
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient oxidation of benzyl alcohol into benzaldehyde catalyzed by graphene oxide and reduced graphene oxide supported bimetallic Au–Sn catalysts†
Lili Liu ,
Xiaojing Zhou,
Chunling Xin,
Baoli Zhang,
Guangman Zhang,
Shanshan Li,
Li Liu* and
Xishi Tai*
,
Xiaojing Zhou,
Chunling Xin,
Baoli Zhang,
Guangman Zhang,
Shanshan Li,
Li Liu* and
Xishi Tai*
School of Chemistry & Chemical Engineering and Environmental Engineering, Weifang University, Weifang 261061, China. E-mail: liuliwfu@126.com; taixs@wfu.edu.cn
First published on 7th August 2023
Abstract
A series of bimetallic and monometallic catalysts comprising Au and Sn nanoparticles loaded on graphene oxide (GO) and reduced graphene oxide (rGO) were prepared using three distinct techniques: two-step immobilization, co-immobilization, and immobilization. X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), energy dispersive X-ray (EDX), and Inductively-coupled plasma optical emission spectroscopy (ICP-OES) were used to characterize the chemical and physical properties of prepared Au–Sn bimetallic and Au or Sn monometallic nanocatalysts. The catalytic performance of the prepared nanocatalysts was evaluated in the selective oxidation of benzyl alcohol (BzOH) to benzaldehyde (BzH) using O2 as an oxidizing agent under moderate conditions. To obtain the optimal BzH yield, the experimental conditions and parameters, including the effects of the reaction time, temperature, pressure, and solvent type on BzOH oxidation, were optimized. Under optimal reaction conditions, bimetallic Au–Sn nanoparticles supported on GO (AuSn/GO-TS, 49.3%) produced a greater yield of BzH than the AuSn/rGO-TS catalysts (35.5%). The Au–Sn bimetallic catalysts were more active than the monometallic catalysts. AuSn/GO-TS and AuSn/rGO-TS prepared by the two-step immobilization method were more active than AuSn/GO-CoIM and AuSn/rGO-CoIM prepared by co-immobilization. In addition, the AuSn/GO-TS and AuSn/rGO-TS catalysts were easily separated from the mixture by centrifugation and reused at least four times without reducing the yield of BzH. These properties make Au–Sn bimetallic nanoparticles supported on GO and rGO particularly attractive for the environmentally friendly synthesis of benzaldehyde.
Introduction
Benzaldehyde (BzH) is an essential industrial starting material or chemical intermediate for producing numerous fundamental fine organic compounds in industries such as pharmaceuticals, agrichemicals, cosmetics, perfumery, and dyestuffs.1–3 Usually, BzH is synthesized by direct toluene oxidation or hydrolysis of benzyl chloride.4,5 The traditional processes for BzH production require costly and environmentally hazardous reagents, such as chromate, potassium permanganate, and hypochlorite, which generate large quantities of toxic acidic waste and consume enormous amounts of energy to maintain the high reaction temperature necessary for the reactions.6,7 The selective oxidation of benzyl alcohol (BzOH) to BzH using O2 as the oxidant is considered an ideal green process route because it eliminates the production of harmful residues and produces only water as a by-product.8–11 Numerous attempts have been made to develop BzOH oxidation systems that employ O2 as an oxidizing agent.2,12,13 Due to their high catalytic activity and selectivity, supported noble metal nanoparticles (NPs) such as Au, Pt, Pd, and Ru have attracted considerable interest among the developed catalysts.14–18 However, their high cost and limited reserves have prevented their widespread industrial application. Therefore, developing inexpensive and effective catalysts for BzOH oxidation is essential. Due to the synergistic effect between the two metals, bimetallic catalysts have greater catalytic activity and selectivity than monometallic catalysts in the selective oxidation of BzOH.19,20 Wu et al.19 reported that Au–Pd NPs supported on foamlike mesoporous silica exhibited a remarkable synergistic effect for the selective oxidation of BzOH. They found that the catalytic activity of bimetallic Au–Pd catalysts (TOF = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–60000 h−1) for BzOH oxidation was much higher than that of monometallic Pd catalyst (TOF = 12500 h−1).19 Yi et al.21 reported that adding Ni to Au NPs catalyst could significantly enhance the O2 activation on Au NPs, which is essential for the oxidation of BzOH using O2 as the oxidant. The Au–Ni bimetallic NPs anchored on the ordered array of extra-large mesopores (EP-FDU-12 support) displayed superior catalytic activity (TOF = 59000 h−1, 240 °C) and space-time yield (STY) of BzH productivity (9.23 kg gAu−1 h−1) in the oxidation of BzOH, which is higher than that of the monometallic Au NPs catalyst (TOF = 15000 h−1 and STY = 1.76 kg gAu−1 h−1).21
000–60000 h−1) for BzOH oxidation was much higher than that of monometallic Pd catalyst (TOF = 12500 h−1).19 Yi et al.21 reported that adding Ni to Au NPs catalyst could significantly enhance the O2 activation on Au NPs, which is essential for the oxidation of BzOH using O2 as the oxidant. The Au–Ni bimetallic NPs anchored on the ordered array of extra-large mesopores (EP-FDU-12 support) displayed superior catalytic activity (TOF = 59000 h−1, 240 °C) and space-time yield (STY) of BzH productivity (9.23 kg gAu−1 h−1) in the oxidation of BzOH, which is higher than that of the monometallic Au NPs catalyst (TOF = 15000 h−1 and STY = 1.76 kg gAu−1 h−1).21
Graphenes are fascinating two-dimensional (2D) nanomaterials that have attracted the interest of scientists and engineers.22–24 Due to their high surface-to-volume aspect ratios, unique electronic properties, excellent physicochemical properties, and high chemical and thermal stability, graphene has been increasingly utilized as a carrier to stabilize and support nanoparticles in recent years.25–28 Moreover, graphene contains carbon vacancy defects and different functional groups, and metal ions can be easily adsorbed onto the graphene surface, forming thermally stable composite materials.29 Guo et al.26 assembled nitrogen-regulated graphene-supported iron nanoparticles using a microwave assembly technique. The 15%Fe/AG(12 h)–W(10) catalyst displayed excellent catalytic performance for Fischer–Tropsch synthesis, with CO conversion and C5+ selectivity reaching 97.2% and 40.0%, respectively, while maintaining mild CO2 selectivity (28.2%).26 Han et al.28 reported that bimetallic Pd–Ag/rGO catalysts synthesized via one-step biosynthesis displayed superior catalytic performance in reducing 4-nitrophenol. The kinetic rate constant of the as-prepared Pd–Ag/rGO for 4-nitrophenol reduction was 0.2413 min−1, which is higher than that reported catalysts for 4-nitrophenol reduction.28 Gao et al.30 reported that a trace amount of SnO2 formed around graphene-Pd and graphene-Pt was beneficial to the enhancement of activity for alcohol oxidation. Wen et al.31 found that the presence of SnO2 may be beneficial to the oxidation of intermediate COads on Pd, leading to the enhancement of electrochemical activity. Sn–W/RGO showed higher catalytic activity than single WO3/graphene catalyst for selective oxidation of alcohols using H2O2 as oxidant without organic solvents and any additives.32 Sn species can induce the formation of hexagonal WO3 with dominant exposed (001) and (200) planes, dramatically increase the specific surface area, change the structural properties and enhance the interactions between Sn–W oxides and RGO.32 Li et al.33 found that Sn–W/RGO presented excellent catalytic properties for the reaction of BzOH to BzH. The BzOH conversion significantly increased, and the BzH selectivity reached 87.38%.33 Ramirez-Barria et al.34 reported that Ru(CO)/NrGO (NrGO:N-doped reduced graphene oxide) displayed excellent BzOH conversions (50%) and BzH selectivity (>99%). The catalyst Ti(SO4)2/GOF (Ti(SO4)2: titanium sulfate; GOF: graphene oxide foam) showed unique catalytic activity (BzOH conversion 91.3%) and selectivity (99.0%) for the oxidation of BzOH to BzH using THF as solvent and H2O2 as oxidant.35 And Ti(SO4)2/GOF could be reused ten times without significant loss of BzOH conversion and selectivity.35
In this study, Au–Sn bimetallic NPs and Au or Sn NPs supported on graphene oxide (GO) and reduced graphene oxide (rGO) were synthesized by two-step immobilization, co-immobilization, and immobilization in the present study. The oxidation of BzOH was used to compare the catalytic properties of the Au–Sn bimetallic- and Au/Sn monometallic catalysts. The solvent type, reaction time, pressure, and temperature of the as-prepared catalysts were optimized for the selective oxidation of BzOH. The effect of the synthetic method, NP size, and support properties was also investigated. In addition, the reusability of the catalysts and their potential mechanisms were investigated in detail. Au–Sn bimetallic catalysts exhibited greater catalytic activity than Au or Sn monometallic catalysts, and AuSn/GO-TS and AuSn/rGO-TS prepared by the two-step immobilization method exhibited greater activity than AuSn/GO-CoIM and AuSn/rGO-CoIM prepared via co-immobilization.
Experimental section
Materials and methods
All the chemicals purchased are of reagent grade and are used without further purification. X-ray diffraction (XRD) measurements were carried out on a Brüker D8 Advance diffractometer (Karlsruhe, Germany) with Cu Kα radiation (λ = 0.154 nm at 40 kV and 40 mA). The microstructures of the catalysts were characterized by transmission electron microscopy (TEM, JEOL-2100F, JEOL Co., Japan) equipped with a high-angle annular dark-field detector (STEM-HAADF). Energy dispersive X-ray spectroscopy (EDS) was performed using an Oxford X-MaxN 80T IE250 instrument (Oxford, UK). The valence states of Au and Sn were analyzed using X-ray photoelectron spectra (XPS, AXIS ULTRADLD, Shimadzu, Japan) equipped with an Al Kα excitation source. Elemental (Au and Sn) analysis was carried out using ICP-OES on an Optima 5300 DV spectrometer (PerkinElmer, USA).Preparation of catalysts
Procedure for catalytic oxidation
The selective oxidation of BzOH to BzH by Au–Sn bimetallic- and Au or Sn monometallic catalysts was studied in a 10 mL high-pressure reactor at 1–8 bar using O2 as the oxidant. In a typical reaction, the catalyst (15.0 mg), BzOH (1.0 mmol, 108.0 mg), and THF (7 mL) were added to the reactor. Then, the mixture was stirred magnetically at different temperatures (80 °C, 90 °C, 100 °C, and 110 °C) for 1–4 h. After the reaction, the reactor was cooled to room temperature, and the reaction mixture was centrifuged for separation. The liquid was analyzed by gas chromatography equipped with an SE-54 capillary column (30 m × 0.25 mm × 0.25 mm) and a flame ionization detector (FID). BzOH and BzH were identified by comparison with known standard samples, and qualitative analysis of the BzOH conversion and BzH selectivity employed a standard external. In the reusability experiment, the catalyst was dried at 100 °C for 5 h under a vacuum after each run.Results and discussion
Characterization of catalysts
GO, and rGO-supported Au–Sn bimetallic and Au/Sn NP catalysts were synthesized using three distinct techniques: co-immobilization, two-step immobilization, and immobilization. The catalysts were fully characterized using XRD, TEM, EDS, XPS, and ICP-OES. The structures of the support and as-prepared catalysts were confirmed using XRD, Fig. 1 shows the XRD patterns of GO, AuSn/GO-CoIM, AuSn/GO-TS, Au/GO, Sn/GO, rGO, AuSn/rGO-CoIM, AuSn/rGO-TS, Au/rGO, and Sn/rGO. The peaks at 11.8°, 21.2°, and 32.4° of GO correspond to the graphitic (002) plane of the graphene structure.36,37 rGO presents a characteristic peak at 21.2°, which is assigned to the graphitic (002) plane of the graphene structure.37 The XRD patterns of AuSn/GO-CoIM, AuSn/GO-TS, Au/GO, Sn/GO, AuSn/rGO-CoIM, AuSn/rGO-TS, Au/rGO, and Sn/rGO changed significantly after loading with Au–Sn, Au, and Sn NPs. The intensity of the peaks at 2θ = 11.8° and 21.2° decreased significantly in the XRD pattern. Five additional peaks at 38.1°, 44.3°, 64.5°, 77.5°, and 81.7° were observed for the Au/GO and Au/rGO catalysts which were attributed to the diffraction peaks of the face-centered cubic (fcc) planes of Au(111), Au(200), Au(220), Au(222), and Au(311) (JCPDS 7440-57-5), respectively.38,39 However, AuSn/GO-CoIM, AuSn/GO-TS, AuSn/rGO-CoIM, and AuSn/rGO-TS catalysts displayed peaks in XRD at 38.2°, 44.5°, 64.7°, 77.6°, and 81.8° corresponding to the fcc planes of Au(111), Au(200), Au(220), Au(222), and Au(311), respectively. The Au(111), Au(200), Au(220), Au(222), and Au(311) peaks of AuSn/GO-CoIM, AuSn/GO-TS, AuSn/rGO-CoIM, and AuSn/rGO-TS shifted slightly toward higher angle theta values, indicating that Sn alloyed with Au.40 The peak intensities at 2θ = 38.2°, 44.5°, 64.7°, 77.6°, and 81.8° for AuSn/GO-CoIM and AuSn/rGO-CoIM were significantly higher than those for AuSn/GO-TS and AuSn/rGO-TS. The co-immobilization method produced larger Au–Sn NPs than the two-step method. Due to the small NP size, no obvious diffractions of Sn crystals on Sn/GO and Sn/rGO were observed.10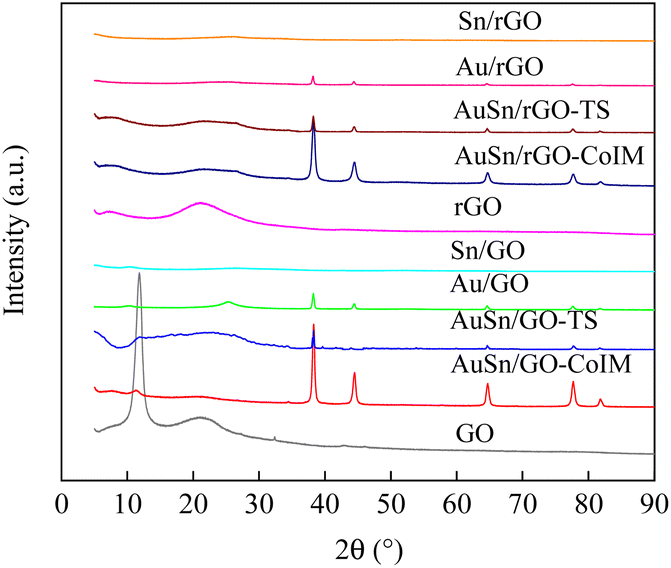 | ||
| Fig. 1 The XRD patterns of GO, AuSn/GO-CoIM, AuSn/GO-TS, Au/GO, Sn/GO, rGO, AuSn/rGO-CoIM, AuSn/rGO-TS, Au/rGO, and Sn/rGO. | ||
XPS measurements revealed the valence states of Au and Sn in the Au–Sn bimetallic and Au or Sn monometallic catalysts (Fig. 2 and S1†). For the AuSn/GO-TS catalyst, two obvious peaks were found in the Au 4f spectra at the binding energy of 83.9 and 87.6 eV, corresponding to the metallic Au0 4f5/2 and Au0 4f7/2, respectively (Fig. 2a).8,41 The Sn 3d XPS spectra of the AuSn/GO-TS catalyst can be briefly fitted with two groups of peaks corresponding to the oxidized Sn2+ and Sn4+ species. The binding energy is located at the position of 496.0, 487.5, 495.3 and 486.9 eV for Sn2+3d5/2, Sn2+3d3/2, Sn4+3d5/2, and Sn4+3d3/2, respectively (Fig. 2b).42 The AuSn/rGO-TS catalyst showed similar Au 4f and Sn 3d XPS spectra. For the AuSn/rGO-TS catalyst, two obvious peaks were obtained in the Au 4f spectra at a binding energy of 83.5 and 87.2 eV, respectively (Fig. 2c). These binding energy values are the characteristic peaks of metallic Au0 4f5/2 and Au0 4f7/2, respectively.8,41 Four Sn 3d signals of AuSn/rGO-TS were found at 496.0, 487.5, 495.3 and 486.9 eV, corresponding to Sn2+3d5/2, Sn2+3d3/2, Sn4+3d5/2, and Sn4+3d3/2, respectively (Fig. 2d).42 For the Au/GO catalyst, two peaks at 84.7 and 88.4 eV were attributed to Au0 4f5/2 and Au0 4f7/2, respectively (Fig. S1a†).8,41 The peaks at 86.8 and 90.5 eV were attributed to Au3+4f5/2 and Au3+4f7/2 thereby suggesting the presence of metallic Au0 and oxidized Au3+ in the Au/GO catalyst, and the ratio of Au0/Au3+ is 2.3 (Fig. S1a†).43 The binding energy peaks located at 84.4 and 88.0 eV in the Au 4f spectra of Au/rGO were attributed to Au0 4f5/2 and Au0 4f7/2, respectively (Fig. S1c†).8,41 The Sn species in Sn/GO and Sn/rGO exist as oxidized Sn2+ and Sn4+, and their binding energies are located at the positions of 496.2, 487.8, 495.5, 487.1 eV, and 496.5, 488.1, 495.8, 487.4 eV for Sn/GO and Sn/rGO, respectively (Fig. S1b and d†).42 The ratio of Sn2+ to Sn4+ can be roughly estimated from the total XPS peak area in each group; the values of Sn2+/Sn4+ are 0.91, 1.09, 0.96, and 1.05 for the AuSn/GO-TS, AuSn/rGO-TS, Sn/GO, and Sn/rGO catalysts, respectively. The Au 4f and Sn 3d binding energies of the bimetallic AuSn/GO-TS and AuSn/rGO-TS catalysts exhibited a negative shift toward lower binding energies than those of the monometallic catalysts, indicating a strong interaction between Au and Sn in the Au–Sn bimetallic catalysts.39
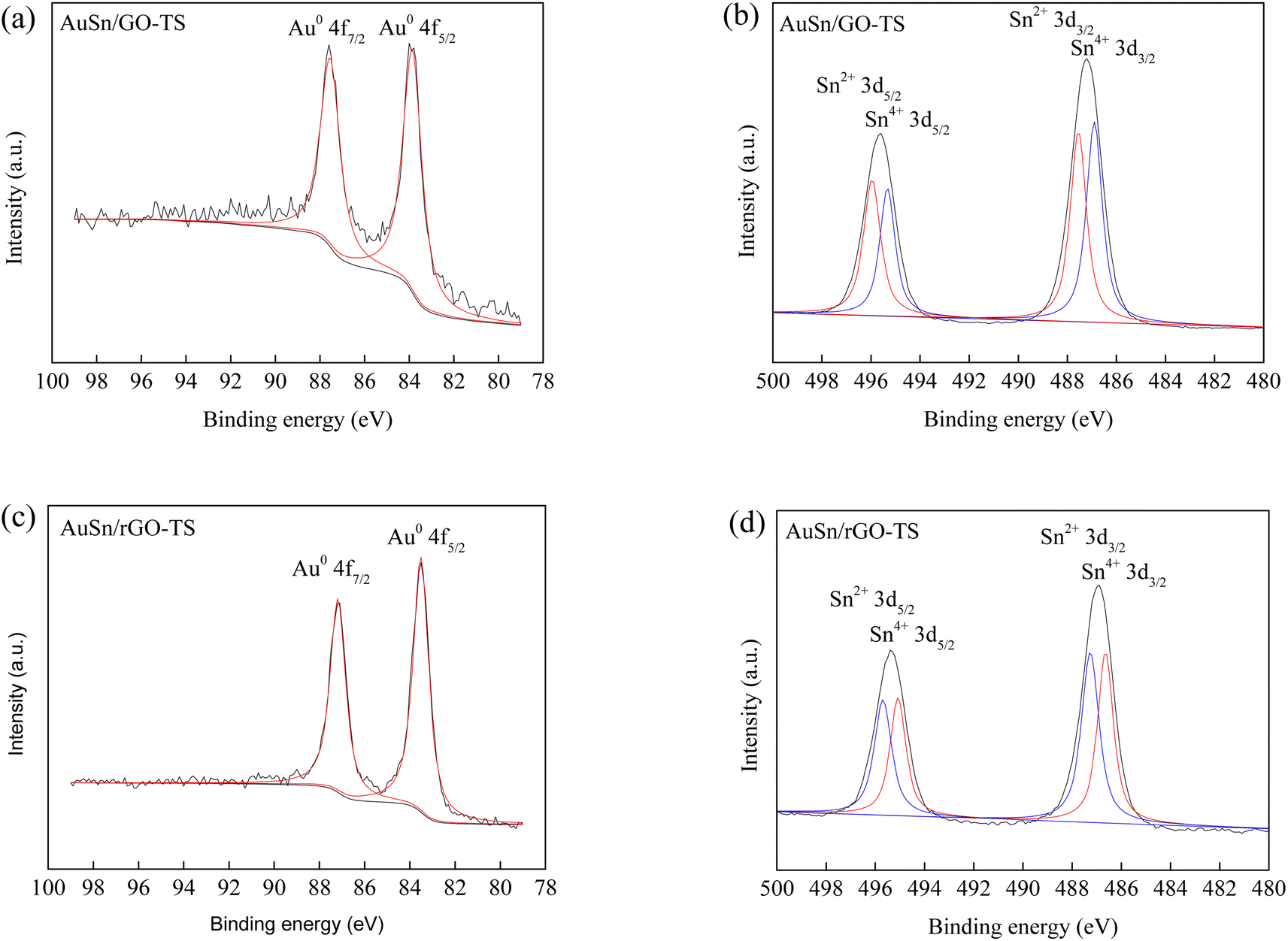 | ||
| Fig. 2 Au 4f and Sn 3d XPS of AuSn/GO-TS (a and b) and AuSn/rGO-TS (c and d). | ||
Fig. 3 and 4, and S2–S7† depict the TEM, HAADF-STEM, and EDS images of the Au–Sn bimetallic and Au or Sn monometallic catalysts, respectively. Au–Sn, Au, and Sn NPs were uniformly distributed on the GO or rGO support, as evidenced by TEM and HAADF-STEM images. The Au–Sn bimetallic particle size distribution histograms for the AuSn/GO-TS, AuSn/GO-CoIM, AuSn/rGO-TS, and AuSn/rGO-CoIM catalysts were obtained through statistical analysis of over 300 Au–Sn particles. The size of the Au–Sn nanoparticles in Au–Sn bimetallic catalysts varies depending on the synthesis method. AuSn/GO-TS and AuSn/rGO-TS catalysts exhibited average Au–Sn NP sizes of 15.2 and 5.2 nm, while AuSn/GO-CoIM and AuSn/rGO-CoIM catalysts exhibited average Au–Sn NP sizes of 73.1 and 43.9 nm, respectively (Fig. 3, 4 and S2, S3†). AuSn/GO-TS and AuSn/rGO-TS synthesized using a two-step immobilization method exhibited smaller Au–Sn particle sizes than AuSn/GO-CoIM and AuSn/rGO-CoIM synthesized using a co-immobilization method. EDS elemental mapping of AuSn/GO-TS, AuSn/GO-CoIM, AuSn/rGO-TS, and AuSn/rGO-CoIM revealed that Au and Sn elements were highly intermixed, indicating the formation of an Au–Sn alloy.8,44 TEM, HAADF-STEM, and EDS images demonstrated that Au or Sn nanoparticles were uniformly distributed throughout the GO or rGO in the Au/GO, Sn/GO, Au/rGO, and Sn/rGO catalyst (Fig. S4–S7†). The average sizes of the Au NPs for the Au/GO and Au/rGO catalysts were 6.5 nm and 10.6 nm, respectively (Fig. S4 and S6†).
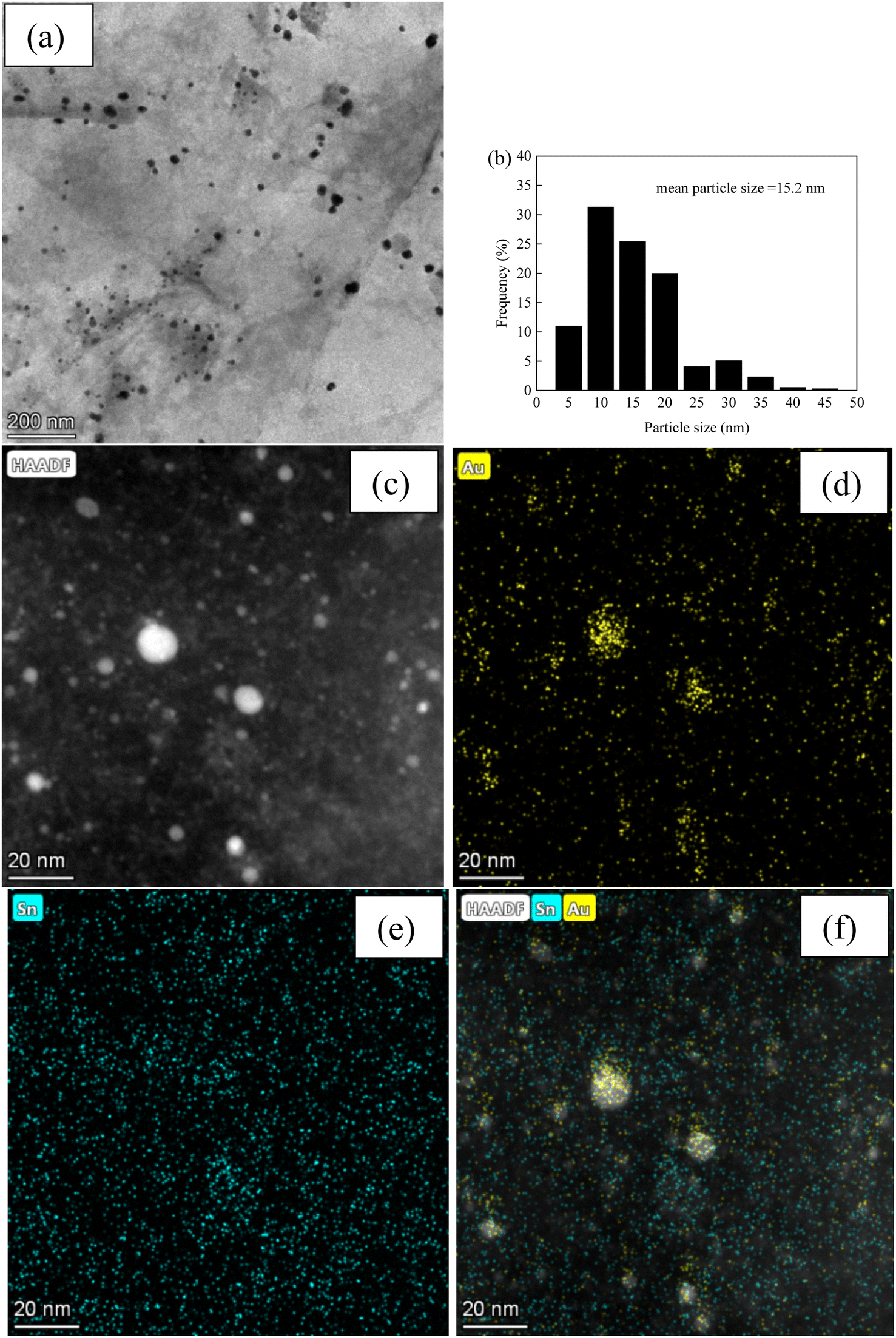 | ||
| Fig. 3 (a) TEM micrograph of AuSn/GO-TS; (b) Au–Sn size distribution of AuSn/GO-TS; (c) HAADF-STEM micrograph; (d–f) EDS elemental mapping images micrographs of AuSn/GO-TS. | ||
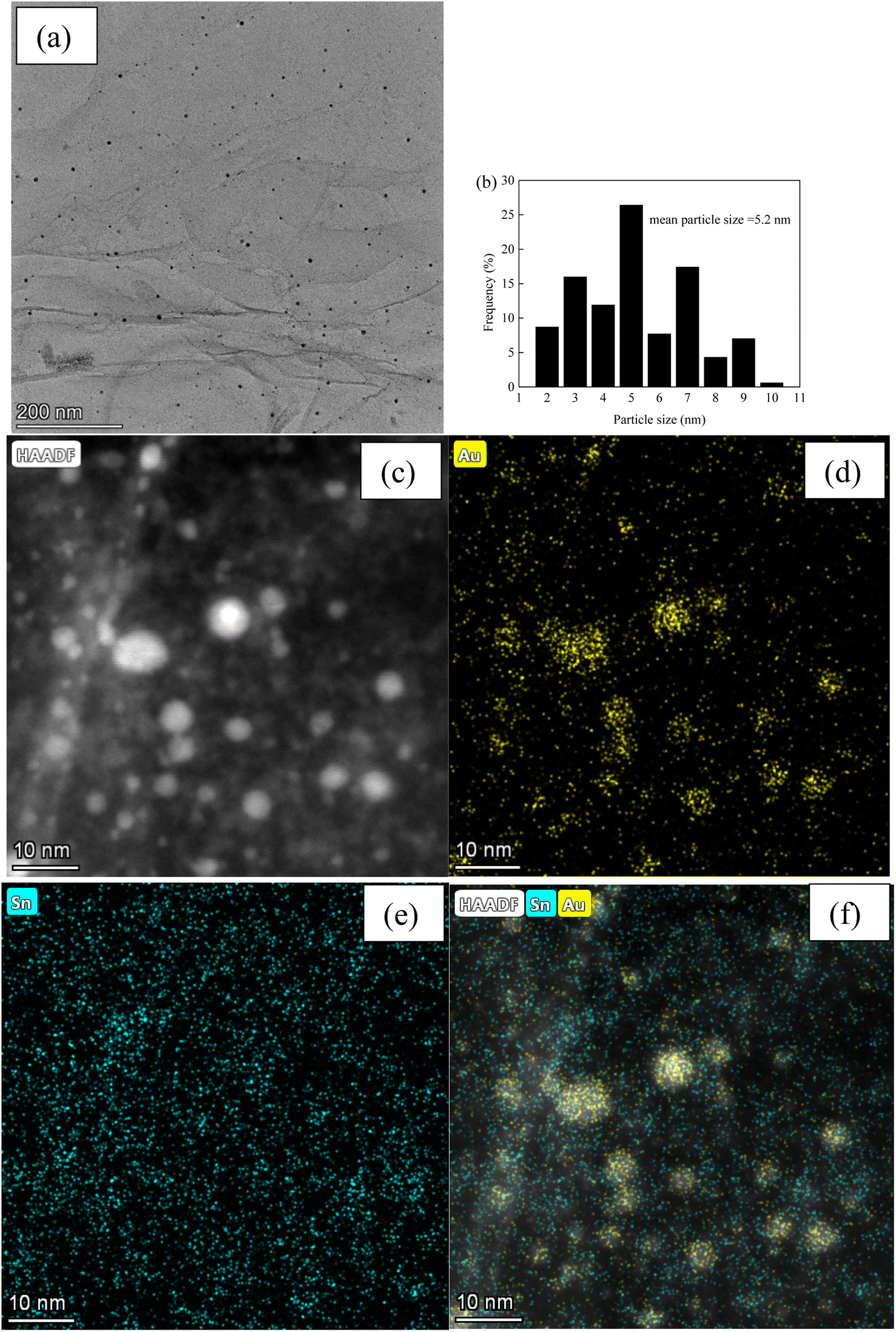 | ||
| Fig. 4 (a) TEM micrograph of AuSn/rGO-TS; (b) Au–Sn size distribution of AuSn/rGO-TS; (c) HAADF-STEM micrograph; (d–f) EDS elemental mapping images micrographs of AuSn/rGO-TS. | ||
The selective oxidation of benzyl alcohol
The catalytic performances of the AuSn/GO-TS, AuSn/GO-CoIM, Au/GO, Sn/GO, AuSn/rGO-TS, AuSn/rGO-CoIM, Au/rGO, and Sn/rGO catalysts were evaluated by the oxidation of BzOH with O2 as a green oxidant, which is a typical aerobic oxidation.45,46 The outcomes are presented in Tables 1, S1 and S2.† Benzoic acid was the primary byproduct of the BzOH oxidation reaction. To prevent the formation of benzoic acid, a series of reactions were conducted to determine the optimal reaction conditions for the synthesis of BzH. The type of solvent, temperature, pressure, and reaction time have a significant impact on the BzOH conversion and BzH selectivity for all catalysts. The GO and rGO samples only converted BzOH by 3% and 1%, respectively. BzOH oxidation was performed in different solvents, such as tetrahydrofuran (THF), acetonitrile, N,N′-dimethylformamide (DMF), and 1,4-dioxane, under the same reaction temperature, pressure, and time (Table 1, entries 1–4, 11–14; Table S1,† entries 1–4, 10–13; Table S2,† entries 1–4, 16–19). The AuSn/GO-TS catalysts exhibited poor catalytic activity in acetonitrile and 1,4-dioxane, with BzOH conversions of 37.6% and 12.6%, respectively (Table 1, entries 2,4). Although AuSn/GO-TS exhibited excellent catalytic activity in DMF, its benzaldehyde selectivity was only 3.0% (Table 1, entry 3). The BzOH conversion, BzH selectivity, and BzH yield were 87.8%, 56.1%, and 49.3% in THF at 90 °C under 3 bar O2 within 2 h, respectively (Table 1, entry 1). AuSn/rGO-TS, AuSn/GO-CoIM, AuSn/rGO-CoIM, Au/GO and Au/rGO all produced identical outcomes. For all catalysts, the best catalytic performance was observed in THF. Temperature, pressure, and time also played crucial roles in BzOH conversion and BzH selectivity. All catalysts exhibited a remarkable increase in the conversion of BzOH as reaction temperature, pressure, and time increased, while the selectivity of BzH decreased significantly due to the oxidation of BzH to BzCOOH (Table 1, entries 5–10, 15–22; Table S1,† entries 1–4, 10–13; Table S2,† entries 1–4, 16–19). The best yields (49.3%, 39.2%, 38.7%) were obtained at 90 °C under 3 bar O2 within 2 h using THF as the solvent for AuSn/GO-TS, AuSn/GO-CoIM, and Au/GO. The highest yields to BzH were observed as 35.5%, 33.5%, and 38.4% for AuSn/rGO-TS, AuSn/rGO-CoIM, and Au/rGO at 100 °C under 3 bar O2 in THF after 1.5 h, 2 h, and 2 h, respectively (Table 1, entry 22; Table S1,† entry 10; Table S2,† entry 16). Hence, the optical reaction temperature, pressure and time were 90 °C, 3 bar of O2, and 2 h for AuSn/GO-TS, AuSn/GO-CoIM, and Au/GO catalysts. The optical reaction conditions for AuSn/rGO-TS, AuSn/rGO-CoIM, and Au/rGO were at 90 °C under 3 bar O2 and using THF as solvent within 1.5 h, 2 h and 2 h, respectively. The Sn/GO and Sn/rGO catalysts presented a very low BzOH conversion (<20%) under the studied reaction conditions (Table S2,† entries 11–15, 26).| Entry | Catalyst | Solvent | T (°C) | Pressure (bar) | Time (h) | Conv. (%) | S (%) | Yield (%) | TOF (h−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: BzOH (1.0 mmoL), solvent (7.0 mL), catalyst (15.0 mg). | |||||||||
| 1 | AuSn/GO-TS | THF | 90 | 3 | 2 | 87.8 | 56.1 | 49.3 | 64.8 |
| 2 | AuSn/GO-TS | Acetonitrile | 90 | 3 | 2 | 37.6 | 14.2 | 5.3 | 27.7 |
| 3 | AuSn/GO-TS | DMF | 90 | 3 | 2 | 100 | 3.0 | 3.0 | 73.8 |
| 4 | AuSn/GO-TS | 1,4-Dioxane | 90 | 3 | 2 | 12.6 | 50.0 | 6.3 | 9.3 |
| 5 | AuSn/GO-TS | THF | 80 | 3 | 2 | 46.9 | 49.8 | 23.4 | 34.6 |
| 6 | AuSn/GO-TS | THF | 100 | 3 | 2 | 96.7 | 12.7 | 12.3 | 71.3 |
| 7 | AuSn/GO-TS | THF | 90 | 1 | 2 | 66.9 | 44.8 | 30.0 | 49.4 |
| 8 | AuSn/GO-TS | THF | 90 | 5 | 2 | 95.7 | 42.7 | 40.9 | 70.6 |
| 9 | AuSn/GO-TS | THF | 90 | 3 | 1 | 28.5 | 85.5 | 24.4 | 42.1 |
| 10 | AuSn/GO-TS | THF | 90 | 3 | 3 | 92.8 | 43.1 | 40.0 | 46.2 |
| 11 | AuSn/rGO-TS | THF | 100 | 3 | 2 | 92.3 | 31.6 | 29.2 | 69.7 |
| 12 | AuSn/rGO-TS | Acetonitrile | 100 | 3 | 2 | 14.8 | 1.3 | 0.2 | 11.2 |
| 13 | AuSn/rGO-TS | DMF | 100 | 3 | 2 | 58.6 | 0 | 0 | 44.2 |
| 14 | AuSn/rGO-TS | 1,4-Dioxane | 100 | 3 | 2 | 20.4 | 22.4 | 4.6 | 15.4 |
| 15 | AuSn/rGO-TS | THF | 80 | 3 | 2 | 25.0 | 100 | 25 | 18.9 |
| 16 | AuSn/rGO-TS | THF | 90 | 3 | 2 | 74.6 | 30.4 | 22.7 | 56.3 |
| 17 | AuSn/rGO-TS | THF | 110 | 3 | 2 | 98.2 | 4.8 | 4.7 | 74.1 |
| 18 | AuSn/rGO-TS | THF | 100 | 1 | 2 | 19.4 | 98.8 | 19.2 | 14.6 |
| 19 | AuSn/rGO-TS | THF | 100 | 5 | 2 | 97.6 | 18.3 | 17.9 | 73.7 |
| 20 | AuSn/rGO-TS | THF | 100 | 3 | 0.5 | 34.3 | 99.5 | 34.1 | 103.6 |
| 21 | AuSn/rGO-TS | THF | 100 | 3 | 1 | 57.0 | 50.3 | 28.7 | 87.0 |
| 22 | AuSn/rGO-TS | THF | 100 | 3 | 1.5 | 88.6 | 40.1 | 35.5 | 89.2 |
The effect of NP size is the most important factor in enhancing the catalytic activity of supported nanocatalysts.47,48 AuSn/GO-TS, AuSn/GO-CoIM, AuSn/rGO-TS, and AuSn/rGO-CoIM catalysts with average Au–Sn NP size of 15.2, 73.1, 5.2, and 43.9 nm, respectively, were prepared using a two-step immobilization and co-immobilization technique (Fig. 1, 2 and S2, S3†). The turnover frequency (TOF) was 64.8, 48.4, 89.2, and 62.5 h−1 based on the total Au content under the optical reaction conditions for AuSn/GO-TS, AuSn/GO-CoIM, AuSn/rGO-TS, and AuSn/rGO-CoIM, respectively (Table 1, entries 1, 22; Table S1,† entries 7, 10). AuSn/GO-TS catalysts with an NP size of 15.2 nm exhibited greater catalytic activity than AuSn/GO-CoIM catalysts (73.1 nm). AuSn/rGO-TS catalysts with an average Au–Sn NP size of 5.2 nm exhibited a greater level of activity than AuSn/rGO-CoIM catalysts (43.9 nm). AuSn/GO-TS and AuSn/rGO-TS catalysts exhibited significantly enhanced activities compared to AuSn/GO-CoIM and AuSn/rGO-CoIM catalysts, which was primarily attributable to the smaller size of the Au–Sn bimetallic NPs produced by the two-step immobilization method. The results of the catalytic oxidation of BzOH are consistent with those published in previous studies.49,50 Liu et al.49 discovered that the BzOH oxidation catalytic performance of gold catalysts with dimensions greater than 5 nm exhibited a size-dependent property. Olmos et al.50 demonstrated that the metal particle size affects the oxidation of BzOH. Wilde et al.51 demonstrated that the activity improved of Rh/AC-based materials by pretreatment with HNO3 while still preserving good selectivity towards BzH, which could be ascribed to the Rh particle size reduced and particle size distribution narrower after pretreatment with HNO3, with the smaller the particle size, the higher the activity.
The support is essential for BzOH oxidation catalysis.45,52,53 TEM results revealed that the size of the Au–Sn bimetallic nanoparticles is also dependent on the support material. AuSn/GO-TS and AuSn/rGO-TS had average Au–Sn nanoparticle size of 15.2 and 5.2 nm, respectively (Fig. 3 and 4). However, the TOF value of AuSn/GO-TS (64.8 h−1) was higher than that of AuSn/rGO-TS (56.3 h−1) at 90 °C under 3 bar of O2 within 2 h. The basal planes of GO contain O doping in the form of functional groups such as hydroxyl and epoxy groups, and the edges of the sheet also contain carboxyl and carbonyl groups.54 The enhancement of the catalytic activity of AuSn/GO-TS may be attributable to the impact of the support properties.
Au–Sn bimetallic catalysts supported on GO and rGO (AuSn/GO-TS and AuSn/rGO-TS) exhibited significantly higher activity than Au or Sn monometallic catalysts (Au/GO, Sn/GO, Au/rGO and Sn/rGO). The TOF values were 64.8, 89.2, 21.5, and 25.3 h−1 based on the total metal content for AuSn/rGO-TS, AuSn/rGO-CoIM, Au/GO, and Au/rGO under the optimal reaction conditions. Sn/GO and Sn/rGO showed very low activity for the BzOH oxidation reaction (TOF < 6.1 h−1). The activity of the AuSn/GO-TS catalyst (64.8 h−1) is three times than that of Au/GO (21.5 h−1) at 90 °C. The TOF value of AuSn/rGO-TS (89.2 h−1) is 3.5 times than that of Au/rGO (25.3 h−1). The activity of AuSn/GO-TS, as well as AuSn/rGO-TS, is even one order of magnitude bigger than that of Sn/GO and Sn/rGO. These remarkable differences demonstrated that Au and Sn had a synergistic effect on the AuSn/GO-TS and AuSn/rGO-TS catalysts. Khawaji et al.55 confirmed the synergic effect of Au–Pd alloys in the oxidation of BzOH. The BzOH oxidation activities of the Au–Pd/Ce-NRSI-R, Au/Ce-NRSI-R, and Pd/Ce-NRSI-R catalysts were tested at 120 °C under 2 bar of O2 within 3 h, and the TOF values were 34747, 1664, and 12300 h−1, respectively. Alshammari et al.51 reported that the BzOH conversion and BzH selectivity of Au–Pd/MgO and Au–Pd/MnO2 were 21.4% and 96.3%, and 24.7% and 96.4%, respectively, at 120 °C under 1 bar of O2 within 2 h for BzOH oxidation reaction. The calculated TOF values were 1501 h−1and 1733 h−1 over Au–Pd/MgO and Au–Pd/MnO2, respectively. Pd@Ni/MWCNT (Pd: 0.2 mmol) catalyst displayed superior BzOH conversion (99%) and BzH selectivity (98%) for the selective oxidation of BzOH with water as a solvent and H2O2 as oxidant at 80 °C.56 The TOF value based on the total Pd content was 5 h−1 for Pd@Ni/MWCNT. The conversion of BzOH given by Au2Pd1@NMOF-Ni was 99% at 120 °C after 9 h of reaction under reflux, and its calculated TOF value reached 30.7 h−1.57 Fu et al.58 found that 2 wt% Ni–Co3/FDU-15 N possessed high BzOH conversion (93.4%) and excellent benzaldehyde selectivity (97.8%) when using air as the oxidant at 110 °C and DMF as the solvent after 7 h with a TOF of 19.5 h−1. PMo@PVM4 (PMo: phosphomolybdate; PVM4: ionic polymers) gave excellent BzOH conversion (99%), BzH selectivity (94%), and yield (93%) for the solvent-free BzOH oxidation reaction at 160°Cunder 1 atm O2 within 16 h.59 The TOF value of PMo@PVM4 was 122.2 h−1 based on the total phosphomolybdate. Wang et al.60 reported that Pd@PDC(0.1) (PDC:ionic copolymers tethered with –COOH group; 0.1: the initial mole ratio of ionic liquid to divinylbenzene) displayed good catalytic activity for the BzOH oxidation at 90°Cfor 5 h using K2CO3 as additive with O2 balloon in water, with a TOF of 151.6 h−1. Pd0.1@PCPMo (PCPMo: porous ionic polymers with the anions partly replaced by [PMo12O40]3−) displayed excellent catalytic performance for the BzOH oxidation at 160 °C under 1 atm O2, the TOF value was 8568 h−1 based on the total Pd contents.61 Based on the above results, the AuSn/GO-TS and AuSn/rGO-TS catalysts displayed better catalytic activity than the Pd@Ni/MWCNT, Au2Pd1@NMOF-Ni, and Ni–Co3/FDU-15-N catalysts. Although the TOF values were smaller than those of Au–Pd/Ce-NRSI-R, Au–Pd/MgO, Au–Pd/MnO2, PMo@PVM4, Pd@PDC(0.1) and Pd0.1@PCPMo, AuSn/GO-TS and AuSn/rGO-TS could oxidize BzOH with superior conversion under mild reaction conditions.
The reusability of catalysts is of great importance in practical applications and green chemistry. The stability of the AuSn/GO-TS and AuSn/rGO-TS catalysts were studied at 90 °C (AuSn/GO-TS) or 100 °C (AuSn/rGO-TS) under 3 bar of O2 within 2 h (Fig. 5). After each reaction, the catalysts were collected by centrifugation, dried at 100 °C under vacuum, and reused for subsequent runs. In the second, third, and fourth reaction runs the BzOH conversions decreased. In contrast, the selectivities of BzH increased during the second, third, and fourth cycles of reaction. In a series of 4 consecutive runs, AuSn/GO-TS and AuSn/rGO-TS catalysts demonstrated stable BzH yields over four consecutive runs (AuSn/GO-TS: 46.8–49.3%; AuSn/rGO-TS: 29.2–30.9%). Fig. 6 showed the XPS spectra of reused AuSn/GO-TS and AuSn/rGO-TS. The Au in the reused AuSn/GO-TS and AuSn/rGO-TS catalysts existed as metallic Au (Au0), indicating that there were no change for the valence states of Au for the reused AuSn/GO-TS and AuSn/rGO-TS during the process of BzOH oxidation reaction.41 However, the ratios of Sn2+ to Sn4+ were increased compared to fresh AuSn/GO-TS (0.91) and AuSn/rGO-TS (1.09). The values of Sn2+/Sn4+ are 1.06 and 1.18 for the reused AuSn/GO-TS and AuSn/rGO-TS, respectively.42 The decrease in conversion and increase in selectivity of the reused AuSn/GO-TS and AuSn/rGO-TS catalysts after each run may be caused by the partial conversion of Sn4+ to Sn2+.
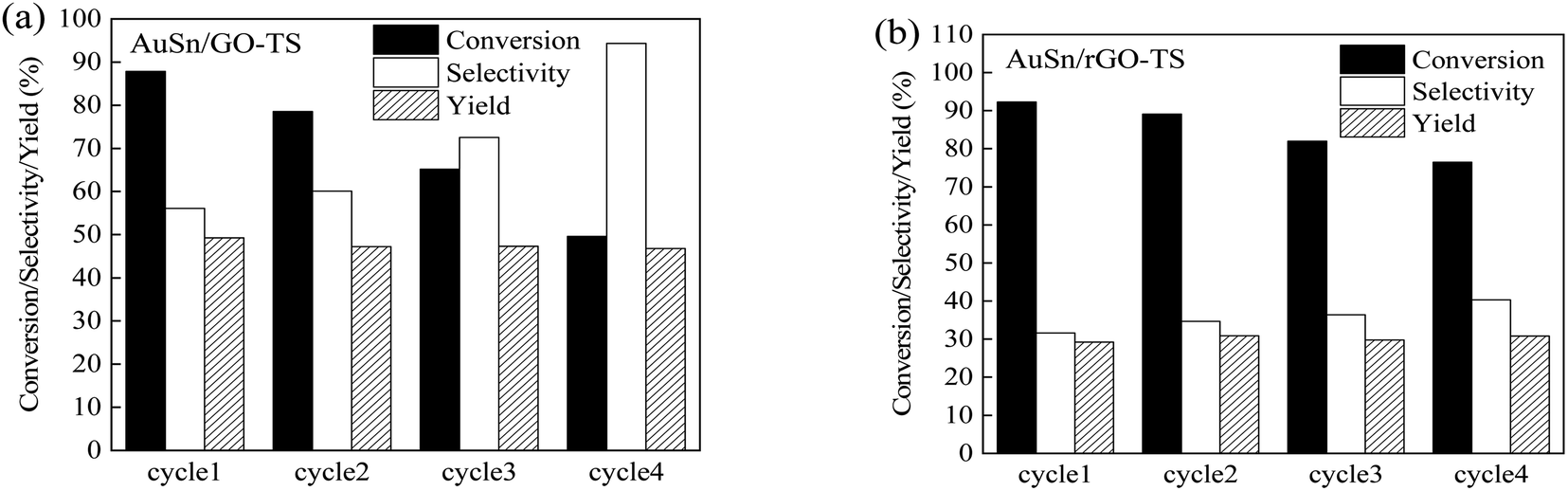 | ||
| Fig. 5 The reusability of AuSn/GO-TS and AuSn/rGO-TS for the oxidation of BzOH (reaction condition: BzOH (1.0 mmoL), THF (7.0 mL), catalyst (15.0 mg), 90 °C (AuSn/GO-TS), 100 °C (AuSn/rGO-TS), 3 bar, 2 h). | ||
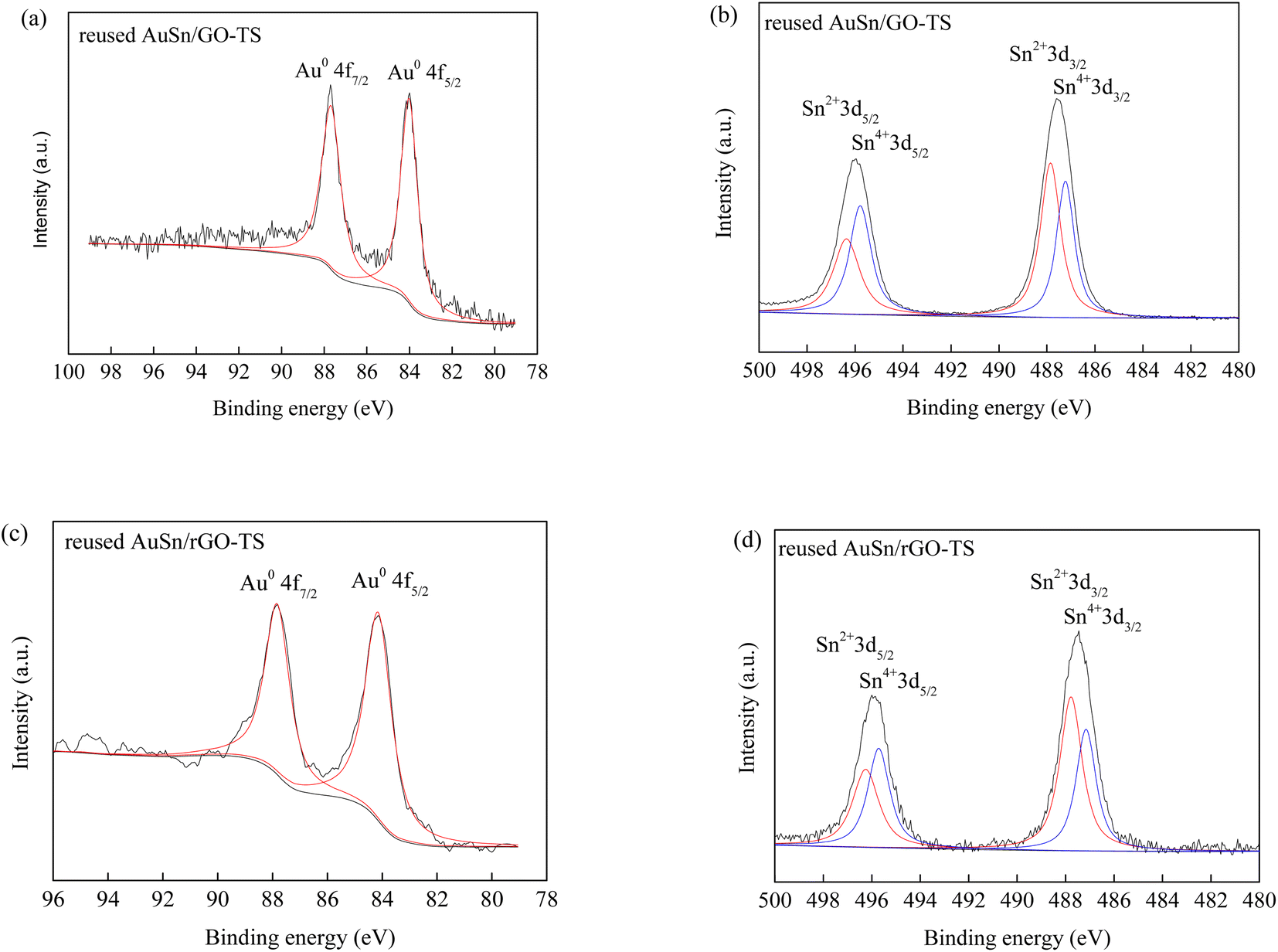 | ||
| Fig. 6 Au 4f and Sn 3d XPS of reused AuSn/GO-TS (a and b) and AuSn/rGO-TS (c and d). | ||
The leaching of AuSn/GO-TS and AuSn/rGO-TS were tested to examine if there were any Au or Sn active species in the reaction solution that could catalyze the BzOH oxidation reaction. The hot filtration tests were carried out by stopping the BzOH oxidation after 1 h under 3 bar of O2 at 90 °C or 100 °C using THF as solvent. The BzOH conversions and BzH selectivities were 28.5% and 85.5%, and 57% and 50.3% for the AuSn/GO-TS and AuSn/rGO-TS, respectively. Then AuSn/GO-TS and AuSn/rGO-TS catalysts were removed from the reaction solution by centrifugation. The reaction solution was transferred to another 10 mL high-pressure reactor and continued to react for 1 h under the same reaction conditions. The BzOH conversions increased by 5.3% and 5.9% for AuSn/GO-TS and AuSn/rGO-TS, respectively. The BzH selectivities decreased by 5.6% and 5.4% over AuSn/GO-TS and AuSn/rGO-TS, respectively. The Au and Sn contents of AuSn/GO-TS and AuSn/rGO-TS after reaction were 8.2wt% and 9.2wt%, 8.3wt% and 9.3wt%, respectively, as determined by ICP-OES. There were some leaching of Au (7.9% and 4.6%) and Sn (6.1% and 4.1%) over AuSn/GO-TS and AuSn/rGO-TS under the reaction conditions. The decline of BzOH conversions in the second, third, and fourth reaction runs was also possibly due to the leaching of Au and Sn under experimental conditions.
Scheme 1 presents a possible catalytic mechanism based on AuSn/GO-TS and AuSn/rGO-TS based on the related reports.8,62,63 We believe that BzOH oxidation over Au–Sn bimetallic catalysts was initiated by oxidative dehydrogenation of BzOH taking place on Au–Sn bimetallic sites. The role of O2 in the oxidation reaction was to remove H from the Au–Sn NP surface and regenerate the catalysts. First, BzOH can be chemisorbed on the Au–Sn bimetallic sites, and then hydrogen is extracted from the –CH2– of BzOH to generate the H–AuSn–alcoholate intermediate. The intermediate underwent β-hydride elimination, releasing the final product, BzH, and forming AuSn–H species. Subsequently, the AuSn–H intermediate reacts with 1/2O2 to produce H2O and regenerates the Au–Sn bimetallic catalysts. The regenerated catalysts can continue to participate in BzOH oxidation and complete the next catalytic cycle.
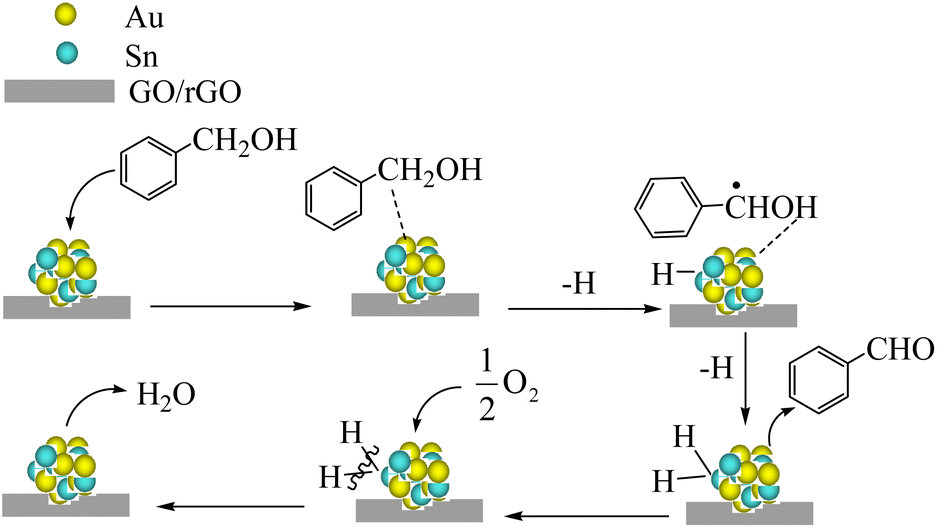 | ||
| Scheme 1 Probable mechanism of BzOH oxidation by AuSn/GO-TS and AuSn/rGO-TS. | ||
Conclusions
Using two-step immobilization, co-immobilization, and immobilization method, a series of bimetallic and monometallic Au and Sn loaded on GO and rGO were prepared. The results of the characterization showed that the average Au–Sn NPs of AuSn/GO-TS and AuSn/rGO-TS prepared by two-step immobilization were smaller than the average Au–Sn NPs of the catalysts prepared by co-immobilization. Upon evaluating the oxidation of BzOH to BzH over the prepared catalysts in the presence of molecular oxygen, the Au–Sn bimetallic catalysts exhibited greater activity than the monometallic catalysts, and the AuSn/GO-TS and AuSn/rGO-TS catalysts performed better than the AuSn/GO-CoIM and AuSn/rGO-CoIM catalysts. Under optimal conditions, the BzOH conversions and BzH yields of AuSn/GO-TS and AuSn/rGO-TS were 87.8% and 49.3%, and 88.6% and 35.5%, respectively. The heterogeneous AuSn/GO-TS and AuSn/rGO-TS catalysts were also stable during the BzOH oxidation reaction. In a series of four consecutive runs, the AuSn/GO-TS and AuSn/rGO-TS catalysts exhibited stable BzH yields. For the oxidation of BzOH to BzH, Au–Sn bimetallic catalysts supported on GO and rGO are efficient and environmentally friendly.Author contributions
Conceptualization, L. L. and X. T.; methodology, L. L. and X. Z.; software, X. Z.; validation, L. L. and C. X.; formal analysis, L. L. and C. X.; investigation, B. Z., G. Z. and S. L.; resources, L. L. and C. X.; data curation, X. Z.; writing—original draft preparation, L. L.; writing—review and editing, L. L. and X. T.; visualization, L. L.; supervision, L. L. and X. T.; project administration, L. L.; funding acquisition, L. L. and X. T. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Natural Science Foundation of China (grant number 21802104) and the Natural Science Foundation of Shandong Province (grant number ZR2017MB056).References
- M. E. Assal, M. R. Shaik, M. Kuniyil, M. Khan, A. Al-Warthan, M. R. H. Siddiqui, S. M. A. Khan, W. Tremel, M. N. Tahir and S. F. Adil, RSC Adv., 2017, 7, 55336–55349 RSC.
- T. R. Chen, Y. X. Wang, W. J. Lee, H. C. Chen and J. D. Chen, Nanotechnology, 2020, 31, 285705–285719 CrossRef CAS PubMed.
- Z. Wang, J. Feng, X. Li, R. Oh and G. Zhang, J. Colloid Interface Sci., 2021, 588, 787–794 CrossRef CAS PubMed.
- H. M. Alshammari, J. R. Humaidi, M. S. Alhumaimess, O. F. Aldosari, M. H. Alotaibi, H. M. A. Hassan and I. Wawata, React. Kinet. Mech. Catal., 2019, 128, 97–108 CrossRef CAS.
- T. Tian, Y. Liu and X. Zhang, Chin. J. Catal., 2015, 36, 1358–1364 CrossRef CAS.
- M. E. Assal, M. R. Shaik, M. Kuniyil, M. Khan, A. Al-Warthan, A. I. Alharthi, R. Varala, M. R. H. Siddiqui and S. F. Adil, Arab. J. Chem., 2019, 12, 54–68 CrossRef CAS.
- M. Kuniyil, J. V. S. Kumar, S. F. Adil, M. E. Assal, M. R. Shaik, M. Khan, A. Al-Warthan, M. R. H. Siddiqui, A. Khan, M. Bilal, H. M. N. Iqbal and W. A. Al-Masry, Catalysts, 2020, 10, 1136–1156 CrossRef CAS.
- L. Liu, X. Tai, X. Zhou, J. Hou and Z. Zhang, J. Alloys Compd., 2019, 790, 326–336 CrossRef CAS.
- L. Liu, X. Zhou, Y. Yan, J. Zhou, W. Zhang and X. Tai, Polymers, 2018, 10, 1089–1104 CrossRef PubMed.
- L. Liu, X. Zhou, L. Liu, S. Jiang, Y. Li, L. Guo, S. Yan and X. Tai, Catalysts, 2019, 9, 538–555 CrossRef.
- R. A. Sheldon and I. W. C. E. Arends, Catal. Today, 2000, 57, 157–166 CrossRef CAS.
- K. Avela, L. Gerard and V. S. Eric, Catal. Lett., 2021, 1–9 Search PubMed.
- O. Alduhaish, S. F. Adil, M. E. Assal, M. R. Shaik, M. Kuniyil, K. M. Manqari, D. Sekou, M. Khan, A. Khan, A. Z. Dewidar, A. Al-Warthan and M. R. H. Siddiqui, Processes, 2020, 8, 910–934 CrossRef CAS.
- K. Jitlada, C. Chanon, K. Pongtanawat, U. Boontida, E. Masahiro and B. Karan, Catalysts, 2021, 11, 720–733 CrossRef.
- S. Hasegawa, S. Takano, K. Harano and T. Tsukuda, JACS Au, 2021, 1, 660–668 CrossRef CAS PubMed.
- W. Zhuang, X. Liu, L. Chen, P. Liu, H. Wen, Y. Zhou and J. Wang, Green Chem., 2020, 12, 4199–4209 RSC.
- H. Song, Z. Liu, Y. Wang, N. Zhang, X. Qu, K. Guo, M. Xiao and H. Gai, Green Energy Environ., 2019, 4, 278–286 CrossRef.
- B. Zadam, D. Obaid, A. Mayoufi, P. Beaunier, F. Launay and F. Z. EI Berrichi, Res. Chem. Intermed., 2019, 45, 1281–1293 CrossRef CAS.
- P. Wu, Y. Cao, L. Zhao, Y. Wang, Z. He, W. Xing, P. Bai, S. Mintova and Z. Yan, J. Catal., 2019, 375, 32–43 CrossRef CAS.
- P. Verma, M. E. Potter, A. E. Oakley, P. M. Mhembere and R. Raja, Nanomaterials, 2021, 11, 350–362 CrossRef CAS PubMed.
- W. Yi, W. Yuan, Y. Meng, S. Zou, Y. Zhou, W. Hong, J. Che, M. Hao, B. Ye, L. Xiao, Y. Wang, H. Kobayashi and J. Fan, ACS Appl. Mater. Interfaces, 2017, 9, 31853–31860 CrossRef CAS PubMed.
- H. Ci and J. Sun, Chin. Sci. Bull., 2019, 64, 3327–3339 Search PubMed.
- J. R. Prekodravac, D. P. Kepića, J. C. Colmenares, D. A. Giannakoudakis and S. P. Jovanović, J. Mater. Chem. C, 2021, 9, 6722–6748 RSC.
- L. Michel, S. Sall, T. Dintzer, C. Robert, A. Demange and V. Caps, Faraday Discuss, 2021, 227, 259–273 RSC.
- Y. B. Chang, C. Zhang, X. L. Lu, W. Zhang and T. B. Lu, Nano Res., 2022, 15(1), 195–201 CrossRef CAS.
- L. Guo, Z. Guo, J. Liang, X. Yong, S. Sun, W. Zhang, J. Sun, T. Zhao, J. Li, Y. Cui, B. Zhang, G. Yang and N. Tsubaki, Chem. Eng. J., 2022, 429, 132063–132071 CrossRef CAS.
- T. Wu, Y. Ma, Z. Qu, J. Fan, Q. Li, P. Shi, Q. Xu and Y. Min, ACS Appl. Mater. Interfaces, 2019, 11, 5136–5145 CrossRef CAS PubMed.
- R. Han, X. Song, Q. Wang, Y. Qi, G. Deng, A. Zhang, Q. Wang, F. Chang, C. Wu and Y. Cheng, J. Chem. Technol. Biotechnol., 2019, 94, 3375–3383 CrossRef CAS.
- A. A. Ali, M. Madkour, F. A. Sagheer, M. I. Zaki and A. A. Zazeer, Catalysts, 2020, 10, 105–119 CrossRef CAS.
- L. Gao, W. Yue, S. Tao and L. Fan, Langmuir, 2013, 29, 957–964 CrossRef CAS PubMed.
- Z. L. Wen, S. D. Yang, Y. Y. Liang, W. He, H. Tong, L. Hao, X. G. Zhang and Q. J. Song, Electrochim. Acta, 2010, 56, 139–144 CrossRef CAS.
- C. Xu, L. Zhang, Y. An, X. Wang, G. Xu, Y. Chen and L. Dai, Appl. Catal., A, 2018, 558, 26–33 CrossRef CAS.
- R. Li, W. Cheng, Y. Yan, Y. Shi, L. Wei, S. Yu and S. Li, SN Appl. Sci., 2021, 3, 483–495 CrossRef CAS.
- C. S. Ramirez-Barria, M. Isaacs, C. Parlett, K. Wilson, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Catal. Today, 2020, 357, 8–14 CrossRef CAS.
- W. Ma, Q. Tong, J. Wang, H. Yang, M. Zhang, H. Jiang, Q. Wang, Y. Liu and M. Cheng, RSC Adv., 2017, 7, 6720–6723 RSC.
- B. A. Yusuf, M. Xie, W. Yaseen, J. Xie and Y. Xu, Int. J. Energy Res., 2021, 1–11 Search PubMed.
- Y. Li, J. Yan, D. Yu, P. Lei, W. Shen, M. Zhong, J. Zhang and S. Guo, Inorg. Chem., 2021, 60, 17635–17640 CrossRef CAS PubMed.
- C. Liu, K. Yang, J. Zhao, Y. Pan and D. Liu, Catal. Commun., 2015, 67, 72–77 CrossRef CAS.
- L. Liu, X. Tai, X. Zhou, L. Liu, X. Zhang, L. Ding and Y. Zhang, J. Taiwan Inst. Chem. Eng., 2020, 114, 220–227 CrossRef CAS.
- P. Mukhi and S. Roy, ChemistrySelect, 2020, 5, 1000–1006 CrossRef CAS.
- L. Liu, X. Zhou, L. Guo, S. Yan, Y. Li, S. Jiang and X. Tai, RSC Adv., 2020, 10, 33417–33427 RSC.
- Z. Xu, R. Xu, Y. Yue, P. Yuan, X. Bao, E. Abou-Hamad, J. M. Basset and H. Zhu, J. Catal., 2019, 374, 391–400 CrossRef CAS.
- L. Liu, X. Tai and X. Zhou, Materials, 2017, 10, 99–111 CrossRef PubMed.
- L. Liu, Z. Han, Y. Lv, C. Xin, X. Zhou, L. Yu and X. Tai, ChemistryOpen, 2022, e202100288 CAS.
- S. Ito, X. Wang, A. Waheed, G. Li, N. Maeda, D. M. Meier, S. Naito and A. Baiker, J. Catal., 2021, 393, 42–50 CrossRef CAS.
- S. S. Moeini, U. P. Laverdura, E. Marconi, N. Lisi, E. Serra, R. Chierchia, I. Luisetto, S. Tuti and D. Tofani, Catalysts, 2022, 12, 20–38 CrossRef CAS.
- J. Lyu, L. Niu, F. Shen, J. Wei, Y. Xiang, Z. Yu, G. Zhang, C. Ding, Y. Huang and X. Li, ACS Omega, 2020, 5, 16865–16874 CrossRef CAS PubMed.
- Y. Song, Y. Lin, X. Chu, J. Tang and S. Xu, J. Mater. Res. Technol., 2020, 9, 2237–2246 CrossRef CAS.
- L. Liu, H. Li, Y. Tan, X. Chen, R. Lin, W. Yang, C. Huang, S. Wang, X. Wang, X. Y. Liu, M. Zhao and Y. Ding, Catalysts, 2020, 10, 107–126 CrossRef CAS.
- C. M. Olmos, L. E. Chinchilla, A. Villa, J. J. Delgado, A. B. Hungría, G. Blanco, L. Prati, J. J. Calvino and X. Chen, J. Catal., 2019, 375, 44–55 CrossRef CAS.
- H. Alshammari, M. Alhumaimess, M. H. Alotaibi and A. S. Alshammari, J. King Saud Univ. Sci., 2017, 29, 561–566 CrossRef.
- G. Nagy, A. Beck, G. Sáfrán, Z. Schay, S. Liu, T. Li, B. Qiao, J. Wang and K. Lázár, React. Kinet. Mech. Catal., 2019, 128, 71–95 CrossRef CAS.
- T. Li, F. Liu, Y. Tang, L. Li, S. Miao, Y. Su, J. Zhang, J. Huang, H. Sun, M. Haruta, A. Wang, B. Qiao, J. Li and T. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7795–7799 CrossRef CAS PubMed.
- N. Syakir, T. Saragi, F. Fitrilawati, Y. Maryati, U. Widyaswari, D. P. Sari and R. Risdiana, Mater. Sci. Forum, 2021, 1028, 296–301 Search PubMed.
- M. Khawaji and D. Chadwick, Catal. Today, 2019, 348, 203–211 CrossRef.
- M. Zhang, Q. Sun, Z. Yan, J. Jing, W. Wei, D. Jiang, J. Xie and M. Chen, Aust. J. Chem., 2013, 66, 564–571 CrossRef CAS.
- T. Guo, S. Bao, G. Jie, C. Wu and L. Wen, Chem. Res. Chin. Univ., 2022, 38, 1344–1348 CrossRef CAS.
- X. Fu, S. Wu, Z. Li, X. Yang, X. Wang, L. Peng, J. Hu, Q. Huo, J. Guan and Q. Kan, RSC Adv., 2016, 6, 57507–57513 RSC.
- Z. Chen, M. Zou, G. Li, X. Liu, Y. Zhou and J. Wang, Mol. Catal., 2022, 532, 112735–112746 CrossRef CAS.
- Q. Wang, X. Cai, Y. Liu, J. Xie, Y. Zhou and J. Wang, Appl. Catal. B Environ., 2016, 189, 242–251 CrossRef CAS.
- S. Li, Z. Chen, X. Ling, J. Cao, Q. Wang, Y. Zhou and J. Wang, Appl. Surf. Sci., 2019, 496, 143650–143660 CrossRef.
- L. Chen, J. Yan, Z. Tong, S. Yu, J. Tang and B. Ou, Microporous Mesoporous Mater., 2018, 266, 126–131 CrossRef CAS.
- W. Cui, Q. Xiao, S. Sarina, W. Ao, M. Xio, H. Zhu and Z. Bao, Catal. Today, 2014, 235, 152–159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ESI figures and tables of the characterizations and catalytic data of the catalysts. See DOI: https://doi.org/10.1039/d3ra03496h |
| This journal is © The Royal Society of Chemistry 2023 |