
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in small molecules for improving mitochondrial disorders
Liying Meng
and
Guanzhao Wu
 *
*
Department of Central Laboratory and Mitochondrial Medicine Laboratory, Qilu Hospital (Qingdao), Cheeloo College of Medicine, Shandong University, Qingdao, China. E-mail: guanzhao.wu@email.sdu.edu.cn
First published on 10th July 2023
Abstract
Mitochondrial disorders are observed in various human diseases, including rare genetic disorders and complex acquired pathologies. Recent advances in molecular biological techniques have dramatically expanded the understanding of multiple pathomechanisms involving mitochondrial disorders. However, the therapeutic methods for mitochondrial disorders are limited. For this reason, there is increasing interest in identifying safe and effective strategies to mitigate mitochondrial impairments. Small-molecule therapies hold promise for improving mitochondrial performance. This review focuses on the latest advances in developing bioactive compounds for treating mitochondrial disease, aiming to provide a broader perspective of fundamental studies that have been carried out to evaluate the effects of small molecules in regulating mitochondrial function. Novel-designed small molecules ameliorating mitochondrial functions are urgent for further research.
 Liying Meng | Dr Liying Meng received her Bachelor's degree from Shandong University in 2015. Dr Meng then obtained her PhD in Integrated Life Sciences (Chemistry) from Peking University in 2020. In September 2020, she joined Qilu Hospital (Qingdao) of Shandong University. Her research interest lies in mitochondrial diseases and chemical biology. |
 Guanzhao Wu | Dr Guanzhao Wu received his Bachelor's degree from the School of Medicine and Pharmacy, Ocean University of China, in 2013. He stayed at the same institution and earned his PhD degree in 2018 under the guidance of Professor Tao Jiang. After graduation, he joined the group of Professor Guigen Li at Texas Tech University, United States as a postdoctoral fellow in 2018. At the end of 2021, he joined Qilu Hospital (Qingdao), Cheeloo College of Medicine, Shandong University, China, as a Professor. His general interests encompass synthesizing neuroprotective agents for treating CNS diseases and chiral macromolecules. |
1. Introduction
Mitochondria are vital organelles that play essential roles in the life and death of the cell. They play a crucial role in energy metabolism and control of stress responses and are a hub for biosynthetic processes.1,2 Mitochondrial diseases are genetically determined metabolic disorders characterized by defects in oxidative phosphorylation (OXPHOS) and caused by mutations in genes in nuclear DNA (nDNA) and mitochondrial DNA (mtDNA) that encode structural mitochondrial proteins or proteins involved in mitochondrial function.3 The mitochondria OXPHOS system is embedded in the mitochondrial inner membrane (MIM) (Fig. 1). It represents the final step in converting nutrients into energy by forming ATP.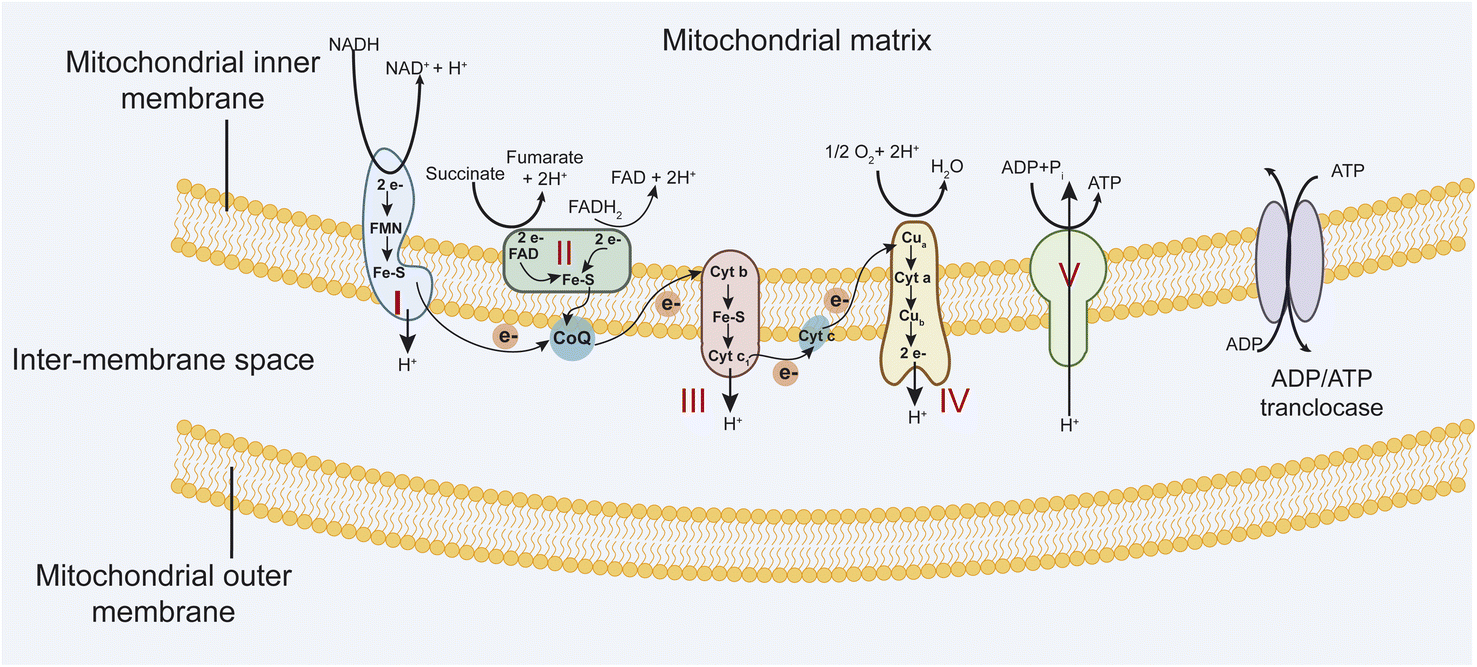 | ||
| Fig. 1 The mitochondrial respiratory chain includes complexes I–IV and complex V, an ATP synthase. | ||
The electron transport chain (ETC) involves four multi-subunit complexes (complex I–complex IV), two mobile electron carriers, coenzyme Q10, and cytochrome c.4 Complex I and II receive electrons from reduced nicotinamide adenine dinucleotide (NADH) and reduced flavin adenine dinucleotide (FADH2), respectively. Afterward, these electrons are transferred to the electron carrier coenzyme Q10, which transports them to complex III. Then, electrons are donated to cytochrome c and transported to complex IV, which transfers electrons to molecular oxygen to form water. At the same time, complexes I, III, and IV generate a transmembrane proton gradient by driving trans-MIM proton (H+) efflux from the mitochondrial matrix. Complex V converts transmembrane electrochemical proton gradient energy into mechanical energy to generate ATP by chemiosmotic coupling.5 These five complexes are assembled from 92 distinct proteins. Complex I is built up of 44 subunits (7 mtDNA- and 37 nDNA-encoded), complex II of 4 subunits, complex III of 11 subunits (1 mtDNA, 10 nDNA), complex IV of 14 subunits (3 mtDNA, 11 nDNA), and complex V of 19 subunits (2 mtDNA, 17 nDNA).6,7 Mutations in mitochondrial DNA (mtDNA) and (or) nuclear-encoded mitochondrial genes (nDNA) that affect OXPHOS function efficiently lead to a diverse group of debilitating conditions (Table 1). Besides, mitochondria are a major source of reactive oxygen species, such as superoxide, because electrons at complex I and III of the respiratory chain are often offloaded to molecular oxygen. Increased ROS production in mitochondrial diseases can result in protein, lipid, and DNA damage, potentially leading to further cellular damage and dysfunction.8,9
| Disease | Gene location | Mutation types | Clinical features |
|---|---|---|---|
| Myoclonus epilepsy with ragged-red fibres (MERRF) | MTTK | A8344G, T8356C, G8361A | Generalized epilepsy, ataxia, and myopathy |
| MTTF | G611A | ||
| Mitochondrial encephalopathy, lactic acidosis, stroke-like episodes (MELAS) | MTTL1 | A3243G, G3244A, A3252G, C3256T, T3271C, T3291C | Epilepsy, encephalopathy, myopathy, severe constipation, failure to thrive |
| MTTV | G1642A | ||
| MTTF | G583A | ||
| MTRNR2 | C3093G | ||
| MTND1 | T3308C, G3376A, G3697A, G3946A, T3949C | ||
| MTND4 | A11084G | ||
| MTND5 | A12770G, A13045C, A13084T, G13513A, A13514G | ||
| MTND6 | G14453A | ||
| MTCYB | 14787Ddel4 | ||
| Kearns–Sayre syndrome (KSS) | MTTL1 | G3249A | Short stature, diabetes mellitus, cardiomyopathy, ataxia |
| Chronic progressive external ophthalmoplegia (CPEO) | MTTL1 | C3254T | Ptosis, muscle weakness |
| MTT1 | T4274C, T4285C, G4298A, G4309A | ||
| MTTA | T5628C | ||
| MTTN | T5692C | ||
| MTTN | G5698A | ||
| MTTN | G5703G | ||
| MTTK | G8342A | ||
| MTTL2 | G12294A, A12308G, T12311C, G12325A | ||
| MTND4 | T11232C | ||
| Neuropathy, ataxia, and retinitis pigmentosa (NARP) | MTATP6 | T8993C, T8993G | Blindness, cerebellar ataxia, seizures, cognitive impairment, and peripheral neuropathy |
| Leigh syndrome(LS) | MTTV | C1624T | Lactic acidosis, failure to thrive, myopathy, bilateral symmetrical lesions in the subcortical brain |
| MTND3 | T10158C | ||
| MTND4 | C11777A | ||
| MTND5 | T12706C | ||
| MTATP6 | T9176C, T9176G, T9185C, T9191C, T8993C |
Mitochondrial dysfunction contributes to numerous health problems, including neurological and muscular degeneration, cardiomyopathies, cancer, diabetes, and aging pathologies (Table 2). Mitochondrial diseases are clinically heterogeneous and can occur at any age. Treatment of mitochondrial disorders has been challenging since multi-organ involvement in various mitochondrial diseases.10 Small molecules play an essential role in drug development.11,12 In this review, we will present small molecules that are beneficial to enhance mitochondrial function and improve primary mitochondrial diseases. We propose that rapid preliminary screening of potential therapeutic compounds in individual patients' fibroblasts could direct and advance personalized medical treatment. Furthermore, novel-designed small molecules ameliorating mitochondrial functions are urgent for further research.
| Neurological characteristics | Central nervous system | Stroke-like episodes, migraine, epilepsy, ataxia, dementia, Parkinsonism, developmental delay, psychiatric or mood disorder, developmental regression |
| Visual system | Ptosis, progressive external ophthalmoplegia, optic atrophy, retinitis pigmentosa | |
| Acoustic system | Sensorineural hearing loss | |
| Skeletal muscle | Myopathy, exercise intolerance | |
| Peripheral nervous system | Peripheral neuropathy | |
| Non-neurological characteristics | Digestive system | Gastrointestinal dysmotility; malabsorption, intestinal pseudo-obstruction, chronic villous atrophy |
| Kidney | Fanconi syndrome, renal tubular acidosis, focal segmental glomerulosclerosis, renal failure, adrenal insufficiency | |
| Metabolic/endocrine apparatus | Lactic acidosis, multiple lipomatosis, short stature, diabetes, hypothyroidism, hypoparathyroidism | |
| Heart | Cardiomyopathy, conduction defect | |
| Hematopoietic system | Sideroblastic anemia |
2. Therapies for mitochondrial disease
2.1 Molecules bypass mitochondrial complex I deficiency
Mitochondrial complex I deficiency is the most prevalent defect in the respiratory chain in pediatric patients and often leads to severe or fatal neurological symptoms, such as Leigh syndrome. Succinate is a mitochondrial substrate that is metabolized through complex II. However, it is not cell membrane-permeable and challenging to be uptaken into cells. Ehinger group reported that several cell membrane-permeable prodrugs 1–3 (Fig. 2) of the complex II substrate succinate increased ATP-linked mitochondrial oxygen consumption in complex I-deficient human blood cells, fibroblasts, and heart fibers. This therapy strategy provided a potential future intervention for patients with metabolic decompensation due to complex I dysfunction.13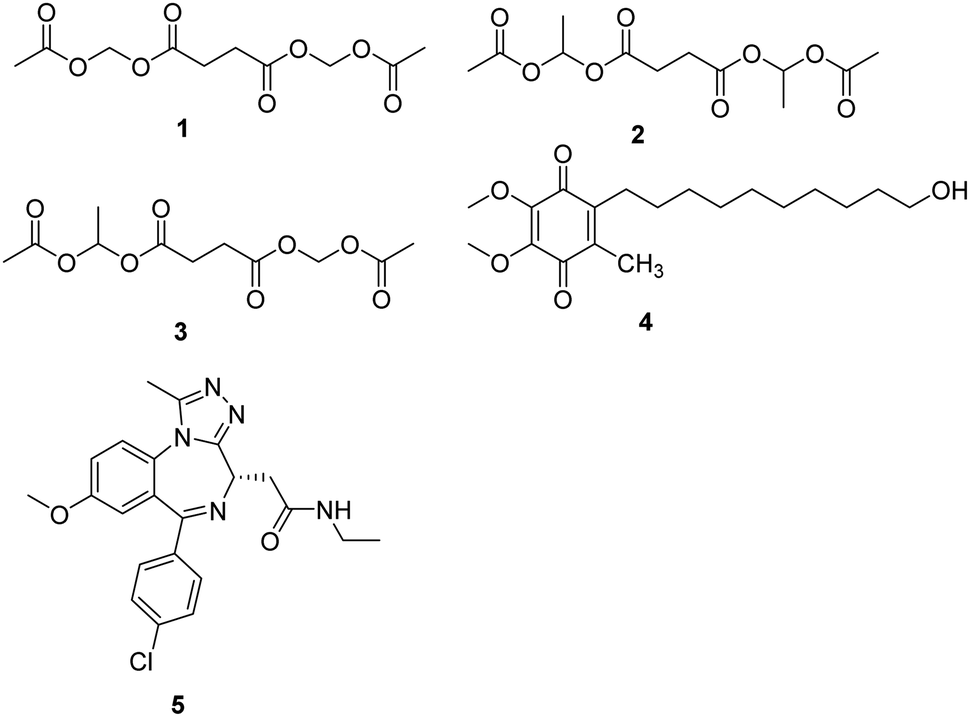 | ||
| Fig. 2 Chemical structures of compounds 1–5. | ||
Idebenone is a well-known compound, developed in the early 1980s by Takeda Pharmaceuticals against cognitive decline/dementia (4, Fig. 2), which has been evaluated in several mitochondrial and neurodegenerative diseases.14,15 Idebenone has the potential to act as an electron carrier in the respiratory chain and as an antioxidant against membrane damage caused by lipid peroxidation. The antioxidant function of idebenone is attributed to the redox cycling between hydroquinone and quinone. NAD(P)H: quinone oxidoreductase 1 (NQO1) and mitochondrial complex III were identified as the major enzymes involved in idebenone activity.16 It has been approved provisionally in Canada for the treatment of Friedreich's ataxia, while withdrawn from the Canadian market in 2013 by Santhera Pharmaceuticals due to lack of efficacy. Leber's hereditary optic neuropathy (LHON), a rare genetic mitochondrial disease that causes rapid and progressive bilateral vision loss, is the only mitochondrial disease for which IDE has been approved by the European Medicine Agency to treat visual impairment in adolescents and adults. Several new insights into the mode action of idebenone were discovered, which may provide a novel indication for this drug that might not have been considered previously.17
Bromodomain-containing protein 4 (BRD4) is a member of the bromodomain and extra terminal domain (BET) family of proteins comprising BRD2–BRD4 and BRDT. BRD4 is a chromatin-bound transcriptional regulator linked to the expression of genes associated with different biological processes, including tumor progression or inflammation.18 A recent study demonstrates that I-BET 525762A (5, Fig. 2), an inhibitor of bromodomain, could remodel the mitochondrial proteome to increase the levels and activity of OXPHOS protein complexes, increase and utilize FADH2, leading to the rescue of the bioenergetic defects and cell death caused by mutations or chemical inhibition of mitochondrial complex I.19
2.2 Agents enhancing electron transfer chain function
Coenzyme Q10 (CoQ10, 6, Fig. 3) is a naturally occurring fat-soluble vitamin-like quinone, which plays a crucial role in mitochondrial oxidative phosphorylation and ATP production.20 CoQ10 is an endogenous antioxidant and a potent free radical scavenger in mitochondrial membranes. CoQ10 exhibits potentially neuroprotective effects in neurodegenerative diseases with excess oxidative stress. However, about 50 clinical studies of CoQ10 revealed a marginal but factual treatment effect.20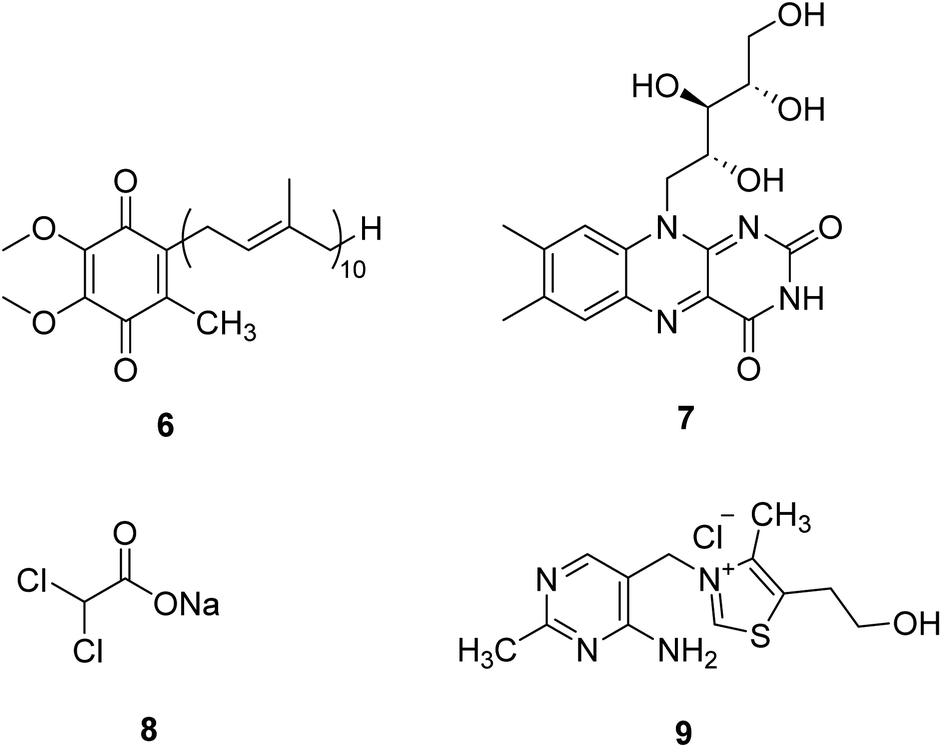 | ||
| Fig. 3 Chemical structures of 6–9. | ||
Riboflavin (7, Fig. 3), a water-soluble vitamin B, is part of the functional group of flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) cofactors and is required for numerous flavoprotein-catalyzed reactions. Riboflavin shows critical antioxidant properties essential for correct cell functioning.21 Riboflavin deficiency has been demonstrated to impair the oxidative state of the body, especially in the nervous system. Riboflavin supplementation treats migraine, Brown–Vialetto–Van Laere syndrome, Fazio–Londe disease, and some mitochondrial diseases.22,23 In the future, riboflavin may be a potential therapeutic intervention in many other neurological disorders.
Dichloroacetate (DCA, 8, Fig. 3) is an analog of pyruvate. DCA activates the E1 (pyruvate decarboxylase) subunit of the PDHC by inhibiting the PDH kinase, which usually phosphorylates and inhibits the enzyme, thus locking the enzyme in the active conformation and promoting the flux of pyruvate into the citric acid cycle.24 It is an investigational drug for the treatment of mitochondrial genetic diseases. Although it can effectively alleviate lactic acidosis in mitochondrial disorders,25 it can also cause peripheral neuropathy in individuals with MELAS syndrome.26
Thiamine (vitamin B1, 9, Fig. 3) can enhance pyruvate dehydrogenase activity, thus increasing the oxidative decomposition of pyruvate and reducing cofactors (NADH and FADH2) generation. Thiamine has been used in mitochondrial diseases individually or with other agents. Supplementation with thiamine in a family with MELAS syndrome and thiamine deficiency can improve the symptoms of myopathy and lactic acidosis and myopathy.27 Combining thiamine with CoQ10, carnitine, and vitamins C and E can improve the clinical symptoms of adult patients with Leigh syndrome with subacute severe brainstem encephalopathy.28
2.3 Agents as antioxidants
Sonlicromanol (KH176, 10, Fig. 4), a chemical entity derivative of the water-soluble form of vitamin E, is a blood–brain barrier permeable ROS-redox modulator. Sonlicromanol hydrochloride is used in the study for mitochondrial disorders. Sonlicromanol hydrochloride maintains microstructural coherence in the brain of Ndufs4−/− mice.29 A clinical research is conducted to evaluate the effect of KH176 in various cognitive domains and the impact of different doses of KH176.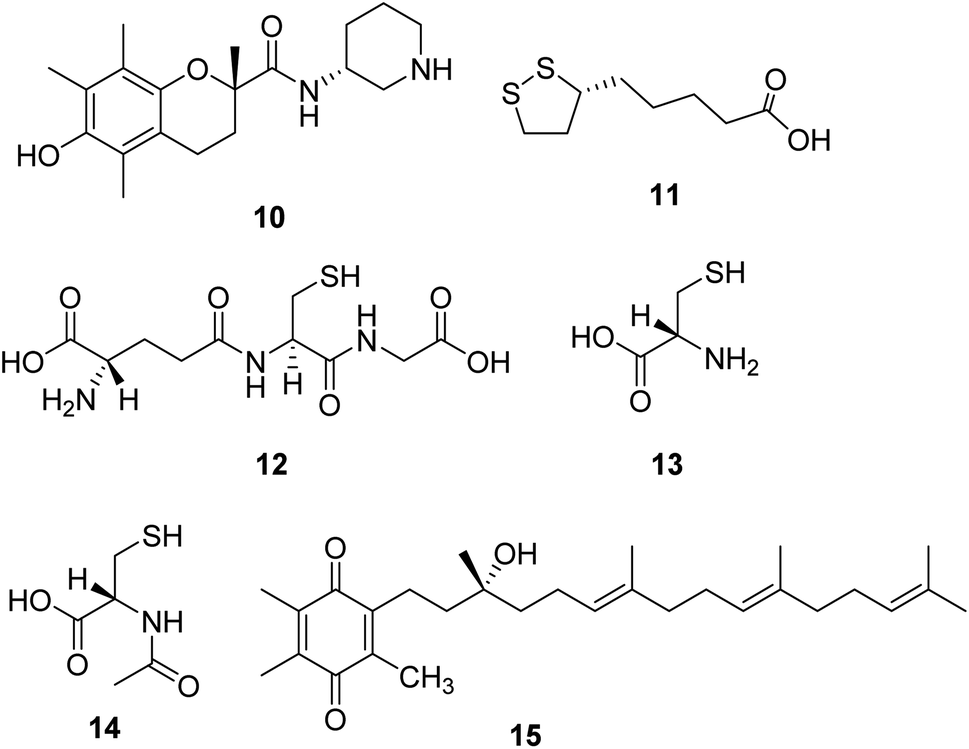 | ||
| Fig. 4 Chemical structures of 10–15. | ||
Lipoic acid (α-LA, 11, Fig. 4) is a natural molecule showing excellent antioxidant and anti-inflammatory properties. It is a coenzyme of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, which plays several roles in the pathogenesis of neurodegenerative diseases.30 One research suggests that the combination of lipoic acid, CoQ10, and creatine monohydrate effectively reduces plasma lactic acid content and oxidative stress markers in urine and improves the symptoms of muscle strength in patients with mitochondrial diseases, which is a beneficial therapeutic strategy for some mitochondrial disorders.31
Glutathione (L-γ-glutamyl-L-cysteinylglycine, 12, Fig. 4) is a crucial intracellular antioxidant that can protect the cell from reactive oxygen species (ROS). Loss of GSH is associated with several mitochondrial diseases. Thus, supplementation with cysteine donors (13, Fig. 4), which can enhance the synthesis starting material of glutathione, can potentially restore glutathione levels and eliminate excessive ROS in mitochondrial diseases.32,33 A study revealed that N-acetylcysteine (14, Fig. 4) could also enhance muscle cysteine and glutathione availability and attenuate fatigue during prolonged exercise in endurance-trained individuals.34
Based on the noticeable results obtained with CoQ10 and idebenone, a novel para-benzoquinone compound EPI-743 (15, Fig. 4) was designed and tested. EPI-743 is 1000 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times more potent than CoQ10 or idebenone in patient fibroblast assays modeling the effects of mitochondrial disease. EPI-743 is now in clinical trials to treat Leigh syndrome and other inherited mitochondrial disorders.35
000 times more potent than CoQ10 or idebenone in patient fibroblast assays modeling the effects of mitochondrial disease. EPI-743 is now in clinical trials to treat Leigh syndrome and other inherited mitochondrial disorders.35
2.4 Agents enhancing mitochondrial biogenesis
Induction of mitochondrial biogenesis through transgenic overexpression of PGC-1α is being developed as a potential treatment for mitochondrial disorders. Bezafibrate (16, Fig. 5) is a pharmacological ligand for the transcriptional cofactor PGC-1α. It shows the most extensive pre-clinical evidence of efficacy in animal models and patient cell lines.36,37 Now, it is in clinical trials to evaluate the safety of inducing mitochondrial biogenesis in patients with the m.3243A>G MTTL1 mutation.38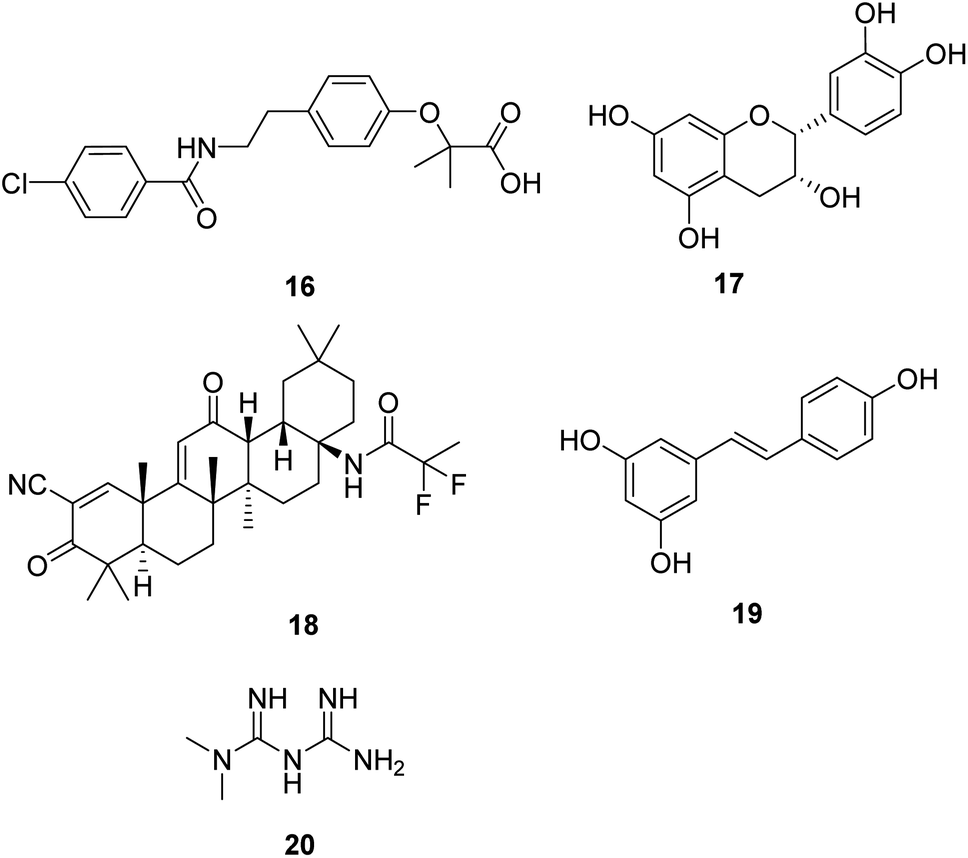 | ||
| Fig. 5 Chemical structures of 16–20. | ||
(−)-Epicatechin (17, Fig. 5) is the main flavonoid present in dark chocolate. Relevant research confirmed that (−)-epicatechin could enhance fatigue resistance and oxidative capacity in mouse muscle, which benefits clinical populations experiencing muscle fatigue.39
Omaveloxolone (RTA-408, 18, Fig. 5) is a novel synthetic oleanane triterpenoid analog. It shows significant cytoprotective effects due to its ability to activate the Nrf2 pathway.40 Studies also proved that the neuroprotective effects of RTA-408 could be attributed to the Keap1 inhibition.41 FDA has approved omaveloxolone as the first treatment for Friedreich's ataxia, a rare, inherited, degenerative disease that damages the nervous system, characterized by impaired coordination and walking.42
Resveratrol (19, Fig. 5), a natural plant polyphenol, increased AMPK and PGC-1α activity, increased mitochondrial number, and improved motor function, which is beneficial for an overall improvement in health and survival.43 As a dietary supplement, it is in clinical studies in patients with mitochondrial myopathies and skeletal muscle fatty acid oxidation disorders.
Metformin (20, Fig. 5) is widely used for treating type 2 diabetes. Studies show that metformin exerts its anti-diabetic effects by inhibiting complex I of the mitochondrial respiratory chain.44 Metformin is also a potential activator of AMPK and the stress-induced transcription factor SKN-1 nuclear factor erythroid 2-related factor 2 (Nrf2), which shows excellent potential in aging-related diseases such as neurodegenerative disease and cancer in humans.45
2.5 Agents regulating NADH/NAD+ ratio
Dysfunction of the mitochondrial oxidative phosphorylation causes an increase in the NADH/NAD+ ratio, which impairs the activity of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in the glycolysis pathway. Treatment with pyruvate (21, Fig. 6) is expected to decrease the ratio and restore glycolysis. Therefore, it is a promising approach for treating mitochondrial diseases.46,47 Phase II clinical trial of sodium pyruvate on lactic acidosis associated with mitochondrial disorders was conducted.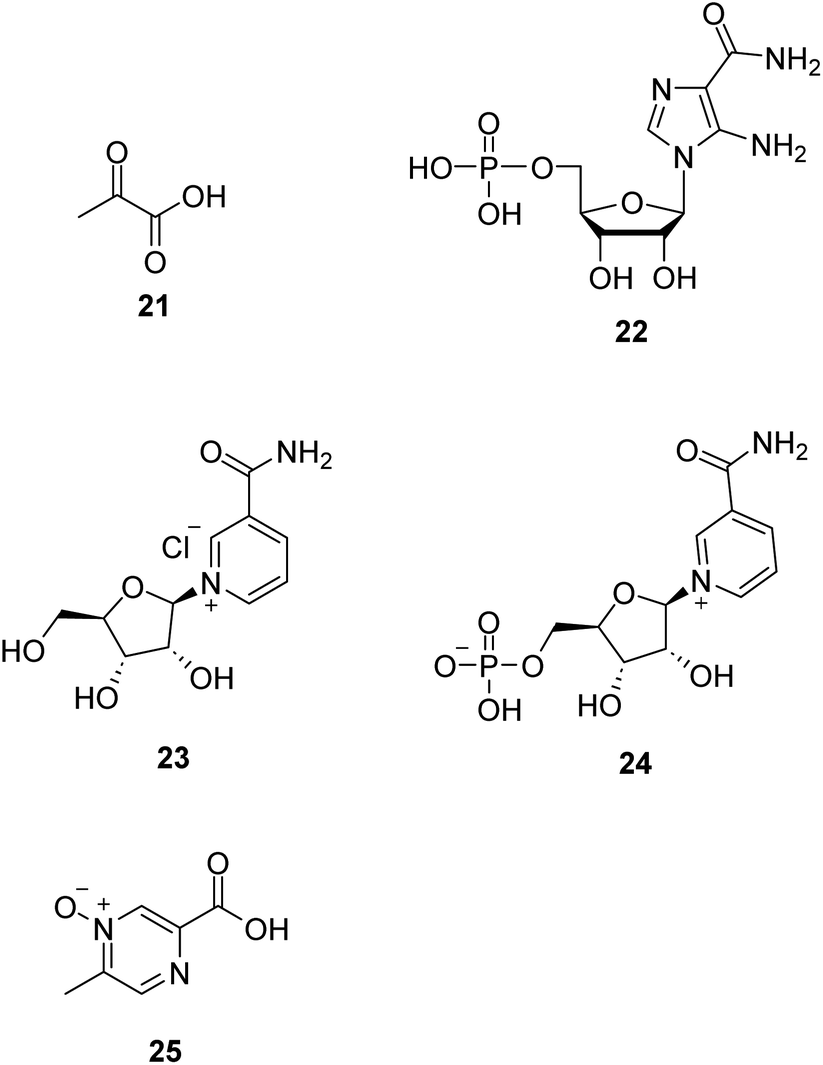 | ||
| Fig. 6 Chemical structures of 21–25. | ||
AMP-activated protein kinase (AMPK) is crucial in regulating energy homeostasis. AMPK regulates energy expenditure by modulating NAD+-dependent-type III deacetylase SIRT1.48 5-Aminoimidazole-4-carboxamide ribotide (22, AICAR, Fig. 6) is a pharmacological activator of AMPK. AICAR could improve growth and ATP content while decreasing ROS production and also increase mitochondrial biogenesis without altering mitochondrial membrane potential.49
Nicotinamide riboside (23, NR, Fig. 6), a vitamin B3 and NAD+ precursor, was previously reported to increase NAD+ levels in mice and induce mitochondrial biogenesis.50 In the mitochondrial myopathy mice model, NR robustly induced mitochondrial mass and function, cured structural abnormalities of mitochondria, and delayed the accumulation of mitochondrial DNA mutations, suggesting a promising treatment strategy for mitochondrial myopathy.51
Nicotinamide mononucleotide (24, NMN, Fig. 6), an NAD+ precursor, increased lifespan by normalizing NAD+ redox imbalance and lowering HIF1α accumulation in Ndufs4-KO skeletal muscle without affecting the brain, and attenuated lactic acidosis in Ndufs4-KO mice.52
Acipimox (25, Fig. 6), a nicotinic acid analog used to treat hyperlipidemia, has been shown to have a direct effect of acipimox on NAD+ levels, mitonuclear protein imbalance, and mitochondrial oxidative capacity and also demonstrates that acipimox can also directly affect skeletal muscle mitochondrial function in humans.53 Now, a randomized, double-blinded, placebo-controlled, adaptive design trial of the efficacy of acipimox in adult patients with mitochondrial myopathy is conducted.54
2.6 Agents restoring nitric oxide production
Nitric monoxide (NO) exerts various physiological functions in the central nervous system. There is growing evidence that NO deficiency in mitochondrial disease can complicate disease pathogenesis, such as MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes).55 NO deficiency can potentially play a significant role in the mechanism of stroke-like episodes observed in MELAS syndrome.56 Both amino acids arginine (26, Fig. 7) and citrulline (27, Fig. 7) potentially act as NO precursors. Their administration may increase NO availability and hence can have therapeutic effects in stroke-like episodes in MELAS syndrome.55 Currently, a clinical study is being conducted to assess if giving arginine or citrulline will increase the formation of nitric oxide in individuals with MELAS. Therefore, if arginine and/or citrulline are shown to increase the formation of nitric oxide, they could be used to prevent or treat strokes in patients with MELAS syndrome.57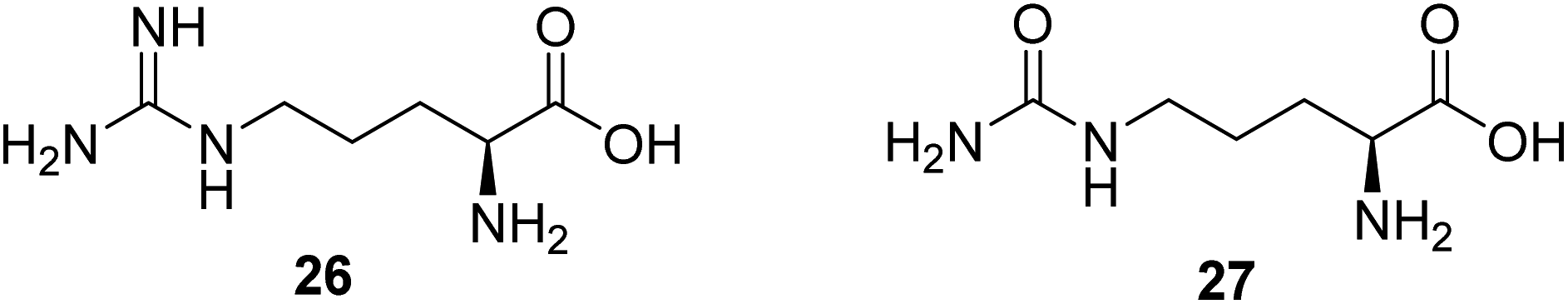 | ||
| Fig. 7 Chemical structures of 26 and 27. | ||
2.7 Agents regulating autophagy
Rapamycin (28, Fig. 8) is a mechanistic target of rapamycin kinase (mTOR) inhibitors. It is demonstrated that inhibition of mTOR improves survival and health in the Ndufs4−/− model of Leigh syndrome, which may offer therapeutic benefits to patients with Leigh syndrome and potentially other mitochondrial disorders.58 However, as a promising compound, the clinical use of rapamycin is limited by concerns about the side effects associated with the drug.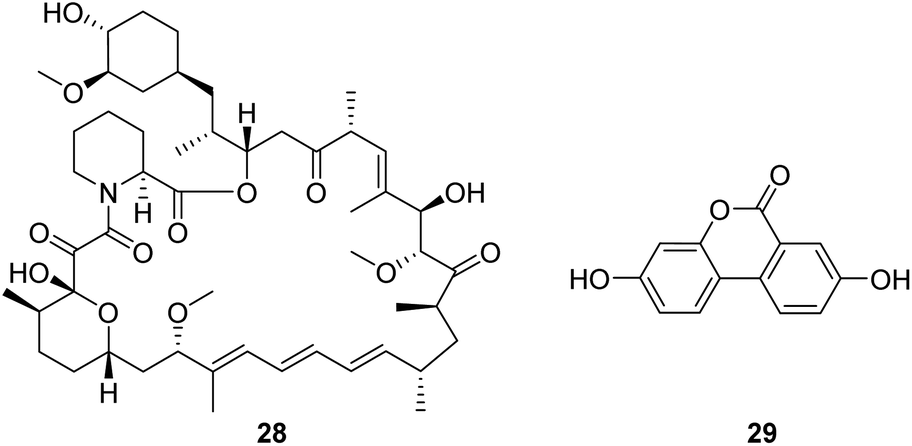 | ||
| Fig. 8 Chemical structures of 28 and 29. | ||
Urolithin A (UA, 29, Fig. 8), a first-in-class natural food metabolite, has been shown to stimulate mitophagy and improve muscle health in old animals and pre-clinical aging models.59 The results of a first-in-human clinical trial show that supplementation with UA as a nutritional intervention is safe, assist in managing the declining mitochondrial function accompanying aging, and promotes healthy muscle function throughout life.60
2.8 Agents as cardiolipin protector
Cardiolipin is a unique phospholipid exclusively expressed on the inner mitochondrial membrane. It plays an essential structural role in cristae formation and the organization of the respiratory complexes into supercomplexes for optimal oxidative phosphorylation. The interaction between cardiolipin and cytochrome c determines whether cytochrome c acts as an electron carrier or peroxidase.61 Cardiolipin has been identified as a target for drug development associated with energy deficiency. Elamipretide (Bendavia, MTP-131, SS-31, 30, Fig. 9) is an aromatic-cationic, cell-permeable tetrapeptide in a new class of mitochondrial-targeted drugs. SS-31 binds selectively to cardiolipin via electrostatic and hydrophobic interactions. By interacting with cardiolipin, SS-31 prevents cardiolipin from converting cytochrome c into a peroxidase while protecting its electron-carrying function.61 Treatment of explanted human hearts with SS-31 improves cardiac mitochondrial function.62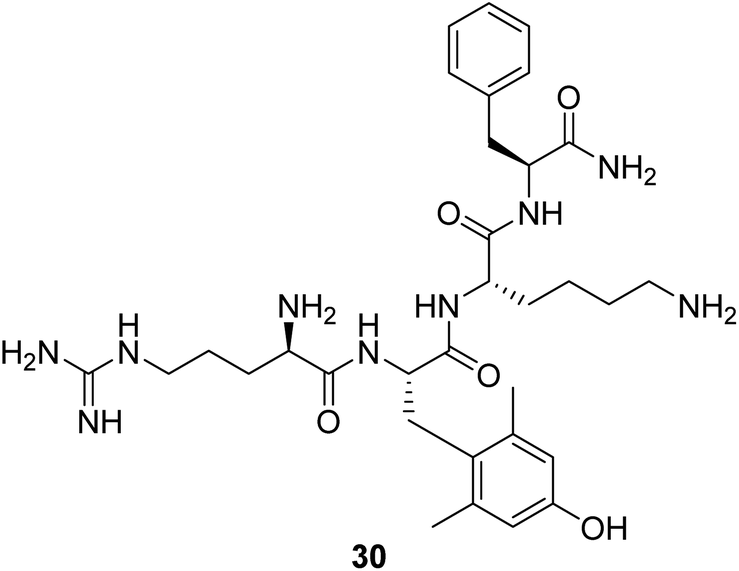 | ||
| Fig. 9 Chemical structure of 30. | ||
2.9 Agents as an energy buffer
Creatine (31, Fig. 10) is a naturally occurring bioenergetic compound. Creatine stabilizes the mitochondrial transition pore and is vital in mitochondrial ATP production. Creatine also plays an essential role in shuttling Pi from the mitochondria into the cytosol to form phosphocreatine (32, Fig. 10) to help maintain cellular bioenergetics.63 A neuroprotective effect of oral creatine was found in several animal models of neurodegenerative diseases. Creatine monohydrate supplementation can improve exercise capacity in some individuals with mitochondrial myopathies.64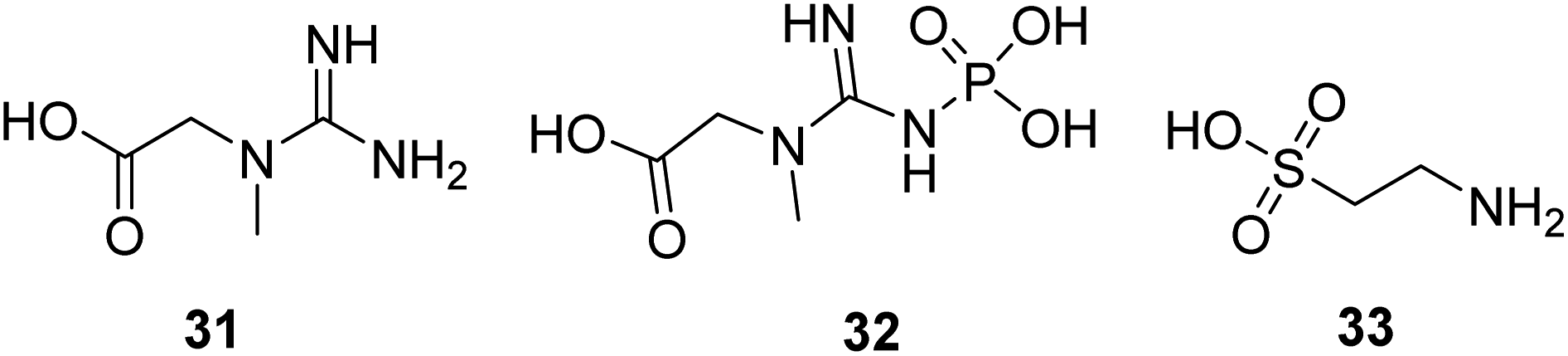 | ||
| Fig. 10 Chemical structures of 31–33. | ||
Taurine (33, Fig. 10) is a naturally occurring sulfur-containing amino acid found abundantly in excitatory tissues, such as the heart, brain, retina, and skeletal muscle. It plays a crucial role in developmental processes, such as brain development, cardiac muscle regulation, and inflammation. Taurine shows protective activity in different neurodegenerative disease models, such as Parkinson's, Alzheimer's, and Huntington's diseases.65–67 Now, it is in a clinical study to treat mitochondrial encephalomyopathy.
3. Conclusion
Overall, there have been several interesting new approaches in the potential development of new drugs to treat mitochondrial diseases. Proteolysis targeting chimera (PROTAC) technology is a novel strategy to develop new drugs with small molecules that can make protein degradation more efficient and specific, thus creating new opportunities in drug development. PROTAC is a small molecule that simultaneously binds a disease-associated protein and a ubiquitin–ligase complex, which uses the ubiquitin–protease system to eliminate mutated, denatured, and harmful cell proteins. It can effectively target and degrade proteins, including proteins that are difficult to identify and bind. Therefore, it has significant implications for drug development and treating mitochondrial diseases.In summary, the current challenges and future goals for treating mitochondrial disease revolve around improving diagnosis, developing targeted therapies, discovering biomarkers, modifying disease progression, and exploring innovative approaches like mitochondrial replacement techniques. Continued research efforts and collaboration among scientists, clinicians, and patients are essential to overcome these challenges and achieve these goals. Encouragingly, there has been remarkable progress in mitochondrial disease over the past decade. The increasing number of clinical trials in mitochondrial disorders aim for more specific and effective therapies. More importantly, the unmet clinical need for treating patients with mitochondrial diseases has stimulated academic and commercial interest in developing new treatments, as has an awareness of mitochondrial involvement in more common diseases. In this review, we discuss the bioactive compounds for treating mitochondrial disorders and focus on different pathways. The efforts in this field to provide a more targeted approach are encouraging.
Author contributions
Liying Meng wrote the manuscript and drew the pictures. Guanzhao Wu is fully responsible for the study design, research fields, drafting, and finalizing of the paper.Conflicts of interest
The authors declare no other conflicts of interest.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (NSFC 22201164), the Qilu Young Scholars Program of Shandong University (SDQLQN2021-01), and the Natural Science Foundation of Shandong Province grant ZR202102200093.References
- M. P. Murphy and R. C. Hartley, Mitochondria as a therapeutic target for common pathologies, Nat. Rev. Drug Discov., 2018, 17, 865–886 CrossRef CAS PubMed.
- F. Kraus, K. Roy, T. J. Pucadyil and M. T. Ryan, Function and regulation of the divisome for mitochondrial fission, Nature, 2021, 590, 57–66 CrossRef CAS PubMed.
- M. R. Duchen, Mitochondria in health and disease: perspectives on a new mitochondrial biology, Mol. Aspect. Med., 2004, 25, 365–451 CrossRef CAS PubMed.
- R. Guo, J. Gu, S. Zong, M. Wu and M. Yang, Structure and mechanism of mitochondrial electron transport chain, Biomed. J., 2018, 41, 9–20 CrossRef PubMed.
- P. Neupane, S. Bhuju, N. Thapa and H. K. Bhattarai, ATP synthase:structure, function and inhibition, Biomol. Concepts, 2019, 10, 1–10 CAS.
- J. Nouws, L. G. J. Nijtmans, J. A. Smeitink and R. O. Vogel, Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: cause, pathology and treatment options, Brain, 2012, 135, 12–22 CrossRef PubMed.
- W. J. H. Koopman, F. Distelmaier, J. A. M. Smeitink and P. H. G. M. Willems, OXPHOS mutations and neurodegeneration, EMBO J., 2013, 32, 9–29 CrossRef CAS PubMed.
- M. P. Murphy, How mitochondria produce reactive oxygen species, Biochem. J., 2009, 417, 1–13 CrossRef CAS PubMed.
- D. C. Wallace, Mitochondrial diseases in man and mouse, Science, 1999, 283, 1482–1488 CrossRef CAS PubMed.
- G. Pfeffer, K. Majamaa, D. M. Turnbull, D. Thorburn and P. F. Chinnery, Treatment for mitochondrial disorders, Cochrane Database Syst. Rev., 2012, 2012, CD004426 Search PubMed.
- A. W. El-Hattab, A. M. Zarante, M. Almannai and F. Scaglia, Therapies for mitochondrial diseases and current clinical trials, Mol. Genet. Metab., 2017, 122, 1–9 CrossRef CAS PubMed.
- O. M. Russell, G. S. Gorman, R. N. Lightowlers and D. M. Turnbull, mitochondrial diseases: hope for the Future, Cell, 2020, 181, 168–188 CrossRef CAS PubMed.
- J. K. Ehinger, S. Piel, R. Ford, M. Karlsson, F. Sjövall, E. Å. Frostner, S. Morota, R. W. Taylor, D. M. Turnbull and C. Cornell, et al., Cell-permeable succinate prodrugs bypass mitochondrial complex I deficiency, Nat. Commun., 2016, 7, 12317 CrossRef CAS PubMed.
- Y. Ikejiri, E. Mori, K. Ishii, K. Nishimoto, M. Yasuda and M. Sasaki, Idebenone improves cerebral mitochondrial oxidative metabolism in a patient with MELAS, Neurology, 1996, 47, 583–585 CrossRef CAS PubMed.
- T. Klopstock, P. Yu-Wai-Man, K. Dimitriadis, J. Rouleau, S. Heck, M. Bailie, A. Atawan, S. Chattopadhyay, M. Schubert and A. Garip, et al., A randomized placebo-controlled trial of idebenone in Leber's hereditary optic neuropathy, Brain, 2011, 134, 2677–2686 CrossRef PubMed.
- C. Varricchio, et al., Discovery of novel 2-aniline-1, 4-naphthoquinones as potential new drug treatment for Leber's hereditary optic neuropathy (LHON), J. Med. Chem., 2020, 63, 13638–13655 CrossRef CAS PubMed.
- N. Gueven, P. Ravishankar, R. Eri and E. Rybalka, Idebenone: When an antioxidant is not an antioxidant, Redox Biol., 2021, 38, 101812 CrossRef CAS PubMed.
- M. G. Baratta, A. C. Schinzel, Y. Zwang, P. Bandopadhayay, C. Bowman-Colin, J. Kutt, J. Curtis, H. Piao, L. C. Wong and A. L. Kung, et al., An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 232–237 CrossRef CAS PubMed.
- J. J. Barrow, E. Balsa, F. Verdeguer, C. D. J. Tavares, M. S. Soustek, L. R. Hollingsworth, M. Jedrychowski, R. Vogel, J. A. Paulo and J. Smeitink, et al., Bromodomain inhibitors correct bioenergetic deficiency caused by mitochondrial disease complex I mutations, Mol. Cell, 2016, 64, 163–175 CrossRef CAS PubMed.
- D. S. Kerr, Treatment of mitochondrial electron transport chain disorders: a review of clinical trials over the past decade, Mol. Genet. Metab., 2010, 99, 246–255 CrossRef CAS PubMed.
- J. T. Pinto and J. Zempleni, Riboflavin, Adv. Nutr., 2016, 7, 973–975 CrossRef CAS PubMed.
- P. L. Bernsen, F. J. Gabreëls, W. Ruitenbeek and H. L. Hamburger, Treatment of complex I deficiency with riboflavin, J. Neurol. Sci., 1993, 118, 181–187 CrossRef CAS PubMed.
- F. Scaglia and J. L. Northrop, The mitochondrial myopathy encephalopathy, lactic acidosis with stroke-like episodes (MELAS) syndrome: a review of treatment options, CNS Drugs, 2006, 20, 443–464 CrossRef CAS PubMed.
- S. Whitehouse, R. H. Cooper and P. J. Randle, Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids, Biochem. J., 1974, 141, 761–774 CrossRef CAS PubMed.
- M. Abdelmalak, A. Lew, R. Ramezani, A. L. Shroads, B. S. Coats, T. Langaee, M. N. Shankar, R. E. Neiberger, S. H. Subramony and P. W. Stacpoole, Long-term safety of dichloroacetate in congenital lactic acidosis, Mol. Genet. Metab., 2013, 109, 139–143 CrossRef CAS PubMed.
- P. Kaufmann, K. Engelstad, Y. Wei, S. Jhung, M. C. Sano, D. C. Shungu, W. S. Millar, X. Hong, C. L. Gooch and X. Mao, et al., Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial, Neurology, 2006, 66, 324–330 CrossRef CAS PubMed.
- Y. Sato, M. Nakagawa, I. Higuchi, M. Osame, E. Naito and K. Oizumi, Mitochondrial myopathy and familial thiamine deficiency, Muscle Nerve, 2000, 23, 1069–1075 CrossRef CAS PubMed.
- C. Mermigkis, I. Bouloukaki, V. Mastorodemos, A. Plaitakis, V. Alogdianakis, N. Siafakas and S. Schiza, Medical treatment with thiamine, coenzyme Q, vitamins E and C, and carnitine improved obstructive sleep apnea in an adult case of Leigh disease, Sleep Breath., 2013, 17, 1129–1135 CrossRef PubMed.
- R. de Haas, D. Das, A. Garanto, H. G. Renkema, R. Greupink, P. van den Broek, J. Pertijs, R. W. J. Collin, P. Willems and J. Beyrath, et al., Therapeutic effects of the mitochondrial ROS-redox modulator KH176 in a mammalian model of Leigh Disease, Sci. Rep., 2017, 7, 11733 CrossRef PubMed.
- K. Marangon, S. Devaraj, O. Tirosh, L. Packer and I. Jialal, Comparison of the effect of alpha-lipoic acid and alpha-tocopherol supplementation on measures of oxidative stress, Free Radic. Biol. Med., 1999, 27, 1114–1121 CrossRef CAS PubMed.
- M. C. Rodriguez, J. R. MacDonald, D. J. Mahoney, G. Parise, M. F. Beal and M. A. Tarnopolsky, Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders, Muscle Nerve, 2007, 35, 235–242 CrossRef CAS PubMed.
- G. M. Enns and T. M. Cowan, Glutathione as a redox biomarker in mitochondrial disease-implications for therapy, J. Clin. Med., 2017, 6, 50 CrossRef PubMed.
- M. Marí, A. Morales, A. Colell, C. García-Ruiz, N. Kaplowitz and J. C. Fernández-Checa, Mitochondrial glutathione: features, regulation and role in disease, Biochim. Biophys. Acta, 2013, 1830, 3317–3328 CrossRef PubMed.
- I. Medved, M. J. Brown, A. R. Bjorksten, K. T. Murphy, A. C. Petersen, S. Sostaric, X. Gong and M. J. McKenna, N-acetylcysteine enhances muscle cysteine and glutathione availability and attenuates fatigue during prolonged exercise in endurance-trained individuals, J. Appl. Physiol., 2004, 97, 1477–1485 CrossRef CAS PubMed.
- D. Martinelli, M. Catteruccia, F. Piemonte, A. Pastore, G. Tozzi, C. Dionisi-Vici, G. Pontrelli, T. Corsetti, S. Livadiotti and V. Kheifets, et al., EPI-743 reverses the progression of the pediatric mitochondrial disease-genetically defined Leigh Syndrome, Mol. Genet. Metab., 2012, 107, 383–388 CrossRef CAS PubMed.
- L. M. Dillon, A. Hida, S. Garcia, T. A. Prolla and C. T. Moraes, Long-term bezafibrate treatment improves skin and spleen phenotypes of the mtDNA mutator mouse, PLoS One, 2012, 7, e44335 CrossRef CAS PubMed.
- A. Hofer, N. Noe, C. Tischner, N. Kladt, V. Lellek, A. Schauß and T. Wenz, Defining the action spectrum of potential PGC-1α activators on a mitochondrial and cellular level in vivo, Hum. Mol. Genet., 2014, 23, 2400–2415 CrossRef CAS PubMed.
- H. Steele, A. Gomez-Duran, A. Pyle, S. Hopton, J. Newman, R. J. Stefanetti, S. J. Charman, J. D. Parikh, L. He and C. Viscomi, et al., Metabolic effects of bezafibrate in mitochondrial disease, EMBO Mol. Med., 2020, 12, e11589 CrossRef CAS PubMed.
- L. Nogueira, I. Ramirez-Sanchez, G. A. Perkins, A. Murphy, P. R. Taub, G. Ceballos, F. J. Villarreal, M. C. Hogan and M. H. Malek, (−)-Epicatechin enhances fatigue resistance and oxidative capacity in mouse muscle, J. Physiol., 2011, 589, 4615–4631 CrossRef CAS PubMed.
- D. C. Goldman, V. Alexeev, E. Lash, C. Guha, U. Rodeck and W. H. Fleming, The triterpenoid RTA 408 is a robust mitigator of hematopoietic acute radiation syndrome in mice, Radiat. Res., 2015, 183, 338–344 CrossRef CAS PubMed.
- T. Shekh-Ahmad, R. Eckel, S. Dayalan Naidu, M. Higgins, M. Yamamoto, A. T. Dinkova-Kostova, S. Kovac, A. Y. Abramov and M. C. Walker, KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy, Brain, 2018, 141, 1390–1403 CrossRef PubMed.
- V. Profeta, K. McIntyre, M. Wells, C. Park and D. R. Lynch, Omaveloxolone: an activator of Nrf2 for the treatment of Friedreich ataxia, Expert Opin. Investig., 2023, 32, 5–16 CrossRef CAS PubMed.
- J. A. Baur, K. J. Pearson, N. L. Price, H. A. Jamieson, C. Lerin, A. Kalra, V. V. Prabhu, J. S. Allard, G. Lopez-Lluch and K. Lewis, et al., Resveratrol improves health and survival of mice on a high-calorie diet, Nature, 2006, 444, 337–342 CrossRef CAS PubMed.
- M. Y. El-Mir, V. Nogueira, E. Fontaine, N. Avéret, M. Rigoulet and X. Leverve, Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I, J. Biol. Chem., 2000, 275, 223–228 CrossRef CAS PubMed.
- A. A. Soukas, H. Hao and L. Wu, Metformin as Anti-Aging Therapy: Is It for Everyone?, Trends Endocrinol. Metab., 2019, 30, 745–755 CrossRef CAS PubMed.
- M. Li, S. Zhou, C. Chen, L. Ma, D. Luo, X. Tian, X. Dong, Y. Zhou, Y. Yang and Y. Cui, Therapeutic potential of pyruvate therapy for patients with mitochondrial diseases: a systematic review, Ther. Adv. Endocrinol. Metab., 2020, 11, 2042018820938240 CAS.
- T. Fujii, F. Nozaki, K. Saito, A. Hayashi, Y. Nishigaki, K. Murayama, M. Tanaka, Y. Koga, I. Hiejima and T. Kumada, Efficacy of pyruvate therapy in patients with mitochondrial disease: a semi-quantitative clinical evaluation study, Mol. Genet. Metab., 2014, 112, 133–138 CrossRef CAS PubMed.
- C. Cantó, Z. Gerhart-Hines, J. N. Feige, M. Lagouge, L. Noriega, J. C. Milne, P. J. Elliott, P. Puigserver and J. Auwerx, AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity, Nature, 2009, 458, 1056–1060 CrossRef PubMed.
- A. Golubitzky, P. Dan, S. Weissman, G. Link, J. D. Wikstrom and A. Saada, Screening for active small molecules in mitochondrial complex I deficient patient's fibroblasts, reveals AICAR as the most beneficial compound, PLoS One, 2011, 6, e26883 CrossRef CAS PubMed.
- C. Cantó, R. H. Houtkooper, E. Pirinen, D. Y. Youn, M. H. Oosterveer, Y. Cen, P. J. Fernandez-Marcos, H. Yamamoto, P. A. Andreux and P. Cettour-Rose, et al., The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity, Cell Metab., 2012, 15, 838–847 CrossRef PubMed.
- N. A. Khan, M. Auranen, I. Paetau, E. Pirinen, L. Euro, S. Forsström, L. Pasila, V. Velagapudi, C. J. Carroll and J. Auwerx, et al., Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3, EMBO Mol. Med., 2014, 6, 721–731 CrossRef CAS PubMed.
- C. F. Lee, A. Caudal, L. Abell, G. A. Nagana Gowda and R. Tian, Targeting NAD+ metabolism as interventions for mitochondrial disease, Sci. Rep., 2019, 9, 3073 CrossRef PubMed.
- T. van de Weijer, E. Phielix, L. Bilet, E. G. Williams, E. R. Ropelle, A. Bierwagen, R. Livingstone, P. Nowotny, L. M. Sparks and S. Paglialunga, et al., Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans, Diabetes, 2015, 64, 1193–1201 CrossRef CAS PubMed.
- A. Abouhajar, L. Alcock, T. Bigirumurame, P. Bradley, L. Brown, I. Campbell, S. Del Din, J. Faitg, G. Falkous and G. S. Gorman, et al., Acipimox in Mitochondrial Myopathy (AIMM): study protocol for a randomised, double-blinded, placebo-controlled, adaptive design trial of the efficacy of acipimox in adult patients with mitochondrial myopathy, Trials, 2022, 23, 789 CrossRef PubMed.
- A. W. El-Hattab, L. T. Emrick, S. Chanprasert, W. J. Craigen and F. Scaglia, Mitochondria: role of citrulline and arginine supplementation in MELAS syndrome, Int. J. Biochem. Cell Biol., 2014, 48, 85–91 CrossRef CAS PubMed.
- Y. Koga, Y. Akita, J. Nishioka, S. Yatsuga, N. Povalko, Y. Tanabe, S. Fujimoto and T. Matsuishi, L-arginine improves the symptoms of strokelike episodes in MELAS, Neurology, 2005, 64, 710–712 CrossRef CAS PubMed.
- A. W. El-Hattab, L. T. Emrick, J. W. Hsu, S. Chanprasert, M. Almannai, W. J. Craigen, F. Jahoor and F. Scaglia, Impaired nitric oxide production in children with MELAS syndrome and the effect of arginine and citrulline supplementation, Mol. Genet. Metab., 2016, 117, 407–412 CrossRef CAS PubMed.
- S. C. Johnson, M. E. Yanos, E.-B. Kayser, A. Quintana, M. Sangesland, A. Castanza, L. Uhde, J. Hui, V. Z. Wall and A. Gagnidze, et al., mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome, Science, 2013, 342, 1524–1528 CrossRef CAS PubMed.
- D. Ryu, L. Mouchiroud, P. A. Andreux, E. Katsyuba, N. Moullan, A. A. Nicolet-Dit-Félix, E. G. Williams, P. Jha, G. Lo Sasso and D. Huzard, et al., Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents, Nat. Med., 2016, 22, 879–888 CrossRef CAS PubMed.
- P. A. Andreux, W. Blanco-Bose, D. Ryu, F. Burdet, M. Ibberson, P. Aebischer, J. Auwerx, A. Singh and C. Rinsch, The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans, Nat. Metab., 2019, 1, 595–603 CrossRef CAS PubMed.
- A. V. Birk, S. Liu, Y. Soong, W. Mills, P. Singh, J. D. Warren, S. V. Seshan, J. D. Pardee and H. H. Szeto, The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin, J. Am. Soc. Nephrol., 2013, 24, 1250–1261 CrossRef CAS PubMed.
- K. C. Chatfield, G. C. Sparagna, S. Chau, E. K. Phillips, A. V. Ambardekar, M. Aftab, M. B. Mitchell, C. C. Sucharov, S. D. Miyamoto and B. L. Stauffer, Elamipretide improves mitochondrial function in the failing human heart, JACC: Basic Transl. Sci., 2019, 4, 147–157 Search PubMed.
- T. Wallimann, M. Tokarska-Schlattner and U. Schlattner, The creatine kinase system and pleiotropic effects of creatine, Amino Acids, 2011, 40, 1271–1296 CrossRef CAS PubMed.
- M. A. Tarnopolsky, Creatine as a therapeutic strategy for myopathies, Amino Acids, 2011, 40, 1397–1407 CrossRef CAS PubMed.
- H. Jang, S. Lee, S. L. Choi, H. Y. Kim, S. Baek and Y. Kim, Taurine directly binds to oligomeric amyloid-β and recovers cognitive deficits in Alzheimer model mice, Adv. Exp. Med. Biol., 2017, 975, 233–241 CrossRef CAS PubMed.
- M. G. Tadros, A. E. Khalifa, A. B. Abdel-Naim and H. M. M. Arafa, Neuroprotective effect of taurine in 3-nitropropionic acid-induced experimental animal model of Huntington's disease phenotype, Pharmacol. Biochem. Behav., 2005, 82, 574–582 CrossRef CAS PubMed.
- J. Menzie, H. Prentice and J.-Y. Wu, Neuroprotective mechanisms of taurine against ischemic stroke, Brain Sci., 2013, 3, 877–907 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |