
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Valence tautomerism in a cobalt–dioxolene complex containing an imidazolic ancillary ligand†‡
Anderson Moledo Vicente Guedes ,
Leandro Sodré de Abreu,
Igor Antunes Vogel Maldonado,
William Silva Fernandes,
Thiago Messias Cardozo,
Rafael A. Allão Cassaro,
Marciela Scarpellini and
Giordano Poneti§
*
,
Leandro Sodré de Abreu,
Igor Antunes Vogel Maldonado,
William Silva Fernandes,
Thiago Messias Cardozo,
Rafael A. Allão Cassaro,
Marciela Scarpellini and
Giordano Poneti§
*
Instituto de Química, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ 21941-909, Brazil. E-mail: giordano.poneti@unitus.it
First published on 4th July 2023
Abstract
This work reports the synthesis, structural, spectroscopic and magnetic investigation of two complexes, [Co(bmimapy)(3,5-DTBCat)]PF6·H2O (1) and [Co(bmimapy)(TCCat)]PF6·H2O (2), where bmimapy is an imidazolic tetradentate ancillary ligand and 3,5-DTBCat and TCCat are the 3,5-di-tert-butyl-catecholate and tetrachlorocatecholate anions, respectively. Their structures have been elucidated using single crystal X-ray diffraction, showing a pseudo-octahedral cobalt ion bound to a chelating dioxolene ligand and the ancillary bmimapy ligand in a folded conformation. Magnetometry displayed an entropy-driven, incomplete, Valence Tautomeric (VT) process for 1 in the 300–380 K temperature range, while 2 displayed a temperature independent, diamagnetic low-spin cobalt(III)–catecholate charge distribution. This behaviour was interpreted on the basis of the cyclic voltammetric analysis, allowing the estimation of the free energy difference associated with the VT interconversion of +8 and +96 kJ mol−1 for 1 and 2, respectively. A DFT analysis of this free energy difference highlighted the ability of the methyl-imidazole pendant arm of bmimapy favouring the onset of the VT phenomenon. This work introduces the imidazolic bmimapy ligand to the scientific community working in the field of valence tautomerism, increasing the library of ancillary ligands to prepare temperature switchable molecular magnetic materials.
Introduction
Molecular systems that can reversibly alter their physical properties under an external stimulus are investigated as potential new materials for different kinds of applications, including treatment1 or storage2–10 of data, sensors11–13 or mechanical actuators.14 Valence tautomeric materials indeed fit into this picture, featuring the possibility to reversibly trigger an intramolecular electron transfer between a metal ion and a non-innocent organic ligand upon heat, pressure, magnetic field or light stimuli,15–23 in the condensed phase,24 solution,25 nanoparticles, thin films26 and even as a self-assembled monolayer on a conductive surface.27 Despite having been observed for different transition metal ions,19 the majority of coordination compounds undergoing Valence Tautomerism (VT) contains a cobalt ion bound to one or two dioxolene (diox, i.e.: ortho-dihydroxo-benzene) ligands; in this case, the intramolecular electron transfer triggers a spin state change of the metal ion, thus defining two different electronic states for the system: low-spin Co(III)Cat (Cat = dianion of the catecholate redox form of the dioxolene ligand – ls-CoIIICat) and high-spin Co(II)SQ (SQ = monoanionic semiquinonate form of the dioxolene – hs-CoIISQ). Such a rearrangement of the intramolecular charge distribution leads to huge variations in the structural, spectroscopic and magnetic properties of the coordination compounds, thereby justifying their utilization in devices incorporating switchable active units.The driving force of the VT equilibrium is the entropy gain of the system on passing from the ls-CoIIICat electronic isomer, with stronger bonds in the first coordination sphere and lower molar enthalpy, to the hs-CoIISQ one, featuring higher density of vibrational states, higher spin multiplicity, and therefore higher molar entropy.24,28–30 For the VT phenomenon to occur, the difference between the free energies of the two electromers involved in the process, ls-CoIIICat and hs-CoIISQ, must be close to the thermal energy at room temperature. Dei and co-workers related this quantity to the difference in the oxidation potential of the metal ion and the dioxolene ligand in the neutral [M(II)(L)(Cat)] species, being L an ancillary, tetradentate ligand, completing the pseudo-octahedral coordination sphere of the metal ion.28,31 Moreover, they showed that chemical variation in the structure of the ancillary ligand can drastically alter the VT behaviour for the family of [Co(Mentpa)(3,5-DTBCat)]+ complexes, where Mentpa are derivatives of the tris-(2-pyridylmethyl)amine bearing methyl-pyridine pendant arms, substituted with n methyl groups in the ortho position (n = 0–3). In particular, for n = 0 and 1 a temperature independent ls-CoIIICat charge distribution was observed, while for n = 3 a hs-CoIISQ one was found, while the [Co(Me2tpa)(diox)]+ displayed VT in solution and in the solid state.21,28,32,33 A similar approach for the chemical control of charge distribution in cobalt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dioxolene complexes of formula [Co(PznPy3−n)(3,5-DTBCat)]+ was achieved by Yu and Li.34 In their work they employed, as ancillary ligand, the PznPy3−n tripodal ligand, featuring a variable number of pyridine (Py) and dimethylated pyrazolyl (Pz) moieties connected to a central tertiary nitrogen atom through a methylene group. They showed that, for n = 1, a ls-CoIIICat charge distribution is the ground state in the 2–300 K temperature range, while for n = 2 and 3 a temperature independent hs-CoIISQ one is found, pointing out that the pyridine moiety is able to stabilize the ls-CoIII ion, while the methylated pyrazolyl groups favour the hs-CoII one. In the case of [Co(diox)2(N–N)] complexes, the role of the diazine N–N ligand was investigated by Pierpont and coworkers, showing how the donating properties35 and the flexibility36 of the diazine ligand affect the VT transition temperature. More recently, Boskovic, Goerigk and co-workers found a linear correlation between the VT transition temperature and the energy of the LUMO of the diazine ligand in a remarkably large temperature interval (150 K) in similar systems.37 Boskovic and co-workers, later on, used the same electrochemical approach to rationalize the presence of multi-step transitions in cobalt complexes with a ditopic dioxolene ligand.38
:dioxolene complexes of formula [Co(PznPy3−n)(3,5-DTBCat)]+ was achieved by Yu and Li.34 In their work they employed, as ancillary ligand, the PznPy3−n tripodal ligand, featuring a variable number of pyridine (Py) and dimethylated pyrazolyl (Pz) moieties connected to a central tertiary nitrogen atom through a methylene group. They showed that, for n = 1, a ls-CoIIICat charge distribution is the ground state in the 2–300 K temperature range, while for n = 2 and 3 a temperature independent hs-CoIISQ one is found, pointing out that the pyridine moiety is able to stabilize the ls-CoIII ion, while the methylated pyrazolyl groups favour the hs-CoII one. In the case of [Co(diox)2(N–N)] complexes, the role of the diazine N–N ligand was investigated by Pierpont and coworkers, showing how the donating properties35 and the flexibility36 of the diazine ligand affect the VT transition temperature. More recently, Boskovic, Goerigk and co-workers found a linear correlation between the VT transition temperature and the energy of the LUMO of the diazine ligand in a remarkably large temperature interval (150 K) in similar systems.37 Boskovic and co-workers, later on, used the same electrochemical approach to rationalize the presence of multi-step transitions in cobalt complexes with a ditopic dioxolene ligand.38
These examples highlight the critical role played by the ancillary ligand in determining the redox properties of the cobalt ion inside the molecular context, and explain the relatively scarce number of these ligands available for the synthesis of VT coordination systems.19 Indeed, for the majority of cobalt–dioxolene complexes showing VT, these ancillary ligands can be pyridine derivatives,39 aromatic40,41 or aliphatic40 diazines, or tetradentate ligands, like macrocyclic derivatives of cyclam (1,4,8,11-tetraazacyclotetradecane)42,43 or tripodal derivatives of tpa (tris(2-pyridylmethyl)amine).17,28,44–46 In this work, we present the ancillary ligand bmimapy ((bis(1-methylimidazol-2-yl)methyl)(2-(pyridyl-2-yl)ethyl)amine)47 (see Chart 1), previously known to bind to the cobalt ion in a folded conformation48 and herein used to synthesize two complexes, [Co(bmimapy)(3,5-DTBCat)]PF6·H2O (1) and [Co(bmimapy)(TCCat)]PF6·H2O (2), with the objective to increase the family of nitrogen-donating, tripodal ligands for the preparation of new valence tautomeric materials.
 | ||
| Chart 1 Molecular structure of the (bis(1-methylimidazol-2-yl)methyl)(2-(pyridyl-2-yl)ethyl)amine ligand (bmimapy). | ||
Results and discussion
Synthesis
The syntheses of complexes 1 and 2 followed the previously employed route used for other 1:1 cobalt:dioxolene complexes featuring a tetradentated ancillary ligand:29,44,45 the Co2+ ion was coordinated by the bmimapy ligand in warm methanol under inert atmosphere, followed by the addition of the dioxolene ligand (3,5-di-tert-butyl-catechol (3,5-DTBCat) or tetrachlorocatechol (TCCat) for 1 and 2, respectively), deprotonated in situ with an excess of triethylamine. The neutral [Co(bmimapy)(diox)] complex was thus oxidized upon air exposure and precipitated with a three-fold excess of aqueous KPF6. Recrystallization in hot methanol yielded single crystals of the desired complexes in moderate yields.
Structural analysis
Single crystal X-ray diffraction experiments were carried out at 130 K for 1 and 290 K for 2 (Fig. 1 and ESI‡). Details of data collection and structure refinement are gathered in Table S1.‡ The structure in both cases contains the cobalt ion hexacoordinated with the tetradentate ancillary ligand adopting a folded conformation around the ion, with the remaining two cis positions occupied by the oxygen atoms of the dioxolenes (see Fig. 1 and S1–S3‡). The coordination environment of cobalt ion is described as slightly distorted octahedral, with the continuous shape measure49 values of 0.396 and 0.407 for 1 and 2, respectively (Table S2‡). Co–O and Co–N bond lengths are in the 1.867–2.052 Å range, indicating a low-spin cobalt(III) ion. Since the bond lengths of the dioxolene ligands change with the oxidation state of the ligand, C–C and C–O bond lengths can be used to calculate the metrical oxidation state (MOS) of the dioxolene.50 The obtained MOS values were −1.9 for both complexes, indicating that the dioxolene ligand is in the catecholate, dinegative form in both compounds. The neutrality of the systems is assured by the presence of one PF6− anion. Selected bond lengths and bond angles are gathered in Table S3.‡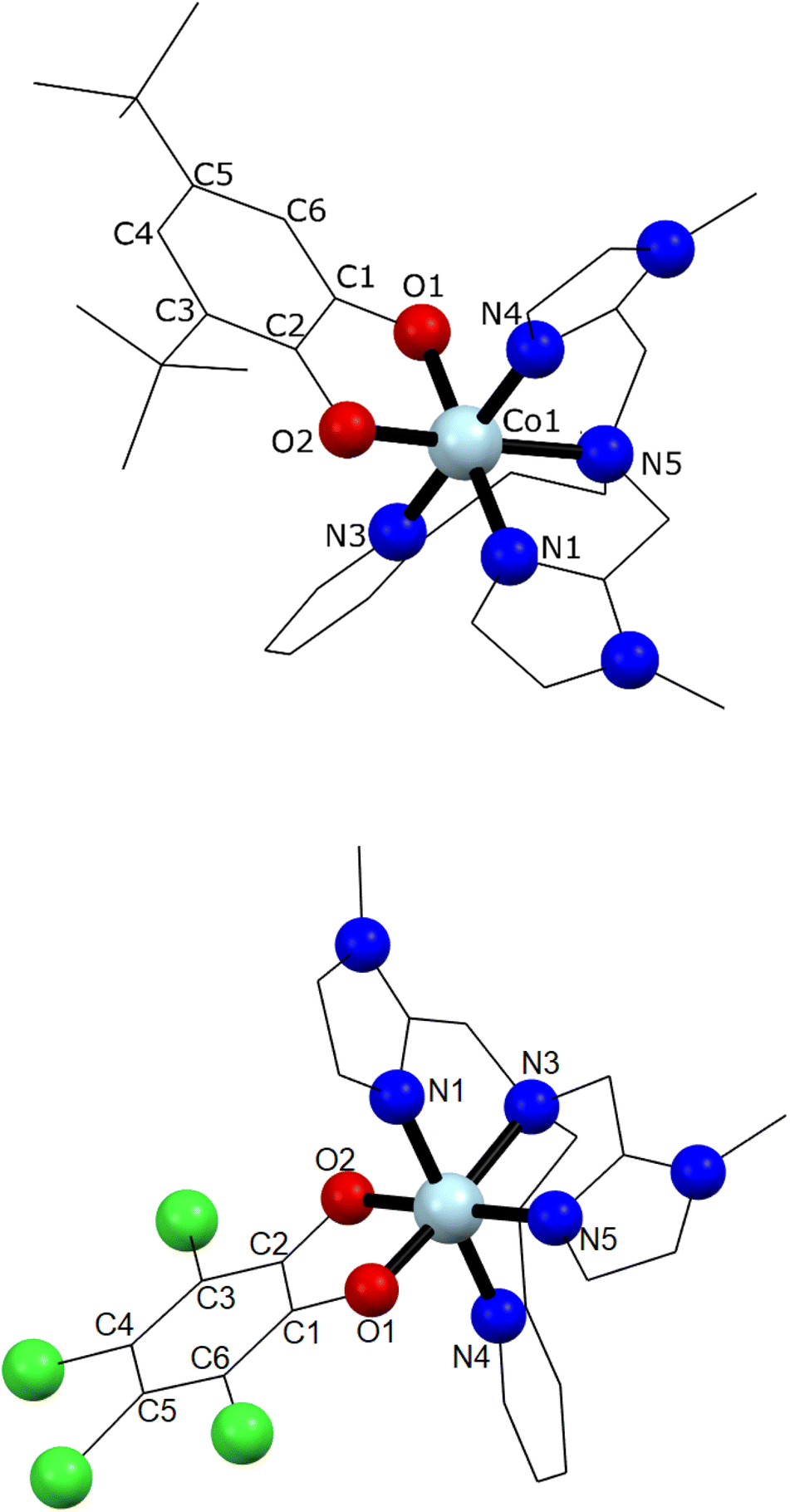 | ||
| Fig. 1 Molecular structures of complexes 1 (top) and 2 (bottom). PF6− ion, lattice solvents, hydrogen and disordered atoms were omitted for clarity. Light blue, red, blue, green, and black colours stand for cobalt(III) ion, oxygen, nitrogen, chlorine and carbon atoms, respectively. | ||
In the structure of 1, the lattice solvent molecules are disordered, and could not be successfully modelled. Therefore, the electron density of disordered solvent molecules was removed using a solvent mask procedure implemented in OLEX2.51 The solvent mask removed a total of 86 electrons per asymmetric unit and a volume of 360 Å3 which can correspond either to one water and four methanol molecules or three methanol and three water molecules. Indeed, powder X-ray diffraction of 1 shows a nearly amorphous diffractogram, presumably due to loss of lattice solvents (Fig. S4‡). The shortest intermolecular distance between metal ions is around 8.5 Å and occurs along the crystallographic direction b. Weak T-shaped π⋯π interactions are observed between the imidazole and pyridine moieties of one molecule and the phenyl rings of the 3,5-DTBCat ligands belonging to two surrounding molecules, yielding C–H⋯centroid distances of 2.53 and 2.87 Å, respectively. π⋯π interactions between pyridine and imidazole moieties (Fig. S2‡) also occur, with a distance between centroids of 3.763 Å and almost parallel rings, the angle between them being 8.3° and the slippage 1.681 Å. In addition, PF6− anions interact with the complex through non-classical hydrogen bonds with intermolecular C–H⋯F distances ranging from 2.31 to 2.48 Å.52 In the structure of 2, one lattice water molecule and TCCat ligand are disordered and were split over two positions. The shortest intermolecular distance between metal ions is around 8.77 Å and occurs along the crystallographic direction c. As observed in 1, weak intermolecular interactions were observed in 2. The packing scheme also reveals C–H⋯π interactions between imidazole–imidazole rings and phenyl-imidazole rings53,54 (Table S4 and Fig. S3‡), with C–H⋯centroid distances of 2.78 and 2.86 Å, as well as non-classical hydrogen bonds interactions with intermolecular C–H⋯F distances ranging from 2.29 to 2.54 Å.
Infrared spectroscopy
Infrared spectroscopy is a useful tool for determining the redox isomeric form of cobalt-based valence tautomers, due to the sensitivity of the C–O and C–C stretching energies on the oxidation state of the dioxolene ligand.45,46,55–57 The infrared absorption spectra of 1 and 2 (Fig. S5‡) display absorptions at 1610 and 1555 cm−1, typical for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–O stretching modes of dinegative catecholato as well as pyridyl ligands.55 The absorption peak at 1440 cm−1 is attributable to a mixing of C–O and CC stretching modes, according to a previous DFT analysis of similar cobalt–dioxolene systems, and agrees with a dinegative catecholato ligands for 1 and 2.55 Similar conclusions can be taken considering the absorptions at 1240 (1247), 1283 (1288) and 1322 (1311) cm−1 for 1 (2), typical for ls-CoIIICat complexes.58 Absorptions related to the semiquinonato form of the dioxolene ligands (expected at about 1580 cm−1) are undetected. These data thus support a low-spin cobalt(III)–catecholate charge distribution for 1 and 2 at room temperature in the solid state, in agreement with the structure determined by single crystal X-ray diffraction. In addition, to corroborate the cationic form of the complex, the P–F stretching and bending modes of the PF6− anion are observed at 840 and 555 cm−1, respectively, for both complexes.
C and C–O stretching modes of dinegative catecholato as well as pyridyl ligands.55 The absorption peak at 1440 cm−1 is attributable to a mixing of C–O and CC stretching modes, according to a previous DFT analysis of similar cobalt–dioxolene systems, and agrees with a dinegative catecholato ligands for 1 and 2.55 Similar conclusions can be taken considering the absorptions at 1240 (1247), 1283 (1288) and 1322 (1311) cm−1 for 1 (2), typical for ls-CoIIICat complexes.58 Absorptions related to the semiquinonato form of the dioxolene ligands (expected at about 1580 cm−1) are undetected. These data thus support a low-spin cobalt(III)–catecholate charge distribution for 1 and 2 at room temperature in the solid state, in agreement with the structure determined by single crystal X-ray diffraction. In addition, to corroborate the cationic form of the complex, the P–F stretching and bending modes of the PF6− anion are observed at 840 and 555 cm−1, respectively, for both complexes.
Magnetometry
In order to quantitatively assess the charge distribution of complexes 1 and 2, the temperature dependence of their molar magnetic susceptibility (χM) has been monitored with SQUID magnetometry in the solid state (Fig. 2).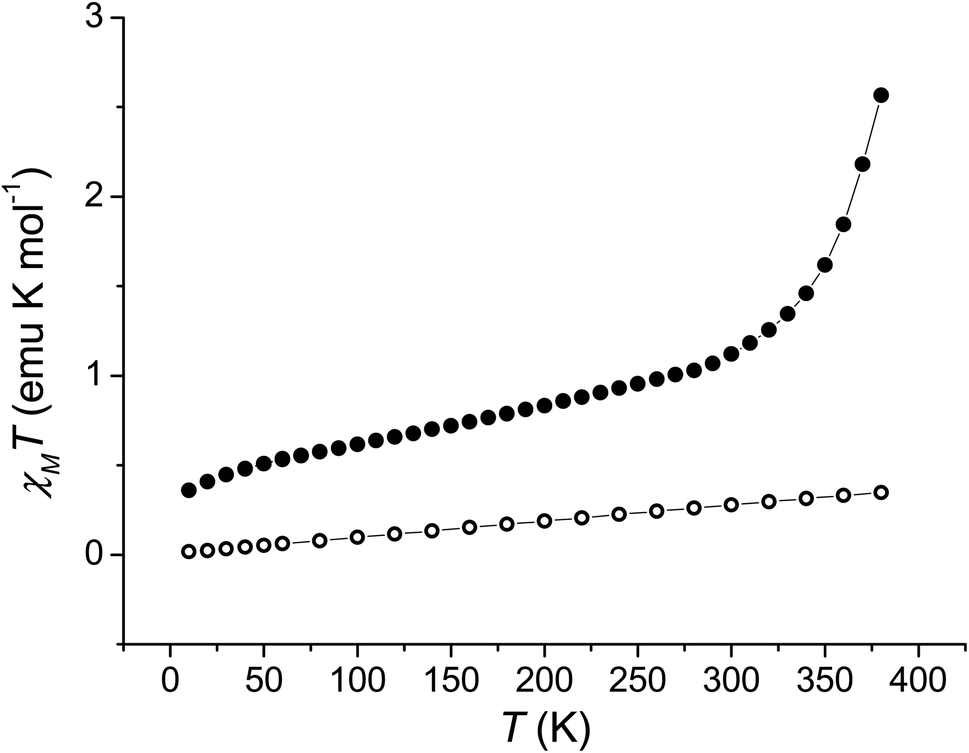 | ||
| Fig. 2 Temperature evolution of the χMT product of 1 (full dots) and 2 (empty dots). | ||
The product of the molar magnetic susceptibility times the temperature (χMT) for 1 takes the 0.36 emu K mol−1 value at 10 K, steadily increasing to reach 1.07 emu K mol−1 at 300 K. Upon heating above room temperature, a steeper increase of the χMT product takes place, reaching the 2.57 emu K mol−1 value at 380 K. This behaviour has been attributed to the presence of a minor hs-CoIISQ, not interconverting, molar fraction (about 7%), and to a temperature independent paramagnetism from low-spin cobalt(III) ion (vide infra), along with a gradual, incomplete, valence tautomeric transition in the 300–380 K temperature range, leading to a molar fraction of the hs-CoIISQ species of about 0.82. This transition profile is typical for VT transitions with low degree of cooperativity.30 Thermogravimetric analysis (Fig. S6‡) points out the partial loss of a solvate water molecule in this temperature window, facilitating the VT transition, as seen for other entropy driven processes.20,59 On the other hand, 2 displayed a linear increase of its χMT product upon heating, passing from 0.02 emu K mol−1 at 10 K to 0.35 emu K mol−1 at 380 K, suggesting the presence of a temperature independent ls-CoIIICat charge distribution, with the diamagnetic ion showing a temperature-independent paramagnetic susceptibility of 9.04 × 10−4 emu mol−1.
Electrochemistry
To understand the different thermal evolution of the charge distribution in complexes 1 and 2, their electrochemical properties have been assessed with cyclic voltammetry of their acetonitrile solutions (Fig. 3).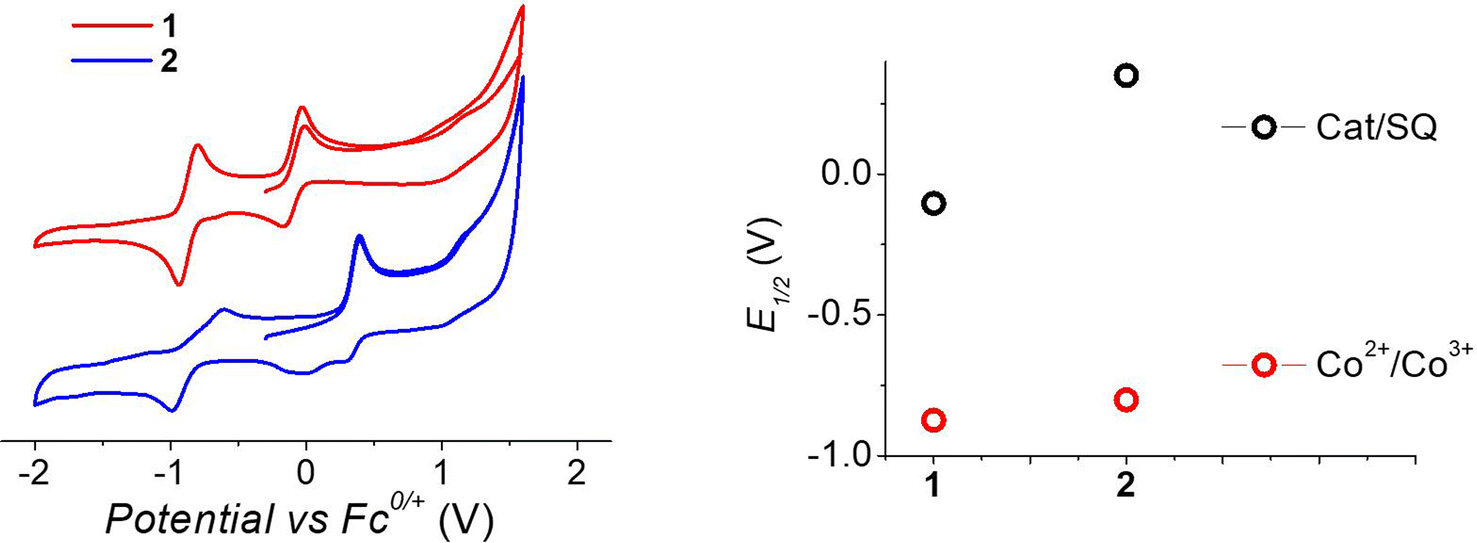 | ||
| Fig. 3 Voltammograms of acetonitrile solutions of the complexes, measured with a 100 mV s−1 sweeping rate, (left) and corresponding potentials (right panel). | ||
In the case of compound 1, the first process in the anodic direction is attributed to the quasi-reversible oxidation of the catecholate ligand of the [Co(bmimapy)(3,5-DTBCat)]+ cation to its correspondent semiquinonate form [Co(bmimapy)(3,5-DTBSQ)]2+, taking place at the potential of −0.09 V (vs. Fc+/Fc redox couple, with a peak to peak separation of 140 mV, see Table S5‡). The other quasi-reversible process, going into the cathodic direction, has been attributed to the metal-centred reduction: [CoIII(bmimapy)(3,5-DTBCat)]+ to [CoII(bmimapy)(3,5-DTBCat)], and features a potential of −0.86 V (vs. Fc+/Fc redox couple), with a peak to peak separation of 140 mV. In the case of compound 2, a similar potential is observed for the Co2+/3+ redox couple (−0.80 V), supporting the interpretation of a metal-centred electrochemical process. The oxidation of the tetrachlorocatechol moiety, on the other hand, occurs at significantly higher potential (+0.35 V), as expected considering the electron-withdrawing nature of the chlorine substituents on the catechol ring.
The metal-centred process can be used to understand the thermodynamic origin of the entropy-driven VT process in 1. It is in fact known that the reduction potential of the 3,5-di-tert-butyl-catecholate ligand in [MII(L)(3,5-DTBSQ)]+/[MII(L)(3,5-DTBCat)] complexes is, to a first approximation, dependent on the oxidation state of the metal ion, but does not depend on its chemical identity, its spin state, as well as on the nature of the ancillary ligand L.28 We can thus assume this reduction potential to be similar to the one observed for the [Co2+(Me3tpa)(3,5-DTBSQ)]+ cation, found to be −0.78 V (vs. Fc+/Fc).28 We can thus estimate the difference between the free energy of the [CoIII(bmimapy)(3,5-DTBCat)]+ and [CoII(bmimapy)(3,5-DTBSQ)]+ redox isomers  of 1 as the difference between the potential of the Co3+/2+ and the SQ/Cat redox couples:28
of 1 as the difference between the potential of the Co3+/2+ and the SQ/Cat redox couples:28
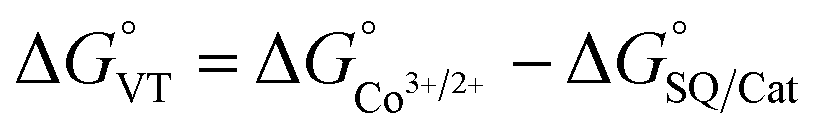
The obtained value of 8 kJ mol−1 for 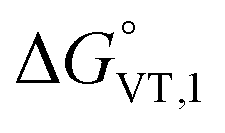 is in the range observed for a system undergoing valence tautomerism.28 Taking the oxidation potential of the [Ni(bdi)(TCCat)] complex60 (+0.2 V, being bdi the N,N′-bis(2,4,6-trimethylphenyl)-2,3-butanediimine) as reference for the TCCat/TCSQ redox couple, we find a
is in the range observed for a system undergoing valence tautomerism.28 Taking the oxidation potential of the [Ni(bdi)(TCCat)] complex60 (+0.2 V, being bdi the N,N′-bis(2,4,6-trimethylphenyl)-2,3-butanediimine) as reference for the TCCat/TCSQ redox couple, we find a  of +96 kJ mol−1 for complex 2, thus justifying its observed temperature independent low-spin cobalt(III)–catecholate charge distribution. In a similar approach, Boskovic and co-workers related the spacing between the metal- and dioxolene-centred electrochemical processes in the same molecule (Δox–red) to the occurrence of VT, identifying a threshold of 740 mV, below which the VT occurs in the 10–300 K range.61 Our data fit fairly well into this treatment, with 1 displaying a Δox–red of 770 mV, corresponding to a VT entropy-driven transition above room temperature.
of +96 kJ mol−1 for complex 2, thus justifying its observed temperature independent low-spin cobalt(III)–catecholate charge distribution. In a similar approach, Boskovic and co-workers related the spacing between the metal- and dioxolene-centred electrochemical processes in the same molecule (Δox–red) to the occurrence of VT, identifying a threshold of 740 mV, below which the VT occurs in the 10–300 K range.61 Our data fit fairly well into this treatment, with 1 displaying a Δox–red of 770 mV, corresponding to a VT entropy-driven transition above room temperature.
DFT analysis
DFT has been previously used to interpret and predict the potential VT behaviour of cobalt complexes.37,61,62 We have employed the TPSSh functional with the def2-TZVP basis set to calculate the relative energy differences (Erel) between ls-CoIIICat and hs-CoIISQ redox isomers for several complexes with the [Co(L)(diox)]PF6 formula, with the aim to clarify the role of the bmimapy ligand in tuning the Co2+/3+ redox potential. Indeed, the TPSSh functional has been successfully employed in the recent past to calculate such energy differences in VT cobalt complexes.38,62,63 Here, the diox ligand alternates between the 3,5-di-tert-butyl-catecholate or the tetrachlorocatecholate ligands, while three ancillary ligands L were chosen to represent the effect of the imidazole binding groups of the bmimapy ligand: PzPy2 (ref. 34) ((3,5-dimethyl-1H-pyrazol-1-yl)-N,N-bis(pyridin-2-ylmethyl)methanamine, known to provide [Co(L)(diox)]+ cations showing the ls-CoIIICat charge distribution), bmimapy and tmima (tris[(1-methyl-2-imidazolyl)methyl]amine),64,65 representing the limit of a tripodal ligand featuring a central nitrogen atom bound to three 1-methyl-imidazole pendant arms. The results of the DFT analysis are reported in Fig. 4, S7 and Table S6.‡ A semi-quantitative evaluation of the solidity of our approach can be obtained observing the results for the [Co(Me3tpa)(3,5-DTBSQ)]+ (ref. 28) and [Co(Me3tpa)(TCSQ)]+ (ref. 29) complexes (Fig. S8‡), which parallel the experimentally observed charge distribution in literature. As such, we expect an uncertainty of about ±4 kJ mol−1 on the Erel value. It must be stressed that Erel does not consider the different vibrational structure of the two electromers and cannot, as such, be considered an estimation of but simply an estimation of the enthalpic difference between the two electromeric forms.61
but simply an estimation of the enthalpic difference between the two electromeric forms.61
 | ||
| Fig. 4 DFT calculated relative energies of the different charge distribution electrometric forms of [Co(L)(diox)]PF6 complexes. | ||
The comparison between the two families of complexes containing the 3,5-DTBCat and TCCat ligands shows a significantly higher Erel for the latter, as expected. Moreover, the calculated energy difference between the 3,5-DTBCat and TCCat derivatives is almost independent of the ancillary ligand L, as previously pointed out, supporting the assumptions we made in the discussion of the electrochemical properties of 1 and 2. The effect of the ancillary ligand, on the other hand, can be understood comparing the trend found for each of these two families of complexes. Considering the 3,5-DTBCat family, the highest value of Erel is found for the complex with the PzPy2 ligand. This result complies with the ls-CoIIICat charge distribution experimentally observed for the [Co(PzPy2)(3,5-DTBCat)]+ complex,34 thus acting as an additional proof of the solidity of our computational results. Moving to the cases of the cobalt complexes of the 3,5-DTBCat dioxolene including the bmimapy and tmima ligands, the calculated Erel values decrease. For the bmimapy derivative (compound 1), the DFT evaluation of Erel (9.4 kJ mol−1) is in very good agreement with the  value extrapolated from the electrochemical analysis, justifying the incipient, high temperature valence tautomeric process observed experimentally. For the tmima derivative, moreover, such a stabilisation leads to a hs-CoIISQ electronic ground state suggested for the [Co(tmima)(3,5-DTBCat)]+ derivative (yet unreported in literature). This comparison highlights that the methyl-imidazole arm of the ligands favours the reduced form of the metal building-block, thus lowering the Erel. This finding cannot be attributed to steric effects, since the imidazole arm generates a five-membered coordination ring, similarly to the methylene-pyridine and methylene-pyrazole arms of the PzPy2 ligand. Moreover, no sterically demanding groups are present in the ortho position of the heterocyclic groups, differently from the case of the pyrazole arm of the PzPy2 ligand, bearing a methyl group in ortho with respect to the donor nitrogen atom. Thus, we attribute this behaviour to the higher π-donation ability of the imidazole ligands, when compared with the pyridine rings. Indeed, pyridines are known to possess low energy π* orbitals able to accept back-donation from the cobalt ion, thus stabilising the higher Co3+ oxidation state.66 In the case of the family of complexes featuring the tetrachlorocatecholate ligand, a similar trend is observed, confirming the relative electrochemical independence of one redox-active building-block (Co(L)) from the other (the dioxolene anion) and corroborating our DFT analysis.
value extrapolated from the electrochemical analysis, justifying the incipient, high temperature valence tautomeric process observed experimentally. For the tmima derivative, moreover, such a stabilisation leads to a hs-CoIISQ electronic ground state suggested for the [Co(tmima)(3,5-DTBCat)]+ derivative (yet unreported in literature). This comparison highlights that the methyl-imidazole arm of the ligands favours the reduced form of the metal building-block, thus lowering the Erel. This finding cannot be attributed to steric effects, since the imidazole arm generates a five-membered coordination ring, similarly to the methylene-pyridine and methylene-pyrazole arms of the PzPy2 ligand. Moreover, no sterically demanding groups are present in the ortho position of the heterocyclic groups, differently from the case of the pyrazole arm of the PzPy2 ligand, bearing a methyl group in ortho with respect to the donor nitrogen atom. Thus, we attribute this behaviour to the higher π-donation ability of the imidazole ligands, when compared with the pyridine rings. Indeed, pyridines are known to possess low energy π* orbitals able to accept back-donation from the cobalt ion, thus stabilising the higher Co3+ oxidation state.66 In the case of the family of complexes featuring the tetrachlorocatecholate ligand, a similar trend is observed, confirming the relative electrochemical independence of one redox-active building-block (Co(L)) from the other (the dioxolene anion) and corroborating our DFT analysis.
Conclusions
This works introduces the tripodal ancillary bmimapy ligand to the community of switchable molecular materials for the preparation of valence tautomeric complexes. Its reactivity is similar to other ancillary ligands used in the literature to prepare heteroleptic cobalt complexes, bonding to the cobalt(II) ion in a folded conformation, thus leaving two cis positions for the coordination of the catecholate chelating ligands. Bmimapy was used to prepare two complexes: [Co(bmimapy)(3,5-DTBCat)]PF6·H2O (1) and [Co(bmimapy)(TCCat)]PF6·H2O (2), where 3,5-DTBCat and TCCat are the 3,5-di-tert-butyl-catecholate and tetrachlorocatecholate anions, respectively. Single crystal X-ray diffraction analysis reported a distorted octahedral coordination environment for the cobalt ions in both complexes, and the bond lengths of the first coordination sphere, including also the MOS analysis of the bond lengths of the catecholate moieties, indicated a ls-CoIIICat charge distribution for both complexes. In the 300–380 K range, however, magnetic susceptibility highlighted an incipient, valence tautomeric process for 1, while 2 retained a ls-CoIIICat charge distribution. Cyclic voltammetry allowed to understand this behaviour, calculating the free energy difference associated to the VT process for both complexes, which resulted in a range accessible with thermal stimula above room temperature for 1 (8 kJ mol−1), while it did not for 2 (96 kJ mol−1). These experimental observations were finally justified by a DFT analysis, highlighting the ability of the methyl-imidazole pendant arm of the bmimapy ligand to stabilise the high-spin Co(II) redox form and favouring the onset of the VT phenomenon in bmimapy containing cobalt complexes. The chemical control of the reduction potential of different metal ions provided by the bmimapy ligand is expected to be a powerful tool to increase the library of valence tautomeric complexes.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
GP and RAAC gratefully acknowledge FAPERJ for funding through grants E-26/202.912/2019, SEI-260003/001167/2020, E-26/010.000978/2019 and E-26/203.033/2018. MS gratefully acknowledges FAPERJ (E-26/26211754/2021) and Programa de Pós-Graduação em Química/IQ/UFRJ (CAPES financial code 001). AMVG, LSA and WSF thank CAPES while IAVM thanks CNPq for the fellowship. The authors thank Prof. Roberta Sessoli (Dipartimento di Chimica “Ugo Schiff”, Università degli Studi di Firenze, Italy) for providing access to the SQUID magnetometer and Prof. Luiz Fernando Brum Malta (Instituto de Química, Universidade Federal do Rio de Janeiro, Brazil) for granting access to the thermogravimetric analyser. The authors acknowledge the support of Núcleo Avançado de Computação de Alto Desempenho (NACAD) of the Instituto Alberto Luiz Coimbra de Pós-Graduação e Pesquisa de Engenharia (COPPE), Universidade Federal do Rio de Janeiro.References
- M. R. Wasielewski, M. D. E. Forbes, N. L. Frank, K. Kowalski, G. D. Scholes, J. Yuen-Zhou, M. A. Baldo, D. E. Freedman, R. H. Goldsmith, T. Goodson, M. L. Kirk, J. K. McCusker, J. P. Ogilvie, D. A. Shultz, S. Stoll and K. B. Whaley, Nat. Rev. Chem., 2020, 4, 490–504 CrossRef CAS PubMed.
- K. S. Kumar and M. Ruben, Angew. Chem., Int. Ed., 2021, 60, 7502–7521 CrossRef CAS PubMed.
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC.
- B. Rösner, M. Milek, A. Witt, B. Gobaut, P. Torelli, R. H. Fink and M. M. Khusniyarov, Angew. Chem., Int. Ed., 2015, 54, 12976–12980 CrossRef PubMed.
- M. A. Halcrow, Spin-Crossover Materials: Properties and Applications, John Wiley & Sons, Chichester, UK 2013 Search PubMed.
- A. Dei and L. Sorace, Appl. Magn. Reson., 2010, 38, 139–153 CrossRef CAS.
- A. Calzolari, Y. Chen, G. F. Lewis, D. B. Dougherty, D. Shultz and M. B. Nardelli, J. Phys. Chem. B, 2012, 116, 13141–13148 CrossRef CAS PubMed.
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed.
- A. Lannes, Y. Suffren, J. B. Tommasino, R. Chiriac, F. Toche, L. Khrouz, F. Molton, C. Duboc, I. Kieffer, J.-L. Hazemann, C. Reber, A. Hauser and D. Luneau, J. Am. Chem. Soc., 2016, 138, 16493–16501 CrossRef CAS PubMed.
- A. Witt, F. W. Heinemann and M. M. Khusniyarov, Chem. Sci., 2015, 6, 4599–4609 RSC.
- M. Mörtel, J. Oschwald, A. Scheurer, T. Drewello and M. M. Khusniyarov, Inorg. Chem., 2021, 60, 14230–14237 CrossRef PubMed.
- I.-R. Jeon, J. G. Park, C. R. Haney and T. D. Harris, Chem. Sci., 2014, 5, 2461–2465 RSC.
- K. Boukheddaden, M. H. Ritti, G. Bouchez, M. Sy, M. M. Dîrtu, M. Parlier, J. Linares and Y. Garcia, J. Phys. Chem. C, 2018, 122, 7597–7604 CrossRef CAS.
- G. Molnar, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Adv. Mater., 2018, 30, 17003862 CrossRef PubMed.
- D. N. Hendrickson and C. G. Pierpont, Spin Crossover in Transition Metal Compounds II, 2004, 234, pp. 63–95 Search PubMed.
- A. Dei, D. Gatteschi, C. Sangregorio and L. Sorace, Acc. Chem. Res., 2004, 37, 827–835 CrossRef CAS PubMed.
- O. Sato, J. Tao and Y. Z. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2152–2187 CrossRef CAS PubMed.
- C. Boskovic, in Spin-Crossover Materials – Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, UK, 2013, ch. 7, pp. 203–224 Search PubMed.
- T. Tezgerevska, K. G. Alley and C. Boskovic, Coord. Chem. Rev., 2014, 268, 23–40 CrossRef CAS.
- O. Drath and C. Boskovic, Coord. Chem. Rev., 2018, 375, 256–266 CrossRef CAS.
- G. Poneti, M. Mannini, L. Sorace, P. Sainctavit, M.-A. Arrio, A. Rogalev, F. Wilhelm and A. Dei, ChemPhysChem, 2009, 10, 2090–2095 CrossRef CAS PubMed.
- C. Fleming, D. Chung, S. Ponce, D. J. R. Brook, J. DaRos, R. Das, A. Ozarowski and S. A. Stoian, Chem. Commun., 2020, 56, 4400–4403 RSC.
- A. Paul and S. Konar, J. Mater. Chem. C, 2022, 10, 4980–4984 RSC.
- D. M. Adams and D. N. Hendrickson, J. Am. Chem. Soc., 1996, 118, 11515–11528 CrossRef CAS.
- E. Evangelio, C. Rodriguez-Blanco, Y. Coppel, D. N. Hendrickson, J. P. Sutter, J. Campo and D. Ruiz-Molina, Solid State Sci., 2009, 11, 793–800 CrossRef CAS.
- N. A. Vazquez-Mera, F. Novio, C. Roscini, C. Bellacanzone, M. Guardingo, J. Hernando and D. Ruiz-Molina, J. Mater. Chem. C, 2016, 4, 5879–5889 RSC.
- G. Poneti, L. Poggini, M. Mannini, B. Cortigiani, L. Sorace, E. Otero, P. Sainctavit, A. Magnani, R. Sessoli and A. Dei, Chem. Sci., 2015, 6, 2268–2274 RSC.
- A. Beni, A. Dei, S. Laschi, M. Rizzitano and L. Sorace, Chem.–Eur. J., 2008, 14, 1804–1813 CrossRef CAS PubMed.
- A. Dei, A. Feis, G. Poneti and L. Sorace, Inorg. Chim. Acta, 2008, 361, 3842–3846 CrossRef CAS.
- A. Dei, G. Poneti and L. Sorace, Inorg. Chem., 2010, 49, 3271–3277 CrossRef CAS PubMed.
- A. Dei, Inorg. Chem., 1993, 32, 5730–5733 CrossRef CAS.
- P. Dapporto, A. Dei, G. Poneti and L. Sorace, Chem.–Eur. J., 2008, 14, 10915–10918 CrossRef CAS PubMed.
- G. Poneti, M. Mannini, L. Sorace, P. Sainctavit, M.-A. Arrio, E. Otero, J. C. Cezar and A. Dei, Angew. Chem., Int. Ed., 2010, 49, 1954–1957 CrossRef CAS PubMed.
- F. Yu and B. Li, Inorg. Chim. Acta, 2012, 392, 199–203 CrossRef CAS.
- O.-S. Jung and C. G. Pierpont, Inorg. Chem., 1994, 33, 2227–2235 CrossRef CAS.
- O.-S. Jung, D. H. Jo, Y.-A. Lee, Y. S. Sohn and C. G. Pierpont, Inorg. Chem., 1998, 37, 5875–5880 CrossRef CAS.
- J. T. Janetzki, F. Z. M. Zahir, R. W. Gable, W. Phonsri, K. S. Murray, L. Goerigk and C. Boskovic, Inorg. Chem., 2021, 60, 14475–14487 CrossRef CAS PubMed.
- G. K. Gransbury, B. N. Livesay, J. T. Janetzki, M. A. Hay, R. W. Gable, M. P. Shores, A. Starikova and C. Boskovic, J. Am. Chem. Soc., 2020, 142, 10692–10704 CrossRef CAS.
- M. Chida, S. Takahashi, R. Konishi, T. Matsumoto, A. Nakada, M. Wakizaka, W. Kosaka, H. Miyasaka and H.-C. Chang, Chem.–Eur. J., 2021, 27, 16354–16366 CrossRef CAS PubMed.
- D. M. Adams, A. Dei, A. L. Rheingold and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 8221–8229 CrossRef CAS.
- S. H. Bodnar, A. Caneschi, A. Dei, D. A. Shultz and L. Sorace, Chem. Commun., 2001, 2150–2151 RSC.
- A. Caneschi, A. Dei, F. F. de Biani, P. Gutlich, V. Ksenofontov, G. Levchenko, A. Hoefer and F. Renz, Chem.–Eur. J., 2001, 7, 3926–3930 CrossRef CAS.
- C. Carbonera, A. Dei, J. F. Letard, C. Sangregorio and L. Sorace, Angew. Chem., Int. Ed., 2004, 43, 3136–3138 CrossRef CAS PubMed.
- Y. Mulyana, G. Poneti, B. Moubaraki, K. S. Murray, B. F. Abrahams, L. Sorace and C. Boskovic, Dalton Trans., 2010, 39, 4757–4767 RSC.
- K. G. Alley, G. Poneti, J. B. Aitken, R. K. Hocking, B. Moubaraki, K. S. Murray, B. F. Abrahams, H. H. Harris, L. Sorace and C. Boskovic, Inorg. Chem., 2012, 51, 3944–3946 CrossRef CAS PubMed.
- K. G. Alley, G. Poneti, P. S. D. Robinson, A. Nafady, B. Moubaraki, J. B. Aitken, S. C. Drew, C. Ritchie, B. F. Abrahams, R. K. Hocking, K. S. Murray, A. M. Bond, H. H. Harris, L. Sorace and C. Boskovic, J. Am. Chem. Soc., 2013, 135, 8304–8323 CrossRef CAS PubMed.
- M. Scarpellini, J. C. Toledo, A. Neves, J. Ellena, E. E. Castellano and D. W. Franco, Inorg. Chim. Acta, 2004, 357, 707–715 CrossRef CAS.
- B. M. Pires, L. C. Giacomin, F. A. V. Castro, A. d. S. Cavalcanti, M. D. Pereira, A. J. Bortoluzzi, R. B. Faria and M. Scarpellini, J. Inorg. Biochem., 2016, 157, 104–113 CrossRef CAS.
- M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575–5582 CrossRef CAS PubMed.
- S. N. Brown, Inorg. Chem., 2012, 51, 1251–1260 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- E. D'Oria and J. J. Novoa, CrystEngComm, 2008, 10, 423–436 RSC.
- A. Di Santo, H. Pérez, G. A. Echeverría, O. E. Piro, R. A. Iglesias, R. E. Carbonio, A. B. Altabef and D. M. Gil, RSC Adv., 2018, 8, 23891–23902 RSC.
- B. Nagaraj, T. Narasimhamurthy, H. S. Yathirajan, P. Nagaraja, R. S. Narasegowda and R. S. Rathore, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o177–o180 CrossRef CAS PubMed.
- P. T. Touceda, S. M. Vazquez, M. Lima, A. Lapini, P. Foggi, A. Dei and R. Righini, Phys. Chem. Chem. Phys., 2012, 14, 1038–1047 RSC.
- G. Poneti, M. Mannini, B. Cortigiani, L. Poggini, L. Sorace, E. Otero, P. Sainctavit, R. Sessoli and A. Dei, Inorg. Chem., 2013, 52, 11798–11805 CrossRef CAS PubMed.
- N. Azzaroli, A. Lapini, M. Di Donato, A. Dei and R. Righini, J. Phys. Chem. B, 2013, 117, 15492–15502 CrossRef CAS PubMed.
- P. T. Touceda, S. M. Vazquez, M. Lima, A. Lapini, P. Foggi, A. Dei and R. Righini, Phys. Chem. Chem. Phys., 2012, 14, 16779–16779 Search PubMed.
- O. Drath, R. W. Gable, G. Poneti, L. Sorace and C. Boskovic, Cryst. Growth Des., 2017, 17, 3156–3162 CrossRef CAS.
- W. W. Kramer, L. A. Cameron, R. A. Zarkesh, J. W. Ziller and A. F. Heyduk, Inorg. Chem., 2014, 53, 8825–8837 CrossRef CAS PubMed.
- G. K. Gransbury, M.-E. Boulon, S. Petrie, R. W. Gable, R. J. Mulder, L. Sorace, R. Stranger and C. Boskovic, Inorg. Chem., 2019, 58, 4230–4243 CrossRef CAS PubMed.
- M. A. Hay, J. T. Janetzki, V. J. Kumar, R. W. Gable, R. Clérac, A. A. Starikova, P. J. Low and C. Boskovic, Inorg. Chem., 2022, 61, 17609–17622 CrossRef CAS PubMed.
- A. A. Starikova, M. G. Chegerev, A. G. Starikov and V. I. Minkin, Comput. Theor. Chem., 2018, 1124, 15–22 CrossRef CAS.
- K. J. Oberhausen, R. J. O'Brien, J. F. Richardson and R. M. Buchanan, Inorg. Chim. Acta, 1990, 173, 145–154 CrossRef CAS.
- S. Durot, C. Policar, F. Cisnetti, F. Lambert, J.-P. Renault, G. Pelosi, G. Blain, H. Korri-Youssoufi and J.-P. Mahy, Eur. J. Inorg. Chem., 2005, 2005, 3513–3523 CrossRef.
- P. J. Steel, Coord. Chem. Rev., 1990, 106, 227–265 CrossRef CAS.
Footnotes |
| † This article is dedicated to Professor Andrea Dei, brilliant teacher and far-sighted chemist. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, additional crystallographic information, powder X-ray diffraction patterns, thermogravimetric analysis. CCDC 2262804 and 2262731 entries contain the supplementary crystallographic data for 1 and 2, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra03235c |
| § Current address: Dipartimento di Scienze Ecologiche e Biologiche, Università degli Studi della Tuscia, Largo dell'Università, 01100, Viterbo, Italy. |
| This journal is © The Royal Society of Chemistry 2023 |