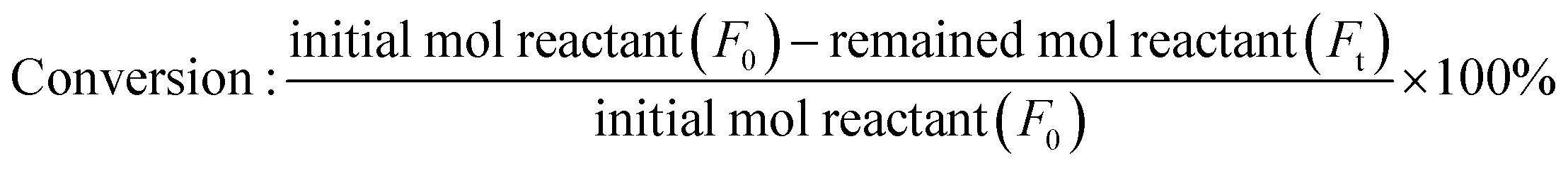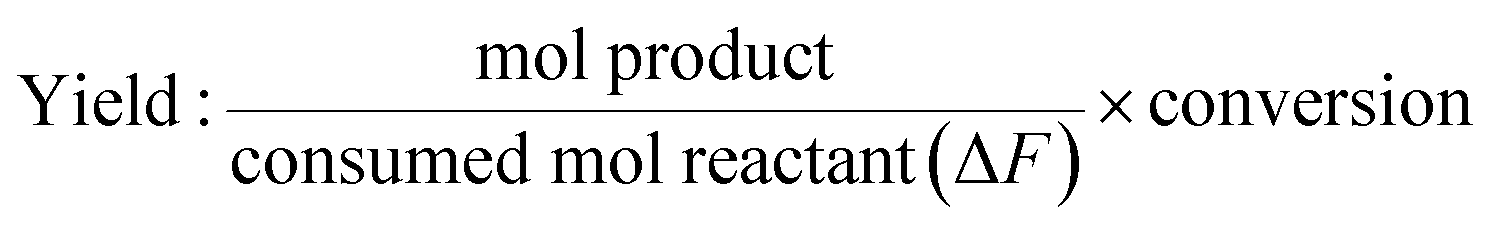DOI:
10.1039/D3RA03642A
(Paper)
RSC Adv., 2023,
13, 21171-21181
Rational design for the fabrication of bulk Ni3Sn2 alloy catalysts for the synthesis of 1,4-pentanediol from biomass-derived furfural without acidic co-catalysts†
Received
31st May 2023
, Accepted 28th June 2023
First published on 13th July 2023
Abstract
This study describes the rational design for the fabrication of bulk Ni3Sn2 alloy catalysts for the de/hydration–hydrogenation of biomass-derived furfural (FFald) to 1,4-pentanediol (1,4-PeD) without the acidic co-catalyst. The presence of both hydration active sites (Brønsted acid sites (Ni–SnOx)) and hydrogenation active sites (Ni0 or Ni–Sn alloy) in Ni3Sn2 alloy could be controlled by changing the pH of Ni–Sn solution during the preparation. Both active sites acted synergistically to catalyse the de/hydration–hydrogenation reactions of FFald to produce a high yield of 1,4-PeD in a batch reaction system at 433 K, 3.0 MPa H2 after 12 h. Bulk Ni3Sn2 obtained at pH of Ni–Sn solution of 8–10, hydrothermal temperature of 423 K for 24 h, and reduction with H2 at 673 K for 1.5 h demonstrated a high yield of 1,4-PeD (81–87%), which is comparable with that from previous work. A 76% yield of 1,4-PeD was also obtained when the reaction was carried out in a fixed-bed reaction system at 433 K, flow rate 0.065 mL min−1, H2 flow rate 70 mL min−1, and 3.29 wt% FFald in H2O/ethanol solution for 12 h. The activity of bulk Ni3Sn2 was maintained with 66% yield of 1,4-PeD even after 52 h reaction on stream. The fabricated bulk Ni3Sn2 alloy catalysts could be the promising heterogeneous Ni–Sn alloy-based catalysts for the catalytic conversion of biomass-derived-furanic compounds (e.g., FFald, furfuryl alcohol (FFalc), and 2-methylfuran (2-MeF)).
Introduction
The utilization of renewable biomass and its derived chemical platforms are involved in more important roles to reduce the dependence on non-renewable fossil fuel-based resources through the production of building block monomers of C5 α,ω-diols, including1,2-pentanediol (1,2-PeD), 1,4-pentanediol (1,4-PeD), and 1,5-pentanediol (1,5-PeD).1,2 The C5 α,ω-diols from renewable feedstocks have attracted much attention as they are promising building blocks for direct replacement of 1,4-butanediol (1,4-BeD) or 1,6-hexanediol (1,6-HeD) from fossil fuel-based in the production of polyesters and thermoplastic polyurethanes.3 Furfural (FFald), a tremendous C5-furan-based chemical platform, is well-known as a substrate for the synthesis of those monomer C5 α,ω-diols using heterogeneous catalysts.4–7 The established approaches for the transformation of FFald into C5 α,ω-diols were devoted to the assertion of the C–O hydrogenolysis of tetrahydrofurfuryl alcohol (THFalc), which can be obtained from total hydrogenation of both C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds of FFald (Fig. 1(#1)). Bimetallic platinum group metals (PGM) (e.g., Pt, Pd, Rh, Ir, Ru, Ni, etc.) in combination with oxophilic metal oxide (e.g., Re, Mo, Ce, Y or W) catalysts have been extensively studied and provided moderate to high yield 1,2- or 1,5-PeD.6,8–15 Another approach is the combination between supported PGM catalyst and acidic metal oxide (e.g., γ-Al2O3, Nb2O5) through the dual ring-opening tautomerisation (hydration/dehydration) and hydrogenation reactions of derived-THFalc pyran or dihydropyran to 1,5-PeD as proposed by Dumesic's group.16,17
C and CO bonds of FFald (Fig. 1(#1)). Bimetallic platinum group metals (PGM) (e.g., Pt, Pd, Rh, Ir, Ru, Ni, etc.) in combination with oxophilic metal oxide (e.g., Re, Mo, Ce, Y or W) catalysts have been extensively studied and provided moderate to high yield 1,2- or 1,5-PeD.6,8–15 Another approach is the combination between supported PGM catalyst and acidic metal oxide (e.g., γ-Al2O3, Nb2O5) through the dual ring-opening tautomerisation (hydration/dehydration) and hydrogenation reactions of derived-THFalc pyran or dihydropyran to 1,5-PeD as proposed by Dumesic's group.16,17
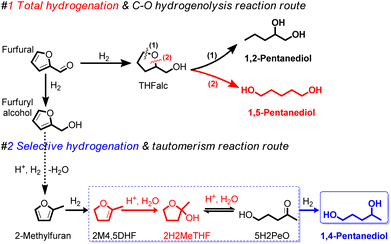 |
| | Fig. 1 Conceivable routes for the synthesis of C5 α,ω-diols of 1,2-PeD, 1,5-PeD, and 1,4-PeD from FFald, FFalc, and 2-MeF platforms using bimetallic Ni–Sn alloy catalysts via dual selective hydrogenation–tautomerism reactions.20,26 PeD = pentanediol. THFalc = tetrahydrofurfuryl alcohol. 2H2MeTHF = 2-hydroxy-2-methyl tetrahydrofuran. 2M4,5DHF = 2-methyl-4,5-dihydrofuran. 5H2PeO = 5-hydroxy-2-pentanone. | |
The catalytic conversion of C5-furan precursors (e.g., FFald, furfuryl alcohol (FFalc)) to 1,4-PeD is less attractive than that of other C5 α,ω-diols due to some shortcomings (e.g., the requirement of a selective and bifunctional-type catalyst, addition of acidic co-catalyst, or assertion of multiple reaction steps).18 Leuck et al. claimed that trace amounts of glacial acetic acid are substantially required to convert FFald to 1,4-PeD (40%) and 1,2,5-(1,4,5)-pentanetriol (∼50%) in the presence of RANEY® Ni catalyst.19 Schniepp et al. reported that a trace amount of formic acid was added to the reaction mixture of 2-methylfuran (2-MeF) in the presence of Ni/Celite catalyst (at H2 8.3 MPa, 423 K for 8 h), affording a mixture of 2-methyltetrahydrofuran (2-MeTHF) (36%) and 1,4-PeD (62%).20 In the presence of ReOx-modified Pd–Ir/SiO2 or Rh–Ir/SiO2 catalyst system, the Brønsted acid sites in the catalysts may facilitate the side reactions such as hydration or dehydration, which are the important reaction steps for catalytic conversion of FFald to 1,4-PeD. However, the selectivity of 1,4-PeD was not so high (23%).21,22 Liu et al. combined Ru/CMK-3 or Ru–FeOx/AC catalysts and Amberlyst-15 as the acidic co-catalyst for the catalytic reaction of FFald and produced a high yield of 1,4-PeD (up to 90%).23,24 Most recently, Cui et al. developed bifunctional ruthenium nanoparticles supported on sulfonated carbon layer coated SBA-15 (Ru/SC-SBA-15) catalysts for conversion of FFald and an 86% selectivity of 1,4-PeD was obtained, which is three times higher than that of unmodified Ru/C-SBA-15 catalyst.25 However, the presence of acidic co-catalysts may lead to the formation of undesired products through over-dehydration or polymerization and severely cause the leaching out of active metal catalysts under severe conditions. Therefore, the development of new heterogenous catalysts with surface acidity (e.g., Brønsted acid sites) without an acidic co-catalyst for the dual hydration–hydrogenation of FFald, FFalc, or 2-methylfuran (2-MeF) into 1,4-PeD is challenging.
The development of heterogeneous bimetallic Ni-based catalysts for the transformation of biomass-derived C5-furan compounds has been a long-standing work in our group. Bulk Ni–Sn(x) alloys (x = 1.5, 2.0, 3.0 and 4.0, Ni/Sn molar ratio) catalysts have shown very good catalytic performance and reusability in the chemoselective hydrogenation of α,β-unsaturated carbonyl compounds to unsaturated alcohol,27 hydrogenation of LA to GVL,28 and the one-pot conversion of FFald to high yield of 1,4-PeD (61–92%) under mild conditions (433 K, 3.0 MPa H2, 12 h) in the ethanol/H2O mixture solvent and without the presence of acidic co-catalyst.26 The best bulk Ni3Sn2 alloy catalyst was obtained at pH 8 of Ni–Sn solution at 423 K for 24 h, and reduction with H2 at 673 K for 1.5 h.
In the present paper, we report an extended work on the fabrication of bulk Ni3Sn2 alloy by revisiting the effect of various synthetic parameters including pH of Ni–Sn solution, additive polyol, hydrothermal time, hydrothermal temperature, and reduction temperature. The bulk Ni3Sn2 alloy catalysts were employed for the conversion of furfural to 1,4-PeD from FFald under identical reaction conditions to those from literature.26,27 We found that bulk Ni3Sn2 alloy catalyst synthesized at pH = 6–10, hydrothermal time of 24–48 h, polyol additives of ethylene glycol (EG), glycerol (G), or 2-methoxy-ethanol (2Me-EtOH), and temperature reduction of 673 K for 1.5 h allowed a high yield of 1,4-PeD (87%), which is comparable with that of previous results over bulk and supported Ni–Sn(x) alloy catalysts (92%).26,29 This catalyst might be served as a bifunctional catalyst, which comprises two active sites of Ni–Sn alloy riched-Ni0 (as Lewis acid) and Ni–SnOx or support (Brønsted acid). The Ni–Sn alloy enriched Ni0 is active and selective for the hydrogenation of CO in FFald to solely FFalc,27 while the Ni–SnOx species or support can serve as Brønsted acid sites for the hydration or hydrogenolysis.30 Both active sites played the key role in the synergistic dual reactions of de/hydration as well as hydrogenation/hydrogenolysis of FFald to 1,4-PeD in acidulated H2O or H2O/ethanol solvent (Fig. 1(#2)).
Results and discussion
Catalytic reaction
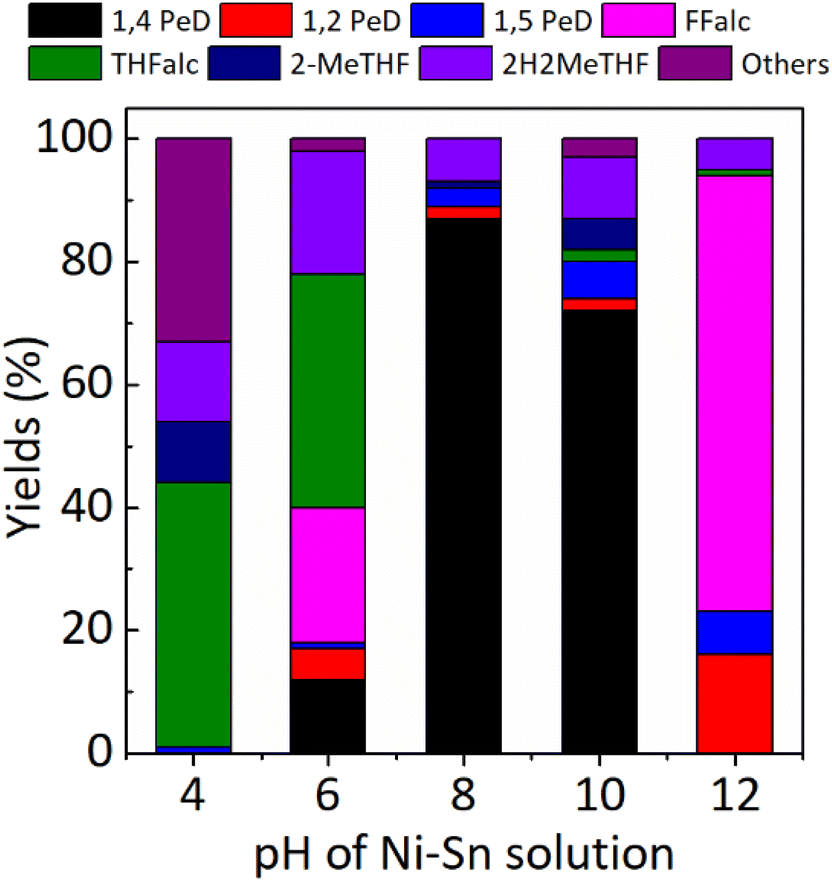
Effect of pH of Ni–Sn solution. In the first set experiment, we evaluated the catalytic performance of various bulk Ni3Sn2 obtained at different pH of Ni–Sn solution for the conversion of biomass-derived FFald to 1,4-PeD at 433 K, 3.0 MPa H2 for 12 h, and the results are shown in Fig. 2.
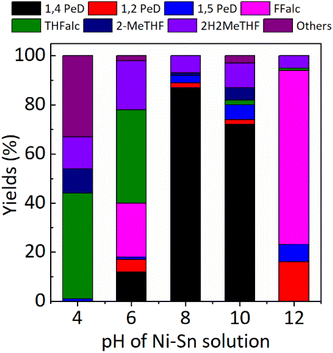 |
| | Fig. 2 Yield of 1,4-PeD obtained over bulk Ni3Sn2 catalysts synthesised at different pH of Ni–Sn solution. Catalysts were reduced with H2 at 673 K for 1.5 h. Reaction conditions: catalyst, 44 mg; substrate, 1.2 mmol; solvent, ethanol/H2O, 3.5 mL (1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.0 volume ratio); 433 K, initial H2 pressure 3.0 MPa, 12 h. :2.0 volume ratio); 433 K, initial H2 pressure 3.0 MPa, 12 h. | |
Using the Ni3Sn2 catalyst obtained at pH = 4, 100% FFald was converted to 1% 1,5-PeD, 43% THFalc, 13% 2H2MeTHF, 10% 2-MeTHF, and 33% others (it may be the condensation product of FFald or FFalc according to GC and GC-MS data) without the formation of 1,4-PeD at full conversion of FFald at 433 K, 3.0 MPa H2 after 12 h. When the pH of Ni–Sn solution was increased to 6 (Ni3Sn2 pH = 6), 12% 1,4-PeD was first observed, however, relatively high yields of FFalc (22%), THFalc (38%), and 2H2MeTHF (20%) were also obtained. Interestingly, a high yield of 1,4-PeD (87%) was obtained over Ni3Sn2 pH = 8 catalyst, whereas the side products were 1,2- and 1,5-PeD (5%), 2H2MeTHF (7%), and 2-MeTHF (1%). This is the highest yield of 1,4-PeD (87%) obtained at present, which is comparable with the previous result using the same Ni–Sn catalysts,26 or with the previous results over Ru-based catalysts in the presence of an acidic co-catalyst.23–25,31 Further increase in pH of Ni–Sn solution to 10 (Ni3Sn2 pH = 10) afforded a quite high yield of 1,4-PeD (72%), however, the side products of 2H2MeTHF, 2-MeTHF, and others remained 10%, 5%, and 3%, respectively. In contrast, the Ni3Sn2 pH = 12 catalyst produced 16% 1,2-PeD, 7% 1,5-PeD, 71% Ffalc, 1% THFalc, and 5% 2H2MeTHF without the formation of 1,4-PeD. A high yield of remaining Ffalc (71%) suggested that the Ni3Sn2 pH = 12 catalyst is selective toward CO hydrogenation of Ffald and inactive to catalyse the dual hydrolysis–hydrogenation reactions to form the final product of 1,4-PeD. To gain more understanding of these results, the catalyst structure–activity relationship upon these catalysts will be discussed later in this paper.
Kinetic studies. To understand more about how the active species of Ni and Ni–Sn and the surface acidity of bulk Ni3Sn2 alloy catalysts synergistically played a prominent role in the hydration–hydrogenation of Ffald to 1,4-PeD, kinetically controlled catalytic reactions were examined for three types of bulk Ni3Sn2 catalysts at different pH (pH = 8, 10, 12) and the results are shown in Fig. 3. At the early reaction time of 0.5–1.0 h, for the Ni3Sn2 pH = 8 catalyst (total acidity = 276 μmol g−1), the FFald conversion was 44–84% to produce only FFalc. At the same time, the conversions of FFald over Ni3Sn2 pH = 10 (total acidity = 129 μmol g−1) and 12 (total acidity = 18 μmol g−1) were 13–55% and 7–12%, respectively. However, at 1.5 h, 100% conversion of FFald was achieved by using Ni3Sn2 pH = 8 catalyst, and at this time, the 1,4-PeD was first formed (3.7%) and then increased to achieve 48.7% as the reaction time was prolonged to 3 h. On the other hand, the amount of FFalc decreased smoothly to reach around 14%, meanwhile, other side products e.g., THFalc, 2-MeTHF, 2H2MeTHF, and 1,2- and 1,5-PeD mixtures, increased slowly. The kinetic profiles and product distributions over the Ni3Sn2 pH = 10 catalyst were identical to those of the Ni3Sn2 pH = 8 catalyst. The similar results are likely due to their similar surface acidities, which not only improve the selectivity towards 1,4-PeD but also the reaction rate for FFalc formation (Fig. 3). In contrast, over the Ni3Sn2 pH = 12 catalyst, the conversion of FFald was 23% after 1.5 h and increased slowly to 76% after 6 h. These results indicate that the Ni3Sn2 pH = 12 catalyst is unsuitable for the hydration–hydrogenation of FFald to 1,4-PeD due to the lower surface acidity. The reaction rate of FFalc formation, as the elementary step of the reaction, over three types of Ni3Sn2 catalysts was roughly calculated at a reaction time of 30 min (0.5 h). Over Ni3Sn2 pH = 8, the reaction rate of FFalc formation was 12.83 10−3 mmol min−1 (43% conversion), while over Ni3Sn2 pH = 10 and pH = 12, the reaction rates were 8.68 10−3 mmol min−1 (13% conversion) and 2.68 10−3 mmol min−1 (7% conversion), respectively.
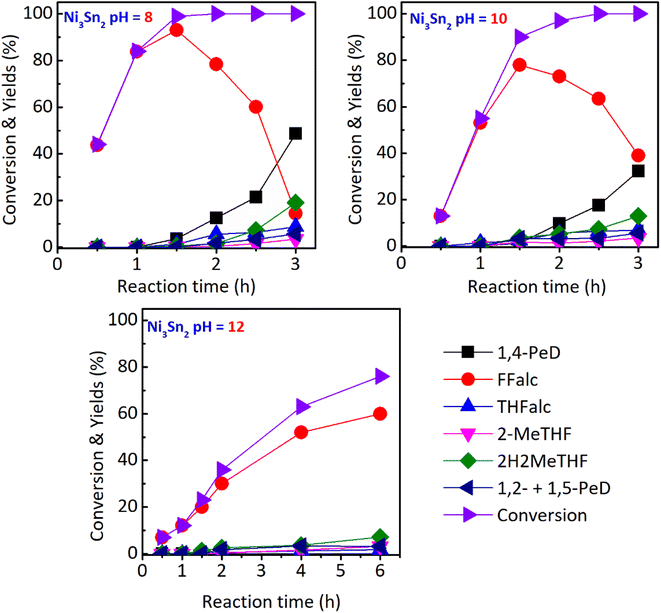 |
| | Fig. 3 Kinetic profiles of three types of bulk Ni3Sn2 catalysts in the hydration–hydrogenation of FFald 1,4-PeD. Reaction conditions: catalyst, 44 mg; substrate, 1.2 mmol; solvent, ethanol/H2O, 3.5 mL (1.5:2.0 volume ratio); 433 K, initial H2 pressure 3.0 MPa, 0–6 h. | |
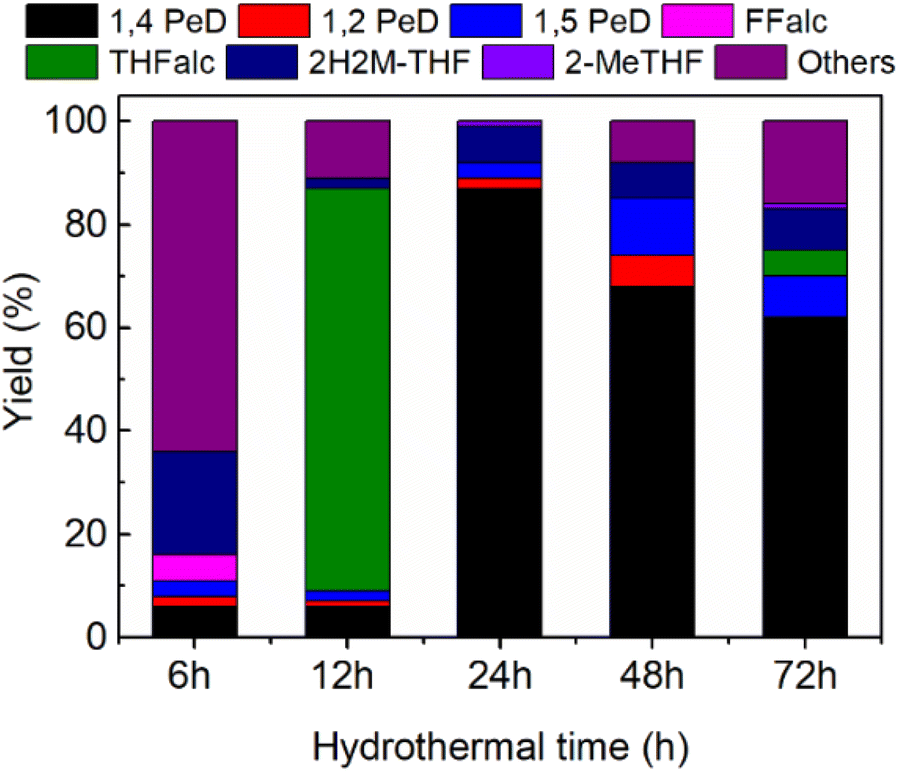
Effect of hydrothermal time. To establish the synthetic procedure of highly selective bulk Ni3Sn2 alloy catalysts towards 1,4-PeD, we prepared bulk Ni3Sn2 alloy at hydrothermal times of 6 h, 12 h, 24 h, 48 h, and 72 h and tested for the synthesis of 1,4-PeD; the results are shown in Fig. 4. Bulk Ni3Sn2 catalyst synthesised at hydrothermal time of 6–12 h afforded only 6% of 1,4-PeD, whereas the main product was others (condensation reaction of FFald (64%)) or THFalc (78%) as the result of total hydrogenation of FFald. The yield of 1,4-PeD remarkably increased to 87% over the bulk Ni3Sn2 catalyst that was obtained at a hydrothermal time of 24 h. When the hydrothermal time was prolonged at 48 h and 72 h, the yield of 1,4-PeD slightly decreased to 68% and 62%, respectively. Over these catalysts, quite a high yield of 1,5-PeD (8–11%) was also observed. Although the XRD patterns of bulk Ni3Sn2 alloy obtained at different hydrothermal times showed no notable differences (Fig. S1 and Table S4, in the ESI†), the reaction results confirmed that the Ni3Sn2 alloy obtained at 24 h of hydrothermal time was the most effective catalyst (Fig. 2–4). Therefore, it can be concluded that the optimised hydrothermal time for the synthesis of bulk Ni3Sn2 catalyst was 24 h, pH adjustment = 8 and 10 or by employing the appropriated support oxides c.a. γ-Al2O3 or commercial-aluminium hydroxide (c-AlOH) containing gibbsite and bayerite structures.32
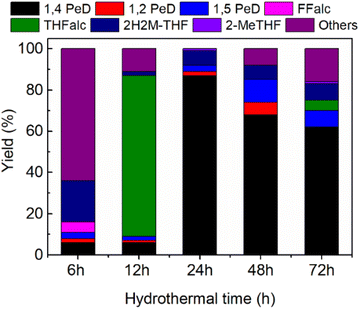 |
| | Fig. 4 Yield of 1,4-PeD obtained over bulk Ni3Sn2 catalysts synthesised at different hydrothermal times. Catalysts were reduced with H2 at 673 K for 1.5 h. Reaction conditions are referred to in Fig. 2. | |
Effect of polyol additives and hydrothermal temperature. To clarify the importance of polyol additives during the synthesis of bulk Ni3Sn2 catalysts, ethylene glycol (EG) and glycerol (G) were employed instead of the 2-methoxy ethanol (Me-EtOH) and without the addition of polyols for comparison and the XRD patterns of the results are shown in Fig. S2 and S3, in the ESI.† The presence of EG, G, or 2-Me-EtOH polyols significantly affected the formation of Ni3Sn2 alloy phases and their crystallinity, whereas upon without polyols resulted in lower crystallinity of Ni3Sn2(102) and Ni3Sn2(110) (Fig. S3 and Table S5, in the ESI†).28,33 The results of 1,4-PeD synthesis from FFald using bulk Ni3Sn2 pH =8 alloy catalysts synthesised at different conditions are summarised in Table 1. Both Ni3Sn2 (EG, pH = 8, 673 K/H2) and (G, pH = 8, 673 K/H2) catalysts gave a high yield of 1,4-PeD, c.a. 82% and 81%, respectively (entries 2 and 3), which are comparable to Ni3Sn2 (2-Me-EtOH, pH = 8; 673 K/H2 (entry 1)). In contrast, the Ni3Sn2 catalyst synthesised in the absence of EG/G or Me-EtOH produced only 14% 1,4-PeD, which is lower than the yield of 1,2-and 1,5-PeD (28%) (entry 4). So that, we then concluded both the pH of the Ni–Sn solution and the presence of polyol additive are important to obtain the selective bulk Ni3Sn2 alloy catalyst for 1,4-PeD synthesis from FFald. Next, we evaluated the effect of hydrothermal temperature (423 K, 473 K, and 523 K for 24 h; the XRD patterns are shown in Fig. S4, in the ESI†) on the 1,4-PeD from FFald hydration–hydrogenation reactions. BuIk Ni3Sn2 synthesised at 423 K exhibited the highest yield of 1,4-PeD, as mentioned above. In contrast, bulk Ni3Sn2 obtained at hydrothermal temperatures of 473 K and 523 K catalysts were not selective towards 1,4-PeD (entries 5 and 6). Additionally, the bulk Ni3Sn2 pH = 8 catalyst was found to be stable even after exposure to the air for 2 months. Obviously, a high yield of 1,4-PeD (79%) was obtained at 100% FFald conversion (entry 7). Therefore, it can be concluded that the optimised hydrothermal temperature was 423 K for 24 h and easy to handle during the preparation as well as the catalytic reaction.
Table 1 Results of the selective synthesis of 1,4-PeD from FFald using bulk Ni3Sn2 pH = 8 alloy catalysts synthesised at different conditionsa
| Entry |
Catalystb |
Conv.c (%) |
Yieldc (%) |
| 1,4-PeD |
1,2-PeD |
1,5-PeD |
FFalc |
THFalc |
2H2MeTHF |
2-MeTHF |
Othersd |
| Reaction conditions: catalyst, 44 mg; substrate, 1.2 mmol; solvent, ethanol/H2O, 3.5 ml (1.5:2.0 volume ratio); initial H2 pressure, 3.0 MPa, 433 K, 12 h. The bulk Ni3Sn2 catalysts were synthesized at various conditions. Conversion of FFald and yield of the product were determined by GC and GC-MS analyses using an internal standard technique. Unknown product may be the condensation product of FFald or FFalc according to GC and GC-MS data. HT = hydrothermal at 473 K and 523 K. The catalyst was stored in a sample bin under exposure to the air for 2 months. Recyclability test after the fourth reaction run in the batch reactor system. The recovered catalyst was reactivated by H2 at 673 K for 1 h before reuse for the next run. |
| 1 |
Ni3Sn2 (2-Me-EtOH; pH = 8; 24 h; 673 K/H2) |
100 |
87 |
2 |
3 |
0 |
0 |
7 |
1 |
0 |
| 2 |
Ni3Sn2 (EG; pH = 8; 24 h; 673 K/H2) |
100 |
82 |
1 |
0 |
0 |
0 |
16 |
1 |
0 |
| 3 |
Ni3Sn2 (G; pH = 8; 24 h; 673 K/H2) |
100 |
81 |
3 |
2 |
0 |
0 |
11 |
3 |
0 |
| 4 |
Ni3Sn2 (without polyol; pH = 8; 673 K/H2) |
100 |
14 |
16 |
12 |
0 |
22 |
36 |
0 |
0 |
| 5e |
Ni3Sn2 (HT 473 K; pH = 8; 673 K/H2) |
100 |
15 |
0 |
1 |
0 |
2 |
35 |
12 |
36 |
| 6e |
Ni3Sn2 (HT 523 K; pH = 8; 673 K/H2) |
100 |
3 |
5 |
3 |
0 |
7 |
22 |
0 |
61 |
| 7f |
Ni3Sn2 (2-Me-EtOH; pH = 8; 24 h; 673 K/H2) |
100 |
79 |
1 |
2 |
0 |
0 |
11 |
0 |
10 |
| 8g |
Ni3Sn2 (2-Me-EtOH; pH = 8; 24 h; 673 K/H2) |
100 |
83 |
0 |
5 |
0 |
0 |
12 |
0 |
0 |
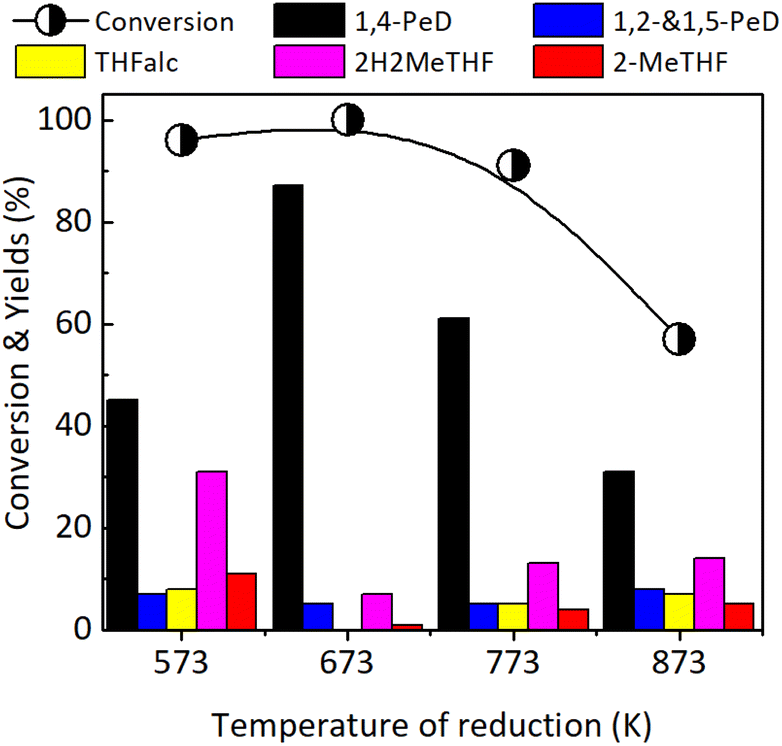
Effect of reduction temperature. The effect of reduction temperature on the FFald conversion and 1,4-PeD yield was examined over the most effective Ni3Sn2 catalyst obtained at pH of 8, with the presence of 2-Me-EtOH, at 423 K for 24 h and the results are shown in Fig. 5. The reduction with H2 was conducted at temperatures of 573–873 K for 1.5 h. Bulk Ni3Sn2 catalyst activated at 573 K converted 96% of FFald and produced 1,4-PeD (45%), 1,2-and 1,5-PeD mixture (7%), THFalc (8%), 2H2MeTHF (31%) and 2-MeTHF (11%). The highest yield of 1,4-PeD (87%) was obtained over the Ni3Sn2 catalyst activated at 673 K for 3 h. When the temperature reduction was furtherly increased to 773 K and 873 K, the conversion of FFald and the yield of 1,4-PeD decreased to 57–91% and 31–61%, respectively. It was found that the conversion of FFald increased as the reduction temperature was increased from 573 K (96%) to 673 K (100%), then gradually decreased as the reduction temperature was increased to 773 K (91%) and 873 K (57% conversion). This volcano-type activity relationship verified that less activity and selectivity were mainly due to the massive aggregation of Ni or Ni–Sn particles at high temperatures. The XRD analysis results confirmed that the diffraction peaks of the Ni3Sn2 alloy phases at 2θ = 30.6, 43.3, and 44.3° corresponding to Ni3Sn2(101), Ni3Sn2(102), and Ni3Sn2(110), respectively, intensified when the temperature reductions were increased to 673–873 K (Fig. S5, in the ESI†). The average crystallite sizes of Ni3Sn2(101) in bulk Ni3Sn2 673 K/H2; 773 K/H2; and 873 K/H2 were 17.0 nm; 26.5 nm; and 28.4 nm, respectively (Tables S1, S2 and S6, in the ESI†). In addition, this suggestion is consistent with the fact that the specific surface area BET (SBET) drastically decreased from 9.7–12.0 m2 g−1 (573 and 673 K) to 1.7–2.9 m2 g−1 after reduction at 773 K and 873 K, respectively.33
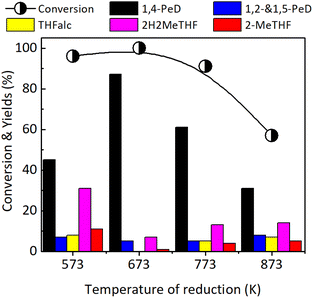 |
| | Fig. 5 Conversion of FFald and yields obtained over bulk Ni3Sn2 catalysts after reduction with H2 at 573–873 K for 1.5 h. Reaction conditions are referred to in Fig. 2. | |
Catalyst structure–activity relationship
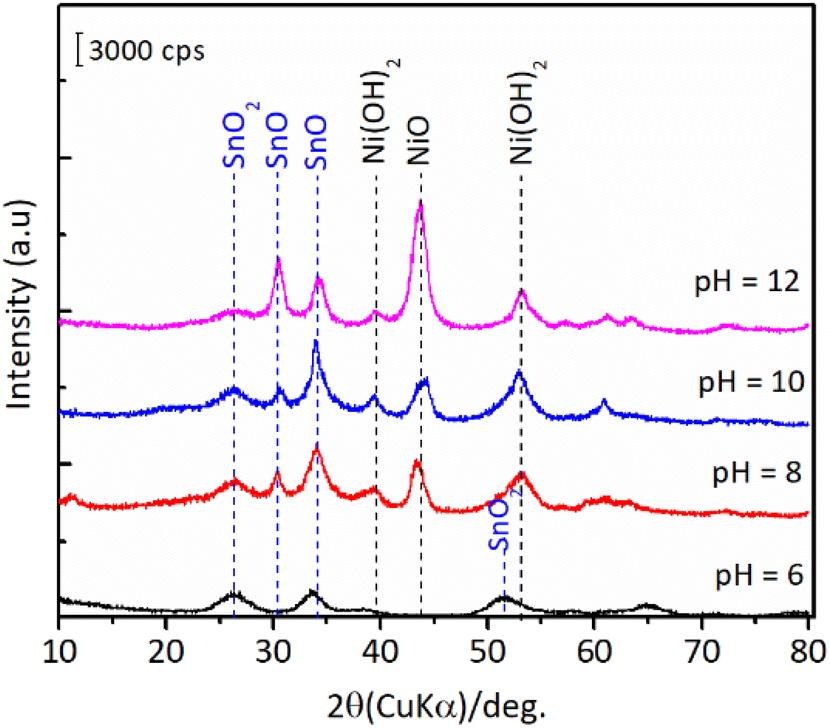
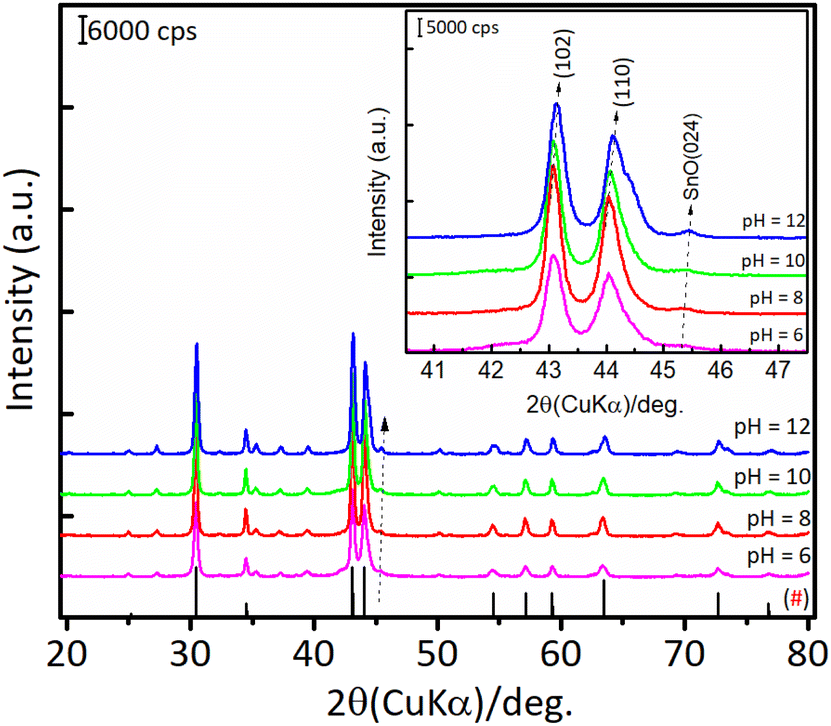
The results of hydration–hydrogenation of FFald to 1,4-PeD over various bulk Ni3Sn2 catalysts synthesised at different synthetic parameters confirmed that the pH of Ni–Sn solution, hydrothermal temperature, the temperature of reduction, and the presence of polyols additives were important for the fabrication of bulk Ni3Sn2 alloy catalysts.34 Fig. 6 shows the XRD patterns of the as-prepared (after hydrothermal processes) and the presence of NiO, Ni(OH)2, SnO, and SnO2 phases were clearly observed in all of the samples. The diffraction peak of SnO (2θ = 30.6°) and NiO (2θ = 43.7°) gradually intensified when the pH of the Ni–Sn solution was increased. On the other hand, the diffraction peak at 2θ = 26.4° that can be assigned to SnO2 was observed in samples of pH = 6–8. Both nickel and tin ions will exist in the forms of metal-oxides or hydroxides, which depend on the pH of the solution system (Pourbaix diagram).35 This might affect the formation of a well-crystallite Ni3Sn2 alloy during the reduction with H2. However, unfortunately, the as-prepared bulk Ni–Sn alloys are inactive for the hydration–hydrogenation of FFald under the current reaction conditions.
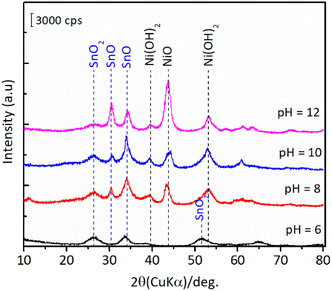 |
| | Fig. 6 XRD patterns of the as-prepared bulk Ni3Sn2 with different pH of nickel–tin solution (pH = 6–12) obtained after hydrothermal treatment at 423 K for 24 h. | |
Furthermore, the as-prepared Ni3Sn2 samples were reduced with H2 at 673 K for 1.5 h, and the XRD patterns of bulk Ni3Sn2 are shown in Fig. 7.
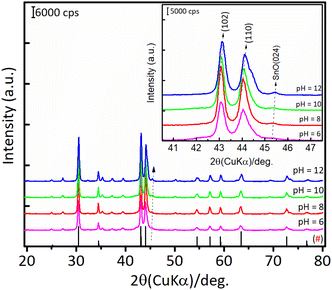 |
| | Fig. 7 XRD patterns of the bulk Ni3Sn2 after reduction with H2 at 673 K for 1.5 h obtained with different pH nickel–tin solution (pH = 6–12). (#) The plotted data were compared with JCPDS card of #06-414 (Ni3Sn2). | |
For Ni3Sn2 at a pH adjustment of 4 (Ni3Sn2 pH = 4), no diffraction peaks of the Ni3Sn2 alloy phase were observed, suggesting that the acidic environment inhibited the formation of metal hydroxides or metal oxides during the hydrothermal processes. A small diffraction peak at 2θ = 51.2° was observed, which can be attributed to the typical diffraction peak of metallic Ni(200)36 (Fig. S6, in the ESI†). This is caused by the main product THFalc obtained over the Ni3Sn2 pH = 4 catalyst as a result of total hydrogenation of CC and CO bonds of FFald. When the pH of Ni–Sn solution of 6 was applied, a series of diffraction peaks of the Ni3Sn2 alloy phase was clearly observed following the JCPDS-6-414 (Ni3Sn2 standard), and the diffraction peaks of the Ni3Sn2 alloy phase intensified as the pH adjustment was increased to 8–12. The increase in pH of Ni–Sn solution from 6 to 12 not only enhanced the crystallite sizes of Ni3Sn2(101) (at 2θ = 30.6°) but also caused a slight shifting in the distance of crystal unit (d) as indicated by the shifting of 2θ to the higher degree (inserted figure). By using a Bragg's law equation, it was found that the d (nm) values of bulk Ni3Sn2 alloy obtained at pH 6, 8, 10, and 12 were 0.2057 nm, 0.2057 nm, 0.2055 nm, and 0.2054 nm, respectively (Table S3, in ESI†). Moreover, a low-intensity peak at 2θ = 45.44° could be assigned to the remaining SnO (JCPDS #13-111). This is slightly intensified as the pH of the Ni–Sn solution was increased. This remaining SnO is consistent with the fact that the SnO in the as-prepared samples may aggregate and transform into another phase during the hydrogen treatment as indicated by the XRD patterns. The systematic studies of the intermetallic microstructure of Ni–Sn reported by Yang et al.37 and Marakatti et al.38 showed that the defect structure of Ni3Sn2 occurred during the preparation or non-stoichiometric alloy composition may strongly affect the geometrical and electronic structure of Ni3Sn2 alloy. Then, we took into account the unique and extraordinary catalytic behavior of Ni3Sn2 pH = 8 by examining their surface acidity using the ammonia-temperature programmed desorption (NH3-TPD), and the profiles are shown in Fig. 8.
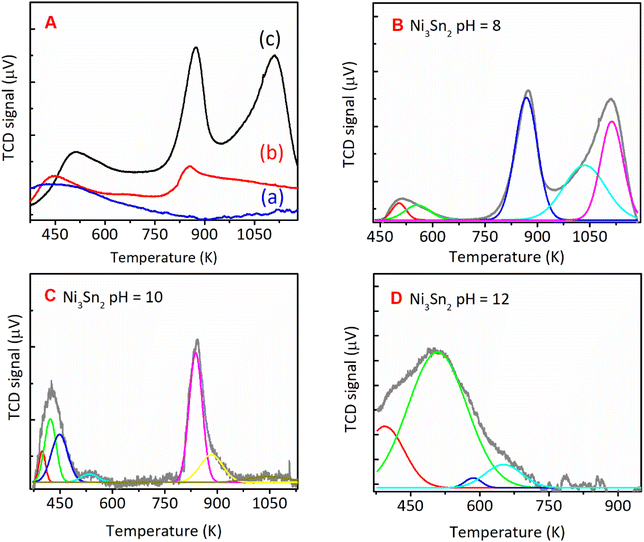 |
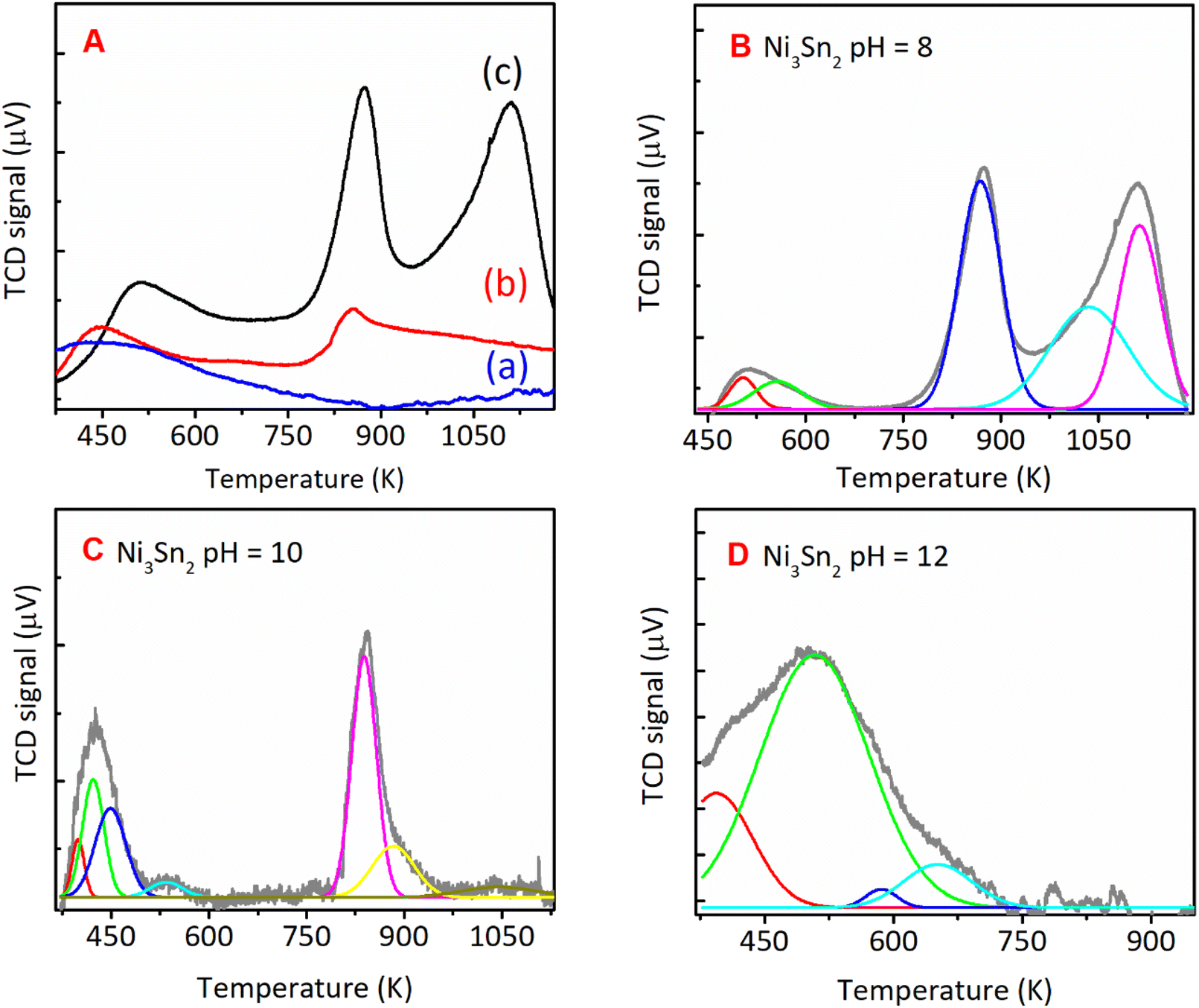
| | Fig. 8 NH3-TPD spectra of (A) the bulk Ni3Sn2 with different pH of nickel–tin solution and deconvoluted NH3-TPD spectra of bulk Ni3Sn2 synthesised with Ni–Sn solution pH of (B) 8, (C) 10, and (D) 12, hydrothermal synthesis at 423 K for 24 h and followed by reduction with H2 at 673 K for 1.5 h. | |
The NH3-TPD profiles were formally divided into three desorption temperature regions to denote three types of acid sites:39–42 (1) weak acid sites, ranging from 373 to 573 K, (2) moderate acid sites, ranging from 573 to 823 K, and (3) strong acid sites, ranging from 823 to 1023 (Table 2). The total acid sites decreased as the pH adjustment was increased from 8, 10, and 12 (Fig. 8). Bulk Ni3Sn2 pH = 8 catalyst has total acid sites of 276 μmol g−1, while Ni3Sn2 pH = 6, Ni3Sn2 pH = 10, and Ni3Sn2 pH = 12 catalysts had the total acid sites of 133 μmol g−1, 129 μmol g−1, and 18 μmol g−1, respectively (Fig. 8 and Table 2). The portion of strong acid sites, as well as the total acid sites, decreased after the pH adjustment was increased from 6 to 8, 10, and 12. Bulk Ni3Sn2 pH = 8 catalyst had 87 μmol g−1 portion of the strong acid sites and produced the highest yield of 1,4-PeD (87%) from FFald (Fig. 2). Ni3Sn2 pH = 10 (63 μmol g−1) and Ni3Sn2 pH = 6 (34 μmol g−1) catalysts produced 72% and 12% 1,4-PeD, respectively (Fig. 8). In contrast, bulk Ni3Sn2 pH = 12 (14 μmol g−1) catalyst did not produce 1,4-PeD, whereas the main product was FFalc without the formation of hydrogenolysed products (e.g., 1,2-or 1,5-PeD) (Fig. 2). These results suggest that the presence of high portion strong acid sites in the bulk Ni3Sn2 pH samples may affect its activity and selectivity during the hydration–hydrogenation of FFald to 1,4-PeD. In the literature, it has been reported that the oxidic tin (in the form of Sn2+, Sn(OH), SnOx) could be generated during the reaction in the aqueous phase and serve as Brønsted acid.30,43–45 The generated Ni0–SnOx species in H2O or H2O/ethanol will synergistically act to hydrogenate the CO of FFald to FFalc on specific Ni0 and the formed FFalc will be protonated and then subsequently dehydrated by the oxidic tin in H2O or H2O/ethanol (in the form of SnOx) to 2-MeF.46,47
Table 2 Physico-chemical properties of the bulk Ni3Sn2 alloy catalysts with different pH adjustments after reduction with H2 at 673 K for 1.5 h
| Entry |
Catalysta |
SBETb (m2 g−1) |
H2 uptakec (μmol g−1) |
Dd (nm) |
Amount of acid sitese (μmol g−1) |
| 373–623 K (w) |
623–823 K (m) |
823–1023 K (s) |
Total |
| The value in the parenthesis is Ni/Sn molar ratio. The composition was determined by using ICP-AES. Determined by N2 adsorption at 77 K. Based on total H2 uptake at 273 K (noted after corrections for physical and chemical adsorption). Average crystallite sizes were calculated by using Scherer's equation at 2θ = 30.37° of Ni3Sn2(101) alloy phase. The amount of acid sites was derived from NH3-TPD spectra and the weak (w), moderate (m), and strong (s) acid sites are referred to the temperature regions of NH3 desorption according to the ref. 42 and 48. |
| 1 |
Ni3Sn2 (2-Me-EtOH; pH = 6; 673 K/H2) |
72.4 |
10 |
10 |
99 |
0 |
34 |
133 |
| 2 |
Ni3Sn2 (2-Me-EtOH; pH = 8; 673 K/H2) |
101.5 |
9.8 |
14 |
90 |
99 |
87 |
276 |
| 3 |
Ni3Sn2 (2-Me-EtOH; pH = 10; 673 K/H2) |
18.6 |
9.3 |
15 |
33 |
33 |
63 |
129 |
| 4 |
Ni3Sn2 (2-Me-EtOH; pH = 12; 673 K/H2) |
12.0 |
8.6 |
17 |
18 |
0.0 |
0.0 |
18 |
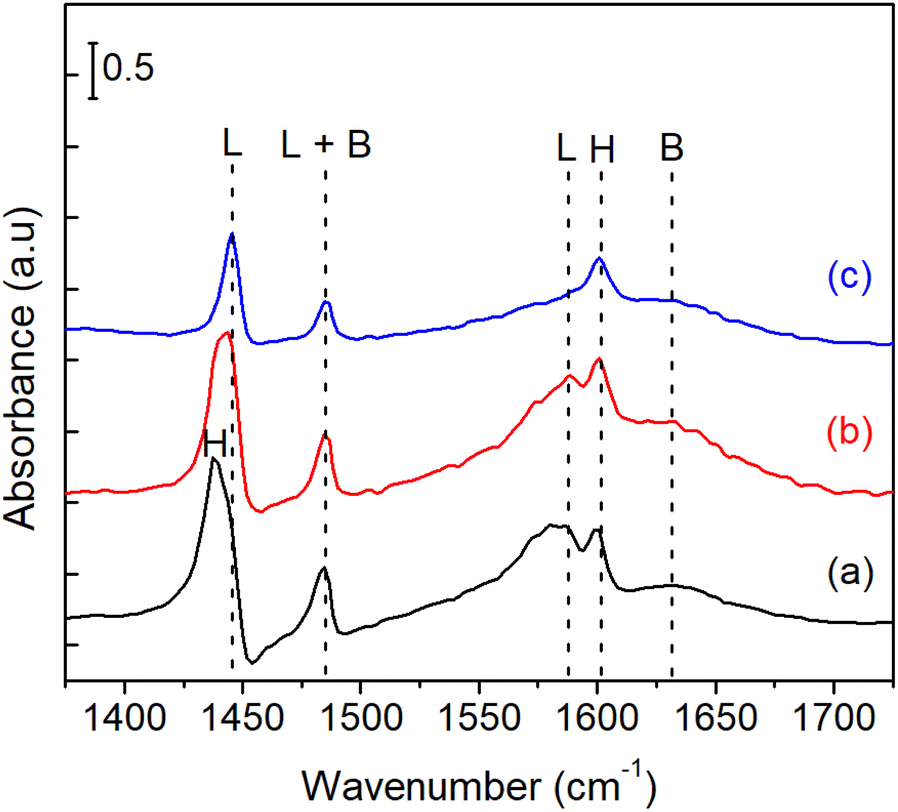
Fig. 9 shows the IR spectra of pyridine adsorbed on bulk Ni3Sn pH = 8 catalyst. The spectra of the adsorbed species were obtained after the introduction of 1–2 mL pyridine at room temperature, followed by purging with N2 flow at 323 K and 423 K until the spectra were stable. According to the literature on pyridine adsorption on Sn-containing catalysts,49,50 the bands are assigned in the following way. The pyridinium ion (PyH+) produced by the reaction of pyridine with Brønsted acid sites (B) shows bands around 1633 cm−1 (v8a). Coordinatively bound pyridine on Lewis acid sites (L) shows bands around 1445 (v19b) and 1575 cm−1. Physisorbed or hydrogen-bonded pyridine (H) shows bands around 1437 and 1599 cm−1. The band around 1490 cm−1 is common to vibrations due to PyH+ (B) and coordinatively bound pyridine (L).51 These results indicate that acid sites of bulk Ni3Sn pH = 8 are both Lewis and Brønsted acidic under the conditions shown in Fig. 8. Additionally, The maximum specific surface area BET (SBET) of the bulk Ni3Sn2 alloy was achieved at pH adjustment of 8 (101.5 m2 g−1), followed by pH adjustment of 6 (72.4 m2 g−1), then drastically decreased to 18.6 m2 g−1 and 12.0 m2 g−1 at the pH adjustment of 10 and 12, respectively (entries 1–4). The average crystallite sizes of Ni3Sn2(101) increased smoothly, while the H2 uptake slightly decreased as the pH adjustment was increased. The total H2 uptake decreased with the increase of pH from 6 to 12 (from 10 mmol g−1 to 9.8 mmol g−1, 9.3 mmol g−1, and 8.6 mmol g−1) and is in good accordance with the increase in the average crystallite sizes of Ni3Sn2(101) alloys (from 10 nm to 14 nm, 15 nm, and 17 nm), respectively (entries 1–4).
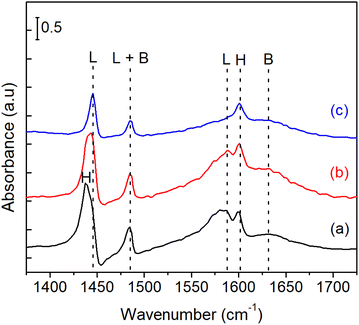 |
| | Fig. 9 Pyridine-FTIR profiles of bulk Ni3Sn pH = 8 after (a) adsorption at room temperature, and degassing at (b) 323 K, and (c) 423 K. L = Lewis acid site; B = Brønsted acid sites; H = physisorption of pyridine. | |
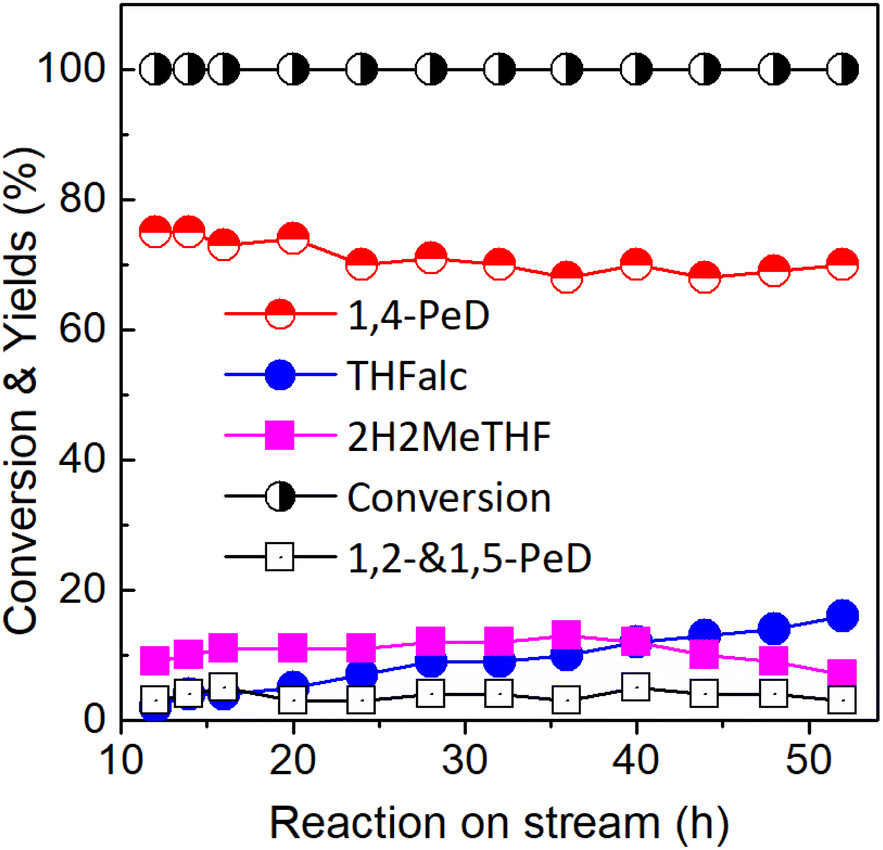
Recyclability and stability tests. The recyclability test of the Ni3Sn2 pH = 8 catalyst was performed in the batch reactor system (TAIATSU Techno Japan). The used bulk Ni alloy was easily separated by either simple centrifugation or filtration after the reaction. The recovered catalyst was reactivated by H2 at 673 K for 1.5 h prior to use in the next reaction run. The yield of 1,4-PeD on the reused catalyst decreased slightly after the second reaction run, but the activity was maintained for at least four consecutive runs (Table 1, entry 8). On the other hand, the stability test was evaluated at a long-term stream under the reaction conditions of flow rate 0.065 mL min−1, H2 flow rate 70 mL min−1, and 3.29 wt% FFald in H2O/ethanol solution, and the results are shown in Fig. 10. The activity of bulk Ni3Sn2 pH = 8 was maintained at ∼100% conversion of FFald with 66% yield of 1,4-PeD even after 52 h reaction on stream. The main side products were THFalc and 2H2MeTHF; THFalc is the result of over hydrogenation of FFald on metallic Ni0, while 2H2MeTHF is a stable intermediate of hydrolysis furan ring of FFalc or 2-MeF on SnOx species. The amounts of metals that leached into the reaction solution were analysed by ICP-AES and were found to be 1.5 mol% (Ni) and 5.0 mol% (Sn) after the fourth run. These results suggested that partial metallic Ni or Sn may leach out into the reaction solution during the reusability test, which can be expected to increase with the number of reaction runs. Moreover, the XRD patterns of the recovered Ni3Sn2 pH = 8 alloy before and after H2 treatment appeared to indicate that no change in the alloy structure occurred (Fig. S6, ESI†).26 Dumesic52 and Linic53 have shown that carbon tolerance of Ni-based catalysts could be improved by synthesising Ni-containing surface alloys with the second electropositive metal of tin (Sn). XRD and Mössbauer spectroscopy studies suggested that Sn migrates into the Ni particles to form intermetallic Ni–Sn.54
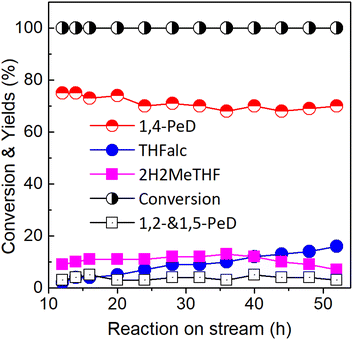 |
| | Fig. 10 Conversion of FFald and Yield of 1,4-PeD as a function of reaction on stream over bulk Ni3Sn2 (G; pH = 8; 673 K/H2) catalyst. Reaction conditions: catalyst, 67 mg; 433 K; flow rate 0.065 mL min−1, H2 flow rate 70 mL min−1 and 3.29 wt% FFald in H2O/ethanol solution. | |
Conclusions
The fabrication of bulk Ni3Sn2 alloy catalysts was examined under different conditions (e.g., pH adjustment of the nickel–tin solution, hydrothermal time, hydrothermal temperature, polyol additives, and reduction temperature). Bulk Ni3Sn2 that was synthesised at pH = 8, hydrothermal time of 24 h, and reduction temperature of 673 K for 1.5 h (Ni3Sn2 pH = 8) catalyst allowed a high yield of 1,4-PeD (87%) in a batch reaction system at 433 K, 3.0 MPa of H2, 12 h without the presence of acidic co-catalyst. A 76% yield of 1,4-PeD was also obtained when the reaction was carried out in a fixed-bed reaction system at 433 K, flow rate 0.065 mL min−1, H2 flow rate 70 mL min−1, and 3.29 wt% FFald in H2O/ethanol solution for 12 h. The activity of bulk Ni3Sn2 was maintained with a 66% yield of 1,4-PeD even after a 52 h reaction on stream. The high yield of 1,4-PeD over this catalyst can be attributed to high specific surface area BET (SBET = 101.5 m2 g−1), H2 uptake, and the portion of strong acid sites (87 mmol g−1). The presence of both hydration active sites (Brønsted acid sites (Ni–SnOx) in Ni3Sn2 alloy) and hydrogenation active sites (Ni0 or Ni–Sn alloy) could be controlled by changing the pH of the Ni–Sn solution during the preparation. Both the active sites acted synergistically to catalyze the hydration–hydrogenation reactions of FFald to produce a high yield of 1,4-PeD in the batch reaction system. The results of hydration–hydrogenation of FFald to 1,4-PeD over various bulk Ni3Sn2 catalysts synthesized at different synthetic parameters confirmed that the pH of Ni–Sn solution, hydrothermal temperature, the temperature of reduction, and the addition of polyols were the important parameters for the fabrication of bulk Ni3Sn2 alloy catalysts. The developed synthetic procedures of Ni3Sn2 alloy catalysts are precisely controlled to ensure the production of promising heterogeneous Ni–Sn alloy-based catalysts for the catalytic conversion of biomass-derived-furanic compounds (e.g., FFald, furfuryl alcohol (FFalc), and 2-methylfuran (2-MeF)).
Experimental
Material and chemicals
Nickel(II) chloride hexahydrate (NiCl2·6H2O), tin(II) chloride dihydrate (SnCl2·2H2O), active carbon (SBET = 815 m2 g−1), MgO, and commercial aluminum hydroxide (c-AlOH) were purchased from WAKO Pure Chemical Industries, Ltd. and used as received unless otherwise stated. g-Al2O3 (SBET = 100 m2 g−1) was purchased from Japan Aerosil Co. Furfural (98%), furfuryl alcohol (97%), 2-methylfuran (98%), and tetrahydrofurfuryl alcohol (99%) were purchased from Tokyo Chemical Industry, Ltd. All organic chemical compounds were purified using standard procedures prior to use.55
Catalysis synthesis
Synthesis of bulk Ni3Sn2. A typical procedure for the synthesis of the bulk Ni3Sn2 alloy catalyst is described as follows:27 NiCl2·6H2O (7.2 mmol) was dissolved in deionised water (denoted as solution A), and SnCl2·2H2O (4.8 mmol) was dissolved in ethanol/2-methoxy ethanol (2:1) (denoted as solution B) at room temperature. Solutions A and B were mixed at room temperature; the temperature was subsequently raised to 323 K and the mixture was stirred for 12 h. The pH of the mixture was adjusted to 12 by the dropwise addition of an aqueous solution of NaOH (3.1 M). The mixture was then placed in a sealed Teflon autoclave for the hydrothermal reaction at 423 K for 24 h. The resulting black precipitate was filtered, washed with distilled water, and then dried under a vacuum overnight. Prior to the catalytic reaction, the obtained black powder was reduced with H2 at 673 K for 1.5 h.
Catalyst characterization
XRD measurements were recorded on a Mac Science M18XHF instrument using monochromatic Cu Kα radiation (λ = 0.15418 nm). The XRD equipment operated at 40 kV and 200 mA with a step width of 0.02° and a scan speed of 4° min−1 (α1 = 0.154057 nm, α2 = 0.154433 nm). The mean crystallite size of Ni–Sn was calculated from the full width at half maximum (FWHM) of the Ni3Sn2(101) alloy diffraction peak at 2θ = 30.4° according to the Scherrer equation.
Inductively coupled plasma (ICP) measurements were performed on an SPS 1800H plasma spectrometer of Seiko Instruments Inc. (Ni: 221.7162 nm and Sn: 189.898 nm). The Brunauer–Emmet–Teller surface area (SBET) and pore volume (Vp) were measured using N2 physisorption at 77 K on a Belsorp Max (BEL Japan). The samples were degassed at 473 K for 2 h to remove physisorbed gases prior to the measurement. The amount of nitrogen adsorbed onto the samples was used to calculate the BET surface area via the BET equation. The pore volume was estimated to be the liquid volume of nitrogen at a relative pressure of 0.995 according to the Barrett–Joyner–Halenda (BJH) approach based on desorption data.
The H2 uptake was determined through irreversible H2 chemisorption. After the catalyst was heated at 393 K under vacuum for 30 min, it was then heated at 673 K under H2 for 30 min. The catalysts were subsequently cooled to room temperature under vacuum for 30 min. The H2 measurement was performed at 273 K, and the H2 uptake was calculated according to a method described in the literature.56
The ammonia-temperature programmed desorption (NH3-TPD) was carried out on a Belsorp Max (BEL Japan). The samples were degassed at an elevated temperature of 373–473 K for 2 h to remove physisorbed gases prior to the measurement. The temperature was then maintained at 473 K for 2 h while flushing with He gas. NH3 gas (balanced NH3, 80% and He, 20%) was introduced at 373 K for 30 min, then treated with helium gas to remove the physisorbed also for 30 min. Finally, temperature-programmed desorption was carried out at a temperature of 273–1073 K and the desorbed NH3 was monitored by TCD.
Catalytic reaction
A typical reaction for the hydration–hydrogenation of FFald was carried out in the following manner.32 The catalyst (44 mg), FFald (1.2 mmol) in ethanol/H2O 1.5/2.0 v/v (3.5 mL) solvent mixture, dodecane (0.25 mmol) as an internal standard, were placed in an autoclave reactor system of Taiatsu Techno (a Pyrex tube was fitted inside of a sus316 jacket to protect the vessel from corrosion in acidic media). After H2 was introduced into the reactor (initial pressure of H2 was 3.0 MPa) at room temperature, the temperature of the reactor was raised to 433 K for 12 h. After 12 h, the conversion of FFald and the yields of 1,4-PeD, FFalc, THFalc, and 2H2MeTHF, 2-MeTHF, 1,2-PeD, and 1,5-PeD were determined by GC analysis using an internal standard technique. The used bulk Ni3Sn2 catalyst was easily separated using either simple centrifugation or filtration, then finally dried overnight under vacuum at room temperature. Prior to the reusability testing, the recovered Ni3Sn2 alloy catalyst was re-reduced with H2 at 673 K for 1 h.
Product analysis
Gas chromatography (GC) analysis of the reactant (FFald) and products (FFalc, THFalc, 1,4-PeD, 1,2-PeD, 1.5-PeD, and 2-MeTHF) was performed on a PerkinElmer Autosystem XL equipped with a flame ionization detector and an Rtx®-BAC PLUS 1 (i.d. 0.32 mm, length of 30 m, and d.f. 1.8 mm) capillary column of RESTEK, US. Gas chromatography-mass spectrometry (GC-MS) was performed on a Shimadzu GC-17B equipped with a thermal conductivity detector and an RT-βDEXsm capillary column. 1H and 13C NMR spectra were obtained on a JNM-AL400 spectrometer at 400 MHz; the samples for NMR analysis were dissolved in chloroform-d1 with TMS as the internal standard. The products were confirmed by a comparison of their GC retention time, mass, 1H, and 13C NMR spectra with those of authentic samples.
The conversion and yield of products were calculated according to the following equations:
| |
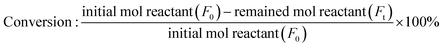 | (1) |
| |
 | (2) |
where
F0 is the introduced mol reactant (FFald),
Ft is the remaining mol reactant, and Δ
F is the consumed mol reactant (introduced mol reactant − remained mol reactant), which are all obtained from GC analysis using an internal standard technique.
Author contributions
Rodiansono: conceptualization, methodology, investigation, writing-original draft, writing-review and editing. Atina Sabila Azzahra, Pathur Razi Ansyah, Sadang Husain: formal analysis, investigation. Shogo Shimadzu: conceptualization and writing-review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by Hibah Penelitian Dasar Kompetitif Nasional (PDKN) FY 2021-2023 under grant number 026/E5/PG.02.00.PT/2023 from the Ministry of Education, Culture, Research and Technology, Indonesian Government.
Notes and references
- J. Zhu and G. Yin, ACS Catal., 2021, 11, 10058–10083 CrossRef CAS.
- A. Gandini, D. Coelho, M. Gomes, B. Reis and A. Silvestre, J. Mater. Chem., 2009, 19, 8656–8664 RSC.
- B. M. Stadler, A. Brandt, A. Kux, H. Beck and J. G. de Vries, ChemSusChem, 2020, 13, 556–563 CrossRef CAS PubMed.
- D. Sun, S. Sato, W. Ueda, A. Primo, H. Garcia and A. Corma, Green Chem., 2016, 18, 2579–2597 RSC.
- X. Li, P. Jia and T. Wang, ACS Catal., 2016, 6, 7621–7640 CrossRef CAS.
- K. Huang, Z. J. Brentzel, K. J. Barnett, J. A. Dumesic, G. W. Huber and C. T. Maravelias, ACS Sustainable Chem. Eng., 2017, 5, 4699–4706 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- S. Koso, I. Furikado, A. Shimao, T. Miyazawa, K. Kunimori and K. Tomishige, Chem. Commun., 2009, 2035–2037 RSC.
- S. Koso, N. Ueda, Y. Shinmi, K. Okumura, T. Kizuka and K. Tomishige, J. Catal., 2009, 267, 89–92 CrossRef CAS.
- Z. Wang, B. Pholjaroen, M. Li, W. Dong, N. Li, A. Wang, X. Wang, Y. Cong and T. Zhang, J. Energy Chem., 2014, 23, 427–434 CrossRef.
- H. W. Wijaya, T. Kojima, T. Hara, N. Ichikuni and S. Shimazu, ChemCatChem, 2017, 9, 2869–2874 CrossRef CAS.
- H. W. Wijaya, T. Hara, N. Ichikuni and S. Shimazu, Chem. Lett., 2018, 47, 103–106 CrossRef CAS.
- E. Soghrati, C. Kok Poh, Y. Du, F. Gao, S. Kawi and A. Borgna, ChemCatChem, 2018, 10, 4652–4664 CrossRef CAS.
- H. W. Wijaya, T. Sato, H. Tange, T. Hara, N. Ichikuni and S. Shimazu, Chem. Lett., 2017, 46, 744–746 CrossRef CAS.
- W. Xu, H. Wang, X. Liu, J. Ren, Y. Wang and G. Lu, Chem. Commun., 2011, 47, 3924–3926 RSC.
- Z. J. Brentzel, K. J. Barnett, K. Huang, C. T. Maravelias, J. A. Dumesic and G. W. Huber, ChemSusChem, 2017, 10, 1351–1355 CrossRef CAS PubMed.
- K. J. Barnett, D. J. McClelland and G. W. Huber, ACS Sustainable Chem. Eng., 2017, 5, 10223–10230 CrossRef CAS.
- H. Mehdi, V. Fábos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horváth, Top. Catal., 2008, 48, 49–54 CrossRef CAS.
- G. J. Leuck, Evanston, J. Pokorny and F. N. Peters Jr, US Pat, 2097493, 1937 Search PubMed.
- L. E. Schniepp, H. H. Geller and R. W. von Korff, J. Am. Chem. Soc., 1947, 69, 672–674 CrossRef CAS.
- S. Liu, Y. Amada, M. Tamura, Y. Nakagawa and K. Tomishige, Green Chem., 2014, 16, 617 RSC.
- S. Liu, Y. Amada, M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2014, 4, 2535–2549 RSC.
- F. Liu, Q. Liu, J. Xu, L. Li, Y. T. Cui, R. Lang, L. Li, Y. Su, S. Miao, H. Sun, B. Qiao, A. Wang, F. Jérôme and T. Zhang, Green Chem., 2018, 20, 1770–1776 RSC.
- Q. Liu, B. Qiao, F. Liu, L. Zhang, Y. Su, A. Wang and T. Zhang, Green Chem., 2020, 22, 3532–3538 RSC.
- K. Cui, W. Qian, Z. Shao, X. Zhao, H. Gong, X. Wei, J. Wang, M. Chen, X. Cao and Z. Hou, Catal. Lett., 2021, 151, 2513–2526 CrossRef CAS.
- R. Rodiansono, M. Dewi Astuti, T. Hara, N. Ichikuni and S. Shimazu, Green Chem., 2019, 21, 2307–2315 RSC.
- R. Rodiansono, S. Khairi, T. Hara, N. Ichikuni and S. Shimazu, Catal. Sci. Technol., 2012, 2, 2139–2145 RSC.
- R. Rodiansono, M. D. Astuti, A. Ghofur and K. C. Sembiring, Bull. Chem. React. Eng. Catal., 2015, 10, 192–200 CrossRef CAS.
- R. Rodiansono, M. D. Astuti, K. Mustikasari, S. Husain and S. Sutomo, Catal. Sci. Technol., 2020, 10, 7768–7778 RSC.
- L. Wang, J. Zhang, X. Wang, B. Zhang, W. Ji, X. Meng, J. Li, D. S. Su, X. Bao and F. S. Xiao, J. Mater. Chem. A, 2014, 2, 3725–3729 RSC.
- M. K. Stones, E. M. J. Banz Chung, I. T. Da Cunha, R. J. Sullivan, P. Soltanipanah, M. Magee, G. J. Umphrey, C. M. Moore, A. D. Sutton and M. Schlaf, ACS Catal., 2020, 10, 2667–2683 CrossRef CAS.
- R. Rodiansono, M. D. Astuti, K. Mustikasari, S. Husain, F. R. Ansyah, T. Hara and S. Shimazu, RSC Adv., 2022, 12, 241–250 RSC.
- R. Rodiansono, A. S. Azzahra, S. Husain and P. R. Ansyah, Bull. Chem. React. Eng. Catal., 2022, 17, 743–754 CrossRef CAS.
- R. E. Cable and R. E. Schaak, Chem. Mater., 2005, 17, 6835–6841 CrossRef CAS.
- B. Beverskog and I. Puigdomenech, Corros. Sci., 1997, 39, 969–980 CrossRef CAS.
- JCPDS-ICDD, Powder diffraction files, JCPDS-International center for diffraction data (JCPDS-ICDD), 1991.
- Y. Yang, L. Chen, Y. Chen, W. Liu, H. Feng, B. Wang, X. Zhang and M. Wei, Green Chem., 2019, 21, 5352–5362 RSC.
- V. S. Marakatti, N. Arora, S. Rai, S. C. Sarma and S. C. Peter, ACS Sustainable Chem. Eng., 2018, 6, 7325–7338 CrossRef CAS.
- E. Dumitriu and V. Hulea, J. Catal., 2003, 218, 249–257 CrossRef CAS.
- J. Wang, P. A. Chernavskii, Y. Wang and A. Y. Khodakov, Fuel, 2013, 103, 1111–1122 CrossRef.
- F. Arena, R. Dario and A. Parmaliana, Appl. Catal., A, 1998, 170, 127–137 CrossRef CAS.
- N. Khandan, M. Kazemeini and M. Aghaziarati, Appl. Catal., A, 2008, 349, 6–12 CrossRef CAS.
- L. Ma, H. Wang, C. Zhu, Q. Liu, J. Tan, C. Wang and Z. Liang, ChemSusChem, 2019, 12, 2154–2160 CrossRef.
- X. Liu, X. Liu, G. Xu, Y. Zhang, C. Wang, Q. Lu and L. Ma, Green Chem., 2019, 21, 5647–5656 RSC.
- S. Xu, D. Yu, T. Ye and P. Tian, RSC Adv., 2017, 7, 1026–1031 RSC.
- N. S. Date, A. M. Hengne, K. W. Huang, R. C. Chikate and C. v. Rode, Green Chem., 2018, 20, 2027–2037 RSC.
- S. Iqbal, X. Liu, O. F. Aldosari, P. J. Miedziak, J. K. Edwards, G. L. Brett, A. Akram, G. M. King, T. E. Davies, D. J. Morgan, D. K. Knight and G. J. Hutchings, Catal. Sci. Technol., 2014, 4, 2280–2286 RSC.
- E. Dumitriu and V. Hulea, J. Catal., 2003, 218, 249–257 CrossRef CAS.
- E. Tututi-Ríos, H. González, D. A. Cabrera-Munguia, A. Gutiérrez-Alejandre and J. L. Rico, Catal. Today, 2022, 394–396, 235–246 CrossRef.
- A. Rezayan, K. Wang, R. Nie, T. Lu, J. Wang, Y. Zhang and C. Charles Xu, Chem. Eng. J., 2022, 429, 132261 CrossRef CAS.
- M. Tamura, K. I. Shimizu and A. Satsuma, Appl. Catal., A, 2012, 433–434, 135–145 CrossRef CAS.
- J. W. Shabaker, G. W. Huber and J. A. Dumesic, J. Catal., 2004, 222, 180–191 CrossRef CAS.
- E. Nikolla, J. Schwank and S. Linic, J. Catal., 2007, 250, 85–93 CrossRef CAS.
- T. B. L. W. Marinelli and V. Ponec, J. Catal., 1995, 156, 51–59 CrossRef CAS.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Elsevier/BH, 6th edn, 2009 Search PubMed.
- C. H. Bartholomew and R. B. Pannell, J. Catal., 1980, 65, 390–401 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abf,
Atina Sabila Azzahrab,
Pathur Razi Ansyahc,
Sadang Husain
*abf,
Atina Sabila Azzahrab,
Pathur Razi Ansyahc,
Sadang Husain