
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of indolizine lactones as anticancer agents and their optimization through late-stage functionalization†
Thiago Sabino da Silva a,
Matheus da Silva Souzab,
Adriano Defini Andricopulob and
Fernando Coelho*a
a,
Matheus da Silva Souzab,
Adriano Defini Andricopulob and
Fernando Coelho*a
aLaboratory of Synthesis of Natural Products and Drugs, Institute of Chemistry, University of Campinas, Rua Monteiro Lobato, S/N, 13083-970, Campinas, São Paulo, Brazil. E-mail: facoelho@unicamp.br
bLaboratory of Medicinal and Computational Chemistry, Institute of Physics of Sao Carlos, University of Sao Paulo – Avenida Joao Dagnone, 1100-13563-120 – Sao Carlos, SP, Brazil
First published on 5th July 2023
Abstract
Indolizines fused with a seven-member lactone ring were identified as a promising scaffold in the search for new anticancer agents. Through a modular synthetic sequence, a library of cis and trans indolizines lactones had their antiproliferative activity evaluated against hormone-refractory prostate DU-145 and triple-negative breast MDA-MB-231 cancer cell lines. A methoxylated analogue was identified as an initial hit against MDA-MB-231 and late-stage functionalization of the indolizine core led to analogues within potencies up to twenty times higher than the parent precursor.
Introduction
Indolizines, which are indole isomers and bioisoesters,1 are present in numerous compounds that exhibit biological activity.2,3 They are considered privileged scaffolds for drug development,4 but no indolizine-based drugs have been marketed so far.1 The best studied indolizines in terms of biological activity were primarily assessed through dipolar cycloadditions involving pyridinium ylides and electron deficient alkenes/alkynes or via the wide-spread Chichibabin reaction from 2-methylpyridinium salts (Scheme 1).1–3,5–15 The mild conditions, the ready access to starting materials and the modular synthetic approach that characterised those methods were some of the reasons leading to their extensive use in the search for indolizines with biological activities. Consequently, indolizines obtained by those methods were typically characterised by a high degree of planarity and by the absence of stereogenic centers, leading to an overall low level of molecular complexity, a property associated with improvements in the physicochemical proprieties, selectivity and safety of compounds during the drug discovery process.16–19 The multi-substituted character of these indolizines, especially at the nucleophilic positions 1 and 3 of the heterocyclic core or/and the presence of electron-withdrawing groups directly attached to them, potentially precluded the evaluation of late-stage functionalization (LSF) strategies as suitable option to increase the drug profile of these molecules. This is another point that should be addressed during the design of new indolizines for biological activity evaluation.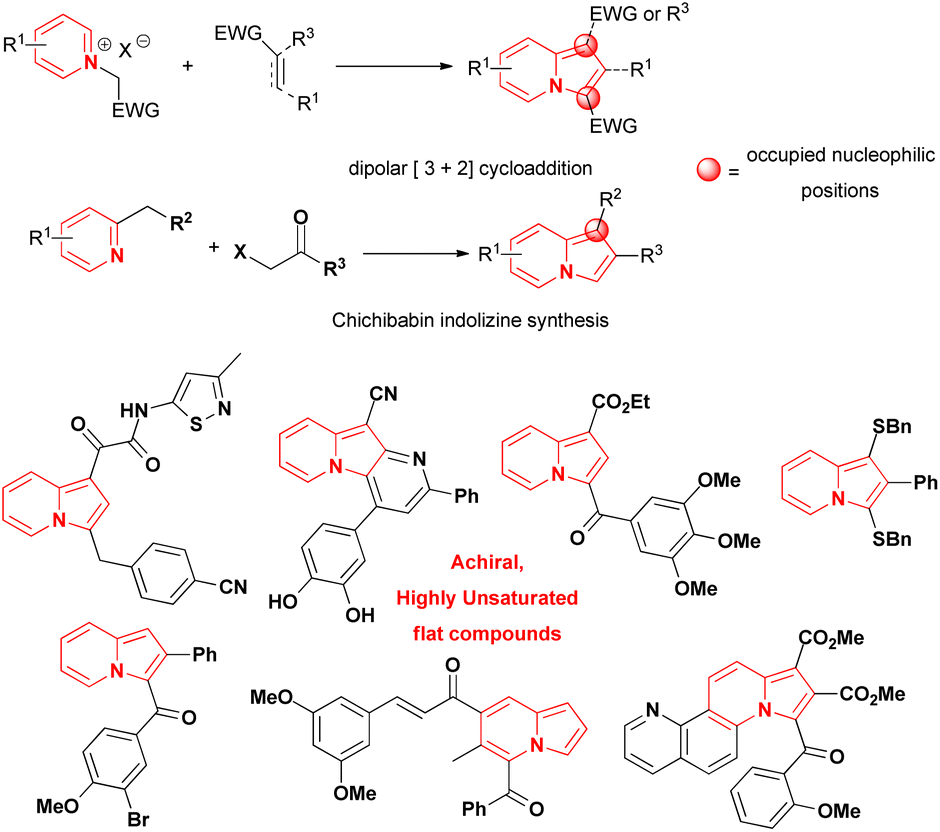 | ||
| Scheme 1 Examples of indolizine motifs founded in compounds with anticancer activity and common strategies for their synthesis.1,10–15 | ||
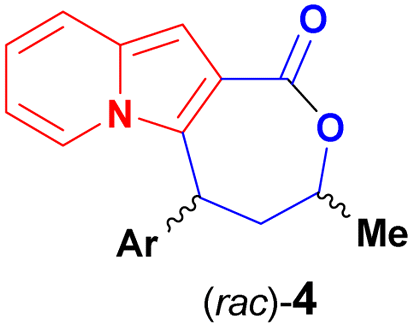
The development of synthetic methods that would lead to more complex indolizines is therefore of great significance, and the introduction of chiral centers, fused rings or increases in the fraction of sp3 carbons (Fsp3 – the number of sp3 hybridised carbons/total carbon count) could be used to assess compounds with different topologies and increased molecular complexity.16 The functionalization of indolizines, rather than their de novo synthesis, appears to represent a suitable way to achieve this goal. Along these lines, we recently described an organocatalytic asymmetric and diastereoselective synthesis of an indolizine fused to a lactone ring bearing two stereocenters, starting from the pre-formed 2-carboxyindolizine core 1 (Scheme 2).20 The synthetic sequence of regioselective conjugate addition of 1 to enone 2, followed by carbonyl reduction/ring closure, offers a modular approach for synthesis of indolizine lactone 4 with different structural patterns, allowing a rapid evaluation of the structure–activity relationship (SAR) towards a biological target. Additionally, the nucleophilic C1 position of the indolizine remains available on 4 and could represent a site for late-stage functionalization after the identification of promising compounds.
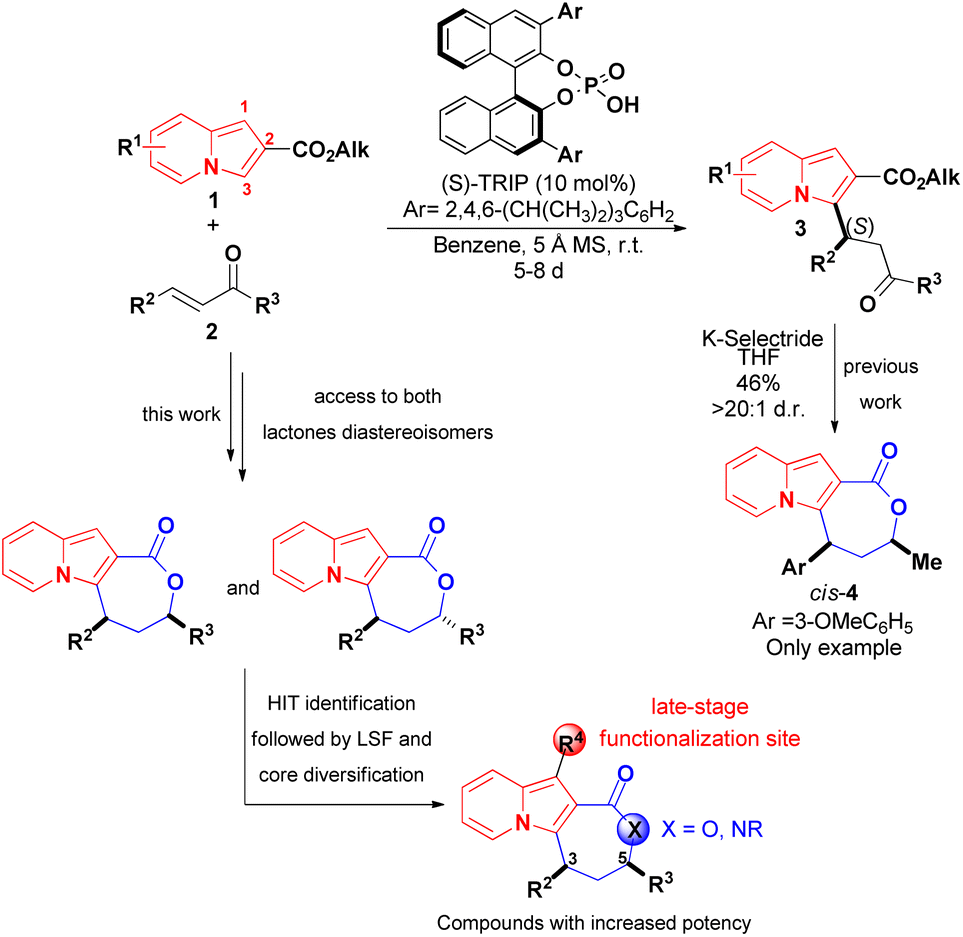 | ||
| Scheme 2 Reported asymmetric synthesis of an indolizine lactone (Coelho and List, 2017)20 and the molecular sites available for further modifications during the investigation of biological activities of 4. | ||
The exquisite indolizine lactone 4 has a seven-membered ring that resembles the one encountered in colchicine, a potent cytotoxic natural product extensively investigated against different types of cancer,21 and its fused heterocyclic–carbocycle motif could be viewed as an ring-expanded version of cyclopenta[b]indoles that display excellent cytotoxicity against multiple cancer cell lines (Fig. 1).22 These factors, associated with the findings that indolizines1,10–15 and lactones23–25 were encountered in compounds with antitumoural proprieties, prompted our evaluation of this new core against triple-negative breast (MDA-MB-231) and hormone-refractory prostate metastatic (DU-145) cancer cells-both of which are metastatic.
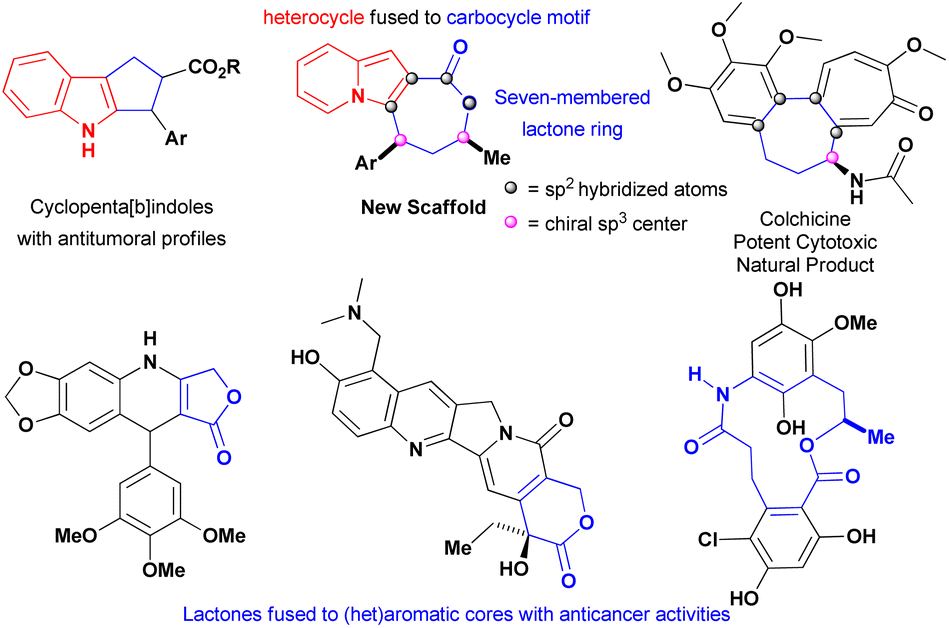 | ||
| Fig. 1 Structural features of indolizines lactones founded in compounds with anticancer proprieties. | ||
We anticipated that the flexible approach used to construct more complex indolizine lactone derivatives, together with the late-stage functionalization strategy, would be fundamental to finding compounds with increased antiproliferative activity.
Results and discussion
Chemistry
Despite the existence of an enantio- and diastereoselective route for the synthesis of cis-indolizine lactones 4, the evaluation of racemates and trans isomers potentially expands the possibilities of finding a hit compound on a first biological screening. Targeted cis and trans (rac)-lactones were obtained using the following 3-step sequence (Scheme 3): (1) diphenyl phosphate (DPP) catalysed conjugate addition between 2-carboxymethyl indolizine (1) and methyl cinnamyl ketones 2; (2) reduction of conjugate adducts 3 using NaBH4 (no lactonization was observed in this step), followed by chromatography separation of the syn and anti-hydroxy esters 5; (3) lactonization of alcohols 5 catalysed by para-toluenesulfonic acid (p-TsOH).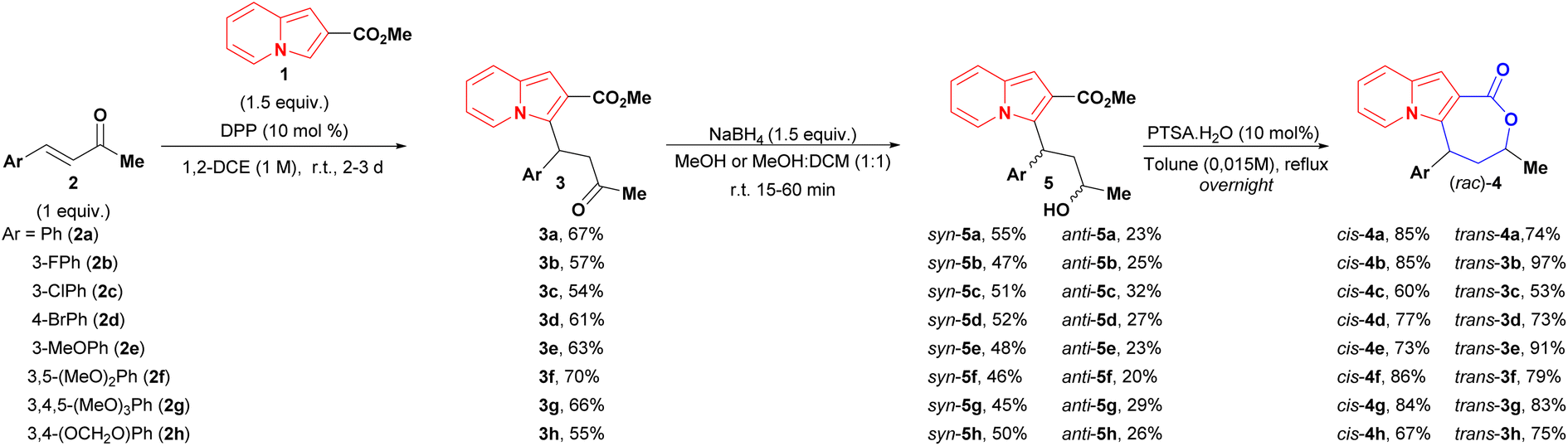 | ||
| Scheme 3 Synthetic sequence to both diastereoisomers of indolizines lactones. aThe relative configurations of 5 were determined after the lactonization step. | ||
As general remarks, the reduction of conjugate adducts 3 using the non-hindered reducing agent NaBH4 favoured the syn isomer, as observed in our previous work.20 It showed moderated stereoselectivity, which allowed the isolation of anti alcohols 5, as well as access to trans lactones 4. Alternatively, the key hydroxy esters could be obtained in a one-pot two-step conjugate addition/reduction way, thereby obviating the need for the one chromatography step, as exemplified in the synthesis of syn and anti 5f analogues in a slightly better global yield of 55% yield (Scheme 4).
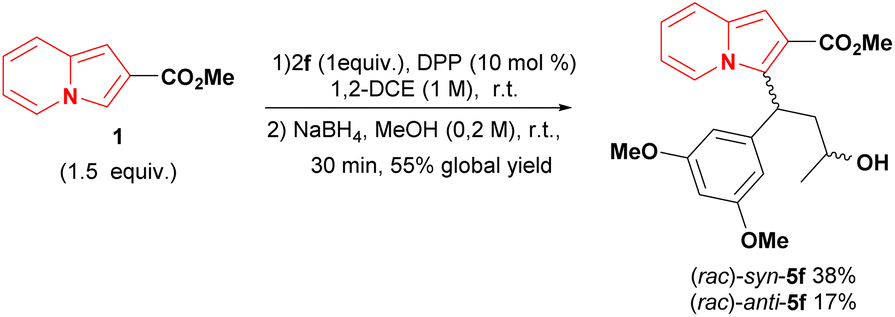 | ||
| Scheme 4 One-pot synthesis of 5f hydroxy esters. | ||
Indolizine lactone cis-4f was identified as an initial hit on the first biological screening (see Table 1) and was selected for further synthetic manipulations. Modifications to the backbone of the seven-membered ring and to the indolizine motif were realised first. Lactam analogues of 4f were synthesised via reductive amination of conjugate adduct 3 to afford a separable mixture of syn and anti aminoesters 6, which cyclized into the corresponding cis and trans lactams (Scheme 5). Intriguingly, the majority product after the reduction step was the anti diastereoisomer 6 rather than the syn diastereoisomer observed after reduction of 3 with NaBH4 or K-Selectride.20
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | IC50 | CC50f | ||
| Cancer cell lines | Non-tumor cell lines (systemic) | ||||
| DU-145b | MDA-MB-231c | FC3Hd | HFF-1e | ||
| a Mean values ± standard error were expressed as the result of three independent experiments. DMSO was used as a negative control and doxorubicin and colchicine were used as positive controls (see ESI for further details).b Human prostate epithelial tumour cells.c Human breast epithelial tumour cells.d Mouse liver fibroblast cells.e Human skin fibroblast cells.f Only molecules with IC50 ≤ 20 μM in cancer cell lines were tested against the nontumour ones.g Values in parentheses are the selectivity indexes (SI = CC50/IC50) towards the HFF-1 cell lines. ND: not determined. | |||||
| 1 | Ph (cis-4a) | >100 | 67.86 ± 6.61 | ND | ND |
| 2 | Ph (trans-4a) | >100 | >100 | ND | ND |
| 3 | 3-FPh (cis-4b) | >100 | 79.51 ± 21.84 | ND | ND |
| 4 | 3-FPh (trans-4b) | >100 | 52.87 ± 0.84 | ND | ND |
| 5 | 3-ClPh (cis-4c) | 54.71 ± 2.59 | 35.99 ± 0.69 | ND | ND |
| 6 | 3-ClPh (trans-4c) | >100 | >100 | ND | ND |
| 7 | 4-BrPh (cis-4d) | 34.41 ± 2.68 (0.03)g | 16.72 ± 0.80 (0.06)g | >100 | 0.92 ± 0.22 |
| 8 | 4-BrPh (trans-4d) | 46.65 ± 7.29 (>2)g | 21.99 ± 3.44 (>5)g | >100 | >100 |
| 9 | 3-MeOPh (cis-4e) | >100 | 21.05 ± 2.22 (>5)g | >100 | >100 |
| 10 | 3-MeOPh (trans-4e) | >100 | 53.03 ± 2.31 | ND | ND |
| 11 | 3,5-MeOPh (cis-4f) | 52.41 ± 0.45 (>2)g | 20.47 ± 0.79 (>5)g | >100 | >100 |
| 12 | 3,5-MeOPh (trans-4f) | >100 | 27.64 ± 0.99 | ND | ND |
| 13 | 3,4,5-MeOPh (cis-4g) | 36.93 ± 2.58 | >100 | ND | ND |
| 14 | 3,4,5-MeOPh (trans-4g) | >100 | >100 | ND | ND |
| 15 | Piperonyl (cis-4h) | 52.28 ± 1.46 | 31.95 ± 2.72 | ND | ND |
| 16 | Piperonyl (trans-4h) | 35.67 ± 2.24 | 29.41 ± 5.46 | ND | ND |
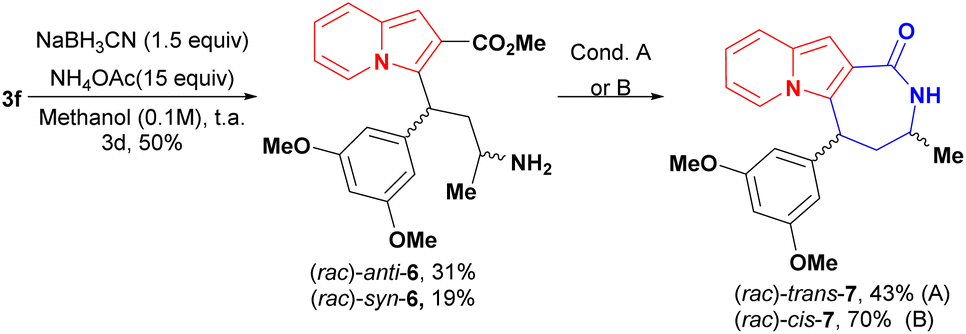 | ||
| Scheme 5 Synthesis of lactams 7 and partial hydrogenation of cis-4f. Condition A: NEt3 (0.022 M) reflux, overnight B: toluene (0.025 M), NEt3 (12 equiv.) reflux, overnight. | ||
Another straightforward modification of the scaffold of cis-4f was achieved through the partial hydrogenation of its indolizine core using platinum oxide (PtO2) as the catalyst. This led to the 5,6,7,8-tetrahydro-indolizine lactone cis-8 in 65% yield (Scheme 6).26
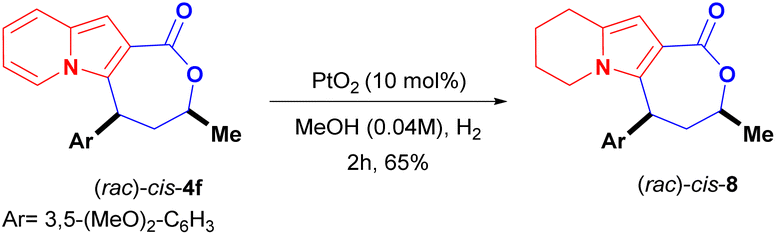 | ||
| Scheme 6 Synthesis of lactams 7 and partial hydrogenation of cis-4f. | ||
In addition to changes to the backbone of cis-4f, late-stage functionalization of the available indolizine C1 position opened up a new chemical space for further investigations. The C1 reactivity of indolizines has scarcely been explored since it is less reactive than C3 and few methods are available for the synthesis of C1-free, C3-substituted indolizines.27 Here, we investigated the reactivity of cis-4f in reactions of thiolation,15,28 trifluoroacetylation and oxidative coupling,29 to further evaluate the impact of these peripheral modifications on the antiproliferative activity (Scheme 7).
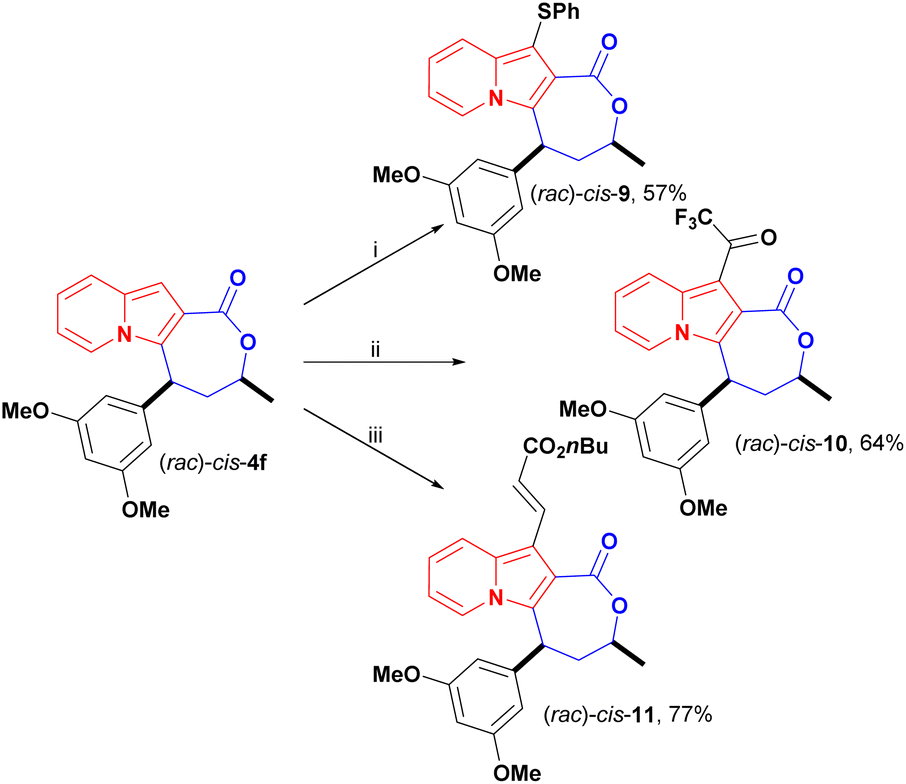 | ||
| Scheme 7 Late-stage functionalization of cis-4f: (i) PhSH (2 equiv.), KI (10%), TBHP (2 equiv.), DMSO (0.1 M), 60 °C, 22 h. (ii) TFFA (1.25 equiv.), NEt3 (1.5 equiv.), DCM (0.02 M), 0 °C, 1 h. (iii) n-butyl acrylate (4 equiv.), [Cp*RhCl2]2 (4 mol%), AgSbF6 (0.16 equiv.), Cu(OAc)2. H2O (2.1 equiv.), DCE (0.1 M), 100 °C. | ||
Thiolation of cis-4f in the presence of thiophenol, tert-butyl hydroperoxide (TBHP), and catalytic potassium iodide (KI) afforded cis-9 in 57% yield (Scheme 7). Previously,28 only unsubstituted 2-aryl indolizines were reported as substrates for this transformation, but indolizine lactone cis-4f proved to be a suitable substrate for this transformation.
Trifluoroacetylation was achieved under mild conditions using trifluoroacetic anhydride (TFFA) and triethylamine. The trifluoromethyl ketone (TFMK) derivative cis-10 was obtained in 64% yield (Scheme 7).
Olefination of cis-4f with n-butyl acrylate was carried out under Rh(II) catalysis using the lactone motif as a directing group for C–H activation at the C1 position, and cis-11 was obtained in good yield, at 77%, in these conditions (Scheme 7). Notably, C–H activation of the C8 and C2 positions of indolizines was previously achieved employing amides, esters, ketones and aldehydes as directing groups.29,30 Here, we successfully expanded this strategy to activate the C-1 position within the carboxyl lactone motif.
Biological activity
The antiproliferative activities of lactones 4 were tested in vitro against two human cancer cell lines (DU-145, ATCC: HTB-81, hormone-refractory prostate cancer; and MDA-MB-231, ATCC: HTB-26, triple-negative breast cancer) purchased from American Type Culture Collection (ATCC, Manassas, Virginia, EUA) or Rio de Janeiro Cell Bank (BCRJ, Rio de Janeiro, Brazil), using resazurin assays. DMSO was used as a negative control, and colchicine and doxorubicin were employed as positive controls (Table 1).Except for the trans lactones 4a, 4c, and 4g, all compounds exhibited antiproliferative activity against at least one of the human cancer cell lines tested. As a general trend, indolizines 4 showed more cytotoxicity towards breast cancer cells than towards the prostate cancer cells. The exception was cis-4g (Table 1, entry 13), which seemed to be promisingly selective for the DU-145 cell lines (IC50 = 36.93 ± 2.58 μM). Fortunately, the diastereoisomer cis was more active than the trans isomers, and could be synthesised diastereoselectively using a hindered reducing agent.20 This difference in activity highlights the important role of chirality in determining biological effectiveness.
The cytotoxic activities of cis halogenated indolizines (Table 1, entries 3, 5 and 7) against MDA-MB-231 cell lines were inversely proportional to the electronegativity of the substituents, suggesting a possible ‘halogen bonding effect’.31,32 The presence of a fluorine atom on cis-4b [IC50 (MDA-MB-231) = 79.51 ± 21.84 μM] had a negative impact on its antiproliferative capacity when compared with unsubstituted phenyl analogue cis-4a [IC50 (MDA-MB-231) = 67.86 ± 6.61 μM]. Conversely, the presence of a bromine atom appeared to increase the antiproliferative activities of both the cis and trans lactones 4d (entries 6 and 7).
Lactone cis-4d (entry 7) exhibited the highest cytotoxicity against both cancer lines [IC50 (DU-145) = 34.41 ± 2.68 μM and IC50 (MDA-MB-231) = 16.72 ± 0.80 μM] in this first evaluation and was almost inactive against non-tumoural mouse liver fibroblast cell lines [CC50 (FC3H, BCRJ: 0082) > 100 μM] (Table 1, entry 7). Unfortunately, cis-4d was more cytotoxic against healthy human skin fibroblast cell lines [CC50 (HFF-1, ATCC: SCRC-1041) = 0.92 ± 0.22 μM] than it was against cancer lines, thereby ruling out further investigations and structural modifications of this compound. The trans isomer of lactone 4d (entry 8) was slightly less potent against cancer cells [IC50 (DU-145) = 46.65 ± 7.29 μM and IC50 (MDA-MB-231) = 21.99 ± 3.44 μM)] than cis-4d; nevertheless, it displayed good selectivity index (SI) values [SI (HFF-1/DU-145) ≥ 2.14 and SI (HFF-1/MDA-MB-231 ≥ 4.55)] when compared with the observed cytotoxicity against both systemic cell types evaluated (CC50 > 100 μM for both cell lines).
Among the alkoxylated analogues (Table 1, entries 9–16), mono-methoxylated cis-4e and bis-methoxylated cis-4f (entries 9 and 11) displayed lower IC50 values against breast cancer cell lines (21.05 ± 2.22 μM and 20.47 ± 0.79 μM, respectively) and good SI values (both compounds showed CC50 values higher than 100 μM for FC3H and HFF-1 cells). Increasing the number of methoxy groups (entries 13 and 14) or using piperonyl derivatives (entries 15 and 16) affected the antiproliferative activity of the lactones.
Indolizine lactone cis-4f showed the best balance between cytotoxicity and selectivity and was therefore selected for further synthetic manipulations that might improve these attributes (Fig. 2). The presence of alkoxylated motifs in a series of natural and synthetic compounds with observed antitumoral activities21,33–36 and in other studies with indolizine derivatives as well,10,11,37,38 reinforced the choice of cis-4f as our first hit.
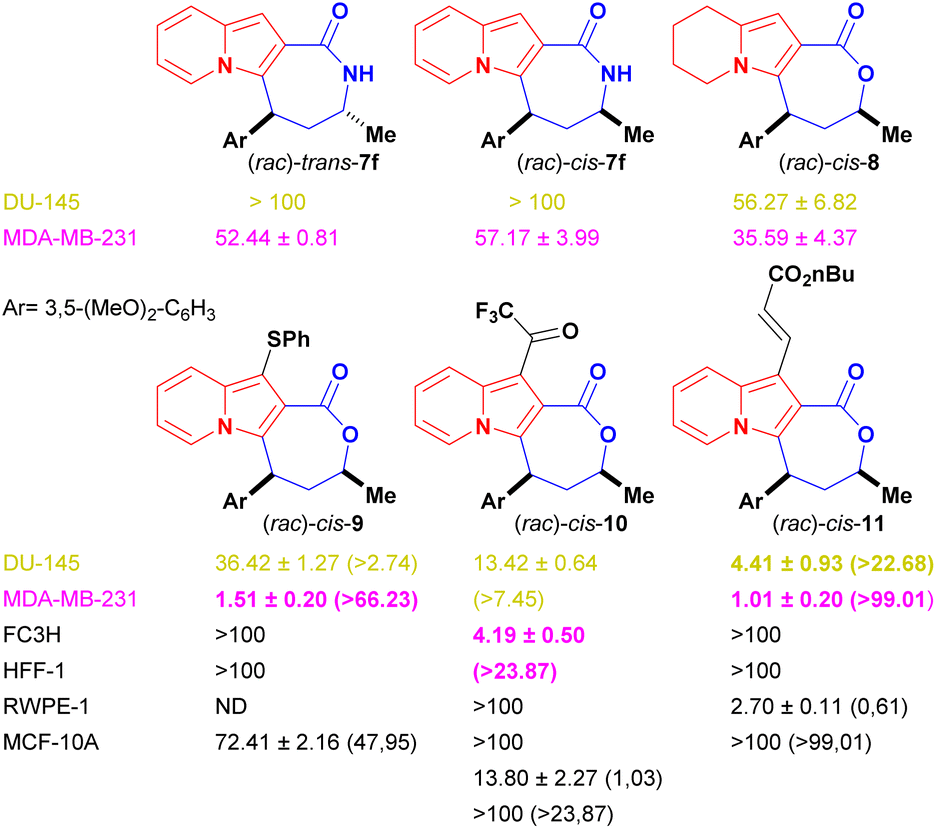 | ||
| Fig. 2 IC50 values (μM) of cis-4f analogues. Selectivity index values (SI = CC50/IC50) between cancer cell lines (DU-154 and MDA-MB-231) and systemic (FC3H and HFF-1) and local non-tumour cells [RWPE-1 (prostate) and MCF-10A (breast)] are listed in parentheses. ND: not determined. | ||
Changes to the backbone of 4f, such as the substitution of the seven-member lactone ring for a lactam ring, could give important insights into the molecular proprieties associated with biological activity. The N–H motif acts as a hydrogen-bond donor and could therefore positively impact the molecular recognition of a compound by a biological target, while also improving its water solubility. In addition, lactams are less susceptible to hydrolysis and might improve compound bioavailability compared with lactones derivatives.39,40 However, the lactam analogues of 4f, cis-7 [IC50 (DU-145) ≥ 100 μM and IC50 (MDA-MB-231) = 57.17 ± 3.99 μM] and trans-7 [IC50 (DU-145) ≥ 100 μM and IC50 (MDA-MB-231) = 52.44 ± 0.81 μM] were considerably less cytotoxic than the parent compound to cancer cell lines (Fig. 2), proving that the presence of oxygen on the seven-membered ring was essential to biological activity.
The role of the fully aromatised indolizine core on the biological outcome was evaluated by synthesising a 5,6,7,8-tetrahydro-indolizine cis-8 (Scheme 6). This new compound possesses a higher fraction of sp3 carbon atoms (where Fsp3 = the number of sp3 hybridised carbons/total carbon count), at Fsp3 = 0.476 versus Fsp3 = 0.285 for the parent compound cis-4f, a property that is used to measure molecular complexity.16,17 Derivative cis-8 was, nevertheless, less active [IC50 (DU-145) ≥ 56.27 ± 6.82 μM and IC50 (MDA-MB-231) = 35.59 ± 4.37 μM] than cis-4f, demonstrating the important role of the indolizine aromatised core (Fig. 2).
Peripherical modifications of cis-4f were then performed through late-stage functionalization of the indolizine C1 position (see Scheme 7). Fortuitously, the thiolated, trifluoroacetylated and olefinated analogues were more cytotoxic than their precursor against both cancer cell lines without being harmful to the systemic non-tumour FC3H and HFF-1 cells (CC50 > 100 μM for both lines) (Fig. 2). Remarkably, all the new analogues were more active against breast cancer cells.
Thiolated cis-9 displayed an impressive 13.5-fold cytotoxicity increase against the breast cancer cells (IC50 = 1.51 ± 0.20 μM) and a modest 1.4-fold against the prostate cancer cells (IC50 = 36.42 ± 1.27 μM) when compared with the activity of the parent compound cis-4f. Decoration of cis-4f with the covalent warheads trifluoromethyl ketone and acrylate resulted in compounds with increased antiproliferative activity (Fig. 2). TFMK cis-10 [IC50 (DU-145) = 13.42 ± 0.64 μM and IC50 (MDA-MB-231) = 4.19 ± 0.50 μM] showed a 4.9-fold increased cytotoxicity against the breast cancer cells and a 3.9-fold increase against the prostate cancer cells. Olefinated analogue cis-11 displayed the highest antiproliferative values against both cancer lines among all compounds tested, with an impressive 12-fold increase against prostate cancer cells (IC50 = 4.41 ± 0.93 μM) and a 20-fold cytotoxicity increase against breast cancer cells (1.01 ± 0.20 μM).
Finally, with the aim of broadening the analysis of the specificity of the selected compounds regarding the site of the primary manifestation of prostate and breast cancer, cis-9 to 11, which displayed IC50 values lower than 20 μM and systemic selectivity indexes higher than 5, had their cytotoxicity evaluated against local tissue non-tumoural RWPE-1 (BCRJ: 0389) and MCF-10A (ATCC: CRL-10317) cells (Fig. 2). Indolizine lactone cis-9, which showed markedly better cytotoxicity against MDA-MB-231 cell lines than against DU-145 lines, displayed a promising local SI value for breast tissues (SI = 47.95). Low or no local selectivity was observed for prostate cells when cis-10 and 11 were evaluated (SI = 1.03 and 0.61 respectively); nevertheless, they showed good to excellent local SI values towards breast cell lines (SI ≥ 24 and ≥99 respectively).
Conclusions
The modular ring synthesis of cis and trans decorated lactones 4 emphasised the role of stereochemistry in biological activity, as the cis compounds were systematically more cytotoxic than the trans compounds for both tested cancer cell lines DU-145 (prostate) and MDA-MB-231 (breast) and were selectively more potent towards breast cancer cells. By screening both diastereoisomers, we doubled the number of identities tested and found bis-methoxylated cis-4f as an initial hit. Late-stage functionalization of cis-4f proved to be a suitable strategy for increasing the antiproliferative activity of the core while still maintaining good selectivity index values. Decoration of the cis-4f motif with covalent warheads, such as trifluoromethyl ketone and acrylate, resulted in up to a 20-fold cytotoxicity increment.Although the introduction of covalent warheads would be expected to lead to an increase in cytotoxicity, we found that the thiolated analogue cis-9 potentially avoided the toxicity issues associated with irreversible covalent inhibitors due their promiscuous reactivity.
Considering the availability of an organocatalytic, asymmetric and diastereoselective synthesis of cis-indolizine lactones 4, we can expect that the evaluation of enantiopure compounds will lead to IC50 values even lower than the ones observed here. The modular synthetic approach towards these cores also offers other sites for further modifications, such as the indolizine pyridine moiety or the thioether substitution pattern of cis-9 derivatives.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financed, in part, by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior–Brasil (CAPES)–Finance Code 001. F. C. thanks also the Brazilian National Council for Science and Development (CNPq) for the research fellowship (process no. 307840/2014-0 and 301330/2018-2) and FAPESP (process no. 2013/07600-3 and 2018/02611-0). T. S. S. thanks CAPES for the fellowship. M. S. S. acknowledge the Brazilian funding agency CNPq (process no. 141245/2019-0) and FAPESP (process no. 2018/25289-7 and 2022/00219-1) for the financial support. The authors would also like to thank the Center for Innovation in Biodiversity and Drug Discovery (CIBFar-CEPID, process no. 2013/07600-3).Notes and references
- D. A. James, K. Koya, H. Li, G. Liang, Z. Xia, W. Ying, Y. Wu and L. Sun, Bioorg. Med. Chem. Lett., 2008, 18, 1784 CrossRef CAS PubMed.
- K. M. Dawood and A. A. Abbas, Expert Opin. Ther. Pat., 2020, 30, 695 CrossRef CAS PubMed.
- G. S. Singh and E. E. Mmatli, Eur. J. Med. Chem., 2011, 46, 5237 CrossRef CAS PubMed.
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS PubMed.
- V. Sharma and V. Kumar, Med. Chem. Res., 2014, 23, 3593 CrossRef CAS.
- C. R. de Souza, A. C. Gonçalves, M. F. Z. J. Amaral, A. A. Dos Santos and G. C. Clososki, Targets Heterocycl. Syst., 2016, 20, 365 CAS.
- B. Sadowski, J. Klajn and D. T. Gryko, Org. Biomol. Chem., 2016, 14, 7804 RSC.
- S. Dong, X. Fu and X. Xu, Asian J. Org. Chem., 2020, 9, 1133 CrossRef CAS.
- J. Hui, Y. Ma, J. Zhao and H. Cao, Org. Biomol. Chem., 2021, 19, 10245 RSC.
- A. Boot, A. Brito, T. van Wezel, H. Morreau, M. Costa and F. Proença, Anticancer Res., 2014, 34, 1673 CAS.
- A. Ghinet, C. M. Abuhaie, P. Gautret, B. Rigo, J. Dubois, A. Farce, D. Belei and E. Bîcu, Eur. J. Med. Chem., 2015, 89, 115 CrossRef CAS PubMed.
- R. Danac, C. M. Al Matarneh, S. Shova, T. Daniloaia, M. Balan and I. I. Mangalagiu, Bioorg. Med. Chem., 2015, 23, 2318 CrossRef CAS PubMed.
- S. Park, E. H. Kim, J. Kim, S. H. Kim and I. Kim, Eur. J. Med. Chem., 2018, 144, 435 CrossRef CAS PubMed.
- A. V. Aksenov, N. A. Arutiunov, N. K. Kirilov, D. A. Aksenov, I. Y. Grishin, N. A. Aksenov, H. Wang, L. Du, T. Betancourt, S. C. Pelly, A. Kornienko and M. Rubin, Org. Biomol. Chem., 2021, 19, 7234 RSC.
- G. Li, X. Wu, P. Sun, Z. Zhang, E. Shao, J. Mao, H. Cao and H. Huang, Biomed. Pharmacother., 2021, 133, 110961 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed.
- F. Lovering, Medchemcomm, 2013, 4, 515 RSC.
- O. Méndez-Lucio and J. L. Medina-Franco, Drug Discovery Today, 2017, 22, 120 CrossRef PubMed.
- S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6699 CrossRef CAS PubMed.
- J. T. M. Correia, B. List and F. Coelho, Angew. Chem., Int. Ed., 2017, 56, 7967 CrossRef CAS PubMed.
- Y. Lu, J. Chen, M. Xiao, W. Li and D. D. Miller, Pharm. Res., 2012, 29, 2943 CrossRef CAS PubMed.
- M. S. Santos, D. C. Fernandes, M. T. Rodrigues, T. Regiani, A. D. Andricopulo, A. L. T. G. Ruiz, D. B. Vendramini-Costa, J. E. de Carvalho, M. N. Eberlin and F. Coelho, J. Org. Chem., 2016, 81, 6626 CrossRef CAS PubMed.
- Y. Hitotsuyanagi, M. Fukuyo, K. Tsuda, M. Kobayashi, A. Ozeki, H. Itokawa and K. Takeya, Bioorg. Med. Chem. Lett., 2000, 10, 315 CrossRef CAS PubMed.
- Y. Pommier, Nat. Rev. Cancer, 2006, 6, 789 CrossRef CAS PubMed.
- M. Wang, G. Shen and B. S. J. Blagg, Bioorg. Med. Chem. Lett., 2006, 16, 2459 CrossRef CAS PubMed.
- B. V. M. Teodoro, J. T. M. Correia and F. Coelho, J. Org. Chem., 2015, 80, 2529 CrossRef CAS PubMed.
- L. A. Zeoly, L. V Acconcia, M. T. Rodrigues, H. Santos, R. A. Cormanich, J. C. Paniagua, A. Moyano and F. Coelho, Org. Biomol. Chem., 2023, 21, 3567 RSC.
- B. Li, Z. Chen, H. Cao and H. Zhao, Org. Lett., 2018, 20, 3291 CrossRef CAS PubMed.
- X. Feng, J. Tian, Y. Sun, H. Hu, M. Lu, Y. Kan, D. Fang and C. Wang, Chin. Chem. Lett., 2021, 32, 470 CrossRef CAS.
- P. P. Jadhav, N. M. Kahar and S. G. Dawande, Eur. J. Org. Chem., 2019, 2019, 7831 CrossRef CAS.
- L. A. Hardegger, B. Kuhn, B. Spinnler, L. Anselm, R. Ecabert, M. Stihle, B. Gsell, R. Thoma, J. Diez, J. Benz, J. M. Plancher, G. Hartmann, D. W. Banner, W. Haap and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 314 CrossRef CAS PubMed.
- Y. Lu, Y. Liu, Z. Xu, H. Li, H. Liu and W. Zhu, Expert Opin. Drug Discovery, 2012, 7, 375 CrossRef CAS PubMed.
- J. M. Andreu, B. Perez-Ramirez, M. J. Gorbunoff, D. Ayala and S. N. Timasheff, Biochemistry, 1998, 37, 8356 CrossRef CAS PubMed.
- M. Niu, J. Qin, C. Tian, X. Yan, F. Dong, Z. Cheng, G. Fida, M. Yang, H. Chen and Y. Gu, Acta Pharmacol. Sin., 2014, 35, 967 CrossRef CAS PubMed.
- L. Li, S. Jiang, X. Li, Y. Liu, J. Su and J. Chen, Eur. J. Med. Chem., 2018, 151, 482 CrossRef CAS PubMed.
- W. Shuai, G. Wang, Y. Zhang, F. Bu, S. Zhang, D. D. Miller, W. Li, L. Ouyang and Y. Wang, J. Med. Chem., 2021, 64, 7963 CrossRef CAS PubMed.
- L. Lucescu, A. Ghinet, D. Belei, B. Rigo, J. Dubois and E. Bîcu, Bioorg. Med. Chem. Lett., 2015, 25, 3975 CrossRef CAS PubMed.
- M.-C. Sardaru, A. M. Craciun, C.-M. Al Matarneh, I. A. Sandu, R. M. Amarandi, L. Popovici, C. I. Ciobanu, D. Peptanariu, M. Pinteala, I. I. Mangalagiu and R. Danac, J. Enzyme Inhib. Med. Chem., 2020, 35, 1581 CrossRef CAS PubMed.
- I. Chourpa, J. M. Millot, G. D. Sockalingum, J. F. Riou and M. Manfait, Biochim. Biophys. Acta, Gen. Subj., 1998, 1379, 353 CrossRef CAS PubMed.
- K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03395c |
| This journal is © The Royal Society of Chemistry 2023 |