000–1009, DOI:10.1086/368163.
- A. Tanaka, S. Nakamura, M. Seki, N. Iwanaga, T. Kajihara, M. Kitano, T. Homma, S. Kurihara, Y. Imamura, T. Miyazaki, K. Izumikawa, H. Kakeya, K. Yanagihara and S. Kohno, The effect of intravenous peramivir, compared with oral oseltamivir, on the outcome of post-influenza pneumococcal pneumonia in mice, Antiviral Ther., 2015, 20(1), 11–19, DOI:10.3851/IMP2744.
- N. Lee and A. C. Hurt, Neuraminidase inhibitor resistance in influenza: a clinical perspective, Curr. Opin. Infect. Dis., 2018, 31(6), 520–526, DOI:10.1097/QCO.0000000000000498.
- J. J. Shie and J. M. Fang, Development of effective anti-influenza drugs: congeners and conjugates – a review, J. Biomed. Sci., 2019, 26(1), 84, DOI:10.1186/s12929-019-0567-0.
- T. Lampejo, Influenza and antiviral resistance: an overview, Eur. J. Clin. Microbiol. Infect. Dis., 2020, 39(7), 1201–1208, DOI:10.1007/s10096-020-03840-9.
- V. M. Lyubchanskaya, L. M. Alekseeva, S. A. Savina and G. V. Granik, Indazolequinones in the Nenitzescu reaction. Synthesis of pyrrolo[2,3-e]- and furo[2,3-e]indazoles, Chem. Heterocycl. Compd., 2000, 36, 1276–1283, DOI:10.1023/A:1017515300244.
- V. M. Lyubchanskaya, L. M. Alekseeva and V. G. Granik, The aza-Nenitzescu reaction. Synthesis of indazole derivatives by condensation of quinones with hydrazones, Chem. Heterocycl. Compd., 1999, 35, 570–574, DOI:10.1007/BF02324640.
- E. Walther, Z. Xu, M. Richter, J. Kirchmair, U. Grienke, J. M. Rollinger, A. Krumbholz, H. P. Saluz, W. Pfister, A. Sauerbrei and M. Schmidtke, Dual acting neuraminidase inhibitors open new opportunities to disrupt the lethal synergism between Streptococcus pneumoniae and influenza virus, Front. Microbiol., 2016, 7, 357, DOI:10.3389/fmicb.2016.0035.
- H. Gut, G. Xu, G. L. Taylor and M. A. Walsh, Structural basis for Streptococcus pneumoniae NanA inhibition by influenza antivirals zanamivir and oseltamivir carboxylate, J. Mol. Biol., 2011, 409(4), 496–503, DOI:10.1016/j.jmb.2011.04.016.
- E. van der Vries, P. J. Collins, S. G. Vachieri, X. Xiong, J. Liu, P. A. Walker, L. F. Haire, A. J. Hay, M. Schutten, A. D. Osterhaus, S. R. Martin, C. A. Boucher, J. J. Skehel and S. J. Gamblin, H1N1 2009 pandemic influenza virus: resistance of the I223R neuraminidase mutant explained by kinetic and structural analysis, PLoS Pathog., 2012, 8(9), e1002914, DOI:10.1371/journal.ppat.1002914.
- V. Zima, C. B. Albiñana, K. Rojíková, J. Pokorná, P. Pachl, P. Řezáčová, J. Hudlicky, V. Navrátil, P. Majer, J. Konvalinka, M. Kožíšek and A. Machara, Investigation of flexibility of neuraminidase 150-loop using tamiflu derivatives in influenza A viruses H1N1 and H5N1, Bioorg. Med. Chem., 2019, 27(13), 2935–2947, DOI:10.1016/j.bmc.2019.05.024.
- J. M. Keil, G. R. Rafn, I. M. Turan, M. A. Aljohani, R. Sahebjam-Atabaki and X. L. Sun, Sialidase inhibitors with different mechanisms, J. Med. Chem., 2022, 65(20), 13574–13593, DOI:10.1021/acs.jmedchem.2c01258.
- D. Parker, G. Soong, P. Planet, J. Brower, A. J. Ratner and A. Prince, The NanA neuraminidase of Streptococcus pneumoniae is involved in biofilm formation, Infect. Immun., 2009, 77(9), 3722–3730, DOI:10.1128/IAI.00228-09.
- J. T. Wren, L. K. Blevins, B. Pang, A. Basu Roy, M. B. Oliver, J. L. Reimche, J. E. Wozniak, M. A. Alexander-Miller and W. E. Swords, Pneumococcal Neuraminidase A (NanA) Promotes Biofilm Formation and Synergizes with Influenza A Virus in Nasal Colonization and Middle Ear Infection, Infect. Immun., 2017, 85(4), e10444, DOI:10.1128/IAI.01044-16.
- E. Walther, M. Richter, Z. Xu, C. Kramer, S. von Grafenstein, J. Kirchmair, U. Grienke, J. M. Rollinger, K. R. Liedl, H. Slevogt, A. Sauerbrei, H. P. Saluz, W. Pfister and M. Schmidtke, Antipneumococcal activity of neuraminidase inhibiting artocarpin, Int. J. Med. Microbiol., 2015, 305(3), 289–297, DOI:10.1016/j.ijmm.2014.12.004.
- U. Grienke, M. Richter, E. Walther, A. Hoffmann, J. Kirchmair, V. Makarov, S. Nietzsche, M. Schmidtke and J. M. Rollinger, Discovery of prenylated flavonoids with dual activity against influenza virus and Streptococcus pneumoniae, Sci. Rep., 2016, 6, 27156, DOI:10.1038/srep27156.
- A. Hoffmann, M. Richter, S. von Grafenstein, E. Walther, Z. Xu, L. Schumann, U. Grienke, C. E. Mair, C. Kramer, J. M. Rollinger, K. R. Liedl, M. Schmidtke and J. Kirchmair, Discovery and characterization of diazenylaryl sulfonic acids as inhibitors of viral and bacterial neuraminidases, Front. Microbiol., 2017, 8, 205, DOI:10.3389/fmicb.2017.00205.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2246679 and 2249815. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra02895j |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2023 |